

Abstract
Protein substrates are targeted to the 26S proteasome through several ubiquitin receptors. One of these receptors, RPN13, is recruited to the proteasome by binding of its N-terminal pleckstrin-like receptor of ubiquitin (PRU) domain to C-terminal residues of the scaffolding protein RPN2. The RPN13 PRU domain is followed by a flexible linker and a C-terminal deubiquitylase adaptor (DEUBAD) domain, which recruits and activates the deubiquitylase UCH37. Both RPN13 and UCH37 have been implicated in human cancers, and inhibitors of the RPN2–RPN13 interaction are being developed as potential therapeutic anticancer agents. Our current study builds on the recognition that a residue central to the RPN2–RPN13 interaction, RPN2 Tyr-950, is phosphorylated in Jurkat cells. We found that the Tyr-950 phosphorylation enhances binding to RPN13. The crystal structure of the RPN2–RPN13 pTyr-950–ubiquitin complex was determined at 1.76-Å resolution and reveals specific interactions with positively charged side chains in RPN13 that explain how phosphorylation increases binding affinity without inducing conformational change. Mutagenesis and quantitative binding assays were then used to validate the crystallographic interface. Our findings support a model in which RPN13 recruitment to the proteasome is enhanced by phosphorylation of RPN2 Tyr-950, have important implications for efforts to develop specific inhibitors of the RPN2–RPN13 interaction, and suggest the existence of a previously unknown stress-response pathway.
Keywords: proteasome, ubiquitin, phosphotyrosine, phosphorylation, protein structure, protein-protein interaction, adhesion regulating molecule 1 (ADRM1), fluorescence polarization, RPN13, RPN2
Introduction
The ubiquitin–proteasome system is a complex network of proteins responsible for the regulated turnover of proteins in eukaryotes (1). Ubiquitin–proteasome system pathways converge on the 26S proteasome, a ∼2.5-MDa complex that selectively degrades targeted cellular proteins. The 26S proteasome comprises a central 20S core particle, which houses the proteolytic sites, and is capped on one or both ends by the 19S regulatory particle, which includes proteins that recognize, process, and translocate targeted proteins into the core particle for degradation (2). Substrates are typically targeted to the proteasome by conjugation to the small protein ubiquitin, which is recognized by the proteasome-associated ubiquitin receptors RPN1, RPN10, and RPN13 (ADRM1 in humans), or by transiently associated ubiquitin shuttles such as RAD23 (HHR23A/B in humans), DSK2 (PLIC2 in humans), and DDI1 (3–9).
The ubiquitin receptor RPN13 is one of the most commonly overexpressed genes in solid tumors (10), and increases in cellular levels of RPN13 have been found in leukemia (11), hepatocellular carcinoma (12), ovarian cancer (13–15), and gastric/colorectal cancers (16–18). RPN13 also recruits and activates the proteasome-associated deubiquitylase UCH37 (UCH-L5) (19–21), which is also found at high levels in esophageal cancer (22), ovarian cancer (23), colorectal cancer (24), multiple myeloma (25), and hepatocellular carcinoma (26). As a result of these observations, RPN13 has become the focus of recent efforts toward development of anticancer therapeutics (27–30), placing additional emphasis on understanding the mechanism and regulation of RPN13 recruitment to the proteasome.
Recent publications describe the structural and biochemical basis of RPN13 recruitment to the proteasome by the scaffolding protein RPN2 (31–34), including details of the interaction between the C-terminal residues of RPN2 (RPN2940–953) and the RPN13 PRU2 domain (residues 1–132; RPN13PRU). Three residues (Pro-945, Phe-948, and Tyr-950) make major contributions to the ∼10 nm binding affinity observed for the RPN2–RPN13 interaction (31). Interestingly, this affinity does not ensure complete occupancy of RPN13 on the proteasome as endogenously purified 26S proteasomes from both yeast and humans possess substoichiometric ratios of RPN13 (33, 35).
When considering the possibility that the RPN2–RPN13 interaction might be regulated by posttranslational modification, we used the publicly accessible PhosphoSitePlus database (www.phosphosite.org)3 (36) to find two curated mass proteomics data sets that report phosphorylation of RPN2 Tyr-950, CST Curation Sets 9181 and 12496. Because Tyr-950 is central to the RPN2–RPN13 interaction and its mutation to alanine reduces affinity by ∼100-fold (31), we hypothesized that its phosphorylation may modulate the affinity of RPN2–RPN13 association. To test this possibility, we performed quantitative binding studies, which revealed an ∼8-fold increase in affinity for RPN2 peptide when phosphorylated on Tyr-950, and determined a crystal structure of the ternary RPN13PRU–RPN2940–952,pTyr950–ubiquitin complex at 1.76-Å resolution, which indicated that the increase in affinity results from interactions of the phosphate group with positively charged RPN13 side chains. Finally, the importance of specific RPN13 residues for the interaction with pTyr-950 peptide was validated by quantification of binding affinity.
Results
Phosphorylation of RPN2 pTyr-950 enhances affinity for RPN13
The RPN2940–952–RPN13PRU binding interaction was quantified using fluorescence polarization (FP) assays. For these studies, RPN13PRU was recombinantly expressed and purified. Peptides corresponding to RPN2940–952 and RPN2940–952,pTyr950were chemically synthesized and C-terminally labeled with fluorescein. Consistent with previous data (31), RPN13 bound the unphosphorylated peptide with a KD of 7.15 ± 0.27 nm (Fig. 1, black). In contrast, the RPN2940–952,pTyr950 peptide bound RPN13PRU with a KD of 0.90 ± 0.07 nm under identical conditions (Fig. 1, red). This indicates that the Tyr-950 phosphate causes an ∼8-fold increase in binding affinity for RPN13PRU.
Figure 1.
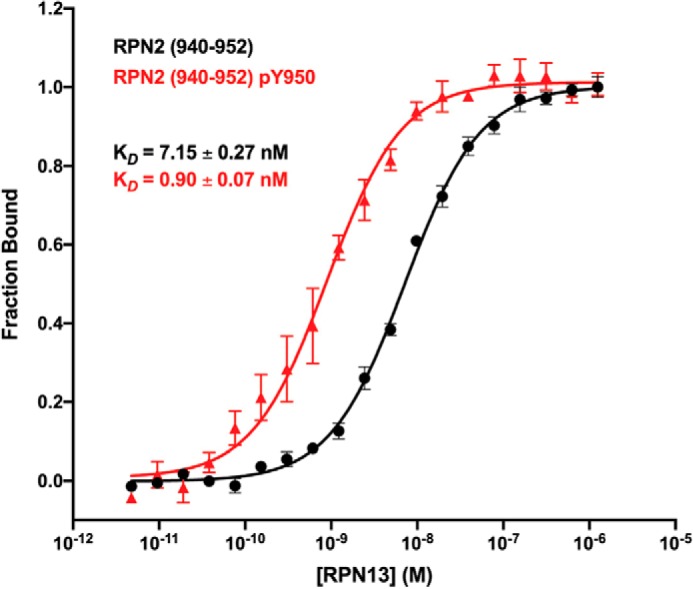
Binding isotherms. Fluorescence polarization binding curves of RPN13PRU binding to RPN2940–952 (black) and to RPN2940–952,pTyr950 (red) are shown. Error bars represent S.E.
Crystal structure of the RPN2940–952,pTyr950–RPN13PRU–ubiquitin complex
To determine the structural basis for the additional binding energy of the phosphotyrosine-containing peptide, we crystallized a ternary RPN2940–952,pTyr950–RPN13PRU–ubiquitin complex and collected X-ray diffraction data to 1.76 Å. Crystals were grown in conditions identical to our previously reported RPN2940–953–RPN13PRU–ubiquitin structure (31), which differs only within the RPN2 peptide. Specifically, the RPN2940–952pTyr-950 peptide used in the current structure lacks one residue, Asp-953, which was unresolved in the earlier structure, and possesses the phosphate on Tyr-950. Interestingly, crystals of the phosphorylated complex grew in a different space group (P31 versus P21), indicating that the two crystal structures are not isomorphous. The structure was determined by molecular replacement using the binary RPN13PRU–ubiquitin complex from the unphosphorylated structure (PDB code 5V1Y) (31) as the search model and was refined to Rwork/Rfree values of 0.173/0.207 (Fig. 2 and Table 1). The two ternary complexes in the asymmetric unit are nearly identical, superimposing with an all-atom root mean square deviation (r.m.s.d.). of 0.45 Å. For both complexes, the main chain is clearly defined for RPN13 residues 21–88, 90–99, and 102–131; RPN2 residues 940–950; and ubiquitin residues 1–75. Residues lacking density in RPN13 are found in mobile loops (Pro-89, Ala-100, and Gly-101) or termini (Ser-19, Asn-20, and Pro-132). Importantly, density was clearly defined for the RPN2 pTyr-950 phosphate in both complexes in the asymmetric unit (Fig. 2B). Due to the similarity between the two complexes, figures and calculations are based on the complex comprising chains B (RPN13PRU), D (ubiquitin), and F (RPN2940–952,pTyr950).
Figure 2.
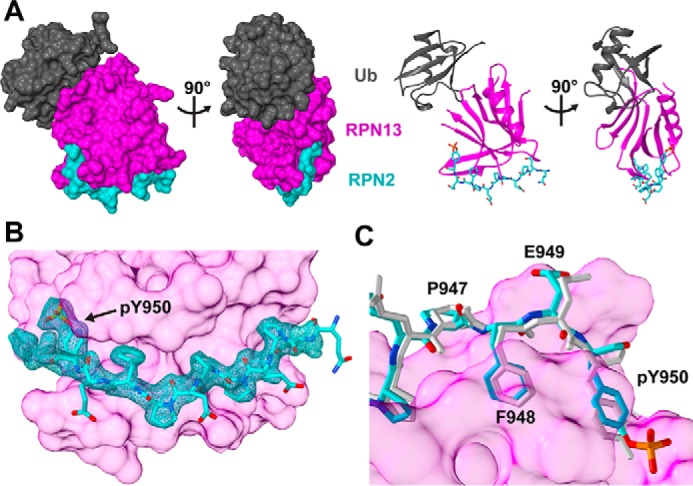
Structure of the RPN2940–952,pTyr950–RPN13PRU–ubiquitin complex. A, orthogonal views of the complex in space-filling (left) and ribbon/stick (right) representations. RPN13PRU, magenta; RPN2940–952,pTyr950, cyan; ubiquitin (Ub), gray. B, detailed view of the RPN2 peptide at the RPN13 interface. Shown is the Fo − Fc map (cyan mesh; 1.88 r.m.s.d.) within 1.8 Å of RPN2 peptide. The map was calculated by removing RPN2 residues (chain F), introducing random shifts to all remaining atoms, and performing a single round of refinement. C, comparison of RPN2 architecture for residues Pro-947 to Tyr-950 in the phosphorylated structure (cyan) and the unphosphorylated structure (white).
Table 1.
Crystallographic statistics
Values in parentheses are for data in the high-resolution shell.
| Data collection | |
|---|---|
| Space group | P31 |
| Cell dimensions | |
| a, b, c (Å) | 99.4, 99.4, 41.7 |
| α, β, γ (°) | 90.0, 90.0, 120.0 |
| Resolution (Å) | 40.0–1.76 (1.82–1.76) |
| Rpim (%) | 6.9 (66.3) |
| I/σI | 12.8 (1.3) |
| Completeness (%) | 0.99 (0.98) |
| Redundancy | 9.2 (7.7) |
| Refinement | |
| Resolution (Å) | 30.0–1.75 (1.81–1.75) |
| No. of reflections | 46,327 (4,392) |
| Rwork/Rfree | 0.173 (0.278)/0.207 (0.263) |
| No. of atoms | |
| Protein | 3,309 |
| Water | 248 |
| B-factors | |
| Protein | 32.7 |
| Water | 40.9 |
| r.m.s.d. | |
| Bond lengths (Å) | 0.011 |
| Bond angles (°) | 1.157 |
Comparison with our previously reported RPN2940–953–RPN13PRU–ubiquitin structure (PDB code 5V1Y) revealed that phosphorylation of RPN2 Tyr-950 does not cause substantial changes to the overall structure. Structural alignment revealed an r.m.s.d. of 0.85 Å over backbone atoms of the 184 residues present in both structures and an r.m.s.d. of 1.36 Å over all atoms, indicating that RPN2 Tyr-950 phosphorylation does not alter the geometry of RPN2–RPN13 association or of ubiquitin binding.
The RPN2 pTyr-950–RPN13 interaction
Only small differences in RPN2 are observed between the phosphorylated and unphosphorylated structures. The largest differences occur near the peptide termini and include an ∼1.2-Å shift of Glu-941 backbone atoms and an ∼1.0-Å shift of Glu-949 and Tyr-950 backbone atoms. These differences include a slight change in the curvature of the RPN2 peptide from Pro-947 to Tyr-950 (Fig. 2C). Nevertheless, the r.m.s.d. of all RPN2 backbone atoms is only 0.53 Å, and the overall structure of the RPN13-bound RPN2 peptide is maintained upon phosphorylation. Hydrogen bonding interactions of backbone atoms of RPN2 Phe-948 with RPN13 Val-48 and of RPN2 Tyr-950 with RPN13 Thr-36 are conserved in the unphosphorylated and phosphorylated structures. Similarly, the salt bridge of RPN2 Glu-941 with RPN13 Arg-64 and hydrogen bonding of RPN2 Glu-949 with RPN13 Thr-36 and Thr-37 are also conserved. Interestingly, a salt bridge between RPN2 Glu-941 and RPN13 Arg-27, which was observed in the unphosphorylated complex, is no longer observed in the phosphorylated structure.
Coordination of the RPN2 pTyr-950 phosphate
Well-defined electron density was observed for the RPN2 pTyr-950 phosphate (Fig. 3A), which is seen to coordinate the side chains of RPN13 Lys-97 and Arg-104, as well as a molecule of ethylene glycol (cryoprotectant). The Lys-97 side chain amine lies within hydrogen-bonding distances of two phosphate oxygens (3.1 and 3.2 Å) (Fig. 3B). The ϵ-amine of RPN13 Arg-104 forms hydrogen bonds with the RPN2 pTyr-950 phosphate and η-oxygens (2.6 and 2.9 Å), and an RPN13 R104 η-amine forms a single hydrogen bond with a pTyr-950 phosphate oxygen (3.2 Å). Our model also suggests that additional phosphate coordination might be provided by RPN13 Lys-83, which is near RPN2 pTyr-950, but lacks observable density beyond the β-carbon (Fig. 3A). Modeling of several lysine rotamers (Fig. 3B) indicates that Lys-83 may occupy states that lie within hydrogen-bonding distance of the RPN2 pTyr-950 phosphate. Moreover, close approach of Lys-83 to the phosphate is prohibited in the crystal by the coordinating ethylene glycol molecule, which would be absent under physiological conditions.
Figure 3.
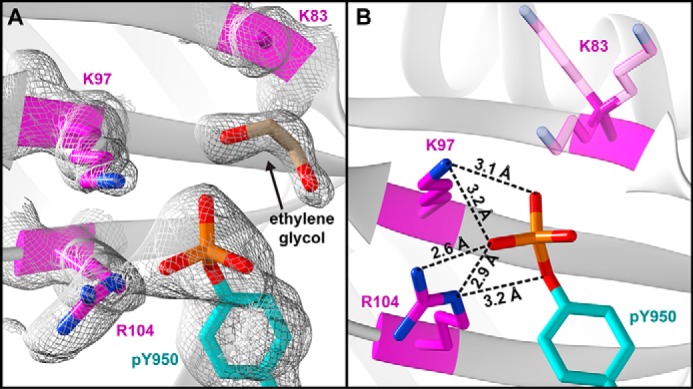
Coordination of pTyr-950. A, electron density (white mesh; 0.8 r.m.s.d.) surrounding pTyr-950. B, coordination distances to phosphate oxygen atoms. RPN13 Lys-83 side chain (transparent) lacks density, but is shown in three frequently observed rotamers.
In addition to the side-chain interactions among RPN2 pTyr-950 and RPN13 Lys-97 and Arg-104, we observed differences in the solvent structure within the RPN2 pTyr-950–binding pocket. Specifically, when unphosphorylated, the side chain of RPN2 Tyr-950 forms a coordinated network with water molecules that bridge its η-hydroxyl group with the side chains of RPN13 residues Lys-97 and Arg-104 (31). In the structure reported here, those waters have been displaced by the phosphate of RPN2 pTyr-950.
Mutation of coordinating residues abrogates preferential binding of RPN2 pTyr-950
To verify our structural model, additional FP assays were performed using RPN13 constructs with mutations in residues that coordinate RPN2 pTyr-950. Specifically, residues Lys-97 and Arg-104 of RPN13 were independently mutated to alanine, aspartic acid, and glutamic acid.
In agreement with the crystal structure, K97A and R104A mutations in RPN13 disrupt the ∼8-fold preferential binding of phosphorylated RPN2 peptide (Table 2). Specifically, RPN13 K97A binds RPN2940–952 with a KD of 18.0 ± 0.07 nm, whereas it binds RPN2940–952,pTyr950 with a KD of 11.2 ± 0.9 nm, thereby displaying a modest ∼1.6-fold preference for the phosphorylated peptide. An even stronger effect was seen for RPN13 R104A, which bound unphosphorylated peptide slightly tighter than RPN2940–952,pTyr950, with observed KD values of 16.8 ± 0.9 and 27.2 ± 0.15 nm, respectively.
Table 2.
Binding statistics
| RPN13 construct | RPN2940–952 |
RPN2940–952,pTyr950 |
pTyr preference (-fold change) | ||
|---|---|---|---|---|---|
| KD | -Fold change | KD | -Fold change | ||
| nm | nm | ||||
| Wildtype | 7.2 ± 0.3 | 0.9 ± 0.09 | 8.0 | ||
| K83A | 8.9 ± 0.3 | 1.2 | 9.4 ± 0.7 | 10.4 | 0.9 |
| K97A | 18.0 ± 0.7 | 2.5 | 11.2 ± 0.9 | 12.4 | 1.6 |
| K97D | 67.0 ± 3.5 | 9.3 | 427.3 ± 72.8 | 474.8 | 0.2 |
| K97E | 21.4 ± 1.3 | 3.0 | 262.5 ± 31.4 | 58.2 | 0.1 |
| R104A | 16.8 ± 0.9 | 2.3 | 27.2 ± 1.5 | 30.2 | 0.6 |
| R104D | 42.6 ± 3.7 | 5.9 | 188.9 ± 26.6 | 209.9 | 0.2 |
| R104E | 66.2 ± 6.0 | 9.2 | 1115 ± 341 | 1238.9 | 0.1 |
To further validate the structural model, the positively charged residues that coordinate RPN2 pTyr-950 were replaced with the negatively charged residues aspartate and glutamate. Each of the K97D, K97E, R104D, and R104E mutants modestly disrupted binding of the unphosphorylated peptide (Table 2, column 3). In every case, however, binding of the phosphorylated peptide was substantially impaired (Table 2, column 5), consistent with the anticipated charge repulsion. The most severe binding defects occurred in RPN13 K97D (KD = 427.3 ± 72.8 nm) and RPN13 R104E (KD = 1115 ± 341 nm) for which binding was disrupted ∼500- and ∼1200-fold, respectively. In addition to greatly weakening binding, these mutations reverse the binding preference such that the unphosphorylated peptide bound ∼5–10-fold more tightly than the phosphorylated peptide (Table 2, column 6).
RPN13 Lys-83 contributes to binding of pTyr-950
In addition to residues that directly coordinate RPN2 pTyr-950 in the crystal structure, we propose that Lys-83 coordinates pTyr-950 under physiological conditions that lack ethylene glycol. To test this possibility, binding studies were performed using the RPN13 K83A mutant, which revealed that the binding of unphosphorylated peptide is essentially unchanged (KD = 8.9 ± 0.5 nm) but that the phosphorylated peptide is no longer preferentially bound (KD = 9.4 ± 0.7 nm) (Table 2). These observations verify that Lys-83 contributes to the additional binding energy observed upon phosphorylation of Tyr-950.
Discussion
Previous work mapped the RPN13-binding epitope within RPN2 (31–34), described the molecular determinants of binding between RPN13PRU and the RPN2 C-terminal peptide (31, 34), and described how RPN13PRU is auto-ubiquitylated and restricts degradation of ubiquitylated protein substrates in impaired proteasomes (38). In this work, we advance the model of RPN13 regulation by describing a phosphorylation “switch” within RPN2 that might regulate recruitment of RPN13 to the proteasome.
Defining the precise physiological roles of RPN13 is complicated by the functional redundancy it shares with the proteasome ubiquitin receptors RPN1 and RPN10 (39–42). Although the down-regulation or loss of RPN13 does not impact the bulk turnover of ubiquitylated proteins, certain physiological substrates of RPN13 have been identified, including the NF-κB inhibitor IκBα (43). NF-κB is best known for its role in inflammation, which fosters multiple hallmarks of cancer (44); impacts most, if not all, stages of tumorigenesis (45); and has been described as the “matchmaker” that connects inflammation to cancers through expression of genes that promote survival, proliferation, and metastasis (46–49). Increased levels of RPN13 result in enhanced rates of IκBα degradation, thus releasing inhibition of the NF-κB signaling pathway. In addition, it has been established that increased cellular levels of RPN13 correlate with the onset and progression of several cancers (11–18, 28).
RPN13 is observed at substoichiometric ratios in endogenously purified proteasomes (33, 35), suggesting that under typical cell culture conditions the RPN13-binding epitope in RPN2 is unsaturated. Thus, Tyr-950 phosphorylation may serve to increase the number of RPN13-associated proteasomes. A related possibility is that phosphorylation decreases the off-rate for RPN13 binding, which may be a factor in optimizing association with substrates that are slow to engage with the proteasome ATPases or are at risk of stalling and premature release. In either case, it is likely that phosphorylation regulates RPN2–RPN13 interactions within the proteasome. Future efforts will be required to definitively validate and quantify this possibility, including quantification of the endogenous pools of free and proteasome-associated RPN2 pTyr-950 as well as the identification of the stress-response pathways, kinases, and phosphatases that modulate it.
Our findings have implications for the development of anticancer therapeutics targeting the RPN2–RPN13 interaction (27–30). In particular, the identification of a specific phosphate-binding site will inform efforts to develop inhibitors mimicking the RPN2 interaction. Moreover, recognizing that phosphorylation enhances binding imposes limits on the affinity required for an inhibitor to effectively block the interaction in vivo. In addition to the inhibitor/drug development interest, important future goals include identifying cellular conditions associated with altered phosphorylation of RPN2 Tyr-950 and determining whether this modification is associated with a cellular pathway that may function in protein quality control, a stress response, or other facet of cellular regulation.
Experimental procedures
Protein expression and purification
All expression plasmids have been submitted to the Addgene plasmid repository (plasmid numbers 73742 and 125823–125829). Recombinant RPN13 proteins were expressed and purified as described previously (31). Briefly, cell pellets were resuspended in GS-BIND buffer (20 mm Tris-HCl, pH 7.5, 50 mm NaCl, 1 mm DTT, 1 mm EDTA) supplemented with protease inhibitors (Sigma) and incubated with 1 mg/ml lysozyme for 1 h at 4 °C prior to sonication. Lysis by sonication was followed by clarification by centrifugation. RPN13 constructs were purified by 1) GSH-Sepharose (GS) resin, 2) on-column HRV3C protease cleavage to remove glutathione S-transferase, and 3) gel-filtration chromatography.
Specifically, clarified lysate was incubated with pre-equilibrated GS resin (GE Healthcare) for 4 h. After washing the resin twice with 10 column volumes of GS-BIND buffer, bound protein was incubated with HRV3C protease in 5 column volumes of GS-BIND buffer at 4 °C overnight before elution. Eluted protein was analyzed by SDS-PAGE, concentrated with a 3,000 molecular weight–cutoff Vivaspin centrifugal concentrator, and loaded onto an SD75 gel filtration column (GE Healthcare) equilibrated with SEC buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm DTT) for final purification. Purified protein was analyzed by SDS-PAGE, concentrated, and stored in SEC buffer with 10% glycerol at −80 °C. Recombinant ubiquitin was expressed and purified as described previously (50).
Peptide synthesis
Peptides were synthesized on a Prelude or Prelude X instrument (Gyros Protein Technologies) using Fmoc (N-(9-fluorenyl)methoxycarbonyl) solid-phase peptide synthesis at 30-μmol scale. Deprotection cycles employed three treatments of 2 ml of 20% piperidine in DMF for 3 min followed by three washes for 30 s using 2 ml of DMF. Coupling cycles consisted of addition of 0.65 ml of 200 mm amino acid in N-methylpyrrolidinone, 0.65 ml of 195 mm HATU in DMF, and 0.5 ml of 600 mm 4-methylmorpholine in DMF. Resin and coupling reagents were then mixed using nitrogen for 25 min at room temperature before being washed three times with 2 ml of DMF. TentaGel R RAM resin (loading density, 0.19 mmol/g) was utilized for the synthesis of C-terminal amides, and 2-chlorotrityl resin (∼0.20 mmol/g) was utilized for C-terminal acids.
N-terminal capping was achieved through the addition of 1 ml of acetic anhydride and 1 ml of 0.6 m 4-methylmorpholine/30 μmol of resin for 15 min. C-terminally labeled peptide was synthesized through the addition of 5-(6)-carboxyfluorescein (Acros Organics) to a Lys ϵ-amine using standard coupling conditions. After completion of syntheses, peptide resins were thoroughly washed with methylene chloride and dried under vacuum.
Cleavage of peptide resins was achieved by 180-min agitation with 4 ml of TFA containing 2.5% water and 2.5% triisopropyl silane/30 μmol of peptide resin. The TFA solution was precipitated into 40 ml of ice-cold diethyl ether and centrifuged at 5,000 × g for 10 min. The supernatant was decanted, and pellets were triturated twice with ether before being dried under vacuum. Peptides were then HPLC-purified before lyophilization for long-term storage.
Formation of RPN2–RPN13 complex for crystallization
The RPN2–RPN13940–952,pTyr950 complex was formed by addition of a slight (1.2:1) molar excess of RPN2 peptide to RPN13 in SEC buffer followed by size-exclusion chromatography. RPN2–RPN13 complex was concentrated with a 3,000 molecular weight–cutoff Vivaspin centrifugal concentrator and loaded onto an SD75 gel filtration column equilibrated with crystallization buffer (20 mm HEPES, pH 7.2, 150 mm NaCl, 1 mm tris(2-carboxyethyl)phosphine, 1 mm EDTA). Eluted fractions were analyzed by SDS-PAGE and MS to confirm the presence of the RPN2 peptide, which is not easily visible by Coomassie staining.
Crystallization and structure determination
Purified RPN2-RPN13 complex was mixed in a 1:1.5 molar ratio with ubiquitin and concentrated to 17 mg/ml (E280 = 20,935 cm−1 m−1). The crystal complex grew at 4 °C from a 0.6-μl drop in a 1:2 ratio of protein and reservoir solutions (0.1 m citric acid, pH 4.6, 20% PEG-6000). Because the pKa2 of tyrosine phosphate is 5.8, crystals were soaked in reservoir solution at pH 6.0 with 25% ethylene glycol prior to plunging into liquid nitrogen to promote a more physiologically relevant state.
Diffraction data were collected on a Rigaku MicroMax-007HF rotating anode generator and RAXIS IV++ detector and processed using HKL2000 (51). Phases were determined by molecular replacement using PHASER (52) using the binary RPN13–ubiquitin complex from our previous structure (PDB code 5V1Y) (31) as the search model. Model building and refinement were performed using Coot (53) and PHENIX (54).
Model geometries were analyzed by MolProbity (55) within PHENIX. 98.4% of residues have favorable backbone dihedrals, and 1.6% fall into allowed regions.
Coordinates and structure factor amplitudes have been deposited at the PDB under the accession code 6OI4. Figures of molecular structures were generated using Chimera (37).
Fluorescence polarization
FP assays were performed on a Biotek Synergy Neo HTS Multi-Mode Microplate Reader using 485/528-nm excitation/emission wavelengths. Dilution series of purified RPN13 were made in FP assay buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1 mm DTT, 1 mg/ml casein, 0.005% Tween 20) in the presence of 100 pm (unphosphorylated complex) or 50 pm (phosphorylated complex) fluorescein-labeled RPN2 peptides. Dissociation constants (KD values) were calculated using GraphPad Prism 8 (GraphPad Software) by fitting raw polarization data to the following equation.
| (Eq. 1) |
Author contributions
C. W. H. and C. P. H. conceptualization; C. W. H. and S. C. H. data curation; C. W. H., S. C. H., and C. P. H. formal analysis; C. W. H. and C. P. H. validation; C. W. H. and C. P. H. investigation; C. W. H. and C. P. H. visualization; C. W. H. and C. P. H. methodology; C. W. H. and C. P. H. writing-original draft; C. W. H., S. C. H., and C. P. H. writing-review and editing; C. P. H. supervision; C. P. H. funding acquisition.
Acknowledgments
We thank Frank Whitby, Heidi Schubert, and Kath Ferrell for technical assistance and James Fulcher, Mark Peterson, and Michael Kay for peptide synthesis. Mass spectrometry validation of proteins was performed at the University of Utah Mass Spectrometry and Proteomics Core Facility, which is supported by National Institutes of Health Grant P30CA042014 from the NCI. Sequencing of constructs was performed at the DNA Sequencing Core Facility, University of Utah.
This work was supported by American Cancer Society Grant PF-16-100-01 (to C. W. H.) and NIGMS, National Institutes of Health Grant R21 CA191929 (to C. P. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 6OI4) have been deposited in the Protein Data Bank (http://wwpdb.org/).
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- PRU
- pleckstrin-like receptor of ubiquitin
- FP
- fluorescence polarization
- PDB
- Protein Data Bank
- r.m.s.d.
- room mean square deviation
- GS
- glutathione-Sepharose
- DMF
- N,N-dimethylformamide
- HATU
- O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate.
References
- 1. Pickart C. M., and Cohen R. E. (2004) Proteasomes and their kin: proteases in the machine age. Nat. Rev. Mol. Cell Biol. 5, 177–187 10.1038/nrm1336 [DOI] [PubMed] [Google Scholar]
- 2. Finley D. (2009) Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 78, 477–513 10.1146/annurev.biochem.78.081507.101607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schreiner P., Chen X., Husnjak K., Randles L., Zhang N., Elsasser S., Finley D., Dikic I., Walters K. J., and Groll M. (2008) Ubiquitin docking at the proteasome through a novel pleckstrin-homology domain interaction. Nature 453, 548–552 10.1038/nature06924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Husnjak K., Elsasser S., Zhang N., Chen X., Randles L., Shi Y., Hofmann K., Walters K. J., Finley D., and Dikic I. (2008) Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488 10.1038/nature06926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Elsasser S., Gali R. R., Schwickart M., Larsen C. N., Leggett D. S., Müller B., Feng M. T., Tübing F., Dittmar G. A., and Finley D. (2002) Proteasome subunit Rpn1 binds ubiquitin-like protein domains. Nat. Cell Biol. 4, 725–730 10.1038/ncb845 [DOI] [PubMed] [Google Scholar]
- 6. Rosenzweig R., Bronner V., Zhang D., Fushman D., and Glickman M. H. (2012) Rpn1 and Rpn2 coordinate ubiquitin processing factors at proteasome. J. Biol. Chem. 287, 14659–14671 10.1074/jbc.M111.316323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fatimababy A. S., Lin Y. L., Usharani R., Radjacommare R., Wang H. T., Tsai H. L., Lee Y., and Fu H. (2010) Cross-species divergence of the major recognition pathways of ubiquitylated substrates for ubiquitin/26S proteasome-mediated proteolysis. FEBS J. 277, 796–816 10.1111/j.1742-4658.2009.07531.x [DOI] [PubMed] [Google Scholar]
- 8. Gomez T. A., Kolawa N., Gee M., Sweredoski M. J., and Deshaies R. J. (2011) Identification of a functional docking site in the Rpn1 LRR domain for the UBA-UBL domain protein Ddi1. BMC Biol. 9, 33 10.1186/1741-7007-9-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saeki Y., Sone T., Toh-e A., and Yokosawa H. (2002) Identification of ubiquitin-like protein-binding subunits of the 26S proteasome. Biochem. Biophys. Res. Commun. 296, 813–819 10.1016/S0006-291X(02)02002-8 [DOI] [PubMed] [Google Scholar]
- 10. Pilarsky C., Wenzig M., Specht T., Saeger H. D., and Grützmann R. (2004) Identification and validation of commonly overexpressed genes in solid tumors by comparison of microarray data. Neoplasia 6, 744–750 10.1593/neo.04277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng X., Guo Y., Chen Y., Chen M., Lin Z., Wu Y., and Chen Y. (2015) Knockdown of adhesion-regulating molecule 1 inhibits proliferation in HL60 cells. Acta Haematol. 134, 88–100 10.1159/000369916 [DOI] [PubMed] [Google Scholar]
- 12. Yang X., Miao X., Wen Y., Hu J., Dai W., and Yin B. (2012) A possible connection between adhesion regulating molecule 1 overexpression and nuclear factor κB activity in hepatocarcinogenesis. Oncol. Rep. 28, 283–290 10.3892/or.2012.1767 [DOI] [PubMed] [Google Scholar]
- 13. Fejzo M. S., Dering J., Ginther C., Anderson L., Ramos L., Walsh C., Karlan B., and Slamon D. J. (2008) Comprehensive analysis of 20q13 genes in ovarian cancer identifies ADRM1 as amplification target. Genes Chromosomes Cancer 47, 873–883 10.1002/gcc.20592 [DOI] [PubMed] [Google Scholar]
- 14. Fejzo M. S., Anderson L., von Euw E. M., Kalous O., Avliyakulov N. K., Haykinson M. J., Konecny G. E., Finn R. S., and Slamon D. J. (2013) Amplification target ADRM1: role as an oncogene and therapeutic target for ovarian cancer. Int. J. Mol. Sci. 14, 3094–3109 10.3390/ijms14023094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fejzo M. S., Ginther C., Dering J., Anderson L., Venkatesan N., Konecny G., Karlan B., and Slamon D. J. (2011) Knockdown of ovarian cancer amplification target ADRM1 leads to downregulation of GIPC1 and upregulation of RECK. Genes Chromosomes Cancer 50, 434–441 10.1002/gcc.20868 [DOI] [PubMed] [Google Scholar]
- 16. Jang S. H., Park J. W., Kim H. R., Seong J. K., and Kim H. K. (2014) ADRM1 gene amplification is a candidate driver for metastatic gastric cancers. Clin. Exp. Metastasis 31, 727–733 10.1007/s10585-014-9663-4 [DOI] [PubMed] [Google Scholar]
- 17. Fejzo M. S., Anderson L., Chen H. W., Anghel A., Zhuo J., Anchoori R., Roden R., and Slamon D. J. (2015) ADRM1-amplified metastasis gene in gastric cancer. Genes Chromosomes Cancer 54, 506–515 10.1002/gcc.22262 [DOI] [PubMed] [Google Scholar]
- 18. Chen W., Hu X. T., Shi Q. L., Zhang F. B., and He C. (2009) Knockdown of the novel proteasome subunit Adrm1 located on the 20q13 amplicon inhibits colorectal cancer cell migration, survival and tumorigenicity. Oncol. Rep. 21, 531–537 10.3892/or_00000254 [DOI] [PubMed] [Google Scholar]
- 19. Yao T., Song L., Xu W., DeMartino G. N., Florens L., Swanson S. K., Washburn M. P., Conaway R. C., Conaway J. W., and Cohen R. E. (2006) Proteasome recruitment and activation of the Uch37 deubiquitinating enzyme by Adrm1. Nat. Cell Biol. 8, 994–1002 10.1038/ncb1460 [DOI] [PubMed] [Google Scholar]
- 20. Vander Linden R. T., Hemmis C. W., Schmitt B., Ndoja A., Whitby F. G., Robinson H., Cohen R. E., Yao T., and Hill C. P. (2015) Structural basis for the activation and inhibition of the UCH37 deubiquitylase. Mol. Cell 57, 901–911 10.1016/j.molcel.2015.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sahtoe D. D., van Dijk W. J., El Oualid F., Ekkebus R., Ovaa H., and Sixma T. K. (2015) Mechanism of UCH-L5 activation and inhibition by DEUBAD domains in RPN13 and INO80G. Mol. Cell 57, 887–900 10.1016/j.molcel.2014.12.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen Y., Fu D., Xi J., Ji Z., Liu T., Ma Y., Zhao Y., Dong L., Wang Q., and Shen X. (2012) Expression and clinical significance of UCH37 in human esophageal squamous cell carcinoma. Dig. Dis. Sci. 57, 2310–2317 10.1007/s10620-012-2181-9 [DOI] [PubMed] [Google Scholar]
- 23. Wang L., Chen Y. J., Xu K., Wang Y. Y., Shen X. Z., and Tu R. Q. (2014) High expression of UCH37 is significantly associated with poor prognosis in human epithelial ovarian cancer. Tumour Biol. 35, 11427–11433 10.1007/s13277-014-2446-3 [DOI] [PubMed] [Google Scholar]
- 24. Arpalahti L., Hagström J., Mustonen H., Lundin M., Haglund C., and Holmberg C. I. (2017) UCHL5 expression associates with improved survival in lymph-node-positive rectal cancer. Tumour Biol. 39, 1010428317716078 10.1177/1010428317716078 [DOI] [PubMed] [Google Scholar]
- 25. Tian Z., D'Arcy P., Wang X., Ray A., Tai Y. T., Hu Y., Carrasco R. D., Richardson P., Linder S., Chauhan D., and Anderson K. C. (2014) A novel small molecule inhibitor of deubiquitylating enzyme USP14 and UCHL5 induces apoptosis in multiple myeloma and overcomes bortezomib resistance. Blood 123, 706–716 10.1182/blood-2013-05-500033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fang Y., Fu D., Tang W., Cai Y., Ma D., Wang H., Xue R., Liu T., Huang X., Dong L., Wu H., and Shen X. (2013) Ubiquitin C-terminal hydrolase 37, a novel predictor for hepatocellular carcinoma recurrence, promotes cell migration and invasion via interacting and deubiquitinating PRP19. Biochim. Biophys. Acta 1833, 559–572 10.1016/j.bbamcr.2012.11.020 [DOI] [PubMed] [Google Scholar]
- 27. Trader D. J., Simanski S., and Kodadek T. (2015) A reversible and highly selective inhibitor of the proteasomal ubiquitin receptor rpn13 is toxic to multiple myeloma cells. J. Am. Chem. Soc. 137, 6312–6319 10.1021/jacs.5b02069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Soong R. S., Anchoori R. K., Yang B., Yang A., Tseng S. H., He L., Tsai Y. C., Roden R. B., and Hung C. F. (2016) RPN13/ADRM1 inhibitor reverses immunosuppression by myeloid-derived suppressor cells. Oncotarget 7, 68489–68502 10.18632/oncotarget.12095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Anchoori R. K., Karanam B., Peng S., Wang J. W., Jiang R., Tanno T., Orlowski R. Z., Matsui W., Zhao M., Rudek M. A., Hung C. F., Chen X., Walters K. J., and Roden R. B. (2013) A bis-benzylidine piperidone targeting proteasome ubiquitin receptor RPN13/ADRM1 as a therapy for cancer. Cancer Cell 24, 791–805 10.1016/j.ccr.2013.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Anchoori R. K., Jiang R., Peng S., Soong R. S., Algethami A., Rudek M. A., Anders N., Hung C. F., Chen X., Lu X., Kayode O., Dyba M., Walters K. J., and Roden R. B. S. (2018) Covalent Rpn13-binding inhibitors for the treatment of ovarian cancer. ACS Omega 3, 11917–11929 10.1021/acsomega.8b01479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. VanderLinden R. T., Hemmis C. W., Yao T., Robinson H., and Hill C. P. (2017) Structure and energetics of pairwise interactions between proteasome subunits RPN2, RPN13, and ubiquitin clarify a substrate recruitment mechanism. J. Biol. Chem. 292, 9493–9504 10.1074/jbc.M117.785287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lu X., Nowicka U., Sridharan V., Liu F., Randles L., Hymel D., Dyba M., Tarasov S. G., Tarasova N. I., Zhao X. Z., Hamazaki J., Murata S., Burke T. R. Jr., and Walters K. J. (2017) Structure of the Rpn13-Rpn2 complex provides insights for Rpn13 and Uch37 as anticancer targets. Nat. Commun. 8, 15540 10.1038/ncomms15540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen X., Lee B. H., Finley D., and Walters K. J. (2010) Structure of proteasome ubiquitin receptor hRpn13 and its activation by the scaffolding protein hRpn2. Mol. Cell 38, 404–415 10.1016/j.molcel.2010.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lu X., Liu F., Durham S. E., Tarasov S. G., and Walters K. J. (2015) A high affinity hRpn2-derived peptide that displaces human Rpn13 from proteasome in 293T cells. PLoS One 10, e0140518 10.1371/journal.pone.0140518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. da Fonseca P. C., He J., and Morris E. P. (2012) Molecular model of the human 26S proteasome. Mol. Cell 46, 54–66 10.1016/j.molcel.2012.03.026 [DOI] [PubMed] [Google Scholar]
- 36. Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., and Skrzypek E. (2015) PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 10.1093/nar/gku1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 38. Besche H. C., Sha Z., Kukushkin N. V., Peth A., Hock E. M., Kim W., Gygi S., Gutierrez J. A., Liao H., Dick L., and Goldberg A. L. (2014) Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. EMBO J. 33, 1159–1176 10.1002/embj.201386906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hamazaki J., Sasaki K., Kawahara H., Hisanaga S., Tanaka K., and Murata S. (2007) Rpn10-mediated degradation of ubiquitinated proteins is essential for mouse development. Mol. Cell. Biol. 27, 6629–6638 10.1128/MCB.00509-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hamazaki J., Hirayama S., and Murata S. (2015) Redundant roles of Rpn10 and Rpn13 in recognition of ubiquitinated proteins and cellular homeostasis. PLoS Genet. 11, e1005401 10.1371/journal.pgen.1005401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szlanka T., Haracska L., Kiss I., Deák P., Kurucz E., Andó I., Virágh E., and Udvardy A. (2003) Deletion of proteasomal subunit S5a/Rpn10/p54 causes lethality, multiple mitotic defects and overexpression of proteasomal genes in Drosophila melanogaster. J. Cell Sci. 116, 1023–1033 10.1242/jcs.00332 [DOI] [PubMed] [Google Scholar]
- 42. Al-Shami A., Jhaver K. G., Vogel P., Wilkins C., Humphries J., Davis J. J., Xu N., Potter D. G., Gerhardt B., Mullinax R., Shirley C. R., Anderson S. J., and Oravecz T. (2010) Regulators of the proteasome pathway, Uch37 and Rpn13, play distinct roles in mouse development. PLoS One 5, e13654 10.1371/journal.pone.0013654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mazumdar T., Gorgun F. M., Sha Y., Tyryshkin A., Zeng S., Hartmann-Petersen R., Jørgensen J. P., Hendil K. B., and Eissa N. T. (2010) Regulation of NF-κB activity and inducible nitric oxide synthase by regulatory particle non-ATPase subunit 13 (Rpn13). Proc. Natl. Acad. Sci. U.S.A. 107, 13854–13859 10.1073/pnas.0913495107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hanahan D., and Weinberg R. A. (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 45. Grivennikov S. I., and Karin M. (2010) Inflammation and oncogenesis: a vicious connection. Curr. Opin. Genet. Dev. 20, 65–71 10.1016/j.gde.2009.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grivennikov S. I., Greten F. R., and Karin M. (2010) Immunity, inflammation, and cancer. Cell 140, 883–899 10.1016/j.cell.2010.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ben-Neriah Y., and Karin M. (2011) Inflammation meets cancer, with NF-κB as the matchmaker. Nat. Immunol. 12, 715–723 10.1038/ni.2060 [DOI] [PubMed] [Google Scholar]
- 48. Karin M., and Greten F. R. (2005) NF-κB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5, 749–759 10.1038/nri1703 [DOI] [PubMed] [Google Scholar]
- 49. Wu Y., Deng J., Rychahou P. G., Qiu S., Evers B. M., and Zhou B. P. (2009) Stabilization of snail by NF-κB is required for inflammation-induced cell migration and invasion. Cancer Cell 15, 416–428 10.1016/j.ccr.2009.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pickart C. M., and Raasi S. (2005) Controlled synthesis of polyubiquitin chains. Methods Enzymol. 399, 21–36 10.1016/S0076-6879(05)99002-2 [DOI] [PubMed] [Google Scholar]
- 51. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 52. McCoy A. J., Grosse-Kunstleve R. W., Storoni L. C., and Read R. J. (2005) Likelihood-enhanced fast translation functions. Acta Crystallogr. D Biol. Crystallogr. 61, 458–464 10.1107/S0907444905001617 [DOI] [PubMed] [Google Scholar]
- 53. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]