

Vibrio cholerae leads a dual life cycle, as it can exist in the aquatic environment and colonize the human small intestine. In both life cycles, V. cholerae encounters a variety of stressful conditions, including fluctuating pH and temperature and exposure to other agents that may negatively affect cell envelope homeostasis. The phage shock protein (Psp) response is required to sense and respond to such insults in other bacteria but has remained unstudied in V. cholerae. Interestingly, the Psp system has protein homologs, principally, PspA, in a number of bacterial clades as well as in archaea and plants. Therefore, our findings not only fill a gap in knowledge about an unstudied extracytoplasmic stress response in V. cholerae, but also may have far-reaching implications.
KEYWORDS: Psp, Vibrio cholerae, cholera, stress response
ABSTRACT
The phage shock protein (Psp) system is a stress response pathway that senses and responds to inner membrane damage. The genetic components of the Psp system are present in several clinically relevant Gram-negative bacteria, including Vibrio cholerae. However, most of the current knowledge about the Psp response stems from in vitro studies in Escherichia coli and Yersinia enterocolitica. In fact, the Psp response in V. cholerae has remained completely uncharacterized. In this study, we demonstrate that V. cholerae does have a functional Psp response system. We found that overexpression of GspD (EpsD), the type II secretion system secretin, induces the Psp response, whereas other V. cholerae secretins do not. In addition, we have identified several environmental conditions that induce this stress response. Our studies on the genetic regulation and induction of the Psp system in V. cholerae suggest that the key regulatory elements are conserved with those of other Gram-negative bacteria. While a psp null strain is fully capable of colonizing the infant mouse intestine, it exhibits a colonization defect in a zebrafish model, indicating that this response may be important for disease transmission in the environment. Overall, these studies provide an initial understanding of a stress response pathway that has not been previously investigated in V. cholerae.
IMPORTANCE Vibrio cholerae leads a dual life cycle, as it can exist in the aquatic environment and colonize the human small intestine. In both life cycles, V. cholerae encounters a variety of stressful conditions, including fluctuating pH and temperature and exposure to other agents that may negatively affect cell envelope homeostasis. The phage shock protein (Psp) response is required to sense and respond to such insults in other bacteria but has remained unstudied in V. cholerae. Interestingly, the Psp system has protein homologs, principally, PspA, in a number of bacterial clades as well as in archaea and plants. Therefore, our findings not only fill a gap in knowledge about an unstudied extracytoplasmic stress response in V. cholerae, but also may have far-reaching implications.
INTRODUCTION
Bacteria have evolved to survive in an astounding number of habitats by monitoring their internal and external environments and modifying their genetic regulation accordingly. The only barriers to separate the interior of the cell from the outside environment are its membranes and periplasmic space, or the cell envelope. Bacteria have complex membranes that define cellular shape, generate energy, and provide protection, while simultaneously maintaining permeability to nutrients, and are the site of numerous other essential cellular processes (1). Gram-negative bacteria are surrounded by two membranes, the inner membrane (IM) and the outer membrane (OM). Any threat to the stability of either membrane could lead to loss of viability. To guard against damage to the cell envelope, bacteria have signal transduction systems called extracytoplasmic stress responses (ESRs) that monitor the integrity of the membrane compartments. Once an ESR has been initiated, proteins are produced that function to restore cell homeostasis (2).
Gram-negative bacteria have a number of characterized ESRs, including the σE, Cpx, Bae, Rcs, and phage shock protein (Psp) responses. The σE, Cpx, Bae, and Rcs pathways involve regulation of a wide array of genes, whereas the Psp response is tightly regulated (2–4). The Psp response was initially discovered by Peter Model and his colleagues when they found that f1 filamentous phage infection of Escherichia coli resulted in production of a 25-kDa protein (5). They subsequently named the protein phage shock protein A (PspA) and determined that its production was induced by the phage gene product pIV (5). However, they later discovered that this response was not limited to phage infection, but that a number of other stressors, such as ethanol, heat, osmotic shock, and stationary-phase growth were also inducers (5, 6). The Psp system includes several genes, with the core set, pspFABC, considered the minimal functional unit in most Gram-negative bacteria (7). Oftentimes, the systems include an uncoupled gene, pspG, and may also include additional less understood genes that play a role in the Psp response. These systems are typically inactive when PspF, the transcriptional activator of the system, is bound in a complex with PspA. However, when membrane disruption occurs, it is sensed by PspB and PspC. PspA then releases PspF and pspABC transcription occurs, resulting in the production of a protective response (8–10). The exact mechanism by which the individual components of the Psp system ameliorate membrane stress is not yet fully understood (11).
Secretins are homomultimeric pores that facilitate the transfer of macromolecules across the outer membranes of bacteria (12). These cylindrical multimers are formed from 12 to 15 monomers and are a major component of 4 different classes of secretion systems: the type II secretion system (T2SS), type III secretion system (T3SS), type IV pilus system (T4PS), and phage extrusion (13). A common theme in the study of Psp systems has been that secretins often induce the response. The phage gene product pIV, produced for phage extrusion out of the cell, is a secretin that localizes to the bacterial outer membrane. However, during phage infection, some amount of this protein often mislocalizes to the inner membrane, stimulating the Psp response (14, 15). When the Psp system was characterized in Yersinia enterocolitica, the T3SS secretins, YsaC and YscC, were also discovered to be inducers of the Psp response (16, 17). Historically, the Psp response has been most thoroughly studied in E. coli and Y. enterocolitica, with limited characterization in a number of other bacteria (18).
Vibrio cholerae is a Gram-negative bacterium that is naturally found in aquatic ecosystems but can also colonize humans, causing the severe diarrheal disease cholera (19). Although there are hundreds of V. cholerae serogroups, only the O1 and O139 serogroups are capable of causing pandemic cholera. The O1 serogroup is subdivided into the classical biotype, which caused the first six cholera pandemics, and the El Tor biotype, which has been the predominant cause of cholera since 1961 (20). Regardless of location, V. cholerae is exposed to numerous stressors that negatively impact membrane integrity (21). While other ESRs have been characterized in V. cholerae, the Psp response has yet to be studied in V. cholerae. Therefore, the aim of this work was to determine if V. cholerae has a functional Psp response and identify its inducers. In the results presented here, we show that V. cholerae does contain a functional Psp system. As anticipated, PspA functions as a negative regulator of the system, whereas PspF, PspB, and PspC are critical for the initiation of the response. In addition, we found that the V. cholerae Psp system is highly induced by overexpression of the T2SS secretin, GspD (EpsD), but not by any of the other encoded secretins. We have also identified several environmental conditions that activate the response, including stationary-phase growth, osmotic shock, SDS treatment, heat, and ethanol stress. Furthermore, the Psp system is important for colonization in the zebrafish model of cholera infection, suggesting that it may be required for environmental transmission of disease.
RESULTS
Genetic organization of the psp genes in V. cholerae.
Previous transcriptomic studies suggested that the V. cholerae Psp system may compensate for the loss of another ESR, the σE response (22; unpublished data). In fact, a similar observation has been made in Salmonella enterica serovar Typhimurium (23). However, before examining the regulatory relationship between the two ESRs, we first wanted to determine whether or not V. cholerae has a functional Psp response. The V. cholerae psp locus was previously predicted to contain the pspFABC and pspG genes during a TBLASTN search examining conservation of Psp systems (7). The amino acid identities between the E. coli and V. cholerae Psp genes from PspF, -A, -B, -C, and -G are as follows: 62%, 58%, 50%, 36%, and 45%, respectively. The N-terminal region of PspA tends to be highly similar between species, and V. cholerae is no exception (Fig. 1B). In PspA, the first 60 of 222 amino acids have 75% identity and 96% similarity to the E. coli PspA. The N-terminal region of PspA is important for membrane binding, and this conservation suggests that the V. cholerae PspA may also retain those characteristics (24). The unlinked pspG is found between 2,750 and 2,390 bp downstream of pspF in E. coli, S. enterica, and Y. enterocolitica; however, pspG is 1,373 bp upstream of pspF in V. cholerae. A subsequent TBLASTN analysis has revealed that V. cholerae also harbors a gene homologous to pspE (Fig. 1A). In E. coli and S. enterica, pspE remains coupled to pspFABCD. Interestingly, in V. cholerae, pspE is unlinked from the core set of psp genes, residing on the second chromosome, and has 42% similarity to E. coli PspE (Fig. 1A). The E. coli PspE is a rhodanese (thiosulfate sulfurtransferase), and structural predictions of V. cholerae PspE show it is also likely to be a rhodanese (data not shown) (25).
FIG 1.

Genetic organization of Psp systems and PspA sequence similarity. (A) Comparison of the genetic organization of the psp genes in V. cholerae with that of three well-studied Gram-negative species, E. coli, S. enterica, and Y. enterocolitica. (B) Alignment of the amino acid sequences of PspA for the four species. Multiple sequence alignment created with ExPASy 3.21 BOXSHADE server (https://sourceforge.net/projects/boxshade/). Black shading or *, fully conserved residues; gray shading or dot, semiconserved residues; white, no conserved residues.
The GspD secretin induces the Psp response.
While the genetic components for a functional Psp system are found in the V. cholerae genome, whether the system is active under any conditions is unknown. Therefore, we first wanted to determine if the Psp response was functional in V. cholerae. To that end, we needed to identify an inducer that activates the system. Previous studies characterizing Psp response systems in other bacteria have shown that secretin proteins are remarkably specific inducers of these systems. This is thought to be a result of secretin mislocalization to the bacterial inner membrane (IM) when these proteins are highly expressed (3, 4). Therefore, we wanted to determine if overexpression of any of the V. cholerae secretins would induce the response, similar to that observed in Y. enterocolitica.
The V. cholerae serogroup O1 classical biotype contains multiple secretion systems: general secretion via the Tat and Sec pathways, T2SS, T4PS, and T6SS, but only the T2SS and the T4PSs contain a secretin protein as a part of their outer membrane machinery. Furthermore, T4PS systems can be divided into two subclasses based on their mode of assembly, type of pilins, and function. T4aPS generally functions in twitching motility and transfer of DNA, whereas T4bPS is usually involved in host colonization in enteric pathogens (26–28). Classical V. cholerae strain O395 harbors three T4PS systems, though only one is fully functional (29, 30). O395 has one T4bPS, the toxin-coregulated pilus system (TCP), which is involved in attachment to mammalian intestines (31). O395 also harbors the mannose-sensitive hemagglutinin complex (MSHA) and pilus (Pil) systems, which have associated secretin proteins. Therefore, we wanted to determine whether all or any of these systems contained secretins that were capable of activating the Psp system when overexpressed. The four secretins identified are as follows: GspD (also known as EpsD; general secretory pathway, T2SS), TcpC (TCP; T4bPS), PilQ (Pil; T4aPS), and MshL (MSHA; T4aPS).
To determine if the system was being induced, we constructed a reporter of pspA promoter activity, as in other studies (16). We generated a chromosomal reporter fusion where the promoter of pspABC was inserted upstream of the V. cholerae endogenous lacZ gene, denoted lacZΩPpspA (see Table S1 in the supplemental material). We also confirmed that pspABC are cotranscribed; therefore, the reporter fusion represents not just pspA transcription but also that of pspB and pspC (see Fig. S1). Each of the four identified V. cholerae secretins were then overexpressed in this background, and pspA promoter activity was assessed using a β-galactosidase assay. We found that only the T2SS secretin, GspD, is capable of inducing the Psp response when overexpressed (Fig. 2A). Additionally, each secretin overexpression construct was designed to contain a C-terminal 6×histidine epitope to measure protein expression levels and stability. To determine if the lack of induction was due to decreased protein levels, we used immunoblotting to detect the relative amounts of the secretins in the cultures. The inability of the T4PS secretins to induce the Psp response does not appear to be due to a defect in expression or stability, as they are expressed at high levels, especially in comparison to the low expression of GspD (Fig. 2B). We also constructed a V. cholerae strain where pspA was tagged with a chromosomal C-terminal 6×histidine epitope for detection. This strain was used to measure native PspA protein levels in the presence of the overexpressed secretins via immunoblotting. Further supporting the transcriptional fusion data, only GspD overexpression was capable of inducing expression of PspA (Fig. 2B).
FIG 2.
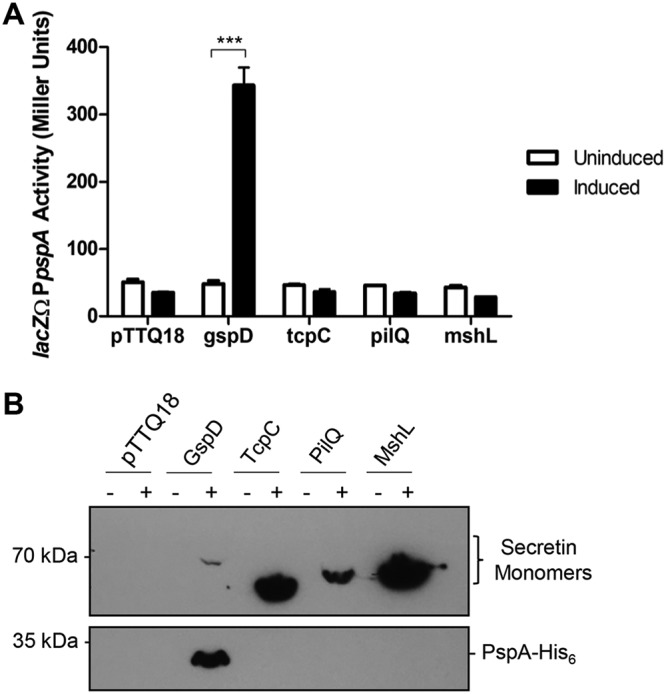
The T2SS secretin, GspD, induces the Psp response in V. cholerae. The four identified secretins of V. cholerae (GspD, TcpC, MshL, and PilQ) were overexpressed from pTTQ18, containing C-terminal 6×His epitopes for detection. (A) Activity of the pspA promoter as detected by β-galactosidase production from a lacZΩPpspA reporter. (B) Chromosomal expression of PspA in response to overexpressed secretins. ***, P < 0.001 by Student's t test. Error bars represent ± standard deviations.
Multimeric stability of secretins determines whether they initiate the Psp response.
Intriguingly, the levels of the monomeric form of the GspD protein were remarkably low in comparison to that of the other T4PS secretins (Fig. 2B). In Y. enterocolitica, secretin mislocalization and multimerization are necessary to induce the Psp response (32). Some secretins, such as TcpC, form stable multimers in SDS sample buffer unless heated above 65°C, whereas phage protein pIV forms stable multimers even after boiling at 100°C in SDS sample buffer (31, 33). It has been reported that if a secretin fails to form SDS- and heat-stable multimers, the Psp system is not induced (32, 33). Therefore, we examined the ability of all four secretins to form high-molecular-weight SDS- and heat-stable multimers by separating them by electrophoresis using a gradient gel. All samples were suspended in sample buffer containing SDS and boiled for 5 min before loading onto the gel. Only GspD formed SDS- and heat-resistant multimers, while the other secretins dissociated into monomers during this treatment (Fig. 3A). This helps to explain our previous observation that GspD protein levels appear to be lower than those of the other overexpressed secretins (Fig. 2B). In fact, the GspD present in the samples was not initially detected since the vast majority were not dissociated into monomers and were running at a very high molecular weight. This result supports Andrew Darwin and colleagues’ hypothesis that only multimeric secretins are sensed to be a threat by the Psp system and result in induction of the response (32). Another part of this theory is that a significant amount of the multimeric secretin complex mislocalizes to the IM. Therefore, we wanted to determine the localization of multimeric GspD in V. cholerae. Total membranes were isolated from V. cholerae overexpressing GspD and subsequently separated into inner membrane (IM) and outer membrane (OM) fractions. OmpT and ToxR were used as OM and IM fractionation controls, respectively. While we observed predominately GspD monomers in the OM fraction, the vast majority of the GspD multimers were found in the IM fraction (Fig. 3B). This supports the hypothesis that overexpressed GspD is forming mislocalized multimers in the IM, which are likely sensed by the Psp system, initiating the response.
FIG 3.
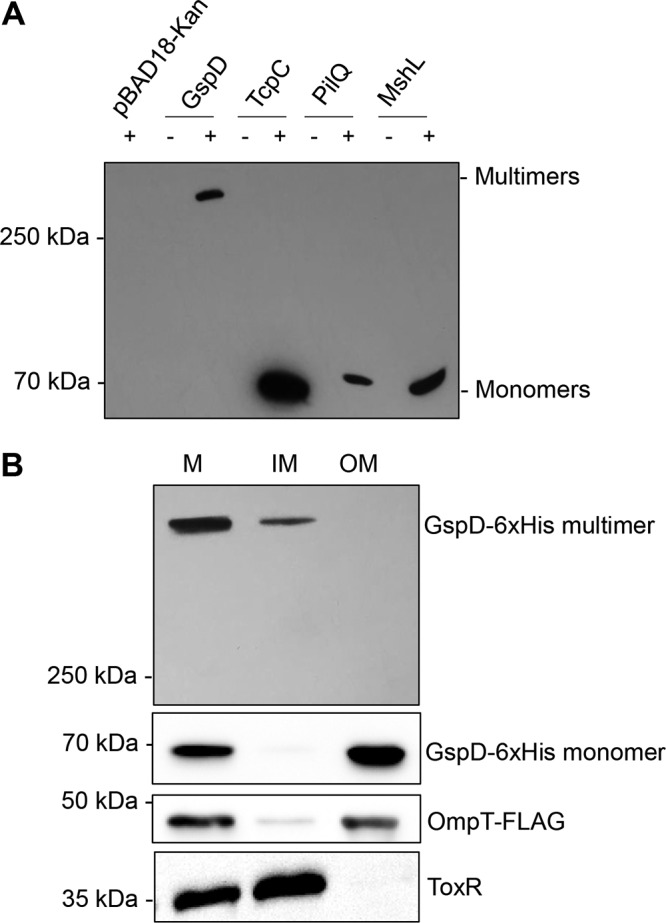
GspD forms heat-resistant multimers that mislocalize to the inner membrane, where the other V. cholerae secretins do not. (A) Cultures were grown to mid-log phase when protein production was induced by the addition of arabinose for 1 h. Samples were resuspended in sample buffer containing SDS and boiled for 5 min before separation by electrophoresis. (B) Induced cultures were fractionated into total membrane (M), inner membrane (IM), and outer membrane (OM) fractions, and GspD multimer and monomers were detected using anti-His antibody. OmpT-FLAG and ToxR were used as outer membrane and inner membrane controls, respectively. Figure is representative of 3 experiments.
GspD overexpression increases expression of other psp genes and causes PspA membrane association.
To gain a more comprehensive understanding of the effect of GspD overexpression on psp gene expression, we examined pspA, pspB, pspC, pspF, pspG, and pspE transcript profiles. V. cholerae was grown with and without overexpressed GspD, and RNA was isolated. After cDNA generation, transcript levels for each of the predicted psp genes were measured using reverse transcription-quantitative PCR (qRT-PCR). Transcript levels were normalized to the housekeeping gene, recA. As expected from the transcriptional reporter results, pspA, pspB, and pspC transcript levels were highly elevated in response to GspD overexpression (Fig. 4A). pspG was also highly expressed in response to secretin overexpression. This is the first evidence that pspG is involved in the Psp response in V. cholerae. In addition, we also found that pspE and pspF expression remained completely unaltered by secretin overexpression (Fig. 4A). PspF expression is negatively autogenously controlled in E. coli through blockage of the pspF promoter by RNA polymerase and PspF itself during pspABCE transcription (34, 35). Therefore, we did not expect to observe elevated levels of this transcript under these conditions. The lack of pspE transcript elevation with secretin overexpression suggests that this gene may not encode a functional member of the Psp response in V. cholerae. However, these results do not rule out the possibility that PspE is induced and functional under other conditions.
FIG 4.

GspD overexpression increases psp transcript levels and leads to PspA membrane association. Cultures were grown to mid-log phase when protein production was induced by the addition of arabinose for 1 h. (A) RNA was harvested and reverse transcribed. Transcript levels were normalized to the housekeeping gene recA. (B) Induced and uninduced cultures were fractionated into soluble and membrane fractions. Crp-FLAG and ToxR were used as cytoplasmic and membrane controls, respectively. Figure is representative of 3 experiments.
In a number of bacteria, PspA has been shown to be localized to both the cytoplasm and membranes, leading to its designation as a membrane-associated protein (5, 36, 37). However, in Y. enterocolitica, secretin overexpression resulted in increased PspA association with the membrane (10, 36). Therefore, we analyzed the effect of GspD overexpression on PspA localization. Without GspD induction, PspA expression is low, and it is primarily localized to the soluble fraction (see Fig. S2). However, upon GspD induction, PspA is highly expressed and predominantly associated with the membrane fraction (Fig. 4B). These results suggest that PspA spatial localization changes with the presence of an inducing stimulus, similar to what was observed in Y. enterocolitica (10).
Regulation of the Psp response.
In other characterized Gram-negative Psp systems, PspA functions as a negative regulator, binding PspF and preventing it from activating transcription from the pspA promoter (11, 16, 38). To determine if Psp regulation is similar in V. cholerae, we made deletions of each psp gene in the strain containing the lacZΩPpspA reporter fusion. Deletion of any of the psp genes did not result in observable growth defects (data not shown). In the absence of induction by secretin overexpression, lacZΩPpspA reporter activity is generally 40 to 50 Miller units. However, when pspA was deleted, there was a 50-fold increase in reporter activity despite the lack of inducer, which is consistent with other systems (Fig. 5A) (16, 38). Overexpression of pspA from a plasmid in the deletion strain dramatically reduced the activity of the reporter. This suggests that PspA is a negative regulator of the Psp response in V. cholerae.
FIG 5.
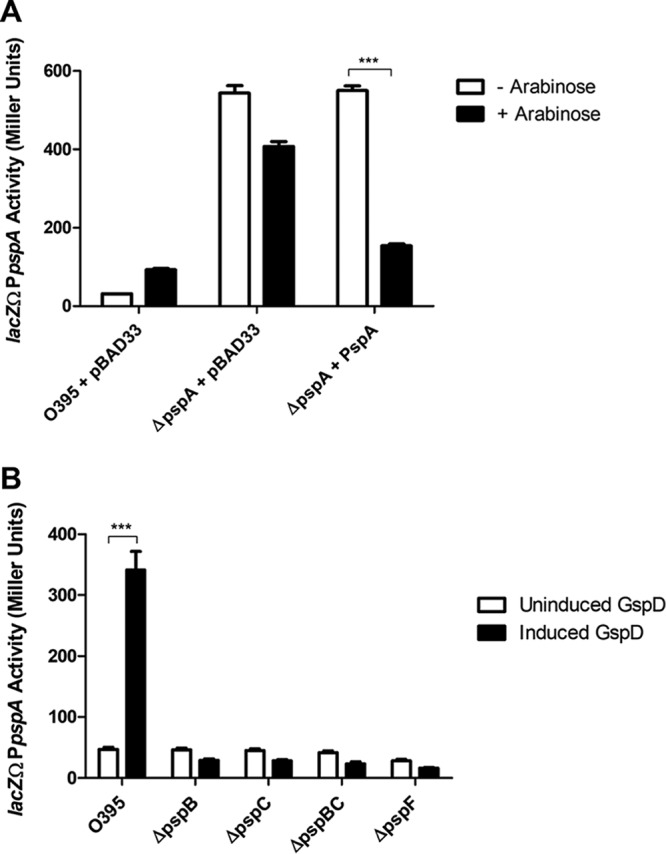
PspA is a negative regulator of the Psp response, and PspB, -C, and -F are positive regulators. Cultures were grown to mid-log phase (4 h). β-Galactosidase activity was produced from a chromosomal promoter fusion of lacZΩPpspA. (A) Loss of pspA leads to a large increase in PpspA activity, which can be reduced by complementation. (B) Loss of pspB, pspC, or pspF results in the inability to induce the Psp response. ***, P < 0.001 by Student’s t test. Error bars represent ± standard deviations.
In the other bacteria where the Psp response has been characterized, PspF, PspB, and PspC are all positive regulators of the system (16, 39). In E. coli, PspF has been shown to be the transcriptional activator of the psp operon (40). PspB and -C are inner membrane proteins that are required to sense membrane damage and bind PspA after Psp induction in Y. enterocolitica (16, 41). To determine whether PspF, PspB, and PspC have conserved positive regulatory roles, we induced the response by GspD overexpression and tested for loss of Psp induction in the mutant strains. When GspD was overexpressed, lacZΩPpspA activity increased 7-fold in the absence of any psp deletion (Fig. 5B). However, in the pspB, pspC, pspBC, and pspF mutants, the secretin was no longer capable of inducing increased activity from the reporter. pspB and pspC were tested individually and in combination, as their sequences partially overlap. Complementation studies resulted in the restoration of activity for each of the mutants; however, these studies were hampered by the necessity of using multiple plasmids and antibiotics, which negatively impacted baseline expression levels. Also, the addition of PspBC or PspF without GspD overexpression resulted in Psp activation, providing further evidence that they are positive regulators (Fig. S3 and data not shown). Overall, these data suggest that PspB, PspC, and PspF play positive regulatory roles in the Psp response in V. cholerae.
Environmental inducers of the Psp response.
A number of environmental conditions, including heat shock, ethanol, osmotic shock, stationary-phase growth, and treatment with the protonophore carbonyl cyanide m-chlorophenylhydrazone (CCCP), were found to increase PspA expression in E. coli (5, 6). In addition, increasing the alkalinity of cultures using NaOH, subjecting bacteria to the detergent SDS, and hyperosmotic shock were shown to be toxic to pspA mutants in Streptomyces lividans, suggesting that the Psp system was needed for survival in the presence of those stressors (37). Due to the varied environmental inducers of the Psp response in other bacteria, we tested a range of possible conditions to determine if any induced the response in V. cholerae. Similar to what was found for E. coli and S. enterica, stationary-phase growth increased PspA expression (Fig. 6A) (6, 23). Exposure to ethanol stress and SDS consistently induced PspA expression in comparison to that in untreated cells (Fig. 6B). Heat shock induces PspA expression over a short period of time, before cellular death begins after 30 min of exposure. Osmotic shock mediated by the addition of salt and increased alkalinity through NaOH treatment modestly induced PspA expression; however, these conditions were less reliable inducers of the response (Fig. 6B). Unlike in E. coli, S. lividans, and S. enterica, treatment with the protonophore CCCP did not reproducibly produce an increase in PspA expression (Fig. 6B). Despite identifying environmental conditions that induced the Psp response, we have not observed significant survival defects when psp mutant strains are exposed to these inducing stressors (data not shown). These results demonstrate that there are specific environmental inducers of the Psp response in V. cholerae and illustrate potential differences in the response between bacterial species.
FIG 6.

The Psp response can be induced by specific environmental conditions. (A) Cultures were grown for 24 h, and 1-ml aliquots were removed at the indicated time points. Cultures were normalized by OD600, and chromosomal PspA-6×His expression was detected by immunoblotting. (B) The indicated stressors were added after 3 h of growth. After 1 h (or the indicated time), cultures were normalized by OD600, and total protein concentration and chromosomal PspA-6×His expression were detected by immunoblotting. Loading control is a cross-reactive protein to ToxR antisera (59). Both panels are representative of three experiments.
A psp null strain shows reduced colonization in the zebrafish model.
The Psp system has been implicated in bacterial virulence in multiple species and is especially well characterized in Y. enterocolitica (42–44). In Y. enterocolitica, a pspC mutant is completely attenuated in a mouse model of infection (42). Therefore, we wanted to determine if the Psp response was required for successful colonization in two different models of V. cholerae infection, namely, the infant mouse and the zebrafish. The infant mouse is the most commonly used model for the study of factors required for V. cholerae colonization. In this model, infant mice are orally infected with a 1:1 mixture of wild-type and mutant strains in order to determine whether the mutant has a competitive disadvantage in colonization of the intestine (45). When we examined the psp null mutant (ΔpspFABC) in this model, we did not observe any significant defect in colonization (Fig. 7A). Additionally, we examined the ability of the psp null strain to colonize the zebrafish intestine (46). Fish are natural hosts for V. cholerae and may play a role in cholera transmission in the environment. Adult zebrafish were incubated in water containing wild-type or mutant bacteria, and colonization was allowed to occur for 6 h. At the 6-h time point, fish were washed and moved to clean sterile water. Both strains survived equivalently in the water over the 6-h interval (wild type, 2.55 × 106 CFU/ml; ΔpspFABC, 2.97 × 106 CFU/ml). After an additional 18 h, the zebrafish intestines were harvested, and colonizing bacteria were enumerated. In comparison to the wild-type strain (O395ΔlacZ), the psp null mutant was significantly less efficient at colonizing the zebrafish intestine (Fig. 7B). In addition, to examine the severity of the disease, mucin production and bacterial excretion were measured. While the reductions in mucin and bacterial levels were not statistically significant, there was an overall trend, suggesting that the psp mutant causes less severe disease in zebrafish (Fig. 7C and D). Overall, these observations indicate that the psp system in V. cholerae may not be required for colonization in mammals but may play a role in the environmental transmission of disease.
FIG 7.

The psp null strain shows reduced colonization in the zebrafish model of cholera infection but no defect in infant mice. (A) Infant mice were orally inoculated with a 1:1 ratio of 106 O395ΔlacZ and O395ΔpspFABC. (B) Zebrafish were incubated in water containing either 108 O395ΔlacZ or O395ΔpspFABC. After overnight infections, intestines were harvested and bacteria were plated for enumeration. Each data point represents data from one fish or mouse. (C) The water was tested for mucin concentration postinfection as a measure of fish diarrhea. The bar diagrams show the mucin level in excreted water after 24 h. (D) Bacterial numbers in the water postinfection were quantified to determine the levels of excreted bacteria. ***, P < 0.001. Error bars represent ± standard error of the means in panel B and standard deviations in panels C and D.
DISCUSSION
In this study, we report for the first time that V. cholerae encodes a functional Psp extracytoplasmic stress response system. We found that overexpression of the secretin, GspD, is a specific inducer of the V. cholerae Psp response. Interestingly, the three other secretins produced by V. cholerae, TcpC, PilQ, and MshL, fail to induce the Psp response. In addition, we found that specific environmental conditions, including ethanol stress and stationary-phase growth, cause an increase in PspA expression, similar to the E. coli Psp response (5, 6). Furthermore, the core set of Psp proteins, PspF, -A, -B, and -C, appear to possess identical regulatory roles to those observed in other bacterial systems (16, 38). Deletion of pspA causes the Psp system to become constitutively active; however, deletion of pspF, pspB, or pspC results in the inability to induce the response. Finally, we found that the Psp system plays a role in zebrafish intestinal colonization, providing a connection to environmental transmission of the organism.
When the Psp system was first discovered, it was theorized that the agents found to induce the Psp response functioned to dissipate the proton motive force (PMF) in the bacterial cell (6, 17, 23). The proton ionophore, CCCP, disrupts the PMF and has been shown to induce the Psp response in E. coli and S. enterica (6, 23). Additionally, the membrane potential component of the PMF is decreased in E. coli psp null strains (6, 9). However, this PMF theory was brought into question by Engl and colleagues in 2011 (47). They showed that dissipation of either the membrane potential or the proton gradient did not induce the Psp response in E. coli. To complicate matters further, some secretins are very specific inducers of the Psp response (5, 16, 48). It has been established that the secretins must mislocalize and multimerize in the inner membrane to activate the Psp cascade (32). The exact damage generated from secretin mislocalization is unknown. It may lead to leakage across the membrane or perhaps it destabilizes the inner membrane and increases membrane-stored curvature elastic stress (11, 49). We also found that secretin multimers of GspD mislocalize to the inner membrane upon overexpression, and we hypothesize that the damage generated is capable of signaling induction of the response in V. cholerae (Fig. 8).
FIG 8.

Model of Psp response in V. cholerae. In the absence of stress, the transcriptional activator PspF is bound by PspA, inhibiting transcription of psp genes. In the presence of stressors, such as mislocalized secretins from the type II secretion system (T2SS), PspA is sequestered to the inner membrane and PspF is free to initiate transcription of the psp genes. The inner membrane proteins PspB and PspC are predicted to aid in sensing damage and binding PspA to the inner membrane.
While V. cholerae is predicted to harbor four different secretin proteins, we found that only one, GspD, can induce the Psp response when overexpressed. Despite the similarity in structure of secretins, there are differences in how they target and insert into the OM. Koo et al. separated secretins into five classes based on localization, stability, and requirement of aide in assembly (50). The classes are as follows: (type 1) auto-assemble and capable of localizing to OM without assistance, (type 2) auto-assemble but need aide to reach the OM, (type 3) auto-assemble and can reach OM without assistance, but do so inefficiently, (type 4) cannot auto-assemble, but can localize to the OM, and (type 5) cannot auto-assemble or reach the OM. GspD in V. cholerae has been classified as a type 3 secretin, which is consistent with our results. In the case of TcpC in V. cholerae, it has been classified as a type 4 secretin, meaning that it cannot assemble by itself and form stable multimers, again, consistent with our findings. The T4aPS secretins, MshL and PilQ, have not been classified to date. Based on our results suggesting that they do not appear to auto-assemble or induce the Psp system, we hypothesize that they are also class 4 secretins, though it is plausible that they could also be categorized as type 5. The T3SS secretin YscC in Y. enterocolitica that causes Psp induction is classified as a type 2 secretin. Interestingly, the class of the first secretin found to induce the Psp response, the filamentous pIV secretin, has not been determined (50). The fact that the Psp system has the ability to differentiate between monomeric and multimeric secretins in the IM highlights its sophistication.
In addition to secretins, a range of other environmental stressors are known to induce the Psp response in the bacterial species where it has been studied. In E. coli, CCCP, heat shock, ethanol, osmotic shock, and stationary-phase growth induced the response. In S. lividans, NaOH and SDS were detrimental to pspA mutant growth and survival (5, 37). Therefore, we tested many of the known inducers to determine if they induced the response in V. cholerae. Ethanol, heat, hyperosmotic shock, and detergent exposure all induced expression of PspA. However, unlike in E. coli, S. enterica, and S. lividans, the proton ionophore CCCP does not reliably increase PspA expression. Again, this brings into question the exact inducing signal that stimulates the Psp response in different bacteria.
Due to the high conservation of the Psp system and the amino acid similarity of the proteins in V. cholerae and E. coli, we anticipated that the individual Psps would maintain similar regulatory roles. We observed that deletion of pspA led to an unchecked Psp response that was constitutively active. In addition, the ability to activate the Psp response was lost in pspF, pspB, and pspC mutants. These results suggest that PspA is a negative regulator of the system, whereas the other proteins are positive regulators. While this is consistent with previous reports, the specific roles of these Psps still need further examination. Based on previous studies and bioinformatic analysis, we hypothesize that PspF is the transcriptional activator of the V. cholerae Psp system, binds sigma-54, and enables RNA polymerase to initiate transcription at the pspABC promoter. In addition, we can predict that PspA binds PspF to inhibit psp transcription, as it has been shown to do in other bacteria. Furthermore, we can postulate that PspB and PspC are inner membrane proteins necessary for sensing the inducing signal and subsequently sequester PspA to the inner membrane. Of note, we discovered that PspC expression is unstable without coexpression of PspB, similarly to that in Y. enterocolitica (51). Interestingly, expression of PspC in E. coli does not depend on PspB coexpression, accentuating differences in PspB and PspC between species. With regard to pspG, we can only conclude at this time that its expression is regulated with the rest of the core psp operon. Further studies are needed to determine if there is a PspF binding site in the pspG promoter. Finally, our studies have not revealed a role for PspE in the V. cholerae Psp response. Further investigation is required to determine if it is connected to the response under different growth conditions.
Since the Psp system has been associated with virulence in other bacteria, we wanted to determine if it played a role in V. cholerae pathogenesis (42–44). We examined this possibility using two different animal models: the infant mouse and zebrafish. With the infant mice, we performed the classical competition between the wild type and a psp null mutant and did not find that the mutant had any competitive defect. In the zebrafish, individual infections with both strains were performed. In this model, we found that the psp mutant had a defect in colonizing the zebrafish intestine in comparison to the wild-type strain. This difference in colonization leads us to hypothesize that the V. cholerae Psp response may play a greater role for survival under environmental stress and may also contribute to transmission of disease in the aquatic environment.
In summary, we have made the first steps in characterizing the Psp response in V. cholerae, including identifying some of the signals that induce its activity. We have identified both specific protein and environmental stressors that initiate the response. Future work will continue to investigate the role of the V. cholerae Psp stress response in survival under environmental stress and in disease transmission.
MATERIALS AND METHODS
Strains, media, and growth conditions.
All V. cholerae strains were derived from classical biotype O395. The Escherichia coli strains JM101 and DH5αλpir were used for generating constructs, and SM10λpir was used for conjugation with V. cholerae. All bacterial strains were grown and maintained at 37°C in Luria-Bertani (LB) medium or on LB agar plates supplemented with appropriate antibiotics. Plasmids used in this study include the suicide vector pKAS32 (52), the arabinose-inducible expression vectors pBAD33, pBAD18-Kan and pBAD30 (53), and the tac-promoter driven, isopropyl-β-d-thiogalactopyranoside (IPTG)-inducible expression vector pTTQ18 (54). E. coli strains were transformed by standard methods (55), plasmid DNA was electroporated into V. cholerae, and pKAS32 was introduced into V. cholerae by conjugation with SM10λpir. Antibiotics were used at the following concentrations unless otherwise indicated: ampicillin, 100 μg/ml; kanamycin, 50 μg/ml; streptomycin, 100 μg/ml; and chloramphenicol, 30 μg/ml (E. coli) or 5 μg/ml (V. cholerae). Expression from pBAD vectors and plasmid pTTQ18 was achieved through the addition of 0.2% l-arabinose and 0.5 mM IPTG, respectively.
Plasmid and strain constructions.
Plasmids and strains used in this study are listed in Table S1 in the supplemental material. Primer sequences are available upon request. All constructs were verified by sequencing.
Chromosomal fusions and deletions were created using splicing by overlap extension PCR (SOE-ing PCR) (56). The lacZΩPpspA chromosomal fusion was generated by amplifying 500 bp upstream and downstream of the start site of lacZ (VC395_2453 [KEGG]) and the promoter region of pspA (VC395_1796) from +210 upstream and −110 downstream of the start site of pspA. The primers encoded 20 bp of homology to the 500-bp segments so that overlap could occur. The SOE-ing construct was isolated through gel excision, ligated into pKAS32, and transformed into SM10λpir. SM10λpir containing the plasmid was mated with O395, and integration and resolution of the cointegrate were selected for as previously described (57). The strain containing a chromosomally 6×His-tagged pspA was created in a similar manner, with the 6×His coding sequence added to the C terminus coding sequence of pspA prior to the stop codon. After the construct was validated by sequencing, it was introduced into O395, and the recombinant was selected for as described above.
β-Galactosidase assays.
Overnight cultures of V. cholerae were subcultured 1:100 in LB broth. The strains were grown for 3 h at 37°C, followed by the addition of either IPTG or arabinose. Duplicate 100-μl aliquots of culture were used to determine β-galactosidase activity using the Miller protocol (58).
Immunoblotting.
Aliquots of cultures were harvested and normalized based on the optical density at 600 nm (OD600). Cell pellets were either stored overnight at −20°C or placed on ice before resuspension in 1:1 water and 2× Laemmli sample buffer (4% sodium dodecyl sulfate [SDS], 20% glycerol, 120 mM Tris-HCl [pH 6.8], 5% 2-mercaptoethanol, 0.02% bromophenol blue). Samples were boiled for 5 min, centrifuged for 1 min at 15,000 rpm, and separated by SDS-PAGE. To detect secretin multimers, 3% to 10% polyacrylamide gradient gels were used, otherwise, all other gels contained 10%, 12.5%, or 15% polyacrylamide. Following electrophoresis, proteins were transferred to nitrocellulose by semidry electroblotting. Membranes were blocked in TBST (20 mM Tris-HCl, 0.5 M HCl, 0.1% Tween) buffer containing 5% milk before incubation with horseradish peroxidase (HRP)-labeled mouse monoclonal IgG1 anti-His (R and D systems), OctA-Probe (H-5) HRP (Santa Cruz Biotechnology), or rabbit ToxR antisera (generously provided by K. Skorupski), followed by incubation with a secondary goat anti-rabbit antibody (Thermo Scientific). The protein fractionation controls used were: Crp-FLAG (cytoplasm) and ToxR (membrane) for soluble and insoluble fractionation and ToxR (inner membrane) and OmpT-FLAG (outer membrane) for membrane subcellular fractionation. ToxR antisera also cross-reacts with another low-molecular-weight protein that can be utilized as a loading control (59). Proteins were visualized by chemiluminescence detection (Clarity Western ECL substrate; Bio-Rad) using film or a Syngene imager.
Subcellular fractionation.
Fractionation into soluble and insoluble fractions was performed as detailed previously with the following modifications (60). O395pspA-6×His containing pBAD30-crp-FLAG and pBAD18-Kan, or pBAD30-crp-FLAG and pBAD18-Kan-gspD-6×His was grown for 4 or 5.5 h. The OD600 was measured for normalization, and a whole-cell lysate fraction was removed and resuspended in 600 μl one-quarter TES (200 mM Tris-HCl [pH 8.0], 0.5 mM EDTA, 0.5 M sucrose). The remaining culture was pelleted via centrifugation, and resuspended in one-quarter TES buffer containing 2 mM phenylmethylsulfonyl fluoride (PMSF). The cells were lysed with 4 freeze-thaw cycles and centrifuged, and 700 μl was removed for centrifugation at 45,000 rpm (125,649 × g) in a TLA-45 Beckman rotor for 45 min. The supernatant was retained as the cytoplasmic fraction. The remaining membrane pellet was washed and resuspended in one-quarter TES. All samples were stored overnight at −20°C before proteins were precipitated by treatment with 23% trichloroacetic acid (TCA) for 30 min on ice. Samples were pelleted and washed twice with cold acetone. Pellets were air dried, resuspended in SDS sample buffer, and boiled for 10 min before immunoblotting.
Subcellular fractionation to isolate whole membrane, inner membrane, and outer membrane compartments was performed as described previously with the following modifications (61). All steps were carried out on ice or at 4°C unless otherwise specified. O395pspA-6×His containing pBAD33-ompT-FLAG and pTTQ18-gspD-6×His was grown for 5.5 h with induction by 0.5 mM IPTG and 0.2% arabinose for the last hour. The OD600 was measured for normalization, and cells were pelleted by centrifugation. The cell pellet was washed with 25 ml Tris-NaCl buffer (10 mM Tris base [pH 7.5], 100 mM NaCl). The pellet was resuspended in 6 ml Tris-NaCl buffer containing 10 μg/ml polymyxin B and incubated for 10 min. The cell suspension was centrifuged, resuspended in 6 ml Tris-NaCl buffer, and stored overnight at −80°C. After thawing on ice, cells were lysed by sonication. The lysed sample was centrifuged for 10 min, and the supernatant was subsequently centrifuged at 42,000 rpm (109,650 × g) in a TLA-45 Beckman rotor for 10 min. Samples were washed with 500 μl Tris buffer (10 mM Tris base [pH 7.5]) two times to remove the cytoplasmic fraction. The samples were rocked for 30 min at room temperature in 360 μl TT buffer (10 mM Tris base [pH 7.5], 2% Triton X-100); 120 μl was removed for the total membrane fraction, and SDS sample buffer was added. The remaining sample was centrifuged at 42,000 rpm for 20 min to pellet the outer membrane fraction; 120 μl of supernatant was removed for the inner membrane fraction, and SDS sample buffer was added. The outer membrane pellet was resuspended in TT and centrifuged for 42,000 rpm for 10 min. The pellet was then washed with Tris buffer and resuspended in 240 μl Tris buffer and SDS sample buffer. Samples were boiled for 5 min before immunoblotting.
RNA isolation and qRT-PCR.
The strains were grown and normalized to an OD600 of 1.75 and pelleted by centrifugation. RNA was extracted using TRIzol (Invitrogen) reagent. Genomic DNA was digested by incubation with DNase at 37°C for 1 h. RNA was purified by ethanol precipitation and resuspended in RNase-free water and Tris-EDTA (TE) buffer. Total RNA was measured using a NanoDrop, and 5 μg was reverse transcribed with Moloney murine leukemia virus reverse transcriptase (Invitrogen). cDNA production (and lack of genomic DNA contamination) was validated using PCR with Taq DNA polymerase (NEB).
qRT-PCR was performed with SYBR green (FastStart Essential DNA Green Master version 04; Roche) using a LightCycler 96 and the following PCR conditions: preincubation for 10 min at 95°C, 3-step amplification with 95°C for 10 s, 51°C for 10 s, and 72°C for 10 s for 45 cycles, and a final melting phase of 95°C for 10 s, 65°C for 60 s, and 97°C for 1 s. Relative quantification was performed using recA as the reference gene, and data were analyzed using the threshold cycle (2−ΔΔCT) method (62).
Zebrafish colonization assay.
The zebrafish colonization assay was performed according to the process described previously (63). Briefly, adult wild-type ZDR zebrafish were housed in an automated recirculating tank system (Aquaneering, CA) using water filtered by reverse osmosis and maintained at pH 7.0 to 7.5. Tank water was conditioned with Instant Ocean salt (Aquarium Systems, OH) to a conductivity of 600 to 700 μS. The fish were fasted for at least 12 h prior to each experiment.
For infection, four to five zebrafish were placed in a 400-ml beaker with a perforated lid containing 200 ml of sterile infection water (autoclaved system water). A V. cholerae culture was grown with aeration in LB broth at 37°C for 16 to 18 h. Then, cells were centrifuged at 10,000 × g for 10 min. The resulting pellet was washed twice with 1× phosphate-buffered saline (PBS; pH ∼7.4) and resuspended in 1× PBS to an estimated concentration of 109 CFU/ml, determined by measuring the optical density at 600 nm. One milliliter of bacterial inoculum was added to the beaker containing fish in 400 ml infection water. The final V. cholerae cell density used was ∼5 × 106 CFU/ml for this study and was verified by plating serial dilutions of the inoculated infection water. The fish were infected for 6 h, washed twice for removal of surface bacteria, and then kept in fresh sterile water for an additional 18 h. The control group included fish that were exposed to 1 ml of 1× PBS only in place of bacterial culture. Each beaker containing fish was placed in a glass-front incubator set at 28°C with a timed light-dark cycle for the duration of the experiment.
Fish were euthanized in 100 ml of 320 μg/ml tricaine methanesulfonate (Tricaine-S, MS-222; Western Chemical, WA) for a minimum of 25 min, and the intestine of each fish was aseptically dissected, placed in homogenization tubes (2.0-ml screw-cap tubes; Sarstedt, Nümbrecht, Germany) with 1.5 g of 1.0-mm glass beads (BioSpec Products, Inc., Bartlesville, OK) and 1 ml of 1× PBS, and held on ice. Homogenization tubes were loaded into a Mini-Beadbeater-24 (BioSpec Products, Inc.) and shaken at maximum speed for two 1-min cycles, with the samples being incubated for 1 min on ice after both cycles. Intestinal homogenates from each fish were diluted and plated for enumeration on LB agar plates with appropriate antibiotics. Plates were incubated overnight at 37°C and CFU were counted. All animal protocols were approved by the Wayne State University IACUC.
Bacterial count and mucin assay from fish excretory water.
Fifty milliliters of fish infection water was removed before the fish colonization assay as a control, in duplicates. For all assays, 50-ml conical tubes were centrifuged at 10,000 rpm for 15 min at 4°C, and the supernatant was decanted, being careful not to disturb the pellet. Each pellet was resuspended in 2 ml of 1× PBS. Unprocessed water samples were stored at 4°C for up to 1 week before analysis.
Excreted water (after infection) was collected as described above, serially diluted, and plated for enumeration on LB agar plates with appropriate antibiotics. Plates were incubated overnight at 37°C and CFU were counted. The mucin content in excreted water was measured as described previously (63, 64). Briefly, prior to the procedure, 1 ml of a 50% (wt/vol) periodic acid (Sigma-Aldrich) stock solution was made. A 96-well plate (Corning Costar, Corning, NY) was loaded with 100 μl/well of the blank (1× PBS), mucin standards, and samples that were loaded in triplicates. A volume of 50 μl/well of fresh 0.1% periodic acid solution (10 μl of the 50% periodic acid stock added to 5 ml of 7% acetic acid, used immediately after making) was added and mixed by pipetting. The plate was covered in plastic wrap and incubated at 37°C for 1 to 1.5 h. After incubation, the plate was cooled to room temperature before adding 100 μl/well Schiff’s reagent (Sigma-Aldrich) and mixing with a pipette. The plate was again covered in plastic wrap and placed on a rocker or shaker for 15 to 40 min or until sufficient color developed. Absorbance was read at 560 nm using a plate reader (Tecan SpectraFluor Plus; Tecan, Männedorf, Switzerland). The effective ODs of test samples were calculated by subtraction of the PBS control (uninfected fish) water OD from the test (infected) fish excreted water OD.
Infant mouse colonization assay.
Four- to five-day-old CD1 mice were inoculated intragastrically with approximately 106 bacteria as previously described (45). Inoculated mice were incubated at 30°C for 16 h, at which time they were sacrificed and their intestines were removed and homogenized. Serial dilutions of the intestinal homogenates were plated for enumeration. The competitive index was calculated as the ratio of the wild type to the mutant in the output divided by the ratio of the wild type to the mutant in the input.
Statistics.
Zebrafish intestinal colonization data were analyzed using randomized block analysis of variance (ANOVA), where the blocks are designated “experiment.” Data analyses were performed in R version 3.5.2 (www.R-project.org/). All other statistical analyses, t tests and two-way ANOVAs, were performed with Prism version 5.03 for Windows (GraphPad Software, La Jolla, CA).
Supplementary Material
ACKNOWLEDGMENTS
We thank Karen Skorupski for generously providing the ToxR antibody. We also thank Sadik Khuder for assistance with statistical analyses.
C.M.D. and J.S.M. were supported by startup funds from the University of Toledo. D.N. and J.H.W. were supported by U.S. Public Health Service grant R01AI27390 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JB.00761-18.
REFERENCES
- 1.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rowley G, Spector M, Kormanec J, Roberts M. 2006. Pushing the envelope: extracytoplasmic stress responses in bacterial pathogens. Nat Rev Microbiol 4:383–394. doi: 10.1038/nrmicro1394. [DOI] [PubMed] [Google Scholar]
- 3.Seo J, Savitzky DC, Ford E, Darwin AJ. 2007. Global analysis of tolerance to secretin-induced stress in Yersinia enterocolitica suggests that the phage-shock-protein system may be a remarkably self-contained stress response. Mol Microbiol 65:714–727. doi: 10.1111/j.1365-2958.2007.05821.x. [DOI] [PubMed] [Google Scholar]
- 4.Lloyd LJ, Jones SE, Jovanovic G, Gyaneshwar P, Rolfe MD, Thompson A, Hinton JC, Buck M. 2004. Identification of a new member of the phage shock protein response in Escherichia coli, the phage shock protein G (PspG). J Biol Chem 279:55707–55714. doi: 10.1074/jbc.M408994200. [DOI] [PubMed] [Google Scholar]
- 5.Brissette JL, Russel M, Weiner L, Model P. 1990. Phage shock protein, a stress protein of Escherichia coli. Proc Natl Acad Sci U S A 87:862–866. doi: 10.1073/pnas.87.3.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weiner L, Model P. 1994. Role of an Escherichia coli stress-response operon in stationary-phase survival. Proc Natl Acad Sci U S A 91:2191–2195. doi: 10.1073/pnas.91.6.2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Darwin AJ. 2005. The phage-shock-protein response. Mol Microbiol 57:621–628. doi: 10.1111/j.1365-2958.2005.04694.x. [DOI] [PubMed] [Google Scholar]
- 8.Weiner L, Brissette JL, Ramani N, Model P. 1995. Analysis of the proteins and cis-acting elements regulating the stress-induced phage shock protein operon. Nucleic Acids Res 23:2030–2036. doi: 10.1093/nar/23.11.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jovanovic G, Lloyd LJ, Stumpf MPH, Mayhew AJ, Buck M. 2006. Induction and function of the phage shock protein extracytoplasmic stress response in Escherichia coli. J Biol Chem 281:21147–21161. doi: 10.1074/jbc.M602323200. [DOI] [PubMed] [Google Scholar]
- 10.Yamaguchi S, Reid DA, Rothenberg E, Darwin AJ. 2013. Changes in Psp protein binding partners, localization and behaviour upon activation of the Yersinia enterocolitica phage shock protein response. Mol Microbiol 87:656–671. doi: 10.1111/mmi.12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flores-Kim J, Darwin AJ. 2016. The phage shock protein response. Annu Rev Microbiol 70:83–101. doi: 10.1146/annurev-micro-102215-095359. [DOI] [PubMed] [Google Scholar]
- 12.Disconzi E, Guilvout I, Chami M, Masi M, Huysmans GHM, Pugsley AP, Bayan N. 2014. Bacterial secretins form constitutively open pores akin to general porins. J Bacteriol 196:121–128. doi: 10.1128/JB.00750-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Korotkov KV, Gonen T, Hol WG. 2011. Secretins: dynamic channels for protein transport across membranes. Trends Biochem Sci 36:433–443. doi: 10.1016/j.tibs.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Russel M, Kaźmierczak B. 1993. Analysis of the structure and subcellular location of filamentous phage pIV. J Bacteriol 175:3998–4007. doi: 10.1128/jb.175.13.3998-4007.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kazmierczak BI, Mielke DL, Russel M, Model P. 1994. pIV, a filamentous phage protein that mediates phage export across the bacterial cell envelope, forms a multimer. J Mol Biol 238:187–198. doi: 10.1006/jmbi.1994.1280. [DOI] [PubMed] [Google Scholar]
- 16.Darwin AJ, Miller VL. 2001. The psp locus of Yersinia enterocolitica is required for virulence and for growth in vitro when the Ysc type III secretion system is produced. Mol Microbiol 39:429–445. doi: 10.1046/j.1365-2958.2001.02235.x. [DOI] [PubMed] [Google Scholar]
- 17.Maxson ME, Darwin AJ. 2004. Identification of inducers of the Yersinia enterocolitica phage shock protein system and comparison to the regulation of the RpoE and Cpx extracytoplasmic stress responses. J Bacteriol 186:4199–4208. doi: 10.1128/JB.186.13.4199-4208.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manganelli R, Gennaro ML. 2017. Protecting from envelope stress: variations on the phage-shock-protein theme. Trends Microbiol 25:205–216. doi: 10.1016/j.tim.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reidl J, Klose KE. 2002. Vibrio cholerae and cholera: out of the water and into the host. FEMS Microbiol Rev 26:125–139. doi: 10.1111/j.1574-6976.2002.tb00605.x. [DOI] [PubMed] [Google Scholar]
- 20.Faruque SM, Albert MJ, Mekalanos JJ. 1998. Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol Mol Biol Rev 62:1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Srinivasan S, Kjelleberg S. 1998. Cycles of famine and feast: the starvation and outgrowth strategies of a marine Vibrio. J Biosci 23:501–511. doi: 10.1007/BF02936144. [DOI] [Google Scholar]
- 22.Ding Y, Davis BM, Waldor MK. 2004. Hfq is essential for Vibrio cholerae virulence and downregulates σE expression. Mol Microbiol 53:345–354. doi: 10.1111/j.1365-2958.2004.04142.x. [DOI] [PubMed] [Google Scholar]
- 23.Becker LA, Bang IS, Crouch ML, Fang FC. 2005. Compensatory role of PspA, a member of the phage shock protein operon, in rpoE mutant Salmonella enterica serovar Typhimurium. Mol Microbiol 56:1004–1016. doi: 10.1111/j.1365-2958.2005.04604.x. [DOI] [PubMed] [Google Scholar]
- 24.Jovanovic G, Mehta P, McDonald C, Davidson AC, Uzdavinys P, Ying L, Buck M. 2014. The N-terminal amphipathic helices determine regulatory and effector functions of phage shock protein A (PspA) in Escherichia coli. J Mol Biol 426:1498–1511. doi: 10.1016/j.jmb.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 25.Adams H, Teertstra W, Koster M, Tommassen J. 2002. PspE (phage-shock protein E) of Escherichia coli is a rhodanese. FEBS Lett 518:173–176. doi: 10.1016/S0014-5793(02)02695-9. [DOI] [PubMed] [Google Scholar]
- 26.Ayers M, Lynne Howell P, Burrows L. 2010. Architecture of the type II secretion and type 4 pilus machineries. Future Microbiol 5:1203–1218. doi: 10.2217/fmb.10.76. [DOI] [PubMed] [Google Scholar]
- 27.Sharma G, Burrows LL, Singer M. 2018. Diversity and evolution of myxobacterial type IV pilus systems. Front Microbiol 9:1630. doi: 10.3389/fmicb.2018.01630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang Y-W, Kjær A, Ortega DR, Kovacikova G, Sutherland JA, Rettberg LA, Taylor RK, Jensen GJ. 2017. Architecture of the Vibrio cholerae toxin-coregulated pilus machine revealed by electron cryotomography. Nat Microbiol 2:16269. doi: 10.1038/nmicrobiol.2016.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meibom KL, Blokesch M, Dolganov NA, Wu CY, Schoolnik GK. 2005. Chitin induces natural competence in Vibrio cholerae. Science 310:1824–1827. doi: 10.1126/science.1120096. [DOI] [PubMed] [Google Scholar]
- 30.Jouravleva EA, McDonald GA, Marsh JW, Taylor RK, Boesman-Finkelstein M, Finkelstein RA. 1998. The Vibrio cholerae mannose-sensitive hemagglutinin is the receptor for a filamentous bacteriophage from V. cholerae O139. Infect Immun 66:2535–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bose N, Taylor RK. 2005. Identification of a TcpC-TcpQ outer membrane complex involved in the biogenesis of the toxin-coregulated pilus of Vibrio cholerae. J Bacteriol 187:2225–2232. doi: 10.1128/JB.187.7.2225-2232.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastava D, Moumene A, Flores-Kim J, Darwin AJ. 2017. Psp stress response proteins form a complex with mislocalized secretins in the Yersinia enterocolitica cytoplasmic membrane. mBio 8:e01088-17. doi: 10.1128/mBio.01088-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Linderoth NA, Model P, Russel M. 1996. Essential role of a sodium dodecyl sulfate-resistant protein IV multimer in assembly-export of filamentous phage. J Bacteriol 178:1962–1970. doi: 10.1128/jb.178.7.1962-1970.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jovanovic G, Dworkin J, Model P. 1997. Autogenous control of PspF, a constitutively active enhancer-binding protein of Escherichia coli. J Bacteriol 179:5232–5237. doi: 10.1128/jb.179.16.5232-5237.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jovanovic G, Model P. 1997. PspF and IHF bind co-operatively in the psp promoter-regulatory region of Escherichia coli. Mol Microbiol 25:473–481. doi: 10.1046/j.1365-2958.1997.4791844.x. [DOI] [PubMed] [Google Scholar]
- 36.Engl C, Jovanovic G, Lloyd LJ, Murray H, Spitaler M, Ying L, Errington J, Buck M. 2009. In vivo localizations of membrane stress controllers PspA and PspG in Escherichia coli. Mol Microbiol 73:382–396. doi: 10.1111/j.1365-2958.2009.06776.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vrancken K, Van Mellaert L, Anné J. 2008. Characterization of the Streptomyces lividans PspA response. J Bacteriol 190:3475–3481. doi: 10.1128/JB.01966-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weiner L, Brissette JL, Model P. 1991. Stress-induced expression of the Escherichia coli phage shock protein operon is dependent on sigma 54 and modulated by positive and negative feedback mechanisms. Genes Dev 5:1912–1923. doi: 10.1101/gad.5.10.1912. [DOI] [PubMed] [Google Scholar]
- 39.Brissette JL, Weiner L, Ripmaster TL, Model P. 1991. Characterization and sequence of the Escherichia coli stress-induced psp operon. J Mol Biol 220:35–48. doi: 10.1016/0022-2836(91)90379-K. [DOI] [PubMed] [Google Scholar]
- 40.Jovanovic G, Weiner L, Model P. 1996. Identification, nucleotide sequence, and characterization of PspF, the transcriptional activator of the Escherichia coli stress-induced psp operon. J Bacteriol 178:1936–1945. doi: 10.1128/jb.178.7.1936-1945.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maxson ME, Darwin AJ. 2006. PspB and PspC of Yersinia enterocolitica are dual function proteins: regulators and effectors of the phage-shock-protein response. Mol Microbiol 59:1610–1623. doi: 10.1111/j.1365-2958.2006.05047.x. [DOI] [PubMed] [Google Scholar]
- 42.Darwin AJ, Miller VL. 1999. Identification of Yersinia enterocolitica genes affecting survival in an animal host using signature-tagged transposon mutagenesis. Mol Microbiol 32:51–62. doi: 10.1046/j.1365-2958.1999.01324.x. [DOI] [PubMed] [Google Scholar]
- 43.Southern SJ, Male A, Milne T, Sarkar-Tyson M, Tavassoli A, Oyston P. 2015. Evaluating the role of phage-shock protein A in Burkholderia pseudomallei. Microbiology 161:2192–2203. doi: 10.1099/mic.0.000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karlinsey JE, Maguire ME, Becker LA, Crouch M-L, Fang FC. 2010. The phage shock protein PspA facilitates divalent metal transport and is required for virulence of Salmonella enterica sv. Typhimurium. Mol Microbiol 78:669–685. doi: 10.1111/j.1365-2958.2010.07357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Matson JS. 2018. Infant mouse model of Vibrio cholerae infection and colonization, p 14–152. In Sikora AE.(ed), Vibrio cholerae: methods in molecular biology, vol 1839 Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- 46.Runft DL, Mitchell KC, Abuaita BH, Allen JP, Bajer S, Ginsburg K, Neely MN, Withey JH. 2014. Zebrafish as a natural host model for Vibrio cholerae colonization and transmission. Appl Environ Microbiol 80:1710–1717. doi: 10.1128/AEM.03580-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Engl C, Beek AT, Bekker M, de Mattos JT, Jovanovic G, Buck M. 2011. Dissipation of proton motive force is not sufficient to induce the phage shock protein response in Escherichia coli. Curr Microbiol 62:1374–1385. doi: 10.1007/s00284-011-9869-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guilvout I, Chami M, Engel A, Pugsley AP, Bayan N. 2006. Bacterial outer membrane secretin PulD assembles and inserts into the inner membrane in the absence of its pilotin. EMBO J 25:5241–5249. doi: 10.1038/sj.emboj.7601402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McDonald C, Jovanovic G, Ces O, Buck M. 2015. Membrane stored curvature elastic stress modulates recruitment of maintenance proteins PspA and Vipp1. mBio 6:e01188-15. doi: 10.1128/mBio.01188-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koo J, Burrows LL, Lynne Howell P. 2012. Decoding the roles of pilotins and accessory proteins in secretin escort services. FEMS Microbiol Lett 328:1–12. doi: 10.1111/j.1574-6968.2011.02464.x. [DOI] [PubMed] [Google Scholar]
- 51.Yamaguchi S, Gueguen E, Horstman NK, Darwin AJ. 2010. Membrane association of PspA depends on activation of the phage-shock-protein response in Yersinia enterocolitica. Mol Microbiol 78:429–443. doi: 10.1111/j.1365-2958.2010.07344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skorupski K, Taylor RK. 1996. Positive selection vectors for allelic exchange. Gene 169:47–52. doi: 10.1016/0378-1119(95)00793-8. [DOI] [PubMed] [Google Scholar]
- 53.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stark M. 1987. Multicopy expression vectors carrying the lac represser gene for regulated high-level expression of genes in Escherichia coli. Gene 51:255–267. doi: 10.1016/0378-1119(87)90314-3. [DOI] [PubMed] [Google Scholar]
- 55.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 56.McPherson MJ, Moller SG. 2006. PCR. THE BASICS (Garland Science), 2nd ed Taylor & Francis, New York, NY. [Google Scholar]
- 57.Teoh WP, Matson JS, DiRita VJ. 2015. Regulated intramembrane proteolysis of the virulence activator TcpP in Vibrio cholerae is initiated by the tail-specific protease (Tsp). Mol Microbiol 97:822–831. doi: 10.1111/mmi.13069. [DOI] [PubMed] [Google Scholar]
- 58.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 59.Mey AR, Craig SA, Payne SM. 2012. Effects of amino acid supplementation on porin expression and ToxR levels in Vibrio cholerae. Infect Immun 80:518–528. doi: 10.1128/IAI.05851-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petiti M, Houot L, Duché D. 2017. Cell fractionation, p 59–64. In Journet L, Cascales E (ed), Bacterial protein secretion systems: methods and protocols. Springer, New York, NY. [Google Scholar]
- 61.Sandrini SM, Haigh R, Freestone PPE. 2014. Fractionation by ultracentrifugation of Gram negative cytoplasmic and membrane proteins. Bio-protocol 4:e1287. [Google Scholar]
- 62.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 63.Nag D, Breen P, Raychaudhuri S, Withey JH. 2018. Glucose metabolism by Escherichia coli inhibits Vibrio cholerae intestinal colonization of zebrafish. Infect Immun 86:e00486-18. doi: 10.1128/IAI.00486-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nag D, Mitchell K, Breen P, Withey JH. 2018. Quantifying Vibrio cholerae colonization and diarrhea in the adult zebrafish model. J Vis Exp 2018:57767. doi: 10.3791/57767. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.