

Abstract
Cardiac troponin C (cTnC) binds intracellular calcium and subsequently cardiac troponin I (cTnI), initiating cardiac muscle contraction. Due to its role in contraction, cTnC has been a therapeutic target in the search for small molecules to treat conditions that interfere with normal muscle contraction like the heritable cardiomyopathies. Structural studies have shown the binding location of small molecules such as bepridil, dfbp-o, 3-methyldiphenylamine (DPA) and W7 to be a hydrophobic pocket in the regulatory domain of cTnC (cNTnC) but have not shown the influence of these small molecules on the energetics of opening this domain. Here we describe an application of an umbrella sampling method used to elucidate the impact these calcium sensitivity modulators have on the free energy of cNTnC hydrophobic patch opening. We found that all these molecules lowered the free energy of opening in the absence of the cTnI, with bepridil facilitating the least endergonic transformation. In the presence of cTnI, however, we saw a stabilization of the open configuration due to DPA and dfbp-o binding, and a destabilization of the open configuration imparted by bepridil and W7. Predicted poor binding molecule NSC34337 left the hydrophobic in under 3 ns in conventional MD simulations suggesting that only hydrophobic patch binders stabilized the open conformation. In conclusion, this study presents a novel approach to study the impact of small molecules on hydrophobic patch opening through umbrella sampling and proposes mechanisms for calcium sensitivity modulation.
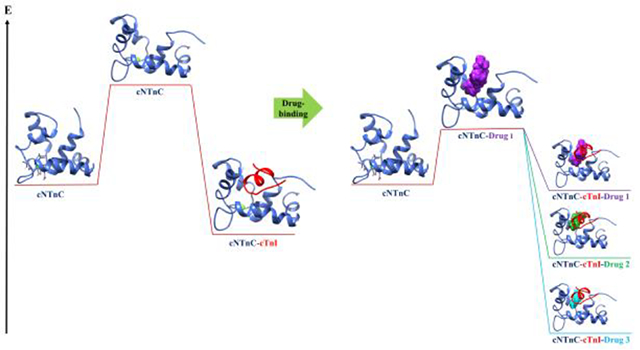
Graphical Abstract
Introduction
Cardiac troponin (cTn) is a three-subunit protein complex that is part of the thin filament in heart muscle and initiates muscle contraction through response to calcium binding.1 The three subunits of cardiac troponin are troponin I (cTnI), troponin T (cTnT), and troponin C (cTnC). Each subunit is named for their role in the complex: cTnI for its inhibitory role in preventing the movement of tropomyosin, cTnT for its interaction with tropomyosin, and cTnC for its calcium-binding properties.2 During contraction, calcium binds to the N-terminal region of cTnC (cNTnC).3 Once calcium is bound, cNTnC, also known as the regulatory domain, is then able to bind the switch-peptide region of cTnI.4 This action causes a sliding of tropomyosin on the actin filament which allows myosin binding to actin permitting contraction to occur.5 The switch-peptide binds to a hydrophobic patch between helix A and B of the regulatory domain. For binding to occur, this hydrophobic patch must be open,6 and the degree of openness is generally defined by an interhelical angle between helices A and B of cNTnC. The cardiac/slow skeletal isoform of TnC (cTnC) is specifically found in cardiac and slow skeletal muscle cells, and only binds one calcium ion in its N-terminal regulatory domain. As a result of its role in muscle contraction, cTnC has implications in heart failure and mutations in the protein have been associated with cardiomyopathies.7 In order to treat these conditions, small molecules that bind to cTnC and act predominantly as calcium sensitizing agents have been reported.8 Small molecules that have been resolved via crystallography or NMR in complex with cNTnC include 3-methyldiphenylamine,9 bepridil,10 dfbp-o,11 trifluoperazine,12 levosimendan-analog i9,13 and W7.14 Additional cNTnC modulators have been identified that do not have experimentally determined structures in complex with cNTnC. NSC147866,15 NSC600285, and NSC61181716 have been found through computational drug screens. The cNTnC–ligand structures show that the small molecule modulators predominately bind to the hydrophobic patch. We are speculating that the presence of small molecules in the hydrophobic patch of cNTnC has an impact on patch opening, affecting cNTnC’s ability to bind cTnI, and ultimately modulating calcium sensitivity. A molecular understanding of this impact is desirable and potentially helpful for drug discovery.
Because of the central role it plays in muscle contraction, there have been a growing number of computational studies on cTnC. These studies have included drug discovery,16 assessing cTnI binding, and investigating calcium binding.17 Studies on cTnI binding to cTnC have been able to show the influence of cardiomyopathy mutations on the relative binding energy. For instance, known hypertrophic cardiomyopathy-associated mutations (A31S) demonstrated a weaker binding interaction compared to wild type cTnC.18 Additionally, the interactions of cTnC and cTnI have been elucidated using MD simulations.19,20,21 Techniques such as umbrella sampling22 and Brownian Dynamics19, 23 have been used to assess the calcium binding of cTnC. For example, Stevens et al.22 assessed calcium binding of cTnC variants in zebrafish at different temperatures by umbrella sampling. These studies have also contributed to our understanding of cTnC function and the impact of mutations and post-translational modifications in modulating these functions. Post-translational modifications on cTnI have been mimicked in MD simulations to probe their impact on cTnC binding.24 The dynamic behavior of cTnC, particularly the exposure of the hydrophobic patch, is key to understanding its functions. Notably, some simulations have shed light on the hydrophobic patch opening, a phenomenon that is difficult to measure through experimental methods. So far, studies of the hydrophobic patch opening, have focused on isolated cTnC,22, 25 sTnC,26 HCM/DCM associated mutations,27 or cNTnC–cTnI systems.28 Notably, simulations have shown that mutations within the regulatory domain heavily influence its dynamic behavior, such as the calcium-sensitizing mutation L48Q27 and the gain-of-function and loss-of-function mutations V44Q and E40A.25 However, simulations have not been performed extensively in the presence of small molecules. There have been conventional molecular dynamic simulations of cTnC bound to bepridil29 focusing on interactions between cTnC–cTnI and binding affinity within the complex. Simulations of the model of cTnC bound to dfbp-o used for drug discovery have been reported as well.9 More rigorous simulations and free energy calculations of the impact of small molecules on cNTnC hydrophobic patch opening have the potential to shed light on this important process at a molecule level.
In this work, we used molecular dynamics simulations and our recently developed umbrella sampling scheme28 to probe the dynamics and energetics of hydrophobic patch opening for small-molecule bound cNTnC systems. Umbrella sampling is a methodology that allows for the exhaustive and targeted simulation of system configurations along a defined reaction coordinate.30 Exploration of these configurations is performed in order to provide energetics that would require prohibitively long timescale simulations utilizing conventional MD on most standard computing systems. Application of umbrella sampling allowed for sampling of the transition between open and closed configurations in cNTnC without the need to simulate the system for hundreds of microseconds. We saw that the presence of the small molecules in the hydrophobic patch lowered the relative free energy of opening, or, in some cases, obviated it altogether. The compounds dfbp-o and 3-methyldiphenylamine also further lowered the relative free energy of opening of cNTnC in the presence of the cTnI-switch peptide compared to cNTnC–cTnI with no small molecule present. In molecular dynamics simulations, weak-binding small molecule NSC34337 left the hydrophobic patch in short (< 3 ns) timeframes.
Methods
Small molecule parameterization
MD simulations were performed with the CHARMM36 additive protein force field developed by MacKerell et al.31 In order to simulate the protein-ligand complexes and perform further umbrella sampling calculations it was necessary to generate MM force field parameters describing each of the small molecules utilized in this study: NSC34337, bepridil, dfbp-o, 3-methyldiphenylamine, and W7 (Scheme 1). Parameters for these small molecules were taken, in part, from the CHARMM general force field for drug-like molecules (CGenFF).32,33 Initial atom typing, assignment of charges, and parameters were obtained with the online CGenFF program interface.34 Each value generated was automatically assigned a penalty score by the CGenFF program to represent deviation from ideality. A penalty score above 50 is generally considered indicative that validation or potential refinement using quantum mechanical methods is necessary.35 In cases where the small molecules’ parameters exceeded penalty scores of 20, we performed refinement and fitting with the Forcefield ToolKit (FFTK) plug-in from VMD36 in conjunction with ab initio calculations utilizing Gaussian 16.37 The general workflow involved derivation of high penalty parameters using methodology self-consistent with the CHARMM force field. To this end, geometry optimization and frequency calculations of the small molecules were performed at the MP2/6–31G* level of theory.38,39 Optimized molecules were verified to be local minima based on vibrational frequency analysis yielding zero imaginary frequencies. The calculated frequencies were also used for defining bond angles and bond lengths of the ligands. Charges were refined by optimizing models of explicit water molecules interacting with the water accessible atoms of the small molecule at the HF/6–31G* level of theory. This simulated the environment of solvation by TIP3P40 parameterized waters utilized during subsequent MD simulations. Finally, parameters for high penalty torsions were derived from relaxed potential energy surface scans of the corresponding dihedral angles calculated at the MP2/6–31G* level of theory.
Scheme I.

Small molecules investigated with umbrella sampling and molecular dynamics simulations. Structures reflect the protonation states utilized during MD simulations.
Molecular dynamics simulations
Starting models:
We used the following structures of small molecules bound to cNTnC as starting models for simulations: cNTnC–bepridil (1LXF41), cNTnC–dfbp-o (2L1R11), 3-methyldiphenylamine (5WCL9), and W7 (2KRD42). One model of a poor docking score molecule, NSC34337, bound to cNTnC was extracted from a previous docking study of small molecules to cNTnC using the software Glide.16 This molecule was chosen based on its poor Glide score, which for NSC34337 was-2.29 kcal/mol. A second criterion for molecule selection was its hydrophilic character with a calculated logP value of 0.51.
System preparation:
Nine systems of the N-terminal regulatory domain of cardiac troponin C were prepared for use in this study. These included cNTnC, without cTnI present, with each of the small molecules: cNTnC–NSC34337, cNTnC–bepridil, cNTnC–dfbp-o, cNTnC–3-methyldiphenylamine, and cNTnC–W7. We also ran MD simulations on four systems with cTnI present: cNTnC–cTnI–bepridil, cNTnC–cTnI–dfbp-o, cNTnC–cTnI–3-methyldiphenylamine, and cNTnC–cTnI–W7. The initial preparation of the systems is described in Lindert et al.25 In short, systems were solvated with explicit TIP3P water molecules and NaCl counterions were incorporated to neutralize the systems and impose an ionic strength of 150 mM. The systems were restrained, and the water molecules were energy minimized for 10000 steps. Energies of the protein and small molecules were then minimized for 10,000 steps. Subsequently, restraints on the protein and small molecule were gradually removed over 190,000 steps during an energy equilibration. A final equilibration of the system was carried out over 10,000 steps.
MD simulations:
The conventional MD simulations of cNTnC–NSC34337, cNTnC–bepridil, cNTnC–dfbp-o, cNTnC–3-methyldiphenylamine, and cNTnC–W7 were carried out using NAMD 2.1243 with the CHARMM3631,44 forcefield under an NPT ensemble at 310 K. Bonds with hydrogen were constrained using the SHAKE algorithm allowing for a timestep of 2 fs. Structures were saved every 2 ps. Production runs were carried out on the Owens Cluster of the Ohio Super Computer (OSC) using 28 processors on 1 node per job, for 50,000,000 timesteps. This corresponded to a runtime of 100 ns.
Steered molecular dynamics and umbrella sampling
Similarly to previously established methods,28 umbrella sampling was utilized to study the free energy of opening of the hydrophobic patch in the presence of several small molecules. We used the interhelical distance as a metric for openness and reaction coordinate in the simulations. The interhelical distance was defined as the distance between the N-terminus of helix A (residue 14) and the C-terminus of helix B (residue 48). To generate the starting frames for the umbrella sampling, steered molecular dynamics simulations were run on the cNTnC–bepridil, cNTnC–dfbp-o, cNTnC–W7, and cNTnC–3-methyldiphenylamine systems. The starting frames were steered from a semi-open configuration (interhelical distance ~22–24 Å) to a closed configuration (interhelical distance ~15 Å) and to an open configuration (interhelical distance ~28 Å) with a force constant of 20 kcal/(mol·Å) in separate simulations. The interhelical angles of the starting models are shown in SI Table 1. The force constant was applied to the backbone atoms of helix A (residues 14–18; atoms C, CA, N) and helix B (residues 45–48; atoms C, CA, N). Additional weak restraints were placed on the small molecules in the steered MD simulations to restrain them within the hydrophobic patch as they were being closed. A force constant of 2 kcal/(mol·Å2) was applied between the center of mass of the small molecules, calculated from all the heavy atoms, and the atoms 1,286 (residue 84, SG), 1,232 (residue 81, SD), 1,215 (residue 80, SD), 1,162 (residue 77, CZ), 928 (residue 60, SD). These restraints were defined in the same collective variables45 (colvars) file that defined the opening. The steered MD simulations were run for 5,000,000 steps corresponding to a simulation time of 10 ns, under NPT conditions at 310 K.
Frames were extracted from the steered MD trajectories every 1000 steps. Interhelical angles and the interhelical distances (between backbone atoms of residue 14 and residue 48) were calculated from these frames to test for correlation. Conformations from the trajectories were chosen based on the correlation between the interhelical angles of 90°−140° in 2.5° increments and the interhelical distances, which corresponded to a ~13 Å range in ~0.5 Å increments. The closed and open interhelical angles were based upon experimentally derived structures of troponin in these states.4, 46 These 21 frames were then used in the umbrella sampling simulations with the distance colvar between residue 14 (atoms 220, 222, 224) and residue 48 (atoms 738, 740, 755) and run for 5,000,000 steps. The interhelical distance was restrained with a force constant of 50 kcal/(mol·Å2). The umbrella sampling simulations did not contain restraints on the small molecules. For each system, these simulations were run in triplicate. The resulting simulations were analyzed via the weighted histogram analysis method (WHAM47). WHAM can derive a potential of mean force from umbrella sampling simulations as a function of the reaction coordinate (in this case the interhelical distance). A biased probability of the system existing at each configuration is calculated by sampling orthogonally at points along the reaction coordinate given a restraining bias potential, the spatial deviation of the ensemble from its ideal restrained position, and the simulation temperature. A detailed description of the method can be found in Kumar et al.48 For the WHAM analysis, the min and max values were dictated by the distance-angle correlations and the calculation was divided among 40 bins. We used a convergence tolerance of 10–7 for this method. This generated free energy profiles for each system, which were averaged amongst the three runs for a final extracted free energy of opening. The data was linearized with the linear fit function of Matplotlib.49
Interhelical angle analysis
The interhelical angles between helix A and helix B were calculated using interhlx (K. Yap, University of Toronto, Ontario, Canada). Interhelical angles were calculated for the steered MD simulations. The helix residue definition for helix A was 14–28 and for helix B 44–48. The frames in the trajectory file were output every 1,000 steps, and the interhelical angle was calculated for each of these frames, resulting in 5,000 calculations per simulation.
Results and Discussion
Known cNTnC binders lower the free energy of opening of cTnC
To explore the impact of known calcium sensitivity modulating small molecules on the free energy landscape of cNTnC hydrophobic patch opening, umbrella sampling was utilized on systems of cNTnC with small molecules bound. The structures of cNTnC bound to the molecules W7, bepridil, 3-methyldiphenylamine, and dfbp-o have been determined by NMR, and were used in the umbrella sampling scheme. Similar to our previously established umbrella sampling scheme,28 these structures were first simulated in steered MD simulations to obtain starting frames for the umbrella sampling simulations. The starting structures were steered from a semi-open configuration to a closed configuration over 10 ns. Additionally, to obtain open structures, the semi-open starting structures were opened over 10 ns. This allowed for a full coverage of interhelical angles between the open to closed states (90° to 140°) which corresponded to interhelical distances of about 13 Å to 28 Å. The correlation of the interhelical angles and interhelical distances for the cNTnC–DPA simulation can be seen in Figure 1A. In these simulations, the closed states were able to accommodate DPA and dfbp-o. Minimal movement was required by bepridil and W7 to be accommodated in the closed state. Frames were then selected for the umbrella sampling simulations based on their correlation between interhelical angle and distance, in 2.5° intervals between 90° and 140° (corresponding to interhelical distances of about 13 Å to 28 Å in 0.5 Å intervals). The umbrella sampling simulations were each run for 10 ns with no restraints on the small molecules. Coverage of the entire interhelical distances for the cNTnC–DPA simulation can be seen in Figure 1B. These simulations were then analyzed with a weighted histogram analysis method (WHAM) to extract a free energy profile of opening. The free energies of opening reported here reflect the difference of potential of mean force values at 130° and 90° degrees as generated by application of WHAM to the respective simulations.
Figure 1.

A) Calculated interhelical angles plotted against interhelical distances from the frames of the steered MD of cNTnC–DPA. The two quantities were strongly correlated suggesting that the interhelical distance can be used as an indirect measure of interhelical angle. B) Interhelical angles and distances were calculated for each of the windows in the umbrella sampling simulations of cNTnC–DPA and plotted against each other. There was sufficient overlap between the angles to ensure adequate sampling.
The previously calculated value of the small molecule-free cNTnC free energy of opening, 13.8 ± 2.2 kcal/mol,28 was used for comparison in this study. As seen in Figure 2, all the small molecules used in this study reduced the free energy of opening the hydrophobic patch region, suggesting a significant destabilization of the closed configuration relative to the open configuration. Since the umbrella sampling protocol provides relative free energies, the absolute extent to which either conformation is stabilized or destabilized cannot be readily extracted and compared among different protein–ligand complexes. In all cases, simulation results were congruent with experimental evidence, most recently summarized by Tikunova et al.50 According to this evidence, it is likely the case that bepridil operates through a stabilization of the open configuration as seen experimentally by an increase in calcium sensitivity when titrated in with cNTnC in the absence of the switch peptide. In silico, bepridil lowered the free energy to the greatest extent, at 1.6 ± 2.8 kcal/mol. This is likely to due to its larger size in comparison to the other small molecules. Dfbp-o lowered the relative free energy of opening to 3.4 ± 0.5 kcal/mol. DPA lowered the free energy of opening to 3.1 ± 0.5 kcal/mol, which was to the same extent as calcium desensitizer W7, at 3.1 ± 0.8 kcal/mol. In experiments similar to those performed with bepridil mentioned above, calcium sensitivity was not seen to increase when DPA analog 3-Cl-DPA is bound suggesting a different operative mechanism than stabilization of the open configuration. Due to the reduced size, the overall lowering in free energy of opening could come as a result of the destabilization of the closed configuration. We are suggesting that the observed overall lowering of the relative free energy of opening is likely the predominant mechanism for the calcium sensitization effects that are imparted by bepridil, dfbp-o, and DPA. By lowering the relative free energy of opening, the cNTnC open conformation can be sampled more readily facilitating more frequent binding to the cTnI-switch peptide. This would modulate calcium-induced contraction without altering the calcium concentration within the cell.
Figure 2.

Umbrella sampling results of cNTnC–bepridil, cNTnC–dfbp-o, cNTnC–DPA, cNTnC–W7 systems. Free energies of opening were determined by WHAM analysis of umbrella sampling simulations. Small molecule-bound forms of cNTnC exhibited a lower relative free energy of opening compared to the corresponding small molecule-free system. These values were calculated from triplicate simulations.
DPA and dfbp-o lower the relative free energy of cNTnC bound to cTnI
As shown above, simulations without the cTnI-switch peptide present elucidated a stabilization of the open configuration compared to the apo (non-small molecule bound) state. The larger molecules, W7 and bepridil, have been shown to interfere with cTnI-switch peptide binding in previous studies.41,42,51 Therefore, simulations with the cTnI-switch peptide present were performed to assess this effect. Models used in the above umbrella sampling scheme have partner structures that have the cTnI-switch peptide bound, and these were used in this study. Steered MD simulations were carried out and then starting frames were chosen based on the correlation between interhelical angle and interhelical distance. The 10 ns umbrella sampling simulations were run on these starting frames and WHAM was utilized to analyze the resulting trajectories. The previously calculated value of the free energy of opening of cNTnC with cTnI-switch peptide present, –6.3 ± 0.2 kcal/mol, was used a comparative value for this analysis.28 Negative relative free energies of opening indicate that the open cTnC conformation is more stable than the closed cTnC conformation as predicted by Stevens et al.18 As seen in Figure 3, DPA lowered the relative free energy of cNTnC bound with the switch-peptide present to the greatest extent, with a measured free energy of –11.8 ± 1.5 kcal/mol, while dfbp-o lowered the relative free energy to –9.9 ± 4.9 kcal/mol. The DPA results in particular agree with the experimental findings of Tikunova et al, while dfbp-o also has a substantially smaller size than bepridil allowing for its accommodation in the hydrophobic pocket of the closed configuration. Bepridil decreased the stability of the cTnC–cTnI-switch peptide complex, compared to the apo form, to –2.6 ± 0.4 kcal/mol. This suggests that in the presence of bepridil the open configuration is still more stable than the closed conformation. However, there are disruptions to the stabilization (an increase from –6.3 to –2.6 kcal/mol) imparted by the interaction of the small molecule with the cTnI-switch peptide. These findings are in agreement with previous work by the Sykes group that demonstrated that the bepridil and cTnI-switch peptide together have a negative cooperativity of binding,41 this would suggest a destabilization of the open configuration of cNTnC. The ability to correctly capture this energetic behavior speaks to the strength of the umbrella sampling methodology to assess the impact of these small molecules. W7, likewise, increased the relative stability of the complex compared to the apo cNTnC–cTnI-switch peptide system to an even greater extent than bepridil, to –1.7 ± 0.7 kcal/mol. Again, this is in agreement with previous work on structure determination of W7 in complex with cNTnC and cTnI-switch peptide which concluded that W7 decreased the affinity for cTnI by 13-fold, perturbed the cNTnC structure, and shifted the cTnI from a preferred binding site.14,51
Figure 3.

Umbrella sampling results of cNTnC–bepridil–cTnI, cNTnC–dfbp-o–cTnI, cNTnC–DPA–cTnI, cNTnC–W7–cTnI systems. Free energies of opening were determined by WHAM analysis of umbrella sampling simulations. Small molecule-bound forms of cNTnC–cTnI-switch peptide complexes exhibited two responses to the presence of the small molecules. DPA and dfbp-o increased the relative stability of the open state of the complex more than cTnI by itself. Bepridil and W7 disrupted the relative stability of the open configuration of the cNTnC–cTnI complex. All values were calculated from triplicate simulations.
Furthermore, the hypothesis that steric clashes between W7 and bepridil with the inhibitory peptide impact the stability of the complex was further investigated by evaluation of the small molecule volumes. Integration of the solvent excluded surface area as calculated by USCF Chimera52 yielded volumes for DPA, dfbp-o, W7, and bepridil of 167, 199, 297, and 368 Å3 respectively. The amount of space occupied by W7 and bepridil was at least 50% higher compared to DPA and dfbp-o, as shown in Figure 4. In addition, we note that W7 and bepridil are the only compounds in the test set that both contain positively charged ammonium groups. It is possible that electrostatic repulsion plays a role in the relative destabilization of these complexes with cTnI compared to the complex without it. In particular, cTnI residues R141 and R161 may contribute to this effect. In PDB ID 2KRD, R141 is in close proximity to W7, though the termini of the truncated inhibitory peptide are noticeably distorted. We speculate that the extreme conformational flexibility of the alkyl chain which terminates in an ammonium functional group and the propensity of this tail to be solvent exposed cause positional binding in a manner that contributes to its overall function as a calcium desensitizer. This is in contrast to bepridil for which the ammonium functionality is less conformationally flexible and could be significantly shielded from unfavorable contacts.
Figure 4:

The volumes occupied by the small molecules, bepridil, dfbp-o, DPA, and W7 bound to cNTnC. All of these small molecules lowered the relative free energy of opening when no cTnI was present in the simulations. Bepridil, with the largest occupied volume, lowered the relative free energy of opening to the greatest extent. The two small molecules that further increased the relative stability of the cNTnC–cTnI complex, dfbp-o and DPA, occupied the least amount of space, whereas bepridil and W7 occupied significantly more space and disrupted the relative stability of the cNTnC–cTnI complex.
Potentially weak binding small molecules escape the cNTnC in conventional MD
We investigated the behavior of small molecules that were predicted as poor binders with molecular dynamics simulations to confirm that not all random small molecules would stabilize the cTnC open configuration as observed for the four known cTnC binders above. One small molecule, NSC34337, was chosen from a docking screen targeting cNTnC and was specifically selected based upon its poor Glide score and hydrophilic character. The starting structures used was its docked pose from the small molecule screen. Models were run in triplicate MD simulations for 100 ns after an energy minimization and equilibration. The resulting trajectories were analyzed by calculating the interhelical angle. NSC34337, after less than 3 ns, left the hydrophobic pocket and began to explore the water box of the simulation. Once having left the pocket, the cNTnC began to adopt the closed configuration, as seen in Figure 5A. In Figure 5B, NSC34337 can be seen moving throughout the water box in the simulations and not returning to the hydrophobic pocket. For comparison, triplicate 100ns conventional MD simulations of the known calcium-sensitivity modulators (DPA, dfbp-o, bepridil and W7) were performed. The known binders did not dissociate from the cNTnC in the simulations. This suggests that the presence of a random small molecule within the hydrophobic pocket is insufficient to stabilize the open configuration. There are key and specific interactions that are being captured within the MD simulations between the known small molecule binders that do not exist with these hydrophilic, predicted poor binders.
Figure 5.

(A) Interhelical angle calculated for triplicate 100 ns molecular dynamics simulations of predicted poor binder NSC34337 bound to cNTnC. In all three simulations, the small molecule left within less than 3 ns of the simulations, allowing cNTnC to close immediately. (B) The center-of-mass distances for cNTnC–NSC34337 over triplicate 100 ns molecular dynamics simulations from the starting position. NSC34337 left the hydrophobic pocket immediately, resulting in a loss of all key binding interactions seen for the known binders
Conclusions
We utilized an umbrella sampling protocol to understand the impact of small molecule calcium sensitivity modulators on cNTnC. This method provided a targeted approach to investigate a potential mechanism of calcium sensitivity modulation. Calcium sensitizers bepridil, dfbp-o, and DPA all lowered the free energy cost of opening of the calcium-bound cNTnC. This suggested a stabilization of an open configuration, allowing the cTnI to bind more readily to cNTnC, which would manifest as an apparent increase in calcium sensitivity. The known calcium desensitizer W7 also lowered the free energy of opening of the calcium-bound cNTnC, implying that its desensitization is conferred through a different mechanism.
Umbrella sampling of the cNTnC–cTnI complex with the small molecules bound revealed the impacts on stability imparted by the known calcium sensitivity modulators. Both dfbp-o and DPA lowered the relative free energy of opening for the cNTnC–cTnI complex to an even larger extent than in the case when cTnI was not present. By lowering the barrier between the open and closed states of this cNTnC–cTnI complex, it is likely that these calcium sensitizers can also sensitize by increasing cTnI affinity to cNTnC, thereby maintaining this interaction. W7, in contrast to dfbp-o and DPA, lowered the stability of the cNTnC–cTnI complex. The open configuration is still favored over the closed configuration, but W7’s destabilization could lead to a decrease in cTnC–cTnI affinity. This is likely dominated by W7 displacing the cTnI from a favorable binding mode, perhaps from highly unfavorable electrostatic interactions between W7 and the inhibitory peptide as discussed above. Bepridil, the known calcium sensitizer, decreased the stability of the cNTnC–cTnI complex to be open. This could be a result of the large size of the bepridil that disrupts the optimal binding of the cTnI to the cNTnC.
Conventional molecular dynamics simulations revealed that the presence of predicted poor binder NSC34337 did not stabilize the open configuration, as seen for the known binding small molecules. This molecule left the hydrophobic patch immediately and allowed the cNTnC to close within nanoseconds. Simply having any small molecule within the hydrophobic patch is insufficient to alter the free energy landscape between open and closed configurations and compound hydrophobicity is necessary to more readily access an open or semi-open configuration.
In summary, we have shown that the umbrella sampling scheme previously used to study the effects of hypertrophic and dilated cardiomyopathy mutations on cNTnC can be extended to probe the effects of known small molecule binders on the free energy of opening. This protocol can be used to probe the proposed mechanisms of calcium sensitivity modulation achieved through small molecule binding and can potentially act as a filter for the possible effects of other small molecules that have not been experimentally tested. Development and application of this methodology has the ability to give insight into the design and creation of specific treatments for cardiomyopathies.
Supplementary Material
Table 1.
The free energy of opening, as determined by WHAM analysis of umbrella sampling simulations, for cNTnC, cNTnC–cTnI, cNTnC–bepridil, cNTnC–cTnI–bepridil, cNTnC–dfbp-o, cNTnC–cTnI–dfbp-o, cNTnC–DPA, cNTnC–cTnI–DPA, cNTnC–W7, cNTnC–cTnI–W7. The free energy values were calculated from triplicate umbrella sampling simulations.
| cTnC complex | Free energy of opening(kcal/mol) |
|---|---|
| Holo cTnC | 13.8 ± 2.2 |
| Tnl-bound Holo | −6.3 ± 0.2 |
| Bepridil | 1.6 ± 2.8 |
| Tnl-bound Bepridil | −2.6 ± 0.4 |
| dFBP-o | 3.4 ± 0.5 |
| Tnl-bound dFBP-o | −9.9 ± 4.9 |
| DPA | 3.1 ± 0.5 |
| Tnl-bound DPA | −11.8 ± 1.5 |
| W7 | 3.1 ± 0.8 |
| Tnl-bound W7 | −1.7 ± 0.7 |
Acknowledgements
We would like to thank members of the Lindert lab for helpful discussions. We would also like to thank the Ohio Supercomputer Center53 for valuable computational resources. This work was supported by NIH (R01 HL137015 to S.L.).
References
- 1.Manning EP; Schwartz SD, A Model of Calcium Activation of the Cardiac Thin Filament. Biochemistry 2011, 50, 7405–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sundaralingam M; Bergstrom R; Strasburg S; Rao ST; Roychowdhury P; Greaser M; Wang BC, Molecular Structure of Troponin C from Chicken Skeletal Muscle at 3-Angstrom Resolution. Science 1985, 227 (4689), 945ߝߝ948. [DOI] [PubMed] [Google Scholar]
- 3.Herzberg O; Moult J; James MNG, A model for the Ca2+-induced conformational transition of troponin C. A trigger for muscle contraction. J. Biol. Chem 1986, 261 (6), 2638––2644. [PubMed] [Google Scholar]
- 4.Spyracopoulos L; Li MX; Sia SK; Gagne S; Chandra M; Solaro RJ; Sykes BD, Calcium-Induced Structural Transition in the Regulatory Domain of Human Cardiac Troponin C. Biochemistry 1997, 36 (40), 12138––12146. [DOI] [PubMed] [Google Scholar]
- 5.Gordon A; Regnier M; Homsher E, Skeletal and cardiac muscle contractile activation: tropomyosin “rocks and rolls”. Physiology 2001, 16 (2), 49–55. [PubMed] [Google Scholar]
- 6.Li MX; Spyracopoulos L; Sykes BD, Binding of cardiac troponin-I147–163induces a structural opening in human cardiac troponin-C. Biochemistry 1999, 38 (26), 8289––8298. [DOI] [PubMed] [Google Scholar]
- 7.Kalyva A; Parthenakis FI; Marketou ME; Kontaraki JE; Vardas PE, Biochemical characterisation of Troponin C mutations causing hypertrophic and dilated cardiomyopathies. J. Muscle Res. Cell Motil 2014, 35 (2), 161––178. [DOI] [PubMed] [Google Scholar]
- 8.Li MX; Hwang PM, Structure and function of cardiac troponin C ( TNNC1 ): Implications for heart failure , cardiomyopathies , and troponin modulating drugs. Gene 2015, 571 (2), 153–−166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai F; Li MX; Pineda-Sanabria SE; Gelozia S; Lindert S; West F; Sykes BD; Hwang PM, Structures reveal details of small molecule binding to cardiac troponin. J. Mol. Cell. Cardiol 2016, 101, 134–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y; Love ML; Putkey JA; Cohen C, Bepridil opens the regulatory N-terminal lobe of cardiac troponin C. Proc. Natl. Acad. Sci 2000, 97 (10), 5140–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robertson IM; Sun Y-B; Li MX; Sykes BD, A structural and functional perspective into the mechanism of Ca2+-sensitizers that target the cardiac troponin complex. J. Mol. Cell. Cardiol 2010, 49 (6), 1031–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Igarashi T; Takeda S; Mori H, Crystal structure of the N-terminal domain of human cardiac troponin C in complex with a calcium-sensitizer; trifluoperazine. J. Mol. Cell. Cardiol 2005, 39 (6), 1016–1016. [Google Scholar]
- 13.Pineda-Sanabria SE; Robertson IM; Sun Y-B; Irving M; Sykes BD, Probing the mechanism of cardiovascular drugs using a covalent levosimendan analog. J. Mol. Cell. Cardiol 2016, 92, 174–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman RMB; Sykes BD, Structure of the Inhibitor W7 Bound to the Regulatory Domain of Cardiac Troponin C. Biochemistry 2009, 48 (24), 5541–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindert S; Li MX; Sykes BD; McCammon JA, Computer-aided drug discovery approach finds calcium sensitizer of cardiac troponin. Chem. Biol. Drug Des 2015, 85 (2), 99––106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aprahamian ML; Tikunova SB; Price MV; Cuesta AF; Davis JP; Lindert S, Successful Identification of Cardiac Troponin Calcium Sensitizers Using a Combination of Virtual Screening and ROC Analysis of Known Troponin C Binders. J. Chem. Inf. Model 2017, 57 (12), 3056––3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dewan S; McCabe KJ; Regnier M; McCulloch AD; Lindert S, Molecular Effects of cTnC DCM Mutations on Calcium Sensitivity and Myofilament Activation-An Integrated Multi-scale Modeling Study. J. Phys. Chem. B 2016, 120 (33), 8264––8275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stevens CM; Kaveh R; Gurpreet S; Bairam L; Tieleman DP; Tibbits GF, Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca2+-sensitizing mutations. J. Biol. Chem 2017, 292 (28), 11915––11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindert S; Kekenes-Huskey PM; Huber G; Pierce L; McCammon JA, Dynamics and calcium association to the N-terminal regulatory domain of human cardiac troponin C: a multiscale computational study. J. of Phys. Chem. B 2012, 116 (29), 8449–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jayasundar JJ; Xing J; Robinson JM; Cheung HC; Dong W-J, Molecular Dynamics Simulations of the Cardiac Troponin Complex Performed with FRET Distances as Restraints. PLoS One 2014, 9 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varughese JF; Chalovich JM; Li Y, Molecular Dynamics Studies on Troponin (TnI-TnT-TnC) Complexes: Insight into the Regulation of Muscle Contraction. J. Biomol. Struc. Dyn 2010, 28 (2), 159––174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stevens CM; Rayani K; Genge CE; Singh G; Liang B; Roller JM; Li C; Li AY; Tieleman DP; Petegem FV; Tibbits GF, Characterization of Zebrafish Cardiac and Slow Skeletal Troponin C Paralogs by MD Simulation and ITC. Biophys. J 2016, 111 (1), 38––49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kekenes-Huskey PM; Lindert S; McCammon JA, Molecular basis of calcium-sensitizing and desensitizing mutations of the human cardiac troponin C regulatory domain: a multi-scale simulation study. PLoS Comput. Biol 2012, 8 (11), e1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng Y; Lindert S; Kekenes-Huskey P; Rao VS; Solaro RJ; Rosevear PR; Amaro R; McCulloch AD; McCammon JA; Regnier M, Computational studies of the effect of the S23D/S24D troponin I mutation on cardiac troponin structural dynamics. Biophys. J 2014, 107 (7), 1675––1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindert S; Kekenes-Huskey PM; McCammon JA, Long-timescale molecular dynamics simulations elucidate the dynamics and kinetics of exposure of the hydrophobic patch in troponin C. Biophys. J 2012, 103 (8), 1784––1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Genchev GZ; Kobayashi T; Lu H, Calcium Induced Regulation of Skeletal Troponin — Computational Insights from Molecular Dynamics Simulations. PloS One 2013, 8 (3), 1––10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stevens CM; Rayani K; Singh G; Lotfalisalmasi B; Tieleman DP; Tibbits GF, Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca2+-sensitizing mutations. J. Biol. Chem 2017, 292 (28), 11915––11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bowman JD; Lindert S, Molecular Dynamics and Umbrella Sampling Simulations Elucidate Differences in Troponin C Isoform and Mutant Hydrophobic Patch Exposure. J. Phys. Chem. B 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varughese JF; Baxley T; Chalovich JM; Li Y, A Computational and Experimental Approach To Investigate Bepridil Binding with Cardiac Troponin. J. Phys. Chem 2011, 115 (10), 2392––2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Torrie GM; Valleau JP, Nonphysical sampling distributions in Monte Carlo free energy estimation: Umbrella sampling. J. Comput. Phys 1977, 23 (2), 187––199. [Google Scholar]
- 31.Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD, Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput 2012, 8 (9), 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD Jr., CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem 2010, 31 (4), 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu W; He X; Vanommeslaeghe K; MacKerell AD Jr., Extension of the CHARMM General Force Field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem 2012, 33 (31), 2451–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vanommeslaeghe K; MacKerell AD Jr, Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 2012, 52 (12), 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanommeslaeghe K; Raman EP; MacKerell AD, Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model 2012, 52 (12), 3155–3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mayne CG; Saam J; Schulten K; Tajkhorshid E; Gumbart JC, Rapid parameterization of small molecules using the force field toolkit. J. Comput. Chem 2013, 34 (32), 2757–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16 Rev. B.01, Wallingford, CT, 2016. [Google Scholar]
- 38.Hariharan PC; Pople JA, The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28 (3), 213–222. [Google Scholar]
- 39.Hehre WJ; Ditchfield R; Pople JA, Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys 1972, 56 (5), 2257–2261. [Google Scholar]
- 40.Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML, Comparison of simple potential functions for simulating liquid water. J. Chem. Phys 1983, 79 (2), 926–935. [Google Scholar]
- 41.Wang X; Li MX; Sykes BD, Structure of the regulatory N-domain of human cardiac troponin C in complex with human cardiac troponin I147–163 and bepridil. J. Biol. Chem 2002, 277 (34), 31124–31133. [DOI] [PubMed] [Google Scholar]
- 42.Oleszczuk M; Robertson IM; Li MX; Sykes BD, Structure of the Regulatory Domain of Human Cardiac Troponin C in Complex with the Switch Region of Cardiac Troponin I and the Drug W7: The Basis of W7 as an Inhibitor of Cardiac Muscle Contraction. Biophys. J 2010, 98 (3), 150a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phillips JC; Braun R; Wang W; Gumbart J; Tajkhorshid E; Villa E; Chipot C; Skeel RD; Kal L; Schulten K, Scalable molecular dynamics with NAMD. J. Comput. Chem 2005, 26 (16), 1781––1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brooks BR; Brooks CL; Mackerell AD; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M, CHARMM: The biomolecular simulation program. J. Comput. Chem 2009, 30 (10), 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fiorin G; Klein ML; Hénin J, Using collective variables to drive molecular dynamics simulations. Mol. Phys 2013, 111 (22–23), 3345––3362. [Google Scholar]
- 46.Soman J; Tao T; Phillips GN, Conformational variation of calcium-bound troponin C. Proteins: Struct., Funct., Genet 1999, 37 (4), 510––511. [DOI] [PubMed] [Google Scholar]
- 47.Grossfield A, WHAM: the weighted histogram analysis method. version 2.0.9, http://membrane.urmc.rochester.edu/wordpress/?page_id=126
- 48.Kumar S; Rosenberg JM; Bouzida D; Swendsen RH; Kollman PA, The weighted histogram analysis method for free‐energy calculations on biomolecules. I. The method. J. Comput. Chem 1992, 13 (8), 1011–1021. [Google Scholar]
- 49.Hunter JD, Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng 2007, 9 (3), 90–95. [Google Scholar]
- 50.Tikunova SB; Cuesta A; Price M; Li MX; Belevych N; Biesiadecki BJ; Reiser PJ; Hwang PM; Davis JP, 3-Chlorodiphenylamine activates cardiac troponin by a mechanism distinct from bepridil or TFP. J. Gen. Physiol 2019, 151 (1), 9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai F; Hwang PM; Sykes BD, Structural Changes Induced by the Binding of the Calcium Desensitizer W7 to Cardiac Troponin. Biochemistry 2018, 57 (46), 6461–6469. [DOI] [PubMed] [Google Scholar]
- 52.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE, UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25 (13), 1605––1612. [DOI] [PubMed] [Google Scholar]
- 53.Ohio Supercomputer Center. 1987.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.