

Abstract
The DNA damage response (DDR) is an evolutionarily conserved process essential for cell survival. Previously, we found that decreased histone expression induces mitochondrial respiration, raising the question whether the DDR also stimulates respiration. Here, using oxygen consumption and ATP assays, RT-qPCR and ChIP-qPCR methods, and dNTP analyses, we show that DDR activation in the budding yeast Saccharomyces cerevisiae, either by genetic manipulation or by growth in the presence of genotoxic chemicals, induces respiration. We observed that this induction is conferred by reduced transcription of histone genes and globally decreased DNA nucleosome occupancy. This globally altered chromatin structure increased the expression of genes encoding enzymes of tricarboxylic acid cycle, electron transport chain, oxidative phosphorylation, elevated oxygen consumption, and ATP synthesis. The elevated ATP levels resulting from DDR-stimulated respiration drove enlargement of dNTP pools; cells with a defect in respiration failed to increase dNTP synthesis and exhibited reduced fitness in the presence of DNA damage. Together, our results reveal an unexpected connection between respiration and the DDR and indicate that the benefit of increased dNTP synthesis in the face of DNA damage outweighs possible cellular damage due to increased oxygen metabolism.
Keywords: DNA damage response, respiration, ATP, histone, chromatin, tricarboxylic acid cycle (TCA cycle) (Krebs cycle), energy metabolism, cell stress, dNTP, oxidative phosphorylation (OXPHOS)
Introduction
All cells face constant challenge to protect their genome integrity. Insults that can directly or indirectly damage DNA arise from inside as well as outside the cell, including normal metabolic processes, DNA replication, UV light and ionizing radiation, and chemical exposure (1–3). Because maintenance of genome stability is crucial for survival, cells have evolved a set of highly-conserved mechanisms to sense and repair damaged DNA, which are collectively referred to as the DNA damage response (DDR)4 (4, 5). A cascade of protein kinases known as the “checkpoint kinases” is the key component of the DDR (Fig. 1 and Table 1) (6–9). In the presence of damaged DNA, the sensor kinases (ATM/ATR in mammals and Tel1/Mec1 in budding yeast) become active and phosphorylate the effector kinases (CHK1 and CHK2 in mammals and Chk1p and Rad53p in budding yeast) (10). Rad53p, the yeast ortholog of CHK2, is an essential intermediate kinase in the checkpoint pathway, as it connects the upstream kinases and downstream effectors to mediate an array of cellular outcomes in response to DNA damage (6, 11, 12). One of the Rad53p targets is another checkpoint kinase Dun1p (13).
Figure 1.
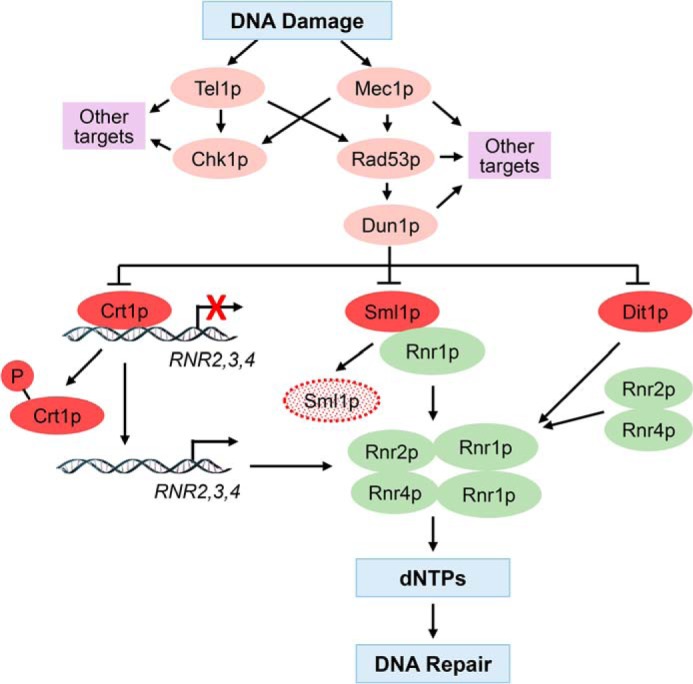
Model depicting the role of checkpoint kinases in DDR, RNR regulation, and synthesis of dNTPs. DNA damage activates the cascade of checkpoint kinases Mec1p, Rad53p, and Dun1p. Dun1p phosphorylates and down-regulates three negative regulators of the RNR complex: Crt1p, Sml1p, and Dif1p. Crt1p is a transcriptional repressor recruited to the RNR2, RNR3, and RNR4 genes. Phosphorylation of Crt1p derepresses RNR2, RNR3, and RNR4 genes by inducing dissociation of Crt1p from the corresponding promoters. Sml1p binds Rnr1p and inhibits RNR activity. Sml1p phosphorylation promotes its ubiquitylation and degradation by the 26S proteasome. Dif1p regulates nucleocytosolic distribution of Rnr2p and Rnr4p. Dun1p-mediated phosphorylation of Dif1p leads to redistribution of Rnr2p and Rnr4p from the nucleus to the cytoplasm, where Rnr1p resides, resulting in assembly of the active RNR complex. The cumulative effect of Dun1p activation is increased RNR assembly and activity and increased synthesis of dNTPs.
Table 1.
Genes/proteins used in this study
| Gene | Function |
|---|---|
| ASF1 | Nucleosome assembly factor and histone chaperone |
| CHK1 | Effector kinase in DNA damage checkpoint |
| CIT1 | Citrate synthase, the first enzyme of the TCA cycle |
| COX1 | Subunit of cytochrome c oxidase, enzyme of the ETC |
| CRT1 | Transcriptional repressor of DNA damage–regulated genes |
| CYC1 | Cytochrome c (isoform 1); component of the ETC |
| CYT1 | Cytochrome c1, component of the ETC |
| DUN1 | Protein kinase involved in DDR; functions downstream of Rad53 |
| HAP1 | Transcription factor regulated by heme; regulates expression of respiratory genes |
| HAP4 | Subunit of the Hap2/3/4/5 transcription complex; regulator of respiratory genes |
| HIR1 | Subunit of the HIR complex; involved in regulation of histone gene transcription |
| HHF1 | Histone H4; one of two identical histone H4 proteins |
| HHF2 | Histone H4; one of two identical histone H4 proteins |
| HHT1 | Histone H3; one of two identical histone H3 proteins |
| HHT2 | Histone H3; one of two identical histone H3 proteins |
| HTA1 | Histone H2A; one of two nearly identical H2A proteins |
| HTA2 | Histone H2A; one of two nearly identical H2A proteins |
| HTB1 | Histone H2B; one of two nearly identical H2B proteins |
| HTB2 | Histone H2B; one of two nearly identical H2B proteins |
| IDH1 | Subunit of isocitrate dehydrogenase; enzyme of the TCA cycle |
| MBP1 | Transcription factor; with Swi6 forms MBF complex |
| MEC1 | Sensor kinase in DNA damage checkpoint; yeast ortholog of ATR |
| QCR7 | Subunit of ubiquinol cytochrome c reductase; component of the ETC |
| RAD52 | Required for DNA double-strand break repair and homologous recombination |
| RAD53 | Effector kinase in DNA damage checkpoint; functions downstream of Mec1 |
| RNR1 | Major isoform of large subunit of the RNR complex |
| RNR2 | Small subunit of the RNR complex |
| RNR3 | Minor isoform of large subunit of the RNR complex |
| RNR4 | Small subunit of the RNR complex |
| SML1 | Inhibitor of the RNR complex |
| SWI6 | Transcription factor; subunit of SBF and MBF complexes |
| TEL1 | Sensor kinase in DNA damage checkpoint; yeast ortholog of ATM |
| TOM1 | E3 ubiquitin ligase involved in degradation of excess histones |
Activation of the checkpoint kinases results in cell cycle arrest, activation of DNA repair, and reprogramming of transcription. One of the key outcomes of the DDR in yeast is the enlargement of the deoxyribonucleoside triphosphate (dNTP) pools, which is a prerequisite for effective DNA repair (Fig. 1) (14, 15). The rate-limiting step of dNTP synthesis is the reduction of ribonucleoside diphosphates into corresponding deoxyribonucleoside diphosphates, catalyzed by ribonucleotide reductase (RNR) (16). In most eukaryotes, RNR enzymes are α2β2 heterotetramers, in which the α2 homodimer and the β2 homodimer represent the large and small subunits, respectively. In yeast, however, the small subunit is a heterodimer of Rnr2p and Rnr4p; the large subunit is a homodimer of Rnr1p. The catalytic site is contained within the large subunit of both mammalian and yeast RNR enzymes. Both mammalian and yeast RNR genes are regulated transcriptionally, and the enzymes are regulated allosterically (17–19). In yeast, transcription of RNR2, RNR3, and RNR4 genes is induced following checkpoint activation and Dun1p-mediated phosphorylation and inactivation of the transcriptional repressor Crt1p (20). Transcription of RNR1 is regulated in a cell cycle–dependent manner by the transcriptional complex MBF and by high mobility group-domain protein Ixr1p, but not by Crt1p (21–24). Dun1p regulates RNR activity and dNTP synthesis by at least two additional mechanisms. Dun1p phosphorylates Dif1p, a protein required for nuclear localization of Rnr2p and Rnr4p. Phosphorylation of Dif1p by Dun1p releases Rnr2p and Rnr4p into the cytoplasm, where they assemble with Rnr1p to form an active RNR enzyme (25–30). During S phase or after DNA damage, Dun1p also phosphorylates and induces degradation of Sml1p, a protein that binds and inhibits the Rnr1p subunit (Fig. 1) (31–34).
Proliferating cells need to maintain a delicate balance between histone and DNA synthesis to ensure correct stoichiometric amounts for chromatin assembly and to avoid genome instability (35, 36). Treatment with genotoxic agents that damage DNA or interfere with DNA replication triggers repression of histone genes (37–39). We have previously shown that a decrease in histone expression induces respiration (40). This poses an intriguing question: does DDR induce mitochondrial respiration? One of the sources of reactive oxygen species (ROS) is the oxidative electron transport chain (ETC) in the mitochondria. It is widely believed that DDR results in down-regulation of respiration to protect DNA from endogenous ROS (41–43). Surprisingly, our data show that DDR and growth in the presence of sublethal concentrations of genotoxic chemicals activate respiration to increase ATP production and to elevate dNTP levels, which are required for efficient DNA repair and cell survival upon DNA damage.
Results
DDR stimulates aerobic respiration
To determine whether DDR stimulates respiration, we used two approaches to introduce DDR. The first approach utilized the genotoxic chemicals bleocin and 4-nitroquinoline 1-oxide (4-NQO). Bleocin belongs to the antibiotic bleomycin family and causes DNA double-strand breaks (44). 4-NQO mimics the effect of UV light and forms DNA adducts (45). Both bleocin and 4-NQO trigger DDR. When compared with control cells, cells grown in the presence of sublethal concentrations of either chemical consumed more oxygen and produced more ATP, two parameters reflecting the activity of aerobic respiration in the mitochondria (Fig. 2, A and B) (40, 46). Oxygen consumption of cells treated with bleocin or 4-NQO increased 1.8- and 1.5-fold, respectively, whereas the cellular ATP level increased 2.6- and 2.0-fold, respectively (Fig. 2, A and B).
Figure 2.

DDR stimulates respiration. A and B, cellular oxygen consumption rate and ATP levels in WT cells (WT, W303-1a) grown in YPD medium in the presence of bleocin at 0, 0.1 and 0.3 μg/ml (A) and 4-NQO at 0, 0.1 and 0.3 μg/ml (B). C, cellular oxygen consumption rate and ATP levels in WT and rad52Δ cells. The experiments were repeated three times, and the results are shown as means ± S.D. Values that are statistically significantly different (p < 0.05) from the WT cells are indicated by an asterisk. The results are expressed relative to the value for the WT strain.
The second approach to introduce DDR employed rad52Δ mutation. RAD52 is required for DNA double-strand break repair and homologous recombination. Inactivation of RAD52 renders cells unable to repair DNA strand breaks and thereby triggers DDR (47). Compared with WT cells, rad52Δ cells consumed 1.6 times more oxygen and displayed 3.2 times increased ATP levels (Fig. 2C).
Checkpoint kinases Mec1p and Rad53p are required for DDR-induced respiration
DDR is mediated through activation of checkpoint kinases and their cellular targets, which coordinate cell cycle arrest and repair of damaged DNA (Fig. 1). To investigate the requirement of the checkpoint kinases Mec1p, Tel1p, Chk1p, Rad53p, and Dun1p for induction of respiration, we introduced the corresponding mutations into rad52Δ cells and determined oxygen consumption (Fig. 3A). The oxygen consumption of tel1Δ cells was elevated compared with WT cells, and introducing the tel1Δ mutation into the rad52Δ cells further increased oxygen consumption above the rad52Δ level. chk1Δ cells consumed less oxygen than WT cells, and the oxygen consumption of the rad52Δchk1Δ cells was attenuated compared with rad52Δ cells. Because mec1Δ and rad53Δ cells are viable only if harboring crt1Δ or sml1Δ mutations (20, 31), we measured oxygen consumption in mec1Δsml1Δ, mec1Δcrt1Δ, rad53Δsml1Δ, and rad53Δcrt1Δ strains. Surprisingly, mec1Δsml1Δ and rad53Δsml1Δ strains displayed increased oxygen consumption compared with WT cells. This increase can be attributed to the sml1Δ mutation, because sml1Δ cells also displayed slightly increased oxygen consumption. This finding is not entirely surprising, because sml1Δ cells display increased copy number of mitochondrial DNA, likely due to the elevated dNTP level (48). The oxygen consumption in rad53Δcrt11Δ was comparable with WT cells, while the oxygen consumption of mec1Δcrt1Δ cells was decreased. Importantly, introducing the rad53Δsml1Δ and rad53Δcrt1Δ mutations into rad52Δ cells completely abrogated the elevated oxygen consumption of rad52Δ cells. Extremely slow growth of rad52Δmec1Δsml1Δ and rad52Δmec1Δcrt1Δ did not allow culturing these cells in sufficient quantity for further analysis. The oxygen consumption of dun1Δ cells was elevated compared with WT cells, and introducing dun1Δ mutation into rad52Δ cells did not diminish the oxygen consumption of rad52Δ cells. This result suggests that induction of respiration in rad52Δ cells is not due to degradation of Sml1p and increased dNTP synthesis, because degradation of Sml1p requires Dun1p. The requirement of Rad53p for DDR-induced respiration is consistent with suppression of the elevated ATP levels in rad52Δ cells by introducing rad53Δsml1Δ and rad53Δcrt1Δ mutations (Fig. 3B).
Figure 3.

Checkpoint kinases Mec1p and Rad53p are required for DDR-induced respiration. A, C, and D, cellular oxygen consumption rates; B, cellular ATP levels in the indicated strains. A, cells were grown in YPD medium, and cellular oxygen consumption was determined in the wildtype (WT, W303-1a), rad52Δ (LG731), tel1Δ (SN159), rad52Δtel1Δ (SN158), mec1Δsml1Δ (SN120), mec1Δcrt1Δ (SN125), chk1Δ (SN136), rad52Δchk1Δ (SN138), rad53Δsml1Δ (LG606), rad53Δcrt1Δ (LG716), rad52Δrad53Δsml1Δ (PB026), rad52Δrad53Δcrt1Δ (PB019), dun1Δ (PB119), rad52Δdun1Δ (PB127), sml1Δ (LG603), and crt1Δ (LG706) cells. B, cells were grown in YPD medium, and cellular ATP levels were determined in the wildtype (WT, W303-1a), rad52Δ (LG731), rad53Δsml1Δ (LG606), rad53Δcrt1Δ (LG716), rad52Δrad53Δsml1Δ (PB026), and rad52Δrad53Δcrt1Δ (PB019) cells. A and B, values that are statistically significantly different (p < 0.05) from each other are indicated by a bracket and an asterisk. C, cells were grown in YPD medium with or without 0.1 μg/ml bleocin, and the cellular oxygen consumption was determined in the WT (W303-1a), tel1Δ (SN159), mec1Δsml1Δ (SN120), mec1Δcrt1Δ (SN125), chk1Δ (SN136), rad53Δsml1Δ (LG606), rad53Δcrt1Δ (LG716), and dun1Δ (PB119) cells. Values that are statistically significantly different (p < 0.05) from the corresponding strain grown in the absence of bleocin are indicated by an asterisk. D, cell cycle arrest of wildtype (WT, W303-1a) and rad52Δ (LG731) cells with α-factor does not affect oxygen consumption. The oxygen consumption of asynchronous and α-factor–treated cells was compared. The α-factor treatment resulted in G1 arrest of more than 90% cells as unbudded cells. A–D, experiments were repeated three times, and the results are shown as means ± S.D. The results are expressed relative to the value for the WT strain.
To corroborate these results, we induced DDR by growing cells bearing deletions of the individual checkpoint kinases in the presence of sublethal concentrations of bleocin. Growth in the presence of bleocin induced respiration in WT, tel1Δ, chk1Δ, and dun1Δ cells. The induction of respiration was completely absent in mec1Δsml1Δ, mec1Δcrt1Δ, rad53Δsml1Δ, and rad53Δcrt1Δ cells (Fig. 3C). We interpret these results to mean that Mec1p and its downstream effector kinase Rad53p are required, whereas Tel1p, Chk1p, and Dun1p are not required for DDR-induced respiration. As a control, we also included the cyt1Δ strain. CYT1 encodes cytochrome c1, and cyt1Δ cells are not able to respire (40). The results show that the ETC is responsible for about 95% of the oxygen consumed by the cells growing in the absence or presence of bleocin (Fig. 3C).
To test the possibility that the increased oxygen consumption in the WT cells grown in the presence of genotoxic chemicals or in rad52Δ cells is caused by a delayed progression through the S phase of the cell cycle, we arrested WT and rad52Δ cells in G1 phase with α-factor and compared oxygen consumption of arrested and asynchronous cells (Fig. 3D). Because the oxygen consumption in the two cell populations does not significantly differ for both WT and rad52Δ cells, we conclude that the DDR induces respiration in a cell cycle–independent manner.
DDR down-regulates histone levels through activation of Rad53p
Because the connection between DDR and respiration is not immediately obvious and to some extent is counterintuitive, we asked the following question. What is the molecular mechanism underlying this phenomenon? We have recently reported that decreased histone expression results in reduced nucleosome occupancy across the genome and altered chromatin structure, which triggers respiration (40). To determine whether a similar mechanism is responsible for DDR-induced respiration, we assessed histone expression in cells grown in the presence of sublethal concentrations of bleocin or 4-NQO. Under these conditions, the expression of all four histone genes as well as the protein levels of histone H3 were markedly decreased (Fig. 4, A–C). A very similar trend of decreased histone gene expression and protein level of histone H3 was observed in rad52Δ cells, where the DDR is induced genetically (Fig. 4, D–H). Furthermore, down-regulation of histone transcripts and protein levels depended on Rad53p, as introducing rad53Δ mutation into rad52Δ cells restored histone transcript and protein levels in rad52Δrad53Δcrt1Δ and rad52Δrad53Δsml1Δ cells to the WT levels (Fig. 4, D–G). Down-regulation of histone transcripts in rad52Δ cells was not suppressed by introducing tel1Δ or dun1Δ mutations into rad52Δ cells and was only partially suppressed by chk1Δ mutation.
Figure 4.

DDR-mediated repression of histone levels requires Rad53p. Transcript levels of core histone genes in WT cells (WT, W303-1a) treated with bleocin at 0, 0.1, and 0.3 μg/ml (A) and with 4-NQO at 0, 0.1, and 0.3 μg/ml (B). A and B, values that are statistically significantly different (p < 0.05) from the WT cells grown in the absence of bleocin or 4-NQO are indicated by an asterisk. C, histone H3 protein levels in WT cells grown in the absence of bleocin or 4-NQO, WT cells grown in the presence of 0.3 μg/ml bleocin, and WT cells grown in the presence of 0.1 μg/ml 4-NQO. Transcript levels of histones HTA1/2 (D), HTB1/2 (E), HHT1/2 (F), and HHF1/2 (G) in the wildtype (WT, W303-1a), rad52Δ (LG731), tel1Δ (SN159), rad52Δtel1Δ (SN158), mec1Δsml1Δ (SN120), mec1Δcrt1Δ (SN125), chk1Δ (SN136), rad52Δchk1Δ (SN138), rad53Δsml1Δ (LG606), rad53Δcrt1Δ (LG716), rad52Δrad53Δsml1Δ (PB026), rad52Δrad53Δcrt1Δ (PB019), dun1Δ (PB119), and rad52Δdun1Δ (PB127) cells. D–G, values that are statistically significantly different (p < 0.05) from each other are indicated by a bracket and an asterisk. H, histone H3 protein levels in WT (W303-1a) and rad52Δ (LG731) cells. A, B, and D–G, experiments were repeated three times, and the results are shown as means ± S.D. The results are expressed relative to the value for the WT strain grown in the absence of bleocin or 4-NQO. C and H, Western blotting analyses were performed three times, and representative results are shown. Pgk1p served as a loading control.
DDR-mediated repression of histone levels alters chromatin structure and induces expression of TCA cycle and ETC genes
Yeast genes can be categorized as growth genes and stress genes, with each group featuring distinct nucleosomal architecture of their promoters (49). The promoter of a growth gene contains a “nucleosome-free region,” whereas the promoter of a stress gene is usually occupied by delocalized nucleosomes. As a result, growth genes are constitutively expressed in contrast to stress genes, which are regulated by factors that affect chromatin structure, including abundance of histone proteins. Considering that respiratory genes in Saccharomyces cerevisiae belong to the stress gene category (50) and that reduced histone expression induces respiration (40), we reasoned that by down-regulating histone expression DDR might affect chromatin structure and induce respiratory genes. To test this possibility, we determined histone H3 and RNA pol II occupancy in the promoters of CIT1, IDH1, and QCR7, genes encoding enzymes of the TCA cycle and ETC. The histone H3 occupancy of CIT1, IDH1, and QCR7 promoters was significantly reduced in cells treated with bleocin or 4-NQO compared with control cells, whereas the occupancy of RNA pol II at the same set of promoters was increased (Fig. 5, A and B). Similarly, the histone H3 occupancy of CIT1, IDH1, and QCR7 promoters was decreased, whereas RNA pol II occupancy at the same promoters was increased in rad52Δ cells (Fig. 5, C and D). The changes in chromatin structure and RNA pol II occupancy were accompanied by increased transcription of the corresponding genes upon treatment with bleocin or 4-NQO and in rad52Δ cells (Fig. 5E). In this analysis, we also included the COX1 gene that is encoded by the mitochondrial genome. As we have shown previously, decreased histone expression and chromatin changes of the nuclear genome affect transcription of genes encoded by the mitochondrial genome. The corresponding mechanism involves elevated expression of nuclear genes RPO41 and MTF1, encoding mitochondrial RNA polymerase and its associated factor, respectively (40).
Figure 5.

DDR-mediated repression of histone levels alters chromatin structure and induces expression of TCA cycle and ETC genes. ChIP-qPCR analysis of histone H3 (A) and RNA polymerase II (B) occupancy at CIT1, IDH1, and QCR7 promoters in WT cells (WT, W303-1a) grown in the absence of bleocin or 4-NQO, WT cells grown in the presence of 0.3 μg/ml bleocin, and WT cells grown in the presence of 0.1 μg/ml 4-NQO is shown. ChIP-qPCR analysis of histone H3 (C) and RNA polymerase II (D) occupancy at CIT1, IDH1, and QCR7 promoters in WT and rad52Δ cells is shown. E, transcript levels of CIT1, IDH1, QCR7, and COX1 genes in WT cells grown in the absence of bleocin or 4-NQO, WT cells grown in the presence of 0.3 μg/ml bleocin, and WT cells grown in the presence of 0.1 μg/ml 4-NQO. The bottom row shows transcript levels of the indicated genes in WT and rad52Δ (LG731) cells. The experiments were repeated three times, and the results are shown as means ± S.D. Values that are statistically significantly different (p < 0.05) from the WT cells are indicated by an asterisk. The results are expressed relative to the value for the WT strain grown in the absence of bleocin or 4-NQO.
Inactivation of RAD52 increased respiration (Fig. 2) and down-regulated histone levels (Fig. 4) in a Rad53p-dependent manner. To determine whether the increased expression of genes required for the TCA cycle, ETC, and OXPHOS in rad52Δ cells also requires Rad53p or other checkpoint kinases, we determined transcript levels for CIT1, IDH1, QCR7, and COX1 genes in rad52Δ cells containing deletions of the individual checkpoint kinases (Fig. 6). We found that only inactivation of RAD53 (in rad53Δsml1Δ and rad53Δcrt1Δ) but not inactivation of TEL1, CHK1, or DUN1 suppressed the elevated expression of CIT1, IDH1, and QCR7 genes in rad52Δ cells.
Figure 6.

DDR-mediated induction of TCA cycle and ETC genes requires Rad53p. Transcript levels of CIT1 (A), IDH1 (B), QCR7 (C), and COX1 (D) in the WT (W303-1a), rad52Δ (LG731), tel1Δ (SN159), rad52Δtel1Δ (SN158), mec1Δsml1Δ (SN120), mec1Δcrt1Δ (SN125), chk1Δ (SN136), rad52Δchk1Δ (SN138), rad53Δsml1Δ (LG606), rad53Δcrt1Δ (LG716), rad52Δrad53Δsml1Δ (PB026), rad52Δrad53Δcrt1Δ (PB019), dun1Δ (PB119), and rad52Δdun1Δ (PB127) cells. The experiments were repeated three times, and the results are shown as means ± S.D. relative to the value for the WT strain. A–C, values that are statistically significantly different (p < 0.05) from each other are indicated by a bracket and an asterisk.
Elevated histone levels suppress respiration in rad52Δ cells
To test whether the decreased histone levels and altered chromatin structure are indeed responsible for induction of respiration when cells grow in the presence of sublethal concentrations of genotoxic chemicals, we elevated histone levels in WT cells by ectopic expression of extra histones. A high copy number plasmid encoding all four core histones significantly reduced oxygen consumption when cells were grown in the presence of bleocin (Fig. 7, A and B). In addition, overexpression of histones suppressed oxygen consumption in rad52Δ cells (Fig. 7C).
Figure 7.

Elevated histone levels suppress DDR-induced respiration. Relative mRNA levels of histone genes (A) and oxygen consumption rate (B) of WT cells (WT, W303-1a) containing either control plasmid or high copy number plasmid expressing all four core histone genes (plasmid pRS426-HTA1/HTB1-HHF1/HHT1). The cells were pre-grown under selection in SC medium and inoculated to an A600 nm of 0.1 into YPD medium containing 0 or 0.3 μg/ml of bleocin and grown for two generations at 30 °C. C, oxygen consumption rate of rad52Δ (LG731) cells containing either control plasmid or plasmid pRS426-HTA1/HTB1-HHF1/HHT1. The cells were pre-grown under selection in SC medium and inoculated to an A600 nm of 0.1 into YPD medium and grown for two generations. D, histone H3 protein levels in the wildtype (WT, W303-1a), rad52Δ (LG731), tom1Δ (LG734), and rad52Δtom1Δ (LG774) cells. Western blotting analyses were performed three times, and representative results are shown. Pgk1p served as a loading control. E, oxygen consumption rate in the WT (W303-1a), rad52Δ (LG731), tom1Δ (LG734), rad52Δtom1Δ (LG774), hir1Δ (MZ700), and rad52Δhir1Δ (PB066) cells. A–C and E, experiments were repeated three times, and the results are shown as means ± S.D. Values that are statistically significantly different (p < 0.05) from each other are indicated by a bracket and an asterisk. The results are expressed relative to the value for the WT strain.
In another approach, we tested whether the histone levels can be increased in rad52Δ cells by introducing tom1Δ or hir1Δ mutations. Tom1p functions in a pathway responsible for degradation of free histones. Free histones that are not assembled into chromatin are degraded in a pathway that depends on phosphorylation by Rad53p and ubiquitylation by Ubc4p, Ubc5p, and Tom1p (51, 52). When TOM1 was deleted in rad52Δ cells, the protein level of histone H3 was restored almost to the WT level, confirming the usefulness of this approach (Fig. 7D). Hir1p is a subunit of the HIR complex that acts as a histone chaperone and a repressor of the majority of histone genes (53, 54). Inactivation of HIR1 induces expression of histone genes (35). Introducing tom1Δ or hir1Δ mutations into rad52Δ cells significantly suppressed oxygen consumption, suggesting that it is indeed the decreased level of histones that is responsible for induction of respiration in rad52Δ cells (Fig. 7E).
DDR-induced respiration activates dNTP synthesis
Does the DDR-induced respiration provide any advantage to yeast cells? One major outcome of DDR is the increase in activity of RNR, the key enzyme that catalyzes the rate-limiting step in dNTP synthesis (16). The complex regulation of RNR activity suggests that the control of the dNTP pools is very important for maintaining genome integrity and cell survival under genotoxic stress. Indeed, the enlargement of dNTP pools is essential for effective DNA repair (14, 15).
In our previous work, we have shown that reduced histone expression and altered chromatin structure induce respiration and significantly elevate cellular ATP levels (40). To determine whether increased respiration and elevated ATP levels can drive up dNTP synthesis, we evaluated the sizes of dNTP pools in swi6Δ and asf1Δ cells (Fig. 8A). Swi6p is the transcriptional activation subunit of the SBF and MBF complexes that regulate histone gene transcription (35, 36). Asf1p is a histone chaperone involved in chromatin assembly. Both swi6Δ and asf1Δ cells display markedly up-regulated oxygen consumption and ATP levels (40). We found that the dNTP pools are significantly increased in swi6Δ and asf1Δ cells, and the increase is abolished in swi6Δcyt1Δ and asf1Δcyt1Δ cells (Fig. 8A). CYT1 encodes the cytochrome c1 subunit, and deletion of CYT1 inactivates the ETC. When we induced DDR in WT cells either chemically or genetically, the dNTP pools were also significantly increased. However, blocking respiration by introducing the cyt1Δ mutation appreciably decreased dNTP levels in rad52Δ cells or WT cells treated with bleocin or 4-NQO (Fig. 8B). These results indicate that decreased histone expression and the defect in chromatin structure or growth in the presence of genotoxic chemicals activate respiration and increase ATP and dNTP levels.
Figure 8.

DDR-induced respiration activates dNTP synthesis and improves cell survival upon DNA damage. A, cellular dNTP levels in the wildtype (WT, W303-1a), cyt1Δ (LG533), swi6Δ (DY5780), swi6Δcyt1Δ (LG567), asf1Δ (MZ576), and asf11Δcyt1Δ (LG570) cells. B, cellular dNTP levels in the wildtype (WT, W303-1a), rad52Δ (LG731), and cyt1Δ (LG533) cells grown in YPD medium and WT cells grown in YPD medium containing 0.3 μg/ml bleocin or 0.1 μg/ml 4-NQO. A and B, experiments were repeated three times, and the results are shown as means ± S.D. Individual dNTP levels are expressed relative to the corresponding dNTP in the WT cells grown in the absence of bleocin or 4-NQO. Strains with all four dNTP levels statistically significantly different from each other are indicated by a bracket and an asterisk. C, 10-fold serial dilutions of the wildtype (WT, W303-1a), cyt1Δ (LG533), sml1Δ (LG603), and sml1Δcyt1Δ (PB061) cells were spotted onto YPD plates, YPD plates containing 0.1 μg/ml bleocin, or YPD plates containing 150 mm hydroxyurea and grown for 3 days. D, electron transport chain is required for suppression of rad53Δ lethality by mbp1Δ mutation. The 10-fold serial dilutions of the wildtype (WT, W303-1a), rad53Δsml1Δ (LG606), rad53Δmbp1Δ (MB051), and rad53Δmbp1Δcyc1Δ (MB054) cells were spotted onto YPD plates and grown for 2 days. C and D, typical results from three independent experiments are shown.
Survival of yeast cells in the presence of DNA damage depends on the availability of dNTPs for effective DNA repair (14, 19). Consistently with this role of increased dNTP pools for effective DNA repair, cyt1Δ cells that cannot up-regulate respiration and are thus unable to effectively enlarge their dNTP pools are more sensitive to bleocin or hydroxyurea (Fig. 8C). Inactivation of SML1 does not significantly change growth on bleocin or hydroxyurea, but sml1Δcyt1Δ cells are more sensitive to bleocin or hydroxyurea than sml1Δ cells. Interestingly, sml1Δcyt1Δ cells appear to be more sensitive to hydroxyurea than cyt1Δ cells. The difference in sensitivity to hydroxyurea between cyt1Δ and sml1Δcyt1Δ cells is quite subtle but reproducible. Because both ATP and Sml1p are allosteric regulators of RNR (55), this observation may indicate that under conditions of direct inhibition of RNR by hydroxyurea, the absence of Sml1p sensitizes RNR to low ATP levels.
To test whether the lethality of rad53Δ mutation can be suppressed by increasing dNTP synthesis by up-regulating respiration and ATP synthesis, we introduced mbp1Δ mutation into rad53Δ cells. MBP1 encodes the DNA-binding subunit of the MBF transcriptional factor. We have shown previously that mbp1Δ cells display increased respiration and ATP levels (40). The rad53Δmbp1Δ cells are viable and grow significantly better than the rad53Δmbp1Δcyc1Δ cells (Fig. 8D). CYC1 encodes cytochrome c, and cyc1Δ cells are not able to respire (56). This result indicates that increasing respiration and presumably ATP and dNTP synthesis represent the major mechanism for suppression of rad53Δ lethality by mbp1Δ mutation.
Only long-term but not acute genotoxic stress induces respiration
Several studies found that DDR represses transcription of genes encoding enzymes of the TCA cycle, ETC, and OXPHOS. These studies evaluated acute exogenous genotoxic stress, typically created by 1–2-h cell exposure to genotoxic chemicals (37, 60). In contrast, our results show that chronic activation of DDR rendered by rad52Δ mutation or by growing WT cells in the presence of sublethal concentrations of genotoxic chemicals activates transcription of the TCA cycle, ETC, and OXPHOS genes and elevates oxygen consumption. To determine whether the duration of the genotoxic stress is responsible for the difference between our results and studies that found the inhibitory effect of DDR on transcription of respiratory genes (37, 60), we performed a time-course experiment and measured oxygen consumption and transcription of CIT1, IDH1, QCR7, and COX1 genes for several hours after addition of bleocin (Fig. 9). Indeed, the most significant increase in oxygen consumption (Fig. 9A) and transcription of the respiratory genes (Fig. 9B) is observed after more than 2 h of growth in the presence of bleocin. These results show that increased respiration is a long-term response to a chronic sublethal genotoxic stress and suggest that long-term survival and growth under chronic genotoxic stress requires increased respiration to support ATP and dNTP synthesis.
Figure 9.
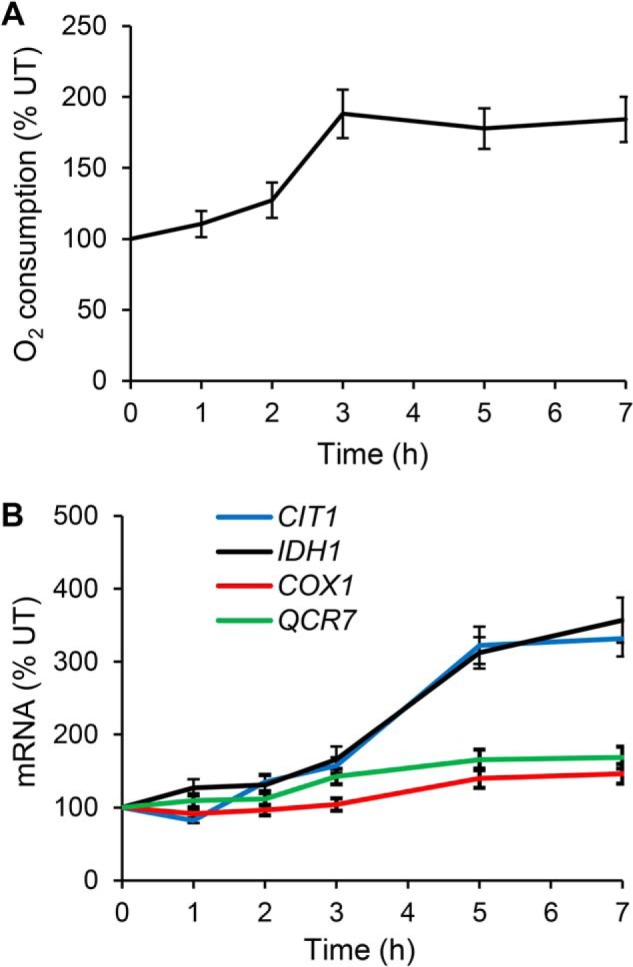
Long-term but not acute genotoxic stress induces respiration. Cellular oxygen consumption rate (A) and transcript levels of CIT1, IDH1, QCR7, and COX1 genes (B) in WT cells (WT, W303-1a) grown in YPD medium containing 0.3 μg/ml bleocin. The culture was maintained in early exponential phase (below A600 nm = 0.6) by diluting with pre-warmed YPD medium containing 0.3 μg/ml bleocin. The experiments were repeated three times, and the results are shown as means ± S.D. The results are expressed relative to the value for untreated (UT) WT strain.
Discussion
The key finding of this study is that the DDR activates respiration to increase ATP production and elevate dNTP levels, which are required for efficient DNA repair. Based on our results, we propose a model in which DDR regulates dNTP synthesis by a bifurcating mechanism (Fig. 10). In one well-established branch of the pathway, Dun1p inactivates Crt1p, Sml1p, and Dif1p, leading to increased RNR activity and dNTP synthesis (31–34). In the second branch of the pathway, Mec1p and Rad53p down-regulate transcription of histone genes (Fig. 10). Decreased histone levels result in altered chromatin structure and induction of TCA cycle and ETC genes required for respiration (40). A direct outcome of elevated respiration is increased production of ATP, a potent allosteric activator for the RNR enzyme (16, 18), increased synthesis of dNTPs, and improved cell survival (Fig. 10). This “histone” branch of the pathway does not require Dun1p and inactivation of Crt1p, Sml1p, and Dif1p, because inducing DDR in dun1Δ cells activates respiration almost to the same level as in the WT cells (Fig. 3, A and C).
Figure 10.
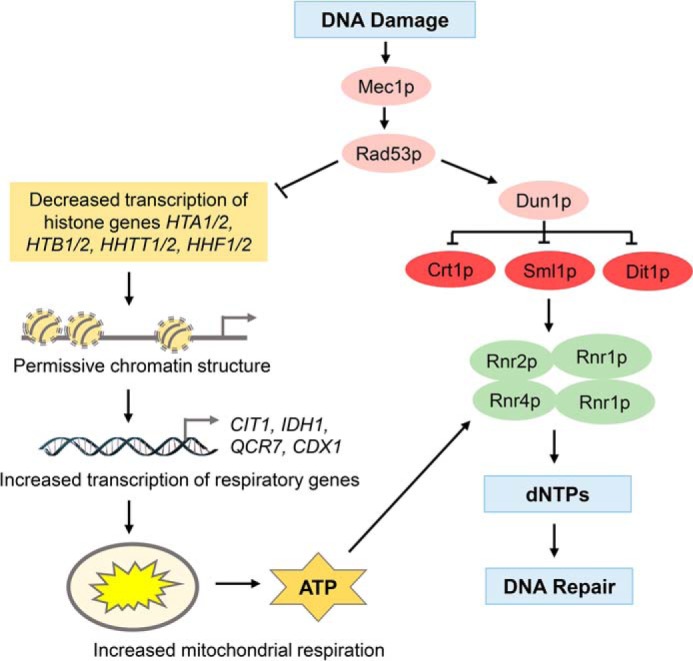
Model depicting DDR-induced respiration. DDR, in a Mec1p- and Rad53p-dependent way, regulates dNTP synthesis by a bifurcating mechanism. In one well-established branch of the pathway, Dun1p inactivates Crt1p, Sml1p, and Dif1p, leading to increased RNR activity and dNTP synthesis. In the second branch of the pathway, Mec1p and Rad53p down-regulate transcription of histone genes. Decreased histone levels result in altered chromatin structure and induction of TCA cycle and ETC genes required for respiration. A direct outcome of elevated respiration is increased production of ATP, a potent allosteric activator for the RNR enzyme, increased synthesis of dNTPs, and improved cell survival.
Based on chromatin architecture, yeast genes belong to one of two broad groups: growth genes and stress genes (49). Growth genes are expressed rather constitutively, and their promoters feature a nucleosome-free region where transcription factors bind upstream of the ORF. Stress genes are expressed at a lower level, and their promoters are dominated by delocalized nucleosomes rather than by the nucleosome-free region. Consequently, stress genes are regulated by factors that affect the structure of chromatin, including histone level. The respiratory genes in S. cerevisiae belong to the stress category, unlike respiratory genes in higher eukaryotes (50). Consequently, reduced histone expression or a defect in chromatin assembly induces respiration by allowing increased activation of the TCA cycle, ETC, and OXPHOS genes by the Hap2/3/4/5p complex (40).
Although it is well-established that DNA replication is coordinated with transcription of histone genes and that DDR represses histone transcription, the role of Rad53p in this process is not fully understood. The expression of histone genes is regulated by two G1/S-specific transcription complexes SBF and MBF, in addition to other transcription regulators (35, 36). Swi4p/Swi6p and Mbp1p/Swi6p form SBF and MBF, respectively. DDR induces Rad53p-dependent phosphorylation of Swi6p, which results in down-regulation of CLN1 and CLN2 transcription and delayed G1 to S progression (58, 59). On the other hand, Rad53p phosphorylates and inactivates Nrm1p, the co-repressor of MBF, which results in activation of MBF targets (23, 24). Systematic phosphoproteomics screen identified Swi6p, Swi4p, and Mbp1p as direct targets of Rad53p (60). Because the transcription of the histone genes is reduced in swi4Δ and mbp1Δ cells (61), the simplest explanation for the role of Rad53p in regulation of histone gene transcription is that Rad53p phosphorylates and down-regulates SBF and MBF complexes.
How does elevated respiration and ATP levels regulate dNTP synthesis? The large subunit of both mammalian and yeast RNR contains the catalytic site as well as two allosteric sites (16, 17, 19). One of the allosteric sites, the “specificity site,” binds dTTP, dGTP, and dATP and regulates the appropriate ratios among the four dNTP pools. The second allosteric site, the “activity site,” binds ATP or dATP and regulates the total dNTP pool size by monitoring the dATP/ATP ratio. When the cellular ATP-to-dATP ratio increases, the binding of ATP to this allosteric site activates RNR, promoting synthesis of all dNTPs. When the dNTP concentration reaches a certain level, the RNR activity is allosterically inhibited by binding of dATP to the “activity site.” DNA replication fidelity requires correct absolute and relative concentrations of the dNTPs (62, 63), and mutations in both the “specificity” and “activity” sites of yeast RNR result in significantly reduced replication fidelity (14, 63). In yeast RNR, the allosteric dATP feedback inhibition is more relaxed, allowing the increase of dNTP pools upon DNA damage (14). The increase in dNTP pools significantly improves survival following DNA damage; however, it also results in higher mutation rates (14, 57, 64). Our results suggest that the DDR-induced expansion of the dNTP pools is partly facilitated by elevated respiration and ATP production.
The relationship between respiratory metabolism and DNA replication and repair is contentious. Leakage of electrons from the ETC is one of the endogenous sources of ROS, which damage cellular structures, including DNA, contributing to the pathogenesis of cardiovascular diseases, inflammatory diseases, and cancer, and a shorter life span (65, 66). However, mitochondria can also function as a cellular antioxidant defense, and increased mitochondrial activity enables more efficient operation of the ETC, limiting ROS production and increasing antioxidant capacity (67). In addition, DNA replication and repair are energetically costly (45), and ETC and OXPHOS generate significantly more ATP than glycolysis. This energetic aspect of DNA repair is evolutionarily conserved, as illustrated by increased fatty acid oxidation, oxidative phosphorylation, and oxygen consumption in response to both chronic endogenous and acute exogenous genotoxic stress in mice (68).
DNA damage induced by methylmethane sulfonate (MMS) was reported to suppress respiration (69), down-regulate yeast AMP-activated protein kinase ortholog Snf1p (43), and suppress transcription of genes regulated by the Hap1p and Hap2/3/4/5p complex (37, 60). Hap1p and Hap2/3/4/5p activate genes encoding enzymes of the TCA cycle, ETC, and OXPHOS. The repression of Hap1p and Hap2/3/4/5p targets was independent of checkpoint kinases and was not observed in cells exposed to ionizing radiation (37). The authors concluded that the MMS-induced repression of Hap1p and Hap2/3/4/5p targets was not specific for DNA damage and was a consequence of oxidative stress or other effect of MMS on Hap1p and Hap2/3/4/5p signaling (37, 60). These results are in agreement with our observations. When we tested different genotoxic chemicals for respiration inducers, we also included MMS. Even with MMS concentrations spanning from 0.0001 to 0.01%, we could not detect any stimulatory effect on oxygen consumption, although we observed consistent and marked stimulation of oxygen consumption in cells treated with bleocin or 4-NQO (Fig. 2). We conclude that the effect of MMS on respiration is not representative of genotoxic chemicals and DDR but rather reflects the particular properties of MMS and/or the specific pathways MMS affects. This conclusion is supported by direct inhibition of respiration in isolated mitochondria by MMS (69).
This study connects respiratory metabolism and DDR, two processes deemed not to be very compatible. We speculate that the benefit of increased ATP and dNTP synthesis for cell survival offsets the deleterious effect of respiratory metabolism on DNA repair.
Materials and methods
Yeast strains, media, and plasmid construction
All yeast strains used in this study are listed in Table 2. Standard genetic techniques were used to manipulate yeast strains and to introduce mutations from non-W303 strains into the W303 background (71). Cells were grown at 30 °C in yeast extract/peptone/dextrose (YPD) medium containing 2% glucose or under selection in synthetic complete medium containing 2% glucose and, when appropriate, lacking specific nutrients to select for a particular genotype. Cell cycle arrest in G1 phase by α-factor was carried out by adding α-factor to 10 μg/ml to cells exponentially growing in YPD medium. Following α-factor addition, the cultures were incubated for 3 h, and the arrest was monitored by examining cell morphology (72). For construction of plasmid pRS426-HTA1/HTB1–HHF1/HHT1, the HTA1/HTB1 locus was amplified by PCR using forward primer 5′-TTCACACGAGCGAATTCTCTGAAG-3′ and reverse primer 5′-AGCAACAGTGCTCGAGGAACCTAA-3′. The HHF1–HHT1 locus was amplified using forward primer 5′-AAATACGAGCTCCGTGTAAGTTACAGAC-3′ and reverse primer 5′-TTTCGAGGGGATCCCCAGGAAAA-3′. The HHF1–HHT1 fragment was digested with SacI and BamHI and ligated into pRS426. The HTB1–HTA1 fragment was digested with EcoRI and XhoI and ligated into the pRS426-HTA1/HTB1 plasmid.
Table 2.
Yeast strains used in this study
| Strain | Genotype | Source/Ref. |
|---|---|---|
| W303-1a | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | R. Rothstein |
| ssd1-d2 can1-100 | ||
| W303-1α | MATα ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | R. Rothstein |
| ssd1-d2 can1-100 | ||
| W303 | MATa/MATα ade2-1/ade2-1 his3-11,15/his3-11,15 | R. Rothstein |
| leu2-3,112/leu2-3,112 trp1-1/trp1-1ura3-1/ura3-1 | ||
| can1-100/can1-100 | ||
| LG716 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad53::KAN crt1::LEU2 | ||
| LG606 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad53::KAN sml1::HYG | ||
| LG731 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP1 | ||
| PB019 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP1 rad53::KAN crt1::LEU2 | ||
| PB026 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP1 rad53::KAN sml1::HYG | ||
| LG734 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 tom1::TRP1 | ||
| MZ700 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 40 |
| ssd1-d2 can1-100 hir1::HIS3 | ||
| LG774 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP tom1::TRP1 | ||
| PB066 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP hir1::HIS3 | ||
| LG533 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 40 |
| ssd1-d2 an1-100 cyt1::KAN | ||
| PB051 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP1 cyt1::KAN | ||
| DY5780 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 70 |
| ssd1-d2 can1-100 swi6::TRP1 | ||
| LG567 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 40 |
| ssd1-d2 can1-100 swi6::TRP1 cyt1::KAN | ||
| MZ576 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 40 |
| ssd1-d2 can1-100 asf1::HIS3 | ||
| LG570 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | 40 |
| ssd1-d2 can1-100 asf1::HIS3 cyt1::KAN | ||
| FY2771 | MATa his3Δ200 ura3Δ0 leu2Δ0 lys2-128δ | 39 |
| mec1::HIS3 sml1::KAN | ||
| SN117 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 mec1::HIS3 sml1::KAN | ||
| SN120 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP1 mec1::HIS3 sml1::KAN | ||
| SN125 | MATα ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 mec1::HIS3 crt1::LEU2 | ||
| SN133 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP1 mec1::HIS3 crt1::LEU2 | ||
| RDKY3731 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl | 41 |
| hom3-10 ade2Δ1 ade8 hxt13::URA3 tel1::HIS3 | ||
| SN159 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 tel1::HIS3 | ||
| SN158 | MATα ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP1 tel1::HIS3 | ||
| RDKY3745 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl | 41 |
| hom3-10 ade2Δ1 ade8 hxt13::URA3 chk1::HIS3 | ||
| SN136 | MATα ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 chk1::HIS3 | ||
| SN138 | MATα ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP1 chk1::HIS3 | ||
| RDKY3739 | MATa ura3-52 leu2Δ1 trp1Δ63 his3Δ200 lys2ΔBgl | 41 |
| hom3-10 ade2Δ1 ade8 hxt13::URA3 dun1::HIS3 | ||
| cyc1::URA3 | MATa ade2-1 his3-1,15 leu2-3,112 trp 1-1 ura3-1 | 56 |
| cyc1::URA3 | ||
| SN102 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 dun1::HIS3 cyt1::KAN | ||
| LG603 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 sml1::HYG | ||
| PB061 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 sml1::HYG cyt1::KAN | ||
| PB119 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 dun1::KAN | ||
| PB127 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad52::TRP1 dun1::KAN | ||
| LG706 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 crt1::LEU2 | ||
| MB051 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad53::KAN mbp1::TRP1 | ||
| MB054 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 | This study |
| ssd1-d2 can1-100 rad53::KAN mbp1::TRP1 cyc1::URA3 |
Oxygen consumption measurement
Oxygen consumption measurements were performed essentially as described (40, 46). Cells were grown to an A600 nm of 0.6 in YPD medium containing 2% glucose, and 3 A600 nm units (9 × 107) of yeast cells were harvested by centrifugation. Cells were resuspended in a buffer containing 10 mm HEPES, 25 mm K2HPO4, pH 7.0, and incubated at 30 °C in an oxygen consumption chamber (Instech Laboratories, Inc.) connected to a NeoFOX fluorescence-sensing detector using NeoFOX software (Ocean Optics, Inc.). Results were calculated as picomoles of O2/106 cells/s and expressed as percentages of the WT value. The oxygen consumption rate in WT cells grown in YPD medium was 5.08 pmol/106 cells/s and was set as 100%.
Cellular ATP assays
Cellular ATP levels were determined as described (40, 46). Cells were grown to an A600 nm of 0.6 in YPD medium containing 2% glucose, and 3 A600 nm units (9 × 107) of yeast cells were harvested, and cells were harvested by centrifugation and lysed in 5% TCA with pre-chilled glass beads. Cell lysate was neutralized to pH 7.5 with 10 m KOH and 2 m Tris-HCl, pH 7.5. ATP levels were measured using the ENLITEN ATP assay (FF2000, Promega) according to the manufacturer's instructions and normalized by the number of cells. The ATP level in WT cells grown on YPD medium was 0.58 μmol/1010 cells and was set as 100%.
RNA extraction and real-time RT-qPCR
The procedures to extract total RNA from yeast cells and perform real-time RT-qPCR were as described previously (73). The primers used are as follows: ACT1 (5′-TATGTGTAAAGCCGGTTTTGC-3′ and 5′-GACAATACCGTGTTCAATTGGG-3′); CIT1 (5′-CAGCGATATTATCAACAACTAGCA-3′ and 5′-TAGTGGCGAGCATTCAATAGTG-3′); IDH1 (5′-TGCTTAACAGAACAATTGCTAAGAG-3′ and 5′-AACACCGTCACCAGGTATCAA-3′); QCR7 (5′-ACGTCTATTGCGAGAATTGGTG-3′ and 5′-AGCCCTAACTTCTTGTAACCTGC-3′); and COX1 (5′-CAACAAATGCAAAAGATATTGCAG-3′ and 5′-AATATTGTGAACCAGGTGCAGC-3′). The histone gene primers recognize both copies of the core histone gene: HTA1/2 (5′-CGGTGGTAAAGGTGGTAAAGC-3′ and 5′-TGGAGCACCAGAACCAATTC-3′); HTB1/2 (5′-CAAAGTTTTGAAGCAAACTCACCC-3′ and 5′-GCCAATTTAGAAGCTTCAGTAGC-3′); HHT1/2 (5′-GAAGCCTCACAGATATAAGCCAG-3′ and 5′-ATCTTGAGCGATTTCTCTGACC-3′); and HHF1/2 (5′-CCAAGCGTCACAGAAAGATTCTA-3′ and 5′-ACCAGAAATACGCTTGACACCA-3′).
Western blotting
Whole-cell lysates were prepared, and Western blotting was performed as described previously (40). Briefly, cells were grown in YPD medium containing 2% glucose to an A600 nm of 0.6. Four A600 nm units (1 A600 nm unit is equal to 3 × 107 cells) of yeast cells were harvested and immediately boiled in SDS sample buffer. Anti-histone H3 polyclonal antibody (Abcam, ab1791) was used at a dilution of 1:1000, and anti-Pgk1p (Invitrogen, 459250) was used at a dilution of 1:3000.
ChIP-qPCR
In vivo chromatin cross-linking and immunoprecipitation were performed essentially as described (73). Immunoprecipitation was performed with ChIP grade anti-histone H3 antibody (Abcam, ab1791) and anti-RNA polymerase II antibody (Abcam, ab817). The primers used for qPCR are as follows: POL1 (5′-TCCTGACAAAGAAGGCAATAGAAG-3′ and 5′-TAAAACACCCTGATCCACCTCTG-3′); CIT1 (5′-CCTTTGGAGCTTTTCCGATA and 5′-GCAAATTTCCCCCTTAAGAC-3′); IDH1 (5′-AGCGATTAAAGGAAGACCCTC-3′ and 5′-CTACGGTAGAGTAAAGAAATC-3′); and QCR7 (5′-ACAGCAGGCCAAAAACCAA-3′ and 5′-AAAGTAAATTGTCAGGCCCCC-3′).
dNTP quantitative analysis
Four A600 nm units (12 × 107) of yeast cells were harvested and lysed in 5% TCA with pre-chilled glass beads. Cell lysate was neutralized to pH 7.5 with 10 m KOH and 2 m Tris-HCl, pH 7.5. The lysate was used immediately for probe-based quantitative PCR analysis of individual dNTP levels or aliquoted and stored at −70 °C until the time of analysis. The procedure for the fluorescence-based dNTP quantitative analysis was essentially as described previously with minor modifications (74). Specifically, both detection templates (DT) 1 and 2 for each dNTP were used, and the results were compared. Fluorescence signal was recorded every 5 min (1 cycle) for a total of 50 min (10 cycles) to monitor the kinetics of individual dNTP incorporation, and the fluorescence signal after a 40-min incubation (8 cycles) was used for calculation of the normalized fluorescence units. Standard curves using appropriate individual dNTPs were established for assay validation.
Spotting assay
Cells were grown to log phase at 30 °C, and 10-fold serial dilutions were spotted on the YPD plates with or without genotoxic chemicals and incubated at 30 °C for 48–72 h.
Statistical analysis
The results represent at least three independent experiments. Numerical results are presented as means ± S.D. Data were analyzed by using an InStat software package (Graphpad, San Diego). Statistical significance was evaluated by one-way analysis of variance, and p < 0.05 was considered significant.
Author contributions
P. B., I. V., and A. V. conceptualization; P. B., S. N., M. B., P. K., I. V., and A. V. data curation; P. B., S. N., M. B., and A. V. formal analysis; P. B. and A. V. supervision; P. B. and A. V. funding acquisition; P. B., S. N., M. B., and A. V. validation; P. B., S. N., M. B., P. K., A. S., J. Z., and A. V. investigation; P. B., S. N., M. B., P. K., A. S., and A. V. methodology; P. B., S. N., M. B., and A. V. writing-original draft; P. B. and A. V. project administration; P. B., I. V., and A. V. writing-review and editing; I. V. visualization.
Acknowledgments
We thank Drs. Barrientos, Chabes, Elledge, Kolodner, Stillman, Tyler, and Winston for strains and plasmids.
This work was supported by National Institutes of Health Grant GM120710 (to A. V). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- DDR
- DNA damage response
- ATM
- ataxia telangiectasia–mutated
- ATR
- ATM and Rad3-related protein
- ChIP
- chromatin immunoprecipitation coupled with quantitative PCR
- ETC
- electron transport chain
- 4-NQO
- 4-nitroquinoline 1-oxide
- OXPHOS
- oxidative phosphorylation
- RNR
- ribonucleotide reductase
- ROS
- reactive oxygen species
- RT-qPCR
- real-time reverse transcription quantitative PCR
- YPD
- yeast extract/peptone/dextrose
- TCA
- tricarboxylic acid
- MMS
- methylmethane sulfonate
- pol
- polymerase.
References
- 1. Kolodner R. D., Putnam C. D., and Myung K. (2002) Maintenance of genome stability in Saccharomyces cerevisiae. Science 297, 552–557 10.1126/science.1075277 [DOI] [PubMed] [Google Scholar]
- 2. Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., and Linn S. (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 10.1146/annurev.biochem.73.011303.073723 [DOI] [PubMed] [Google Scholar]
- 3. Ciccia A., and Elledge S. J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell 40, 179–204 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Longhese M. P., Foiani M., Muzi-Falconi M., Lucchini G., and Plevani P. (1998) DNA damage checkpoint in budding yeast. EMBO J. 17, 5525–5528 10.1093/emboj/17.19.5525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Putnam C. D., and Kolodner R. D. (2017) Pathways and mechanisms that prevent genome instability in Saccharomyces cerevisiae. Genetics 206, 1187–1225 10.1534/genetics.112.145805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Branzei D., and Foiani M. (2006) The Rad53 signal transduction pathway: replication fork stabilization, DNA repair, and adaptation. Exp. Cell Res. 312, 2654–2659 10.1016/j.yexcr.2006.06.012 [DOI] [PubMed] [Google Scholar]
- 7. Putnam C. D., Jaehnig E. J., and Kolodner R. D. (2009) Perspectives on the DNA damage and replication checkpoint responses in Saccharomyces cerevisiae. DNA Repair 8, 974–982 10.1016/j.dnarep.2009.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Polo S. E., and Jackson S. P. (2011) Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25, 409–433 10.1101/gad.2021311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhou C., Elia A. E., Naylor M. L., Dephoure N., Ballif B. A., Goel G., Xu Q., Ng A., Chou D. M., Xavier R. J., Gygi S. P., and Elledge S. J. (2016) Profiling DNA damage-induced phosphorylation in budding yeast reveals diverse signaling networks. Proc. Natl. Acad. Sci. U.S.A. 113, E3667–E3675 10.1073/pnas.1602827113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Connor M. J. (2015) Targeting the DNA damage response in cancer. Mol. Cell 60, 547–560 10.1016/j.molcel.2015.10.040 [DOI] [PubMed] [Google Scholar]
- 11. Bashkirov V. I., Bashkirova E. V., Haghnazari E., and Heyer W. D. (2003) Direct kinase-to-kinase signaling mediated by the FHA phosphoprotein recognition domain of the Dun1 DNA damage checkpoint kinase. Mol. Cell. Biol. 23, 1441–1452 10.1128/MCB.23.4.1441-1452.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pellicioli A., and Foiani M. (2005) Signal transduction: how Rad53 kinase is activated. Curr. Biol. 15, R769–771 10.1016/j.cub.2005.08.057 [DOI] [PubMed] [Google Scholar]
- 13. Zhou Z., and Elledge S. J. (1993) DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell 75, 1119–1127 10.1016/0092-8674(93)90321-G [DOI] [PubMed] [Google Scholar]
- 14. Chabes A., Georgieva B., Domkin V., Zhao X., Rothstein R., and Thelander L. (2003) Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112, 391–401 10.1016/S0092-8674(03)00075-8 [DOI] [PubMed] [Google Scholar]
- 15. Sabouri N., Viberg J., Goyal D. K., Johansson E., and Chabes A. (2008) Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res. 36, 5660–5667 10.1093/nar/gkn555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nordlund P., and Reichard P. (2006) Ribonucleotide reductases. Annu. Rev. Biochem. 75, 681–706 10.1146/annurev.biochem.75.103004.142443 [DOI] [PubMed] [Google Scholar]
- 17. Hofer A., Crona M., Logan D. T., and Sjöberg B.-M. (2012) DNA building blocks: keeping control of manufacture. Crit. Rev. Biochem. Mol. Biol. 47, 50–63 10.3109/10409238.2011.630372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sanvisens N., de Llanos R., and Puig S. (2013) Function and regulation of yeast ribonucleotide reductase: cell cycle, genotoxic stress, and iron bioavailability. Biomed. J. 36, 51–58 10.4103/2319-4170.110398 [DOI] [PubMed] [Google Scholar]
- 19. Mathews C. K. (2015) Deoxyribonucleotide metabolism, mutagenesis and cancer. Nat. Rev. Cancer 15, 528–539 10.1038/nrc3981 [DOI] [PubMed] [Google Scholar]
- 20. Huang M., Zhou Z., and Elledge S. J. (1998) The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 94, 595–605 10.1016/S0092-8674(00)81601-3 [DOI] [PubMed] [Google Scholar]
- 21. Workman C. T., Mak H. C., McCuine S., Tagne J.-B., Agarwal M., Ozier O., Begley T. J., Samson L. D., and Ideker T. (2006) A systems approach to mapping DNA damage response pathways. Science 312, 1054–1059 10.1126/science.1122088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tsaponina O., Barsoum E., Aström S. U., and Chabes A. (2011) Ixr1 is required for the expression of the ribonucleotide reductase Rnr1 and maintenance of dNTP pools. PLoS Genet. 7, e1002061 10.1371/journal.pgen.1002061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Travesa A., Kuo D., de Bruin R. A., Kalashnikova T. I., Guaderrama M., Thai K., Aslanian A., Smolka M. B., Yates J. R. 3rd., Ideker T., and Wittenberg C. (2012) DNA replication stress differentially regulates G1/S genes via Rad53-dependent inactivation of Nrm1. EMBO J. 31, 1811–1822 10.1038/emboj.2012.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bastos de Oliveira F. M., Harris M. R., Brazauskas P., de Bruin R. A., and Smolka M. B. (2012) Linking DNA replication checkpoint to MBF cell-cycle transcription reveals a distinct class of G1/S genes. EMBO J. 31, 1798–1810 10.1038/emboj.2012.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yao R., Zhang Z., An X., Bucci B., Perlstein D. L., Stubbe J., and Huang M. (2003) Subcellular localization of yeast ribonucleotide reductase regulated by the DNA replication and damage checkpoint pathways. Proc. Natl. Acad. Sci. U.S.A. 100, 6628–6633 10.1073/pnas.1131932100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee Y. D., and Elledge S. J. (2006) Control of ribonucleotide reductase localization through an anchoring mechanism involving Wtm1. Genes Dev. 20, 334–344 10.1101/gad.1380506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang Z., An X., Yang K., Perlstein D. L., Hicks L., Kelleher N., Stubbe J., and Huang M. (2006) Nuclear localization of the Saccharomyces cerevisiae ribonucleotide reductase small subunit requires a karyopherin and a WD40 repeat protein. Proc. Natl. Acad. Sci. U.S.A. 103, 1422–1427 10.1073/pnas.0510516103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee Y. D., Wang J., Stubbe J., and Elledge S. J. (2008) Dif1 is a DNA-damage-regulated facilitator of nuclear import for ribonucleotide reductase. Mol. Cell 32, 70–80 10.1016/j.molcel.2008.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu X., and Huang M. (2008) Dif1 controls subcellular localization of ribonucleotide reductase by mediating nuclear import of the R2 subunit. Mol. Cell. Biol. 28, 7156–7167 10.1128/MCB.01388-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sanvisens N., Romero A. M., Zhang C., Wu X., An X., Huang M., and Puig S. (2016) Yeast Dun1 kinase regulates ribonucleotide reductase small subunit localization in response to iron deficiency. J. Biol. Chem. 291, 9807–9817 10.1074/jbc.M116.720862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao X., Muller E. G., and Rothstein R. (1998) A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell 2, 329–340 10.1016/S1097-2765(00)80277-4 [DOI] [PubMed] [Google Scholar]
- 32. Zhao X., Chabes A., Domkin V., Thelander L., and Rothstein R. (2001) The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J. 20, 3544–3553 10.1093/emboj/20.13.3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhao X., and Rothstein R. (2002) The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc. Natl. Acad. Sci. U.S.A. 99, 3746–3751 10.1073/pnas.062502299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Andreson B. L., Gupta A., Georgieva B. P., and Rothstein R. (2010) The ribonucleotide reductase inhibitor, Sml1, is sequentially phosphorylated, ubiquitylated and degraded in response to DNA damage. Nucleic Acids Res. 38, 6490–6501 10.1093/nar/gkq552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eriksson P. R., Ganguli D., Nagarajavel V., and Clark D. J. (2012) Regulation of histone gene expression in budding yeast. Genetics 191, 7–20 10.1534/genetics.112.140145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kurat C. F., Recht J., Radovani E., Durbic T., Andrews B., and Fillingham J. (2014) Regulation of histone gene transcription in yeast. Cell. Mol. Life Sci. 71, 599–613 10.1007/s00018.013-1443-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gasch A. P., Huang M., Metzner S., Botstein D., Elledge S. J., and Brown P. O. (2001) Genomic expression responses to DNA-damaging agents and the regulatory role of the yeast ATR homolog Mec1p. Mol. Biol. Cell 12, 2987–3003 10.1091/mbc.12.10.2987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Su C., Gao G., Schneider S., Helt C., Weiss C., O'Reilly M. A., Bohmann D., and Zhao J. (2004) DNA damage induces downregulation of histone gene expression through the G1 checkpoint pathway. EMBO J. 23, 1133–1143 10.1038/sj.emboj.7600120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Libuda D. E., and Winston F. (2010) Alterations in DNA replication and histone levels promote histone gene amplification in Saccharomyces cerevisiae. Genetics 184, 985–997 10.1534/genetics.109.113662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Galdieri L., Zhang T., Rogerson D., and Vancura A. (2016) Reduced histone expression or a defect in chromatin assembly induces respiration. Mol. Cell. Biol. 36, 1064–1077 10.1128/MCB.00770-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huang M. E., and Kolodner R. D. (2005) A biological network in Saccharomyces cerevisiae prevents the deleterious effects of endogenous oxidative DNA damage. Mol. Cell 17, 709–720 10.1016/j.molcel.2005.02.008 [DOI] [PubMed] [Google Scholar]
- 42. Ragu S., Faye G., Iraqui I., Masurel-Heneman A., Kolodner R. D., and Huang M. (2007) Oxygen metabolism and reactive oxygen species cause chromosomal rearrangements and cell death. Proc. Natl. Acad. Sci. U.S.A. 104, 9747–9752 10.1073/pnas.0703192104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simpson-Lavy K. J., Bronstein A., Kupiec M., and Johnston M. (2015) Cross-talk between carbon metabolism and the DNA damage response in S. cerevisiae. Cell Rep. 12, 1865–1875 10.1016/j.celrep.2015.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bolzán A. D., and Bianchi M. S. (2018) DNA and chromosome damage induced by bleomycin in mammalian cells: an update. Mutat. Res. 775, 51–62 10.1016/j.mrrev.2018.02.003 [DOI] [PubMed] [Google Scholar]
- 45. Friedberg E. C. (1995) Out of the shadows and into the light: the emergence of DNA repair. Trends Biochem. Sci. 20, 381 10.1016/S0968-0004(00)89082-9 [DOI] [PubMed] [Google Scholar]
- 46. Zhang T., Bu P., Zeng J., and Vancura A. (2017) Increased heme synthesis in yeast induces a metabolic switch from fermentation to respiration even under conditions of glucose repression. J. Biol. Chem. 292, 16942–16954 10.1074/jbc.M117.790923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mortensen U. H., Lisby M., and Rothstein R. (2009) Rad52. Curr. Biol. 19, R676–R677 10.1016/j.cub.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 48. Taylor S. D., Zhang H., Eaton J. S., Rodeheffer M. S., Lebedeva M. A., O'rourke T. W., Siede W., and Shadel G. S. (2005) The conserved Mec1/rRd53 nuclear checkpoint pathway regulates mitochondrial DNA copy number in Saccharomyces cerevisiae. Mol. Biol. Cell 16, 3010–3018 10.1091/mbc.e05-01-0053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rando O. J., and Winston F. (2012) Chromatin and transcription in yeast. Genetics 190, 351–387 10.1534/genetics.111.132266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tsankov A. M., Thompson D. A., Socha A., Regev A., and Rando O. J. (2010) The role of nucleosomes positioning in the evolution of gene regulation. PLoS Biol. 8, e1000414 10.1371/journal.pbio.1000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gunjan A., and Verreault A. (2003) A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 115, 537–549 10.1016/S0092-8674(03)00896-1 [DOI] [PubMed] [Google Scholar]
- 52. Singh R. K., Kabbaj M. H., Paik J., and Gunjan A. (2009) Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat. Cell Biol. 11, 925–933 10.1038/ncb1903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Osley M. A., and Lycan D. (1987) Trans-acting regulatory mutations that alter transcription of Saccharomyces cerevisiae histone genes. Mol. Cell. Biol. 7, 4204–4210 10.1128/MCB.7.12.4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Xu H., Kim U. J., Schuster T., and Grunstein M. (1992) Identification of a new set of cell cycle-regulatory genes that regulate S-phase transcription of histone genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 12, 5249–5259 10.1128/MCB.12.11.5249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Misko T. A., Wijerathna S. R., Radivoyevitch T., Berdis A. J., Ahmad M. F., Harris M. E., and Dealwis C. G. (2016) Inhibition of yeast ribonucleotide reductase by Sml1 depends on the allosteric state of the enzyme. FEBS Lett. 590, 1704–1712 10.1002/1873-3468.12207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ocampo A., Liu J., Schroeder E. A., Shadel G. S., and Barrientos A. (2012) Mitochondrial respiratory tresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab. 16, 55–67 10.1016/j.cmet.2012.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chabes A., and Stillman B. (2007) Constitutively high dNTP concentration inhibits cell cycle progression and the DNA damage checkpoint in yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 104, 1183–1188 10.1073/pnas.0610585104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sidorova J. M., and Breeden L. L. (1997) Rad53-dependent phosphorylation of Swi6 and down-regulation of CLN1 and CLN2 transcription occur in response to DNA damage in Saccharomyces cerevisiae. Genes Dev. 11, 3032–3045 10.1101/gad.11.22.3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Sidorova J. M., and Breeden L. L. (2003) Rad53 checkpoint kinase phosphorylation site preference identified in the Swi6 protein of Saccharomyces cerevisiae. Mol. Cell. Biol. 23, 3405–3416 10.1128/MCB.23.10.3405-3416.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Jaehnig E. J., Kuo D., Hombauer H., Ideker T. G., and Kolodner R. D. (2013) Checkpoint kinases regulate a global network of transcription factors in response to DNA damage. Cell Rep. 4, 174–188 10.1016/j.celrep.2013.05.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hess D., and Winston F. (2005) Evidence that Spt10 and Spt21 of Saccharomyces cerevisiae play distinct roles in vivo and functionally interact with MCB-binding factor, SCB-binding factor and Snf1. Genetics 170, 87–94 10.1534/genetics.104.039214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yao N. Y., Schroeder J. W., Yurieva O., Simmons L. A., and O'Donnell M. E. (2013) Cost of rNTP/dNTP pool imbalance at the replication fork. Proc. Natl. Acad. Sci. U.S.A. 110, 12942–12947 10.1073/pnas.1309506110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Watt D. L., Buckland R. J., Lujan S. A., Kunkel T. A., and Chabes A. (2016) Genome-wide analysis of the specificity and mechanisms of replication infidelity driven by imbalanced dNTP pools. Nucleic Acids Res. 44, 1669–1680 10.1093/nar/gkv1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schmidt T. T., Reyes G., Gries K., Ceylan C. Ü., Sharma S., Meurer M., Knop M., Chabes A., and Hombauer H. (2017) Alterations in cellular metabolism triggered by URA7 or GLN3 inactivation cause imbalanced dNTP pools and increased mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 114, E4442–E4451 10.1073/pnas.1618714114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Finkel T. (2003) Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 15, 247–254 10.1016/S0955-0674(03)00002-4 [DOI] [PubMed] [Google Scholar]
- 66. Balaban R. S., Nemoto S., and Finkel T. (2005) Mitochondria, oxidants, and aging. Cell 120, 483–495 10.1016/j.cell.2005.02.001 [DOI] [PubMed] [Google Scholar]
- 67. Barros M. H., bandy B., Tahara E. B., and Kowaltowski A. J. (2004) Higher respiratory activity decreases mitochondrial reactive oxygen release and increases life span in Saccharomyces cerevisiae. J. Biol. Chem. 279, 49883–49888 10.1074/jbc.M408918200 [DOI] [PubMed] [Google Scholar]
- 68. Brace L. E., Vose S. C., Stanya K., Gathungu R. M., Marur V. R., Longchamp A., Trevino-Villarreal H., Mejia P., Vargas D., Inouye K., Bronson R. T., Lee C. H., Neilan E., Kristal B. S., and Mitchell J. R. (2016) Increased oxidative phosphorylation in response to acute and chronic DNA damage. NPJ Aging Mech. Dis. 2, 16022 10.1038/npjamd.2016.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kitanovic A., Walther T., Loret M. O., Holzwarth J., Kitanovic I., Bonowski F., Van Bui N., Francois J. M., and Wölfl S. (2009) Metabolic response to MMS-mediated DNA damage in Saccharomyces cerevisiae is dependent on the glucose concentration in the medium. FEMS Yeast Res. 9, 535–551 10.1111/j.1567-1364.2009.00505.x [DOI] [PubMed] [Google Scholar]
- 70. Yu Y., Eriksson P., and Stillman D. J. (2000) Architectural transcription factors and the SAGA complex function in parallel pathways to activate transcription. Mol. Cell. Biol. 20, 2350–2357 10.1128/MCB.20.7.2350-2357.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sherman F. (1991) Getting started with yeast. Methods Enzymol. 194, 3–21 10.1016/0076-6879(91)94004-V [DOI] [PubMed] [Google Scholar]
- 72. Stuart D., and Wittenberg C. (1995) CLN3, not positive feedback, determines the timing of CLN2 transcription in cycling cells. Genes Dev. 9, 2780–2794 10.1101/gad.9.22.2780 [DOI] [PubMed] [Google Scholar]
- 73. Galdieri L., and Vancura A. (2012) Acetyl-CoA carboxylase regulates global histone acetylation. J. Biol. Chem. 287, 23865–23876 10.1074/jbc.M112.380519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wilson P. M., Labonte M. J., Russell J., Louie S., Ghobrial A. A., and Ladner R. D. (2011) A novel fluorescence-based assay for the rapid detection and quantification of cellular deoxyribonucleoside triphosphates. Nucleic Acids Res. 39, e112 10.1093/nar/gkr350 [DOI] [PMC free article] [PubMed] [Google Scholar]