

Abstract
HMGB1 is a multifunctional nuclear protein, probably known best as a prototypical alarmin or damage-associated molecular pattern (DAMP) molecule when released from cells. However, HMGB1 has multiple functions that depend on its location in the nucleus, in the cytosol, or extracellularly after either active release from cells, or passive release upon lytic cell death. Movement of HMGB1 between cellular compartments is a dynamic process induced by a variety of cell stresses and disease processes, including sepsis, trauma and hemorrhagic shock. Location of HMGB1 is intricately linked with its function and is regulated by a series of post-translational modifications. HMGB1 function is also regulated by the redox status of critical cysteine residues within the protein, and is cell-type dependent. This review highlights some of the mechanisms that contribute to location and functions of HMGB1, and focuses on some recent insights on important intracellular effects of HMGB1 during sepsis and trauma.
Keywords: DAMPs, caspase-11, AIM2, autophagy, pyroptosis
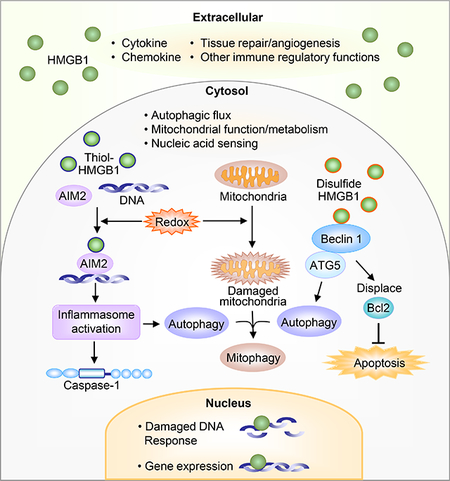
Graphical abstract
Schematic representation of location-specific functions of HMGB1 in extracellular space, cytosol, and nucleus.
Introduction
High mobility group box 1 (HMGB1) was first discovered in the 1960s as a nonhistone chromatin-binding protein that showed high electrophoretic mobility when run on polyacrylamide gels[1, 2]. HMGB1 is highly conserved in evolution and there is a 99% homology between rodent and human[3–5]. In the nucleus, HMGB1 is known to play important roles in the regulation of a wide range of processes, including transcription, replication, DNA repair and nucleosome formation. All these processes are important in maintaining homeostatic cellular function[6]. More recently HMGB1 has been found to play multiple roles in regulating inflammation and responses to cell and tissue stress when it is mobilized from the nucleus into the cell cytoplasm, or released into the extracellular space either by active or passive mechanisms. In these contexts, HMGB1 has been described as a prototypical damage-associated molecular pattern (DAMP) molecule[6–9] (often also described as an alarmin[7, 8]). Importantly, HMGB1 has been shown to contribute to the pathogenesis of various diseases including sepsis, traumatic shock, autoimmune diseases, cancer, as well as hepatic steatosis and fatty liver disease[8, 10–13]. In this review, we will focus on some recent insights into the mechanisms of cytosolic HMGB1 regulation of inflammasome activation after trauma and hemorrhagic shock, and how HMGB1 facilitates LPS-uptake into macrophages and their subsequent pyroptotic, inflammatory cell death in models of sepsis.
Nuclear-to-cytoplasm shuttling of HMGB1
HMGB1 consists of 215 amino acid (aa) residues, and contains two HMG DNA-binding domains, designated as A and B boxes, together with a negatively charged C-terminal acidic region[14]. Two nuclear localization sites (NLSs), one located in the A box (aa 28–44) and one in the B box (aa 179–185), control the nuclear localization of HMGB1 under homeostatic states[14]. Posttranslational modifications of NLS sites, including acetylation, phosphorylation and methylation, regulate the ability of HMGB1 to translocate to the cytoplasm during cellular stress. Once in the cytoplasm HMGB1 can affect multiple inflammatory responses, and can be actively released into the extracellular space and circulation.
HMGB1 is predominantly anchored inside the nucleus under physiological conditions through its binding, via NLS sites, with nuclear cargo carrier proteins. Acetylation, phosphorylation, or methylation at multiple amino acid residues within the NLS result in neutralization of the usual positive NLS charge, or result in addition of negative charge within the NLS, which loosens the association of HMGB1 with carrier proteins[15–17]. This is therefore critical for translocation of HMGB1 from the nucleus to the cytoplasm[18]. There are four conserved lysine residues in NLS1, and five in NLS2. Bonaldi et al. first described hyperacetylation of lysines within NLS sites as critical for HMGB1 translocation from the nucleus to the cytoplasm in monocytes after stimulation with LPS, TNF, or IL-1β [15]. Acetylation of lysines within the NLS of HMGB1 is controlled by histone deacetylases (HDACs), important enzymes that remove acetyl groups and control the acetylation state (and function) of histones as well as some additional intracellular proteins [15, 19, 20]. Pharmacological inhibition of HDACs, or mutations of all lysines in NLS, inhibits HMGB1 hyperacetylation and translocation from the nucleus to the cytoplasm [15]. Acetylation of NLS lysines also appears to block the re-entry of HMGB1 into the nucleus, allowing accumulation within the cytoplasm [15]. Notably, the effect of hyperacetylation on HMGB1 translocation from the nucleus to the cytoplasm is not limited to immune cells. We have shown that HMGB1 acetylation and cytoplasmic accumulation in hepatocytes is also dependent on the activities of HDACs during liver ischemia/reperfusion (I/R) injury[21], or in hypoxic primary hepatocytes in vitro[21]. We and others have also shown that the acetylation and cytoplasmic translocation of HMGB1 is mediated via the type 1 and type 2 interferons (IFN)/janus kinase (JAK)/signal transducer and activator of transcription 1 (STAT1) signaling pathways[22, 23]. Researchers within our group have shown that interferon regulatory factor 1 (IRF1) interacts with the nuclear histone acetyltransferase enzyme p300, and this can regulate HMGB1 acetylation in hepatocytes during liver I/R injury[23]. As IRF1 is known to be a key signaling factor downstream of JAK/STAT1 in immune cells, it is one potential mechanism by which JAK/STAT1 could regulate HMGB1 acetylation. However, the downstream mechanism by which JAK/STAT1 mediates HMGB1 acetylation is not fully understood.
Phosphorylation and methylation of HMGB1 NLS sites have also been shown to be important in allowing HMGB1 to translocate to the cytoplasm. Youn et al. demonstrated that HMGB1 is phosphorylated and translocates into the cytoplasm in murine macrophages after TNF stimulation [24]. However, the upstream signaling pathway that regulates phosphorylation within NLS sites of HMGB1 is not fully understood. Work from our group has shown one potential upstream pathway of HMGB1-NLS phosphorylation involving calcium/calmodulin-dependent protein kinase (CaMK) IV. We showed that translocation and release of HMGB1 from LPS-stimulated macrophages[17], as well as from hepatocytes during I/R injury[25] is regulated by CAMK IV activity. However, currently we have not determined whether CaMK IV directly mediates HMGB1 serine phosphorylation within NLS sites in order to allow translocation. HMGB1 can also be monomethylated at lysine 42 (within NLS1) in what appears to be a neutrophil specific manner [16]. Methylation at this site weakens DNA-binding activity of HMGB1 leading to passive diffusion of HMGB1 out of the nucleus[16]. It remains to be determined whether this is a neutrophil-specific mechanism, and whether this translocation of HMGB1 from the nucleus contributes to neutrophil extracellular trap (NET) formation in sepsis.
Getting HMGB1 out of cells and into the circulation
Extracellular HMGB1 functions as an alarmin or a pro-inflammatory cytokine/chemokine and perpetuates inflammatory responses in a wide variety of pathologies, including sepsis and trauma[10]. Circulating HMGB1 has varying cell type-dependent effects, and can regulate the release of cytokines, neutrophil recruitment, vascular permeability, as well as cell death[26]. HMGB1 was first identified as an inflammatory mediator in lethal sepsis shock in 1999[27], and it is now well-established that circulating HMGB1 levels are significantly elevated in experimental sepsis models[27, 28] and in septic patients[29–31]. Circulating HMGB1 levels are elevated significantly later than other acute phase inflammatory cytokines, such as TNF and IL-6, and reach a plateau 16 to 32h after the onset of sepsis in both cecal ligation and puncture (CLP)-induced polymicrobial Gram-negative murine sepsis[27] and in septic patients[31]. Circulating HMGB1 levels also remain elevated for weeks after CLP-induced sepsis[31, 32] and are a late predictor of mortality during sepsis in patients[32].
Circulating HMGB1 arises from a combination of both passive release and active secretion mechanisms from cells. HMGB1 can be released by many cell types, including macrophages and monocytes[27, 33], endothelial cells[34], enterocytes[35], and hepatocytes[25, 28] during sepsis, trauma and I/R injury. During lytic cell death, such as necrosis, pyroptosis, and necroptosis, HMGB1 can be passively released into the extracellular space through rupture of the cell membrane[10]. The majority of HMGB1 released passively is HMGB1 that was still located within the nucleus, which is therefore non-acetylated and has also been shown to be fully reduced[10, 18], which affects HMGB1 function and pro-inflammatory potential (see section on redox changes of HMGB1 below). Lack of acetylation has been used in studies to try to identify the proportion of HMGB1 that is passively released from damaged/dead cells, as opposed to actively released as part of the inflammatory process[18]. Aerobic glycolysis (PMID: 25019241 and PMID: 27779186) and lipid peroxidation (PMID: 29937272) has been shown to promote HMGB1 release through activation of inflammasomes in sepsis. However, it is unclear whether such metabolic stress induces passive or active release of HMGB1. Identifying lytic versus active release of HMGB1 may be important when considering potential effectiveness of novel therapeutics designed to attenuate either lytic cell death, or active HMGB1 release.
Active secretion of HMGB1 can occur from cells under a wide range of cell stresses, or from inflammatory cells in response to signaling by pathogen-associated molecular patterns (PAMPs), DAMPs or pro-inflammatory cytokines[10, 18]. A first step in active secretion is translocation of HMGB1 from the nucleus to the cytoplasm, which requires posttranslational modifications, such as acetylation and phosphorylation, as detailed above. The mechanisms behind the movement of cytoplasmic HMGB1 out of the cell, however, are much less well understood, and may be cell, tissue and stimulus-dependent. Active secretion of HMGB1 has been shown to occur via exocytosis of HMGB1-containing secretory lysosomes, microparticles and exosomes[33, 35–37]. However, the details of the signaling pathways leading to HMGB1 exocytosis, including targeting to specific vesicles, remain elusive.
Macrophages can actively release HMGB1 after LPS stimulation via CD14/TLR4/NFκB-dependent mechanisms[38, 39]. Similarly, we have shown that LPS can stimulate active HMGB1 release from hepatocytes. The signaling pathways involve TLR4-dependent uptake of LPS into hepatocytes[28], followed by LPS release into the cytoplasm by a currently unknown mechanism. This allows cytoplasmic LPS to be detected by caspase-11, the recently identified intracellular receptor for LPS[40, 41]. HMGB1 release from hepatocytes is dependent on activation of caspase-11[28], and our data also suggest dependence on activation/cleavage of gasdermin D, the main substrate for caspase-11[28]. Interestingly, gasdermin D activation in hepatocytes does not result in pyroptosis and lytic cell death of these cells, and so the role of gasdermin D in HMGB1 release is still under investigation. (Figure 1)
Figure 1: Intracellular and extracellular roles for HMGB1 in LPS-mediated infection and sepsis.

LPS uptake into hepatocytes triggers HMGB1 mobilization from the nucleus, and active release via a caspase-11/gasdermin D-mediated pathway. Extracellular HMGB1 then facilitates LPS uptake into macrophages, endosomal/lysosomal rupture and release of HMGB1 and LPS into the cell cytosol, inducing caspase-11 and caspase-1 signaling and macrophage pyroptosis.
We have recently demonstrated that hepatocytes are the main source of circulating HMGB1 in LPS-induced endotoxemia and CLP-induced polymicrobial sepsis[28], as well as hemorrhagic shock (MJS, unpublished data). Cell-specific depletion of HMGB1 in hepatocytes dramatically reduces circulating HMGB1 level and confers protection from sepsis lethality in mice[28], as well as organ damage in response to hemorrhagic shock. Understanding the active release mechanism of HMGB1 from liver/hepatocytes is therefore important in both sepsis and trauma. Regulating HMGB1 release from the liver may subsequently regulate circulating HMGB1 levels and so may be a viable therapeutic strategy to reduce inflammation and organ damage associated with sepsis and trauma.
Redox regulation of HMGB1 function
There are three cysteines in HMGB1: C23 and C45 located in the A box, which can form a disulfide bond under partially oxidizing conditions, and C106 located in the B box[42]. The redox state of these cysteines alters the biological function and activities of HMGB1[18, 43]. Extracellular fully reduced, all thiol HMGB1 - where all three cysteines have reduced -SH thiol groups – is able to associate with stromal cell-derived factor 1 (SDF1 or CXCL12) to synergistically promote migration of immune cells via the cognate chemokine receptor C-X-C chemokine receptor type 4 (CXCR4) and the receptor for advanced glycation end products (RAGE)[44]. Besides the known chemoattractant effect, we have recently shown that the all thiol, fully reduced form of HMGB1 is vital to delivery of LPS into the cytoplasm of inflammatory macrophages via RAGE[28]. Cytosolic LPS is then able to activate the non-canonical caspase-11 inflammasome to induce gasdermin D-mediated pyroptosis of immune cells[28], which results in increased levels of inflammation and DAMP release. (Figure 1)
The two cysteines in the A box of HMGB1, C23 and C45, are close enough within the tertiary structure to enable disulfide bond formation between them under mild oxidizing conditions[43]. The third cysteine remains reduced, however. Disulfide HMGB1 has the most potent inflammatory effects, and acts as a DAMP when released from cells in this form. Disulfide HMGB1 is able to trigger massive inflammatory cytokine production from immune cells through multiple signaling pathways involving cell surface pattern recognition receptors, such as RAGE and TLR4[43, 45]. Interestingly, it is the B box of HMGB1 and not the disulfide bond within the A box that has been shown to be the main driver of proinflammatory responses to HMGB1 through the induction of cytokine production in immune cells[46, 47]. This may be because the reduced cysteine in the B-box is important for binding and signaling through TLR4/Myeloid Differentiation factor 2 (MD2) [46, 47]. Furthermore, a recent study has indicated that the A-box binds to TLR4 with high affinity but also a relatively high dissociation rate, while the B-box binds to MD2 with low affinity but a very slow dissociation rate[48]. Importantly, formation of disulfide HMGB1 is reversible, and as the redox environment changes HMGB1 can convert back to the all-thiol, reduced form[49]. If oxidative stress continues to increase, however, any or all of the cysteines in HMGB1 can become fully oxidized to form sulfonyl groups, and this is irreversible[45]. Fully oxidized, sulfonyl HMGB1 has no chemokine or cytokine activities. Oxidized HMGB1 has some RAGE binding capacity[45] and can regulate autophagy in cancer cells through RAGE [50]. Furthermore, oxidized HMGB1 can neutralize its proinflammatory activity resulting in induction of immunological tolerance[51] and attenuation of liver ischemia and reperfusion injury[52]. Similarly, HMGB1 can also undergo ADP-ribosylation in tumor cells to affect autophagy, although it is not known if this regulatory pathway is also effective in sepsis/trauma[53]. The function of fully oxidized HMGB1 in setting of sepsis and trauma remains elusive, but may represent a mechanism to try to reduce inflammation, or promote resolution of inflammation.
We recently identified an additional role for the different redox states of HMGB1, this time within the cell. Our recent work has identified cytoplasmic HMGB1 as an important redox regulator of AIM2 inflammasome activation in hepatocytes in conditions of redox stress, such as hemorrhagic shock with resuscitation, or hypoxia with reoxygenation[54]. Cytoplasmic HMGB1 binds directly to AIM2 to help initiate inflammasome signaling in hepatocytes during redox stress[54], and this is a protective mechanism that induces mitophagy and clearance of oxidative stress-inducing damaged mitochondria[54]. Binding of HMGB1 to AIM2 is strongest when HMGB1 is in the all-thiol, reduced state, and is weaker once the disulfide bond is able to form. Binding strength also corresponds to inflammasome activation. This suggests HMGB1 is an important factor in redox-mediated regulation of AIM2-inflammasome activation. We showed that HMGB1 binds to a separate site from the DNA binding site of AIM2[54], suggesting that HMGB1 may not only act as a chaperone for double-stranded DNA to activate AIM2, but may facilitate structural opening of AIM2 to enable DNA binding (Figure 2). A recent study has shown that AIM2 inflammasome-mediated HMGB1 release can induce PD-L1 expression in tumor cells and results in immunosuppression in tumor microenvironment [55]. However, it remains to be determined whether HMGB1 can also induce PD-L1 expression in some cell types during sepsis and trauma.
Figure 2: Regulation of autophagy/mitophagy by intracellular HMGB1.

HMBG1 interacts with AIM2 and double stranded DNA in response to redox stress and activates inflammasome and caspase-1, inducing autophagy/mitophagy via beclin1-mediated pathways. HMGB1 also displaces Bcl2 from its association with beclin1, allowing autophagy initiation. Displaced Bcl2 acts as a apoptosis inhibitor. Unassociated beclin1 can be more easily cleaved/degraded by calpain and other cleavage enzymes which increases apoptosis.
Intracellular, endogenous disulfide HMGB1 has also been shown to have important protective effects in response to redox stress by increasing autophagy and inhibiting apoptosis[56–59]. We have shown that lack of hepatocyte HMGB1 exacerbates mitochondrial instability and damage, consequently leading to increased cell death during liver ischemia and reperfusion [59]. Cytoplasmic HMGB1 is able to bind directly to beclin1 [60], an important autophagy initiator, allowing it to form autophagy initiation complexes, which can remove harmful oxidative stresses (e.g. from mitochondria)[58]. HMGB1 is able to bind beclin1 by displacing Bcl2, an apoptosis inhibitor, which once dissociated from beclin1 can perform its role in preventing programmed apoptotic cell death[61]. Binding of HMGB1 to beclin1 occurs in the area of C23 and C45 and mutation of these cysteines to prevent disulfide bridge formation also prevents beclin1 association and diminishes autophagy[62]. (Figure 2) Furthermore, HMGB1 interacts with beclin1 and ATG5 to prevent calpain-mediated cleavage of these proteins, allowing autophagy to proceed [49] (Figure 2). Intracellular HMGB1 can also be degraded through proteolytic cleavage[63], as well as through cleavage by caspase-1[64] during sepsis, suggesting there are regulatory pathways to limit HMGB1 intracellular effects.
Location, location, location: the three guiding influences for HMGB1 function
The three critical factors that determine the function and activity of HMGB1 are: 1. Location of post-translational modifications of HMGB1 (acetylation, phosphorylation, methylation, and redox status of cysteines); 2. Location of HMGB1 within the cell (nucleus or cytoplasm) or release outside the cell; 3. Location of action of HMGB1 on specific cell types (e.g. macrophages, neutrophils) or in specific tissues and organs (e.g. gut). (Table 1) Each factor allows HMGB1 to have multifactorial effects and HMGB1 movement between each location (or group of locations) changes the overall effects of HMGB1 on inflammation in multiple diseases, including sepsis and trauma.
Table:
Location specific functions of HMGB1 in sepsis and trauma
| Cell type | Posttranslational modification | Intracellular | Extracellular | |
|---|---|---|---|---|
| Modification | Functions | |||
| Monocyte & Macrophage | Acetylation with NLS site | Nucleus to the cytoplasm translocation [12] | Induce autophagy preventing macrophage cell death from endotoxemia and bacterial infection by mediating [67] | 1. Induce cytokines and chemokine production [56,57] 2. Deliver LPS into the cytoplasm [25] 3. Induce pyroptosis [25,58] |
| Phosphorylation with NLS site | Nucleus to the cytoplasm translocation [14, 21] | |||
| Fully reduced, all thiol | Chemotaxis [41] Delivery of LPS into the cytoplasm [25] |
|||
| Disulfide | Inflammatory response [40,42–45] | |||
| Hepatocyte | Acetylation with NLS site | Nucleus to the cytoplasm translocation [18] | 1. Stabilize nucleus and mitochondria preventing cell death during liver ischemia and reperfusion [50] 2. Activate Aim2 inflammasome [46] |
|
| Phosphorylation with NLS site | Nucleus to the cytoplasm translocation [22] | |||
| Fully reduced, all thiol | Activate Aim2 inflammasome [46] | |||
| Neutrophil | Methylation at lysine 42 | Passive diffusion out of the nucleus [23] | 1. Inflammatory response [60] 2. NADPH oxidase dysfunction [61] 3. Stimulate neutrophil extracellular trap formation [62] |
|
| Vascular endothelial cells | 1. Inflammatory response [44,63] 2. Induce loss of endothelial integrity [66–68] |
|||
| Intestinal epithelia cells | Bind to beclin initiating autophagy [49] | Induce mucosal barrier dysfunction [70] | ||
| Platelet | Activate platelets in sepsis [71] | Activate platelet and thrombosis in trauma [72] | ||
Monocytes and macrophages are key players in host defense against pathogen invasion in sepsis, and key producers of cytokines in response to hemorrhagic shock and trauma[65, 66]. Intracellular HMGB1 in macrophages has been shown to prevent macrophage cell death from endotoxemia and bacterial infection by mediating autophagy[67]. Extracellular disulfide HMGB1 is a known activator of monocytes and macrophages to induce TNF release (and other cytokine and chemokine production) when signaling via the TLR4/MD2/MyD88/NFκB pathway[68, 69]. Interestingly, Wang et al. reported that administration of nontoxic quantities of HMGB1 together with nontoxic doses of LPS synergistically induced lethality in mice[27]. The mechanisms underlying this synergistic effect of HMGB1 and LPS had not been elucidated until recently when we reported that fully reduced HMGB1 delivers LPS into macrophages and endothelial cells via RAGE[28]. We had previously shown that HMGB1 acting through RAGE was able to induce inflammasome and caspase-1 activation in macrophages, leading to subsequent pyroptosis[70]. At that time we showed that internalized RAGE:HMGB1 contributed to the breakdown of lysosomes and release of cathepsins into the cytosol, which stimulated inflammasome activation in macrophages[70]. We have subsequently shown that HMGB1 bound to LPS is internalized by macrophages in the same RAGE-mediated pathway, and that HMGB1 is able to open the phospholipid bilayer of lysosomes once inside the lysosomal acidic environment. This leads to the release of LPS into the cytosol and subsequent activation of caspase-11-dependent pyroptosis[28] and non-canonical caspase-1 activation. It was recently confirmed that activation of caspase-11 inflammasome is required for lethality in sepsis[28, 40, 41], so indicating an essential role for HMGB1 on macrophages in mediating caspase-11-induced sepsis lethality. (Figure 1)
Neutrophils are key executors of bacterial clearance in sepsis, and have been implicated in organ damage in both sepsis and trauma[71]. HMGB1 aggravates sepsis-induced organ dysfunction and mortality via inhibition of neutrophil ability to clear bacteria, and so induction of persistent inflammation. HMGB1, acting via RAGE-dependent signaling decreases NADPH oxidase activity in neutrophils[72], which is vital for bacterial killing during sepsis. HMGB1:RAGE-mediated neutrophil NADPH oxidase dysfunction occurs in both mouse and human neutrophils, and is detected in patients who survive septic shock[73]. Treatment of CLP sepsis mice with anti-HMGB1 Ab significantly diminished sepsis-induced dysfunction of neutrophil NADPH oxidase activity and improved bacterial clearance[73]. Interestingly, extracellular HMGB1:TLR4 signaling activated neutrophil NADPH oxidase activity in a model of hemorrhagic shock[74]. Similarly, Zhou et al.[75]showed that HMGB1 in platelets was crucial for platelet activation and subsequently induced ROS production from neutrophils in a mouse model of sepsis. These conflicting data suggest differential function of HMGB1 when acting on different cell types through either TLR4 or RAGE, and the balance of these interactions may be crucial in determining protective or detrimental effects in sepsis and trauma. Some of this receptor specificity may be driven by the redox status of HMGB1, and so disease or animal model dependent, and also dependent on the severity or timing of the disease process. Yet again, these complex functions of HMGB1 suggest that the ability to target anti-HMGB1 therapy to a specific cell type or to a specific redox structure within HMGB1 may prove useful in treating a multifactorial disease such as sepsis.
Furthermore, HMGB1 released by injured hepatocytes during ischemia and reperfusion injury has been shown to stimulate NET formation through Toll-like receptor (TLR4)- and TLR9-MyD88 signaling pathways [76]. A recent study has also shown that platelet derived HMGB1 can stimulate NET formation and thus induce thrombosis in a mouse deep vein thrombosis model [77]. However, whether HMGB1 can stimulate NET formation in sepsis is unclear. NET formation can be an active process independent of cell death, or a passive process resulting from “NETosis”, a form of programmed, necrotic cell death in neutrophils[78]. It remains to be determined whether HMGB1 induces NET formation in an active or passive fashion during sepsis and trauma.
Vascular endothelial cells form an important barrier preventing circulatory and inflammatory components, as well as fluid, from leaking into surrounding organs and tissues[79]. Loss of endothelial cell monolayer integrity can therefore lead to exacerbation of organ damage, and this is prevalent in sepsis and after severe trauma[79]. HMGB1 binding to endothelial cells can dose-dependently upregulate endothelial expression of adhesion molecules such as ICAM-1, VCAM-1 and E-selectin, which can increase binding and movement of inflammatory cells across the vascular endothelium[80]. Endothelial cells also increase the production of proinflammatory IL-8 and G-CSF in response to HMGB1[47, 81]. In addition to direct effects on proinflammatory mediator production and attraction of immune cells, HMGB1 also plays an important role in regulating endothelial cell cytoskeletal rearrangement and vascular permeability[82]. High levels of HMGB1 in sera from patients with sepsis induce loss of vascular endothelial monolayer integrity, elicit formation of endothelial F‑actin stress fibers and initiate VE‑cadherin redistribution[82]. HMGB1-induced loss of endothelial integrity is dose and time-dependent, as shown by decreases in transendothelial electrical resistance in endothelial cells[83], and is mediated by signaling through RAGE and p38 mitogen-activated protein kinase (MAPK)[83, 84]. The interaction between HMGB1 and endothelial cells can be limited by proteolytic cleavage of HMGB1 by thrombin-thrombomodulin complexes[85], suggesting there are regulatory pathways to limit the effect of HMGB1 on endothelial cells.
Intestinal epithelium represents another important physical and physiological barrier within the body. Loss of intestinal epithelium integrity propagates uncontrolled immune responses and causes multiple organ failure during sepsis, severe trauma and hemorrhagic shock. High levels of HMGB1 in bile lead to mucosal barrier dysfunction in rats during endotoxemia potentially via down-regulation of expression of mucosal tight junction proteins (e.g. occludin, claudin-1 and ZO-1) in intestinal epithelial cells[86]. Ghrelin, an orexigenic hormone, has been used experimentally in mice as a treatment for sepsis, and has been shown to reduce serum HMGB1 levels and ameliorate gut barrier dysfunction via vagus nerve activation[87]. However, it is unclear which cell types are affected by grehlin-mediated vagus activation and how this regulates HMGB1 release.
Platelet-derived HMGB1 plays important roles in both sepsis and trauma[75, 88]. Deletion of HMGB1 specifically in platelets, allowed our group to show that platelet HMGB1 is essential for platelet activation and thrombosis[88], as well as platelet α granule degranulation and release of neutrophil chemoattractants, such as PF4 and CCL5 (RANTES)[75]. Platelet derived HMGB1 can also induce NET formation and subsequently promote thrombosis in a mouse model of deep vein thrombosis[77]. We recently also showed that platelet HMGB1 is required for efficient bacterial clearance in a mouse model of CLP sepsis, and this occurs via regulation of neutrophil recruitment and ROS production[75]. Extracellular HMGB1 can also affect platelet function through interactions with TLR4. During hemorrhagic shock with resuscitation, platelet TLR4 signaling regulates platelet function, can contribute to coagulation abnormalities associated with trauma and worsens organ injury[89].
Concluding remarks
HMGB1 is a multi-functional protein with major homeostatic roles, as well as important roles as a DAMP mediator of inflammation. The role HMGB1 plays is dependent largely on its location, and it can have both protective and detrimental roles during the pathogenesis of sepsis and trauma/hemorrhagic shock. Given these potentially different functions of HMGB1 any attempt to target HMGB1 in any disease, but especially in sepsis and trauma, will need to be extremely specific. Cell type specific targeting of inhibitors for HMGB1 will likely be necessary, and inhibition may also need to be specific to redox status. Some cell-targeting technology is being developed and is going to be of great importance to anyone in the field who is hoping to make therapeutic use of HMGB1 inhibitors and have them be effective. This usefulness of potential anti-HMGB1 therapeutics is not in doubt, however.
Acknowledgements
This review was supported by grants from the NIH: R01-GM102146 (MJS), R35-GM127027 (TRB), NIH HL079669 (JF), NIH HL139547 (JF), NIH HL076179 (JF), VA Merit Award BX002729 (JF), and BLR&D Research Career Scientist Award BX004211 (JF).
Abbreviations
- AIM2
absent in melanoma 2
- Bcl2
B-cell lymphoma 2
- CaMK
calcium calmodulin kinase
- CCL
CC chemokine
- CD14
cluster of differentiation 14
- CXCL
CXC chemokine
- CXCR
CXC chemokine receptor
- DAMP
damage-associated molecular pattern
- G-CSF
granulocyte colony forming factor
- HDAC
histone deacetylase
- HMGB1
high mobility group box 1
- ICAM1
intercellular adhesion molecule
- IFN
interferon
- IL
interleukin
- I/R
ischemia/reperfusion
- IRF
interferon regulatory factor
- JAK
janus kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-associated protein kinase
- MyD88
myeloid differentiation factor 88
- NET
neutrophil extracellular traps
- NLS
nuclear localization sequences
- PAMP
pathogen-associated molecular pattern
- RAGE
receptor for advanced glycation end products
- SDF1
stromal differentiation factor 1
- STAT
signal transduction and activator of transcription
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- VCAM1
vascular cell adhesion molecule 1
- ZO1
zonula occludens 1
Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
References
- 1.Einck L and Bustin M (1985) The intracellular distribution and function of the high mobility group chromosomal proteins. Exp Cell Res 156, 295–310. [DOI] [PubMed] [Google Scholar]
- 2.Javaherian K, Liu JF, Wang JC (1978) Nonhistone proteins HMG1 and HMG2 change the DNA helical structure. Science 199, 1345–6. [DOI] [PubMed] [Google Scholar]
- 3.Ferrari S, Ronfani L, Calogero S, Bianchi ME (1994) The mouse gene coding for high mobility group 1 protein (HMG1). J Biol Chem 269, 28803–8. [PubMed] [Google Scholar]
- 4.Gariboldi M, De Gregorio L, Ferrari S, Manenti G, Pierotti MA, Bianchi ME, Dragani TA (1995) Mapping of the Hmg1 gene and of seven related sequences in the mouse. Mamm Genome 6, 581–5. [DOI] [PubMed] [Google Scholar]
- 5.Wen L, Huang JK, Johnson BH, Reeck GR (1989) A human placental cDNA clone that encodes nonhistone chromosomal protein HMG-1. Nucleic Acids Res 17, 1197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lotze MT and Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5, 331–42. [DOI] [PubMed] [Google Scholar]
- 7.Klune JR, Dhupar R, Cardinal J, Billiar TR, Tsung A (2008) HMGB1: endogenous danger signaling. Mol Med 14, 476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tang D, Kang R, Zeh HJ 3rd, Lotze MT (2011) High-mobility group box 1, oxidative stress, and disease. Antioxid Redox Signal 14, 1315–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, Sun X, Wang H, Wang Q, Tsung A, Billiar TR, Zeh HJ 3rd, Lotze MT, Tang D (2014) HMGB1 in health and disease. Mol Aspects Med 40, 1–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersson U, Yang H, Harris H (2018) High-mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol. [DOI] [PubMed] [Google Scholar]
- 11.Okamoto K, Tamura T, Sawatsubashi Y (2016) Sepsis and disseminated intravascular coagulation. J Intensive Care 4, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME (2016) HMGB1 as biomarker and drug target. Pharmacol Res 111, 534–544. [DOI] [PubMed] [Google Scholar]
- 13.Yu Y, Tang D, Kang R (2015) Oxidative stress-mediated HMGB1 biology. Front Physiol 6, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schulman IG, Wang T, Wu M, Bowen J, Cook RG, Gorovsky MA, Allis CD (1991) Macronuclei and micronuclei in Tetrahymena thermophila contain high-mobility-group-like chromosomal proteins containing a highly conserved eleven-amino-acid putative DNA-binding sequence. Mol Cell Biol 11, 166–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22, 5551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ito I, Fukazawa J, Yoshida M (2007) Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J Biol Chem 282, 16336–44. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Wheeler D, Tang Y, Guo L, Shapiro RA, Ribar TJ, Means AR, Billiar TR, Angus DC, Rosengart MR (2008) Calcium/calmodulin-dependent protein kinase (CaMK) IV mediates nucleocytoplasmic shuttling and release of HMGB1 during lipopolysaccharide stimulation of macrophages. J Immunol 181, 5015–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang Y, Zhao X, Antoine D, Xiao X, Wang H, Andersson U, Billiar TR, Tracey KJ, Lu B (2016) Regulation of Posttranslational Modifications of HMGB1 During Immune Responses. Antioxid Redox Signal 24, 620–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen L, Fischle W, Verdin E, Greene WC (2001) Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 293, 1653–7. [DOI] [PubMed] [Google Scholar]
- 20.Gu W and Roeder RG (1997) Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90, 595–606. [DOI] [PubMed] [Google Scholar]
- 21.Evankovich J, Cho SW, Zhang R, Cardinal J, Dhupar R, Zhang L, Klune JR, Zlotnicki J, Billiar T, Tsung A (2010) High mobility group box 1 release from hepatocytes during ischemia and reperfusion injury is mediated by decreased histone deacetylase activity. J Biol Chem 285, 39888–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu B, Antoine DJ, Kwan K, Lundback P, Wahamaa H, Schierbeck H, Robinson M, Van Zoelen MA, Yang H, Li J, Erlandsson-Harris H, Chavan SS, Wang H, Andersson U, Tracey KJ (2014) JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc Natl Acad Sci U S A 111, 3068–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dhupar R, Klune JR, Evankovich J, Cardinal J, Zhang M, Ross M, Murase N, Geller DA, Billiar TR, Tsung A (2011) Interferon regulatory factor 1 mediates acetylation and release of high mobility group box 1 from hepatocytes during murine liver ischemia-reperfusion injury. Shock 35, 293–301. [DOI] [PubMed] [Google Scholar]
- 24.Youn JH and Shin JS (2006) Nucleocytoplasmic shuttling of HMGB1 is regulated by phosphorylation that redirects it toward secretion. J Immunol 177, 7889–97. [DOI] [PubMed] [Google Scholar]
- 25.Tsung A, Klune JR, Zhang X, Jeyabalan G, Cao Z, Peng X, Stolz DB, Geller DA, Rosengart MR, Billiar TR (2007) HMGB1 release induced by liver ischemia involves Toll-like receptor 4 dependent reactive oxygen species production and calcium-mediated signaling. J Exp Med 204, 2913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Zoelen MA, Yang H, Florquin S, Meijers JC, Akira S, Arnold B, Nawroth PP, Bierhaus A, Tracey KJ, van der Poll T (2009) Role of toll-like receptors 2 and 4, and the receptor for advanced glycation end products in high-mobility group box 1-induced inflammation in vivo. Shock 31, 280–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285, 248–51. [DOI] [PubMed] [Google Scholar]
- 28.Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, Zhao X, Liu J, Tang C, Liu Z, Huang Y, Peng H, Xiao L, Tang D, Scott MJ, Wang Q, Liu J, Xiao X, Watkins S, Li J, Yang H, Wang H, Chen F, Tracey KJ, Billiar TR, Lu B (2018) The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49, 740–753 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angus DC, Yang L, Kong L, Kellum JA, Delude RL, Tracey KJ, Weissfeld L, Gen IMSI (2007) Circulating high-mobility group box 1 (HMGB1) concentrations are elevated in both uncomplicated pneumonia and pneumonia with severe sepsis. Crit Care Med 35, 1061–7. [DOI] [PubMed] [Google Scholar]
- 30.Gaini S, Pedersen SS, Koldkjaer OG, Pedersen C, Moller HJ (2007) High mobility group box-1 protein in patients with suspected community-acquired infections and sepsis: a prospective study. Crit Care 11, R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sunden-Cullberg J, Norrby-Teglund A, Rouhiainen A, Rauvala H, Herman G, Tracey KJ, Lee ML, Andersson J, Tokics L, Treutiger CJ (2005) Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med 33, 564–73. [DOI] [PubMed] [Google Scholar]
- 32.Chavan SS, Huerta PT, Robbiati S, Valdes-Ferrer SI, Ochani M, Dancho M, Frankfurt M, Volpe BT, Tracey KJ, Diamond B (2012) HMGB1 mediates cognitive impairment in sepsis survivors. Mol Med 18, 930–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, Rubartelli A (2002) The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep 3, 995–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mullins GE, Sunden-Cullberg J, Johansson AS, Rouhiainen A, Erlandsson-Harris H, Yang H, Tracey KJ, Rauvala H, Palmblad J, Andersson J, Treutiger CJ (2004) Activation of human umbilical vein endothelial cells leads to relocation and release of high-mobility group box chromosomal protein 1. Scand J Immunol 60, 566–73. [DOI] [PubMed] [Google Scholar]
- 35.Liu S, Stolz DB, Sappington PL, Macias CA, Killeen ME, Tenhunen JJ, Delude RL, Fink MP (2006) HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. Am J Physiol Cell Physiol 290, C990–9. [DOI] [PubMed] [Google Scholar]
- 36.Sheller-Miller S, Urrabaz-Garza R, Saade G, Menon R (2017) Damage-Associated molecular pattern markers HMGB1 and cell-Free fetal telomere fragments in oxidative-Stressed amnion epithelial cell-Derived exosomes. J Reprod Immunol 123, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maugeri N, Capobianco A, Rovere-Querini P, Ramirez GA, Tombetti E, Valle PD, Monno A, D’Alberti V, Gasparri AM, Franchini S, D’Angelo A, Bianchi ME, Manfredi AA (2018) Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci Transl Med 10. [DOI] [PubMed] [Google Scholar]
- 38.Chen G, Li J, Ochani M, Rendon-Mitchell B, Qiang X, Susarla S, Ulloa L, Yang H, Fan S, Goyert SM, Wang P, Tracey KJ, Sama AE, Wang H (2004) Bacterial endotoxin stimulates macrophages to release HMGB1 partly through CD14- and TNF-dependent mechanisms. J Leukoc Biol 76, 994–1001. [DOI] [PubMed] [Google Scholar]
- 39.Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, Al-Abed Y, Wang H, Metz C, Miller EJ, Tracey KJ, Ulloa L (2004) Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med 10, 1216–21. [DOI] [PubMed] [Google Scholar]
- 40.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–92. [DOI] [PubMed] [Google Scholar]
- 41.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose-Girma M, Dixit VM (2011) Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–21. [DOI] [PubMed] [Google Scholar]
- 42.Antoine DJ, Harris HE, Andersson U, Tracey KJ, Bianchi ME (2014) A systematic nomenclature for the redox states of high mobility group box (HMGB) proteins. Mol Med 20, 135–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Venereau E, Casalgrandi M, Schiraldi M, Antoine DJ, Cattaneo A, De Marchis F, Liu J, Antonelli A, Preti A, Raeli L, Shams SS, Yang H, Varani L, Andersson U, Tracey KJ, Bachi A, Uguccioni M, Bianchi ME (2012) Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med 209, 1519–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schiraldi M, Raucci A, Munoz LM, Livoti E, Celona B, Venereau E, Apuzzo T, De Marchis F, Pedotti M, Bachi A, Thelen M, Varani L, Mellado M, Proudfoot A, Bianchi ME, Uguccioni M (2012) HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J Exp Med 209, 551–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang H, Antoine DJ, Andersson U, Tracey KJ (2013) The many faces of HMGB1: molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J Leukoc Biol 93, 865–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li J, Kokkola R, Tabibzadeh S, Yang R, Ochani M, Qiang X, Harris HE, Czura CJ, Wang H, Ulloa L, Wang H, Warren HS, Moldawer LL, Fink MP, Andersson U, Tracey KJ, Yang H (2003) Structural basis for the proinflammatory cytokine activity of high mobility group box 1. Mol Med 9, 37–45. [PMC free article] [PubMed] [Google Scholar]
- 47.Treutiger CJ, Mullins GE, Johansson AS, Rouhiainen A, Rauvala HM, Erlandsson-Harris H, Andersson U, Yang H, Tracey KJ, Andersson J, Palmblad JE (2003) High mobility group 1 B-box mediates activation of human endothelium. J Intern Med 254, 375–85. [DOI] [PubMed] [Google Scholar]
- 48.He M, Bianchi ME, Coleman TR, Tracey KJ, Al-Abed Y (2018) Exploring the biological functional mechanism of the HMGB1/TLR4/MD-2 complex by surface plasmon resonance. Mol Med 24, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang H, Lundback P, Ottosson L, Erlandsson-Harris H, Venereau E, Bianchi ME, Al-Abed Y, Andersson U, Tracey KJ, Antoine DJ (2012) Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med 18, 250–9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 50.Tang D, Kang R, Cheh CW, Livesey KM, Liang X, Schapiro NE, Benschop R, Sparvero LJ, Amoscato AA, Tracey KJ, Zeh HJ, Lotze MT (2010) HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 29, 5299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kazama H, Ricci JE, Herndon JM, Hoppe G, Green DR, Ferguson TA (2008) Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29, 21–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu A, Fang H, Dirsch O, Jin H, Dahmen U (2012) Oxidation of HMGB1 causes attenuation of its pro-inflammatory activity and occurs during liver ischemia and reperfusion. PLoS One 7, e35379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang M, Liu L, Xie M, Sun X, Yu Y, Kang R, Yang L, Zhu S, Cao L, Tang D (2015) Poly-ADP-ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy 11, 214–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun Q, Loughran P, Shapiro R, Shrivastava IH, Antoine DJ, Li T, Yan Z, Fan J, Billiar TR, Scott MJ (2017) Redox-dependent regulation of hepatocyte absent in melanoma 2 inflammasome activation in sterile liver injury in mice. Hepatology 65, 253–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li C, Zhang Y, Cheng X, Yuan H, Zhu S, Liu J, Wen Q, Xie Y, Liu J, Kroemer G, Klionsky DJ, Lotze MT, Zeh HJ, Kang R, Tang D (2018) PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev Cell 46, 441–455 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang D, Kang R, Livesey KM, Kroemer G, Billiar TR, Van Houten B, Zeh HJ 3rd, Lotze MT (2011) High-mobility group box 1 is essential for mitochondrial quality control. Cell Metab 13, 701–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang D, Kang R, Livesey KM, Zeh HJ 3rd, Lotze MT (2011) High mobility group box 1 (HMGB1) activates an autophagic response to oxidative stress. Antioxid Redox Signal 15, 2185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu X, Messer JS, Wang Y, Lin F, Cham CM, Chang J, Billiar TR, Lotze MT, Boone DL, Chang EB (2015) Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J Clin Invest 125, 1098–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang H, Nace GW, McDonald KA, Tai S, Klune JR, Rosborough BR, Ding Q, Loughran P, Zhu X, Beer-Stolz D, Chang EB, Billiar T, Tsung A (2014) Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high-mobility group box 1 in cellular protection. Hepatology 59, 1984–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd, Lotze MT (2010) Endogenous HMGB1 regulates autophagy. J Cell Biol 190, 881–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ranjan K and Pathak C (2016) Expression of cFLIPL Determines the Basal Interaction of Bcl-2 With Beclin-1 and Regulates p53 Dependent Ubiquitination of Beclin-1 During Autophagic Stress. J Cell Biochem 117, 1757–68. [DOI] [PubMed] [Google Scholar]
- 62.Kang R, Livesey KM, Zeh HJ, Loze MT, Tang D (2010) HMGB1: a novel Beclin 1-binding protein active in autophagy. Autophagy 6, 1209–11. [DOI] [PubMed] [Google Scholar]
- 63.Yu H, Schwarzer K, Forster M, Kniemeyer O, Forsbach-Birk V, Straube E, Rodel J (2010) Role of high-mobility group box 1 protein and poly(ADP-ribose) polymerase 1 degradation in Chlamydia trachomatis-induced cytopathicity. Infect Immun 78, 3288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.LeBlanc PM, Doggett TA, Choi J, Hancock MA, Durocher Y, Frank F, Nagar B, Ferguson TA, Saleh M (2014) An immunogenic peptide in the A-box of HMGB1 protein reverses apoptosis-induced tolerance through RAGE receptor. J Biol Chem 289, 7777–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peiseler M and Kubes P (2018) Macrophages play an essential role in trauma-induced sterile inflammation and tissue repair. Eur J Trauma Emerg Surg 44, 335–349. [DOI] [PubMed] [Google Scholar]
- 66.Delano MJ and Ward PA (2016) Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest 126, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yanai H, Matsuda A, An J, Koshiba R, Nishio J, Negishi H, Ikushima H, Onoe T, Ohdan H, Yoshida N, Taniguchi T (2013) Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci U S A 110, 20699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang H, Wang H, Ju Z, Ragab AA, Lundback P, Long W, Valdes-Ferrer SI, He M, Pribis JP, Li J, Lu B, Gero D, Szabo C, Antoine DJ, Harris HE, Golenbock DT, Meng J, Roth J, Chavan SS, Andersson U, Billiar TR, Tracey KJ, Al-Abed Y (2015) MD-2 is required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med 212, 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yu M, Wang H, Ding A, Golenbock DT, Latz E, Czura CJ, Fenton MJ, Tracey KJ, Yang H (2006) HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 26, 174–9. [DOI] [PubMed] [Google Scholar]
- 70.Xu J, Jiang Y, Wang J, Shi X, Liu Q, Liu Z, Li Y, Scott MJ, Xiao G, Li S, Fan L, Billiar TR, Wilson MA, Fan J (2014) Macrophage endocytosis of high-mobility group box 1 triggers pyroptosis. Cell Death Differ 21, 1229–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McDonald B (2018) Neutrophils in critical illness. Cell Tissue Res 371, 607–615. [DOI] [PubMed] [Google Scholar]
- 72.Tadie JM, Bae HB, Banerjee S, Zmijewski JW, Abraham E (2012) Differential activation of RAGE by HMGB1 modulates neutrophil-associated NADPH oxidase activity and bacterial killing. Am J Physiol Cell Physiol 302, C249–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gregoire M, Tadie JM, Uhel F, Gacouin A, Piau C, Bone N, Le Tulzo Y, Abraham E, Tarte K, Zmijewski JW (2017) Frontline Science: HMGB1 induces neutrophil dysfunction in experimental sepsis and in patients who survive septic shock. J Leukoc Biol 101, 1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fan J, Li Y, Levy RM, Fan JJ, Hackam DJ, Vodovotz Y, Yang H, Tracey KJ, Billiar TR, Wilson MA (2007) Hemorrhagic shock induces NAD(P)H oxidase activation in neutrophils: role of HMGB1-TLR4 signaling. J Immunol 178, 6573–80. [DOI] [PubMed] [Google Scholar]
- 75.Zhou H, Deng M, Liu Y, Yang C, Hoffman R, Zhou J, Loughran PA, Scott MJ, Neal MD, Billiar TR (2018) Platelet HMGB1 is required for efficient bacterial clearance in intra-abdominal bacterial sepsis in mice. Blood Adv 2, 638–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L, Wang S, Kim J, Billiar T, Wang Y, Tsung A (2015) Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology 62, 600–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P, Tsung A, Zuckerbraun BS, Simmons RL, Neal MD (2018) Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci Rep 8, 2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yousefi S, Stojkov D, Germic N, Simon D, Wang X, Benarafa C, Simon HU (2019) Untangling “NETosis” from NETs. Eur J Immunol. [DOI] [PubMed] [Google Scholar]
- 79.Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY (2009) Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev Mol Med 11, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nawaz MI and Mohammad G (2015) Role of high-mobility group box-1 protein in disruption of vascular barriers and regulation of leukocyte-endothelial interactions. J Recept Signal Transduct Res 35, 340–5. [DOI] [PubMed] [Google Scholar]
- 81.Fiuza C, Bustin M, Talwar S, Tropea M, Gerstenberger E, Shelhamer JH, Suffredini AF (2003) Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood 101, 2652–60. [DOI] [PubMed] [Google Scholar]
- 82.Zheng YJ, Xu WP, Ding G, Gao YH, Wang HR, Pan SM (2016) Expression of HMGB1 in septic serum induces vascular endothelial hyperpermeability. Mol Med Rep 13, 513–21. [DOI] [PubMed] [Google Scholar]
- 83.Shao M, Tang ST, Liu B, Zhu HQ (2016) Rac1 mediates HMGB1induced hyperpermeability in pulmonary microvascular endothelial cells via MAPK signal transduction. Mol Med Rep 13, 529–35. [DOI] [PubMed] [Google Scholar]
- 84.Huang W, Liu Y, Li L, Zhang R, Liu W, Wu J, Mao E, Tang Y (2012) HMGB1 increases permeability of the endothelial cell monolayer via RAGE and Src family tyrosine kinase pathways. Inflammation 35, 350–62. [DOI] [PubMed] [Google Scholar]
- 85.Ito T, Kawahara K, Okamoto K, Yamada S, Yasuda M, Imaizumi H, Nawa Y, Meng X, Shrestha B, Hashiguchi T, Maruyama I (2008) Proteolytic cleavage of high mobility group box 1 protein by thrombin-thrombomodulin complexes. Arterioscler Thromb Vasc Biol 28, 1825–30. [DOI] [PubMed] [Google Scholar]
- 86.Yang R, Miki K, Oksala N, Nakao A, Lindgren L, Killeen ME, Mennander A, Fink MP, Tenhunen J (2009) Bile high-mobility group box 1 contributes to gut barrier dysfunction in experimental endotoxemia. Am J Physiol Regul Integr Comp Physiol 297, R362–9. [DOI] [PubMed] [Google Scholar]
- 87.Wu R, Dong W, Qiang X, Wang H, Blau SA, Ravikumar TS, Wang P (2009) Orexigenic hormone ghrelin ameliorates gut barrier dysfunction in sepsis in rats. Crit Care Med 37, 2421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vogel S, Bodenstein R, Chen Q, Feil S, Feil R, Rheinlaender J, Schaffer TE, Bohn E, Frick JS, Borst O, Munzer P, Walker B, Markel J, Csanyi G, Pagano PJ, Loughran P, Jessup ME, Watkins SC, Bullock GC, Sperry JL, Zuckerbraun BS, Billiar TR, Lotze MT, Gawaz M, Neal MD (2015) Platelet-derived HMGB1 is a critical mediator of thrombosis. J Clin Invest 125, 4638–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ding N, Chen G, Hoffman R, Loughran PA, Sodhi CP, Hackam DJ, Billiar TR, Neal MD (2014) Toll-like receptor 4 regulates platelet function and contributes to coagulation abnormality and organ injury in hemorrhagic shock and resuscitation. Circ Cardiovasc Genet 7, 615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]