

Abstract
Cellular metabolism is a means of generating ATP to provide energy for key cellular functions. However, recent research shows that citric acid cycle intermediates target vital cellular functions of the innate immune system. Succinate, itaconate, citrate and fumarate have been shown to mediate or regulate important myeloid cell functions during infection and inflammation. This review covers the regulatory functions of citric acid cycle intermediates in myeloid cells and discusses potential translational applications, key mechanistic questions and future research directions.
Keywords: Metabolic reprogramming, succinate, itaconate, citrate, fumarate
Summary Sentence:
This review outlines current understanding of inflammation-induced alterations in leukocyte metabolism and implications for leukocyte function and novel therapeutic development.
Graphical Abstract
Introduction
Traditionally, the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) have been recognized as dynamic cellular functions that support cellular energy demands. Interestingly, metabolic intermediates of the TCA cycle have recently been shown to play an important role in regulating innate immune cell responses [1, 2]. Activation of innate leukocytes, such as macrophages and monocytes, with pro-inflammatory stimuli, such as lipopolysaccharide (LPS), leads to remodeling of the TCA cycle and intracellular accumulation of metabolic intermediates such as succinate, itaconate, citrate and fumarate, which link cellular metabolism to innate leukocyte responses [3, 4]. The major focus of this review is to highlight the role of TCA cycle metabolites, namely succinate, itaconate, citrate and fumarate in rewiring innate immune cell metabolism, to discuss their translational relevance, and provide novel hypotheses for targeted therapeutics in inflammatory conditions. The following sections will describe in detail the role of each of these metabolites in leukocyte regulation and function.
Succinate
Succinate undergoes oxidation to fumarate via SDH in the TCA cycle and functions as a key intermediate of the mitochondrial electron transport chain complex II, thus providing a link between the TCA cycle and the mitochondrial electron transport chain (Figure 1). Intracellular accumulation of succinate has been shown to be a hallmark of activated macrophages. Tannahill et al., demonstrated that LPS stimulation leads to significant succinate accumulation within macrophages, with associated enhancement of IL-1β production [1]. Succinate has been shown to reduce the expression of interleukin-1 receptor antagonist (IL-1RA) and IL-10, in LPS-treated macrophages [5]. The major mechanisms postulated for succinate’s pro-inflammatory effects include stabilization of HIF-1α, enhancement of mitochondrial reactive oxygen species (mROS) generation, post-translational modification of other proteins (via succinylation) and signaling via its G-protein coupled receptor.
Figure 1. Role of succinate and itaconate in modulating myeloid cell functions.

Inflammatory activation on myeloid cells leads to accumulation of succinate and itaconate in the mitochondria. High oxidation levels of succinate in TCA cycle favors reverse electron transport, from complex II (CII or SDH) to complex I (CI), generating elevated mitochondrial reactive oxygen species (mROS). Mitochondrial succinate is transported into the cytosol, where it stabilizes HIF-1α along with mROS. HIF-1α acts to increase glycolysis and proinflammatory cytokine secretion including IL-1β and IL-6. Succinate also causes post-translational modification of proteins (succinylation). Succinate is also transported into extracellularly, where it can act on its receptor, SUCNR1, which further fuels inflammatory response. Citrate is diverted away from TCA cycle to produce Itaconate via immunoregulatory gene 1 (IRG1) enzyme through the intermediate, cis-aconitate. This is accomplished via a disruption in the TCA cycle at the level of α-ketoglutarate synthesis. Itaconate inhibits SDH and limits the proinflammatory effects of succinate. Itaconate is also transported into cytosol via an unknown carrier protein, where it activates Nrf2 via alkylation of Keap1. Itaconate can also be transported extracellularly via unknown mechanisms, where it can kill pathogens and improve microbial clearance. Aspartate arginosuccinate shunt fuels the TCA cycle through replenishing fumarate levels.
(i). Succinate induced HIF-1α stabilization
HIF-1α is recognized as a key mediator of a wide range of cellular responses to hypoxia and its signaling pathway affects numerous processes including metabolic adaptation, angiogenesis, erythropoiesis, cell growth and survival [6]. In terms of metabolic reprogramming of activated myeloid cells, HIF-1α facilitates a metabolic shift from oxidative phosphorylation to glycolysis and sustains glycolytic capacity through induction of key target genes [7]. Absence of HIF-1α in myeloid cells results in profound metabolic defects leading to impaired aggregation, motility, invasiveness, and bacterial killing capacity [7]. HIF-1α is normally degraded under normoxic conditions, owing to hydroxylation by cytosolic prolyl hydroxylase (PHD). PHD hydroxylate the conserved proline residues on the HIF-1α subunit and mark it for ubiquitin-proteasome mediated degradation. During hypoxic conditions, prolyl hydroxylation of HIF-1α residues is suppressed, and HIF-1α is protected from further degradation. Accumulated HIF-α then dimerizes with HIF-1β, which translocates to the nucleus, leading to transcription of target genes required for metabolic adaptation [8]. HIF-1α stabilization is an important adaptive mechanism for myeloid cell functions at the sites of inflammation where oxygen concentrations are low, and cells undergo a metabolic switch to glycolysis. Treatment of macrophages with LPS results in significant succinate accumulation along with an increased expression of HIF-1α protein [1]. During inflammatory activation of myeloid cells, increased HIF-1α expression also drives increased expression of IL-1β and promotes inflammation [9]. Thus, succinate stabilizes HIF-1α even under normoxic conditions in myeloid and acts as an important inflammatory signal (Figure 1). HIF-1α has also been shown to be increased in macrophages derived from inflamed synovium of rheumatoid arthritis patients, showing the importance of HIF-1α activation in other inflammatory contexts as well [10].
It is important to note the differential effect of succinate on immune cell cytokine responses. In LPS treated macrophages, succinate reduces expression of anti-inflammatory cytokines, interleukin-1 receptor antagonist (IL-1RA) and IL-10 [1, 11]. This perplexing finding demonstrates the specificity of succinate for increasing LPS-induced IL-1β, without affecting IL-6 and TNF-α production. Production of IL-1β is usually a two-step process, whereby a pathogen associated molecular patterns (PAMP) such as LPS induces pro-IL-1β, which is further cleaved by caspase-1 to IL-1β. Caspase-1 activation is mediated by cytosolic inflammasomes, which are intracellular pattern recognition receptors (PRR). Interestingly, succinate alone does not induce either pro-IL-1β or IL-1β, rather it enhances LPS-induced production of IL-1β. Taken together, it is reasonable to hypothesize that succinate may be acting on intracellular PRR to differentially regulate intracellular IL-1β response. These findings bring to light a thought-provoking concept that although TCA cycle intermediates, such as succinate, do not directly act through PRR’s, they may still elicit pronounced intracellular responses when administered with a PAMP. The exact mechanism for the differential cytokine response induced by succinate is not yet clear and opens an interesting avenue for future studies. It is also important to note that succinate can act through its own cell surface receptor, SUCNR1 (G protein-coupled receptor, also termed as GPR91).
(ii). Role of succinate dehydrogenase (SDH) and mitochondrial ROS (mROS)
Mills et al., demonstrated that intact function of SDH is critical to sustain the pro-inflammatory response of LPS-activated macrophages [11]. Treatment with a specific SDH inhibitor, dimethyl malonate (DMM), increased succinate levels, decreased HIF-1α expression, and inhibited the expression of genes associated with inflammation. DMM boosted the expression of anti-inflammatory genes, both in vitro (LPS- stimulated macrophages) and in vivo (LPS- treated mice). DMM is a cell permeable precursor of malonate and it is rapidly metabolized within the cells to malonate, which is a competitive inhibitor of SDH [12]. These findings are in contrast to those reported by Tannahill et al., which showed that the ‘SDH inhibitor’, diethylbutylmalonate, increased LPS-induced HIF-1α and IL-1β expression. However, diethylbutylmalonate blocks mitochondrial succinate transport, rather than directly inhibiting SDH, thereby increasing intracellular succinate levels. Mills et al., further showed that LPS treatment leads to increased mitochondrial membrane potential in macrophages. Increased mitochondrial membrane potential and SDH activity collectively enhance ROS production [13]. LPS treatment led to a significant increase in mitochondrial ROS production, which was inhibited by DMM, but boosted by succinate [11]. The same study demonstrated the importance of mROS for succinate-induced IL-1β production because mROS scavengers inhibited the effect of succinate on LPS-induced pro-IL-1β and HIF-1α. Furthermore, the complex I inhibitor rotenone significantly decreased LPS-induced ROS and IL-1β mRNA expression, implying a role for complex I in ROS generation. Increased oxidation of accumulated succinate to fumarate by SDH along with higher mitochondrial membrane potential drives increased mROS generation through reverse electron transport (RET) from complex II to complex I (Figure 1). Rapid oxidation of succinate by SDH causes over-reduction of coenzyme Q (CoQ), limiting its availability to accept forward electrons from SDH (complex II), which then flow backwards (RET) towards complex I, leading to increased generation of superoxide radicals [12]. Mills et al., further showed macrophages overexpressing alternative oxidases (AOX) have impaired mROS production and reduced LPS-induced IL-1β and HIF-1α production upon treatment with succinate [11]. AOX quenches excess electrons generated from rapid succinate oxidation by SDH, thereby limiting RET and mROS generation. Importantly, ROS has also been shown to indirectly stabilize HIF-1α by limiting PHD activity [14]. Garaude et al., reported that inhibition of SDH with 3-nitorpropionic acid not only impairs bactericidal activity of bone marrow-derived macrophages in vitro, but also impairs systemic bacterial clearance in a model of E. coli peritonitis [15]. These results show that LPS-induced and mROS-generated redox signals in macrophages caused by high succinate oxidation are pivotal for generation of the inflammatory macrophage phenotype. Mitochondria are the major sites of cellular ROS generation, which may aid in anti-bacterial responses of myeloid cells. In line with this, TLR signaling has been shown to recruit mitochondria to macrophage phagosomes and to augment mROS production, which facilitates killing of phagocytosed microbes [16]. It is reasonable to hypothesize that active SDH in the setting of high intracellular succinate accumulation upon LPS stimulation will support generation of high levels of mROS via RET. It has been shown that ischemic injury leads to high levels of succinate accumulation in major tissues including liver, heart, kidney and brain [12]. The accumulated succinate drives excessive mROS production during reperfusion via its SDH oxidation which drives RET at complex I. Pharmacological inhibition of succinate accumulation ameliorated mROS generation and injury in mouse models of stroke and myocardial infarction. It is also important to note that complex I plays a critical role in generating mROS during RET, which depends on high succinate accumulation [12]. However, detailed in vivo studies analyzing the impact of SDH on ROS generation and consequent immunomodulation in animal models of inflammation are currently lacking. It is reasonable to hypothesize that intact SDH and complex I activity would have a significant impact on myeloid cell functions and would play a critical role in immunomodulation.
(iii). Succinylation
Succinylation is a protein post-translational modification (PTM), first described by Zhang et al., to modify the isocitrate dehydrogenase enzyme of the TCA cycle [17]. The sirtuin enzyme, SIRT5, is responsible for desuccinylation [18]. Since then, other proteins impacting cellular metabolism including pyruvate kinase, pyruvate dehydrogenase complex, and succinate dehydrogenase have been shown to be succinylated, but the specific mechanism of succinylation remains unclear [18]. Tannahill et al., showed that treatment of macrophages with LPS leads to succinylation of malate dehydrogenase, glutamate carrier 1, L-lactate dehydrogenase A chain and transaldolase, but the functional impact of such a PTM in relation to LPS induced inflammatory cytokine secretion is not yet clear [1]. It is possible that succinylation of these and other proteins may play a direct or indirect role in regulating immune cell responses. A unique example of succinylation in relation of inflammatory cytokines is the role of Pyruvate Kinase M2 (PKM2) enzyme and SIRT5. [18]. Wang et al., demonstrated that LPS stimulation of SIRT5 deficient macrophages induced high levels of pro-inflammatory cytokine transcripts including IL-1β, IL-6 and TNF-α, along with hyper-succinylation of PKM2. Pharmacological activation of PKM2 in SIRT5 KO macrophages lead to inhibition of LPS-induced IL-1β, with no effect on IL-6 and TNF-α [19]. LPS-induced succinylation of PKM2 promotes its translocation to the nucleus, where it formed a complex that directly bound the IL-1β promoter and induced its transcription [9, 19]. Furthermore, Tannahill et al., showed that LPS treatment increased intracellular succinate accumulation and downregulatesdSIRT5 expression, which may lead to increased succinylation of endogenous target proteins, such as PKM2, leading to its inactivation [1]. Therefore, in the case of PKM2, it is reasonable to hypothesize that LPS treatment hyper-succinylates PKM2 and predominantly induces its inactive monomeric form due to reduction in SIRT5 activity, leading to a pro-inflammatory macrophage phenotype. Currently, there are no studies which demonstrate cytokines as a direct target for succinylation. This opens a novel field of investigation which needs thoughtful experimental analysis. Overall increases in succinylation due to increased succinate accumulation and decreased SIRT5 activity, may be important for sustaining LPS-induced metabolic adaptation in activated macrophages. In contrast, Park et al., demonstrated that succinylation favors increased activity of pyruvate dehydrogenase and succinate dehydrogenase leading to increased mitochondrial respiration, as SIRT5 knock-down resulted in enhanced succinylation and activity of these enzymes [18]. However, it must be noted that studies conducted by Park et al., measured SDH activity in SIRT5 knock-out mouse embryonic fibroblast cells and hepatocytes, without LPS stimulation. On the other hand, studies demonstrating LPS-induced decline in SDH activity have focused on macrophages in which itaconate production is induced leading to direct inhibition of SDH activity [2]. Our recent studies show that the TLR4 agonist, MPLA (24 hours post-stimulation) suppresses OXPHOS followed by significant recovery over time (three days post-stimulation) [20]. Based on these findings, it can be hypothesized that stimulation with TLR4 agonists initially suppresses mitochondrial respiration, but increased succinylation of target proteins such as SDH along with modulation of itaconate levels and desuccinylation of PKM2 via recovery of SIRT5 may play a role in delated recovery of OXPHOS.
(iv). Role of succinate signaling via succinate receptor, SUCNR1
SUCNR1 is a G protein-coupled receptor, also termed GPR91 [21]. SUCNR1 is expressed in various tissues (liver, spleen, kidney, small intestine) and cells (dendritic cells and macrophages) [21–23]. Apart from demonstrating the expression and functional significance of SUCNR1 in macrophages and dendritic cells, few studies have elucidated the exact expression levels of SUCNR1 in other immune cells. Rubic et al., showed that SUCNR1 mRNA is expressed at a very low or undetectable level in human T cells, B cells and CD14+ monocytes [22]. Succinate administration has been shown to increase the number of circulating neutrophils and platelets, presumably via activation of SUCNR1 on hematopoietic progenitor cells leading to their proliferation [24]. Apart from this finding, expression of SUCNR1 on neutrophils has not been studied.
Rubic et al., demonstrated that monocyte-derived dendritic cells express high levels of SUCNR1 and succinate administration activates extracellular signal-regulated kinases Erk1 and Erk2 [22]. Rubic et al., further showed that succinate synergizes with TLR3 and TLR7 ligands to increase production of pro-inflammatory cytokines such as IL-1β and TNF-α from dendritic cells. In non-immune cells, it has been shown that signaling through SUCNR1 could be mediated by either multiple intracellular G protein mediators including Gi, Go or Gs depending on the cell type involved. For example, in kidney cells, succinate increases intracellular calcium, ERK 1/2 activation and a decrease in cyclic AMP levels (effects of Gi or Gq signaling), whereas in cardiomyocytes it increases cyclic AMP levels suggesting involvement of Gs [25]. Detailed description of these signaling pathways is beyond the scope of this review article. The exact downstream signaling mechanism of SUCNR1 in macrophages remains to be investigated. Rubic et al., demonstrated that succinate induces dose-dependent alterations in dendritic cell function including chemotaxis, pro-inflammatory cytokine production in synergy with TLR3 (poly I:C) and TLR7 (imiquimod) agonists, and enhanced T cell activation [22]. LPS treatment of macrophages leads to significant release of succinate into the extracellular milieu [23]. Extracellular succinate potentiated LPS-induced HIF-1α expression and IL-1β production, whereas macrophages lacking SUCNR1 showed attenuated IL-1β release upon LPS stimulation [23]. Using a mouse arthritis model, this study also showed that succinate accumulates in high levels within synovial fluid and joint resident macrophages expressing SUCNR1 play a pivotal role in IL-1β secretion and inflammation [23]. SUCNR1 deficiency led to a significant decrease in knee swelling. It is reasonable to hypothesize that under conditions of systemic and local inflammation/infection, succinate released from activated macrophages can act in both an autocrine and paracrine manner to promote inflammation. Therefore, development of SUCNR1 antagonists might be a novel approach to treat inflammatory conditions. Bhuniya et al., have identified selective antagonists of SUCNR1 and a compound called ‘4c’ has been shown to be the most effective antagonist [26]. Furthermore, ‘5g’ and ‘7e’, compounds, structurally related to 4c, are active upon oral administration. Nevertheless, the effectiveness of SUCNR1 antagonists in inflammatory conditions requires further investigation.
Itaconate
Itaconate (or itaconic acid) is also known as methylene succinic acid is an organic, unsaturated dicarboxylic acid. It was first discovered by the Swiss chemist Samuel Baup during distillation of citric acid in 1836, followed by Crasso’s discovery of its synthesis via decarboxylation of cis-aconitic acid in 1940 [27]. Michelucci et al., demonstrated that production of itaconate from cis-aconitate is catalyzed by immunoresponsive gene 1 (Irg1), also known as aconitate decarboxylase 1 (Acod1), in mouse and human leukocytes [28]. Irg1 is highly upregulated upon inflammatory activation of peritoneal macrophages, and itaconate is present in blood of human sepsis patients [29]. Recent studies have shown itaconate plays an important role during metabolic reprogramming of myeloid cells and it also possesses antimicrobial properties (Figure 1).
(i). Itaconate production during leukocyte metabolic reprogramming
Jha et al., reported specific break points in the TCA cycle of activated M1 macrophages, specifically a seven-fold downregulation of isocitrate dehydrogenase 1 (IDH1) and increased accumulation of citrate [30]. IDH1 is responsible for conversion of citrate to alpha-ketoglutarate (αKG). Therefore, activation of macrophages leads to accumulation of citrate which is then diverted towards fatty acid biosynthesis and production of itaconate, through the intermediate metabolite, cis-aconitate, which is decarboxylated by IRG1/Acod1 to synthesize itaconate. Itaconate represents one of the most abundant metabolites accumulated intracellularly upon LPS treatment of M1 macrophages and dendritic cells [2, 28, 31, 32]. A time course study by Micchelucci et al., using RAW 264.7 cells showed that significant levels of intracellular itaconate were measurable at 6 hours after LPS stimulation. Itaconate levels continued to increase reaching a peak concentration of approximately 8mM at 6 to10 hours, followed by a gradual decline to approximately 3.5mM after 18 hours of LPS stimulation [28]. Cordes et al., demonstrated that LPS stimulation of RAW 264.7 macrophages led to a gradual intracellular accumulation of itaconate and succinate in parallel, as measured hourly over 6 hours, without a change in citrate, malate, fumarate, and αKG concentrations [33]. The maximum concentrations of itaconate and succinate at 6 hours after stimulation reached approximately 5mM and 1.5mM respectively. A recent study by Zhu et al., using THP-1 cells demonstrated that itaconate levels peak at 8 hours after LPS stimulation, remain elevated up to 24 hours and gradually decline until 96 hours [34]. Long term temporal alterations in itaconate production during inflammatory conditions such as systemic infections is currently lacking. Our knowledge regarding the role of itaconate in immune cell responses is currently limited to macrophages alone. Further studies are needed to understand if itaconate also plays in role in inflammatory and anti-microbial functions of other immune cells such as neutrophils, dendritic cells, and other organ systems.
(ii). Anti-inflammatory mechanisms of itaconate
Recent studies show that itaconate acts as an anti-inflammatory metabolite and opposes the inflammatory actions of succinate [2]. Lampropoulou et al., showed that pre-treatment of LPS-activated macrophages with dimethyl itaconate (the presumed cell permeable form of itaconate), suppressed pro-inflammatory mediators including nitric oxide, IL-6, IL-1β, IL-18, and also downregulated a spectrum of pro-inflammatory transcripts along with decreased expression of NLRP3 and ASC inflammasome components. These effects were found to be absent in Irg1 deficient macrophages [2]. Li et al., showed that induction of IRG1 significantly inhibited the LPS-driven production of the inflammatory cytokines including TNF-α, IL-6, and IFN-β in LPS-tolerized macrophages [35]. Furthermore, the Li and colleagues reported that the inhibitory effect of IRG1 on pro-inflammatory cytokine expression is through induction of ROS mediated expression of A20 protein, thereby implicating A20 as a crucial mediator of IRG1 function in LPS-tolerized macrophages. A20 protein has been shown to negatively regulate NLRP3 inflammasome activation and IL-1β secretion. Walle et al., demonstrated that LPS stimulation of macrophages lacking A20 produce significantly higher NLRP3 inflammasome mediated IL-1β [36]. Another study by Voet et al., also showed that A20 deficiency in microglia, hyperactivates NLRP3 inflammasome causing enhanced IL-1β secretion and central nervous system inflammation upon LPS administration in mice [37]. Therefore, it is reasonable hypothesize that IRG1 induction inhibits inflammasome activation through negative regulation mediated by A20.
Studies by Jha et al., demonstrated that macrophages deficient in IRG1 have lower levels of succinate accumulation after LPS stimulation, as compared to wild type macrophages [30]. Cordes et al., showed that exogenous itaconate treatment dose dependently inhibits SDH activity leading to increased succinate accumulation in LPS-treated macrophages [33]. Lampropoulou et al., also showed itaconate blocks SDH activity in LPS-activated macrophages and reasoned that SDH inhibition decreases oxidation of accumulated succinate, thereby limiting RET from SDH to complex I [2]. These findings implicate inhibition of SDH as an important mechanism through which itaconate acts to inhibit the pro-inflammatory effects induced by accumulated succinate (Figure 1).
A second important mechanism underlying the anti-inflammatory action of itaconate is the activation of the transcription factor, nuclear respiratory factor 2 (Nrf2, also known as NFE2L2) (Figure 1). Under basal conditions, KEAP1 protein associates with Nrf2 and leads to its degradation. Mills et al., showed that treatment of macrophages with cell permeable 4-octyl itaconate (OI) significantly increased LPS-induced Nrf2 levels via alkylation of key cysteine residues of the KEAP1 protein [38]. This was associated with increased expression of downstream anti-inflammatory target genes including heme oxygenase 1 (HMOX1). OI also stimulated the synthesis of glutathione (a major anti-oxidant). Importantly, Nrf2 activation was shown to be independent of SDH inhibition, as dimethyl malonate (a potent SDH inhibitor) did not affect Nrf2 levels. Nrf2 stabilization by OI also lead to decreased production of LPS-induced IL-1β transcript, pro-IL-1β, HIF-1α and IL-10 protein levels. Interestingly, OI also decreased IL-1β and TNF-α in human PBMCs. In another study, Bambouskova et al., showed that dimethyl itaconate (DMI) treatment induced both Nrf2 dependent and independent anti-inflammatory responses along with induction of electrophilic and xenobiotic stress pathways [39]. DMI treatment induced Nrf2 levels and downstream targets of Nrf2, including increased expression of HMOX1 and Nqo1 proteins. LPS treatment of macrophages also increased endogenous itaconate levels and Nrf2 expression, and this effect was limited in Irg1 knockout macrophages. Another important mechanism uncovered by this study demonstrated that DMI prevented the induction of IκBζ and its target genes, via inhibition of IκBζ translation by inactivating the initiation factor eIF2α. Bambouskova et al., identified ATF3 as a critical mediator of Nrf2-independent effects of itaconate, as DMI failed to block IκBζ and IL-6 induction in ATF3-deficient macrophages, despite inducing Nrf2. Overall, these studies lay the foundation for further exploration of the mechanistic basis for the anti-inflammatory potential of itaconate.
(iii). Antimicrobial properties of itaconate
Itaconate is known to inhibit the glyoxylate shunt. The glyoxalate shunt is necessary for bacteria to survive under glucose limiting conditions such as infection [27, 40]. The glyoxylate shunt is thought to serve as an alternative metabolic pathway to enable bypass of TCA cycle metabolism under conditions of limited carbon source availability [41]. Isocitrate lyase (ICL) is the first rate limiting enzyme in the glyoxylate shunt, and ICL converts isocitrate to succinate and glyoxylate, which ultimately are converted to malate and oxaloacetate for gluconeogenesis [41]. Itaconate acts as a competitive inhibitor of ICL, effectively shutting down the function of the glyoxylate shunt and inhibiting bacterial growth [27, 42]. Within the context of infection, a study by Shin et al., showed the presence of itaconate in the lung tissue of Mycobacterium tuberculosis infected mice, but failed to identify the exact source of itaconate (host vs bacteria) [43]. Michelucci et al., showed that IRG1-deficient macrophages have reduced intracellular itaconate levels and severely compromised antibacterial activity against Salmonella enteretica [28]. They also showed that addition of itaconate to the growth medium significantly affected the growth of Salmonella enteretica and Mycobacterium tuberculosis in a dose dependent manner. Exogenous itaconate has been shown to inhibit the growth of other pathogens including Staphylococcus aureus, Legionella pneumophilia, and Acinetobacter baumannii in in vitro studies [44]. Interestingly, certain pathogens such as Yersinia pestis and Pseudomonas aeruginosa can degrade and detoxify itaconate [45].
The dose of itaconate used for studying bactericidal action in vitro varies among studies and it is important to correlate these doses with the actual cellular concentrations that can be achieved upon cellular stimulation. Intracellular concentration of itaconate in LPS-treated macrophages have been observed in the range of 40 μM to 8 mM; whereas itaconate in the concentration of 5 mM to 100 mM has been used to evaluate its bactericidal effect in in vitro studies [46]. The average concentration of itaconate which most effectively inhibited growth of organisms was 10 mM, which is in close proximity to levels achieved within macrophages. Naujoks et al., showed that overexpressed IRG1 localizes to the mitochondria, which are closely associated with intracellular Legionella pneumophilia-containing vacuoles of macrophages [44]. It is reasonable to hypothesize that itaconate might be actively transported out of the mitochondria into the restricted pathogen-containing vacuoles, thereby achieving high itaconate concentrations, which can effectively restrict pathogen survival. Sugimoto et al., showed that LPS stimulation of RAW 264.7 macrophages releases itaconate into extracellular milieu, implying that itaconate is actively secreted by activated cells [47]. Therefore, in addition to intracellular accumulation, activated macrophages also secrete itaconate extracellularly. In the context of systemic infections, this might have significant implications as extracellular itaconate might play a role in restricting pathogen burden, owing to its antimicrobial action.
Shin et al., measured itaconate levels in the whole lung tissue at 4 weeks after Mtb infection [43]. However, the source of itaconate was not evaluated in this study. Innate leukocytes have been shown to be the major producers of itaconate and production of itaconate by lung parenchymal cells is yet to be evaluated. Nair et al., demonstrated that Irg1−/− deletion in a mouse model of Mtb led to increased bacterial burden and pro-inflammatory cytokine concentrations in the lung, severe lung injury and increased lethality [48]. Interestingly, the defect in controlling Mtb infection in Irg−/− mice is independent of isocitrate lyase 1 activity and a functional glyoxalate shunt. This study highlighted that increased number of infiltrating neutrophils in Irg−/− mice produced excessive mROS and iNOS, indicating their heightened inflammatory state due to the absence of Irg1. Importantly, in vivo depletion of neutrophils in Irg1−/− mice reduced lung bacterial burden and lung injury, attenuated pro-inflammatory cytokine responses and prolonged survival after Mtb infection. Therefore, in a mouse model of Mtb infection it seems that Irg1 and itaconate act to dampen the neutrophil-mediated inflammation in the lung and better control the infection. Micchelucci et al., showed that human monocyte derived macrophages produce less itaconate as compared to mouse macrophages (60μM vs 8mM respectively) [28]. Itaconate production by neutrophils has not been evaluated. It is possible that in case of clinical Mtb infection in humans, sufficient levels of itaconate might not be attained which is required to effectively contain active and latent infection. Therefore, novel strategies to augment endogenous IRG1 activity to promote itaconate production might mitigate inflammatory pathology in infections such as Mtb. The clinical outcome will also depend on the immune status of the patient and severity of the infection.
Citrate
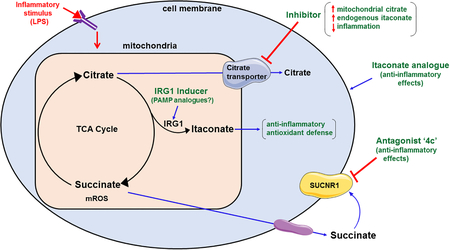
Citrate is produced in the TCA cycle via combination of oxaloacetate and acetyl CoA (Figure 2). Citrate is normally converted into isocitrate through the intermediate cis-aconitate by aconitase. Isocitrate is then decarboxylated to generate α-ketoglutarate (αKG) via IDH. As shown in Figure 2, citrate can be transported from the mitochondria into the cytosol via the mitochondrial citrate carrier (CIC, also known as Slc25a1), in exchange for malate [49]. In the cytosol, citrate can be metabolized to generate acetyl-CoA and oxaloacetate via the action of cytosolic ATP citrate lyase enzyme [49]. Jha et al., reported that activated macrophages exhibit significant downregulation of IDH, and showed significant intracellular accumulation of citrate [30], which is important for metabolic adaptation of activated myeloid cells [50]. Accumulated citrate serves as the precursor to itaconate, which then orchestrates numerous changes during metabolic reprogramming (Figure 1).
Figure 2. Role of Citrate in metabolic reprogramming of myeloid cells.

Citrate accumulates intracellularly upon myeloid cell activation. Accumulated citrate is transported into the cytosol via mitochondrial citrate carrier (CIC). In the cytosol, citrate is broken down into oxaloacetate and acetyl-CoA. Oxaloacetate is converted to malate via and malate is further converted to pyruvate leading to generation of NADPH as a by-product. NADPH is utilized by NADPH oxidase and inducible nitric oxide synthase to generate reactive oxygen and nitrogen species, and PGE2. The majority of accumulated pyruvate is transported back into the mitochondria to generate citrate for fueling the TCA cycle. Malate can also be transported back into the mitochondria later during inflammation to replenish mitochondrial stores of oxaloacetate and citrate. Furthermore, acetyl-CoA derived from citrate acts either to acetylate substrates or is utilized for fatty acid synthesis in the cytosol.
Infantino et al., demonstrated that treatment of macrophages with LPS significantly upregulated the expression of CIC mRNA and protein levels [51]. This was associated with an increased release of pro-inflammatory mediators including nitric oxide, ROS and prostaglandin E2 (PGE2), an effect that was significantly inhibited in CIC deficient cells or cells treated with a CIC inhibitor. In further studies, Infantino et al., showed that stimulation of macrophages with pro-inflammatory cytokines such as TNF-α and IFNγ, also upregulated CIC expression and increased cytosolic citrate concentrations, along with increased nitric oxide and PGE2 levels [52]. Importantly, they demonstrated that citrate exported from the mitochondria via CIC and its subsequent cytosolic metabolism to acetyl-CoA is required for the pro-inflammatory response of activated macrophages [52]. Infantino et al, hypothesized that CIC inhibition blocks the transport of mitochondria solutes, which limits the availability of the precursors (citrate-derived acetyl-CoA) needed to synthesize pro-inflammatory mediators, while addition of exogenous acetate rescues the effect of CIC inhibition [51, 52].
Our studies have also implicated increased mitochondrial citrate export as an important metabolic adaptation during metabolic reprogramming of activated macrophages. To investigate the metabolic reprogramming induced by TLR4 ligands, we treated mouse bone marrow derived macrophages with MPLA for 24 hours (24h) and assessed their metabolic profile 3 days later (3dp). Our results showed that treatment with MPLA increases citrate export from the mitochondria to the cytosol in activated macrophages [20]. In the cytosol, citrate can be metabolized to generate acetyl-CoA and oxaloacetate via the action of cytosolic ATP citrate lyase enzyme (ACLY) [49]. We observed that 24 hours after MPLA priming, macrophages recycled all the cytosolic oxaloacetate to malate, which is then converted to pyruvate through the action of malic enzyme 1. On the contrary, at 72 hours after MPLA priming, macrophages significantly upregulated malate transport back into the mitochondria to replenish the mitochondrial pool of oxaloacetate, which was further converted into citrate via mitochondrial citrate synthase. This is followed by an eventual increase in TCA cycle flux and citrate transport back into the cytosol, potentially creating a positive feed forward loop. Our study shows that it is important to study the time course of metabolic changes occurring in leukocytes in correlation to inflammatory events. It is reasonable to hypothesize that although the initial increase in citrate accumulation upon TLR4 agonist stimulation is associated with a pro-inflammatory phenotype for myeloid cells, increased citrate flux during inflammation resolution might fuel anti-inflammatory responses. Detailed time course studies are currently lacking and the exact role of citrate metabolism in immune cell responses needs to be further investigated.
Fumarate
Stimulation of macrophages with LPS or β-glucan induces fumarate accumulation, but the exact role of fumarate during inflammation is yet to be investigated. Jha et al., reported that LPS treatment of macrophages leads to significant increase in flux through the aspartate-arginosuccinate shunt, which connects with the TCA cycle at fumarate [30]. They showed that inflammatory macrophage activation leads to a break in the TCA cycle at SDH, leading to inefficient conversion of succinate to fumarate. Therefore, increased intracellular fumarate accumulation resulting from the balancing mechanism of the activated aspartate-arginosuccinate shunt fuels and maintains the TCA cycle flux in activated macrophages. Importantly, inhibition of the aspartate-arginosuccinate shunt decreased the pro-inflammatory mediators including nitric oxide and IL-6, along with abolition of the Warburg effect, which is characteristic of activated macrophages. Therefore, perturbation in the mechanisms replenishing fumarate might be associated with impaired metabolic reprogramming of macrophages during inflammation.
An important mechanism of action for accumulated fumarate in activated macrophages might be generation of antioxidant responses through activation of Nrf2 dependent signaling, similar to itaconate. The cell permeable fumarate analog, dimethyl fumarate (BG-12, Tecfidera), is a fumaric acid ester, and has been in limited clinical use for psoriasis and multiple sclerosis (MS) owing to its observed immunomodulatory and anti-inflammatory properties [53, 54]. A clinical study by Hammer et al., involving MS patients, showed that DMF treatment activated the Nrf2 transcriptional pathway and facilitated a shift towards regulatory immune cell subsets [55]. Treatment with dimethyl fumarate (DMF) has been shown to be protective in a mouse model of cardiac ischemia-reperfusion (IR) injury via Nrf2-dependent upregulation of antioxidant target genes and it was not associated with HIF-1α stabilization [56]. In contrast to Nrf2-dependent effects of DMF, a recent study by McGuire et al., demonstrated that treatment of bone marrow derived macrophages with DMF inhibits the transcription of key pro-inflammatory cytokines including TNFα and IL-6 independent of Nrf2 signaling [57]. Their findings showed that DMF blocks cytokine secretion in macrophages via inhibition of the NF-κB and MAP kinase signaling pathways. However, based on this study, we can only make limited conclusions, as a detailed time course of DMF effects was not studied and macrophages were pre-incubated with DMF for 4 hours, followed by 8 hours of LPS stimulation, before cytokine measurements. In line with this, another study by Gillard et al., showed that DMF treatment inhibited the pro-inflammatory cytokine production in both LPS-stimulated mouse splenocytes and human PBMCs, independent of the Nrf2 signaling mechanism [58]. In a mouse model of autoimmune encephalitis, Schulze-Topphoff et al., showed that treatment with DMF promoted immune modulation and provided equal clinical benefit in both wild type and Nrf2 knockout mice, suggesting DMF effects are independent of Nrf2 [59]. Therefore, DMF has been shown to act both via Nrf2 pathways and independent of Nrf2 pathways, depending on the cell type and disease conditions studied. The contrasting results might very well be due to differences in dose and duration of DMF treatment, along with different cell phenotypes. As opposed to other studies, it is important to note that Ashrafian et al., showed that fumarate induced cardioprotection against IR injury via Nrf2-dependent mechanism [56]. Therefore, Nrf2-dependent protection might be a critical mechanism for fumarate-induced anti-inflammatory responses in immune cells. Further studies need to be conducted to evaluate the exact role for fumarate in metabolic reprogramming and immune cell activation.
TCA cycle metabolites as clinical biomarkers of inflammation
Inflammatory activation of innate leukocytes, especially macrophages, leads not only to significant intracellular accumulation, but also to extracellular transport of TCA cycle metabolites. These findings lead to an important translational question of whether any of these metabolites could serve as specific biomarkers of human inflammatory conditions such as systemic infections, sepsis and autoimmune disorders. Accurate measurement of citric acid cycle intermediates relies on mass spectrometry-based techniques, which are rapid, sensitive, and reproducible techniques that could easily be standardized for clinical use [60].
Itaconate is known to be released from activated macrophages [2, 47] and it is reasonable to hypothesize that itaconate might be detected in blood of patients with systemic inflammation such as sepsis and thus serve as a clinical biomarker. However, a recent study by Meiser et al., demonstrated that itaconate was not detected in plasma or urine samples of septic patients, of which all were elderly, and the majority were diagnosed with pneumonia [61]. The minimum level of itaconate found in serum and plasma of healthy control subjects was approximately 0.2–0.5 μM, whereas no itaconate was detected in plasma of septic patients and only one septic patient was shown to have a serum concentration of approximately 0.4 μM. Itaconate was detected at an extremely low concentration in the urine sample of only one septic patient who was suffering from genitourinary sepsis, but absent in a corresponding plasma sample. Importantly, very low levels of itaconate signal were detected in the cells pelleted from broncho-alveolar lavage fluid of patients suffering from lung infection and this correlated with C-reactive protein levels. Therefore, the authors concluded that itaconate does not qualify as a measurable biomarker in systemic fluids during sepsis, but serves as a pro-inflammatory marker only in localized infections [61]. However, further detailed clinical studies are needed before completely negating itaconate as a potential biomarker. The above study performed a snapshot of itaconate at a single time point and fails to mention the exact time points when the body fluids were collected after sepsis diagnosis. No information on the clinical condition of patients at the time of sample collection, timing of sample collection in relation to antibiotic treatment was provided. In a mouse model of scrub typhus infection, Chao et al., demonstrated that itaconate was detectable in serum at an extremely low but detectable levels as early as 6 hours after infection, which was increased 60 folds higher than control at day 10 after infection [62]. Ideally, it will be important to perform the metabolite measurements as soon as signs and symptoms of sepsis are evident and before starting specific antibiotic therapy, followed by periodic measurements over the course of disease progression. It is likely that itaconate is elevated and within detectable limits during the early stages of sepsis and might decline later owing to immunosuppression, which is frequently observed in septic patients [63, 64].
Succinate has been shown to accumulate at high concentrations in synovial fluid of patients suffering from rheumatoid arthritis [23]. In a study involving patients suffering from chronic kidney disease, urinary excretion of succinate, citrate, cis-aconitate and isocitrate were significantly reduced, and this correlated with the renal expression of genes regulating these metabolites [65]. Citric acid was observed to be slightly lower in plasma of bacteremic sepsis cases as compared to controls in a patient population studied in the emergency room [66]. Succinate and citrate have also been shown to be significantly decreased among urinary metabolites in patients suffering from inflammatory bowel diseases, as compared to healthy individuals [67]. These limited clinical studies show that it is possible to measure the TCA cycle intermediates in easily accessible body fluids of patients suffering from different inflammatory conditions. Succinate, citrate, itaconate and fumarate stand out as candidate metabolites to be measured in clinical studies for potential role as biomarkers of inflammation in different disease conditions, of which systemic infections and sepsis are the most appealing. It is important to understand that metabolic intermediates have a relatively short half-life as they are in a constant metabolic flux within the cells. Careful time course studies in large numbers of patients need to be conducted before drawing broad conclusions regarding the clinical utility of any metabolic intermediate as a biomarker for a disease.
Translational relevance for developing novel drugs targeting citric acid cycle intermediates for the treatment of inflammatory conditions
Emerging understanding of the leukocyte metabolic profile induced by inflammation has uncovered novel therapeutic targets that could benefit patients with infection and sepsis, inflammatory and autoimmune diseases, transplantation and diabetes as outlined briefly in the following section.
1. SUCNR1 –
Succinate has been shown to act via its receptor, SUCNR1, to promote inflammation and disease severity in autoimmune conditions such as rheumatoid arthritis [23]. Therefore, succinate receptor antagonists represent an important class of new molecules that could be developed for therapeutic application in various inflammatory conditions. Recent studies have demonstrated the utility of a few SUCNR1 antagonists, namely the ‘4c’ compound [26]. However, this area of research is still in its infancy and needs to be further developed to harness its potential. Furthermore, the exact mechanism of action of exogenously administered itaconate analogs, DMI and 4-octyl itaconate, is not clearly understood. It is postulated that these molecules might bind to the SUCNR1 receptor or other unknown cell surface receptor, leading to the observed effects, [38, 68].
2. Irg1 and Itaconate –
Treatment with exogenous itaconate decreases production of pro-inflammatory cytokines and activates antioxidant protective pathways through inhibition of SDH and activation of Nrf2 [2, 38]. Therefore, itaconate represents a novel therapeutic for intervention in inflammatory conditions. However, the charged itaconate molecule will not cross the plasma membrane freely. Furthermore, a study by El Azzouny et al., showed that cell permeable DMI is not metabolized to itaconate by mouse bone marrow derived macrophages and treatment with 13C labelled DMI elevated intracellular succinate only, with no evidence of intracellular itaconate uptake [68]. Therefore, suitable molecules aimed at modulating intracellular itaconate levels remain to be developed. Mills et al., showed that a novel itaconate analog, 4-octyl itaconate, recapitulates most of the in vitro protective effects observed with DMI [38]. Irg1, which catalyzes the production of itaconate in the mitochondria, might be another novel therapeutic target. Discovering ways to augment IRG1 activity could increase endogenous levels of itaconate and would be an ideal way to modulate the protective effects of itaconate.
Mammalian immune cells do not possess glyoxalase shunt or any such pathway employing isocitrate lyase enzyme for energy production. Therefore, it is unlikely that intracellular accumulation of itaconate would be lethal to human leukocytes. Stimulation of murine macrophages and human monocyte derived macrophages with LPS for 6 hours resulted in 8mM and 60μM intracellular concentration of itaconate respectively [28]. Concentration in mM range is quite high for an intracellular metabolite and even at these intracellular levels was not toxic. However, this does not negate the fact that long term over-accumulation of itaconate such as under conditions of severe infections or long-term inflammation, might have adverse implications for leukocyte function. Itaconate has been shown to alkylate protein cysteine residues of Keap1, thereby activating the protective role of Nrf2 in the short-term [38]. If intracellular concentrations of itaconate are left unchecked at high concentrations for a longer duration, it might negatively affect other protein sulfhydryl groups, affecting protein activity, leading to derangement of overall immune functions. The majority of the current studies measuring itaconate accumulation upon LPS stimulation have focused only on early time points ranging from 1 to 24 hours, and none of the studies have evaluated the consequences of long term itaconate accumulation. Therefore, while considering the therapeutic utility of itaconate it will also be important to perform time course and dose response studies.
With respect to the anti-inflammatory effects of itaconate, its use as therapeutic raises an important question if it could impair host immune responses. LPS stimulation of macrophages increases itaconate production, without any discernible adverse effects on cell viability [28, 33]. A study by Widdrington et al., and our published studies show that although TLR agonist-priming of macrophages decreases their capacity to produce pro-inflammatory cytokines, it augments cellular metabolism, phagocytic capacity and respiratory burst activity [69, 70]. We also observed that priming with TLR agonists protected mice from a subsequent lethal infection with Pseudomonas aeruginosa, despite an attenuated pro-inflammatory response [70]. However, a direct role of itaconate in these studies still needs to be determined. Using a mouse model of mycobacterium tuberculosis infection, Nair et al., showed that absence of itaconate due to IRG1 deletion increases neutrophil infiltration in lungs causing them to transform into a hyper-inflammatory phenotype which then drives lung injury and lethality. Taken together, it is reasonable to hypothesize that even if itaconate is produced at high levels during an inflammatory response, it might act to augment immune responses in favor of host protection. Furthermore, itaconate by itself is an anti-microbial compound, which can effectively kill a wide variety of microbes [27]. However, in-depth dose and time response studies employing direct treatment with itaconate in in vivo infection models have yet to be conducted to conclusively evaluate its therapeutic utility.
3. Citrate and mitochondrial citrate carrier (CIC) –
Intracellular accumulation of citrate and its transport into cytosol via CIC represents an important phenomenon during metabolic reprogramming of macrophages [50]. Inhibition of CIC in LPS-stimulated macrophages decreases production of pro-inflammatory cytokines [51]. Further studies are needed to delineate the detailed role of CIC in metabolic reprogramming of immune cells during inflammation. CIC also represents an emerging therapeutic target in inflammatory conditions.
4. Fumarate –
Exogenous treatment with the fumarate analog, dimethyl fumarate, has been shown to be protective in inflammatory conditions and is clinically used for psoriasis and multiple sclerosis [53, 54], which acts via both Nrf2-dependent and -independent pathways. However, further studies are needed to implicate its direct role in modulating immune cell responses during inflammatory conditions such as infections.
5. Combination therapy –
It is important to note that a single approach may not be the answer to achieve the desired results during inflammatory conditions. Therefore, a combination of therapies using the above-mentioned strategies should also be explored and hold potential for the future of drug development for treating a myriad of inflammatory conditions. For example, a strategy to augment endogenous IRG1 activity along with inhibition of CIC, might prove a highly potent approach, as it will significantly boost the levels of endogenous itaconate through mitochondrial accumulation of its precursor, citrate.
Future considerations
Some important considerations for future studies include the following:
While considering the therapeutic utility of TCA cycle metabolites, it is also important to understand that the majority of such metabolic intermediates have a short-lifespan and metabolic reprogramming induced by them may not translate into a sustained therapeutic effect. Time course studies employing metabolic intermediates are currently lacking, and most studies only focus on the alterations in glycolysis, OXPHOS and metabolite levels during the initial 24 hours after stimulation. Long term therapeutic benefit of metabolic intermediates needs further evaluation. Interestingly, published findings from our laboratory have shown that MPLA-induced TLR4-driven resistance to infection in mice is sustained up to 15 days [70]. MPLA priming has been shown to induce sustained dynamic metabolic reprogramming of macrophages with increased glycolytic and OXPHOS capacity [20, 70]. Therefore, TLR4 agonist-induced metabolic reprogramming could translate into a sustained anti-microbial therapeutic benefit. The role of TCA cycle intermediates in MPLA induced protection is currently under investigation in our laboratory and it is reasonable to hypothesize that TLR4 driven alterations in metabolic intermediates play a significant role in sustaining long term innate immune cell memory effects.
Much of the studies evaluating the changes in TCA cycle metabolites during metabolic reprogramming have been performed only in macrophages. Future studies using other leukocyte populations such as neutrophils, dendritic cells, T cell and even non-immune tissue parenchymal cells will advance our understanding of metabolic reprogramming and provide a broader view for integrating drug development.
The field of metabolic reprogramming is upon a frontier of exploration, requiring further description and processes to better corelate in vitro observations with in vivo models of inflammatory diseases. For example, Mills et al., evaluated the effect of 4-octyl itaconate on survival in an acute (24 hour) mouse model of LPS-induced inflammation and observed only a slight benefit in prolonging survival with none of the treated mice surviving at the end of study. Future studies evaluating the therapeutic effect of itaconate in slowly progressive, clinically relevant models of infection are needed, along with dose response studies. Furthermore, it will be imperative to study the phenotype of immune cells upon direct isolation from the animal, such as from spleen, lymph nodes, bone marrow stem cells and circulating peripheral immune cells.
The role for other TCA cycle intermediates such as alpha-ketoglutarate, malate, oxaloacetate also needs to be investigated. Preliminary studies from our laboratory show that, activation of macrophages leads to initial conversion of cytosolic oxaloacetate to pyruvate through the intermediate malate, followed later in the course by complete recycling of malate into the mitochondria to replenish mitochondrial oxaloacetate and citrate stores [20]. Thus, the dynamic alterations in other TCA cycle intermediates needs further investigation.
Most previous studies have not demonstrated a time course of alterations in mitochondrial metabolism after the initial inflammatory stimuli. Our studies show that MPLA induces dynamic changes in TCA cycle activity over the course of days ultimately resulting in. a sustained increased in glycolysis and OXPHOS in association with endotoxin tolerance, increased phagocytic capacity and mitochondrial biogenesis. The exact signaling mechanisms leading to this unique phenotype need to be evaluated to define the signaling mechanisms controlling mitochondrial biogenesis and their correlation with alterations in TCA cycle intermediates.
Alterations in mitochondrial metabolism might have significant impact not only mitochondrial health but also on overall cellular functions. A recent study by Widdrington et al., demonstrated that LPS stimulation of THP-1 cells leads to a distinct simultaneous cellular response comprised of autophagy induced mitochondrial degradation as well as increased mitochondrial mass via biogenesis, with a net result of replenishment of any degraded mitochondria and maintenance of its levels, along with increased mitochondrial respiration [69]. Furthermore, these changes did not affect cell viability and increased phagocytic capacity. In contrast, in another study using RAW 264.7 cells, Deng et al., showed that LPS dose- (10–500 ng/mL) and time-dependently triggered mitochondrial membrane depolarization, mitochondrial structural damage and ATP depletion [71]. Therefore, it is critical to note that the effects of inflammatory stimulus on mitochondria and cellular functions depend not only on the intensity of stimulus but also the cell type studied. In the case of severe infections such as sepsis, immune cells would be exposed to a very high levels of LPS or other PRRs, cytokines and other microbial products which could eventually lead to mitochondrial damage and cell death.
Garaude et al., demonstrated that the architecture of the mitochondrial ETC complexes was altered in response to exposure of macrophages to viable bacteria due to a decrease in the abundance of assembled complex I subunits [15]. Using heat-killed bacteria, this study also confirmed many of the metabolic changes that have been described previously with the use of LPS stimulation. However, some major alterations such as induction of SDH activity and destabilization of ETC were only observed upon exposure to viable bacteria. This study showed that changes in leukocyte responses observed with exposure to a single (PAMP such as LPS, might be very different from those observed with exposure to intact microbes, which possess multiple PAMPs. Future studies should be designed to further explore the immune cell responses to live microbes and a combination of multiple PAMPs.
Future studies must extend the findings observed in mouse macrophages to that of human cells, such as peripheral blood monocytes, which can be differentiated to macrophages ex vivo. Although many studies have shown strong correlation between mouse and human findings, further detailed studies will provide novel insights into metabolic adaptation of immune cells during inflammation.
Conclusions
Mitochondrial metabolism is no longer considered to be just an ATP producing mechanism, but dynamically affects numerous cell signaling pathways through modulating the levels of TCA cycle intermediates. Succinate, itaconate, citrate and fumarate play a critical role in affecting innate immune cell functions during metabolic reprogramming of immune cells. The role of other TCA cycle intermediates is still an open field of investigation and will potentially yield ground breaking results in the near future.
Acknowledgements
N.K.P is supported by a National Institutes of Health (NIH) T32 grant, 5T32GM108554–05, and this work is also supported by the NIH grants RO1 GM104306 and R01 GM119197 to ERS, K08 GM123345 to AH, and R01 GM121711 to JKB.
Abbreviations
- ATP
Adenosine triphosphate
- ACLY
ATP citrate lyase enzyme
- AOX
Alternative oxidase
- CIC
Mitochondrial citrate carrier
- DMI
Dimethyl itaconate
- DMM
Dimethyl malonate
- HIF-1α
Hypoxia inducible factor - 1α
- IL-1β
Interleukin-1β
- Irg1
Immunoresponsive gene 1
- IRG1
Immunoresponsive gene 1 protein
- IDH
Isocitrate dehydrogenase
- IL-6
Interlukin-6
- KEAP1
Kelch-like ECH-associated protein 1
- LPS
Lipopolysaccharide
- mROS
Mitochondrial reactive oxygen species
- MPLA
Monophosphoryl lipid A
- Nrf2
Nuclear respiratory factor 2
- OXPHOS
Oxidative phosphorylation
- PRR
Pattern recognition receptor
- PHD
prolyl hydroxylase enzymes
- PAMP
Pathogen associated molecular pattern
- ROS
Reactive oxygen species
- RET
Reverse electron transport
- SUCNR1
Succinate receptor
- SDH
Succinate dehydrogenase
- TCA
Tricarboxylic acid cycle
- TLR
Toll-like receptor
- TNF-α
Tumor necrosis factor – α
Footnotes
Disclosure of Conflicts of Interest
Authors declare no conflict of interest.
References
- 1.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O’Neill LA (2013) Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC, Griss T, Weinheimer CJ, Khader S, Randolph GJ, Pearce EJ, Jones RG, Diwan A, Diamond MS, Artyomov MN (2016) Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab 24, 158–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Murphy MP and O’Neill LAJ (2018) Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 174, 780–784. [DOI] [PubMed] [Google Scholar]
- 4.Kelly B and O’Neill LA (2015) Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell research 25, 771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majmundar AJ, Wong WJ, Simon MC. Hypoxia‐inducible factors and the response to hypoxic stress. Mol Cell 2010; 40: 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majmundar AJ, Wong WJ, Simon MC (2010) Hypoxia-inducible factors and the response to hypoxic stress. Molecular cell 40, 294–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS (2003) HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weidemann A and Johnson RS (2008) Biology of HIF-1alpha. Cell Death Differ 15, 621–7. [DOI] [PubMed] [Google Scholar]
- 9.Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ, O’Neill LA (2015) Pyruvate kinase M2 regulates Hif-1alpha activity and IL-1beta induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab 21, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollander AP, Corke KP, Freemont AJ, Lewis CE (2001) Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum 44, 1540–4. [DOI] [PubMed] [Google Scholar]
- 11.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA (2016) Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP (2016) A Unifying Mechanism for Mitochondrial Superoxide Production during Ischemia-Reperfusion Injury. Cell Metab 23, 254–63. [DOI] [PubMed] [Google Scholar]
- 14.Kietzmann T and Gorlach A (2005) Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin Cell Dev Biol 16, 474–86. [DOI] [PubMed] [Google Scholar]
- 15.Garaude J, Acin-Perez R, Martinez-Cano S, Enamorado M, Ugolini M, Nistal-Villan E, Hervas-Stubbs S, Pelegrin P, Sander LE, Enriquez JA, Sancho D (2016) Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nature immunology 17, 1037–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S (2011) TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Z, Tan M, Xie Z, Dai L, Chen Y, Zhao Y (2011) Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol 7, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J, Chen Y, Tishkoff DX, Peng C, Tan M, Dai L, Xie Z, Zhang Y, Zwaans BM, Skinner ME, Lombard DB, Zhao Y (2013) SIRT5-mediated lysine desuccinylation impacts diverse metabolic pathways. Molecular cell 50, 919–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang F, Wang K, Xu W, Zhao S, Ye D, Wang Y, Xu Y, Zhou L, Chu Y, Zhang C, Qin X, Yang P, Yu H (2017) SIRT5 Desuccinylates and Activates Pyruvate Kinase M2 to Block Macrophage IL-1beta Production and to Prevent DSS-Induced Colitis in Mice. Cell Rep 19, 2331–2344. [DOI] [PubMed] [Google Scholar]
- 20.Fensterheim BA, Young JD, Luan L, Kleinbard RR, Stothers CL, Patil NK, McAtee-Pereira AG, Guo Y, Trenary I, Hernandez A, Fults JB, Williams DL, Sherwood ER, Bohannon JK (2018) The TLR4 Agonist Monophosphoryl Lipid A Drives Broad Resistance to Infection via Dynamic Reprogramming of Macrophage Metabolism. Journal of immunology 200, 3777–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L (2004) Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429, 188–93. [DOI] [PubMed] [Google Scholar]
- 22.Rubic T, Lametschwandtner G, Jost S, Hinteregger S, Kund J, Carballido-Perrig N, Schwarzler C, Junt T, Voshol H, Meingassner JG, Mao X, Werner G, Rot A, Carballido JM (2008) Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nature immunology 9, 1261–9. [DOI] [PubMed] [Google Scholar]
- 23.Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, Muller A, Tigani B, Kneuer R, Patel S, Valeaux S, Gommermann N, Rubic-Schneider T, Junt T, Carballido JM (2016) GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. The Journal of experimental medicine 213, 1655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hakak Y, Lehmann-Bruinsma K, Phillips S, Le T, Liaw C, Connolly DT, Behan DP (2009) The role of the GPR91 ligand succinate in hematopoiesis. Journal of leukocyte biology 85, 837–43. [DOI] [PubMed] [Google Scholar]
- 25.de Castro Fonseca M, Aguiar CJ, da Rocha Franco JA, Gingold RN, Leite MF (2016) GPR91: expanding the frontiers of Krebs cycle intermediates. Cell Commun Signal 14, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhuniya D, Umrani D, Dave B, Salunke D, Kukreja G, Gundu J, Naykodi M, Shaikh NS, Shitole P, Kurhade S, De S, Majumdar S, Reddy SB, Tambe S, Shejul Y, Chugh A, Palle VP, Mookhtiar KA, Cully D, Vacca J, Chakravarty PK, Nargund RP, Wright SD, Graziano MP, Singh SB, Roy S, Cai TQ (2011) Discovery of a potent and selective small molecule hGPR91 antagonist. Bioorg Med Chem Lett 21, 3596–602. [DOI] [PubMed] [Google Scholar]
- 27.Cordes T, Michelucci A, Hiller K (2015) Itaconic Acid: The Surprising Role of an Industrial Compound as a Mammalian Antimicrobial Metabolite. Annu Rev Nutr 35, 451–73. [DOI] [PubMed] [Google Scholar]
- 28.Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, Buttini M, Linster CL, Medina E, Balling R, Hiller K (2013) Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proceedings of the National Academy of Sciences of the United States of America 110, 7820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee CG, Jenkins NA, Gilbert DJ, Copeland NG, O’Brien WE (1995) Cloning and analysis of gene regulation of a novel LPS-inducible cDNA. Immunogenetics 41, 263–70. [DOI] [PubMed] [Google Scholar]
- 30.Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ, Driggers EM, Artyomov MN (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–30. [DOI] [PubMed] [Google Scholar]
- 31.Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, Redmann V, Freitas TC, Blagih J, van der Windt GJ, Artyomov MN, Jones RG, Pearce EL, Pearce EJ (2014) TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nature immunology 15, 323–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meiser J, Kramer L, Sapcariu SC, Battello N, Ghelfi J, D’Herouel AF, Skupin A, Hiller K (2016) Pro-inflammatory Macrophages Sustain Pyruvate Oxidation through Pyruvate Dehydrogenase for the Synthesis of Itaconate and to Enable Cytokine Expression. J Biol Chem 291, 3932–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cordes T, Wallace M, Michelucci A, Divakaruni AS, Sapcariu SC, Sousa C, Koseki H, Cabrales P, Murphy AN, Hiller K, Metallo CM (2016) Immunoresponsive Gene 1 and Itaconate Inhibit Succinate Dehydrogenase to Modulate Intracellular Succinate Levels. J Biol Chem 291, 14274–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu X, Meyers A, Long D, Ingram B, Liu T, Yoza BK, Vachharajani V, McCall CE (2019) Frontline Science: Monocytes sequentially rewire metabolism and bioenergetics during an acute inflammatory response. Journal of leukocyte biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Zhang P, Wang C, Han C, Meng J, Liu X, Xu S, Li N, Wang Q, Shi X, Cao X (2013) Immune responsive gene 1 (IRG1) promotes endotoxin tolerance by increasing A20 expression in macrophages through reactive oxygen species. J Biol Chem 288, 16225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, Beyaert R, Elewaut D, Kanneganti TD, van Loo G, Lamkanfi M (2014) Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 512, 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voet S, Mc Guire C, Hagemeyer N, Martens A, Schroeder A, Wieghofer P, Daems C, Staszewski O, Vande Walle L, Jordao MJC, Sze M, Vikkula HK, Demeestere D, Van Imschoot G, Scott CL, Hoste E, Goncalves A, Guilliams M, Lippens S, Libert C, Vandenbroucke RE, Kim KW, Jung S, Callaerts-Vegh Z, Callaerts P, de Wit J, Lamkanfi M, Prinz M, van Loo G (2018) A20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat Commun 9, 2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, Szpyt J, Runtsch MC, King MS, McGouran JF, Fischer R, Kessler BM, McGettrick AF, Hughes MM, Carroll RG, Booty LM, Knatko EV, Meakin PJ, Ashford MLJ, Modis LK, Brunori G, Sevin DC, Fallon PG, Caldwell ST, Kunji ERS, Chouchani ET, Frezza C, Dinkova-Kostova AT, Hartley RC, Murphy MP, O’Neill LA (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, Korenfeld D, Mathyer ME, Kim H, Huang LH, Duncan D, Bregman H, Keskin A, Santeford A, Apte RS, Sehgal R, Johnson B, Amarasinghe GK, Soares MP, Satoh T, Akira S, Hai T, de Guzman Strong C, Auclair K, Roddy TP, Biller SA, Jovanovic M, Klechevsky E, Stewart KM, Randolph GJ, Artyomov MN (2018) Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis. Nature 556, 501–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McFadden BA and Purohit S (1977) Itaconate, an isocitrate lyase-directed inhibitor in Pseudomonas indigofera. J Bacteriol 131, 136–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dolan SK and Welch M (2018) The Glyoxylate Shunt, 60 Years On. Annu Rev Microbiol 72, 309–330. [DOI] [PubMed] [Google Scholar]
- 42.Williams JO, Roche TE, McFadden BA (1971) Mechanism of action of isocitrate lyase from Pseudomonas indigofera. Biochemistry 10, 1384–90. [DOI] [PubMed] [Google Scholar]
- 43.Shin JH, Yang JY, Jeon BY, Yoon YJ, Cho SN, Kang YH, Ryu DH, Hwang GS (2011) (1)H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J Proteome Res 10, 2238–47. [DOI] [PubMed] [Google Scholar]
- 44.Naujoks J, Tabeling C, Dill BD, Hoffmann C, Brown AS, Kunze M, Kempa S, Peter A, Mollenkopf HJ, Dorhoi A, Kershaw O, Gruber AD, Sander LE, Witzenrath M, Herold S, Nerlich A, Hocke AC, van Driel I, Suttorp N, Bedoui S, Hilbi H, Trost M, Opitz B (2016) IFNs Modify the Proteome of Legionella-Containing Vacuoles and Restrict Infection Via IRG1-Derived Itaconic Acid. PLoS Pathog 12, e1005408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sasikaran J, Ziemski M, Zadora PK, Fleig A, Berg IA (2014) Bacterial itaconate degradation promotes pathogenicity. Nat Chem Biol 10, 371–7. [DOI] [PubMed] [Google Scholar]
- 46.Luan HH and Medzhitov R (2016) Food Fight: Role of Itaconate and Other Metabolites in Antimicrobial Defense. Cell Metab 24, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sugimoto M, Sakagami H, Yokote Y, Onuma H, Kaneko M, Mori M, Sakaguchi Y, Soga T, Tomita M (2012) Non-targeted metabolite profiling in activated macrophage secretion. Metabolomics 8, 624–633. [Google Scholar]
- 48.Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, Boon ACM, Schwarzkopf EA, Bradstreet TR, Edelson BT, Artyomov MN, Stallings CL, Diamond MS (2018) Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. The Journal of experimental medicine 215, 1035–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palmieri F (2004) The mitochondrial transporter family (SLC25): physiological and pathological implications. Pflugers Arch 447, 689–709. [DOI] [PubMed] [Google Scholar]
- 50.Williams NC and O’Neill LAJ (2018) A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front Immunol 9, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, Palmieri F, Iacobazzi V (2011) The mitochondrial citrate carrier: a new player in inflammation. Biochem J 438, 433–6. [DOI] [PubMed] [Google Scholar]
- 52.Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F (2014) A key role of the mitochondrial citrate carrier (SLC25A1) in TNFalpha- and IFNgamma-triggered inflammation. Biochim Biophys Acta 1839, 1217–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moharregh-Khiabani D, Linker RA, Gold R, Stangel M (2009) Fumaric Acid and its esters: an emerging treatment for multiple sclerosis. Curr Neuropharmacol 7, 60–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mrowietz U, Christophers E, Altmeyer P (1999) Treatment of severe psoriasis with fumaric acid esters: scientific background and guidelines for therapeutic use. The German Fumaric Acid Ester Consensus Conference. Br J Dermatol 141, 424–9. [DOI] [PubMed] [Google Scholar]
- 55.Hammer A, Waschbisch A, Kuhbandner K, Bayas A, Lee DH, Duscha A, Haghikia A, Gold R, Linker RA (2018) The NRF2 pathway as potential biomarker for dimethyl fumarate treatment in multiple sclerosis. Ann Clin Transl Neurol 5, 668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ashrafian H, Czibik G, Bellahcene M, Aksentijevic D, Smith AC, Mitchell SJ, Dodd MS, Kirwan J, Byrne JJ, Ludwig C, Isackson H, Yavari A, Stottrup NB, Contractor H, Cahill TJ, Sahgal N, Ball DR, Birkler RI, Hargreaves I, Tennant DA, Land J, Lygate CA, Johannsen M, Kharbanda RK, Neubauer S, Redwood C, de Cabo R, Ahmet I, Talan M, Gunther UL, Robinson AJ, Viant MR, Pollard PJ, Tyler DJ, Watkins H (2012) Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metab 15, 361–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McGuire VA, Ruiz-Zorrilla Diez T, Emmerich CH, Strickson S, Ritorto MS, Sutavani RV, Weibeta A, Houslay KF, Knebel A, Meakin PJ, Phair IR, Ashford ML, Trost M, Arthur JS (2016) Dimethyl fumarate blocks pro-inflammatory cytokine production via inhibition of TLR induced M1 and K63 ubiquitin chain formation. Sci Rep 6, 31159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gillard GO, Collette B, Anderson J, Chao J, Scannevin RH, Huss DJ, Fontenot JD (2015) DMF, but not other fumarates, inhibits NF-kappaB activity in vitro in an Nrf2-independent manner. J Neuroimmunol 283, 74–85. [DOI] [PubMed] [Google Scholar]
- 59.Schulze-Topphoff U, Varrin-Doyer M, Pekarek K, Spencer CM, Shetty A, Sagan SA, Cree BA, Sobel RA, Wipke BT, Steinman L, Scannevin RH, Zamvil SS (2016) Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proceedings of the National Academy of Sciences of the United States of America 113, 4777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tan B, Lu Z, Dong S, Zhao G, Kuo MS (2014) Derivatization of the tricarboxylic acid intermediates with O-benzylhydroxylamine for liquid chromatography-tandem mass spectrometry detection. Anal Biochem 465, 134–47. [DOI] [PubMed] [Google Scholar]
- 61.Meiser J, Kraemer L, Jaeger C, Madry H, Link A, Lepper PM, Hiller K, Schneider JG (2018) Itaconic acid indicates cellular but not systemic immune system activation. Oncotarget 9, 32098–32107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chao CC, Ingram BO, Lurchachaiwong W, Ching WM (2018) Metabolic characterization of serum from mice challenged with Orientia tsutsugamushi-infected mites. New Microbes New Infect 23, 70–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patil NK, Bohannon JK, Sherwood ER (2016) Immunotherapy: A promising approach to reverse sepsis-induced immunosuppression. Pharmacol Res 111, 688–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hotchkiss RS, Monneret G, Payen D (2013) Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis 13, 260–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hallan S, Afkarian M, Zelnick LR, Kestenbaum B, Sharma S, Saito R, Darshi M, Barding G, Raftery D, Ju W, Kretzler M, Sharma K, de Boer IH (2017) Metabolomics and Gene Expression Analysis Reveal Down-regulation of the Citric Acid (TCA) Cycle in Non-diabetic CKD Patients. EBioMedicine 26, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kauppi AM, Edin A, Ziegler I, Molling P, Sjostedt A, Gylfe A, Stralin K, Johansson A (2016) Metabolites in Blood for Prediction of Bacteremic Sepsis in the Emergency Room. PloS one 11, e0147670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stephens NS, Siffledeen J, Su X, Murdoch TB, Fedorak RN, Slupsky CM (2013) Urinary NMR metabolomic profiles discriminate inflammatory bowel disease from healthy. J Crohns Colitis 7, e42–8. [DOI] [PubMed] [Google Scholar]
- 68.ElAzzouny M, Tom CT, Evans CR, Olson LL, Tanga MJ, Gallagher KA, Martin BR, Burant CF (2017) Dimethyl Itaconate Is Not Metabolized into Itaconate Intracellularly. J Biol Chem 292, 4766–4769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Widdrington JD, Gomez-Duran A, Pyle A, Ruchaud-Sparagano MH, Scott J, Baudouin SV, Rostron AJ, Lovat PE, Chinnery PF, Simpson AJ (2018) Exposure of Monocytic Cells to Lipopolysaccharide Induces Coordinated Endotoxin Tolerance, Mitochondrial Biogenesis, Mitophagy, and Antioxidant Defenses. Front Immunol 9, 2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fensterheim BA, Guo Y, Sherwood ER, Bohannon JK (2017) The Cytokine Response to Lipopolysaccharide Does Not Predict the Host Response to Infection. Journal of immunology 198, 3264–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Deng SY, Zhang LM, Ai YH, Pan PH, Zhao SP, Su XL, Wu DD, Tan HY, Zhang LN, Tsung A (2017) Role of interferon regulatory factor-1 in lipopolysaccharide-induced mitochondrial damage and oxidative stress responses in macrophages. Int J Mol Med 40, 1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]