

Abstract
Molecular docking is the key ingredient of virtual drug screening, a promising and costeffective approach for finding new drugs. A critical limitation of this approach is the inadequate sampling efficiency of both ligand and/or receptor conformations for finding the lowest energy bound state. To circumvent this limitation, we develop a protein-ligand docking methodology capable of incorporating structural constraints, experimentally derived or theoretically predicted, to improve accuracy and efficiency. We develop a web server with a user-friendly online graphical interface as a platform for accurate and efficient protein-ligand molecule docking.
Graphical Abstract
INTRODUCTION
The discovery of biologically active compounds is often a long and expensive trial-anderror process1 that does not meet the needs of modern drug development. Even with modern automated high-throughput screening technologies, development of a typical smallmolecule drug takes years and costs millions of dollars2,3. Computational molecular docking provides an efficient and relatively inexpensive way to identify the lead compounds and to estimate their relative binding affinities and binding modes4,5. Improving the docking accuracy is critical to the success rate of virtual drug screening and to using virtual drug screening in drug discovery pipeline. Recent advances in the field of computational drug screening have led to major improvements in both scoring and sampling algorithms6–12, resulting in increased accuracy and reliability of predictions. There are, however, several fundamental limitations to the conventional molecular docking approach. First, exploring both the receptor and the ligand flexibility4,5,13, which is critical for simulating induced-fit phenomenon and improving the reliability of the predictions, is not computationally feasible for the ligands with a large number of rotatable bonds. Second, physically based scoring functions, designed to quantify inter-atomic interactions and predict affinity of the ligand-receptor complex during molecular docking, strongly rely on force field parameterization14. More accurate descriptions of such interactions are computationally costly and prohibitive for a significant virtual screening campaign. Thus, current molecular docking methods balance accuracy and efficiency.
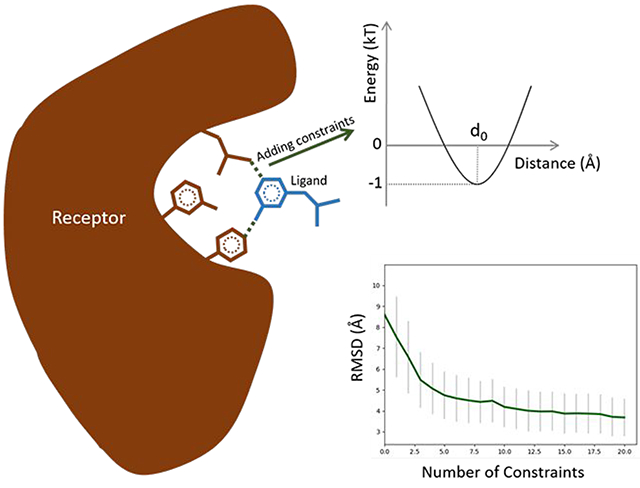
To address these limitations, we have developed a method that accounts for externally derived constraints during molecular docking, increasing the accuracy of the predictions without compromising computational efficiency. The method is based on our previously proposed flexible protein-ligand docking method, MedusaDock4,13,15,16. A combination of structural constraints (derived from structural information obtained from X-ray crystallography, NMR spectroscopy, Cryo-EM, or cheminformatics) with the physicalbased MedusaScore17 force field enables more efficient sampling, eliminated unrealistic conformations, and provides a faster, more accurate convergence to a native-like pose than does the original version of MedusaDock. Since we have incorporated extensive changes and improvements to MedusaDock algorithm, hence we will refer old and new versions as MedusaDock 1.0 and MedusaDock 2.0 respectively. The workflow of MedusaDock 2.0 is presented in Figure 1A. Apart from the existing energy contributions that include van der Waals, solvation, and hydrogen bond energy terms in MedusaScore energy function, we add a weighted energy term to favor the satisfaction of structural constraints.
Figure 1.

(A) A simplified diagram of MedusaDock based docking algorithm (Methods). (B and C) Docking accuracy in terms of RMSD/lRMSD as a function of the number of constraints. The RMSD and lRMSDs are averages calculated over the 30 attempts performed in each case. The grey bars represent the standard deviations of the averages. (D and E) The comparison of the RMSD/lRMSD distribution after docking with no constraint and with one constraint.
Although constraints-based docking is widely used by docking algorithms such as GOLD18, SwissDock19, DOCK12, FlexX20, HADDOCK21, ICM22, AutoDock23, and RosettaLigand24, we compare MedusaDock 2.0 with two commonly used methods: AutoDock23 and RosettaLigand24. We evaluate the performance of MedusaDock, AutoDock, and RosettaLigand algorithms in (a) presence and (b) absence of native-structure derived constraints. The benchmarking analysis suggests that MedusaDock 2.0 outperforms AutoDock and RosettaLigand. We also find that the incorporation of constraints does not always yield better results. In few instances, we find that the inclusion of constraints generated poor docking results. Hence, we propose here an improved strategy for utilizing constraints specifically in MedusaDock. Finally, we build an efficient, fast, and userfriendly online web server MedusaDock (http://MedusaDock.dokhlab.org) for proteinligand docking.
RESULTS
Validation of the efficiency and docking accuracy of MedusaDock 2.0
To evaluate the performance of MedusaDock 2.0, we compile a benchmark protein-small molecule dataset containing 100 complexes with known native poses. For each native pose, we extract a series of ligand-receptor inter-atomic distances that is used as external constraints during the docking simulations. Using this dataset, we demonstrate that the incorporation of structural constraints improves the docking accuracy. We find that the RMSD between predicted and native poses of the ligands decreases rapidly when the number of incorporated constraints is increased (Figure 1B, Table S1). The constraints are only introduced to confine some inter-atom distances between the receptor and the ligand, so it is intriguing to observe that (Figure 1C, Table S2) the lRMSD (Methods) also decreases rapidly with the increase of the number of constraints. Providing even one constraint to the new docking algorithm is sufficient to significantly improve the accuracy of the predictions (Figure 1D, 1E). An analysis of RMSD versus the number of rotatable bonds using no constraints and one constraint further demonstrates the importance of constraints (Supporting Information; Figure S3). Apart from self-docking on the 100 complexes dataset, we also perform cross-docking on dataset obtained in our previous work4. RMSDs in cross-docking also decrease with the increase of the number of constraints (Figure S1, Table S3).
Since the incorporation of constraints would usually increase the docking time because of the extra time spent on calculating energies related to constraints, we carefully analyze the procedure of MedusaDock 1.0 and then optimize the whole program to improve the efficiency. We analyze the duration of the coarse and the fine docking stages of MedusaDock3 and find that fine docking is the most time consuming process (Figure S2A).
During fine docking, we observe that the RMSD initially rapidly decreases but later reaches a plateau (Figure S2B). This fact allows us to significantly optimize the original MedusaDock algorithm by reducing the number of steps of fine docking, which reduces the running time (Figure 2A) without compromising the docking accuracy (Figure 2B).
Figure 2.

(A) Comparison of the docking time of MedusaDock 1.0 and MedusaDock 2.0. (B) Comparison of the RMSD between predicted ligand poses and native ligand poses of MedusaDock 1.0 and MedusaDock 2.0. No constraints are imposed in the comparison. (C) The comparison of the docking times of Autodock23, RosettaLigand5, and MedusaDock 2.0. The suffix /0 refers to docking no constraints. The suffix /2 refers to docking with 2 constraints. (D) The comparison of the RMSDs of Autodock23, RosettaLigand5, and MedusaDock 2.0.
We then conduct time and accuracy comparison between MedusaDock 2.0 and other prevailing docking tools (AutoDock23 and RosettaLigand24) that account for both receptor and ligand flexibility. We find that the optimization makes MedusaDock significantly faster than the other two tools (Figure 2C; Table S4), and the addition of two constraints during the docking results in notably docking accuracy improvement (Figure 2D; Table S4).
Correlation between docking accuracy and nature of residues or atoms involved in constraints
Interestingly, we find that different constraints have diverse influences on the docking (Figure 3). Most constraints improve the docking results (Figure 3A); these are referred to as the positive constraints. Some, however, cause deterioration of the results (Figure 3C), and these are referred to as the negative constraints. We demonstrate this phenomenon by choosing 100 different constraints from the tertiary structure of the CXCR4 chemokine receptor in complex with small molecule antagonist IT1t25 (PDB ID: 3ODU). We perform several docking attempts for CXCR4/IT1t complex with each of the 100 selected constraints. We differentiate these 100 constraints into positive and negative based on the docking outputs (Figure 3D).
Figure 3.

A test of how residue types in the distance constraints affect the docking. (A) Comparison of the distance distribution between the results of 3ODU with no constraints and with one constraint. (B) A constraint in 3ODU between two atoms pulls the two atoms from a large distance (8.2 Å, yellow) to a smaller distance (3.4 Å, brown) that is much closer to the native distance (2.7 Å, cyan). (C) The green line specifies the RMSD of 3ODU obtained by docking with no constraints. The grey dots represent docking results obtained by using different constraints. (D) Positive constraints of 3ODU are in green and negative constraints are in red. (E) The relationship between the net RMSD and residue types obtained from test results on the 100 complexes dataset. The net RMSD is equal to RMSD with constraints minus the RMSD without constraint. (F) The relationship between the net RMSD and atom types obtained from test results on the 100 complexes dataset.
Based on the docking predictions from 100 complexes dataset, we analyze the relationship (Figure 3E) between the types of receptor residues participating in constraints and the influences of constraints to avoid the usage of negative constraints. We find that residue types involved in positive constraints are six non-polar amino acids (glycine, alanine, valine, cysteine, isoleucine, and methionine), two polar amino acids (threonine and asparagine), and one negatively charged amino acid (aspartic acid) (Figure 3E). Interestingly, we observe that serine is only one residue that is nearly always involved in negative constraints. We also study the relationship between atom types and constraints (Figure 3F) and find that five atom types (CZ2, CZ3, CE3, NE1, OG) are always observed in negative constraints.
Based on the relationship between residue types and the influences of constraints, we see no noticeable correlation between the influences of constraints and any single property of residues, such as polarity, hydrophobicity, and structural complexity. Instead, the relation between residue types and constraints is mainly dependent on several crucial factors, such as the structural complexity of residues, rotamer library of residue side-chains, hydrogen bond forming capability, and the conformational space of a ligand. However, the correlation between residue types and constraints is different for other docking methods due to alterations in residue rotamer library and the conformational space of a ligand. In addition, the relationship between atom types and constraints is dependent on factors, such as the position of the atom in the residue, the type of the atom, and the type of the residue. Overall, the analysis of the relationship between residue/atom types and constraints facilitates users to make better use of MedusaDock by utilizing positive constraints and avoiding negative constraints deduced from above mentioned types of residues/atoms.
Selecting robust docking results based on MedusaScore
In virtual drug screening, typically numerous rounds of docking attempts are performed to generate an ensemble of complex conformations. Complexes in the ensemble are then scored and clustered to facilitate the selection of a best-scoring conformation as the final docking result. Since adding constraints and conducting multiple docking attempts can both expand the conformational space to be explored for near-native conformations, we analyze the significance of incorporating constraints when multiple docking attempts are to be performed. We perform 2000 rounds of MedusaDock 2.0 docking attempts for Endothiapepsin in complex with ritonavir (PDB ID: 3PRS) with no constraints and with one constraint, respectively. As shown in Figure 4A and 4C, the application of constraints decreases both the RMSD and the lRMSD in nearly every round of the simulation, indicating that the incorporation of constraints significantly improves docking accuracy in comparison to multiple docking attempts.
Figure 4.

A test of multiple rounds of docking: the case of 3PRS. (A) RMSD with no constraints varies when multiple rounds of docking are performed, and RMSD with one constraint is nearly always lower than that with no constraints. (B) Distribution of RMSDs with no constraints and one constraint. (C) Comparison of RMSD distribution after docking with no constraints and with one constraint. (D) Comparison of lRMSD distribution after docking with no constraints and with one constraint. (E) The relationship between MedusaScore calculated free energy and RMSD. (F) The relationship between MedusaScore calculated free energy and lRMSD.
In order to test the performance of MedusaScore, we evaluate the interaction energies of MedusaDock 2.0 generated Endothiapepsin-ritonavir complexes using MedusaScore. We find that RMSD and interaction energy are positively correlated with each other (Figure 4E), which suggests that MedusaScore is able to select a near-native pose. However, the correlation coefficient of the linear fitting of RMSD and the energy is only 0.8, indicating that the scoring function could be further improved. Since MedusaScore is primarily devised for evaluating the conformations of receptor-ligand interacting interfaces and lRMSD only considers ligand conformation, we found nearly no correlation between MedusaScore evaluated energy and lRMSD (Figure 4F).
Webserver of MedusaDock 2.0
Finally, we build a web-based MedusaDock server (https://MedusaDock.dokhlab.org) that provides a user-friendly interface for submission and management of protein-small molecule docking calculations with or without constraints. In order to decrease the risk of using negative constraints, web server users are recommended to use constraint types that are concluded to be positive. We anticipate that improved MedusaDock will be appreciated by the community as a platform for accurate and efficient protein-ligand molecule docking.
METHODS
Dataset
The generation of the dataset is based on the refined set of the PDBbind 2017 database26,27, which provides a comprehensive collection of experimentally measured binding affinity data for the biomolecule complexes in the Protein Data Bank28 (PDB). We download the whole PDBbind refined set containing 4154 complexes. Complexes with more than one ligand in the dataset are then removed. To identify protein-ligand complexes those are difficult to handle with MedusaDock, we perform docking attempts for all complexes and remove complexes with RMSDs less than 2 Å, leaving 729 complexes. Ligands with fewer rotatable bonds are much easier to dock than those with a large number of rotatable bonds, thus complexes having a small number of rotatable bonds cannot be used to effectively assess the effect of adding constraints. For this reason, most ligands with fewer than eight rotatable bonds are removed. The final dataset include 100 protein-small molecule complexes with ligands having averagely 22 rotatable bonds; the smallest number of rotatable bonds is eight and the largest number was 39. Apart from the 100 complexes dataset, we have also considered the dataset with 36 complexes from our previous work to test MedusaDock 2.0 in terms of cross-docking.
Optimization of MedusaDock
The MedusaDock workflow is divided into three stages (Figure 1A): stochastic rotamer library of ligands (STROLL) generation, coarse docking, and fine docking. In the STROLL generation stage, numerous ligand rotamers are generated by randomly rotating rotatable chemical bonds. All rotamers in STROLL are clustered and each of the centroids is then subjected to the coarse docking. During the coarse docking, rigid docking and receptor side chain repacking are performed alternately and iteratively. After coarse docking, all the poses are clustered and each of the centroids is then subjected to fine docking. Unlike coarse docking, the rotamer pose adjusting and the receptor side chain repacking are performed simultaneously in fine docking. After fine docking, the ligand rotamer that has the lowest binding free energy with the receptor is selected as the final candidate. Fine docking is the most time-consuming step (Figure S2A), requiring approximately 5-fold more time than coarse docking. Ligand RMSD decreases dramatically during coarse docking but decreases very little in fine docking (Figure S2B). In MedusaDock 2.0, we thoroughly optimize the source codes by using new C++ programming techniques. For instance, we encapsulate the return values of most functions with “std::move” function in C++ 11 in order to avoid redundant copies of the return values. We leverage new smart pointers in C++ 11 to further reduce the possibility of copying bulky objects. We also utilize non-copyable classes to store global variables to avoid redundant copies. Additionally, we adjust internal parameters to reduce the number of fine docking steps, and it turns out that the optimization effectively decreases the running time (Figure 2A) without adversely altering the docking results (Figure 2B).
Structural constraints
The structural constraints utilized in the tests are all distance constraints (i.e., the distance between two atoms). We also implement in-house supports for angle constraints (i.e., the angle between two bonds) and dihedral constraints (i.e., the dihedral of three bonds in the complex structure), but they are not tested in this work. Either an exact distance value or a distance range consisting of the minimum and maximum values of the distance between two atoms can be specified as constraints. In both circumstances, an additional constraintsrelated energy item (Figure 5B) is added to the total MedusaScore energy, which originally contains van der Waals energy, solvation energy, and hydrogen bonding energy (Figure 5B). The van der Waals energy is calculated by Lennard-Jones potential, the solvation energy is calculated by Lazaridis-Karplus model29 and the hydrogen bonding energy is derived from the statistical potential model (Figure 5A). In this work, the distance constraint is mainly introduced to confine the relative position of the receptor and the ligand, but theoretically, it could also confine two atoms when they are both in the receptor or the ligand so as to restrain the flexible conformation of the loop in the interface of the receptor or to restrain the ligand itself.
Figure 5.

(A) The hydrogen bonding energy is derived from the distribution of dHA, θDA, θHX by using the statistical energy function. Pobs is the observation frequencies; Pran is the random frequencies; D: Donor atom; H: Hydrogen atom; A: Acceptor atom; X: The heavy atom bonded with A. (B) The constraints energy is of the spring potential if a definite distance is given as the constraint, otherwise it is –k from d1 to d2 if the range is given. k is calculated by . The receptor is colored by red and the ligand is colored by blue. d1 and d2 are the lower and the upper bound of the user-provided distance range.
RMSD calculation
The RMSD refers to the ligand RMSD calculated by aligning the receptors before and after the docking, which considers the composite deviation of not only the position and the orientation of the ligand but also the rotation status of interior rotatable bonds in the ligand. Constraints definitely restrain the rigid conformation (i.e., the position and the orientation) of the ligand. To determine whether constraints influence the flexible conformation (i.e., the rotation status of interior rotatable bonds) of the ligand, we calculated the ligand RMSD by aligning the ligands themselves; this is referred to as the lRMSD.
Test
For each of the 100 complexes in the dataset, we utilize MedusaDock to generate nonnative complexes as the inputs for the following test. Apart from van der Waals repulsive energy terms, we first set weights of all other energy terms in MedusaScore to zero. We subsequently perform a short period time (500 steps in MedusaDock) of docking attempts for all the complexes with MedusaDock by using this reduced version of MedusaScore. The results are then used as the inputs for further tests, where the average RMSD of the inputs is 10.8 Å. We efficiently sample conformations of ligand, receptor backbone, and receptor sidechain during docking. The sampling is conducted in a cubic pocket centered at the mass center of the native ligand conformation. The side length of the cubic is 20 Å. After we generate all the inputs, we reset the weights of all the energy terms of MedusaScore to their default values.
For each of the 100 complexes in the dataset, we extract all the ligand-receptor inter-atomic distances that are less than 4 Å. We randomly pick 30 times 1, 2, …, and 20 distances as the constraints from all the extracted distances, respectively. For each number (1–20) of constraints, we perform 30 dockings attempts by using the 30 different constraints sets. The results are shown in Figure 1B–E and Table S1–2. We efficiently sample conformations of ligand along with receptor backbone and sidechain conformations during the docking. The search space is still a cubic box with a side length of 20 Å centered at the designated binding site.
We then perform docking attempts for these 100 complexes without constraints by using MedusaDock before and after optimization, respectively. We perform 30 rounds of docking attempts for each case to determine the average RMSD and running time (Figure 2A, 2B).
We then compare the performances of AutoDock, RosettaLigand, MedusaDock 2.0 with no constraints and with two constraints, respectively. The two constraints are randomly selected from native structure derived constraints. We perform 30 docking attempts for each of the 100 complexes (Figure 2C, 2D; Table S4). The sampling space in all three methods utilizes a cubic box with dimensions 20 Å × 20 Å × 20 Å. The flexible region of AutoDock and RosettaLigand are set to the chains that are involved in the binding pocket. The flexible region of MedusaDock involves the residues that are in the proximity to the binding pocket (residues within 12 Å around the binding pocket). Conformations of the ligand along with the receptor side chains and backbones are all sampled in RosettaLigand and MedusaDock 2.0. Only conformations of the ligand and the receptor side chains are sampled in AutoDock.
Website
The front-end of the website is based on Vue.js 2.0 framework. The source code of the front-end is deposited on BitBucket (https://bitbucket.org/dokhlab/medusadock-web/src).
The back-end of the website is built upon the native PHP supported by the Apache HTTP server running in a Linux server.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully thank Dr. Venkata R. Chirasani and Dr. Konstantin I. Popov for their proofreading of the manuscript.
FUNDING
This work is supported by NIH grants R01GM114015, R01GM064803, and R01GM123247.
Footnotes
Availability: http://MedusaDock.dokhlab.org
REFERENCES
- (1).Aherne GW; McDonald E; Workman P Finding the Needle in the Haystack: Why High-Throughput Screening Is Good for Your Health. Breast Cancer Res. 2002, 4, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Convertino M; Das J; Dokholyan NV Pharmacological Chaperones: Design and Development of New Therapeutic Strategies for the Treatment of Conformational Diseases. ACS Chem. Biol 2016, 11, 1471–1489. 10.1021/acschembio.6b00195. [DOI] [PubMed] [Google Scholar]
- (3).DiMasi JA; Hansen RW; Grabowski HG The Price of Innovation: New Estimates of Drug Development Costs. J. Health Econ. 2003, 22, 151–185. [DOI] [PubMed] [Google Scholar]
- (4).Ding F; Yin S; Dokholyan NV Rapid Flexible Docking Using a Stochastic Rotamer Library of Ligands. J. Chem. Inf. Model 2010, 50, 1623–1632. 10.1021/ci100218t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Meiler J; Baker D ROSETTALIGAND: Protein-Small Molecule Docking with Full Side-Chain Flexibility. Proteins Struct. Funct. Bioinforma 2006, 65, 538–548. 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- (6).Proctor EA; Dokholyan NV Applications of Discrete Molecular Dynamics in Biology and Medicine. Curr. Opin. Struct. Biol 2016, 37, 9–13. 10.1016/j.sbi.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Allen SE; Dokholyan N V; Bowers, A. A. Dynamic Docking of Conformationally Constrained Macrocycles: Methods and Applications. ACS Chem. Biol 2016, 11, 10–24. 10.1021/acschembio.5b00663. [DOI] [PubMed] [Google Scholar]
- (8).Proctor EA; Yin S; Tropsha A; Dokholyan NV Discrete Molecular Dynamics Distinguishes Nativelike Binding Poses from Decoys in Difficult Targets. Biophys. J 2012, 102, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dagliyan O; Proctor EA; D’Auria KM; Ding F; Dokholyan NV Structural and Dynamic Determinants of Protein-Peptide Recognition. Structure 2011, 19, 1837–1845. 10.1016/j.str.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kitchen DB; Decornez H; Furr JR; Bajorath J Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications. Nat. Rev. Drug Discov 2004, 3, 935 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- (11).Ferreira LG; dos Santos RN; Oliva G; Andricopulo AD Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. 10.3390/molecules200713384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Allen WJ; Balius TE; Mukherjee S; Brozell SR; Moustakas DT; Lang PT; Case DA; Kuntz ID; Rizzo RC DOCK 6: Impact of New Features and Current Docking Performance. J. Comput. Chem 2015, 36, 1132–1156. 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ding F; Dokholyan NV Incorporating Backbone Flexibility in MedusaDock Improves Ligand-Binding Pose Prediction in the CSAR2011 Docking Benchmark. J. Chem. Inf. Model 2013, 53, 1871–1879. 10.1021/ci300478y. [DOI] [PubMed] [Google Scholar]
- (14).Khatun J; Khare SD; Dokholyan NV Can Contact Potentials Reliably Predict Stability of Proteins? J. Mol. Biol 2004, 336, 1223–1238. [DOI] [PubMed] [Google Scholar]
- (15).Hsieh J-H; Yin S; Wang XS; Liu S; Dokholyan NV; Tropsha A Cheminformatics Meets Molecular Mechanics: A Combined Application of Knowledge-Based Pose Scoring and Physical Force Field-Based Hit Scoring Functions Improves the Accuracy of Structure-Based Virtual Screening. J. Chem. Inf. Model 2012, 52, 16–28. 10.1021/ci2002507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Politi R; Convertino M; Popov K; Dokholyan NV; Tropsha A Docking and Scoring with Target-Specific Pose Classifier Succeeds in Native-Like Pose Identification But Not Binding Affinity Prediction in the CSAR 2014 Benchmark Exercise. J. Chem. Inf. Model 2016, 56, 1032–1041. 10.1021/acs.jcim.5b00751. [DOI] [PubMed] [Google Scholar]
- (17).Yin S; Biedermannova L; Vondrasek J; Dokholyan NV MedusaScore: An Accurate Force Field-Based Scoring Function for Virtual Drug Screening. J. Chem. Inf. Model 2008, 48, 1656–1662. 10.1021/ci8001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Verdonk ML; Cole JC; Hartshorn MJ; Murray CW; Taylor RD Improved Protein–ligand Docking Using GOLD. Proteins Struct. Funct. Bioinforma 2003, 52, 609–623. [DOI] [PubMed] [Google Scholar]
- (19).Grosdidier A; Zoete V; Michielin O SwissDock, a Protein-Small Molecule Docking Web Service Based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Rarey M; Kramer B; Lengauer T; Klebe G A Fast Flexible Docking Method Using an Incremental Construction Algorithm. J. Mol. Biol 1996, 261, 470–489. [DOI] [PubMed] [Google Scholar]
- (21).Dominguez C; Boelens R; Bonvin AMJJ HADDOCK: A Protein− Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc 2003, 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- (22).Totrov M; Abagyan R Flexible Protein–ligand Docking by Global Energy Optimization in Internal Coordinates. Proteins Struct. Funct. Bioinforma 1997, 29, 215–220. [DOI] [PubMed] [Google Scholar]
- (23).Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility; NIH Public Access, 2009; Vol. 30, pp 2785–2791. 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Davis IW; Baker D RosettaLigand Docking with Full Ligand and Receptor Flexibility. J. Mol. Biol 2009, 385, 381–392. [DOI] [PubMed] [Google Scholar]
- (25).Wu B; Chien EYT; Mol CD; Fenalti G; Liu W; Katritch V; Abagyan R; Brooun A; Wells P; Bi FC; Hamel DJ; Kuhn P; Handel TM; Cherezov V; Stevens RC Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Liu Z; Su M; Han L; Liu J; Yang Q; Li Y; Wang R Forging the Basis for Developing Protein-Ligand Interaction Scoring Functions. Acc. Chem. Res 2017, 50, 302. [DOI] [PubMed] [Google Scholar]
- (27).Liu Z; Li Y; Han L; Li J; Liu J; Zhao Z; Nie W; Liu Y; Wang R PDBWide Collection of Binding Data: Current Status of the PDBbind Database. Bioinformatics 2015, 31, 405. [DOI] [PubMed] [Google Scholar]
- (28).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. https://doi.org/gkd090 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lazaridis T; Karplus M Effective Energy Function for Proteins in Solution. Proteins Struct. Funct. Bioinforma 1999, 35, 133–152. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.