

Abstract
Healthy kidney structure and environment rely on epithelial integrity and interactions between epithelial cells and other kidney cells. The Ser/Thr kinase 90 kDa ribosomal protein S6 kinase 1 (p90RSK) belongs to a protein family that regulates many cellular processes, including cell motility and survival. p90RSK is predominantly expressed in the kidney, but its possible role in chronic kidney disease (CKD) remains largely unknown. Here, we found that p90RSK expression is dramatically activated in a classic mouse obstructive chronic kidney disease model, largely in the interstitial FSP-1–positive fibroblasts. We generated FSP-1–specific p90RSK transgenic mouse (RSK-Tg) and discovered that these mice, after obstructive injury, display significantly increased fibrosis and enhanced tubular epithelial damage compared with their wt littermates (RSK-wt), indicating a role of p90RSK in fibroblast–epithelial communication. We established an in vitro fibroblast–epithelial coculture system with primary kidney fibroblasts from RSK-Tg and RSK-wt mice and found that RSK-Tg fibroblasts consistently produce excessive H2O2 causing epithelial oxidative stress and inducing nuclear translocation of the signaling protein β-catenin. Epithelial accumulation of β-catenin, in turn, promoted epithelial apoptosis by activating the transcription factor forkhead box class O1 (FOXO1). Of note, blockade of reactive oxygen species (ROS) or β-catenin or FOXO1 activity abolished fibroblast p90RSK-mediated epithelial apoptosis. These results make it clear that p90RSK promotes kidney fibrosis by inducing fibroblast-mediated epithelial apoptosis through ROS-mediated activation of β-catenin/FOXO1 signaling pathway.
Keywords: RSK, kidney, fibrosis, FOXO, beta-catenin (β-catenin), reactive oxygen species (ROS), 90 kDa ribosomal protein S6 kinase 1 (p90RSK), cell-cell crosstalk, fibroblast-epithelial communication, forkhead box class O1 (FOXO1), RPS6KA1, chronic kidney disease (CKD)
Introduction
Regardless of the etiology, interstitial fibrosis, characterized by extensive interstitial fibroblast activation and excessive extracellular matrix deposition, is a hallmark of chronic kidney disease (CKD)4 (1–3). The extent of interstitial fibrosis generally predicts the prognosis of patients with CKD (1, 2, 4). Both interstitial fibroblasts and tubular epithelial cells play essential roles in fibrogenesis and CKD progression. Interstitial fibroblasts are considered to be the primary matrix-producing cells and principal mediators of renal fibrosis associated with progressive renal failure (2, 5, 6). In response to injury, epithelial cells, especially proximal tubular epithelial cells, not only initiate inflammatory response by producing proinflammatory chemokines, but also undergo apoptotic death, leading to kidney parenchymal destruction. Structurally, fibroblasts reside in the renal interstitium surrounding the tubules formed by epithelial cells. This proximity facilitates interstitial–epithelial communication and interactions that are fundamental in maintaining the integrity of the kidney structure and environment, as well as fine-regulated process of adaption to pathogenic cues (7, 8). However, the exact mechanisms of these interactions and their roles in CKD are largely unknown.
The 90 kDa ribosomal S6 kinases (RSKs) are a group of serine/threonine kinases that regulate diverse cellular process, such as cell growth, cell motility, and cell survival (9, 10). There are four RSK isoforms (RSK1–4), of which RSK1 is also designated as p90RSK and is predominantly expressed in the kidney (9, 10), suggesting that p90RSK may play a unique role in the pathogenesis of CKD. Recently, p90RSK has been shown to promote endothelial cell dysfunction and atherosclerosis in a diabetes model (11). However, the role of p90RSK in CKD development and progression remains unknown.
In the present study, we investigated the role of p90RSK in CKD pathogenesis in a novel FSP-1–specific p90RSK transgenic mouse. Our data demonstrated that p90RSK promotes kidney fibrosis by inducing fibroblast-mediated epithelial apoptosis through a novel signaling mechanism involving reactive oxygen species (ROS), β-catenin, and forkhead box class O1 (FOXO1).
Results
p90RSK is activated during chronic kidney disease
To evaluate the activation of p90RSK during CKD, we examined the phosphorylation of p90RSK in the fibrotic kidneys from C57BL/6J mice with unilateral ureter obstruction (UUO), a classic CKD model, for 3, 7, and 14 days. We found that with the progression of kidney fibrosis, as indicated by increased fibronectin deposition (Fig. 1, A and C), p90RSK phosphorylation was dramatically induced (Fig. 1, A and B). However, there was little change of total-p90RSK abundance during kidney fibrosis (Fig. 1, A and B). Of note, phospho-p90RSK was largely induced in the interstitial FSP-1–positive cells (Fig. 1D).
Figure 1.

Induction of p90RSK phosphorylation correlates with degree of kidney fibrosis. A, C57BL/6 mice were subjected to UUO for 3, 7, and 14 days, followed by Western blotting for phospho- and total p90RSK, fibronectin (Fn), and GAPDH. Numbers indicate individual mouse. B, relative abundance (-fold increase) of phospho- and total p90RSK. *, p < 0.05; ***, p < 0.001 versus control; n = 5 mice/group. C, fibronectin abundance. **, p < 0.01; ***, p < 0.001 versus control; n = 5 mice/group. D, immunofluorescence staining of phospho-p90RSK (green) and FSP-1 (red) in mouse kidneys. Scale bar, 20 μm. CLK, control kidney; T, tubular epithelial cells. Error bars, S.E.
FSP-1–specific p90RSK accelerates kidney fibrosis
Based on the above finding that p90RSK is activated, primarily in FSP-1–positive interstitial cells, during CKD (Fig. 1), we generated FSP-1–specific p90RSK transgenic mice (RSK-Tg) by cross-breeding p90RSK-Tgflox mice (11) and S100A4 (FSP-1)-Cre mice (The Jackson Laboratory). Although other types of cells, such as macrophages (12) and podocytes (13), also express FSP-1, it has been verified in various organ systems, including kidneys, that S100A4 (FSP-1)-Cre specifically targets fibroblasts (14–17). In particular, Inoue et al (14), using enhanced GFP reporter mice, have further confirmed that the FSP-1 promoter only drives Cre recombinase expression in interstitial fibroblasts in kidneys. Consistently, we also confirmed the overexpression of p90RSK in primary kidney fibroblasts from these transgenic mice (Fig. 2A) compared with their littermates (RSK-wt), and RSK-Tg mice displayed dramatically increased phosphorylation of p90RSK in FSP-1–positive fibroblasts after fibrotic kidney injury (Fig. 2B). Under physiological condition, RSK-Tg mice had a phenotype similar to RSK-wt mice (data not shown). However, after receiving UUO for 7 days, RSK-Tg mice displayed significantly more severe tubular epithelial damage (Fig. 2, C and D) and increased interstitial fibronectin and collagen deposition (Fig. 2, E–G).
Figure 2.
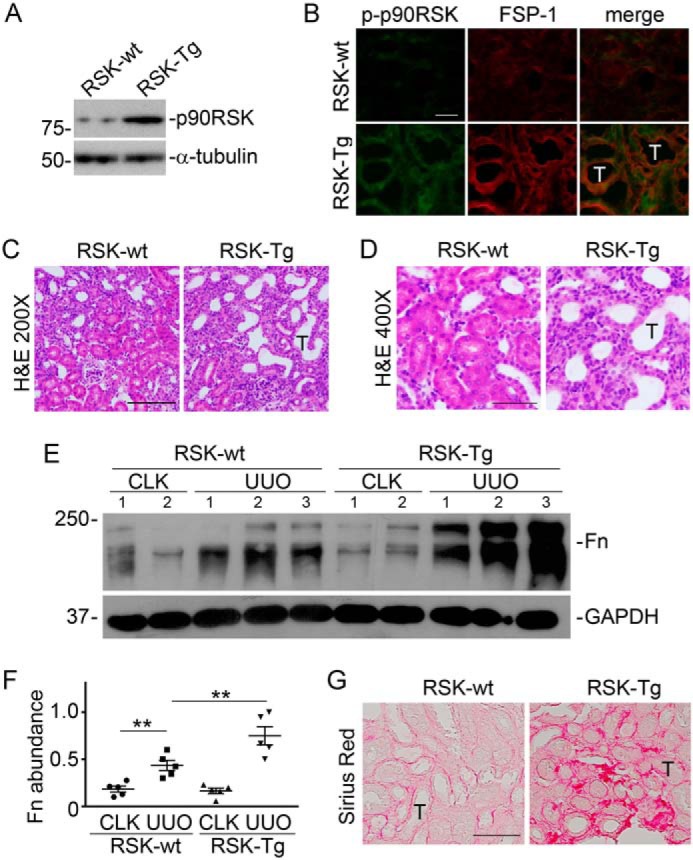
p90RSK promotes kidney fibrosis. FSP-1–specific p90RSK transgenic mice (RSK-Tg) and their littermate controls (RSK-wt) were subjected to UUO for 7 days. A, Western blotting for p90RSK and tubulin in mouse primary kidney fibroblasts. B, immunofluorescence staining of phospho-p90RSK (green) and FSP-1 (red) in mouse kidneys with UUO, 400×. Scale bar, 50 μm. C, H&E staining of UUO kidneys, 200×. Scale bar, 100 μm. D, H&E staining of UUO kidneys, 400×. Scale bar, 50 μm. E, Western blotting for fibronectin (Fn) and GAPDH. Numbers indicate individual mouse. F, quantitation of fibronectin abundance. **, p < 0.01; n = 5 mice/group. G, Sirius Red staining of UUO kidneys. Scale bar, 50 μm. CLK, control kidney; T, tubular epithelial cells. Error bars, S.E.
p90RSK induces fibroblast-mediated tubular epithelial apoptosis
We further evaluated apoptosis in the fibrotic kidneys from RSK-Tg and RSK-wt mice using TUNEL and cleaved caspase-3 immune staining. It was shown that there were more apoptotic cells in the obstructed kidneys from RSK-Tg mice, and these apoptotic cells were almost exclusively lectin-positive proximal tubular epithelial cells (Fig. 3, A–C), consistent with the histological assessment of epithelial damage (Fig. 2, C and D). Because FSP-1–positive cells are largely fibroblasts in the kidneys, it is presumable that fibroblast-specific p90RSK mediates epithelial damage. We established an in vitro fibroblast–epithelial coculture system to test our hypothesis (Fig. 3D). Briefly, primary kidney fibroblasts extracted from RSK-Tg and RSK-wt mice were seeded onto Transwell inserts in a 6-well plate with HKC-8 cells on the bottom. After serum-free overnight, low concentration of two classical apoptosis inducers, staurosporine and H2O2, were added to the lower chamber for additional 4 or 16 h, respectively, and followed by apoptosis assay. Consistent with our in vivo studies (Fig. 3, A–C), RSK-Tg fibroblasts dramatically enhanced epithelial apoptosis compared with RSK-wt fibroblasts (Fig. 3, E–H), suggesting a pathogenic role of p90RSK in mediating fibroblast-induced epithelial damage.
Figure 3.

p90RSK induces fibroblast-mediated tubular epithelial apoptosis in vivo and in vitro. A, obstructed kidney sections from FSP-1–specific p90RSK transgenic mice (RSK-Tg) and their littermates (RSK-wt) were subjected to immunofluorescence staining of cleaved caspase-3 (red) and lectin (green, proximal tubular epithelial marker), 200×. Scale bar, 50 μm. B, immune staining of TUNEL (green) and lectin (red), 400×. Scale bar, 20 μm. C, quantitation of apoptotic epithelial cells. ***, p < 0.001; n = 5 microscopic fields per mouse × 5 mice/group. D, illustration of primary fibroblast (RSK-Tg and RSK-wt) and epithelial cell (HKC-8) coculture. After serum starvation overnight in the coculture, 50 nm staurosporine or 150 μm H2O2 was added to the lower chamber for additional 4 or 16 h, respectively. Then, HKC-8 cells were harvested and subjected to Western blotting for cleaved caspase-3 and GAPDH. E, Western blotting of cleaved caspase-3 after staurosporine treatment. F, quantitation of cleaved caspase-3 abundance. *, p < 0.05; **, p < 0.01; n = 3 experiments. G, Western blotting of cleaved caspase-3 after H2O2 treatment. H, quantitation of cleaved caspase-3 abundance. **, p < 0.01; n = 3 experiments. CLK, control kidney; staur, staurosporine. Error bars, S.E.
p90RSK induces fibroblast-mediated oxidative stress and triggers tubular epithelial apoptosis
To clarify the underlying mechanism, we examined the level of 4-hydroxynonenal (4-HNE), a specific marker for oxidative stress, in the fibrotic kidneys from RSK-Tg mice and their littermates. We found that there was remarkably increased 4-HNE expression largely in tubular epithelial cells of the fibrotic kidneys of RSK-Tg mice compared with control littermates (Fig. 4A). Notably, positive 4-HNE staining concentrated at the basal aspect of tubular epithelial cells, as well as in the interstitium (Fig. 4A), strongly implicating an interstitial origin of oxidative stress. We also measured the ROS level in mouse kidney homogenates and found that obstruction-induced renal ROS production was dramatically increased in RSK-Tg mice (Fig. 4B). To confirm the role of oxidative stress in fibroblast-mediated epithelial damage, we further measured the H2O2 content in the medium of our coculture system. We found that RSK-Tg fibroblasts consistently released remarkably higher levels of H2O2 into the coculture medium (Fig. 4C). To clarify whether ROS is necessary to fibroblast p90RSK-induced epithelial cell death, ROS-specific inhibitor YCG063 was added into the coculture. We found that YCG063 remarkably decreased the effects of fibroblast-specific p90RSK in both staurosporine- and H2O2-induced epithelial apoptosis (Fig. 4, D–G). Thus, it is clear that ROS mediates fibroblast-specific p90RSK-induced epithelial cell death.
Figure 4.

ROS mediates fibroblast p90RSK-induced tubular epithelial apoptosis. A, obstructed kidneys were subjected to immune staining for 4-HNE. Scale bar, 50 μm. B, quantitation of ROS level in the obstructed kidneys. **, p < 0.01; ***, p < 0.001; n = 5 mice/group. Primary fibroblasts from FSP-1–specific p90RSK transgenic mice (RSK-Tg) or littermates (RSK-wt) were in coculture with HKC-8 cells for 4 h. C, then, H2O2 level in the coculture medium were measured. **, p < 0.01; ***, p < 0.001; RSK-Tg versus RSK-wt, n = 3 experiments. D, 50 nm staurosporine and 50 nm ROS-specific inhibitor YCG063 were added into the coculture for 4 h, followed by Western blotting for cleaved caspase-3 and GAPDH in HKC-8 lysates. E, quantitation of epithelial cleaved caspase-3 abundance. *, p < 0.05; **, p < 0.01; n = 3 experiments. F, 150 μm H2O2 and 50 nm YCG063 were added into the coculture for 16 h, followed by Western blotting for cleaved caspase-3 and GAPDH. G, quantitation of cleaved caspase-3 abundance in epithelial cells. **, p < 0.01; n = 3 experiments. Staur, staurosporine; T, tubular epithelial cells. Error bars, S.E.
β-Catenin acts as a downstream effector of ROS and mediates fibroblast p90RSK-induced tubular epithelial apoptosis
We further found that H2O2, at a concentration comparable with that in the medium of fibroblast–epithelial coculture system, was able to induce epithelial β-catenin nuclear translocation (Fig. 5A). We also uncovered that ROS mediated epithelial induction of β-catenin, because YCG063, a specific ROS inhibitor, largely abolished fibroblast-specific p90RSK-induced epithelial β-catenin accumulation (Fig. 5, B and C), as well as active β-catenin (Fig. 5D), in our fibroblast–epithelial coculture system. Moreover, knockdown of epithelial β-catenin by siRNA, as indicated by Western blotting in HKC-8 nuclear extracts (Fig. 5E), almost completely abolished p90RSK-enhanced epithelial apoptosis (Fig. 5, E and F). Thus, we conclude that ROS-activated epithelial β-catenin mediates fibroblast-specific p90RSK-induced tubular epithelial apoptosis.
Figure 5.

ROS-activated β-catenin mediates fibroblast p90RSK-induced tubular epithelial apoptosis. A, HKC-8 cells were incubated with 1.5 μm H2O2 for 1 h, followed by immunofluorescence staining of β-catenin (green) and phalloidin (red). Scale bar, 25 μm. B, ROS-specific inhibitor YCG063 (50 nm) was added into the fibroblast–epithelial coculture, followed by Western blotting for β-catenin and GAPDH in epithelial lysates. C, quantitation of β-catenin abundance. *, p < 0.05; **, p < 0.01; n = 3 experiments. D, Western blotting for active β-catenin and GAPDH in epithelial lysates. E, HKC-8 cells were transfected with control or β-catenin siRNAs, followed by coculture with RSK-Tg or RSK-wt fibroblasts with or without 50 nm staurosporine for 4 h. HKC-8 lysates were subjected to Western blotting for cleaved caspase-3 and GAPDH. Nuclear extracts of HKC-8 were probed with β-catenin and PCNA. F, quantitation of cleaved caspase-3 abundance in epithelial lysates. ***, p < 0.001; n = 3 experiments. staur, staurosporine; consiRNA, control siRNA; ctnsiRNA, β-catenin siRNA. Error bars, S.E.
FOXO1 mediates fibroblast p90RSK-induced tubular epithelial apoptosis
FOXO1, a downstream transcription factor of oxidative stress pathway (18, 19), has been shown to bind to β-catenin (20) and mediate TGF-β1–induced myofibroblast activation (21, 22). Given the prominent role of TGF-β1 and β-catenin in kidney fibrosis, we further examined the role of FOXO1 in fibroblast-specific p90RSK-induced epithelial apoptosis. First, we checked the effect of H2O2, at a concentration comparable to that in the medium of fibroblast–epithelial coculture system, on epithelial FOXO1 activity. We found that H2O2 dramatically induced FOXO1 nuclear translocation in epithelial cells (Fig. 6A). We further examined the epithelial FOXO1 activation level in our fibroblast–epithelial coculture system using two complementary approaches such as measuring FOXO1 abundance in epithelial nuclear extracts (Fig. 6, B and C) and evaluating FOXO1 activity by a TransAM FKHR Transcription Factor ELISA Kit (Fig. 6D). We found that RSK-Tg fibroblasts triggered a dramatically higher level of epithelial FOXO1 activation than RSK-wt fibroblasts (Fig. 6, B–D). Intriguingly, we also discovered that FOXO1 was dramatically up-regulated, predominantly in tubular epithelial nuclei, in the fibrotic kidneys from RSK-Tg mice (Fig. 6E), indicating an in vivo significance of FOXO1 in fibroblast–epithelial communication and kidney fibrosis. We further found that ROS-specific inhibitor YCG063 eliminated RSK-Tg fibroblast-induced FOXO1 activation in epithelial cells (Fig. 6F), suggesting that FOXO1 acted in the downstream of ROS pathway. Additionally, FOXO1-specific inhibitor AS1842856 (Fig. 6, G and H) or siRNA (Fig. 6, I–K) abolished fibroblast-specific p90RSK-induced epithelial apoptosis in fibroblast–epithelial coculture system. Thus, FOXO1 clearly mediates fibroblast-specific p90RSK-induced tubular epithelial death.
Figure 6.

ROS-activated FOXO1 mediates fibroblast p90RSK-induced tubular epithelial apoptosis. A, HKC-8 cells were incubated with 1.5 μm H2O2 for 4 h, followed by immunofluorescence staining of FOXO1 (green) and phalloidin (red). Scale bar, 25 μm. B, HKC-8 cells were in coculture with primary RSK-Tg or RSK-wt fibroblasts for the period as indicated, then the nuclear extracts from HKC-8 cells were subjected to Western blotting for FOXO1 and PCNA. C, quantitation of nuclear FOXO1 abundance. ***, p < 0.001; RSK-Tg versus RSK-wt; n = 3 experiments. D, FOXO1 activity was also evaluated by FKHR ELISA assay. *, p < 0.05; **, p < 0.01; n = 3 experiments. E, immune staining of FOXO1 in the obstructed kidneys from FSP-1–specific p90RSK transgenic mice (RSK-Tg) and their littermates (RSK-wt). Scale bar, 50 μm. F, 50 nm YCG063 (ROS-specific inhibitor) was added into the coculture for 24 h, followed by FKHR ELISA assay in HKC-8 nuclear extracts. *, p < 0.05; **, p < 0.01; n = 3 experiments. G, 1 μm FOXO1-specific inhibitor AS1842856 and staurosporine (50 nm) were added into the coculture for 4 h, followed by Western blotting for cleaved caspase-3 and GAPDH in HKC-8 lysates. H, quantitation of cleaved caspase-3 abundance. ***, p < 0.001; n = 3 experiments. I, HKC-8 cells were transfected with FOXO1 siRNA, followed by Western blotting for FOXO1 and GAPDH. J, HKC-8 cells were transfected with 80 nm control or FOXO1 siRNAs, followed by coculture with RSK-Tg or RSK-wt fibroblasts with or without 50 nm staurosporine for 4 h, and Western blotting for cleaved caspase-3 and GAPDH. K, quantitation of cleaved caspase-3 abundance. ***, p < 0.001; n = 3 experiments. Staur, staurosporine; consiRNA, control siRNA; FOXsiRNA, FOXO1 siRNA; T, tubular epithelial cells. Error bars, S.E.
Discussion
Although p90RSK has been shown to be predominantly expressed in the kidney (9, 10), its in vivo role in CKD pathogenesis remains unknown. Our previous in vitro work has demonstrated that p90RSK is a key control point that regulates the size of the interstitial fibroblast population (23–25), which largely determines the outcome in patients with chronic kidney injury (1, 2, 4, 6, 26, 27). p90RSK plays an important role in the Ras mitogen-activated protein kinase (MAPK) signaling cascade and is the direct downstream effector of Ras-Erk1/2 signaling. Erk1/2 activation directly phosphorylates and activates p90RSK (9, 10, 28), which in turn activates various signaling events through selection of different phosphorylation substrates including GSK3β and BAD (9, 10), suggesting a pivotal role of p90RSK in tissue fibrosis. This notion is supported by recent findings that p90RSK is involved in the pathogenesis of atherosclerosis of various causes (11, 29, 30). In the present study, we found that the abundance of total p90RSK was little changed during progressive CKD. However, phosphorylation or activation of p90RSK was dramatically induced and correlated with the extent of fibrotic injury (Fig. 1, A and B), suggesting that activation status of p90RSK, rather than its abundance, determines the severity of kidney fibrosis. Consistently, RSK-Tg mice, under physiological condition (lower p90RSK activation level), had a similar phenotype as their littermates, whereas, after obstructive injury (higher p90RSK activation level), these mice displayed a significantly enhanced fibrosis (Fig. 2). It is known that other types of cells, such as macrophages and podocytes, also express FSP-1. Previous work strongly indicates that FSP-1–Cre–driven cells are primarily fibroblasts in various organs, including kidneys (14–17). Particularly, recent work from Inoue et al. (14) specifically confirms that the promoter for FSP-1 driving Cre recombinase only expresses in the interstitial fibroblasts in the same obstructive CKD model using enhanced GFP reporter mice. However, we cannot exclude the possible contribution from macrophages to p90RSK-induced epithelial apoptosis because some kidney macrophages also express FSP-1. Future investigations in this aspect are warranted. In summary, we have found that fibroblast-specific p90RSK induces tubular epithelial apoptosis and promotes kidney fibrosis. As summarized in Fig. 7, fibroblasts with p90RSK overexpression produce and release excessive H2O2, causing ROS accumulation and β-catenin nuclear translocation in the surrounding epithelial cells. Excessive β-catenin interacts with transcription factor FOXO1 to promote tubular epithelial apoptosis, leading to kidney structural destruction and eventually fibrosis.
Figure 7.

Schematic illustration of fibroblast p90RSK-mediated epithelial injury. p90RSK-transgenic fibroblasts surrounding the tubular epithelial cells generate excessive H2O2 and trigger epithelial ROS induction, which causes cytosol accumulation and increased nuclear translocation of β-catenin. Then, β-catenin directly binds to and activates FOXO1 to induce epithelial apoptosis by activating pro-apoptotic proteins including BIM and BAD, resulting in accelerated kidney fibrosis.
The current results have illustrated a novel mechanism and the pivotal role of p90RSK-mediated fibroblast–epithelial communications in CKD development and progression. The juxtaposition of fibroblasts and epithelium facilitates their interactions. Under physiological condition, interstitial–epithelial communication plays a fundamental role in maintaining the integrity of the kidney structure and environment (7, 8). As an essential part of wound healing process, renal interstitial cells, including fibroblasts, also provide a supportive environment to promote tubular epithelial regeneration in response to transient injury (31). However, persistent pathogenic stimuli will cause extensive activation of signaling mediators, such as p90RSK (Fig. 1), and disrupt the delicate healing process, resulting in progressive tissue destruction and loss of function. The present results have shown that RSK-Tg fibroblasts, after chronic obstructive injury, acquire substantially enhanced ability to generate sustained H2O2 that not only induces epithelial injury (Fig. 7), but also further triggers the activation of p90RSK (32, 33) and forms a vicious loop of amplification, eventually leading to irreversible kidney fibrosis. Several factors, including mitochondria and NADPH oxidases, may contribute to the generation of ROS (34). It is possible that p90RSK induces oxidative stress through modulating the functions of these factors. However, future investigations are required to elucidate the exact mechanisms.
The FOXO family of transcription factors controls multiple cellular processes, including cell cycle, survival, and metabolism (18, 35). There are four FOXOs, namely FOXO1, FOXO3, FOXO4, and FOXO6, of which FOXO1 and FOXO3 are the most studied members (21). Generally, FOXO activity is regulated by two evolutionarily conserved signaling pathways: canonical insulin signaling and oxidative stress pathway (18, 19). FOXO1 has been shown to mediate TGF-β1–induced myofibroblast activation (21, 22) and plays an important role in diabetes and its complications (36–38). In the present study, we have found that FOXO1, a transcription factor dramatically induced in RSK-Tg mice with obstructive injury (Fig. 6E), mediates ROS/β-catenin–induced epithelial apoptosis (Figs. 4–6). β-catenin directly binds to the C-terminal of FOXO1 through its armadillo repeats 1 to 8 and activates FOXO1 transcriptional activity (39), which subsequently promotes apoptosis by activating pro-apoptotic proteins including BIM and BAD (40). Our result is consistent with a recent report that FOXO1 is induced after acute kidney injury (41), a disease condition characterized by extensive tubular epithelial apoptosis. Generally, epithelial regeneration follows after apoptosis. However, sustaining injury cues trigger persistent activation of fibroblast p90RSK, which forms a vicious environment for tubular regeneration and differentiation, resulting in structural destruction and fibrotic scar formation. Thus, it is likely that p90RSK-mediated fibroblast–epithelial communication plays a decisive role in driving kidney fibrosis.
In summary, our results demonstrate that p90RSK plays an essential role in promoting kidney fibrosis through a novel mechanism of fibroblast–epithelial communication involving ROS, β-catenin, and FOXO1. These finding indicate that p90RSK may be a promising therapeutic target for CKD treatment.
Experimental procedures
Antibodies and reagents
The antibodies against phospho-specific (9344) and total p90RSK (8408), FOXO1 (2880), GAPDH (2118), and cleaved caspase-3 (9661) were purchased from Cell Signaling Technology (Danvers, MA). Rabbit anti-fibronectin antibody (F3648) was obtained from Sigma. Rabbit anti-PCNA antibody (sc-7907) was purchased from Santa Cruz Biotechnology (Dallas, TX). Polyclonal rabbit anti-human S100A4 antibody (A5114) was purchased from Dako Denmark A/S (Glostrup, Denmark). Rabbit anti-4 HNE (ab46545) and anti-β-catenin (ab2365) antibodies were bought from Abcam (Cambridge, MA). Anti-active β-catenin was purchased from EMD Millipore (Billerica, MA). Picrosirius Red Stain Kit was from Polysciences Inc. (Warrington, PA). The biotinylated anti-rabbit antibody (BA1000) was obtained from Vector Laboratories (Burlingame, CA). The secondary HRP-conjugated antibodies, Alexa Fluor 488– and Alexa Fluor 594–coupled goat anti-rabbit or anti-mouse IgG, FBS, and supplements were obtained from Fisher Scientific. Dulbecco's modified Eagle's medium and minimum Eagle's medium were obtained from American Type Culture Collection (ATCC, Manassas, VA). The Lipofectamine 2000 Reagent was purchased from Invitrogen. All other chemicals of analytic grade were obtained from Fisher Scientific unless otherwise indicated.
Animal model
Animal studies were performed using a protocol (45872) approved by the Institutional Animal Care and Use Committee at the Pennsylvania State University College of Medicine. FSP-1–specific p90RSK transgenic mice (RSK-Tg) and their littermates (RSK-wt) were generated by cross-breeding p90RSK-Tgflox mice (11) and S100A4 (FSP-1)-Cre mice (The Jackson Laboratory). The original p90RSK-Tgflox and FSP-1–Cre mice had been backcrossed for at least 10 generations to the C57BL/6 background. UUO was performed in 20–22 g sex-matched mice (five animals per group) using established procedures described previously (42–44). The right unobstructed kidneys served as controls. At day 7 after UUO, mice were sacrificed and kidneys were harvested for analyses.
TUNEL and immunofluorescence staining
Snap-frozen kidney was cryosectioned at 5-μm thickness and subjected to TUNEL or immunofluorescence staining as described previously (25, 45). TUNEL was performed using Roche Cell Death Detection kit. Fluorescence-conjugated lectins from Tetragonolobus purpureas (BioWorld and Vector Laboratories) were used to localize the proximal tubules. Images were acquired by an Olympus IX81 fluorescence microscope (Olympus America Inc.). Sections stained without primary antibodies served as negative controls. TUNEL-positive epithelial cells were counted from five non-overlapping 400× microscopic fields per slide, and the percentage of TUNEL-positive epithelial cells in total epithelial cell population was calculated, five mice per group.
Immunohistochemistry and Sirius Red staining
Paraffin-embedded kidney tissue was sectioned at 4 μm and then subjected to Sirius Red or immune staining (23). Briefly, tissue sections were deparaffinized, hydrated, and antigen-retrieved, followed by Sirius Red staining using Picrosirius Red Stain kit or by incubation with primary and secondary antibodies for immune staining. Sections stained without primary antibodies served as negative controls. Sections were then incubated with ABC reagents, followed by NovaRED or DAB staining, and counterstained with methyl green (Vector Laboratories).
Fibroblast–epithelial coculture
Human kidney proximal tubular epithelial cells (HKC-8) were maintained as described previously (48). Primary kidney fibroblasts were isolated from fibroblast-specific p90RSK transgenic mice (RSK-Tg) and WT (RSK-wt) mice and maintained as reported (49). Primary fibroblasts were validated by morphology and positive staining of FSP-1 and vimentin. An in vitro fibroblast–epithelial coculture system was established using above primary kidney fibroblasts and HKC-8 cells. Briefly, primary fibroblasts (3 × 105 cells) were seeded onto Transwell inserts (0.4 mm PET, 4.5 cm2) in a 6-well plate with HKC-8 cells (3 × 105 cells) on the bottom. After serum-free overnight, 50 nm staurosporine or 150 μm H2O2 was added into the lower chamber for an additional 4 or 16 h, respectively, followed by apoptosis assay. In some experiments, epithelial cells were treated with various chemical inhibitors, such as FOXO1 inhibitor (AS1842856, 1 μm, EMD Millipore, Billerica, MA) (46) or ROS inhibitor (YCG063, 50 nm, Calbiochem).
RNAi
HKC-8 cells were transfected with control siRNA, β-catenin siRNA, or FOXO1 siRNA (Santa Cruz Biotechnology, Dallas, TX) at a final concentration of 40 nm or 80 nm using Lipofectamine 2000 reagent (Invitrogen) as described previous (23, 47). Three days later, cells were co-cultured with primary fibroblasts.
Western blot analysis
Tissue or cell lysates were prepared and separated on SDS-polyacrylamide gels as described previously (23, 25, 45). The PVDF membrane with transferred proteins was probed with various antibodies, and the signals on the membrane were visualized by the SuperSignal West Pico Chemiluminescent Substrate kit (Fisher Scientific).
FOXO1 activity
Primary kidney fibroblasts were cocultured with HKC-8 cells in serum-free medium for 24 h, then HKC-8 cells were harvested at 1, 2, 4, 12, and 24 h. In some experiments, 50 nm ROS-specific inhibitor YCG063 was added into the coculture medium for 24 h. Then, nuclear fractions were isolated from cells using a TransAM Nuclear Extract Kit (Active Motif, Carlsbad, CA), and FOXO1 activities were determined using a TransAM FKHR Transcription Factor ELISA Kit (Active Motif, Carlsbad, CA).
Oxidative stress
Mouse kidney ROS level was assessed by a Mouse Reactive Oxygen Species ELISA kit (Neo Scientific). Hydrogen peroxide level in coculture medium was evaluated by a hydrogen peroxide assay kit from Bioassay Systems.
Hydrogen peroxide production assay
HKC-8 and fibroblasts were cocultured in serum-free medium for 4 h, followed by measurement of hydrogen peroxide content in the coculture medium by a hydrogen peroxide assay kit (Bioassay Systems, Hayward, CA).
Statistical analysis
All the experimental data were presented as mean ± S.E. Statistical analysis of data were performed using SigmaStat 3.5 software (Jandel Scientific Software). Comparison between multiple groups was performed by using one-way analysis of variance (ANOVA) followed by the Student–Newman–Keuls test or Student's t test between two groups. A p value of less than 0.05 was considered statistically significant.
Author contributions
L. L., N.-T. L., J.-I. A., and K. H. resources; L. L., C. S., Z. S., and K. H. data curation; L. L., C. S., and K. H. formal analysis; L. L. and K. H. supervision; L. L. validation; L. L., C. S., and K. H. investigation; L. L., C. S., and K. H. methodology; L. L. writing-original draft; K. H. conceptualization; K. H. funding acquisition; K. H. project administration; K. H. writing-review and editing.
Acknowledgment
We thank Samantha White for technical assistance.
This work was supported by National Institutes of Health Grant DK102624 (to K. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- CKD
- chronic kidney disease
- 4-HNE
- 4-hydroxynonenal
- ROS
- reactive oxygen species
- RSK
- ribosomal S6 kinase
- Tg
- transgenic
- UUO
- unilateral ureter obstruction.
References
- 1. Grande M. T., and López-Novoa J. M. (2009) Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat. Rev. Nephrol. 5, 319–328 10.1038/nrneph.2009.74 [DOI] [PubMed] [Google Scholar]
- 2. Strutz F., and Zeisberg M. (2006) Renal fibroblasts and myofibroblasts in chronic kidney disease. J. Am. Soc. Nephrol. 17, 2992–2998 10.1681/ASN.2006050420 [DOI] [PubMed] [Google Scholar]
- 3. Kaissling B., and Le Hir M. (2008) The renal cortical interstitium: Morphological and functional aspects. Histochem. Cell Biol. 130, 247–262 10.1007/s00418-008-0452-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bohle A., Strutz F., and Müller G. A. (1994) On the pathogenesis of chronic renal failure in primary glomerulopathies: A view from the interstitium. Exp. Nephrol. 2, 205–210 [PubMed] [Google Scholar]
- 5. Neilson E. G. (2006) Mechanisms of disease: Fibroblasts–a new look at an old problem. Nat. Clin. Pract. Nephrol. 2, 101–108 10.1038/ncpneph0093 [DOI] [PubMed] [Google Scholar]
- 6. Qi W., Chen X., Poronnik P., and Pollock C. A. (2006) The renal cortical fibroblast in renal tubulointerstitial fibrosis. Int. J. Biochem. Cell Biol. 38, 1–5 10.1016/j.biocel.2005.09.005 [DOI] [PubMed] [Google Scholar]
- 7. El-Achkar T. M., and Dagher P. C. (2015) Tubular cross talk in acute kidney injury: A story of sense and sensibility. Am. J. Physiol. Renal. Physiol. 308, F1317–F1323 10.1152/ajprenal.00030.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Borges F. T., Melo S. A., Özdemir B. C., Kato N., Revuelta I., Miller C. A., Gattone V. H. 2nd, LeBleu V. S., and Kalluri R. (2013) TGF-β1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J. Am. Soc. Nephrol. 24, 385–392 10.1681/ASN.2012101031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Anjum R., and Blenis J. (2008) The RSK family of kinases: Emerging roles in cellular signalling. Nat. Rev. Mol. Cell Biol. 9, 747–758 10.1038/nrm2509 [DOI] [PubMed] [Google Scholar]
- 10. Romeo Y., Zhang X., and Roux P. P. (2012) Regulation and function of the RSK family of protein kinases. Biochem. J. 441, 553–569 10.1042/BJ20110289 [DOI] [PubMed] [Google Scholar]
- 11. Le N. T., Heo K. S., Takei Y., Lee H., Woo C. H., Chang E., McClain C., Hurley C., Wang X., Li F., Xu H., Morrell C., Sullivan M. A., Cohen M. S., Serafimova I. M., et al. (2013) A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation 127, 486–499 10.1161/CIRCULATIONAHA.112.116988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Österreicher C. H., Penz-Österreicher M., Grivennikov S. I., Guma M., Koltsova E. K., Datz C., Sasik R., Hardiman G., Karin M., and Brenner D. A. (2011) Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. U.S.A. 108, 308–313 10.1073/pnas.1017547108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morikawa Y., Takahashi N., Kamiyama K., Nishimori K., Nishikawa Y., Morita S., Kobayashi M., Fukushima S., Yokoi S., Mikami D., Kimura H., Kasuno K., Yashiki T., Naiki H., Hara M., and Iwano M. (2019) Elevated levels of urinary extracellular vesicle fibroblast-specific protein 1 in patients with active crescentic glomerulonephritis. Nephron 141, 177–187 10.1159/000495217 [DOI] [PubMed] [Google Scholar]
- 14. Inoue T., Takenaka T., Hayashi M., Monkawa T., Yoshino J., Shimoda K., Neilson E. G., Suzuki H., and Okada H. (2010) Fibroblast expression of an IκB dominant-negative transgene attenuates renal fibrosis. J. Am. Soc. Nephrol. 21, 2047–2052 10.1681/ASN.2010010003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhowmick N. A., Chytil A., Plieth D., Gorska A. E., Dumont N., Shappell S., Washington M. K., Neilson E. G., and Moses H. L. (2004) TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303, 848–851 10.1126/science.1090922 [DOI] [PubMed] [Google Scholar]
- 16. Trimboli A. J., Cantemir-Stone C. Z., Li F., Wallace J. A., Merchant A., Creasap N., Thompson J. C., Caserta E., Wang H., Chong J. L., Naidu S., Wei G., Sharma S. M., Stephens J. A., Fernandez S. A., et al. (2009) Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature 461, 1084–1091 10.1038/nature08486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsutsumi R., Xie C., Wei X., Zhang M., Zhang X., Flick L. M., Schwarz E. M., and O'Keefe R. J. (2009) PGE2 signaling through the EP4 receptor on fibroblasts upregulates RANKL and stimulates osteolysis. J. Bone Miner. Res. 24, 1753–1762 10.1359/jbmr.090412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eijkelenboom A., and Burgering B. M. (2013) FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14, 83–97 10.1038/nrm3507 [DOI] [PubMed] [Google Scholar]
- 19. Klotz L. O., Sánchez-Ramos C., Prieto-Arroyo I., Urbánek P., Steinbrenner H., and Monsalve M. (2015) Redox regulation of FoxO transcription factors. Redox Biol. 6, 51–72 10.1016/j.redox.2015.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu H., Fergusson M. M., Wu J. J., Rovira I. I., Liu J., Gavrilova O., Lu T., Bao J., Han D., Sack M. N., and Finkel T. (2011) Wnt signaling regulates hepatic metabolism. Sci. Signal. 4, ra6 10.1126/scisignal.2001249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Norambuena-Soto I., Núñez-Soto C., Sanhueza-Olivares F., Cancino-Arenas N., Mondaca-Ruff D., Vivar R., Díaz-Araya G., Mellado R., and Chiong M. (2017) Transforming growth factor-beta and Forkhead box O transcription factors as cardiac fibroblast regulators. Biosci. Trends 11, 154–162 10.5582/bst.2017.01017 [DOI] [PubMed] [Google Scholar]
- 22. Vivar R., Humeres C., Muñoz C., Boza P., Bolivar S., Tapia F., Lavandero S., Chiong M., and Diaz-Araya G. (2016) FoxO1 mediates TGF-β1-dependent cardiac myofibroblast differentiation. Biochim. Biophys. Acta 1863, 128–138 10.1016/j.bbamcr.2015.10.019 [DOI] [PubMed] [Google Scholar]
- 23. Hu K., Lin L., Tan X., Yang J., Bu G., Mars W. M., and Liu Y. (2008) tPA protects renal interstitial fibroblasts and myofibroblasts from apoptosis. J. Am. Soc. Nephrol. 19, 503–514 10.1681/ASN.2007030300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hu K., Mars W. M., and Liu Y. (2008) Novel actions of tissue-type plasminogen activator in chronic kidney disease. Front. Biosci. 13, 5174–5186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin L., Bu G., Mars W. M., Reeves W. B., Tanaka S., and Hu K. (2010) tPA activates mitogenic signaling involving LDL receptor-related protein 1-mediated p90RSK and GSK3β pathway. Am. J. Pathol. 177, 1687–1696 10.2353/ajpath.2010.100213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Müller G. A., Zeisberg M., and Strutz F. (2000) The importance of tubulointerstitial damage in progressive renal disease. Nephrol. Dial. Transplant. 15, Suppl. 6, 76–77 10.1093/ndt/15.suppl_6.76 [DOI] [PubMed] [Google Scholar]
- 27. Zeisberg M., Strutz F., and Müller G. A. (2000) Role of fibroblast activation in inducing interstitial fibrosis. J. Nephrol. 13, Suppl. 3, S111–S120 [PubMed] [Google Scholar]
- 28. Carriere A., Ray H., Blenis J., and Roux P. P. (2008) The RSK factors of activating the Ras/MAPK signaling cascade. Front. Biosci. 13, 4258–4275 [DOI] [PubMed] [Google Scholar]
- 29. Abe J., and Berk B. C. (2014) Novel mechanisms of endothelial mechanotransduction. Arterioscler. Thromb. Vasc. Biol. 34, 2378–2386 10.1161/ATVBAHA.114.303428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Singh M. V., Kotla S., Le N. T., Ae Ko K., Heo K. S., Wang Y., Fujii Y., Thi Vu H., McBeath E., Thomas T. N., Jin Gi Y., Tao Y., Medina J. L., Taunton J., Carson N., et al. (2019) Senescent phenotype induced by p90RSK-NRF2 signaling sensitizes monocytes and macrophages to oxidative stress in HIV-positive individuals. Circulation 139, 1199–1216 10.1161/CIRCULATIONAHA.118.036232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schiessl I. M., Grill A., Fremter K., Steppan D., Hellmuth M. K., and Castrop H. (2018) Renal interstitial platelet-derived growth factor receptor-β cells support proximal tubular regeneration. J. Am. Soc. Nephrol. 29, 1383–1396 10.1681/ASN.2017101069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sauer H., Klimm B., Hescheler J., and Wartenberg M. (2001) Activation of p90RSK and growth stimulation of multicellular tumor spheroids are dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. FASEB J. 15, 2539–2541 10.1096/fj.01-0360fje [DOI] [PubMed] [Google Scholar]
- 33. Takeishi Y., Abe Ji, Lee J. D., Kawakatsu H., Walsh R. A., and Berk B. C. (1999) Differential regulation of p90 ribosomal S6 kinase and big mitogen-activated protein kinase 1 by ischemia/reperfusion and oxidative stress in perfused guinea pig hearts. Circ. Res. 85, 1164–1172 10.1161/01.RES.85.12.1164 [DOI] [PubMed] [Google Scholar]
- 34. Coughlan M. T., and Sharma K. (2016) Challenging the dogma of mitochondrial reactive oxygen species overproduction in diabetic kidney disease. Kidney Int. 90, 272–279 10.1016/j.kint.2016.02.043 [DOI] [PubMed] [Google Scholar]
- 35. Lam E. W., Brosens J. J., Gomes A. R., and Koo C. Y. (2013) Forkhead box proteins: Tuning forks for transcriptional harmony. Nat. Rev. Cancer 13, 482–495 10.1038/nrc3539 [DOI] [PubMed] [Google Scholar]
- 36. Tsuchiya K., and Ogawa Y. (2017) Forkhead box class O family member proteins: The biology and pathophysiological roles in diabetes. J. Diabetes Investig. 8, 726–734 10.1111/jdi.12651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kandula V., Kosuru R., Li H., Yan D., Zhu Q., Lian Q., Ge R. S., Xia Z., and Irwin M. G. (2016) Forkhead box transcription factor 1: Role in the pathogenesis of diabetic cardiomyopathy. Cardiovasc. Diabetol. 15, 44 10.1186/s12933-016-0361-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hameedaldeen A., Liu J., Batres A., Graves G. S., and Graves D. T. (2014) FOXO1, TGF-β regulation and wound healing. Int. J. Mol. Sci. 15, 16257–16269 10.3390/ijms150916257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Essers M. A., de Vries-Smits L. M., Barker N., Polderman P. E., Burgering B. M., and Korswagen H. C. (2005) Functional interaction between beta-catenin and FOXO in oxidative stress signaling. Science 308, 1181–1184 10.1126/science.1109083 [DOI] [PubMed] [Google Scholar]
- 40. Zhang X., Tang N., Hadden T. J., and Rishi A. K. (2011) Akt, FoxO and regulation of apoptosis. Biochim. Biophys. Acta 1813, 1978–1986 10.1016/j.bbamcr.2011.03.010 [DOI] [PubMed] [Google Scholar]
- 41. Dang L. T. H., Aburatani T., Marsh G. A., Johnson B. G., Alimperti S., Yoon C. J., Huang A., Szak S., Nakagawa N., Gomez I., Ren S., Read S. K., Sparages C., Aplin A. C., Nicosia R. F., Chen C., Ligresti G., and Duffield J. S. (2017) Hyperactive FOXO1 results in lack of tip stalk identity and deficient microvascular regeneration during kidney injury. Biomaterials 141, 314–329 10.1016/j.biomaterials.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin L., and Hu K. (2017) Tissue-type plasminogen activator modulates macrophage M2 to M1 phenotypic change through annexin A2-mediated NF-κB pathway. Oncotarget 8, 88094–88103 10.18632/oncotarget.21510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lin L., Jin Y., and Hu K. (2015) Tissue-type plasminogen activator (tPA) promotes M1 macrophage survival through p90 ribosomal S6 kinase (RSK) and p38 mitogen-activated protein kinase (MAPK) pathway. J. Biol. Chem. 290, 7910–7917 10.1074/jbc.M114.599688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lin L., Jin Y., Mars W. M., Reeves W. B., and Hu K. (2014) Myeloid-derived tissue-type plasminogen activator promotes macrophage motility through FAK, Rac1, and NF-κB pathways. Am. J. Pathol. 184, 2757–2767 10.1016/j.ajpath.2014.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hu K., Wu C., Mars W. M., and Liu Y. (2007) Tissue-type plasminogen activator promotes murine myofibroblast activation through LDL receptor-related protein 1-mediated integrin signaling. J. Clin. Invest. 117, 3821–3832 10.1172/JCI32301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Damayanti D. S., Utomo D. H., and Kusuma C. (2016) Revealing the potency of Annona muricata leaves extract as FOXO1 inhibitor for diabetes mellitus treatment through computational study. In Silico Pharmacol. 5, 3 10.1007/s40203-017-0023-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu K., Yang J., Tanaka S., Gonias S. L., Mars W. M., and Liu Y. (2006) Tissue-type plasminogen activator acts as a cytokine that triggers intracellular signal transduction and induces matrix metalloproteinase-9 gene expression. J. Biol. Chem. 281, 2120–2127 10.1074/jbc.M504988200 [DOI] [PubMed] [Google Scholar]
- 48. Racusen L. C., Monteil C., Sgrignoli A., Lucskay M., Marouillat S., Rhim J. G., and Morin J. P. (1997) Cell lines with extended in vitro growth potential from human renal proximal tubule: characterization, response to inducers, and comparison with established cell lines. J. Lab. Clin. Med. 129, 318–329 10.1016/S0022-2143(97)90180-3 [DOI] [PubMed] [Google Scholar]
- 49. Nakagawa N., Yuhki K., Kawabe J., Fujino T., Takahata O., Kabara M., Abe K., Kojima F., Kashiwagi H., Hasebe N., Kikuchi K., Sugimoto Y., Narumiya S., and Ushikubi F. (2012) The intrinsic prostaglandin E2-EP4 system of the renal tubular epithelium limits the development of tubulointerstitial fibrosis in mice. Kidney Int. 82, 158–171 10.1038/ki.2012.115 [DOI] [PubMed] [Google Scholar]