

Abstract
The divergent sequences, protein structures, and catalytic mechanisms of serine- and metallo-β- lactamases hamper the development of wide-spectrum β-lactamase inhibitors that can block both types of enzymes. The O-aryloxycarbonyl hydroxamate inactivators of Enterobacter cloacae P99 class C serine-β-lactamase are unusual covalent inhibitors in that they target both active-site Ser and Lys residues, resulting in a crosslink consisting of only two atoms. Many clinically-relevant metallo-β-lactamases have an analogous active-site Lys residue used to bind β-lactam substrates, suggesting a common site to target with covalent inhibitors. Here, we demonstrate that an O-aryloxycarbonyl hydroxamate inactivator of serine β-lactamases can also serve as a classical affinity label for New Delhi metallo-β-lactamase-1 (NDM-1). Rapid dilution assays, site-directed mutagenesis, and global kinetic fitting are used to map covalent modification at Lys211 and determine KI (140 μM) and kinact (0.045 min−1) values. Mass spectrometry of the intact protein and the use of ultraviolet photodissociation (UVPD) for extensive fragmentation confirm stoichiometric covalent labeling that occurs specifically at Lys211. A 2.0 Å resolution X-ray crystal structure of inactivated NDM-1 reveals that the covalent adduct is bound at the substrate-binding site, but is not directly coordinated to the active-site zinc cluster. These results indicate that Lys-targeted affinity labels might be a successful strategy for developing compounds that can inactivate both serine and metallo-β-lactamases.
Graphical Abstract
INTRODUCTION
β-lactam drugs comprise a large proportion of therapeutics regularly used to treat bacterial infections, but their usefulness is threatened by emergence of resistant bacteria that often produce β-lactamase enzymes that degrade these drugs.1 Co-treatment with β-lactamase inhibitors can effectively extend the lifetime and usefulness of β-lactam drugs. However, one of the challenges in designing wide-spectrum β-lactamase inhibitors is the different catalytic mechanisms used by metallo-β-lactamases and serine-β-lactamases. Clinically-used β-lactamase inhibitor co-drugs such as avibactam work by mimicking the normal substrate and stabilizing a covalent reaction intermediate formed at the active-site Ser of serine-β-lactamases.2 Therefore, these co-drugs can not effectively inhibit metallo-β-lactamases that instead use a hydroxide nucleophile that is bound non-covalently at a dinuclear zinc ion site between zinc-1 (ligated by 3 His residues) and zinc-2 (ligated by Cys, His and Asp residues) (Figure 1).3, 4
Figure 1.

Proposed mechanisms of inactivation and turnover. A) Proposed mechanism for inactivation of AmpC β-lactamase by an O-aryloxycarbonyl hydroxamate (1), with the phenol leaving-group shown in red, the hydroxamate ‘arm’ in blue (3), and the crosslinking carbonyl in green (4). B) Proposed mechanism of chromacef turnover by NDM-1, showing Michaelis complex (5), anionic intermediate (6), and product complex (7), all noncovalently bound near zinc-1 (Zn1) and zinc-2 (Zn2) of the active site dinuclear zinc cluster. Each enzyme has a Lys residue (A, K315; B, K211) that serves a similar function in binding β-lactam substrates.
Pratt and co-workers previously described a novel type of covalent inhibitor for serine β-lactamases that possibly represents an alternative strategy (Figure 1A).5–9 These O-aryloxycarbonyl hydroxamates (e.g. 1), originally designed to mimic β-lactamase substrates, react covalently with the active site Ser of serine-β-lactamases (2), as do typical substrates. However, after the initial covalent adduct is formed, the same carbonyl carbon that was first attacked by the active-site Ser is subsequently attacked by a neighboring Lys, which otherwise serves as a binding partner for the carboxylate of β-lactam substrates.5, 10 The resulting product left behind (4) consists of only two atoms - a single carbonyl group - that covalently crosslinks two active-site residues, thereby abrogating activity.5 Although metallo-β-lactamases lack a nucleophilic Ser, many contain an analogous Lys residue that helps zinc-2 anchor the carboxylate of β-lactam substrates (Figure 1B).11 Because the O-aryloxycarbonyl hydroxamates were designed as substrate mimics, because they can target other β-lactam-binding enzymes including the serine β-lactamases TEM-2 and OXA-1, the R39 DD-peptidase from Actinomadura, and pencillin acylase, and because many metallo-β-lactamases share an analogous Lys residue that serves the same binding function, we hypothesized that the same affinity label designed for serine-β-lactamases might also work with metallo-β-lactamases.6, 12 Here, we show that N-(benzyloxycarbonyl)-O-[(phenoxycarbonyl)]hydroxylamine (1) also acts as a classical affinity label of New Delhi metallo-β-lactamase-1 (NDM-1) and specifically targets the active-site Lys211 for covalent modification. Although further optimization will be required, this example serves as a proof of principle that Lys-targeted affinity labels can be used to target both mechanistically distinct classes of serine- and metallo-β-lactamase enzymes.
EXPERIMENTAL PROCEDURES
Purified NDM-1.
For the experiments described below, a purified NDM-1 truncation missing the initial 35 amino acids was used, as described previously.13 This truncation corresponds to the most predominant soluble fragment found in overexpression cultures of full-length NDM-1, and removes the site of posttranslational lipid modification that otherwise leads to membrane association.
Time-dependent Inactivation of NDM-1 and NDM variants.
To monitor time-and concentration-dependent inactivation of NDM-1, we preincubated NDM-1 (2 μM) with various concentrations of 1 (0 – 2.0 mM). Stock solutions of 15 (40 mM) were prepared in dry acetonitrile (ACROS organic), with a final co-solvent concentration in preincubation and assay solutions of 5% (v/v). At increasing time points, aliquots of the preincubation mixture were rapidly diluted (100-fold) into saturating amounts of competing substrate for subsequent determination of remaining enzyme activity by determining initial rates. Conditions and procedures used were the same as those described previously (50 mM HEPES, pH 7.0, 25 °C)14, except that we added 0.02% Tween-20 and substituted chromacef15 (20 μM, a generous gift from Prof. L. Sutton, Benedictine College, Atchison, KS) in the place of nitrocefin as the reporter substrate, using Δε442nm= 14500 M−1cm−1 as described previously.16 The purified NDM-1 and its C208D, and K211A variants were described earlier.14, 17 To compensate for differences in kcat and KM values and zinc affinity among NDM-1 mutants, different concentrations of C208D NDM-1 (15 μM; this mutant has lower kcat and KM values than wild-type and binds zinc-2 more weakly) and K211A NDM-1 (0.2 μM; this mutant has higher kcat and KM values than wild-type) were used in the preincubation mixtures, and the concentrations of ZnSO4 (10 μM) and inhibitor (1 mM) were increased in comparison to similar experiments using wild-type NDM-1.
Test for Inactivation by Product.
To test whether a reaction product of NDM-1 and 1 was responsible for the observed inactivation, we used the procedure above to monitor inactivation of NDM-1 (2 μM) by 1 (500 μM) during a 90 min incubation, and then a second aliquot (1 μL) of a fresh stock of uninhibited NDM-1 (1 mM, ~2 μM final concentration added) was added and the rapid dilution assay continued for an additional 60 min.
Non-Enzymatic Hydrolysis of 1.
Under the conditions used for enzyme labeling, 1 undergoes non-enzymatic hydrolysis to release phenol.5 The largest absorbance difference between 1 and phenol stock solutions occurs at 278 nm, so we used dilutions of a phenol standard solution (Ricca Chemical Company, Arlington, TX) to construct a linear standard curve that relates absorption at 278 nm to phenol concentration in the same buffer solution used for enzyme inactivation. A time-dependent increase in absorbance at 278 nm was then monitored upon dilution of 1 (0.4 mM) in assay buffer in the absence of enzyme, and the standard curve used to convert the change in absorption into a graph depicting phenol formation over time.
Global Kinetic Fitting.
Data for non-enzymatic 1 hydrolysis and the time- and concentration-dependent inactivation of NDM-1 by 1 were loaded into KinTek Global Kinetic Explorer Version 8 (Kintek Co., Snow Shoe, PA).18, 19 A kinetic model was entered to describe the non-enzymatic decay of 1 to an inactive product, as well as a two-step mechanism for inactivation of NDM-1 that includes an initial reversible binding step, followed by an irreversible inactivation. The on-rate for binding was fixed at a typical diffusion-controlled rate (109 M−1 s−1), the off-rate and inactivation rates were kept variable, and the other experimental values (see above) were entered as starting conditions. The non-enzymatic decay rate constant, the rate constant for 1 dissociation and the NDM-1 inactivation rate constant were then determined by global fitting and reported with the standard errors given by Global Kinetic Explorer for the best fits.
Mass Spectrometry.
NDM-1 (16 μM) was incubated with 1 (2 mM) for 20 h in HEPES buffer (50 mM) pH 7.0. An untreated NDM-1 solution was used as a control. Prior to MS analysis, a centrifugal 10 kDa molecular weight cut-off filter was used to exchange buffer for water and to concentrate each protein solution. This samples were diluted to make ~15 μM protein solutions in 59.25:39.25:0.5 acetonitrile:water:formic acid (v/v/v). The solutions were then infused into a Thermo Fisher Scientific Elite Orbitrap Mass Spectrometer equipped with a Coherent ExciStar ArF (193 nm, 500 Hz pulse rate) excimer laser (Santa Clara, CA) at a rate of 1.20 μL/min with a spray voltage of +3.5kV. MS1 spectra for intact proteins were collected at 120000 resolution at m/z 400. The 28+ charge state of NDM-1 inactivated by 1 was selected for UVPD and activated using a single 2.0 mJ laser pulse. UVPD was performed in the higher collision energy dissociation (HCD) cell located at the back end of the Orbitrap mass spectrometer as described previously,20 with the pressure adjusted to 5 mTorr in the HCD cell. The resulting MS/MS spectra from UVPD were collected at 240000 resolution at m/z 400 and averaged for 1000 scans. Both the ESI mass spectra and the UVPD MS/MS spectra were deconvolved using Xtract (Thermo Fisher Scientific) with a signal-to-noise cut-off of 2. MS/MS fragments were assigned using a modified version of ProSightPC to accommodate UVPD fragment ions with a fragment mass tolerance of 10 ppm.
Crystallization of 1-treated NDM-1.
12 mg/mL purified NDM-1 (with the initial 35 residues truncated) was preincubated with 1. Crystals of the resulting complex were grown at room temperature by vapor diffusion using the sitting drop method from 0.2 M calcium chloride dihydrate, 20% polyethylene glycol (PEG) 3350.
X-ray data collection and processing.
A crystal resulting from 1-treated NDM-1 was removed from its drop using a nylon loop, and flash frozen in liquid nitrogen. Diffraction data were collected at 100 K at the Advanced Light Source beamline 5.0.3 at the Lawrence Berkeley National Laboratory with the assistance of the Berkeley Center for Structural Biology. Data were processed using HKL2000.21
Structure determination.
Cell parameters of the crystal described above suggested that the asymmetric unit might contain two protein molecules. The solution of two NDM-1 molecules (each lacking the initial 35 residues) in the asymmetric unit was solved by molecular replacement with Phaser22 using the structure of NDM1 with the initial 46 residues truncated23 (PDB accession code 3S0Z) as the search model.
Model building was carried out using Coot.24 Refinement of models was done using PHENIX.25 There were several rounds of refinement followed by manual rebuilding of the model. To facilitate manual rebuilding, difference maps and a 2Fo – Fc map, σA-weighted to eliminate bias from the model,26 were prepared. A portion (5%) of the diffraction data was set aside throughout refinement for cross-validation.27 MolProbity28 was used to determine areas of poor geometry and to make Ramachandran plots. The final model does not include side chain atoms for which there was no observed electron density. Coordinates and structure factors were deposited in the Protein Data Bank (accession code 6OVZ).
RESULTS AND DISCUSSION
Affinity Labeling of NDM-1 by 1.
To test for affinity labeling, we first incubated NDM-1 with an excess of 1, removed aliquots at successive time points, and diluted each into saturating amounts of excess substrate to test for remaining activity. Substrate is expected to outcompete any inhibitor that is bound non-covalently to the active site and result in fully recovered activity. However, covalent labeling is expected to be irreversible and result in a time-dependent loss in activity. 1 causes a time-dependent loss in NDM-1 activity that is irreversible to dilution (Figure 2A), consistent with covalent bond formation. However, one alternative explanation is the slow accumulation of a product that can instead serve as an inhibitor.29 For example, hydrolysis of 1 can yield a hydroxamic acid (3), which is a common metal-binding pharmacophore that could possibly inactivate NDM-1 by metal ion removal.30 We tested for inactivation by accumulating product by inactivating NDM-1, and then adding a second, fresh aliquot of uninhibited NDM-1 to the same preincubation solution. If accumulating product was responsible for inhibition, faster inactivation rates are expected after the second aliquot. However, the observed inactivation rate after the second addition of NDM-1 (0.007 ± 0.001 min−1) was less than after the first addition (0.012 ± 0.001 min−1), and is therefore inconsistent with the proposal of an accumulating product as the inactivating species (Figure 2B).
Figure 2.

Time-dependent inactivation of NDM variants. A) Dilution assays indicate time-dependent irreversible inactivation of NDM-1 by 1 ( , 10 μM), in contrast to a control incubation that omits the inactivator (
, 10 μM), in contrast to a control incubation that omits the inactivator ( ). B) Inactivation of NDM-1 by 1 (
). B) Inactivation of NDM-1 by 1 ( , 10 μM) is followed by addition of a second aliquot of fresh enzyme to the same preincubation tube (
, 10 μM) is followed by addition of a second aliquot of fresh enzyme to the same preincubation tube ( ), with the activity immediately after addition reset to 100%. A control incubation omits the inactivator (
), with the activity immediately after addition reset to 100%. A control incubation omits the inactivator ( ). C) Dilution assays indicate time-dependent irreversible inactivation of C208D NDM-1 by 1 (
). C) Dilution assays indicate time-dependent irreversible inactivation of C208D NDM-1 by 1 ( , 10 μM). The control incubation with inactivator omitted (
, 10 μM). The control incubation with inactivator omitted ( ), indicates lower stability of the C208D NDM-1 variant that wild type NDM-1. D) Dilution assays indicate no time-dependent irreversible inactivation of K211A NDM-1 by 1 (
), indicates lower stability of the C208D NDM-1 variant that wild type NDM-1. D) Dilution assays indicate no time-dependent irreversible inactivation of K211A NDM-1 by 1 ( , 10 μM), in comparison to a control incubation that omits the inactivator (
, 10 μM), in comparison to a control incubation that omits the inactivator ( ). Fitting errors from incubations that showed appreciable inactivation were <15 %.
). Fitting errors from incubations that showed appreciable inactivation were <15 %.
A few different types of covalent inhibitors for NDM-1 have previously been reported, and these inactivators target either Cys208, which serves as a ligand for zinc-2, or they target Lys211, which helps to bind the carboxylic acid found in most β-lactam substrates.14, 17, 31–33 We previously reported and characterized NDM-1 mutants at each of these sites: C208D, which removes all Cys residues from our soluble NDM-1 construct yet still supports zinc-2 binding, and K211A, which removes only one of eight total Lys residues, all of which, except for Lys125, are solvent accessible.17 Each of these two mutant NDM-1 enzymes (C208D and K211A) retains β-lactamase activity, so we tested each to see if a change in either side chain a could prevent time-dependent inactivation by 1. The C208D mutation of NDM-1 can still be inactivated by 1, suggesting that Cys208 is not targeted for covalent modification, nor is otherwise essential for inactivation (Figure 2C). In contrast, the K211A mutation of NDM-1 prevents inactivation by 1, indicating that either Lys211 is a nucleophile targeted for covalent modification and thereby enzyme inactivation, or that Lys211 is otherwise essential for 1 to bind or react (Figure 2D).
Because 1 and similar compounds are known to readily undergo hydrolysis,5 we sought to determine the rate constant for non-enzymatic hydrolysis under the same experimental conditions used for enzyme inactivation by quantifying the time-dependent release of phenol (Figures 3B, 3C). We also measured the time- and concentration-dependence of 1 inactivation of NDM-1 (Figures 3A, 3C). Under these conditions, the magnitude of the rate constant for nonenzymatic hydrolysis of 1 is similar to that for NDM-1 inactivation (see below). Therefore, we did not use a Kitz-Wilson analysis or Tsou-plot to determine inactivation parameters because the concentration of inactivator would not remain constant over the incubation time, but instead used a global simulation method to simultaneously fit both 1 hydrolysis and NDM-1 inactivation kinetics using the minimal kinetic mechanism shown in Figure 3A.18, 19, 34, 35 This mechanism proposes a one-step non-enzymatic hydrolysis of 1 to release phenol, and a two-step inactivation mechanism for NDM-1 that proceeds through rapid formation of an initial reversible non-covalent complex followed by an irreversible inactivation step. Using these assumptions, fits to the experimental data (Figure 3B, 3C) gave values for knon-enz (0.010 ± 0.001 min−1), k−1 ((8.6 ± 0.7) × 106 min−1)), and kinact (0.045 ± 0.004 min−1), which enabled the further calculation of KI (140 ± 10 μM) and kinact/KI (5.4 ± 0.9 M−1s−1) values. Use of a two-step model for enzyme inactivation that includes an initial non-covalent binding step is necessary to achieve good fits. For inactivation of serine-β-lactamases, the addition of an enzyme-catalyzed partitioning step (from the non-covalent EI complex) to form a non-inhibitory product is required to achieve good fits.5 Addition of a similar partitioning step to the kinetic mechanism for NDM-1 inactivation can also be accommodated, but does not greatly improve the fits, and so is not included in our proposed minimal kinetic mechanism (not shown). The second order inactivation rate constants (kinact/KI) of 1 for NDM-1 is approximately 1000-fold smaller than that for the serine-β-lactamase P99 (6100 M−1s−1),6 and approximately 2×106 -fold larger than for reaction with water (estimated by knon-enz / 55.5 M water). Taken together, these results are consistent with the proposal that 1 is a classical affinity label with moderate potency for NDM-1.
Figure 3.

Global kinetic fitting. a) Kinetic mechanism used to model non-enzymatic hydrolysis of 1 (top) and affinity labeling of NDM-1 (below). b) Non-enzymic hydrolysis of 1 (0.4 mM) was monitored by determining time-dependent production of phenol, and c) time- and concentration-dependent inactivation of NDM-1 was monitored by a dilution assay (see Methods), using 0 (black), 50 (red), 100 (orange), 250 (yellow), 500 (green), 1000 (blue), and 2000 (purple) μM 1. Kintek Global Kinetic Explorer was used to determine global fits of data in panels b and c, and are shown in dotted lines of corresponding color.
Mass Spectrometry.
To confirm and characterize covalent labeling, we used mass spectrometry to compare NDM-1 before and after incubation with 1. To determine the mass addition and stoichometry of any covalent adduct(s), we first used ESI-MS to characterize intact protein. The deconvolved mass spectra for intact NDM-1 before inactivation indicates a monoisotopic mass (24823.29 Da) that is increased by 193.11 Da in samples that have been incubated with excess 1 (25016.40 Da) (Figure 4). A very minor species is also observed at 25209.41 Da, indicating the presence of a small amount of double labeling by this ~193 Da covalent adduct. To determine which particular amino acid is covalently modified, we further characterized the 1-treated NDM-1 protein by using UVPD fragmentation of the precursor (28+ charge state) to obtain a very extensive sequence map (Figure 5), wherein backbone cleavages occur between nearly every pair of amino acids. This UVPD sequence map provides unambiguous identification of the major covalent modification site of 1 as residue Lys211, and also confirms the amino acid sequence of NDM-1 that is encoded in the blaNDM gene of our expression vector. The covalent +193.11 Da adduct matches very accurately the mass of the predicted adduct formed by attack of Lys211 on the carbonate carbon of 1, followed by loss of phenol and an additional proton from the attacking Lys residue (193.04 Da, see below). These results support the proposal that 1 inactivates NDM-1 specifically by a single covalent modification of a Lys residue found at the active-site of this enzyme.
Figure 4.

Deconvoluted mass spectra of intact NDM-1 before and after inactivation by 1. a) NDM-1 before inactivation showing the dominant species marked by E at 24823.29 Da. b) NDM-1 after inactivation, revealing a major species marked by E-I at 25016.40 Da, which indicates an adduct of 193.11 Da. A minor species is also observed, marked by E-I2 at 25209.41 Da, and is discussed in the main text.
Figure 5.

Sequence map of the covalent adduct to Lys211. Identified fragments from UVPD fragmentation of the 28+ species of 1-treated NDM-1 using 1 pulse at 2.0 mJ. 1000 scans were averaged at 240000 resolution at 400 m/z. 10 ppm mass accuracy constraint was used for fragment identification. The P-score was 8.45E-142. Highlighted K indicates the amino acid site of mass addition after reaction with 1 (+193.04 Da). Cys208 (unmodified) is colored in yellow.
Structural Determination.
To obtain more information about how the affinity label 1 binds and reacts with NDM-1, we determined an X-ray crystal structure of the inactivated complex to 2.0 Å resolution (Table 1). Although soluble constructs of NDM-1 are active as monomers in solution,4, 13, 23, 36 NDM is commonly observed to engage in homo-oligomeric interactions in crystals, often making interactions through a β-hairpin loop (Tyr64-Ala74) that is used to bind substrates.37 Here, the individual NDM-1 monomers are found to have an overall fold and dinuclear zinc site structure that can be closely superimposed with that of previously reported NDM-1 structures (not shown). A crystallographic dimer is again observed, formed in part through an intertwining of the substrate-binding β-hairpin loops of neighboring monomers (Figures 6A, 6B). These same loops also interact with the bound affinity label, although the interaction is not symmetrical. The exact positioning of the loop, and the structures of the bound ligands differ between the intertwined monomers, with notable differences described below.
Table 1.
Crystallographic Data
| 1-treated NDM-1 | |
|---|---|
| Space group | P212121 |
| Cell constants (Å) | a=39.0, b=73.9, c=145.6 |
| Resolution (Å) (outer shell) | 50.−2.02 (2.05–2.02) |
| Rmerge (%) (outer shell) | 0.126 (0.545) |
| <I/σI> (outer shell) | 5.7 (2.3) |
| Completeness (%) (outer shell) | 99.8 (98.1) |
| Unique reflections | 28,507 |
| Redundancy | 7.0 (5.6) |
| # of residues | 454 |
| # of protein atoms | 3320 |
| # of ligand atoms | 29 |
| # of solvent atoms | 221 |
| # of metal atoms | 8 |
| Rworking | 0.183 |
| Rfree | 0.231 |
| Average B factor for protein atoms (Å2) | 18.8 |
| Average B factor for ligand atoms (Å2) | 35.9 |
| Average B factor for solvent atoms (Å2) | 21.3 |
| Average B factor for metal atoms (Å2) | 27.4 |
| rms deviation from ideality | |
| bonds(Å) | 0.006 |
| angles (°) | 0.81 |
| Ramachandran plot | |
| % of residues in most favored region | 98.0 |
| % of residues in additional allowed region | 2.0 |
Figure 6.

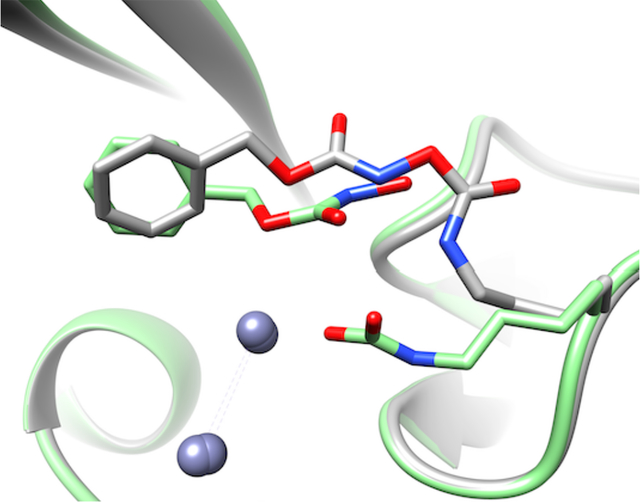
X-ray crystal structure of NDM-1 inactivated by 1. a) A surface coated representation of the crystallographic dimer interface of NDM-1 with one monomer in green, the other in grey, and the inactivator in blue; b) Ribbon representation of the crystallographic dimer interface and active-sites of each monomer, with selected residues shown as sticks, and waters (red) and zinc ions (blue grey) as spheres. The F70 residues of the β-hairpin substrate-binding loops are intertwined. Chain A (grey) shows covalent bonding of the inactivator (colored in blue and by heteroatoms) to K211 as the 193 Da adduct (Figure 7: 9), and Chain B (green) shows the hydrolysis product of a non-covalently bound hydroxamic acid fragment (Figure 7: 10), and a carbamylated K211 in H-bonding distance with a water molecule coordinated to zinc-2 (zinc-1 is labeled with a 2; zinc-1 is labeled with a 1). c & d) The Fo-Fc electron density maps calculated using models that omit either the side chain of Nε-carbamylated K211 and the non-covalent product 10 (c), or the side chain of K211 with the covalent adduct 9 (d) are shown in blue, contoured at 2σ.
Extra electron density is observed in each active site that is not fit by the protein’s amino acids, zinc ions, components used to promote crystallography (e.g. PEG), or water molecules Figures 6B, 6C, 6D). In one monomer (Chain A), the extra density is well fit by a covalent adduct attached to the e-amino group of Lys211, with a proposed structure matching that expected to form upon attack of the carbonate carbon of 1 by the Lys211 side chain and subsequent loss of phenol (the +193 Da adduct, see below). This result confirms the selective covalent modification of Lys211 detected by MS and provides additional details about the conformation that the covalent adduct adopts after inactivation. In the second monomer (Chain B), the extra electron density is better fit by two ligands, a covalent Nε-carbamylation of Lys211 (now repositioned) and a non-covalently bound hydroxamic acid. The stabilization of a carbamylated Lys by H-bonding instead of metal ion coordination has precedence in the case of the class D serine-β-lactamase OXA10.38 In the NDM-1 complex described here, the Lys211 carbamylation modification may be analogously stabilized through H-bonding to the water molecule ligated to zinc-2 and the backbone carbonyl oxygen of Cys208. The two ligands observed in Chain B can be formed by hydrolysis of a precursor adduct that matches the structure of the single +193 Da adduct observed in Chain A. Because the MS analysis did not indicate a significant +43 Da product (for Lys carbamylation) and because the non-covalently bound hydroxamate product is not coordinated to the active-site zinc cluster, it is likely that the two separate ligands observed in Chain B are formed by degradation of a single +193 Da adduct during or following crystallization rather than formation through independent binding mechanisms.
In contrast to the differences in covalent ligand attachments, the phenyl ring of each 1-derived ligand is bound in a similar manner by each monomer (Abstract Graphic). Key interactions in both monomers include packing the face of the phenyl ring against the side chain of Val73, which forms part of a conserved hydrophobic patch found the substrate-binding β-hairpin loop. For Chain A, the Phe70 sidechain found at the apex of the same loop closes over the inactivator and makes an edge-to-face interaction with the inactivator’s phenyl ring. Another Phe70 from the β-hairpin loop of the neighboring monomer adds a third hydrophobic surface (a tilted edge-to-face orientation) forming a pocket akin to a “C-clamp” that binds the ligand’s phenyl ring. In Chain B, the Phe70 of the hairpin loop is more distant and does not make a direct interaction. Rather, the Phe70 of the neighboring monomer’s β-hairpin loop and the adjacent Cα of Gly69 complete the “C-clamp.” When compared to the structure of NDM-1 bound to hydrolyzed benzylpenicillin (PDB accession code 4EYF)39, the phenyl ring of the 1-derived adduct does not bind in the same pocket as the benzyl substituent of benzylpenicillin, but is instead positioned closer to the binding site for the thiazolidine ring of the β-lactam. Also unlike previously described product-bound NDM-1 structures (e.g. ref. 39), the adducts formed from 1 are not observed to make any direct coordination to the dinuclear zinc active site.
Although determination of the crystal structure of 1-treated NDM-1 reveals specific binding interactions made with the resulting covalent adduct and its hydrolysis products, there are some limitations of this experiment. Some of the observed binding interactions may be the same as those formed in the initial non-covalent binding complex of 1 and NDM-1, but others may only be formed after inactivation occurs. Conversely, some binding conformations leading up to inactivation may be lost after phenol release and would not be visualized in the structure. Additionally, interactions with the neighboring β-hairpin loop are likely a result of crystallization because NDM-1 can function as a monomer in solution,4, 13, 23, 36 and the intertwining of these loops in the crystal may obscure interactions that could otherwise predominate in solution.
Proposed Mechanism of Inactivation.
By combining the results described above, we can propose a mechanism for NDM-1 inactivation by 1 (Figure 7), and can classify 1 as a classical affinity label for this protein.40 Fits to a minimal kinetic mechanism are consistent with formation of an initial reversible binding complex (KI = 140 μM), which provides selectivity for modification and fulfills one of the criteria for classical affinity labeling. Since crystallography only reports on the structure after inactivation, the conformation of the initial binding complex is undefined and several possibilities could be considered. The phenyl ring might mimic the bicyclic rings of β-lactam substrates and bind at the hydrophobic base of the substrate-binding β-hairpin loop, inducing closure of Phe70 at the apex of this loop. The carbamate carbonyl might coordinate to one of the active-site zinc ions, similar to the proposed Michaelis complex for β-lactam substrates. Alternatively, the low pKa of 1 (6.8) might allow this compound to mimic the anionic reaction intermediate of NDM-1 (6), although deprotonation to form the anion makes 1 more resistant to hydrolysis.6 The design of compounds that mimic substrates, intermediates, or products has been a successful strategy for NDM-1 inhibitor development41 and similarities found in this O-aryloxycarbonylhydroxamate may facilitate initial binding. After formation of the non-covalent complex, the sidechain amine of the active-site Lys211 is proposed to attack the carbonate electrophile of 1, which is held adjacent to this residue. The resulting high effective concentration (rather than a depressed Lys pKa) likely drives the reaction, leading to loss of phenol, formation of the +193 Da adduct (9) and adoption of the conformation observed in Chain A. The lower kinact/KI values of NDM-1 may represent the poorer nucleophilicity of Lys211, particularly at pH 7, when compared to the active site Ser of P99 β-lactamase.6 Further degradation of the adduct during crystallography can form the Nε-carbamylated Lys211 and hydroxamic acid 10. The irreversible covalent modification of Lys211 (9) blocks substrate binding and results in the observed NDM-1 inactivation. Therefore, we find that the O-aryloxycarbonyl hydroxamate 1 can specifically and covalently label functionally-analogous Lys residues found in both serine and metallo-β-lactamases, but that labeling of NDM-1 occurs through a direct attack of Lys on bound 1, whereas labeling of P99 β-lactamase occurs by Lys attack of a preceding covalent adduct formed at the active-site Ser (Figures 1A, 7).6
Figure 7.

Proposed affinity labeling mechanism of NDM-1 by 1. One possible non-covalent binding complex is depicted (8) and others discussed in the text. Formation of a non-covalent complex provides a high effective molarity of the electrophile, promoting nucleophilic attack by the side chain amine of Lys211 and loss of phenol. The resulting +193 Da adduct (9) results in enzyme inactivation by blocking substrate binding at the active-site, and is irreversible to dilution in excess substrate. During crystallography, 9 can degrade to form an Nε-carbamylated K211 and a non-covalently bound hydroxamic acid (10).
CONCLUSIONS
Although serine- and metallo-β-lactamases differ drastically in sequence, protein fold, and catalytic mechanism, these differences might be bridged by common features that have emerged through convergent evolution. Here, we show that an O-aryloxycarbonyl hydroxamate affinity label that targets a specific Lys residue (Lys315) used for substrate binding in the serine-β-lactamase P99 can also specifically label a specific Lys residue (Lys211) in NDM-1 that serves a similar purpose. The inactivation mechanisms for these two targets differ considerably, but they each involve an attack on the same carbonyl carbon of the inactivator to result in covalent Lys modification. Previous design efforts to develop wide-spectrum β-lactamase inhibitors have also relied on the shared function of these divergent enzymes to bind β-lactams, but have not previously targeted Lys for covalent modification.42–49 This particular Lys residue is conserved in most other B1-subclass metallo-β-lactamases (e.g. NDM, IMP, CcrA, BcII, SPM), but not in VIMs, which have a more distant Arg residue that serves an analogous function.11, 50–54 Although the instability of 1 precludes application as an antibacterial agent in its current form, this Lys-targeted affinity label can selectively modify the active-sites of clinically-relevant serine- and metallo-β-lactamases, suggesting a new strategy for the development of covalent, wide-spectrum β-lactamase inhibitors.
ACKNOWLEDGMENT
We thank Ken Johnson (University of Texas at Austin) for the generous gift of the KinTek Explorer software package. Assistance was provided by the Macromolecular Crystallography Facility, with financial support from the College of Natural Sciences, the Office of the Executive Vice President and Provost, and the Institute for Cellular and Molecular Biology at the University of Texas at Austin. The Berkeley Center for Structural Biology is supported in part by the National Institutes of Health, National Institute of General Medical Sciences, and the Howard Hughes Medical Institute. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02–05CH11231.
Funding
This work was supported in part by the National Institutes of Health (GM111926 to WF), the National Science Foundation (CHE-1402753 to JSB) and the Robert A. Welch Foundation (F-1572 to WF and F-1155 to JSB).
Footnotes
Supporting Information. None.
The authors declare no competing financial interest.
REFERENCES
- [1].Drawz SM, and Bonomo RA (2010) Three decades of beta-lactamase inhibitors, Clin. Microbiol. Rev 23, 160–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, and Fisher SL (2012) Avibactam is a covalent, reversible, non-beta-lactam beta-lactamase inhibitor, Proc. Natl. Acad. Sci. USA 109, 11663–11668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lohans CT, Brem J, and Schofield CJ (2017) New Delhi Metallo-beta-Lactamase 1 Catalyzes Avibactam and Aztreonam Hydrolysis, Antimicrob. Agents. Chemother 61, e01224–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yang H, Aitha M, Hetrick AM, Richmond TK, Tierney DL, and Crowder MW (2012) Mechanistic and spectroscopic studies of metallo-beta-lactamase NDM-1, Biochemistry 51, 3839–3847. [DOI] [PubMed] [Google Scholar]
- [5].Wyrembak PN, Babaoglu K, Pelto RB, Shoichet BK, and Pratt RF (2007) O-aryloxycarbonyl hydroxamates: new beta-lactamase inhibitors that cross-link the active site, J. Am. Chem. Soc 129, 9548–9549. [DOI] [PubMed] [Google Scholar]
- [6].Pelto RB, and Pratt RF (2008) Kinetics and mechanism of inhibition of a serine beta-lactamase by O-aryloxycarbonyl hydroxamates, Biochemistry 47, 12037–12046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Tilvawala R, and Pratt RF (2013) Covalent inhibition of serine beta-lactamases by novel hydroxamic acid derivatives, Biochemistry 52, 3712–3720. [DOI] [PubMed] [Google Scholar]
- [8].Tilvawala R, Cammarata M, Adediran SA, Brodbelt JS, and Pratt RF (2015) A New Covalent Inhibitor of Class C beta-Lactamases Reveals Extended Active Site Specificity, Biochemistry 54, 7375–7384. [DOI] [PubMed] [Google Scholar]
- [9].Malico AA, Dave K, Adediran SA, and Pratt RF (2019) Specificity of extended O-aryloxycarbonyl hydroxamates as inhibitors of a class C beta-lactamase, Bioorg. Med. Chem 27, 1430–1436. [DOI] [PubMed] [Google Scholar]
- [10].Lobkovsky E, Moews PC, Liu H, Zhao H, Frere JM, and Knox JR (1993) Evolution of an enzyme activity: crystallographic structure at 2-Å resolution of cephalosporinase from the ampC gene of Enterobacter cloacae P99 and comparison with a class A penicillinase, Proc. Natl. Acad. Sci. USA 90, 11257–11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang H, and Hao Q (2011) Crystal structure of NDM-1 reveals a common beta-lactam hydrolysis mechanism, FASEB J 25, 2574–2582. [DOI] [PubMed] [Google Scholar]
- [12].Adediran SA, and Pratt RF (2017) Penicillin acylase and O-aryloxycarbonyl hydroxamates: Two acyl-enzymes, one leading to hydrolysis, the other to inactivation, Arch. Biochem. Biophys 614, 65–71. [DOI] [PubMed] [Google Scholar]
- [13].Thomas PW, Zheng M, Wu S, Guo H, Liu D, Xu D, and Fast W (2011) Characterization of purified New Delhi metallo-beta-lactamase-1, Biochemistry 50, 10102–10113. [DOI] [PubMed] [Google Scholar]
- [14].Thomas PW, Cammarata M, Brodbelt JS, and Fast W (2014) Covalent inhibition of New Delhi metallo-beta-lactamase-1 (NDM-1) by cefaclor, ChemBioChem 15, 2541–2548. [DOI] [PubMed] [Google Scholar]
- [15].Yu S, Vosbeek A, Corbella K, Severson J, Schesser J, and Sutton LD (2012) A chromogenic cephalosporin for beta-lactamase inhibitor screening assays, Anal. Biochem 428, 96–98. [DOI] [PubMed] [Google Scholar]
- [16].Chen AY, Thomas PW, Stewart AC, Bergstrom A, Cheng Z, Miller C, Bethel CR, Marshall SH, Credille CV, Riley CL, Page RC, Bonomo RA, Crowder MW, Tierney DL, Fast W, and Cohen SM (2017) Dipicolinic Acid Derivatives as Inhibitors of New Delhi Metallo-beta-lactamase-1, J. Med. Chem 60, 7267–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Thomas PW, Spicer T, Cammarata M, Brodbelt JS, Hodder P, and Fast W (2013) An altered zinc-binding site confers resistance to a covalent inactivator of New Delhi metallo-beta-lactamase-1 (NDM-1) discovered by high-throughput screening, Bioorg. Med. Chem 21, 3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Johnson KA (2009) Fitting enzyme kinetic data with KinTek Global Kinetic Explorer, Methods Enzymol 467, 601–626. [DOI] [PubMed] [Google Scholar]
- [19].Johnson KA, Simpson ZB, and Blom T (2009) Global kinetic explorer: a new computer program for dynamic simulation and fitting of kinetic data, Anal. Biochem 387, 20–29. [DOI] [PubMed] [Google Scholar]
- [20].Han SW, Lee SW, Bahar O, Schwessinger B, Robinson MR, Shaw JB, Madsen JA, Brodbelt JS, and Ronald PC (2012) Tyrosine sulfation in a Gram-negative bacterium, Nat. Commun 3, 1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Otwinowski Z, and Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode, Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- [22].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software, J. Appl. Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Guo Y, Wang J, Niu G, Shui W, Sun Y, Zhou H, Zhang Y, Yang C, Lou Z, and Rao Z (2011) A structural view of the antibiotic degradation enzyme NDM-1 from a superbug, Protein Cell 2, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot, Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Read RJ (1986) Improved Fourier Coefficients for Maps Using Phases from Partial Structures with Errors, Acta Crystallogr. A 42, 140–149. [Google Scholar]
- [27].Brunger AT (1993) Assessment of phase accuracy by cross validation: the free R value. Methods and applications, Acta Crystallogr. D Biol. Crystallogr 49, 24–36. [DOI] [PubMed] [Google Scholar]
- [28].Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB 3rd, Snoeyink J, Richardson JS, and Richardson DC (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids, Nucleic Acids Res 35, W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Silverman RB (1995) Mechanism-based enzyme inactivators, Methods Enzymol 249, 240–283. [DOI] [PubMed] [Google Scholar]
- [30].Kawai K, and Nagata N (2012) Metal-ligand interactions: an analysis of zinc binding groups using the Protein Data Bank, Eur. J. Med. Chem 51, 271–276. [DOI] [PubMed] [Google Scholar]
- [31].Chiou J, Wan S, Chan KF, So PK, He D, Chan EW, Chan TH, Wong KY, Tao J, and Chen S (2015) Ebselen as a potent covalent inhibitor of New Delhi metallo-beta-lactamase (NDM-1), Chem. Commun 51, 9543–9546. [DOI] [PubMed] [Google Scholar]
- [32].Christopeit T, Albert A, and Leiros HS (2016) Discovery of a novel covalent non-beta-lactam inhibitor of the metallo-beta-lactamase NDM-1, Bioorg. Med. Chem 24, 2947–2953. [DOI] [PubMed] [Google Scholar]
- [33].Su J, Liu J, Chen C, Zhang Y, and Yang K (2019) Ebsulfur as a potent scaffold for inhibition and labelling of New Delhi metallo-beta-lactamase-1 in vitro and in vivo, Bioorg. Chem 84, 192–201. [DOI] [PubMed] [Google Scholar]
- [34].Kitz R, and Wilson IB (1962) Esters of methanesulfonic acid as irreversible inhibitors of acetylcholinesterase, J. Biol. Chem 237, 3245–3249. [PubMed] [Google Scholar]
- [35].Tsou CL (1988) Kinetics of substrate reaction during irreversible modification of enzyme activity, Adv. Enzymol. Relat. Areas Mol. Biol 61, 381–436. [DOI] [PubMed] [Google Scholar]
- [36].Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, and Walsh TR (2009) Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India, Antimicrob. Agents Chemother 53, 5046–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Fast W, and Sutton LD (2013) Metallo-beta-lactamase: inhibitors and reporter substrates, Biochim. Biophys. Acta 1834, 1648–1659. [DOI] [PubMed] [Google Scholar]
- [38].Maveyraud L, Golemi D, Kotra LP, Tranier S, Vakulenko S, Mobashery S, and Samama JP (2000) Insights into class D beta-lactamases are revealed by the crystal structure of the OXA10 enzyme from Pseudomonas aeruginosa, Structure 8, 1289–1298. [DOI] [PubMed] [Google Scholar]
- [39].King DT, Worrall LJ, Gruninger R, and Strynadka NC (2012) New Delhi metallo-beta-lactamase: structural insights into beta-lactam recognition and inhibition, J. Am. Chem. Soc 134, 11362–11365. [DOI] [PubMed] [Google Scholar]
- [40].Tuley A, and Fast W (2018) The Taxonomy of Covalent Inhibitors, Biochemistry 57, 3326–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ju LC, Cheng Z, Fast W, Bonomo RA, and Crowder MW (2018) The Continuing Challenge of Metallo-beta-Lactamase Inhibition: Mechanism Matters, Trends Pharmacol. Sci 39, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bush K, Macalintal C, Rasmussen BA, Lee VJ, and Yang Y (1993) Kinetic interactions of tazobactam with beta-lactamases from all major structural classes, Antimicrob. Agents Chemother 37, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nagano R, Adachi Y, Imamura H, Yamada K, Hashizume T, and Morishima H (1999) Carbapenem derivatives as potential inhibitors of various beta-lactamases, including class B metallo-beta-lactamases, Antimicrob. Agents Chemother 43, 2497–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Buynak JD, Chen H, Vogeti L, Gadhachanda VR, Buchanan CA, Palzkill T, Shaw RW, Spencer J, and Walsh TR (2004) Penicillin-derived inhibitors that simultaneously target both metallo- and serine-beta-lactamases, Bioorg. Med. Chem. Lett 14, 1299–1304. [DOI] [PubMed] [Google Scholar]
- [45].Venkatesan AM, Agarwal A, Abe T, Ushirogochi H, Yamamura I, Kumagai T, Petersen PJ, Weiss WJ, Lenoy E, Yang Y, Shlaes DM, Ryan JL, and Mansour TS (2004) Novel imidazole substituted 6-methylidene-penems as broad-spectrum beta-lactamase inhibitors, Bioorg. Med. Chem 12, 5807–5817. [DOI] [PubMed] [Google Scholar]
- [46].Ganta SR, Perumal S, Pagadala SR, Samuelsen O, Spencer J, Pratt RF, and Buynak JD (2009) Approaches to the simultaneous inactivation of metallo- and serine-beta-lactamases, Bioorg. Med. Chem. Lett 19, 1618–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Johnson JW, Gretes M, Goodfellow VJ, Marrone L, Heynen ML, Strynadka NC, and Dmitrienko GI (2010) Cyclobutanone analogues of beta-lactams revisited: insights into conformational requirements for inhibition of serine- and metallo-beta-lactamases, J. Am. Chem. Soc 132, 2558–2560. [DOI] [PubMed] [Google Scholar]
- [48].Brem J, Cain R, Cahill S, McDonough MA, Clifton IJ, Jimenez-Castellanos JC, Avison MB, Spencer J, Fishwick CW, and Schofield CJ (2016) Structural basis of metallo-beta-lactamase, serine-beta-lactamase and penicillin-binding protein inhibition by cyclic boronates, Nat. Commun 7, 12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Torelli NJ, Akhtar A, DeFrees K, Jaishankar P, Pemberton OA, Zhang X, Johnson C, Renslo AR, and Chen Y (2019) Active-Site Druggability of Carbapenemases and Broad-Spectrum Inhibitor Discovery, ACS Infect. Dis in press: doi: 10.1021/acsinfecdis.9b00052 [DOI] [PubMed] [Google Scholar]
- [50].Concha NO, Janson CA, Rowling P, Pearson S, Cheever CA, Clarke BP, Lewis C, Galleni M, Frere JM, Payne DJ, Bateson JH, and Abdel-Meguid SS (2000) Crystal structure of the IMP-1 metallo beta-lactamase from Pseudomonas aeruginosa and its complex with a mercaptocarboxylate inhibitor: binding determinants of a potent, broad-spectrum inhibitor, Biochemistry 39, 4288–4298. [DOI] [PubMed] [Google Scholar]
- [51].Concha NO, Rasmussen BA, Bush K, and Herzberg O (1996) Crystal structure of the wide-spectrum binuclear zinc beta-lactamase from Bacteroides fragilis, Structure 4, 823–836. [DOI] [PubMed] [Google Scholar]
- [52].Carfi A, Pares S, Duee E, Galleni M, Duez C, Frere JM, and Dideberg O (1995) The 3-D structure of a zinc metallo-beta-lactamase from Bacillus cereus reveals a new type of protein fold, EMBO J 14, 4914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Murphy TA, Catto LE, Halford SE, Hadfield AT, Minor W, Walsh TR, and Spencer J (2006) Crystal structure of Pseudomonas aeruginosa SPM-1 provides insights into variable zinc affinity of metallo-beta-lactamases, J. Mol. Biol 357, 890–903. [DOI] [PubMed] [Google Scholar]
- [54].Garcia-Saez I, Docquier JD, Rossolini GM, and Dideberg O (2008) The three-dimensional structure of VIM-2, a Zn-beta-lactamase from Pseudomonas aeruginosa in its reduced and oxidised form, J. Mol. Biol 375, 604–611. [DOI] [PubMed] [Google Scholar]