

Abstract
Hyperactivation of the canonical Wnt-signaling pathway is a prominent feature of a number of human malignancies. Transcriptional activation of this signaling cascade depends on the formation of the β-catenin–B-cell CLL/lymphoma 9 (BCL9)–pygopus (PYGO) family plant homeodomain finger 1 complex, yet how the assembly of this complex is regulated remains to be investigated. Here, using MCF-7, HeLa, HEK293T, MDA–MB-231, and Sf9 cells, along with immunoblotting and immunofluorescence, nano-HPLC–MS/MS, deubiquitination, immunoprecipitation, and chromatin immunoprecipitation (ChIP) assays, we report that BCL9 physically associates with a protein deubiquitinase, ubiquitin-specific peptidase 9, X-linked (USP9X), and that USP9X removes Lys-63–linked polyubiquitin on Lys-212 of BCL9. Importantly, the USP9X-mediated BCL9 deubiquitination facilitated the formation of the β-catenin–BCL9–PYGO complex, thereby potentiating the transcriptional activation of Wnt/β-catenin target genes. We also show that USP9X-mediated BCL9 deubiquitination promotes the proliferation and invasion of breast cancer cells. Together, these results uncover USP9X as a deubiquitinase of BCL9, implicating USP9X in Wnt/β-catenin signaling and breast carcinogenesis.
Keywords: deubiquitylation (deubiquitination); breast cancer; cell signaling; carcinogenesis; Wnt pathway; B-cell CLL/lymphoma 9 (BCL9); Lys-63–linked polyubiquitins; ubiquitin-specific peptidase 9, X-linked (USP9X)
Introduction
Wingless-type (Wnt)3 signaling is an important molecular cascade regulating embryonic development and adult homeostasis (1, 2), and its dysregulation has been implicated in the pathogenesis of a number of disease states, including various human malignancies such as colorectal cancer, prostate cancer, and breast cancer (3–7). In the absence of the canonical Wnt signaling, free cytoplasmic β-catenin is rapidly targeted for ubiquitination and proteasomal degradation by the destruction complex (Axin/adenomatous polyposis coli/glycogen synthase kinase 3β (GSK3β)) (8, 9). The absence of β-catenin defines the inactive state of Wnt-induced genes, which are silenced by a repressive complex containing T-cell factor/lymphoid enhancer factor (TCF/LEF), transducing-like enhancer protein (TLE/Groucho), and histone deacetylases (5, 10, 11). Activation of Wnt signaling leads to the relief of this repression by blocking the degradation of β-catenin, which translocates into the nucleus and converts the Wnt-responsive genes into an active state (2). This de-repression process involves the displacement of the TLE/Groucho complex and the formation of a β-catenin–nucleated transcription permissive complex (12–16), in which β-catenin, together with PYGO and B-cell lymphoma 9 (BCL9), forms a quaternary complex with TCF/LEF to drive transcriptional activation of Wnt signaling target genes (17–20).
Human BCL9 was originally identified as an oncoprotein associated with precursor B-cell acute lymphoblastic leukemia (21, 22), and its overexpression has been documented in a number of malignancies, including colorectal cancer (23) and breast cancer (24). BCL9 functions as a critical adaptor between PYGO and β-catenin by binding through its homology domain 1 (HD1) to the plant homeodomain finger in the C terminus of PYGO and through its homology domain 2 (HD2) to the Armadillo repeat domain of β-catenin (25, 26). In support of the importance of this molecular engagement, targeted disruption of the BCL9/β-catenin complex via synthetic peptides is shown to suppress Wnt signaling and inhibit its oncogenic potential (27, 28). Nevertheless, how the formation of the β-catenin/BCL9/PYGO complex is regulated remains to be investigated.
Ubiquitination, a major type of post-translational modification featured with conjugation of the highly-conserved 76-residue protein ubiquitin, is essential for protein stabilization, localization, as well as signal transduction in all eukaryotes (29–31). Ubiquitin linkages, ranging from a single ubiquitin molecule to complex polymeric chains, often elicit distinct outcomes (29). The ubiquitin-specific peptidase 9, X-linked (USP9X, also known as FAM for Drosophila fat facets), is a C19-peptidase family protein with known deubiquitinase activity (32). It cleaves Lys-48- and Lys-63–linked ubiquitin species to promote substrate stability and to favor/disfavor signal transduction, respectively (33, 34). Through targeting distinct components of multiple signaling pathways, including Notch (35, 36), EGF (37, 38), and mTOR (39, 40), USP9X regulates a number of cellular activities, including trafficking/endocytosis, cell growth, and migration, supporting a critical role for USP9X in coordinating cellular responses to multiple signaling inputs. However, its role in Wnt signaling remains to be explored.
In this study, we report that USP9X physically interacts with and functionally deubiquitinates BCL9. We showed that USP9X augments the interaction of BCL9 with β-catenin and PYGO1, thereby promoting transcriptional activation of Wnt/β-catenin–responsive genes and the proliferation and invasion of breast cancer cells.
Results
BCL9 is physically associated with USP9X
To further understand how Wnt/β-catenin signaling is regulated, we employed affinity purification and MS to identify proteins that are associated with BCL9, an essential co-activator of β-catenin–mediated transcription, in vivo. Specifically, we generated HeLa cells with doxycycline (Dox)-inducible expression of stably integrated FLAG–BCL9. Whole-cell extracts from these cells were prepared and subjected to affinity purification using an anti-FLAG affinity column. After extensive washing, the bound proteins were eluted with excess FLAG peptides, resolved on SDS-PAGE, and visualized by silver staining. The protein bands on the gel were recovered by trypsinization and analyzed by MS. The results revealed that BCL9 was co-purified with a number of proteins, including β-catenin, PYGO1, and PYGO2, all known components of the Wnt-signaling pathway (Fig. 1A and Table S1). Interestingly, USP9X, a member of the protein deubiquitinases, was also identified in the BCL9-containing protein complex (Fig. 1A). The presence of these proteins in the BCL9-associated protein complex was confirmed by Western blotting of the column eluates (Fig. 1B).
Figure 1.

BCL9 is physically associated with USP9X. A, immunoaffinity purification and MS analysis of BCL9-containing protein complexes. HeLa cells with Dox-inducible expression of stably integrated FLAG–BCL9 were collected. Cellular extracts were immunopurified with anti-FLAG–affinity beads and eluted with FLAG peptide. The eluates were resolved on SDS-PAGE and silver-stained followed by MS analysis. Detailed results from the mass spectrometric analysis are provided as shown in Table S1. B, column-bound proteins were analyzed by Western blotting with antibodies against the indicated proteins. C, whole-cell lysates from HeLa or MCF-7 cells were immunoprecipitated (IP) followed by immunoblotting (IB) with antibodies against the indicated proteins. D, cellular extracts from HeLa cells were fractionated on Superose 6 size-exclusion columns. Chromatographic elution profiles (top panel) and Western blot analysis (bottom panel) of the chromatographic fractions with antibodies against the indicated proteins are shown. The elution positions of calibration proteins with known molecular masses are indicated, and an equal volume from each fraction was analyzed. E, pulldown analysis of the molecular interface involved in the interaction between BCL9 and USP9X with in vitro-transcribed/translated full-length BCL9 and His-tagged deletion mutants of USP9X purified from Sf9 cells. The conserved domains of USP9X were determined by the SMART program (Simple Modular Architecture Research Tool, http://smart.embl-heidelberg.de. Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.). The purple rectangle represents α-α super-helix in the N-terminal region of USP9X (amino acids 249–610), and the red rectangle represents the peptidase C19 domain in the C-terminal region of USP9X (amino acids 1,554–1,953). F, pulldown analysis of the molecular interface involved in the interaction between BCL9 and USP9X with in vitro-transcribed/translated full-length BCL9 and His-tagged deletion mutants M1, M2, or M3 of USP9X purified from Sf9 cells.
To confirm the in vivo association of BCL9 with USP9X, co-immunoprecipitation experiments were performed with HeLa cell extracts. Immunoprecipitation (IP) with antibodies against BCL9 followed by immunoblotting (IB) with antibodies against USP9X demonstrated that USP9X was efficiently co-immunoprecipitated with BCL9 (Fig. 1C). Reciprocally, IP with antibodies against USP9X and IB with antibodies against BCL9 also revealed that BCL9 was efficiently co-immunoprecipitated with USP9X. The association between BCL9 and USP9X was also detected in MCF-7 cells (Fig. 1C) and HCT116 cells (Fig. S1) by co-immunoprecipitation. Furthermore, co-IP analysis also showed that β-catenin was physically associated with USP9X (Fig. 1C). To gain further support of the physical interaction between BCL9 and USP9X, protein fractionation experiments were carried out by fast protein LC (FPLC) with Superose 6 columns and a size-exclusion approach. The results indicated that the elution pattern of USP9X largely overlapped with that of BCL9, β-catenin, and PYGO1 (Fig. 1D).
To further support the interaction between BCL9 and USP9X and to gain a molecular insight into this interaction, His-tagged domain deletion mutants of USP9X were generated and expressed in Sf9 cells. In vitro pulldown analysis with these mutants and in vitro transcribed/translated FLAG-tagged full-length BCL9 revealed that a sub-middle region spanning amino acids 1,101 to 1,553 of USP9X is responsible for the direct interaction of USP9X with BCL9 (Fig. 1, E and F).
USP9X deubiquitinates BCL9
To address the functional significance of the physical interaction between BCL9 and USP9X, we next investigated whether or not USP9X could target BCL9 for deubiquitination. To this end, HeLa cells stably expressing FLAG–BCL9 were co-transfected with HA-tagged ubiquitin (HA–Ub) and control siRNA or USP9X siRNAs. Immunoprecipitation (IP) of cellular lysates with anti-FLAG followed by IB with anti-HA showed that knockdown of USP9X resulted in evident increases in ubiquitinated BCL9 species (Fig. 2A), suggesting that USP9X acts to oppose BCL9 ubiquitination. To support this notion, cellular extracts from HeLa cells expressing FLAG–BCL9 and HA–Ub were collected and sequentially purified with anti-FLAG and anti-HA affinity gels to enrich HA–Ub-conjugated BCL9. After trypsinization, the retrieved peptides were subjected to MS analysis. The results showed that three lysine residues, Lys-23, Lys-212, and Lys-906, carry di-glycine remnants (Fig. 2B), indicating that these residues were subjected to ubiquitination. We thus generated BCL9 mutants with each of the lysine residues at 23, 212, and 906 individually replaced by arginine (K23R, K212R, and K906R), and we examined the polyubiquitination levels of these mutants in USP9X-depleted cells. In vivo deubiquitination assays showed that although the association of USP9X with all BCL9 mutants was comparable with that of USP9X with WT BCL9 (BCL9/WT) (Fig. S2), increased poly-ubiquitination level was only observed for K23R and K906R but not K212R in USP9X-deficient cells (Fig. 2C), suggesting that USP9X targets BCL9/Lys-212 for deubiquitination.
Figure 2.

USP9X deubiquitinates BCL9. A, HeLa cells with Dox-inducible expression of stably integrated FLAG–BCL9 were co-transfected with control siRNA or USP9X siRNAs together with HA–Ub as indicated. Cellular extracts were prepared for co-immunoprecipitation assays with anti-FLAG followed by IB with anti-HA. B, mass spectrometry analysis of BCL9 ubiquitin conjugation sites. HeLa cells stably expressing FLAG–BCL9 were co-transfected with HA–Ub. Cellular extracts were collected and sequentially purified with anti-FLAG affinity gel and HA affinity gel to enrich HA–Ub-conjugated BCL9. After trypsinization, the retrieved peptides were subjected to MS analysis. Fragmentation spectrums and parameters of the identified BCL9 peptides with the di-glycine remnants are shown. C, HeLa cells with Dox-inducible expression of stably integrated FLAG–BCL9/K23R, FLAG–BCL9/K212R, or FLAG–BCL9/K906R were co-transfected with control siRNA or USP9X siRNAs together with HA–Ub as indicated. Cellular extracts were prepared for co-immunoprecipitation assays with anti-FLAG followed by IB with anti-HA. D, HeLa cells with Dox-inducible expression of stably integrated FLAG–BCL9 were co-transfected with control siRNA or USP9X siRNAs together with HA–Ub/Lys-48–only or HA–Ub/Lys-63–only as indicated. Cellular extracts were prepared for co-immunoprecipitation assays with anti-FLAG followed by IB with anti-HA. E, HeLa cells with Dox-inducible expression of stably integrated FLAG–BCL9/K212R were co-transfected with control siRNA or USP9X siRNAs together with HA–Ub/Lys-63–only as indicated. Cellular extracts were prepared for co-immunoprecipitation assays with anti-FLAG followed by IB with anti-HA.
To differentiate the ubiquitin moieties of polyubiquitinated BCL9 that are cleaved by USP9X, we generated HA-tagged ubiquitin mutants with all lysine residues replaced by arginine except Lys-48 (Lys-48–only) or Lys-63 (Lys-63–only). These ubiquitin mutants were then co-transfected with USP9X siRNAs into FLAG–BCL9-expressing HeLa cells. In vivo deubiquitination assays demonstrated that USP9X depletion was associated with a dramatic increase in the level of Lys-63–linked, but not Lys-48–linked, polyubiquitin moieties of BCL9 (Fig. 2D). In addition, we showed that USP9X depletion had no effect on the Lys-63–linked polyubiquitination level of FLAG–BCL9/K212R (Fig. 2E). Together, these observations support a notion that USP9X targets BCL9/Lys-212 for Lys-63–conjugated ubiquitin removal.
We also examined the effect of USP9X on the expression of BCL9. Western blot analysis revealed that, although the level of CEP131 markedly decreased as reported previously (41), the expression of BCL9 was essentially unchanged upon USP9X depletion by distinct siRNAs in MCF-7 and HeLa cells (Fig. S3, A and B). These findings are consistent with the observation that USP9X prefers to cleave Lys-63–linked, but not Lys-48–linked, ubiquitin moieties on BCL9. In addition, although it has been reported that β-catenin is stabilized in human cells (42, 43) but destabilized in mouse cells (44) by USP9X, our experiments indicate that the expression levels of β-catenin and PYGO1 are not affected by USP9X depletion (Fig. S3, A and B).
USP9X-promoted BCL9 deubiquitination potentiates the formation of the β-catenin/BCL9/PYGO1 complex
It is proposed that Lys-63–linked polyubiquitin plays a critical role in the assembly/disassembly of protein complexes (29). To understand the functional significance of USP9X-targeted removal of Lys-63–conjugated ubiquitin of BCL9/Lys-212, we next asked the question whether USP9X-targeted BCL9 deubiquitination affects in any way the formation of the β-catenin/BCL9/PYGO1 complex. To test this, HeLa cells stably expressing different sets of USP9X shRNAs and FLAG–BCL9/WT (WT) or FLAG–BCL9/K212R were established. Co-immunoprecipitation analysis in these cells showed that USP9X knockdown was associated with an overt reduction of the interaction between β-catenin and BCL9/WT, whereas the impact of USP9X depletion on the interaction between β-catenin and BCL9/K212R was marginal (Fig. 3A). Interestingly, the K212R mutation enhanced, although modestly, the binding of BCL9 to β-catenin (Fig. 3A). Additionally, USP9X knockdown impaired the interaction of PYGO1 with endogenous BCL9 (Fig. 3B) but not with BCL9/K212R (Fig. 3C). Furthermore, the efficient binding between PYGO1 and β-catenin also requires the presence of USP9X, whereas overexpression of BCL9/K212R overcomes, to certain extent, this requirement (Fig. 3, B and C). Collectively, these results indicate that BCL9/Lys-212 ubiquitination inhibits the formation of the β-catenin/BCL9/PYGO1 complex and that BCL9 deubiquitination by USP9X, in contrast, promotes the formation of this complex.
Figure 3.

USP9X-promoted BCL9 deubiquitination controls the formation of β-catenin/BCL9/PYGO1 complex. A, HeLa cells stably expressing different sets of USP9X shRNAs and FLAG–BCL9/WT or FLAG–BCL9/K212R were collected. The cellular extracts were prepared and analyzed with IP followed by IB with antibodies against the indicated proteins. B, HeLa cells stably expressing different sets of USP9X shRNAs were transfected with FLAG–PYGO1. The cellular extracts were prepared and analyzed with IP followed by IB with antibodies against the indicated proteins. C, HeLa cells stably expressing different sets of USP9X shRNAs were transfected with Myc–PYGO1 and FLAG–BCL9/K212R. The cellular extracts were prepared and analyzed with IP followed by IB with antibodies against the indicated proteins. D, immunostaining and confocal microscopy analysis of USP9X and BCL9 subcellular localization in USP9X-depleted HeLa cells (upper panel). Immunostaining and confocal microscopy analysis of the subcellular localization of FLAG–BCL9/WT and FLAG–BCL9/K212R in HeLa cells (lower panel) are shown. Scale bar, 10 μm.
It has also been proposed that Lys-63–linked ubiquitination may affect the cellular distribution of targeted proteins (29). Thus, we next examined the influence of USP9X on the subcellular localization of BCL9. Immunostaining followed by confocal microscopy analysis revealed that BCL9 predominantly resides in the nucleus even in the absence of USP9X (Fig. 3D), and the cellular localization of BCL9/WT and BCL9/K212R was similar (Fig. 3D). These observations argue against the possibility that impaired β-catenin/BCL9/PYGO1 complex formation upon USP9X depletion might be due to subcellular redistribution of BCL9.
USP9X-promoted BCL9 deubiquitination impacts the transcriptional response to the Wnt signaling
To further explore the functional significance of USP9X-targeted BCL9 deubiquitination, we then tested whether USP9X is involved in the transcriptional response to Wnt signaling. To this end, pGL3–TopFlash reporter containing eight tandem repeats of TCF/LEF-binding elements or its counterpart pGL3–FopFlash reporter containing mutated TCF/LEF-binding elements (Fig. 4A) was co-transfected into HEK293T cells together with USP9X siRNA and Renilla luciferase vector. Cells were cultured in the absence or presence of LiCl, which inhibits GSK3β and thus stabilizes β-catenin to activate the canonical Wnt pathway (7, 45). Reporter assays showed that USP9X depletion resulted in a marked reduction of TCF/LEF reporter activity in responding to LiCl stimulation (Fig. 4A), although the expression level of β-catenin was not affected, even in the presence of LiCl (Fig. 4B). Measurement of the expression of the endogenous Wnt/β-catenin target genes AXIN2, c-Myc, and LGR5 (9) using quantitative reverse transcription (qRT)-PCR, revealed that knockdown of USP9X in MCF-7 cells led to a strong inhibition of LiCl-stimulated expression of endogenous Wnt/β-catenin target genes (Fig. 4C). Similar results were obtained in HeLa cells (Fig. 4C) and HCT116 cells (Fig. S4). Together, these results support an important role for USP9X in the transcriptional response to Wnt signaling.
Figure 4.

USP9X-promoted BCL9 deubiquitination is required for transcriptional Wnt responses. A, schematic diagrams of the TopFlash/FopFlash-luciferase reporter constructs are as shown (upper panel). For reporter assays, HEK293T cells were co-transfected with control siRNA or USP9X siRNA together with Renilla and pGL3–TopFlash or pGL3–FopFlash. These cells were cultured in the absence or presence of 25 mm LiCl for 6 h before measuring luciferase activity. The relative luciferase activity was determined by sequential normalization with values of Renilla and pGL3–FopFlash activity. Each bar represents the mean ± S.D. for three biological experiments. **, p < 0.01, one-way ANOVA. B, cellular extracts from cells used in A were analyzed by Western blotting. C, MCF-7 (left panel) or HeLa (right panel) cells transfected with control siRNA or USP9X siRNAs were cultured in the absence or presence of 25 mm LiCl for 6 h and collected for qRT-PCR analysis with USP9X, AXIN2, c-Myc, and LGR5 primers. Each bar represents the mean ± S.D. for three biological experiments. *, p < 0.05; **, p < 0.01, one-way ANOVA. D, HEK293T cells expressing FLAG–BCL9/WT or FLAG–BCL9/K212R were co-transfected with control siRNA or USP9X siRNA together with Renilla and pGL3–TopFlash or pGL3–FopFlash. These cells were cultured in the absence or presence of 25 mm LiCl for 6 h before measuring luciferase activity. The relative luciferase activity was determined by sequential normalization as shown in A and then normalized with the control treatment. Each bar represents the mean ± S.D. for three biological experiments. **, p < 0.01, one-way ANOVA. E, cellular extracts for reporter assays in D were analyzed by Western blotting.
To investigate whether the influence of USP9X on the transcriptional activity of the Wnt/β-catenin signaling was through targeting BCL9 ubiquitination, FLAG–BCL9/WT and FLAG–BCL9/K212R were transfected into USP9X-depleted HEK293T cells. Luciferase reporter assays showed that USP9X depletion-associated inhibition of reporter activity could be rescued, at least partially, by FLAG–BCL9/K212R, but not FLAG–BCL9/WT (Fig. 4D). The knockdown and overexpression efficiency was verified by Western blot analysis (Fig. 4E). Collectively, these results support a notion that USP9X-promoted BCL9 deubiquitination affects the transcriptional response to the Wnt signaling.
USP9X promotes the binding of BCL9 to Wnt/β-catenin target genes
To further strengthen the functional engagement of USP9X-promoted BCL9 deubiquitination in the transcription regulation by the Wnt/β–catenin pathway, we then investigated the effect of USP9X on the recruitment of BCL9 to Wnt/β–catenin target gene promoters by chromatin immunoprecipitation (ChIP) assays. To this end, soluble chromatin from MCF-7 cells stably expressing FLAG–BCL9 were immunoprecipitated with the antibody against FLAG followed by quantitative PCR analysis of the precipitated DNAs. The results showed that FLAG–BCL9 was recruited to the promoter regions of AXIN2 and c-Myc genes under LiCl stimulation in control cells, whereas in USP9X-depleted cells, the recruitment of FLAG–BCL9 on these promoters was significantly reduced (Fig. 5A). Moreover, the recruitment of p300, a histone acetyltransferase known to be involved in the transcriptional response to the Wnt/β-catenin signaling (16, 46), on the promoter of c-Myc was also impaired upon USP9X depletion (Fig. 5B). These results support an argument that USP9X affects the recruitment of the components of the Wnt/β-catenin signaling-initiated transcription complex, including BCL9 and p300 to target gene promoters, reinforcing the notion that USP9X affects the transcription regulation by the Wnt/β-catenin pathway.
Figure 5.

USP9X controls the recruitment of BCL9 and p300 to Wnt-signaling target gene promoters. A, MCF-7 cells with Dox-inducible expression of stably integrated FLAG–BCL9/WT were transfected with control siRNA or USP9X siRNA, and the cells were treated with 25 mm LiCl for 2 h. Soluble chromatins were collected for ChIP analysis using antibodies against FLAG followed by qPCR with primers covering the promoter of the indicated genes. Each bar represents the mean ± S.D. for three biological experiments. *, p < 0.05; **, p < 0.01, one-way ANOVA. B, experiments analogous to A were performed with antibodies against p300. Each bar represents the mean ± S.D. for three biological experiments. *, p < 0.05; **, p < 0.01, one-way ANOVA. C, MCF-7 cells with Dox-inducible expression of stably integrated FLAG–BCL9/K212R were transfected with control siRNA or USP9X siRNA, and the cells were treated with 25 mm LiCl for 2 h. Soluble chromatins were collected for ChIP analysis using antibodies against FLAG followed by qPCR with primers covering the promoter of the indicated genes. Each bar represents the mean ± S.D. for three biological experiments. p value is determined by one-way ANOVA. D, experiments analogous to C were performed with antibodies against p300. Each bar represents the mean ± S.D. for three biological experiments. p value is determined by one-way ANOVA.
To ask whether USP9X-targeted BCL9 deubiquitination is involved in this process, MCF-7 cells stably expressing FLAG–BCL9/K212R were generated. These cells were transfected with control siRNA or USP9X siRNA and cultured in the absence or presence of LiCl. ChIP assays showed that knockdown of USP9X had a limited effect on the binding of FLAG–BCL9/K212R to the promoters of AXIN2 and c-Myc, although BCL9/K212R was recruited more efficiently than BCL9/WT upon LiCl treatment in control cells (Fig. 5C). The recruitment of p300 was also comparable on the promoter of c-Myc in control cells versus USP9X-depleted cells (Fig. 5D). These results support the notion that USP9X-promoted BCL9 deubiquitination is involved in the recruitment of BCL9 and p300 to the promoters of Wnt target genes.
USP9X promotes breast cancer cell proliferation and invasion through targeting BCL9 for deubiquitination
It has been well-documented that the Wnt/β-catenin signaling pathway is implicated in cell proliferation, migration, invasion, and metastasis in various types of cancer (3, 7), and we reported previously that USP9X is up-regulated in breast carcinoma (41). Thus, we next investigated whether USP9X-targeted BCL9 deubiquitination contributes in any way to breast carcinogenesis. To this end, we developed MCF-7 cells stably expressing USP9X shRNA and FLAG–BCL9/WT or FLAG–BCL9/K212R. Colony formation assays showed that USP9X knockdown was associated with a reduced colony number of MCF-7 cells, an effect that could be ameliorated by overexpression of FLAG–BCL9/K212R but not FLAG–BCL9/WT (Fig. 6A). Similar results were obtained when the growth of USP9X-depleted MCF-7 cells expressed FLAG–BCL9/WT or FLAG–BCL9/K212R (Fig. 6, B and C). These results support an argument that USP9X promotes breast cancer cell proliferation through targeting BCL9.
Figure 6.

USP9X promotes breast cancer cell proliferation and invasion through targeting BCL9. A, colony formation assays with MCF-7 cells stably expressing control shRNA or USP9X shRNA and FLAG–BCL9/WT or FLAG–BCL9/K212R. Representative images from three biological experiments are shown. Each bar represents the mean ± S.D. for three biological experiments. **, p < 0.01, one-way ANOVA. B, MCF-7 cells stably expressing FLAG–BCL9/WT or FLAG–BCL9/K212R were transfected with control siRNA or USP9X siRNA and then subjected to growth viability assay. Each bar represents the mean ± S.D. for three biological experiments. *, p < 0.05, two-way ANOVA. C, cellular extracts from cells used in B were collected and analyzed by Western blotting. D, MDA–MB-231 cells stably expressing FLAG–BCL9/WT or FLAG–BCL9/K212R were transfected with control siRNA or USP9X siRNA followed by transwell invasion assays. Each bar represents the mean ± S.D. for three biological experiments. **, p < 0.01, one-way ANOVA. Scale bar, 50 μm. E, MDA–MB-231 cells stably expressing FLAG–BCL9 or FLAG–BCL9/K212R were transfected with control siRNA or USP9X siRNA and then subjected to wound healing scratch assays. Each bar represents the mean ± S.D. for three biological experiments. **, p < 0.01, one-way ANOVA. Scale bar, 50 μm. F, cellular extracts from cells used in D and E were collected and analyzed by Western blotting.
We also generated MDA–MB-231 cells stably expressing FLAG–BCL9/WT or FLAG–BCL9/K212R. These cells were then transfected with control siRNA or USP9X siRNA. Transwell invasion assays showed that USP9X depletion inhibited the invasion of MDA–MB-231 cells, an effect that could be rescued, at least partially, by overexpression of FLAG–BCL9/K212R but not FLAG–BCL9/WT (Fig. 6D). In addition, wound healing scratch assays further consolidated the functional link between USP9X-promoted BCL9 deubiquitination and breast cancer cell migration (Fig. 6, E and F), supporting a notion that USP9X promotes breast cancer cell invasion through targeting BCL9. Because cell growth and invasive potential are often affected by the off-target effect of siRNA/shRNA, we have verified the specificity of RNAi by performing the rescue experiments with an RNAi-resistant USP9X exogene. Colony formation assays and transwell invasion assays demonstrated that forced expression of RNAi-resistant USP9X could successfully offset the growth or invasion defects resulted from USP9X depletion (Fig. S5, A and B). The expression of endogenous and exogenous USP9X was examined by Western blotting (Fig. S5, A and B).
Discussion
In this study, we report that USP9X plays an important role in regulating the Wnt/β-catenin signaling. Specifically, we found USP9X physically interacts with and functionally deubiquitinates BCL9. We showed that USP9X, in doing so, influences the formation of the β-catenin/BCL9/PYGO1 complex and the activation of Wnt/β-catenin target genes to promote the proliferation and invasion of breast cancer cells (Fig. 7).
Figure 7.
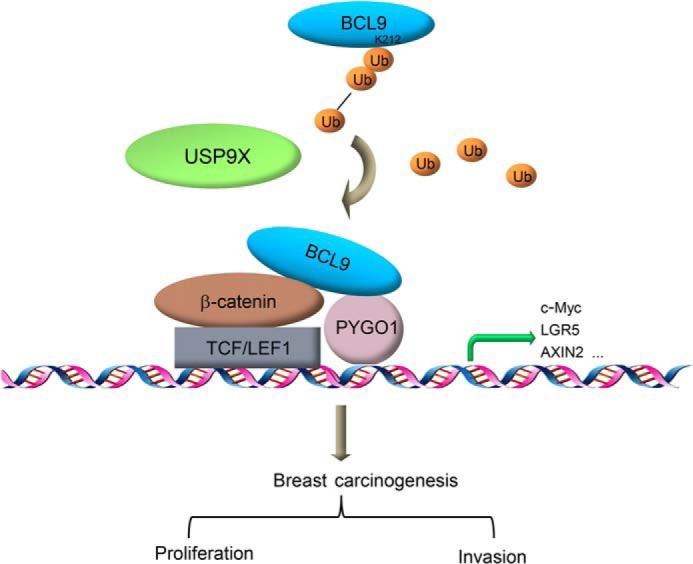
USP9X in Wnt/β-catenin signaling and breast carcinogenesis. The protein deubiquitinase USP9X interacts with BCL9 and acts to remove Lys-63–linked polyubiquitin on lysine 212 of BCL9. Then, USP9X-targeted BCL9 deubiquitination facilitates the formation of the β-catenin/BCL9/PYGO complex, thereby potentiating the transcriptional activation of Wnt/β-catenin target genes and promoting the proliferation and invasion of breast cancer cells.
Extensive studies have led to the identification of additional factors that are involved in the Wnt pathway. These components include the Wnt secretory machinery, Wnt co-receptors, components of the β-catenin destruction complex, and its nuclear co-factors (5). In the past years, evident is accumulating to indicate that dysregulation or mutations in the Wnt pathway occur frequently in human cancers (5, 7). Accordingly, extensive efforts have been focused on the pursuit of the components of this pathway for diagnostic and therapeutic values. For example, the β-catenin–nucleated transcriptional complex is considered as a pharmacologically attractive target with a high priority because of its pathologic role in colorectal cancer (28, 47), multiple myeloma (45, 48–50), and breast cancer (51, 52). In addition, because the formation of the β-catenin/BCL9/PYGO is critical for TCF/LEF-directed Wnt transcriptional activity, targeted disruption of the β-catenin/BCL9 complex has been shown to selectively suppress oncogenic Wnt signaling (27, 28). We report in this study that the protein deubiquitinase USP9X is a regulatory component of the Wnt/β-catenin pathway by interacting with the β-catenin/BCL9/PYGO complex. We showed that USP9X preferentially catalyzes Lys-63–linked ubiquitin chain removal from BCL9 to facilitate the formation of the β-catenin/BCL9/PYGO complex and thus the activation of transcriptional response. Interestingly, Lys-212 of BCL9 targeted by USP9X locates between the HD1 and HD2 domains, which are required to bind PYGO1 and β-catenin, respectively (25, 53). Thus, it is possible that Lys-63–linked polyubiquitin conjugates on Lys-212 disfavors the association of BCL9 with β-catenin or PYGO via steric hindrance by the modification itself or reader proteins. If this is the case, the importance of USP9X in the control of the canonical Wnt signaling cannot be overlooked.
Pygo and Legless in Drosophila melanogaster are orthologs of PYGO and BCL9 in Homo sapiens, respectively. These genes were initially discovered in flies and defined as key Wnt signaling components that are essential for Armadillo (β-catenin in3 mammals)-mediated transcription during normal development, although the paralogs of these genes (PYGO1 and PYGO2, or BCL9 and BCL9L) in mammals may participate in different fate-determining processes (54–58). Interestingly, USP9X also displays an extraordinarily high level of sequence conservation from Drosophila to mammals (32). The first USP9X ortholog identified was the Drosophila gene fat facets (faf), found in a mutagenesis screen and shown to be required for the embryo development (59). Across vertebrates, the level of conservation of USP9X is almost equivalent to that of β-catenin (32), consistent with the functional link between USP9X and Wnt/β-catenin signaling reported in this study, although it remains to be investigated whether the functional link between USP9X and β-catenin/BCL9/PYGO complex extends to other vertebrates.
Enzymatically, USP9X is able to cleave monoubiquitin from substrates and distinct types of ubiquitin linkages, including Lys-29-, Lys-23-, Lys-48-, and Lys-63–linked ubiquitin moieties (33, 37, 60, 61). Whether other types of ubiquitin linkages conjugated to BCL9 could be cleaved by USP9X remains to be examined. Although the enzyme that is responsible for BCL9 ubiquitination is still unclear, we demonstrated that USP9X-targeted BCL9 deubiquitination activates the expression of Wnt/β-catenin target genes AXIN2 and c-Myc and promotes the proliferation and invasion of breast cancer cells. It is possible that inhibition of the enzymatic activity of USP9X could potentially interfere with Wnt signaling and benefit the treatment of breast cancer.
Actually, USP9X has been involved in a number of cellular processes through targeting components of Notch (35, 36)-, EGF (37, 38)-, mTOR (39, 40)-, and transforming growth factor β (61, 62)-signaling pathways that are fundamental to many aspects of development and disease. Also, previous studies from our lab and from others reported that USP9X promotes breast cancer cell proliferation and invasion through targeting, at least, the centrosome proteins CEP131 (41) and SMAD4 (61), respectively. Here, we demonstrated that forced expression of USP9X could successfully offset the growth or invasion defects resulting from USP9X depletion, whereas overexpression of FLAG–BCL9/K212R could not fully restore USP9X depletion–associated effects. These results suggest that USP9X-promoted tumorigenesis also relies on targeting other substrates or influencing pathways other than Wnt/β-catenin, although USP9X-targeted BCL9 deubiquitination elicits profound effects on Wnt response and cellular activity. Considering that there is an increasingly apparent cross-talk of Wnt/β-catenin with other tumorigenic signaling pathways (64, 65), it is possible that BCL9/K212R may contribute to the proliferation and invasion of cancer cells via potentiating or re-wiring other signaling cascades that are disrupted in USP9X-deficient cells. This could be the reason why overexpression of BCL9/K212R alone could efficiently, albeit not fully, restore the effects induced by USP9X depletion. Therefore, we propose that USP9X-targeted BCL9 deubiquitination and thus Wnt activation play a non-negligible role in breast cancer cell proliferation and invasion, although the exact contribution of Wnt/β-catenin in USP9X-promoted tumorigenesis remains to be investigated.
The USP9X-encoding gene is located on the X chromosome, and it escapes X-inactivation in females (66) leading to a sexually dimorphic and sex-dependent expression feature (67). In both males and females, it is without a doubt that the expression of USP9X must be strictly regulated. However, it is conceivable that under certain conditions in the development of tumors, the expression of USP9X is dysregulated at either the genomic, transcriptional/post-transcriptional, or translational/post-translational level, leading to overexpression of this protein. For example, mutations or copy number variations in USP9X were found in 53 types of the 86 cancer types listed in cBioPortal for cancer genomics, with the frequency up to 13% for the alterations of USP9X within a single cancer type (68, 69). For Wnt-driven cancer, aberrant expression of USP9X might result from genomic amplification of USP9X, although on the other side, it might be possible that aberrant firing of Wnt signaling may consequently promote the expression or enzymatic activity of USP9X, which, in turn, according to our model, further activates the transcriptional response of Wnt/β-catenin and advances the development or progression of cancer.
In summary, we report in this study that the protein deubiquitinase USP9X interacts with BCL9, and USP9X acts to remove Lys-63–linked polyubiquitin on lysine 212 of BCL9. We demonstrated that USP9X-targeted BCL9 deubiquitination facilitates the formation of the β-catenin/BCL9/PYGO complex, thereby potentiating the transcriptional activation of Wnt/β-catenin target genes. We showed that USP9X-targeted BCL9 deubiquitination promotes the proliferation and invasion of breast cancer cells. Although it is still unclear how the expression or function of USP9X is up-regulated and it remains to be defined the contribution of USP9X-promoted BCL9 deubiquitination in cancers associated with USP9X dysregulation, our results identify USP9X as a deubiquitinase of BCL9 and implicate USP9X in Wnt/β-catenin signaling and breast carcinogenesis.
Materials and methods
Antibodies and reagents
The sources of antibodies against the following proteins were: HA (sc-805, 0.4 μg/ml for WB) from Santa Cruz Biotechnology; β-actin (A1978, 0.2 μg/ml for WB) and FLAG (F3165, 2 μg/ml for IP and 0.38 μg/ml for WB) from Sigma; USP9X (55054-1-AP, 2 μg/ml for IP, 0.4 μg/ml for WB, and 2 μg/ml for IF), BCL9 (22947-1-AP, 2 μg/ml for IP, 0.44 μg/ml for WB, and 2.2 μg/ml for IF), β-catenin (51067-2-AP, 0.23 μg/ml for WB) from Proteintech; Myc (M047-3, 0.2 μg/ml for WB) from MBL; CEP131 (ab99379, 0.5 μg/ml for WB) and PYGO1 (ab95072, 1 μg/ml for WB) from Abcam. Anti-FLAG M2 affinity gel (A2220), 3×FLAG peptide (F4799), anti-HA affinity gel (E6779), and doxycycline (D9891) were purchased from Sigma.
Plasmids
The FLAG-tagged BCL9 was carried by pLenti-Tight-Puro vector, pLenti-Hygro vector, or pcDNA3.1 vector (for in vitro transcription/translation). The FLAG-tagged K23R, K212R, and K906R of BCL9 mutants carried by pLenti-Tight-Puro vector or pLenti-Hygro vector were constructed by QuikChange strategy using the point mutation kit from Stratagene. The truncation mutants of USP9X expressed in insect cells were carried by pFastBac-HTA vector. The FLAG-tagged or Myc-tagged PYGO1 was carried by pLenti-Hygro vector. TopFlash and FopFlash plasmids were gifts from Dr. Xuyu Zhou (Chinese Academy of Sciences, Beijing, China). HA-tagged ubiquitin Lys-48–only (plasmid no. 17605, Addgene) and Lys-63–only (plasmid no. 17606, Addgene) were gifts from Dr. Ted Dawson (Johns Hopkins University School of Medicine, Baltimore, MD).
Cell culture
MCF-7, HeLa, HEK293T, MDA–MB-231, HCT116, and Sf9 cells were purchased from the American Type Culture Collection (Manassas, VA) and cultured according to the manufacturer's instructions. Cells that allow protein expression under doxycycline treatment were created in two steps. First, cells were infected with lentivirus carrying rtTA and subjected to neomycin selection. Subsequently, the established rtTA cells were infected with virus carrying pLenti-Tight-Puro vector that encodes BCL9, followed by puromycin selection. All of the cells integrated with rtTA were cultured in Tet-approved FBS and medium from Clontech. All of the cells were authenticated by examination of morphology and growth characteristics and were confirmed to be mycoplasma-free.
Western blotting
Whole-cell lysates were harvested from treated cells followed by resuspending in 5× SDS-PAGE loading buffer. The boiled protein samples were then subjected to SDS-PAGE followed by immunoblotting with the appropriate primary antibodies and secondary antibodies.
Immunopurification and silver staining
Lysates from HeLa cells stably expressing FLAG–BCL9 were prepared by incubating the cells in lysis buffer containing protease inhibitor mixture (Roche Applied Science). Anti-FLAG immunoaffinity columns were prepared using anti-FLAG M2 affinity gel (Sigma) following the manufacturer's suggestions. Cell lysates were obtained from about 5 × 108 cells and applied to an equilibrated FLAG column of 1-ml bed volume to allow for adsorption of the protein complex to the column resin. After binding, the column was washed with cold PBS plus 0.2% Nonidet P-40. FLAG peptide (Sigma) was applied to the column to elute the FLAG protein complex as described by the vendor. The eluents were collected and visualized on NuPAGE 4–12% BisTris gel (Invitrogen) followed by silver staining with a silver staining kit (Pierce). The distinct protein bands were retrieved and analyzed by LC-MS/MS.
Nano-HPLC-MS/MS analysis of BCL9-containing protein complex
To identify proteins associated with FLAG–BCL9, LC-MS/MS analysis was performed using a Thermo Finnigan LTQ linear ion trap mass spectrometer in line with a Thermo Finnigan Surveyor MS Pump Plus HPLC system. Tryptic peptides generated were loaded onto a trap column (300SB-C18, 5 × 0.3 mm, 5-μm particle; Agilent Technologies, Santa Clara, CA), which was connected through a zero dead volume union to the self-packed analytical column (C18, 100 μm inner diameter × 100 mm, 3-μm particle; SunChrom, Germany). The peptides were then eluted over a gradient (0–45% B in 55 min, 45–100% B in 10 min, where B = 80% acetonitrile, 0.1% formic acid) at a flow rate of 500 nl min−1 and introduced online into the linear ion trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) using nano-electrospray ionization. Data-dependent scanning was incorporated to select the five most abundant ions (one microscan per spectra; precursor isolation width 1.0 m/z; 35% collision energy; 30-ms ion activation; exclusion duration, 90 s; repeat count, 1) from a full-scan mass spectrum for fragmentation by collision-induced dissociation. MS data were analyzed using SEQUEST (version 28) against NCBI Human Protein Database (downloaded December 14, 2011, 33,256 entries), and the results were filtered, sorted, and displayed using Bioworks 3.2. Peptides (individual spectra) with Preliminary Score (Sp) ≧500; Rank of Sp (RSp) ≦5; and peptides with +1, +2, or +3 charge states were accepted if they were fully enzymatic and had a cross-correlation (Xcorr) of 1.90, >2.75, and >3.50, respectively. The following residue modifications were allowed in the search: carbamidomethylation on cysteine as fix modification and oxidation on methionine as variable modification. Peptide sequences were searched using trypsin specificity and allowing a maximum of two missed cleavages. Sequest was searched with a peptide tolerance of 3.0 Da and a fragment ion tolerance of 1.0 Da.
LC-MS/MS analysis of BCL9 ubiquitination sites
FLAG–BCL-conjugated ubiquitin bands were excised from the gel and subjected to in-gel tryptic digestion. Resulting peptides were separated by reverse-phase LC on an easy-nLC 1000 system (Thermo Fisher Scientific) and directly sprayed into a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific). The MS analysis was carried out in a data-dependent mode with an automatic switch between a full MS and an MS/MS scan in the obitrap. For full MS survey scan, automatic gain control target was 3e-6, and scan range was from 350 to 1800 with a resolution of 70,000. The 10 most intense peaks with charge state of ≥2 were selected for fragmentation by higher-energy collision dissociation with normalized collision energy of 27%. The MS2 spectra were acquired with 17, 500 resolution. The exclusion window was set at ±1.6 Da. All MS/MS spectra were searched against the Uniport–Human protein sequence database by using Proteome Discoverer software (version 1.4) with an overall false discovery rate for peptides of less than 1%. Peptide sequences were searched using trypsin specificity and allowing a maximum of two missed cleavages. Carbamidomethylation on Cys was specified as a fixed modification. Gly–Gly on lysine, oxidation of methionine, and acetylation on the peptide N terminus were fixed as variable modifications. Mass tolerances for precursor ions were set at ±10 ppm for precursor ions and ±0.02 Da for MS/MS. The interesting MS/MS spectra were manually verified.
FPLC
Cellular extracts from HeLa cells were applied to a Superose 6 size-exclusion column (GE Healthcare) that had been equilibrated with DTT-containing buffer and calibrated with protein standards (Amersham Biosciences). The column was eluted at a flow rate of 0.5 ml/min, and fractions were collected every 2 min.
Immunoprecipitation
Cellular lysates were prepared by incubating the cells in NETN buffer (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.2% Nonidet P-40, 2 mm EDTA) in the presence of protease inhibitor cocktails (Roche Applied Science) for 20 min at 4 °C followed by centrifugation at 14,000 × g for 15 min at 4 °C. For immunoprecipitation, about 500 μg of protein were incubated with control or specific antibodies (1–2 μg) for 12 h at 4 °C with constant rotation; 50 μl of 50% protein G magnetic beads (Invitrogen) was then added, and the incubation was continued for an additional 2 h. Beads were then washed five times using the lysis buffer. Between washes, the beads were collected by a magnetic stand (Invitrogen) at 4 °C. The precipitated proteins were eluted from the beads by resuspending the beads in 2× SDS-PAGE loading buffer and boiling for 5 min. The boiled immune complexes were subjected to SDS-PAGE followed by immunoblotting with appropriate antibodies.
Recombinant protein purification
Recombinant baculovirus carrying mutants of USP9X was generated with the Bac-to-Bac System (Invitrogen). Infected Sf9 cells were grown in spinner culture for 48–96 h at 27 °C and lysed by ultrasonicator in Equilibration buffer (50 mm sodium phosphate, 0.3 m sodium chloride, 10 mm imidazole, and 10 mm Tris-HCl, pH 8.0). His-tagged proteins were purified using Ni2+-nitrilotriacetic acid–agarose (Invitrogen) according to standard procedures.
In vivo deubiquitination assay
Cells with different treatments were lysed in RIPA buffer in the presence of protease inhibitors at 4 °C for 30 min with rotation and centrifuged at 20,000 × g for 15 min. About 0.5–1.5 mg of cellular extracts were immunoprecipitated with anti-FLAG affinity gel for 2 h. The beads were then washed five times with RIPA buffer, boiled in SDS loading buffer, and subjected to SDS-PAGE followed by immunoblotting.
RNA interference
All siRNA transfections were performed using Lipofectamine RNAiMAX (Invitrogen) following the manufacturer's recommendations. The final concentration of the siRNA molecules is 10 nm, and cells were harvested 72 or 96 h later according to the purposes of the experiments. Control siRNA (ON-TARGETplus nontargeting pool, D-001810-10) and USP9X siRNA (ON-TARGETplus, L-006099, set of 4 and pool) were purchased from Dharmacon, and USP9X siRNA-5 (5′-CTGTGATTCAGCAACTCTA-3′) was chemically synthesized by Sigma (Shanghai, China). The short hairpin RNAs (shRNAs) against USP9X in pLKO vectors were purchased from Sigma. The sequences of shRNAs are provided in Table S2.
qRT-PCR
Total cellular RNAs were isolated with TRIzol reagent (Invitrogen) and used for first strand cDNA synthesis with the reverse transcription system (Roche Applied Science). Quantitation of all gene transcripts was done by qPCR using a Power SYBR Green PCR Master Mix (Roche Applied Science) and an ABI PRISM 7500 sequence detection system (Applied Biosystems) with the expression of ACTB as the internal control. The primers used were listed in Table S3.
Chromatin immunoprecipitation
ChIP experiments were performed according to the procedure described previously (63, 70, 71). About 10 million cells were cross-linked with 1% formaldehyde for 10 min at room temperature and quenched by the addition of glycine to a final concentration of 125 mm for 5 min. The fixed cells were resuspended in SDS lysis buffer (1% SDS, 5 mm EDTA, and 50 mm Tris-HCl, pH 8.1) in the presence of protease inhibitors and subjected three times for 10 cycles (30 s on and 30 s off) of sonication (Bioruptor, Diagenode) to generate chromatin fragments of ∼300 bp in length. Lysates were diluted in buffer containing 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, and 150 mm NaCl. For immunoprecipitation, the diluted chromatin was incubated with control or specific antibodies (2 μg) for 12 h at 4 °C with constant rotation; 50 μl of 50% (v/v) protein G magnetic beads was then added, and the incubation was continued for an additional 2 h. Beads were then washed with the following buffers: TSE I (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, and 150 mm NaCl); TSE II (0.1% SDS, 1% Triton X-100, 2 mm EDTA, 20 mm Tris-HCl, pH 8.1, and 500 mm NaCl); buffer III (0.25 m LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mm EDTA, and 10 mm Tris-HCl, pH 8.1); and Tris/EDTA buffer. Between washes, the beads were collected by a magnetic stand at 4 °C. Then the pulldown chromatin complex together with input was de-cross–linked at 70 °C for 2 h in elution buffer (1% SDS, 5 mm EDTA, 20 mm Tris-HCl, pH 8.1, 50 mm NaCl, and 0.1 mg/ml proteinase K). Eluted DNA was purified with a PCR purification kit (Qiagen) and analyzed by qPCR using primers described in Table S4.
Lentivirus production
The shRNAs targeting USP9X or vectors encoding rtTA, USP9X, and BCL9, the mutant PYGO1, and three assistant vectors pMDLg/pRRE, pRSV-REV, and pVSVG were transiently transfected into HEK293T cells. Viral supernatants were collected 48 h later, clarified by filtration, and concentrated by ultracentrifugation.
Immunofluorescence
HeLa cells on glass coverslips (BD Biosciences) were fixed with 2% paraformaldehyde and permeabilized with 0.2% Triton X-100 in PBS. Samples were then blocked in 5% donkey serum in the presence of 0.1% Triton X-100 and stained with the appropriate primary and secondary antibodies coupled to AlexaFluor 488 or 594 (Invitrogen). Confocal images were captured on FluoView1000 Olympus using a ×60 oil objective. To avoid bleed-through effects in double-staining experiments, each dye was scanned independently in a multitracking mode.
Colony formation assay
MCF-7 cells stably expressing the indicated genes and/or shRNAs were maintained in culture media for 14 days. After 14 days, the cells were washed with PBS, fixed with methyl alcohol for 10 min, and stained with crystal violet (0.5% w/v) for 20 min. The number of colonies per well was counted.
Cell invasion assay
Transwell chamber filters (Chemicon Inc.) were coated with Matrigel. After transfection, cells were resuspended in serum-free media, and 2.5 × 104 cells in 0.5 ml of serum-free media were placed in the upper chamber of the transwell. The chamber was then transferred to a well containing 500 μl of media containing 10% FBS. Cells were incubated for 18 h at 37 °C. Cells in the upper well were removed by wiping the top of the membrane with cotton swabs. The membranes were then stained, and the remaining cells were counted. Representative fields were captured and counted for each membrane.
Statistical analysis
Data from three biological experiments are presented with error bars as mean ± S.D. Two-tailed unpaired Student's t test was used for comparing two groups of data. Analysis of variance (ANOVA) with Bonferroni's correction was used to compare multiple groups of data. A p value of less than 0.05 was considered significant. All of the statistical testing results were determined by SPSS 20.0 software.
Author contributions
Z. S., N. Y., and S. Y. conceptualization; Z. S., J. Z., K. Z., N. Y., and S. Y. data curation; Z. S., J. Z., Q. Z., K. Z., N. Y., and S. Y. formal analysis; Z. S., J. Z., Q. Z., L. L., and S. Y. validation; Z. S., J. Z., Q. Z., C. C., S. T., N. Y., and S. Y. investigation; Z. S. visualization; Z. S. and K. Z. methodology; Z. S., N. Y., and S. Y. writing-original draft; Z. S., J. Z., Q. Z., C. C., S. T., L. L., N. Y., and S. Y. project administration; Z. S., L. S., N. Y., and S. Y. writing-review and editing; L. S. resources; L. S. and N. Y. funding acquisition; N. Y. supervision.
Supplementary Material
This work was supported by Grant 81602511 from the National Natural Science Foundation of China (to N. Y.) and a grant from Excellent Talent Project of Tianjin Medical University(to L. S.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S5 and Tables S1–S4.
- Wnt
- Wingless-type
- TCF/LEF
- T-cell factor/lymphoid enhancer factor
- TLE
- transducing-like enhancer protein
- ANOVA
- analysis of variance
- FBS
- fetal bovine serum
- PYGO
- Pygopus
- qPCR
- quantitative PCR
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- WB
- Western blot
- IP
- immunoprecipitation
- IB
- immunoblot
- Ub
- ubiquitin
- shRNA
- short hairpin RNA
- Dox
- doxycycline
- EGF
- epidermal growth factor
- mTOR
- mammalian target of rapamycin
- IF
- immunofluorescence.
References
- 1. MacDonald B. T., Tamai K., and He X. (2009) Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev. Cell 17, 9–26 10.1016/j.devcel.2009.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Valenta T., Hausmann G., and Basler K. (2012) The many faces and functions of β-catenin. EMBO J. 31, 2714–2736 10.1038/emboj.2012.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mani M., Carrasco D. E., Zhang Y., Takada K., Gatt M. E., Dutta-Simmons J., Ikeda H., Diaz-Griffero F., Pena-Cruz V., Bertagnolli M., Myeroff L. L., Markowitz S. D., Anderson K. C., and Carrasco D. R. (2009) BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res. 69, 7577–7586 10.1158/0008-5472.CAN-09-0773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pohl S. G., Brook N., Agostino M., Arfuso F., Kumar A. P., and Dharmarajan A. (2017) Wnt signaling in triple-negative breast cancer. Oncogenesis 6, e310 10.1038/oncsis.2017.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhan T., Rindtorff N., and Boutros M. (2017) Wnt signaling in cancer. Oncogene 36, 1461–1473 10.1038/onc.2016.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Logan C. Y., and Nusse R. (2004) The Wnt-signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 10.1146/annurev.cellbio.20.010403.113126 [DOI] [PubMed] [Google Scholar]
- 7. Klaus A., and Birchmeier W. (2008) Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 8, 387–398 10.1038/nrc2389 [DOI] [PubMed] [Google Scholar]
- 8. Latres E., Chiaur D. S., and Pagano M. (1999) The human F box protein β-Trcp associates with the Cul1/Skp1 complex and regulates the stability of β-catenin. Oncogene 18, 849–854 10.1038/sj.onc.1202653 [DOI] [PubMed] [Google Scholar]
- 9. Clevers H. (2006) Wnt/β-catenin signaling in development and disease. Cell 127, 469–480 10.1016/j.cell.2006.10.018 [DOI] [PubMed] [Google Scholar]
- 10. Debeb B. G., Lacerda L., Xu W., Larson R., Solley T., Atkinson R., Sulman E. P., Ueno N. T., Krishnamurthy S., Reuben J. M., Buchholz T. A., and Woodward W. A. (2012) Histone deacetylase inhibitors stimulate dedifferentiation of human breast cancer cells through WNT/β-catenin signaling. Stem Cells 30, 2366–2377 10.1002/stem.1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sekiya T., and Zaret K. S. (2007) Repression by Groucho/TLE/Grg proteins: genomic site recruitment generates compacted chromatin in vitro and impairs activator binding in vivo. Mol. Cell 28, 291–303 10.1016/j.molcel.2007.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gough N. R. (2012) Focus issue: Wnt and β-catenin signaling in development and disease. Sci. signal. 5, eg2 10.1126/scisignal.2002806 [DOI] [PubMed] [Google Scholar]
- 13. Li J., Chen X., Ding X., Cheng Y., Zhao B., Lai Z. C., Al Hezaimi K., Hakem R., Guan K. L., and Wang C. Y. (2013) LATS2 suppresses oncogenic Wnt signaling by disrupting β-Catenin/BCL9 interaction. Cell Rep. 5, 1650–1663 10.1016/j.celrep.2013.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lien W. H., and Fuchs E. (2014) Wnt some lose some: transcriptional governance of stem cells by Wnt/β-catenin signaling. Genes Dev. 28, 1517–1532 10.1101/gad.244772.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gang E. J., Hsieh Y. T., Pham J., Zhao Y., Nguyen C., Huantes S., Park E., Naing K., Klemm L., Swaminathan S., Conway E. M., Pelus L. M., Crispino J., Mullighan C. G., McMillan M., et al. (2014) Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene 33, 2169–2178 10.1038/onc.2013.169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hecht A., Vleminckx K., Stemmler M. P., van Roy F., and Kemler R. (2000) The p300/CBP acetyltransferases function as transcriptional co-activators of β-catenin in vertebrates. EMBO J. 19, 1839–1850 10.1093/emboj/19.8.1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Thompson B. J. (2004) A complex of armadillo, legless, and pygopus co-activates dTCF to activate wingless target genes. Curr. Biol. 14, 458–466 10.1016/j.cub.2004.02.026 [DOI] [PubMed] [Google Scholar]
- 18. Townsley F. M., Cliffe A., and Bienz M. (2004) Pygopus and legless target armadillo/β-catenin to the nucleus to enable its transcriptional co-activator function. Nat. Cell Biol. 6, 626–633 10.1038/ncb1141 [DOI] [PubMed] [Google Scholar]
- 19. Mieszczanek J., de la Roche M., and Bienz M. (2008) A role of Pygopus as an anti-repressor in facilitating Wnt-dependent transcription. Proc. Natl. Acad. Sci. U.S.A. 105, 19324–19329 10.1073/pnas.0806098105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hoffmans R., Städeli R., and Basler K. (2005) Pygopus and legless provide essential transcriptional co-activator functions to armadillo/β-catenin. Curr. Biol. 15, 1207–1211 10.1016/j.cub.2005.05.054 [DOI] [PubMed] [Google Scholar]
- 21. Willis T. G., Zalcberg I. R., Coignet L. J., Wlodarska I., Stul M., Jadayel D. M., Bastard C., Treleaven J. G., Catovsky D., Silva M. L., and Dyer M. J. (1998) Molecular cloning of translocation t(1;14)(q21;q32) defines a novel gene (BCL9) at chromosome 1q21. Blood 91, 1873–1881 [PubMed] [Google Scholar]
- 22. Beroukhim R., Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J. S., Dobson J., Urashima M., Mc Henry K. T., Pinchback R. M., Ligon A. H., Cho Y. J., Haery L., et al. (2010) The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 10.1038/nature08822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moor A. E., Anderle P., Cantù C., Rodriguez P., Wiedemann N., Baruthio F., Deka J., André S., Valenta T., Moor M. B., Gyorffy B., Barras D., Delorenzi M., Basler K., and Aguet M. (2015) BCL9/9L–β-catenin signaling is associated with poor outcome in colorectal cancer. EBioMedicine 2, 1932–1943 10.1016/j.ebiom.2015.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Elsarraj H. S., Hong Y., Valdez K. E., Michaels W., Hook M., Smith W. P., Chien J., Herschkowitz J. I., Troester M. A., Beck M., Inciardi M., Gatewood J., May L., Cusick T., McGinness M., et al. (2015) Expression profiling of in vivo ductal carcinoma in situ progression models identified B-cell lymphoma-9 as a molecular driver of breast cancer invasion. Breast Cancer Res. 17, 128 10.1186/s13058-015-0630-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fiedler M., Sánchez-Barrena M. J., Nekrasov M., Mieszczanek J., Rybin V., Müller J., Evans P., and Bienz M. (2008) Decoding of methylated histone H3 tail by the Pygo–BCL9 Wnt signaling complex. Mol. Cell 30, 507–518 10.1016/j.molcel.2008.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kramps T., Peter O., Brunner E., Nellen D., Froesch B., Chatterjee S., Murone M., Züllig S., and Basler K. (2002) Wnt/wingless signaling requires BCL9/legless-mediated recruitment of pygopus to the nuclear β-catenin–TCF complex. Cell 109, 47–60 10.1016/S0092-8674(02)00679-7 [DOI] [PubMed] [Google Scholar]
- 27. de la Roche M., Rutherford T. J., Gupta D., Veprintsev D. B., Saxty B., Freund S. M., and Bienz M. (2012) An intrinsically labile α-helix abutting the BCL9-binding site of β-catenin is required for its inhibition by carnosic acid. Nat. Commun. 3, 680 10.1038/ncomms1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takada K., Zhu D., Bird G. H., Sukhdeo K., Zhao J. J., Mani M., Lemieux M., Carrasco D. E., Ryan J., Horst D., Fulciniti M., Munshi N. C., Xu W., Kung A. L., Shivdasani R. A., et al. (2012) Targeted disruption of the BCL9/β-catenin complex inhibits oncogenic Wnt signaling. Sci. Transl. Med. 4, 148ra117 10.1126/scitranslmed.3003808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yau R., and Rape M. (2016) The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586 10.1038/ncb3358 [DOI] [PubMed] [Google Scholar]
- 30. Yang S., Liu L., Cao C., Song N., Wang Y., Ma S., Zhang Q., Yu N., Ding X., Yang F., Tian S., Zhang K., Sun T., Yang J., Yao Z., Wu S., and Shi L. (2018) USP52 acts as a deubiquitinase and promotes histone chaperone ASF1A stabilization. Nat. Commun. 9, 1285 10.1038/s41467-018-03588-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Su D., Ma S., Shan L., Wang Y., Wang Y., Cao C., Liu B., Yang C., Wang L., Tian S., Ding X., Liu X., Yu N., Song N., Liu L., et al. (2018) Ubiquitin-specific protease 7 sustains DNA damage response and promotes cervical carcinogenesis. J. Clin. Invest. 128, 4280–4296 10.1172/JCI120518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murtaza M., Jolly L. A., Gecz J., and Wood S. A. (2015) La FAM fatale: USP9X in development and disease. Cell. Mol. Life Sci. 72, 2075–2089 10.1007/s00018-015-1851-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schwickart M., Huang X., Lill J. R., Liu J., Ferrando R., French D. M., Maecker H., O'Rourke K., Bazan F., Eastham-Anderson J., Yue P., Dornan D., Huang D. C., and Dixit V. M. (2010) Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature 463, 103–107 10.1038/nature08646 [DOI] [PubMed] [Google Scholar]
- 34. Das A., Qian J., and Tsang W. Y. (2017) USP9X counteracts differential ubiquitination of NPHP5 by MARCH7 and BBS11 to regulate ciliogenesis. PLoS Genet. 13, e1006791 10.1371/journal.pgen.1006791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Izrailit J., Jaiswal A., Zheng W., Moran M. F., and Reedijk M. (2017) Cellular stress induces TRB3/USP9x-dependent Notch activation in cancer. Oncogene 36, 1048–1057 10.1038/onc.2016.276 [DOI] [PubMed] [Google Scholar]
- 36. Overstreet E., Fitch E., and Fischer J. A. (2004) Fat facets and liquid facets promote Delta endocytosis and Delta signaling in the signaling cells. Development 131, 5355–5366 10.1242/dev.01434 [DOI] [PubMed] [Google Scholar]
- 37. Marx C., Held J. M., Gibson B. W., and Benz C. C. (2010) ErbB2 trafficking and degradation associated with Lys-48 and Lys-63 polyubiquitination. Cancer Res. 70, 3709–3717 10.1158/0008-5472.CAN-09-3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Savio M. G., Wollscheid N., Cavallaro E., Algisi V., Di Fiore P. P., Sigismund S., Maspero E., and Polo S. (2016) USP9X controls EGFR fate by deubiquitinating the endocytic adaptor Eps15. Curr. Biol. 26, 173–183 10.1016/j.cub.2015.11.050 [DOI] [PubMed] [Google Scholar]
- 39. Bridges C. R., Tan M. C., Premarathne S., Nanayakkara D., Bellette B., Zencak D., Domingo D., Gecz J., Murtaza M., Jolly L. A., and Wood S. A. (2017) USP9X deubiquitylating enzyme maintains RAPTOR protein levels, mTORC1 signalling and proliferation in neural progenitors. Sci. Rep. 7, 391 10.1038/s41598-017-00149-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Agrawal P., Chen Y. T., Schilling B., Gibson B. W., and Hughes R. E. (2012) Ubiquitin-specific peptidase 9, X-linked (USP9X) modulates activity of mammalian target of rapamycin (mTOR). J. Biol. Chem. 287, 21164–21175 10.1074/jbc.M111.328021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li X., Song N., Liu L., Liu X. H., Ding X., Song X., Yang S. D., Shan L., Zhou X., Su D. X., Wang Y., Zhang Q., Cao C., Ma S., Yu N., et al. (2017) USP9X regulates centrosome duplication and promotes breast carcinogenesis. Nat. Commun. 8, 14866 10.1038/ncomms14866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ouyang W., Zhang S., Yang B., Yang C., Zhang J., Zhou F., and Xie C. (2016) β-Catenin is regulated by USP9x and mediates resistance to TRAIL-induced apoptosis in breast cancer. Oncol. Rep. 35, 717–724 10.3892/or.2015.4463 [DOI] [PubMed] [Google Scholar]
- 43. Murray R. Z., Jolly L. A., and Wood S. A. (2004) The FAM deubiquitylating enzyme localizes to multiple points of protein trafficking in epithelia, where it associates with E-cadherin and β-catenin. Mol. Biol. Cell 15, 1591–1599 10.1091/mbc.e03-08-0630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Premarathne S., Murtaza M., Matigian N., Jolly L. A., and Wood S. A. (2017) Loss of Usp9x disrupts cell adhesion, and components of the Wnt and Notch signaling pathways in neural progenitors. Sci. Rep. 7, 8109 10.1038/s41598-017-05451-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Derksen P. W., Tjin E., Meijer H. P., Klok M. D., MacGillavry H. D., van Oers M. H., Lokhorst H. M., Bloem A. C., Clevers H., Nusse R., van der Neut R., Spaargaren M., and Pals S. T. (2004) Illegitimate WNT signaling promotes proliferation of multiple myeloma cells. Proc. Natl. Acad. Sci. U.S.A. 101, 6122–6127 10.1073/pnas.0305855101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun Y., Kolligs F. T., Hottiger M. O., Mosavin R., Fearon E. R., and Nabel G. J. (2000) Regulation of β-catenin transformation by the p300 transcriptional co-activator. Proc. Natl. Acad. Sci. U.S.A. 97, 12613–12618 10.1073/pnas.220158597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Van der Flier L. G., Sabates-Bellver J., Oving I., Haegebarth A., De Palo M., Anti M., Van Gijn M. E., Suijkerbuijk S., Van de Wetering M., Marra G., and Clevers H. (2007) The intestinal Wnt/TCF signature. Gastroenterology 132, 628–632 10.1053/j.gastro.2006.08.039 [DOI] [PubMed] [Google Scholar]
- 48. Edwards C. M. (2008) Wnt signaling: bone's defense against myeloma. Blood 112, 216–217 10.1182/blood-2008-04-149278 [DOI] [PubMed] [Google Scholar]
- 49. Chim C. S., Pang R., Fung T. K., Choi C. L., and Liang R. (2007) Epigenetic dysregulation of Wnt-signaling pathway in multiple myeloma. Leukemia 21, 2527–2536 10.1038/sj.leu.2404939 [DOI] [PubMed] [Google Scholar]
- 50. van Andel H., Ren Z., Koopmans I., Joosten S. P., Kocemba K. A., de Lau W., Kersten M. J., de Bruin A. M., Guikema J. E., Clevers H., Spaargaren M., and Pals S. T. (2017) Aberrantly expressed LGR4 empowers Wnt signaling in multiple myeloma by hijacking osteoblast-derived R-spondins. Proc. Natl. Acad. Sci. U.S.A. 114, 376–381 10.1073/pnas.1618650114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li Y., Jin K., van Pelt G. W., van Dam H., Yu X., Mesker W. E., Ten Dijke P., Zhou F., and Zhang L. (2016) c-Myb enhances breast cancer invasion and metastasis through the Wnt/β-catenin/Axin2 pathway. Cancer Res. 76, 3364–3375 10.1158/0008-5472.CAN-15-2302 [DOI] [PubMed] [Google Scholar]
- 52. Jang G. B., Hong I. S., Kim R. J., Lee S. Y., Park S. J., Lee E. S., Park J. H., Yun C. H., Chung J. U., Lee K. J., Lee H. Y., and Nam J. S. (2015) Wnt/β-catenin small-molecule inhibitor CWP232228 preferentially inhibits the growth of breast cancer stem-like cells. Cancer Res. 75, 1691–1702 10.1158/0008-5472.CAN-14-2041 [DOI] [PubMed] [Google Scholar]
- 53. Sampietro J., Dahlberg C. L., Cho U. S., Hinds T. R., Kimelman D., and Xu W. (2006) Crystal structure of a β-catenin/BCL9/Tcf4 complex. Mol. Cell 24, 293–300 10.1016/j.molcel.2006.09.001 [DOI] [PubMed] [Google Scholar]
- 54. Miller T. C., Rutherford T. J., Johnson C. M., Fiedler M., and Bienz M. (2010) Allosteric remodelling of the histone H3 binding pocket in the Pygo2 PHD finger triggered by its binding to the B9L/BCL9 co-factor. J. Mol. Biol. 401, 969–984 10.1016/j.jmb.2010.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Thompson B., Townsley F., Rosin-Arbesfeld R., Musisi H., and Bienz M. (2002) A new nuclear component of the Wnt signalling pathway. Nat. Cell Biol. 4, 367–373 10.1038/ncb786 [DOI] [PubMed] [Google Scholar]
- 56. Adachi S., Jigami T., Yasui T., Nakano T., Ohwada S., Omori Y., Sugano S., Ohkawara B., Shibuya H., Nakamura T., and Akiyama T. (2004) Role of a BCL9-related β-catenin-binding protein, B9L, in tumorigenesis induced by aberrant activation of Wnt signaling. Cancer Res. 64, 8496–8501 10.1158/0008-5472.CAN-04-2254 [DOI] [PubMed] [Google Scholar]
- 57. Brembeck F. H., Schwarz-Romond T., Bakkers J., Wilhelm S., Hammerschmidt M., and Birchmeier W. (2004) Essential role of BCL9–2 in the switch between β-catenin's adhesive and transcriptional functions. Genes Dev. 18, 2225–2230 10.1101/gad.317604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. de la Roche M., Worm J., and Bienz M. (2008) The function of BCL9 in Wnt/β-catenin signaling and colorectal cancer cells. BMC Cancer 8, 199 10.1186/1471-2407-8-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fischer-Vize J. A., Rubin G. M., and Lehmann R. (1992) The fat facets gene is required for Drosophila eye and embryo development. Development 116, 985–1000 [DOI] [PubMed] [Google Scholar]
- 60. Al-Hakim A. K., Zagorska A., Chapman L., Deak M., Peggie M., and Alessi D. R. (2008) Control of AMPK-related kinases by USP9X and atypical Lys(29)/Lys(33)-linked polyubiquitin chains. Biochem. J. 411, 249–260 10.1042/BJ20080067 [DOI] [PubMed] [Google Scholar]
- 61. Dupont S., Mamidi A., Cordenonsi M., Montagner M., Zacchigna L., Adorno M., Martello G., Stinchfield M. J., Soligo S., Morsut L., Inui M., Moro S., Modena N., Argenton F., Newfeld S. J., and Piccolo S. (2009) FAM/USP9x, a deubiquitinating enzyme essential for TGFβ signaling, controls Smad4 monoubiquitination. Cell 136, 123–135 10.1016/j.cell.2008.10.051 [DOI] [PubMed] [Google Scholar]
- 62. Wu Y., Yu X., Yi X., Wu K., Dwabe S., Atefi M., Elshimali Y., Kemp K. T. 2nd, Bhat K., Haro J., Sarkissyan M., and Vadgama J. V. (2017) Aberrant phosphorylation of SMAD4 Thr277-mediated USP9x-SMAD4 interaction by free fatty acids promotes breast cancer metastasis. Cancer Res. 77, 1383–1394 10.1158/0008-5472.CAN-16-2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li X., Liu L., Yang S., Song N., Zhou X., Gao J., Yu N., Shan L., Wang Q., Liang J., Xuan C., Wang Y., Shang Y., and Shi L. (2014) Histone demethylase KDM5B is a key regulator of genome stability. Proc. Natl. Acad. Sci. U.S.A. 111, 7096–7101 10.1073/pnas.1324036111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nusse R., and Clevers H. (2017) Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 169, 985–999 10.1016/j.cell.2017.05.016 [DOI] [PubMed] [Google Scholar]
- 65. Clevers H., and Nusse R. (2012) Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 10.1016/j.cell.2012.05.012 [DOI] [PubMed] [Google Scholar]
- 66. Xu J., Burgoyne P. S., and Arnold A. P. (2002) Sex differences in sex chromosome gene expression in mouse brain. Hum. Mol. Genet. 11, 1409–1419 10.1093/hmg/11.12.1409 [DOI] [PubMed] [Google Scholar]
- 67. Xu J., Taya S., Kaibuchi K., and Arnold A. P. (2005) Sexually dimorphic expression of Usp9x is related to sex chromosome complement in adult mouse brain. Eur. J. Neurosci. 21, 3017–3022 10.1111/j.1460-9568.2005.04134.x [DOI] [PubMed] [Google Scholar]
- 68. Cerami E., Gao J., Dogrusoz U., Gross B. E., Sumer S. O., Aksoy B. A., Jacobsen A., Byrne C. J., Heuer M. L., Larsson E., Antipin Y., Reva B., Goldberg A. P., Sander C., and Schultz N. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., and Schultz N. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 10.1126/scisignal.2004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wang Y., Zhang H., Chen Y., Sun Y., Yang F., Yu W., Liang J., Sun L., Yang X., Shi L., Li R., Li Y., Zhang Y., Li Q., Yi X., and Shang Y. (2009) LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138, 660–672 10.1016/j.cell.2009.05.050 [DOI] [PubMed] [Google Scholar]
- 71. Zhang H., Yi X., Sun X., Yin N., Shi B., Wu H., Wang D., Wu G., and Shang Y. (2004) Differential gene regulation by the SRC family of co-activators. Genes Dev. 18, 1753–1765 10.1101/gad.1194704 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.