

Abstract
Brucellosis, caused by the intracellular bacterial pathogen Brucella, is a globally important zoonotic disease for which arthritis is the most common focal complication in humans. Wild-type mice infected systemically with Brucella typically do not exhibit arthritis, but mice lacking IFN-γ develop arthritis regardless of the route of Brucella infection. Here we investigated mechanisms by which IFN-γ suppresses Brucella-induced arthritis. Several cell types, including innate lymphoid cells, contributed to IFN-γ production and suppression of joint swelling. IFN-γ deficiency resulted in elevated joint IL-1β levels, and severe joint inflammation that was entirely inflammasome dependent, and in particular, reliant on the NLRP3 inflammasome. IFN-γ was vital for induction of the nitric oxide producing enzyme, iNOS, in infected joints, and nitric oxide directly inhibited IL-1β production and inflammasome activation in Brucella-infected macrophages in vitro. During in vivo infection, iNOS deficiency resulted in an increase in IL-1β and inflammation in Brucella infected joints. Collectively, this data indicate that IFN-γ prevents arthritis both by limiting Brucella infection, and by inhibiting excessive inflammasome activation through the induction of nitric oxide.
Keywords: brucellosis, innate lymphoid cell, NLRP3, caspase-1
Summary Sentence: IFN-γ suppresses inflammation both by limiting Brucella infection, and by inhibiting excessive inflammasome activation through the induction of nitric oxide.
Graphical Abstract
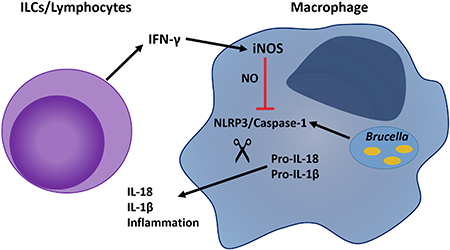
Introduction
Brucellosis, caused by the genus Brucella, is one of the most common zoonotic infections worldwide infecting over 500,000 individuals each year.1,2 Transmission typically occurs through consumption of unpasteurized dairy goods or inhalation of aerosols from contaminated animal products.3,4 Osteoarticular and/or musculoskeletal inflammation are the most common focal complications of brucellosis, with an incidence of 40–80% in infected patients.5,6 Active infection of the joints is thought to be required for arthritis development, as viable brucellae are found within the synovial fluid of infected patients along with polymorphonuclear and mononuclear leukocytes.6–9 Brucella induced arthritis is typically treated with prolonged antibiotic therapy; however, the time to resolution is often extensive, and disease can relapse.6,10
Wild-type (WT) mice infected with Brucella through systemic routes typically do not develop arthritis, however we reported IFN-γ−/− mice develop arthritis and musculoskeletal inflammation regardless of the route of Brucella infection.11 It is currently unknown if IFN-γ prevents arthritis by limiting Brucella dissemination to the joints, and/or if IFN-γ acts locally in the joint to restrict infection and inflammation.12 Many studies, including our own11,12, have demonstrated IFN-γ is needed for resistance to brucellosis, however it is not clear what innate cells contribute to early IFN-γ production, particularly at focal sites of infection such as the joint. In addition to T cells and NK cells, there has been an increase in reports indicating tissue resident cells, such as innate lymphoid cells (ILCs), can rapidly produce IFN-γ to protect the host against infection.13,14
Recently we reported that inflammasomes induce joint inflammation, but also contribute to control of infection during Brucella-induced arthritis.15 Inflammasomes are multi-protein structures that use sensors, such as NLRP3 and AIM2, to detect intracellular, cytosolic threats.16 Upon sensor activation, canonical inflammasomes recruit pro-caspase-1, and cleave it into its functional form, caspase-1. Activated caspase-1 proteolyticly activates IL-1β and IL-18 into their functional/secreted forms, and also induces pyroptosis, a pro-inflammatory cell death.16 We recently showed that Brucella is also recognized by the non-canonical, inflammasome, caspase-11, which is activated by cytosolic LPS.15,17 Caspase-11 does not directly cleave IL-1β or IL-18 into their active forms, but like caspase-1, can induce pyroptosis.18 While inflammasomes can restrict infection, unregulated inflammasome activation can lead to immunopathology.15,19 Here, we investigated cell types that contribute to the protective effects of IFN-γ within the joint, and examined mechanisms by which inflammasome-dependent pathology is regulated by IFN-γ.
Materials and Methods
Bacteria
Brucella melitensis 16M was grown on brucella agar (Ba) at 37°C (Becton Dickinson). Colonies were picked from Ba plates and cultured in brucella broth (Becton Dickinson) overnight at 37°C. Overnight Brucella concentration was estimated by measuring optical density at 600 nm, and inoculum was diluted to the appropriate concentration in sterile phosphate-buffered saline (PBS). Actual viable titer was confirmed by dilution of inoculum onto Ba.
Mice
Experiments were conducted using 6- to 12-week-old age- and sex-matched mice on a C57BL/6J background. Rag1−/−, Caspase-1/11−/−, NLRP3−/−, AIM2−/−, Caspase-11−/−, and NOS2−/− mice were obtained from Jackson Laboratory. IL-1R−/−/IL-18−/− mice were obtained from the University of North Carolina. Mice were infected in each rear footpad with 50 μl of PBS containing 1×105 CFU of Brucella/footpad,20 or intraperitoneally (i.p.) with 1×105 CFU of Brucella in 200 μl of PBS.11,15 All studies were conducted in accordance with University of Missouri Animal Care and Use Committee guidelines. To neutralize IFN-γ during footpad infection, mice were treated i.p. with 0.5 mg anti-IFN-γ (clone XMG1.2, BioXCell) 1 day prior to, and 3 days after infection. Control mice received Rat IgG (Southern Biotech). To neutralize IFN-γ during i.p. infection, mice were treated i.p. with 0.25 mg anti-IFN-γ 1 day prior to infection, and 3 times a week thereafter.12 Rag1−/− mice were treated with 0.2 mg of anti-NK1.1 (clone PK136) or anti-CD90.2 (clone 30H12), on days −1, 2, and 5 in relation to infection,21 to deplete these mice of NK cells, or ILCs respectively.
Joint processing for bacterial burdens and cytokine measurements
Spleens and joints (following removal of skin) were mechanically ground in PBS.15 Serial dilutions of homogenates were plated onto Ba and CFUs/tissue calculated. Cytokines were measured via Luminex (Millipore) or ELISA (Invitrogen) according to manufacturer’s instructions. Cytokine data was normalized to total protein by BCA (Thermo Scientific).
Macrophage infections
Bone marrow derived macrophages (BMDMs) were generated with M-CSF in complete media (CM: RPMI 1640 containing HEPES, sodium pyruvate, non-essential amino acids, and 10% FBS).15 For Western blots, BMDMs were infected in CM with 2% FBS, while all other infections utilized CM with 10% FBS. BMDMs were infected with B. melitensis at a multiplicity of infection (MOI) of 100 for 6 hours, washed, incubated in CM with 50 μg/ml gentamicin for 0.5 hours, washed, and then incubated in CM containing 2.5 μg/ml of gentamicin for the remainder of the experiment. To determine bacterial burdens, BMDMs were washed, lysed in H2O and plated onto Ba.
Immunoblots
24 hours after infection, BMDMs were lysed in RIPA buffer (Thermo), and total protein normalized using BCA. Supernatants and lysates were probed with anti-Caspase-1 p20 (casper-1, Adipogen) and then peroxidase-conjugate Goat Anti-Mouse IgG (Jackson Immuno Research). Detection was performed with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo).
RT-PCR
Joints were homogenized in TRI reagent, and RNA was isolated according to manufacturer’s instructions (Sigma). RNA was further purified on an RNeasy column (Qiagen). cDNA was generated using the superscript III First Strand Synthesis System (Invitrogen) using oligodT primers. Relative iNOS mRNA in relation to GAPDH was quantified by measuring SYBR green incorporation using the comparative threshold method.22
Assessment of Pathology
Basal joint measurements were made prior to infection. Joint swelling was determined by collective measurement of tibiotarsal joints following footpad infection, or by collective measurement of tibiotarsal and radiocarpal joints for i.p. infections, in relation to basal values. For histology, H&E sections from tibiotarsal joints were scored from 0–4 as previously detailed.[20]
Flow cytometry
Rear ankle joints were processed with collagenase/DNAse as described15 and cells were acquired on a CyAn ADP Analyzer (Beckman Coulter) or LSR Fortessa (Becton Dickinson). FlowJo (Tree Star) software was used for analysis. Immunofluorescence staining was performed using the following fluorochrome-labeled monoclonal antibodies from eBioscience or BioLegend: F4/80 (BM8), Ly-6G (1A8), CD11b (M1/70), CD45.2 (104), CD90.2 (30H12), NK1.1 (PK136), TCRγδ (UC7–13D5).
Statistics
Statistical analysis of the difference between 2 mean values was conducted using a two-tailed Student t test with significance at P ≤ .05, while comparisons of ≥3 mean values was done using ANOVA, followed by Tukey’s test with significance at P ≤ .05. Differences in histology scores were determined by ANOVA on ranks.
Online supplemental material
Supplementary Figure 1 depicts the effects of; IFN-γ deficiency on neutrophil recruitment, NK cell or γδ T cell depletion on joint swelling, IFN-γ production, and control of infection, and the effect of ILC depletion on iNOS transcription in infected joints. Confirmation of depletion of NK cells, γδ T cells, and ILCs is also shown in Supplementary Figure 1. The effects of IL-18/IL-1R, AIM2, Caspase-11, and the NLRP3 inhibitor, MCC950, on joint swelling in IFN-γ deficient mice is shown in Supplementary Figure 2. In addition, Supplementary Figure 2, shows iNOS-dependent NO production by Brucella-infected macrophages stimulated with IFN-γ, NO levels in SNAP treated macrophage cultures, and the effect of the iNOS inhibitor, AGHS on joint inflammation.
Results and Discussion
ILCs are required for IFN-γ production and suppression of joint swelling in Brucella infected mice
We previously reported Brucella infection in IFN-γ-deficient mice results in arthritis regardless of the route of infection.11 However, it was unknown if IFN-γ prevented arthritis by solely limiting Brucella dissemination to the joints, and/or if IFN-γ acted locally in the joint to restrict infection and inflammation. To determine if IFN-γ plays a local role during the development of Brucella- induced arthritis, we utilized a footpad model of Brucella infection.20 Using this model, we found that Brucella infection in WT mice resulted in a robust increase in myeloid cells, and in particular neutrophils, within the joint (Figure S1A–B). However, neutralization of IFN-γ in footpad infected WT mice enhanced neutrophil recruitment ~10 fold (Figure S1A–B), indicating that local IFN-γ within the joint contributes to suppression of arthritis during Brucella infection.
Previously, we demonstrated lymphocytes were required for resolution of chronic joint infection and inflammation in mice infected in the footpad with Brucella.20 To investigate whether lymphocytes contribute to the effect of IFN-γ during Brucella-induced arthritis, we footpad infected WT and Rag1−/− (lymphocyte deficient) mice and treated them with an IFN-γ neutralizing antibody or an isotype control. Joint swelling was measured and bacterial burdens were assayed at day 7 post-infection (Figure 1A–B). Lymphocytes did contribute to control of infection as IgG-treated Rag1−/− mice had increased joint Brucella burdens compared to IgG-treated WT animals (Figure 1A). The effect of lymphocytes appears to require IFN-γ, as Rag1−/− mice treated with anti-IFN-γ have similar bacterial burdens as WT-anti-IFN-γ treated mice (Figure 1A). While the effect of lymphocytes on control of infection was IFN-γ-dependent, cells other than lymphocytes contribute to the IFN-γ response in the joint, as neutralization of IFN-γ from Rag1−/− mice resulted in increased joint bacterial burdens and swelling compared to Rag1−/− mice treated with IgG (Figure 1A–B), and IFN-γ was still detected in Rag1−/− joints treated with IgG (Figure S1D). Anti-IFN-γ treated Rag1−/− mice also had reduced joint swelling relative to anti-IFN-γ treated WT mice (Figure 1B), indicating that lymphocytes partially contribute to the joint swelling and inflammation that is observed in the absence of IFN-γ.
Figure 1. Innate lymphoid cells restrict Brucella-induced joint swelling and contribute to IFN-γ production.

WT and Rag1−/− mice (n=4–5/group) were treated with anti-IFN-γ or an isotype control, then infected in both rear footpads with 1×105 B. melitensis 16M A-B). Mice were euthanized at day 7 and CFUs were enumerated A) while joint swelling was measured throughout the experimental time course B). Error bars depict S.D. of the mean. Means at the same timepoint with the same letter are not statistically different from each other as determined by ANOVA followed by Tukey’s test. Rag1−/− mice (n=5/group) were treated with anti-NK1.1 or anti-CD90, then infected in both rear footpads with 1×105 B. melitensis 16M C-E). Joint swelling was measured over time C), and on day 7 mice were euthanized and joint IFN-γ and tissue CFUs were measured D-E). Error bars depict S.D. of the mean. *P<0.05 as compared to Rag1−/−, anti-NK1.1 treated mice. Data in C-E) is representative of 2 independent experiments.
We next investigated the role of innate sources of IFN-γ. While both NK cells and γδ T cells can rapidly produce IFN-γ in response to infection,23,24 we found that depletion of NK cells or γδ T cells from WT mice did not alter joint swelling, IFN-γ levels, or Brucella burdens (Figure S1E-G, I,J). Therefore, we investigated the effect of ILCs, which can be tissue resident innate sources of IFN-γ.25 To do this, we treated Rag-1−/− mice with anti-NK1.1, to deplete NK cells, or with anti-CD90, to deplete all ILCs including NK cells.21 ILC depletion was confirmed via flow cytometry (Figure S1K). By day 7 post-infection joint swelling in Rag1−/− mice depleted of ILCs was ~3 times higher than in NK cell-depleted Rag1−/− mice (Figure 1C). In addition, joint IFN-γ levels (Figure 1D) and mRNA expression of the nitric oxide producing enzyme iNOS (Figure S1H) were significantly reduced by ILC depletion. Surprisingly, despite having increased joint swelling, Rag1−/−, anti-CD90 treated mice actually had slightly reduced joint and spleen bacterial burdens compared to Rag1−/−, anti-NK1.1 treated animals (Figure 1E). These data suggest that while IFN-γ protects the host by restricting Brucella infection, IFN-γ may also suppress inflammation by mechanisms that are independent of bacterial clearance. Three major ILC populations have been described, (ILC1, ILC2, and ILC3).25 While IFN-γ production is a signature of ILC1 cells, recent studies have shown that ILC3 cells can also protect the host by producing IFN-γ.26 Future work will determine the relative contribution of ILC subsets to IFN-γ production, and control of infection and arthritis during brucellosis.
IFN-γ deficiency results in caspase-1/11-dependent joint inflammation
Brucella-induced IL-1β production requires caspase-1,27 and in an earlier study, we determined that both caspase-1 and caspase-11 contribute to joint inflammation during Brucella infection.15 As we found that IFN-γ deficiency resulted in markedly increased joint IL-1β, (Figure S1C), we investigated whether IFN-γ restrains caspase-1/11 dependent inflammation. WT and caspase1/11−/− mice were footpad infected with Brucella and treated with anti-IFN-γ or an isotype control. At day 7 post infection, we found mice deficient in either IFN-γ or caspases-1 and 11 had increased Brucella burdens (Figure 2A). However, caspase-1/11−/− mice neutralized of IFN-γ had significantly higher Brucella joint colonization than caspase-1/11−/− mice treated with IgG, indicating the protective effect of IFN-γ on bacterial clearance is not entirely inflammasome dependent.
Figure 2. IFN-γ deficiency results in severe caspase-1/11 dependent inflammation.

WT and caspase-1/11−/− mice were treated with anti-IFN-γ or an isotype control, then infected in both rear footpads with 1×105 B. melitensis 16M A-E). Mice were euthanized at day 7 and joint CFUs and IL-1β levels were enumerated A,E). Joint swelling was measured throughout the experimental time course B). At day 7 post-infection, H&E staining on joint sections was conducted and slides were scored for inflammation C&D). Depicted are representative images (100X) from each group with amplified boxed regions (400X) displayed to their right C). Data are representative of 3 independent experiments each with 3–5 mice/group except for D), in which data was combined from 2 experiments with 10–13 mice/group. Error bars depict S.D. of the mean. Means with the same letter at the same timepoint are not statistically different from each other as determined by ANOVA followed by Tukey’s test in A,B, and E) or ANOVA on Ranks D).
Neutralization of IFN-γ from WT mice resulted in markedly enhanced joint swelling by day 3 post-infection and everyday thereafter (Figure 2B). In contrast, IFN-γ neutralization in caspase-1/11−/− mice did not result in enhanced joint swelling (Figure 2B), or inflammation (Figure 2C–D), despite the fact that joints from anti-IFN-γ treated caspase-1/11−/− mice had Brucella loads ~50 fold higher than joints from IgG-treated caspase-1/11−/− mice (Figure 2A). The finding that IFN-γ neutralization enhances inflammation in WT, but not caspase-1/11−/− mice, indicates that the increased inflammation caused by IFN-γ deficiency requires inflammasomes. In addition, neutralization of IFN-γ from WT, but not caspase-1/11−/− mice resulted in increased joint IL-1β levels (Figure 2E), suggesting that IFN-γ deficiency results in increased inflammasome activation. Enhanced caspase-1-dependent production of IL-1 and IL-18 in IFN-γ deficient animals likely contributes to joint inflammation, as IL-18/IL-1R signaling appeared to play a larger role in joint swelling in IFN-γ deficient animals than did caspase-11 (Figure S2A–B).
Brucella is known to activate caspase-1 via the AIM2 and NLRP3 inflammasomes.27,28 In order to identify what inflammasome components contribute to arthritis, and if these phenotypes are altered by IFN-γ deficiency, we treated WT and inflammasome deficient mice with anti-IFN-γ or an isotype control during Brucella footpad infection. The AIM2 inflammasome was dispensable for joint swelling (Figure S2C) and control of infection (data not shown) in the presence or absence of IFN-γ. NLRP3 contributed transiently to joint swelling in the presence of IFN-γ, with NLRP3−/− mice displaying a reduction in swelling only at day 2 post-infection (Figure 3A). Interestingly, in the absence of IFN-γ, NLRP3 deficiency significantly reduced joint swelling at every timepoint measured (Figure 3B), without affecting Brucella burdens in the joint (data not shown). In addition, we found that treatment of IFN-γ deficient animals with the NLRP3-specific inhibitor, MCC950,29 resulted in decreased joint swelling in Brucella-infected mice (Figure S2D). Thus, these data indicate that the inflammasome-dependent joint pathology that occurs in the absence of IFN-γ requires NLRP3.
Figure 3. IFN-γ deficiency results in NLRP3 dependent arthritis following focal, or systemic infection.

WT and NLRP3−/− mice (n=4–5/group) were treated with an isotype control or anti-IFN-γ, then infected in their rear footpads with 1×105 B. melitensis 16M. Rear ankle joint swelling was measured over time A-B). *P<0.05 as compared to WT mice. Error bars depict S.D. of the mean. Data are representative of 2 independent experiments. WT, caspase-1/11−/− and NLRP3−/− mice (n=10–11/group) were treated with anti-IFN-γ then infected i.p. with 1×105 B. melitensis 16M C-D). Joint swelling was measured over the time course of the experiment C), and on day 27 post-infection bacterial burdens in joint and spleen were enumerated D). Error bars depict S.D. of the mean. Means with the same letter at the same timepoint are not statistically different from each other as determined by ANOVA followed by Tukey’s test.
To determine if the effect of inflammasomes/NLRP3 was limited to footpad infection, we measured arthritis in IFN-γ neutralized WT, caspase-1/11−/− and NLRP3−/− mice infected i.p. with B. melitensis. NLRP3 and caspase1/11 deficiency resulted in significantly reduced joint swelling, while no differences in Brucella burdens in spleens or joints were observed at day 27 post-infection (Figure 3C–D). Thus, these data indicate that IFN-γ prevents inflammasome/NLRP3 dependent joint pathology regardless of the route of infection.
While humans produce IFN-γ during Brucella infection, chronic human brucellosis patients display an impaired IFN-γ response to Brucella relative to patients at the onset of disease.30 Human brucellosis patients are also more likely to have IFN-γ gene polymorphisms associated with reduced IFN-γ production.31 Thus, impaired IFN-γ production in these patients could lead to inflammasome-dependent pathology. Unregulated inflammasome activation in the absence of IFN-γ could also lead to mortality. Indeed, one study in mice demonstrated T cell or IFN-γ deficiency resulted in similarly elevated Brucella burdens following pulmonary infection; however, IFN-γ deficient mice developed neutrophilia and succumbed to infection while T cell deficient mice do not.32 This, coupled with our own data, suggests disease severity and mortality in IFN-γ deficient mice is due to elevated bacterial burdens, but also due to overwhelming pathology, possibly caused by an inability to regulate inflammasome induced inflammation.
IFN-γ-Dependent Nitric Oxide Suppresses Brucella-induced inflammasome activation
Enhanced inflammasome-dependent pathology in the absence of IFN-γ is likely due in part to higher bacterial loads in IFN-γ deficient animals, which results in increased inflammasome activation. However, to explain why mice lacking ILCs had enhanced joint swelling, despite lower levels of Brucella and IFN-γ (Figure 1C–E), we considered IFN-γ, or effectors downstream of IFN-γ, may be negatively regulating inflammasome activation. We found reduced iNOS transcript in ILC deficient mice (Figure S1H), which was of interest as IFN-γ induced nitric oxide (NO) s-nitrosylates NLRP3 to suppress inflammasome activation during Mycobacterium tuberculosis infection.19 As NLRP3 played a major role in joint pathology in Brucella-infected, IFN-γ deficient mice (Figure 3B&C), we investigated the ability of IFN-γ-dependent NO to suppress inflammasome activation and inflammation during Brucella infection.
IFN-γ neutralization decreased iNOS mRNA transcript in Brucella-infected joints at day 2 post-infection (Figure 4A) and at later timepoints (data not shown). Addition of recombinant IFN-γ to Brucella infected macrophages also resulted in NO production that could be blocked by the addition of the iNOS inhibitor, aminoguanidine hemisulfate salt (AGHS) (Figure S2E). To determine if iNOS was protective against Brucella-induced arthritis, we administered AGHS, to the animals’ drinking water, and compared these mice to those that received untreated water.19 Mice receiving AGHS displayed increased joint inflammation, IL-1β, and Brucella burdens relative to animals receiving regular drinking water (Figure S2F–H). Next, we evaluated the relative ability of iNOS and IFN-γ to control infection and suppress inflammasome-dependent IL-1β production in the joint. To do this, we footpad-infected, IgG-treated WT and iNOS deficient mice (NOS2−/−), and WT mice neutralized of IFN-γ. As early as day 3 post-infection, IgG-treated NOS2−/− mice displayed increased joint swelling relative to WT, IgG-treated mice (Figure 4B). iNOS deficiency also resulted in slightly, but significantly increased joint Brucella burdens relative to WT animals at day 7 post-infection (Figure 4C). iNOS deficiency did not increase joint bacterial levels to the same degree as IFN-γ deficiency. However, joint IL-1β levels were similarly elevated in NOS2−/− mice and IFN-γ neutralized mice (Figure 4D) despite bacterial loads being ~100-fold lower in NOS2−/− animals relative to IFN-γ deficient mice. These data indicate NO limits inflammasome-dependent inflammation and IL-1β production by mechanisms beyond simply restricting Brucella infection.
Figure 4. IFN-γ-Dependent Nitric Oxide Suppreses Brucella-induced inflammasome activation.

WT mice (n=3–4/group) were treated with either anti-IFN-γ or an isotype control, then infected in the rear footpads with 1×105 B. melitensis 16M. Mice were euthanized at day 2 post-infection along with naïve animals and RNA was isolated from ankle joints and relative iNOS transcript was measured A). Error bars depict S.D. of the mean. * P<0.05 compared to IgG-treated mice. In a separate experiment, WT mice received anti-IFN-γ while other WT mice along with NOS2−/− mice received IgG. Mice were infected in the rear footpads with 1×105 B. melitensis 16M B-D). Joint swelling was monitored B) and mice were euthanized at day 7 post-infection and joint CFUs and IL-1β were enumerated C-D). Data are from 4–5 mice/group. Means with the same letter at the same timepoint are not statistically different from each other as determined by ANOVA followed by Tukey’s test. Error bars depict S.D. of the mean. WT bone marrow derived macrophages were infected with B. melitensis 16M at an MOI of 100 for 6 hours E-G). Media containing increasing doses of SNAP was added after washing of the cells and addition of gentamicin containing media. 24 hours post-infection intracellular Brucella burdens were enumerated and IL-1β levels in cell supernatants were determined E-F). Cell supernatants and cell lysates were also subjected to Western blot G). Data is representative of 2 independent experiments with 3–5 wells/group. Means with the same letter are not statistically different from each other as determined by ANOVA followed by Tukey’s test. Error bars depict S.D. of the mean.
Brucella can infect a variety of phagocytic and non-phagocytic cells,33 however within the joint we have found myeloid cells, and in particular macrophages are the major target of Brucella (unpublished observation). Therefore, we utilized bone-marrow derived macrophages to further elucidate the relationship between NO, IFN-γ and inflammasome activation. To determine if NO can directly inhibit inflammasome activation during Brucella infection, BMDMs were treated with increasing doses of the NO donor, S-nitroso-N-acetyl-penicillamine (SNAP). NO is potently bactericidal against Brucella.34 Therefore, we treated macrophages with SNAP only after Brucella infection. Under these conditions, we found intracellular Brucella burdens were similar in vehicle and SNAP treated wells when cell lysates and supernatants were harvested 24 hours post-infection (Figure 4E). Administration of SNAP resulted in NO production (Figure S2I) and suppressed Brucella-induced IL-1β release into cell supernatant (Figure 4F). Western blots were then performed to confirm this effect was due to inhibition of caspase-1 activation. We demonstrated caspase-1 is activated by B. melitensis infection, as cleaved caspase-1 was found in the cell supernatant of infected, but not uninfected macrophages (Figure 4G). In addition, administration of SNAP reduced Brucella-induced caspase-1 activation, as supernatants from Brucella infected cells treated with SNAP contained reduced active caspase-1 relative to vehicle treated, infected cells (Figure 4G).
While it has long been known that IFN-γ restricts Brucella infection, our data here show IFN-γ also limits inflammation by inducing nitric oxide that in turn suppresses inflammasome activation and pathology. Human macrophages do not produce NO in response to IFN-γ as robustly as do murine macrophages.35 Thus, enhanced NO production relative to humans could be one of the factors that contributes to the resistance of wild-type mice to Brucella-induced arthritis. NO donors are well tolerated in humans.36 In addition, NO is antimicrobial against Brucella37 and we show here that NO can inhibit unregulated inflammasome-dependent pathology. Thus, NO donors and/or NLRP3 inhibitors, in combination with antibiotics, might be potential complementary therapeutic candidates to treat brucellosis, particularly in patients with impaired IFN-γ production.
Supplementary Material
Acknowledgements:
This work was supported in part by the National Institutes of Health (NIH R21AI119634) and funding from the University of Missouri College of Veterinary Medicine and Research Board.. IL-1R−/−/IL-18−/− mice were a gift from Dr. Edward Miao at the University of North Carolina.
Abbreviations:
- AGHS
aminoguanidine hemisulfate salt
- AIM2
absent in melanoma 2
- Ba
brucella agar
- BCA
bicinchoninic acid assay
- BMDMs
bone marrow derived macrophages
- CFU
colony forming unit
- CM
complete media
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- ILCs
innate lymphoid cells
- iNOS
inducible nitric oxide synthase
- i.p.
intraperitoneal
- LPS
lipopolysaccharide
- MOI
multiplicity of infection
- NLRP3
NACHT, LLR and PYD domains-containing protein 3
- NO
nitric oxide
- PBS
phosphate buffered saline
- SD
standard deviation
- SNAP
S-nitroso-N-acetyl-penicillamine
- WT
wild-type
Footnotes
Conflict of Interest Disclosure: The authors declare no conflicts of interest
References
- 1.Pappas G, Papadimitriou P, Akritidis N, Christou L, Tsianos EV 2006. The new global map of human brucellosis. Lancet. Infect Dis. 6:91–9. [DOI] [PubMed] [Google Scholar]
- 2.Colmenero JD, Reguera JM, Fernandez-Nebro A, Cabrera-Franquelo F 1991. Osteoarticular complications of brucellosis. Ann. Rheum. Dis 50:23–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zinsstag J, Roth F, Orkhon D, Chimed-Ochir G, Nansalmaa M, Kolar J, Vounatsou P 2005. A model of animal–human brucellosis transmission in Mongolia. Prev. Vet. Med 69:77–95. [DOI] [PubMed] [Google Scholar]
- 4.Kaufmann AF, Fox MD, Boyce JM, Anderson DC, Potter ME, Martone WJ, Patton CM 1980. Airborne spread of brucellosis. Ann. NY Acad. Sci 353:105–14. [DOI] [PubMed] [Google Scholar]
- 5.Rajapakse CN 1995. Bacterial infections: osteoarticular brucellosis. Bailliere’s Clin. Rheum 9:161–77. [DOI] [PubMed] [Google Scholar]
- 6.Gotuzzo E, Alarcon GS, Bocanegra TS, Carrillo C, Guerra JC, Rolando I, Espinoza LR 1982. Articular involvement in human brucellosis: a retrospective analysis of 304 cases. Semin. Arthritis. Rheum 112:245–55. [DOI] [PubMed] [Google Scholar]
- 7.Martin-Hernandez CB-JJ, Espallargas-Doñate T, Fuertes-Vallcorba A. 2009. Arthroscopic synovectomy, an alternative in the treatment of brucellar arthritis of the knee with prolonged course. A report of two cases. Int. J Orthopedic. Surg 13. [Google Scholar]
- 8.Khateeb MI, Araj GF, Majeed SA, Lulu AR 1990. Brucella arthritis: a study of 96 cases in Kuwait. Ann. Rheum. Dis 49:994–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Press J, Peled N, Buskila D, Yagupsky P 2002. Leukocyte count in the synovial fluid of children with culture-proven brucellar arthritis. Clin. Rheum 21:191–3. [DOI] [PubMed] [Google Scholar]
- 10.Ayaşlıoğlu E, Özlük Ö, Kılıç D, Kaygusuz S, Kara S, Aydın G, Çokca F, Tekeli E A case of brucellar septic arthritis of the knee with a prolonged clinical course. Rheumatol. Internat 25:69–71. [DOI] [PubMed] [Google Scholar]
- 11.Skyberg JA, Thornburg T, Kochetkova I, Layton W, Callis G, Rollins MF, Riccardi C, Becker T, Golden S, Pascual DW 2012. IFN-gamma-deficient mice develop IL-1-dependent cutaneous and musculoskeletal inflammation during experimental brucellosis. J. Leukoc. Biol 92:375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lacey CA, Keleher LL, Mitchell WJ, Brown CR, Skyberg JA 2016. CXCR2 Mediates Brucella-Induced Arthritis in interferon gamma-Deficient Mice. J.Infect. Dis 214:151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weizman O-E, Adams NM, Schuster IS, Krishna C, Pritykin Y, Lau C, Degli-Esposti MA, Leslie CS, Sun JC, O’Sullivan TE 2017. ILC1 confer early host protection at initial sites of viral infection. Cell. 171:795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abt MC, Lewis BB, Caballero S, Xiong H, Carter RA, Sušac B, Ling L, Leiner I, Pamer EG 2015. Innate immune defenses mediated by two ILC subsets are critical for protection against acute Clostridium difficile infection. Cell Host Microb. 18:27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lacey CA, Mitchell WJ, Dadelahi AS, Skyberg JA (2018) 2018. Caspase-1 and caspase-11 mediate pyroptosis, inflammation, and control of Brucella joint infection. Infect. Immun 86(9). e00361–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rathinam VA and Fitzgerald KA 2016. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell. 165:792–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA (2013) 2013. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science 341:1250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kayagaki N, Warming S, Lamkanfi M, Walle LV, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S 2011. Non-canonical inflammasome activation targets caspase-11. Nature. 479:117. [DOI] [PubMed] [Google Scholar]
- 19.Mishra BB, Rathinam VA, Martens GW, Martinot AJ, Kornfeld H, Fitzgerald KA, Sassetti CM 2013. Nitric oxide controls the immunopathology of tuberculosis by inhibiting NLRP3 inflammasome-dependent processing of IL-1beta. Nat. Immunol 14:52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lacey CA, Mitchell WJ, Brown CR, Skyberg JA 2017. Temporal Role for MyD88 in a model of Brucella-induced arthritis and musculoskeletal inflammation. Infect. Immun 85(3)e00961–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T 2011. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol 12:1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmittgen TD and Livak KJ 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc 3:1101. [DOI] [PubMed] [Google Scholar]
- 23.Skyberg JA, Thornburg T, Rollins M, Huarte E, Jutila MA, Pascual DW 2011. Murine and bovine gammadelta T cells enhance innate immunity against Brucella abortus infections. PloS One. 6:e21978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP 1999. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Ann. Rev. Immunol 17:189–220. [DOI] [PubMed] [Google Scholar]
- 25.Klose CS and Artis D 2016. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol 17:765–774. [DOI] [PubMed] [Google Scholar]
- 26.Seo GY, Shui JW, Takahashi D, Song C, Wang Q, Kim K, Mikulski Z, Chandra S, Giles DA, Zahner S Kim PH, Cheroutre H, Colonna M, Kronenberg M 2018. LIGHT-HVEM signaling in innate lymphoid cell subsets protects against enteric bacterial infection. Cell Host Microbe. 24:249–60. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomes MT, Campos PC, Oliveira FS, Corsetti PP, Bortoluci KR, Cunha LD, Zamboni DS, Oliveira SC 2013. Critical role of ASC inflammasomes and bacterial type IV secretion system in caspase-1 activation and host innate resistance to Brucella abortus infection. J. Immunol 190:3629–3638. [DOI] [PubMed] [Google Scholar]
- 28.Marim FM, Franco MMC, Gomes MTR, Miraglia MC, Giambartolomei GH, Oliveira SC 2017. The role of NLRP3 and AIM2 in inflammasome activation during Brucella abortus infection. Semin. Immunopath 39: 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Periasamy S, Le HT, Duffy EB, Chin H, Harton JA 2016. Inflammasome-independent NLRP3 restriction of a protective early neutrophil response to pulmonary tularemia. PLoS Path. 12, e1006059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rafiei A, Ardestani SK, Kariminia A, Keyhani A, Mohraz M, Amirkhani A 2006. Dominant Th1 cytokine production in early onset of human brucellosis followed by switching towards Th2 along prolongation of disease. J. Infect 54:315–324. [DOI] [PubMed] [Google Scholar]
- 31.Eskandari-Nasab E, Moghadampour M, Hasani SS, Hadadi-fishani M, Mirghanizadeh-Bafghi SA, Asadi-Saghandi A, Zare F, Sadeghi-Kalani B, Ghazali-bina M 2013. Relationship between gamma-interferon gene polymorphisms and susceptibility to brucellosis infection. Microbiol. Immunol 57:785–91. [DOI] [PubMed] [Google Scholar]
- 32.Hanot Mambres D, Machelart A, Potemberg G, De Trez C, Ryffel B, Letesson JJ, Muraille E 2016. Identification of immune effectors essential to the control of primary and secondary intranasal infection with Brucella melitensis in mice. J. Immunol 196:3780–93. [DOI] [PubMed] [Google Scholar]
- 33.Corbel MJ 1997. Brucellosis: an overview. Emerg. Infect. Dis 3:213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gross A, Spiesser S, Terraza A, Rouot B, Caron E, Dornand J 1998. Expression and bactericidal activity of nitric oxide synthase in Brucella suis-infected murine macrophages. Infect. Immun 66:1309–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Albina JE 1995. On the expression of nitric oxide synthase by human macrophages. Why no NO? J. Leukoc. Biol 58:643–9. [DOI] [PubMed] [Google Scholar]
- 36.Knox CD, de Kam PJ, Azer K, Wong P, Ederveen AG, Shevell D, Morabito C, Meehan AG, Liu W, Reynders T, Denef JF, Mitselos A, Jonathan D, Gutstein DE, Mitra K, Sun SY, Lo MM, Cully D, Ali A 2016. Discovery and clinical evaluation of MK‐8150, a novel nitric oxide donor with a unique mechanism of nitric oxide release. J. Am. Heart. Assoc 2016;5:e003493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang X, Leonard B, Benson R, Baldwin CL 1993. Macrophage control of Brucella abortus: role of reactive oxygen intermediates and nitric oxide. Cell. Immunol. 151:309–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.