

Abstract
The oral cavity is a unique environment containing teeth juxtaposed with soft tissues, all of which are constantly bathed in microbial products and host-derived factors. While microbial dysbiosis in the oral cavity clearly leads to oral inflammatory disease, recent advances find that endogenous danger-associated molecular patterns (DAMPs) released from oral and salivary tissue also contribute to the progression of inflammatory and autoimmune disease, respectively. In contrast, DAMPs produced during oral fungal infection actually promote the resolution of infection. Here we present a review of the literature suggesting a role for signaling by DAMPs, which may intersect with pathogen-associated molecular pattern (PAMP) signaling, in diseases that manifest in the oral cavity, specifically periodontal disease, oropharyngeal candidiasis and Sjögren’s Syndrome.
Keywords: Periodontal disease, Sjögren’s syndrome, candidiasis, DAMP, PAMP
Summary sentence:
Review of how activation of the immune system through signaling of danger signals contributes to the progression or resolution of oral diseases.
GRAPHICAL ABSTRACT
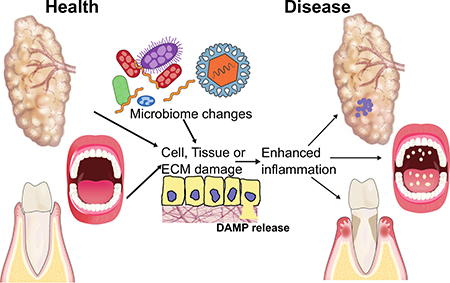
Introduction
The innate immune system is fundamental to host survival and overall health, being the first line of defense against pathogens as well as fundamental in maintaining tissue homeostasis. This is especially true in the complex environment of the oral cavity, which is a unique anatomical structure characterized by the juxtaposition of soft and hard tissues constantly bathed in microbial products and host-derived factors. Many ecological niches are present in the oral cavity reflecting the variety of tissues present including teeth and multiple keratinized and non-keratinized epithelial surfaces. Each niche has varying microenvironments due to differing immune capacity and physiological conditions: one distinct feature of the oral cavity is the presence of hard non-shedding tooth enamel surfaces which are ideal to promote the growth and maintenance of complex microbial biofilms. The oral mucosa, comprised of epithelium and underlying connective tissue, generally functions as an effective mechanical barrier despite being constantly exposed to, and in direct interaction with, environmental stimuli. However, despite continuous turnover of superficial oral epithelial layers, these mucosal surfaces are colonized abundantly with organisms [1].
The mucosa immediately adjacent to the teeth, termed the gingival sulcus, normally forms a tight seal against the teeth that prevents microbial invasion. This periodontal tissue is highly vascular and thus the gingival sulcus is bathed in the serum-like exudate termed gingival crevicular fluid (GCF) which carries components such as complement, antibodies and immune cells that function to maintain homeostasis at the biofilm-gingival sulcus interface [1].
The mucosal tissues and tooth surfaces of the oral cavity are also constantly bathed in saliva, a watery substance produced by the salivary glands. Saliva functions to provide lubrication but also serves as source of nutrients and proteins for initial formation and succession of the oral biofilm community. Saliva is a key component of the oral defense system as it contains a plethora of innate components that aim to maintain bacterial symbiosis. The importance of saliva is illustrated in patients with salivary hypofunction, including the autoimmune disease Sjögren’s syndrome (SS). In this disease, the salivary glands display immune-mediated dysfunction and patients often lose salivary flow, leading to rampant dental decay [2]. Thus, saliva plays a key role in oral homeostasis and alterations in the amount produced or the composition has profound effects on the integrity of the oral tissues.
The oral cavity is under constant microbial pressure, with a diverse microbiome composed of bacteria, viruses, fungi and protozoa. The bacterial community in the mouth is highly complex, with over 1000 potential species present; it is second only to the gut in microbial diversity in the human body [3]. Likewise, a vast diversity in the fungal microbiome is observed [4]. The continual presence of microbes in the oral cavity, even in health, necessitates constant immune surveillance to maintain a characteristic homeostatic steady-state. A rich immunological cellular and cytokine network is present at the gingival tissue interface and supporting oral mucosa [5, 6] and a large number of neutrophils are recruited to the gingival crevice in health, which are significantly increased during inflammation. These neutrophils play a role in initial microbial surveillance as well as orchestrating the overall immune response to maintain oral health. Mononuclear populations in the gingiva are comprised of resident macrophages and dendritic cells with increased recruitment and activation of these cells along with monocytes in response to microbial dysbiosis during oral disease progression [7]. These cells are involved in directly defending the tissue barrier against bacterial insults as well as promoting immune regulation through cross-talk with adaptive immune cells and their antigen presentation functions. In health, gingival lymphocyte populations consist of minimal B-cells and a prominent resident T-cell infiltrate with increased proportions of various T- and B-cell subsets in disease, where some subsets, such as Th17 cells, may drive pathogenesis while others such as Treg, may mediate a protective response [6].
Like the gingival crevice, other oral mucosal tissues including the tongue, palate and buccal surface have similar immune constituents, however slight differences in cellular proportions and subsets may occur in different mucosal areas reflecting regional differences. Generally, the salivary glands are a sterile location with tightly regulated immune responses. In addition to IgA-producing plasma cells, macrophages, dendritic cells, innate lymphoid cells, and T-cells are present in salivary gland tissue to contribute to protection of oral tissues. In pathoses such as SS, increased infiltration and hyperactivity of these immune cell populations contribute to disease [8].
It is well known that the immune response involves complex signaling systems that 1) recognize pathogens and tolerate commensals while initiating defense responses to control infection and 2) properly recognize self versus non-self under homeostatic conditions. The first line of immune defense involves detection of conserved molecular motifs expressed by members of the microbiome referred to as pathogen-associated molecular patterns (PAMPs) or more generally microbial-associated molecular patterns (MAMPs). Conversely, danger-associated molecular patterns (DAMPs) are endogenous factors released upon cellular damage or tissue disruption [9]. These PAMPs and DAMPs are recognized by a variety of germ-line encoded membrane-bound and cytoplasmic sensors termed pattern-recognition receptors (PRRs). PRRs are expressed on tissue resident cells and infiltrating immune cells and include Toll-like receptors (TLRs), C-type-lectin receptors, nucleotide-binding and oligomerization domain (NOD1 and NOD2), NOD-like receptors (NLR), receptor for advanced glycation end-products (RAGE), retinoic acid-inducible gene-I (RIG-I) receptors and the inflammasome, among others [10, 11]. Signaling through PRRs induces downstream activation of signaling pathways to modulate generally pro-inflammatory responses, though polymorphisms can alter the signaling responses from PRRs. Indeed, recent studies have begun to associate links between polymorphisms in PRRs and the development of periodontal disease and SS [12–14].
It is well established that microbial products contribute to oral disease. However, we have also recently come to appreciate that DAMPs activate PRRs expressed by host cells, thereby also serving as potent inflammatory stimuli that can help promote a robust immune response in order to clear the pathogen, or promote excessive inflammation and exacerbate disease. Here we present a review of the literature suggesting a role for DAMPs in diseases that manifest in the oral cavity, specifically periodontal disease, oropharyngeal candidiasis and SS (Figure 1 and 2). Modulation of DAMPs themselves or pathways activated by DAMPs may serve as novel therapeutic strategies for these debilitating diseases.
Figure 1:

Immunity at oral mucosal surfaces. Mucosal surfaces such as the tongue and periodontium are sites of rich immune networks due to the constant presence of a diverse microbiome. Tongue epithelium is colonized with normally commensal organisms including the dimorphic fungi Candida albicans. If overgrowth persists in a susceptible host, hyphae can invade the epithelium and the host normally mounts an effective cellular immune response through release of DAMPs. In health, the periodontium tissue surrounding the tooth and associated gingival crevice is in homeostatic balance while dysbiosis of the bacterial community drives destructive cellular inflammation with activation of DAMP signaling. DAMP production through matrix degradation and cellular damage can help to enhance inflammation and promote characteristic tissue destruction of periodontal disease.
Figure 2:

DAMPs associated with SS-related inflammation in salivary tissue. DAMPs implicated in SS and the innate and adaptive immune cells that may be activated as a result of DAMP interactions are shown. SGECs = Salivary Gland Epithelial Cells.
Periodontal Disease
Periodontal disease is the most prevalent chronic inflammatory diseases, with nearly 50% of those over 30 years of age in the US being affected [15]. While periodontal disease is a multifactorial disease with development dependent on oral care, environmental triggers and host genetic makeup, it is well understood that overgrowth and transformation of the subgingival oral microbiome into a dysbiotic state is ultimately required to initiate and sustain chronic inflammation; this chronic inflammation in turn is the principal driver of tissue destruction and disease [16, 17]. The involvement of the oral microbial community leads to a largely PAMP-driven inflammatory response and disease development [18]. While largely extracellular [10], there is evidence of intracellular PAMP responses during the development of disease.
Levels of IL-1β, the prototypical cytokine released upon intracellular PAMP and DAMP recognition and inflammasome activation [19], are linked with periodontal disease progression [20, 21], as is the expression of the NLRP3 inflammasome [22, 23]. A number of periodontal pathogens, including Porphyromonas gingivalis, Fusobacterium nucleatum, Aggregatibacter actinomycetemcomitans, Treponema denticola and Tannerella forsythia are capable of eliciting an Il-1β response from immune cells [24–26] as well as from non-immune cells including gingival epithelial cells and fibroblasts [27, 28]. In addition to whole bacterial interactions, recent studies have found outer membrane vesicles (OMVs) from P. gingivalis, T. denticola and T. forsythia are capable of inducing the inflammasome and IL-1β release [29, 30]. Although the majority of studies indicate signaling through the intracellular NLRP3 inflammasome is required [31], the exact bacterial components required to fully activate complement and IL-1β release, or if DAMPs such as ATP are additionally required [27, 32], remains uncertain.
P. gingivalis, the most extensively studied oral pathogenic bacterium, has a large number of pathogenic factors contributing to its ability to disrupt both the oral microbiota and the immune system, to initiate dysbiosis and promote an environment within which it can flourish [16]. There have been conflicting reports suggesting P. gingivalis may have a unique ability to inhibit the activation of the inflammasome and release of IL-1β, even when in the presence of other inflammasome activating oral pathogens [33, 34]. Indeed, there is precedent for this pathogen interfering with IL-8 production by releasing into the cytoplasm of invaded epithelial cells a phosphatase to reduce NFκB RelA phosphorylation [35]. However, recent studies suggest that P. gingivalis activates the inflammasome, but that the presence of bacterial-produced proteases (gingipains) are responsible for destroying much of the cytokine produced [36, 37].
A number of oral pathogens, including P. gingivalis, are able to survive within both immune and non-immune cells [28, 38, 39]. While the exact mechanisms of survival are still unclear, an ability to survive within mammalian cells provides a potential means for transport of bacterial components into the cytoplasm leading to internal PAMP and DAMP signaling [40]. Further, though much focus has been on the role of oral microbes with a traditional pathogenic phenotype, recent evidence highlights a potential role for traditionally commensal oral microbes, including Streptococcus sp. and Enterococcus sp. in the development of disease [41]. While there is an essential role for the adherence of the traditional pathogens such as P. gingivalis to the commensal flora [42], there is emerging evidence for commensal-host immune cell interactions changing during inflammation, with increased survival within phagocytes potentially leading to increased PAMP signaling within the phagocyte cytoplasm [43].
Extensive tissue destruction is a hallmark of periodontal disease and therefore tissue-released DAMPs may contribute to disease (Figure 1). T. denticola, P. gingivalis, T. forsythia and F. nucleatum have been implicated in inducing the release of DAMPs, including ATP, HSP60, fibronectin and HMGB1 from mammalian cells [28, 44]. ATP can function as a secondary internal signal through the P2X7 receptor (P2X7R) to promote the activation of the inflammasome of cells when in the presence of oral pathogens [27]. Whether such signaling is required in vivo for periodontal disease development is currently unclear, however P. gingivalis can secrete a nucleoside diphosphate kinase-like enzyme that can remove ATP and inhibit such signaling [45]. In addition, recent studies have begun to indicate a role for the DAMPs serum amyloid A and HMGB1 in periodontal disease, as these are increased in tissue and GCF [46–48] and can activate TLR signaling in mouse models of periodontal disease [48, 49]. Systemic administration of anti-HMGB1 inhibited periodontal inflammation and bone loss in a bacterial induced murine model, highlighting a potential role in periodontal progression [50].
Finally, neutrophil extracellular traps (NETs) can be induced in the oral cavity and may be involved in maintaining oral health, as their impaired formation due to genetic mutations or bacterial DNAse production may exacerbate periodontal disease. Similarly, exaggerated NET production occurs in the periodontal pocket, obstructing removal of cellular debris and potentially PAMPs and DAMPs by preventing efficient flushing of the pocket by GCF flow leading to bystander tissue damage (reviewed in [51]). While DAMPS such as HGMB1 and histones released by injured liver hepatocytes can induce NET formation through TLR activation [52], and NETs with associated IL-1B are linked with active ulcerative colitis [53], to our knowledge the involvement of DAMPs in NET formation within the oral cavity is currently unknown.
Sjögren’s Syndrome (SS)
SS is an autoimmune disease that primarily affects women [2]. The disease is characterized by loss of saliva and tear production, although patients may also exhibit many serious extra-glandular disease manifestations such as B-cell lymphoma [2]. Like many other autoimmune diseases, the cause of SS is likely multifactorial. While PRRs and PRR-mediated signaling pathways are clearly dysregulated in SS, adult SS patients do not appear to be particularly susceptible to microbial infections [54] and so it is unlikely that the activating ligands in SS are derived from exogenous sources. Thus, it is probable that PRR activation is mediated by endogenous triggers, as is the case for other autoimmune diseases [55].
Evidence in both SS models and patients shows that DAMPs may be released through the pathologic degradation of exocrine tissue [56–58] (Figure 2). Indeed, extracts from SS salivary biopsy tissue showed elevated proteolysis of extra-cellular matrix molecules (ECM) proteins, such as fibronectin and laminin [57]. Moreover, fibronectin is dysregulated in salivary tissue from SS mice and is also elevated in saliva from SS patients [59, 60]. Studies show the ECM proteins decorin and biglycan are degraded by saliva derived from an SS mouse model [58]. Importantly, each of these ECM molecules can activate TLRs [61–63]. In addition, the ECM molecule versican is increased in salivary gland epithelial cell lines derived from SS patients as compared to those from healthy controls [64]. Versican activates TLR2/6 heterodimers [55] and is regulated by type I interferon (IFN) in lung tissue [65]. This finding may have particular relevance to SS pathogenesis, since a subset of SS patients display an IFN gene signature [14]. Significantly, 2 separate GWAS studies revealed a suggestive association between variants in the TNFAIP3 gene locus with SS [14]. While the functional consequence of this is unclear at present, TNFAIP3 encodes a negative regulator of TLR signaling and dysregulation of TLR-mediated networks could contribute to DAMP-driven inflammation in disease.
In addition to ECM molecules, elevated levels of the DAMP S100A8/A9 (calprotectin) are seen in the sera, salivary tissue, and saliva of SS patients [66–69]. Treatment of PBMCs from SS patients with S100A8/A9 causes significant TNFα secretion [68]. Studies in an infection model demonstrate S100A9 activates TLR4 [70]. Aberrant mucin localization may also activate TLR4 in SS. Of note, acinar cells in SS display loss of polarity and this has profound effects on cell function [71]. As a consequence of this loss of polarity, salivary mucins are secreted into the ECM at the basolateral aspect of the cell rather than from the apical pole [71]. Studies in HSG cells revealed that the mucins MUC5B and MUC7 activate TLR4 signaling [71], suggesting that ligation of epithelial cell-derived TLR4 by mucins may provide a continuous endogenous source of unremitting salivary inflammation in SS.
Finally, recent work to identify factors that upregulate IFN in autoimmunity identified a family of endogenous virus-like genomic retroelements, termed long interspersed nuclear element 1 (LINE-1 or L1) that are increased in minor salivary gland (MSG) tissue from SS patients [72]. L1 contains 2 open reading frames, and inappropriate expression of this retroelement activates innate immunity [72]. Significantly, L1 transcripts correlate with type I IFN in MSG tissue of SS patients [72]. Expression of L1 in pDCs or monocytes results in upregulation of IFNα and inhibition of TLR-7/TLR-8 reduced this expression in pDCs [72]. Significantly, L1 may also contribute to the female disease predilection observed in SS, as these sequences are located on the X chromosome and are known to escape X-chromosome inactivation [73]. Thus, endogenous genetic elements may serve as DAMPs in SS through activation of endosomal TLRs. Altogether, these studies provide evidence that host-derived mediators of inflammation may stimulate TLRs in SS patients, thereby precipitating and/or exacerbating disease.
In addition to TLR agonism, there is evidence that the P2X7R is activated by ATP in SS. ATP may be released from host cells as a result of necrosis or apoptosis. This results in inflammasome activation and subsequent secretion of pro-inflammatory cytokines including IL-1β and IL-18 [74]. Indeed, P2X7R activation causes salivary gland inflammation and studies in a murine model of SS show that blockade of this receptor reduces Il-1β expression [75]. Corroborative work using human samples found that P2X7R expression was higher in salivary tissue and PBMCs from SS patients compared with controls [76, 77]. In addition, they found that inflammasome components and IL-18 were elevated in the salivary tissue and saliva of SS patients, respectively [76]. Moreover, studies using CD14+ PBMCs from SS patients found that ATP treatment resulted in upregulation of P2X7R expression [78]. Taken together, these data suggest that activation of the P2X7R may contribute to both local and systemic inflammation in SS.
Oropharyngeal Candidiasis
Candida albicans is normally commensal in many healthy individuals, however, C. albicans is a significant cause of morbidity in individuals immunocompromised due to AIDS, neutropenia, diabetes mellitus or immunosuppressive drug treatment [79]. In addition, many primary immune deficiencies result in increased susceptibility to fungal infection, including neutropenia and patients with deficiencies in NADPH oxidase complex reactive oxygen production, leukocyte adhesion, and IL-17-related immunity [80]. Production of IL-1 cytokines is also critical to anti-fungal defenses [81]. Recent evidence also indicates there may also be a role for IL-1 cytokines, and their production by DAMPs, in distinguishing commensal yeast colonization from pathogenic invasion by immune cells [82].
The ability of C. albicans to escape from the phagosome of phagocytes is clear [83, 84]. This is achieved through filamentation and elongation of the yeast, an essential component of C. albicans pathogenicity [85]. Cell wall changes that accompany this filamentation are then responsible for the activation of the NLRC4 and NLRP3 inflammasomes that are also required for effective immunity [86, 87]. Importantly however, before significant interactions with phagocytes occur, as it is normally present in the oral cavity as a commensal, the yeast to hyphal transition allows C. albicans to invade the epithelium and grow within the host [88]. This invasion of the epithelial cells can cause the release of a number of endogenous DAMPs, including IL-1α, S100A8 and S100A9 [89, 90] (Figure 1). In the case of mucosal candidiasis, including OPC, release of DAMPs and internal signaling from DAMPs and PAMPs are important in mounting an effective immune response and in contrast to the above examples of periodontal disease and SS, we are unaware of any evidence suggesting cellular DAMP signaling induces increased Candida virulence.
Concluding Remarks
The oral cavity is a complex tissue environment with a variety of bacterial-mediated and autoimmune conditions that can occur. While immune responses to maintain tissue homeostasis and respond to dysbiotic bacterial communities at other body sites are documented, knowledge of immune responses in the unique oral environment are not as well understood. Emerging evidence suggests endogenous signals play an important role in disease progression and effective immune responses in the oral cavity. However, further studies are needed to understand the way in which these DAMPs are generated, the subsequent intracellular signals that result, and whether pharmacologic modulation of DAMP-dependent pathways has therapeutic efficacy.
Acknowledgments
We apologize to those who’s papers we were unable to reference due to limited space. The research was supported by NIH grants R03DE025062 to JGK, R56DE025218 to JMK and R03DE024769 and R01DE027073 to MBV.
Abbreviations
- DAMP
Damage-associated molecular pattern
- ECM
Extracellular matrix
- GCF
Gingival crevicular fluid
- HMGB1
High mobility group box 1
- LINE-1 or L1
Long interspersed nuclear element 1
- MAMP
Microbial-associated molecular patterns
- MSG
Minor salivary gland
- NET
Neutrophil extracellular trap
- NLR
NOD-like receptors
- NOD
Nucleotide-binding and oligomerization doma
- PAMP
Pathogen-associated molecular pattern
- RAGE
Receptor for advanced glycation end-products
- RIG-I
Retinoic acid-inducible gene-I
- SS
Sjögren’s syndrome
- SSA
Serum amyloid A
- TLR
Toll-like receptor
Footnotes
Conflict of Interest Disclosure
The authors declare no conflict of interest.
References
- 1.Proctor DM and Relman DA (2017) The Landscape Ecology and Microbiota of the Human Nose, Mouth, and Throat. Cell Host & Microbe 21, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mavragani CP and Moutsopoulos HM (2014) Sjögren’s syndrome. Annual review of pathology 9, 273–285. [DOI] [PubMed] [Google Scholar]
- 3.Chen T, Yu W-H, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE (2010) The Human Oral Microbiome Database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database : the journal of biological databases and curation 2010, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghannoum MA, Jurevic RJ, Mukherjee PK, Cui F, Sikaroodi M, Naqvi A, Gillevet PM (2010) Characterization of the oral fungal microbiome (mycobiome) in healthy individuals. PLoS Pathogens 6, e1000713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dutzan N, Konkel JE, Greenwell-Wild T, Moutsopoulos NM (2016) Characterization of the human immune cell network at the gingival barrier. Mucosal immunology 9, 1163–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moutsopoulos NM and Konkel JE (2018) Tissue-Specific Immunity at the Oral Mucosal Barrier. Trends in Immunology 39, 276–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delima AJ and Van Dyke TE (2003) Origin and function of the cellular components in gingival crevice fluid. Periodontology 2000 31, 55–76. [DOI] [PubMed] [Google Scholar]
- 8.Wu R-Q, Zhang D-F, Tu E, Chen Q-M, Chen W (2014) The mucosal immune system in the oral cavity-an orchestra of T cell diversity. International journal of oral science 6, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Lorenzo G, Ferrari S, Cervone F, Okun E (2018) Extracellular DAMPs in Plants and Mammals: Immunity, Tissue Damage and Repair. Trends in Immunology 39, 937–950. [DOI] [PubMed] [Google Scholar]
- 10.Wallet SM, Puri V, Gibson FC (2018) Linkage of Infection to Adverse Systemic Complications: Periodontal Disease, Toll-Like Receptors, and Other Pattern Recognition Systems. Vaccines 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franz KM and Kagan JC (2017) Innate Immune Receptors as Competitive Determinants of Cell Fate. Molecular Cell 66, 750–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leite FRM, Enevold C, Bendtzen K, Baelum V, Lopez R (2018) Pattern recognition receptor polymorphisms in early periodontitis. J Periodontol. [DOI] [PubMed] [Google Scholar]
- 13.Gursoy UK, He Q, Pussinen P, Huumonen S, Kononen E (2016) Alveolar bone loss in relation to toll-like receptor 4 and 9 genotypes and Porphyromonas gingivalis carriage. Eur J Clin Microbiol Infect Dis 35, 1871–1876. [DOI] [PubMed] [Google Scholar]
- 14.Teos LY and Alevizos I (2017) Genetics of Sjogren’s syndrome. Clin Immunol 182, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eke PI, Dye BA, Wei L, Slade GD, Thornton-Evans GO, Borgnakke WS, Taylor GW, Page RC, Beck JD, Genco RJ (2015) Update on Prevalence of Periodontitis in Adults in the United States: NHANES 2009 to 2012. Journal of periodontology 86, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hajishengallis G and Korostoff JM (2017) Revisiting the Page & Schroeder model: the good, the bad and the unknowns in the periodontal host response 40 years later. Periodontology 2000 75, 116–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mark Bartold P and Van Dyke TE (2017) Host modulation: controlling the inflammation to control the infection. Periodontology 2000 75, 317–329. [DOI] [PubMed] [Google Scholar]
- 18.Lamont RJ, Koo H, Hajishengallis G (2018) The oral microbiota: dynamic communities and host interactions. Nature Reviews Microbiology 7, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Netea MG, van de Veerdonk FL, van der Meer JWM, Dinarello CA, Joosten LAB (2015) Inflammasome-independent regulation of IL-1-family cytokines. Annual review of immunology 33, 49–77. [DOI] [PubMed] [Google Scholar]
- 20.Jandinski JJ, Stashenko P, Feder LS, Leung CC, Peros WJ, Rynar JE, Deasy MJ (1991) Localization of interleukin-1 beta in human periodontal tissue. Journal of periodontology 62, 36–43. [DOI] [PubMed] [Google Scholar]
- 21.Kinane DF, Winstanley FP, Adonogianaki E, Moughal NA (1992) Bioassay of interleukin 1 (IL-1) in human gingival crevicular fluid during experimental gingivitis. Archives of Oral Biology 37, 153–156. [DOI] [PubMed] [Google Scholar]
- 22.Xue F, Shu R, Xie Y (2015) The expression of NLRP3, NLRP1 and AIM2 in the gingival tissue of periodontitis patients: RT-PCR study and immunohistochemistry. Arch Oral Biol 60, 948–958. [DOI] [PubMed] [Google Scholar]
- 23.Isaza-Guzmán DM, Medina-Piedrahíta VM, Gutiérrez-Henao C, Tobón-Arroyave SI (2017) Salivary Levels of NLRP3 Inflammasome-Related Proteins as Potential Biomarkers of Periodontal Clinical Status. Journal of periodontology 88, 1329–1338. [DOI] [PubMed] [Google Scholar]
- 24.Visser MB, Koh A, Glogauer M, Ellen RP (2011) Treponema denticola major outer sheath protein induces actin assembly at free barbed ends by a PIP2-dependent uncapping mechanism in fibroblasts. PLoS ONE 6, e23736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hanazawa S, Nakada K, Ohmori Y, Miyoshi T, Amano S, Kitano S (1985) Functional role of interleukin 1 in periodontal disease: induction of interleukin 1 production by Bacteroides gingivalis lipopolysaccharide in peritoneal macrophages from C3H/HeN and C3H/HeJ mice. Infection and Immunity 50, 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varanat M, Haase EM, Kay JG, Scannapieco FA (2017) Activation of the TREM-1 pathway in human monocytes by periodontal pathogens and oral commensal bacteria. Molecular oral microbiology 32, 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yilmaz Ö, Sater AA, Yao L, Koutouzis T, Pettengill M, Ojcius DM (2010) ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cellular Microbiology 12, 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bui FQ, Johnson L, Roberts J, Hung S-C, Lee J, Atanasova KR, Huang P-R, Yilmaz Ö, Ojcius DM (2016) Fusobacterium nucleatum infection of gingival epithelial cells leads to NLRP3 inflammasome-dependent secretion of IL-1β and the danger signals ASC and HMGB1. Cellular Microbiology 18, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cecil JD, O' Brien-Simpson ,NM, Lenzo JC, Holden JA, Singleton W, Perez-Gonzalez A, Mansell A, Reynolds EC (2017) Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Frontiers in immunology 8, 346–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fleetwood AJ, Lee MKS, Singleton W, Achuthan A, Lee M-C, O' Brien-Simpson NM, Cook AD, Murphy AJ, Dashper SG, Reynolds EC, Hamilton JA (2017) Metabolic Remodeling, Inflammasome Activation, and Pyroptosis in Macrophages Stimulated by Porphyromonas gingivalis and Its Outer Membrane Vesicles. Frontiers in Cellular and Infection Microbiology 7, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shibata K-I (2018) Historical aspects of studies on roles of the inflammasome in the pathogenesis of periodontal diseases. Molecular oral microbiology. [DOI] [PubMed] [Google Scholar]
- 32.Ramos-Junior ES, Morandini AC, Almeida-da-Silva CLC, Franco EJ, Potempa J, Nguyen KA, Oliveira AC, Zamboni DS, Ojcius DM, Scharfstein J, Coutinho-Silva R (2015) A Dual Role for P2X7 Receptor during Porphyromonas gingivalis Infection. Journal of Dental Research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taxman DJ, Swanson KV, Broglie PM, Wen H, Holley-Guthrie E, Huang MT-H, Callaway JB, Eitas TK, Duncan JA, Ting JPY (2012) Porphyromonas gingivalis mediates inflammasome repression in polymicrobial cultures through a novel mechanism involving reduced endocytosis. Journal of Biological Chemistry 287, 32791–32799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park E, Na HS, Song Y-R, Shin SY, Kim Y-M, Chung J (2014) Activation of NLRP3 and AIM2 inflammasomes by Porphyromonas gingivalis infection. Infection and Immunity 82, 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeuchi H, Hirano T, Whitmore SE, Morisaki I, Amano A, Lamont RJ (2013) The serine phosphatase SerB of Porphyromonas gingivalis suppresses IL-8 production by dephosphorylation of NF-κB RelA/p65. PLoS Pathogens 9, e1003326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okano T, Ashida H, Suzuki S, Shoji M, Nakayama K, Suzuki T (2018) Porphyromonas gingivalistriggers NLRP3-mediated inflammasome activation in macrophages in a bacterial gingipains-independent manner. European Journal of Immunology, 1–28. [DOI] [PubMed] [Google Scholar]
- 37.Bodet C, Chandad F, Grenier D (2005) Modulation of cytokine production by Porphyromonas gingivalis in a macrophage and epithelial cell co-culture model. Microbes and infection / Institut Pasteur 7, 448–456. [DOI] [PubMed] [Google Scholar]
- 38.El-Awady AR, Miles B, Scisci E, Kurago ZB, Palani CD, Arce RM, Waller JL, Genco CA, Slocum C, Manning M, Schoenlein PV, Cutler CW (2015) Porphyromonas gingivalis Evasion of Autophagy and Intracellular Killing by Human Myeloid Dendritic Cells Involves DC-SIGN-TLR2 Crosstalk. PLoS Pathogens 10, e1004647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jenkinson HF and Lamont RJ (2005) Oral microbial communities in sickness and in health. Trends in Microbiology 13, 589–595. [DOI] [PubMed] [Google Scholar]
- 40.Stewart MK and Cookson BT (2016) Evasion and interference: intracellular pathogens modulate caspase-dependent inflammatory responses. Nature Reviews Microbiology 14, 346–359. [DOI] [PubMed] [Google Scholar]
- 41.Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, Darveau RP, Curtis MA (2011) Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host & Microbe 10, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daep CA, Novak EA, Lamont RJ, Demuth DR (2011) Structural dissection and in vivo effectiveness of a peptide inhibitor of Porphyromonas gingivalis adherence to Streptococcus gordonii. Infection and Immunity 79, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Croft AJ, Metcalfe S, Honma K, Kay JG (2018) Macrophage Polarization Alters Postphagocytosis Survivability of the Commensal Streptococcus gordonii. Infection and Immunity 86, e00858–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jun H-K, Jung Y-J, Choi B-K (2017) Treponema denticola, Porphyromonas gingivalis, and Tannerella forsythia induce cell death and release of endogenous danger signals. Arch Oral Biol 73, 72–78. [DOI] [PubMed] [Google Scholar]
- 45.Choi CH, Spooner R, DeGuzman J, Koutouzis T, Ojcius DM, Yilmaz Ö (2013) Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cellular Microbiology 15, 961–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ebe N, Hara-Yokoyama M, Iwasaki K, Iseki S, Okuhara S, Podyma-Inoue KA, Terasawa K, Watanabe A, Akizuki T, Watanabe H, Yanagishita M, Izumi Y (2011) Pocket epithelium in the pathological setting for HMGB1 release. J Dent Res 90, 235–40. [DOI] [PubMed] [Google Scholar]
- 47.Luo L, Xie P, Gong P, Tang XH, Ding Y, Deng LX (2011) Expression of HMGB1 and HMGN2 in gingival tissues, GCF and PICF of periodontitis patients and peri-implantitis. Arch Oral Biol 56, 1106–11. [DOI] [PubMed] [Google Scholar]
- 48.Hirai K, Furusho H, Kawashima N, Xu S, de Beer MC, Battaglino R, Van Dyke T, Stashenko P, Sasaki H (2018) Serum Amyloid A Contributes to Chronic Apical Periodontitis via TLR2 and TLR4. Journal of Dental Research 191, 22034518796456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Johnson L, Almeida-da-Silva CLC, Takiya CM, Figliuolo V, Rocha GM, Weissmüller G, Scharfstein J, Coutinho-Silva R, Ojcius DM (2018) Oral infection of mice with Fusobacterium nucleatum results in macrophage recruitment to the dental pulp and bone resorption. Biomedical Journal 41, 184–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yoshihara-Hirata C, Yamashiro K, Yamamoto T, Aoyagi H, Ideguchi H, Kawamura M, Suzuki R, Ono M, Wake H, Nishibori M, Takashiba S (2018) Anti-HMGB1 neutralizing antibody attenuates periodontal inflammation and bone resorption in a murine periodontitis model. Infection and Immunity, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vitkov L, Hartl D, Minnich B, Hannig M (2017) Janus-Faced Neutrophil Extracellular Traps in Periodontitis. Front Immunol 8, 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y, Tsung A (2015) Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 62, 600–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Angelidou I, Chrysanthopoulou A, Mitsios A, Arelaki S, Arampatzioglou A, Kambas K, Ritis D, Tsironidou V, Moschos I, Dalla V, Stakos D, Kouklakis G, Mitroulis I, Ritis K, Skendros P (2018) REDD1/Autophagy Pathway Is Associated with Neutrophil-Driven IL-1beta Inflammatory Response in Active Ulcerative Colitis. J Immunol 200, 3950–3961. [DOI] [PubMed] [Google Scholar]
- 54.Flaitz CM (2001) Parotitis as the initial sign of juvenile Sjögren’s syndrome. Pediatric dentistry 23, 140–142. [PubMed] [Google Scholar]
- 55.Bryant CE, Gay NJ, Heymans S, Sacre S, Schaefer L, Midwood KS (2015) Advances in Toll-like receptor biology: Modes of activation by diverse stimuli. Critical reviews in biochemistry and molecular biology 50, 359–379. [DOI] [PubMed] [Google Scholar]
- 56.Schenke-Layland K, Xie J, Angelis E, Starcher B, Wu K, Riemann I, MacLellan WR, Hamm-Alvarez SF (2008) Increased degradation of extracellular matrix structures of lacrimal glands implicated in the pathogenesis of Sjögren’s syndrome. Matrix biology : journal of the International Society for Matrix Biology 27, 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goicovich E, Molina C, Pérez P, Aguilera S, Fernández J, Olea N, Alliende C, Leyton C, Romo R, Leyton L, González M-J (2003) Enhanced degradation of proteins of the basal lamina and stroma by matrix metalloproteinases from the salivary glands of Sjögren’s syndrome patients: correlation with reduced structural integrity of acini and ducts. Arthritis and rheumatism 48, 2573–2584. [DOI] [PubMed] [Google Scholar]
- 58.Yamachika S, Brayer J, Oxford GE, Peck AB, Humphreys-Beher MG (2000) Aberrant proteolytic digestion of biglycan and decorin by saliva and exocrine gland lysates from the NOD mouse model for autoimmune exocrinopathy. Clin Exp Rheumatol 18, 233–240. [PubMed] [Google Scholar]
- 59.Enger TB, Samad-Zadeh A, Bouchie MP, Skarstein K, Galtung HK, Mera T, Walker J, Menko AS, Varelas X, Faustman DL, Jensen JL, Kukuruzinska MA (2013) The Hippo signaling pathway is required for salivary gland development and its dysregulation is associated with Sjogren’s syndrome. Laboratory investigation; a journal of technical methods and pathology 93, 1203–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silvestre FJ, Puente A, Bagán JV, Castell JV (2009) Presence of fibronectin peptides in saliva of patients with Sjögren’s syndrome: a potential indicator of salivary gland destruction. Medicina oral, patologia oral y cirugia bucal 14, e365–70. [PubMed] [Google Scholar]
- 61.Kramer JM (2014) Early events in Sjögren’s Syndrome pathogenesis: the importance of innate immunity in disease initiation. Cytokine 67, 92–101. [DOI] [PubMed] [Google Scholar]
- 62.Midwood KS, Piccinini AM, Sacre S (2009) Targeting Toll-like receptors in autoimmunity. Current drug targets 10, 1139–1155. [DOI] [PubMed] [Google Scholar]
- 63.Schaefer L (2014) Complexity of danger: the diverse nature of damage-associated molecular patterns. Journal of Biological Chemistry 289, 35237–35245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vakrakou AG, Polyzos A, Kapsogeorgou EK, Thanos D, Manoussakis MN (2018) Perturbation of transcriptome in non-neoplastic salivary gland epithelial cell lines derived from patients with primary Sjögren’s syndrome. Data in brief 17, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chang MY, Kang I, Gale M, Manicone AM, Kinsella MG, Braun KR, Wigmosta T, Parks WC, Altemeier WA, Wight TN, Frevert CW (2017) Versican is produced by Trif- and type I interferon-dependent signaling in macrophages and contributes to fine control of innate immunity in lungs. American journal of physiology. Lung cellular and molecular physiology 313, L1069–L1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nordal HH, Brun JG, Halse A-K, Madland TM, Fagerhol MK, Jonsson R (2014) Calprotectin (S100A8/A9), S100A12, and EDTA-resistant S100A12 complexes (ERAC) in primary Sjögren’s syndrome. Scandinavian journal of rheumatology 43, 76–78. [DOI] [PubMed] [Google Scholar]
- 67.Cuida M, Halse A-K, Johannessen AC, Tynning T, Jonsson R (1997) Indicators of salivary gland inflammation in primary Sjogren’s syndrome. European journal of oral sciences 105, 228–233. [DOI] [PubMed] [Google Scholar]
- 68.Nicaise C, Weichselbaum L, Schandene L, Gangji V, Dehavay F, Bouchat J, Balau B, Vogl T, Soyfoo MS (2017) Phagocyte-specific S100A8/A9 is upregulated in primary Sjögren’s syndrome and triggers the secretion of pro-inflammatory cytokines in vitro. Clin Exp Rheumatol 35, 129–136. [PubMed] [Google Scholar]
- 69.Jazzar AA, Shirlaw PJ, Carpenter GH, Challacombe SJ, Proctor GB (2018) Salivary S100A8/A9 in Sjögren’s syndrome accompanied by lymphoma. J Oral Pathol Med 47, 900–906. [DOI] [PubMed] [Google Scholar]
- 70.Tsai S-Y, Segovia JA, Chang T-H, Morris IR, Berton MT, Tessier PA, Tardif MR, Cesaro A, Bose S (2014) DAMP molecule S100A9 acts as a molecular pattern to enhance inflammation during influenza A virus infection: role of DDX21-TRIF-TLR4-MyD88 pathway. PLoS Pathogens 10, e1003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barrera M-J, Aguilera S, Veerman E, Quest AFG, Díaz-Jiménez D, Urzúa U, Cortés J, González S, Castro I, Molina C, Bahamondes V, Leyton C, Hermoso MA, González M-J (2015) Salivary mucins induce a Toll-like receptor 4-mediated pro-inflammatory response in human submandibular salivary cells: are mucins involved in Sjögren’s syndrome? Rheumatology (Oxford, England) 54, 1518–1527. [DOI] [PubMed] [Google Scholar]
- 72.Mavragani CP, Sagalovskiy I, Guo Q, Nezos A, Kapsogeorgou EK, Lu P, Liang Zhou J, Kirou KA, Seshan SV, Moutsopoulos HM, Crow MK (2016) Expression of Long Interspersed Nuclear Element 1 Retroelements and Induction of Type I Interferon in Patients With Systemic Autoimmune Disease. Arthritis & rheumatology (Hoboken, N.J.) 68, 2686–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mougeot J-L, Noll BD, Bahrani Mougeot FK (2018) Sjögren’s syndrome X-chromosome dose effect: An epigenetic perspective. Oral diseases 36, 59. [DOI] [PubMed] [Google Scholar]
- 74.Vénéreau E, Ceriotti C, Bianchi ME (2015) DAMPs from Cell Death to New Life. Frontiers in immunology 6, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Khalafalla MG, Woods LT, Camden JM, Khan AA, Limesand KH, Petris MJ, Erb L, Weisman GA (2017) P2X7 receptor antagonism prevents IL-1β release from salivary epithelial cells and reduces inflammation in a mouse model of autoimmune exocrinopathy. Journal of Biological Chemistry 292, 16626–16637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baldini C, Rossi C, Ferro F, Santini E, Seccia V, Donati V, Solini A (2013) The P2X7 receptor-inflammasome complex has a role in modulating the inflammatory response in primary Sjögren’s syndrome. Journal of internal medicine 274, 480–489. [DOI] [PubMed] [Google Scholar]
- 77.Yu J, Chen Y, Li M, Gao Q, Peng Y, Gong Q, Zhang Z, Wu X (2015) Paeoniflorin down-regulates ATP-induced inflammatory cytokine production and P2X7R expression on peripheral blood mononuclear cells from patients with primary Sjögren’s syndrome. International Immunopharmacology 28, 115–120. [DOI] [PubMed] [Google Scholar]
- 78.Xie B, Chen Y, Zhang S, Wu X, Zhang Z, Peng Y, Huang X (2014) The expression of P2X7 receptors on peripheral blood mononuclear cells in patients with primary Sjögren’s syndrome and its correlation with anxiety and depression. Clin Exp Rheumatol 32, 354–360. [PubMed] [Google Scholar]
- 79.Salvatori O, Puri S, Tati S, Edgerton M (2016) Innate Immunity and Saliva in Candida albicans-mediated Oral Diseases. Journal of Dental Research 95, 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Romani L (2011) Immunity to fungal infections. Nature Reviews Immunology 11, 275–288. [DOI] [PubMed] [Google Scholar]
- 81.Bellocchio S, Montagnoli C, Bozza S, Gaziano R, Rossi G, Mambula SS, Vecchi A, Mantovani A, Levitz SM, Romani L (2004) The contribution of the Toll-like/IL-1 receptor superfamily to innate and adaptive immunity to fungal pathogens in vivo. Journal of immunology (Baltimore, Md. : 1950) 172, 3059–3069. [DOI] [PubMed] [Google Scholar]
- 82.Caffrey AK and Obar JJ (2016) Alarmin(g) the innate immune system to invasive fungal infections. Current Opinion in Microbiology 32, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.McKenzie CGJ, Koser U, Lewis LE, Bain JM, Mora-Montes HM, Barker RN, Gow NAR, Erwig L-P (2010) Contribution of Candida albicans cell wall components to recognition by and escape from murine macrophages. Infection and Immunity 78, 1650–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Salvatori O, Pathirana RU, Kay JG, Edgerton M (2018) Candida albicans Ras1 inactivation increases resistance to phagosomal killing by human neutrophils. Infection and Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mayer FL, Wilson D, Hube B (2013) Candida albicans pathogenicity mechanisms. Virulence 4, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hise AG, Tomalka J, Ganesan S, Patel K, Hall BA, Brown GD, Fitzgerald KA (2009) An essential role for the NLRP3 inflammasome in host defense against the human fungal pathogen Candida albicans. Cell Host & Microbe 5, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tomalka J, Ganesan S, Azodi E, Patel K, Majmudar P, Hall BA, Fitzgerald KA, Hise AG (2011) A novel role for the NLRC4 inflammasome in mucosal defenses against the fungal pathogen Candida albicans. PLoS Pathogens 7, e1002379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thompson DS, Carlisle PL, Kadosh D (2011) Coevolution of morphology and virulence in Candida species. Eukaryotic Cell 10, 1173–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jayatilake JAMS, Samaranayake LP, Lu Q, Jin LJ (2007) IL-1alpha, IL-1ra and IL-8 are differentially induced by Candida in experimental oral candidiasis. Oral diseases 13, 426–433. [DOI] [PubMed] [Google Scholar]
- 90.Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, Filler SG, Masso-Welch P, Edgerton M, Gaffen SL (2009) Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. Journal of Experimental Medicine 206, 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]