

Abstract
Mouse studies linking adropin, a peptide hormone encoded by the energy homeostasis–associated (ENHO) gene, to biological clocks and to glucose and lipid metabolism suggest a potential therapeutic target for managing diseases of metabolism. However, adropin's roles in human metabolism are unclear. In silico expression profiling in a nonhuman primate diurnal transcriptome atlas (GSE98965) revealed a dynamic and diurnal pattern of ENHO expression. ENHO expression is abundant in brain, including ventromedial and lateral hypothalamic nuclei regulating appetite and autonomic function. Lower ENHO expression is present in liver, lung, kidney, ileum, and some endocrine glands. Hepatic ENHO expression associates with genes involved in glucose and lipid metabolism. Unsupervised hierarchical clustering identified 426 genes co-regulated with ENHO in liver, ileum, kidney medulla, and lung. Gene Ontology analysis of this cluster revealed enrichment for epigenetic silencing by histone H3K27 trimethylation and biological processes related to neural function. Dietary intervention experiments with 59 adult male rhesus macaques indicated low plasma adropin concentrations were positively correlated with fasting glucose, plasma leptin, and apolipoprotein C3 (APOC3) concentrations. During consumption of a high-sugar (fructose) diet, which induced 10% weight gain, animals with low adropin had larger increases of plasma leptin and more severe hyperglycemia. Declining adropin concentrations were correlated with increases of plasma APOC3 and triglycerides. In summary, peripheral ENHO expression associates with pathways related to epigenetic and neural functions, and carbohydrate and lipid metabolism, suggesting co-regulation in nonhuman primates. Low circulating adropin predicts increased weight gain and metabolic dysregulation during consumption of a high-sugar diet.
Keywords: apolipoprotein, circadian, type 2 diabetes, epigenetics, glucose metabolism, insulin resistance, leptin, dyslipidemia, obesity, nutrition
Introduction
The energy homeostasis–associated (ENHO) gene contains a single highly-conserved ORF (adropin1–76). In silico analysis indicates a secreted domain (adropin34–76) released from a secretory signal (adropin1–33) by proteolysis (1, 2). Adropin34–76 has bioactivity that influences carbohydrate and lipid metabolism when administered to rodents and cultured cells (1, 3–12). Adropin immunoreactivity has been measured in the circulation of humans, nonhuman primates, and mice (1, 8, 11, 13). However, retention of adropin1–76 in plasma membrane fractions has been observed (14). The secretory processes and protein structure of adropin thus remain under investigation.
Experiments performed in male C57BL/6J (B6) mice implicate a role for adropin in metabolic homeostasis. Treating diet-induced obese (DIO)4 B6 mice with exogenous adropin34–76 reduce adiposity (1). Conversely, adropin deficiency increases adiposity without altering food intake (11). In mice and cultured cells, adropin34–76 increases activity of the pyruvate dehydrogenase complex, enhancing the coupling of glycolysis with the tricarboxylic acid cycle and oxidative glucose metabolism (1, 3, 9, 10, 12). Treatment with adropin34–76 also enhances insulin signaling, glucose tolerance, and whole-body oxidative glucose disposal in DIO B6 mice independently of weight loss (1, 9). Male B6 mice lacking either one or both functional copies of the Enho gene exhibit insulin resistance and impaired glucose tolerance (11, 15). Partial loss of adropin signaling is thus sufficient to result in impaired glucose homeostasis.
The initial studies examining regulation of hepatic adropin expression in mice indicated rapid effects of fasting and intake of dietary macronutrients (1, 16–19). More recent studies indicate rhythmicity of hepatic adropin expression under the control of core elements of the biological clock (8). Diurnal cycles of transcriptional activation or repression by retinoic acid–related orphan receptors (ROR) or REV-ERB contribute to the rhythmicity of the biological clock (20–22) and to hepatic Enho expression (8). Members of the ROR (α, β, and γ) and REV-ERB (α and β) families bind competitively to common genomic elements, regulating transcription of genes involved in carbohydrate and lipid metabolism; these nuclear receptors also possess ligand-binding domains responsive to cellular lipid and redox conditions (20). Involvement of ROR and REV-ERB in regulating ENHO transcription provides a plausible mechanism linking adropin expression to biological clocks. As adropin regulates glucose and fat oxidation (3, 9, 10, 12), it may have a role in translating cues from biological clock genes into rhythms in carbohydrate and lipid metabolism.
The relationships between adropin and metabolism in humans are less clear and remain under investigation. Several studies have reported that fasting plasma adropin concentrations are lower in humans with insulin resistance (23–26). Relationships have been observed between circulating adropin concentrations and fasting glucose levels in children with Prader-Willi syndrome, as well as circulating levels of the gut peptide obestatin in children (27). A more recent study using diverse age groups observed an inverse association between plasma adropin concentrations and indices of atherogenic hypercholesterolemia (total cholesterol, LDL-C, and nonHDL-C) in males possibly mediated by suppression of ENHO expression by cholesterol (8). In adults, plasma adropin concentrations are positively correlated with dietary fat intake and inversely with carbohydrate intake (26, 28) and increase during fructose consumption (29). Differences in dietary preferences (fat versus carbohydrates) could be a confounding variable in cross-sectional human studies examining the relationship between circulating adropin levels and indices of cardio-metabolic risk.
Here, we report the results from two studies investigating adropin physiology in nonhuman primates. Nonhuman primates are useful models for translating preclinical data obtained using rodents to clinical studies in humans (30–32). Other advantages to using nonhuman primate models include minimizing variability from diet and personal history and access to a number of tissues that cannot normally be obtained in humans. Baboons (Papio anubis) and rhesus macaques (Macaca mulatta) belong to a closely related tribe (Papionini) that diverged from lineages leading to the evolution of humans and great apes 25–28 million years ago (33). We used the transcriptome atlas of a nonhuman primate (baboon) (34) to profile diurnal ENHO expression. We then investigated the relationships between plasma adropin concentrations and indices and risk factors for metabolic dysregulation in a well-characterized rhesus macaque model of a high-sugar (fructose) diet–induced obesity, insulin resistance, and dyslipidemia (13, 31, 35).
Results
ENHO expression profile in baboon tissues
Comparisons of FPKM data averaged over 24 h between baboon tissues indicate abundant ENHO expression in the central nervous system, particularly in the amygdala and lateral and ventromedial areas of the hypothalamus (VMH) (Fig. 1A). Lower expression levels are observed in liver, kidney (medulla and cortex), ileum, lung, and testes and the pituitary, pineal, and adrenal glands (Fig. 1A). ENHO expression exhibits a predominantly diurnal pattern in most neural and peripheral tissues (Fig. 1B). However, in two areas of the brain exhibiting high expression (amygdala and VMH), expression was observed during both the light and dark periods.
Figure 1.
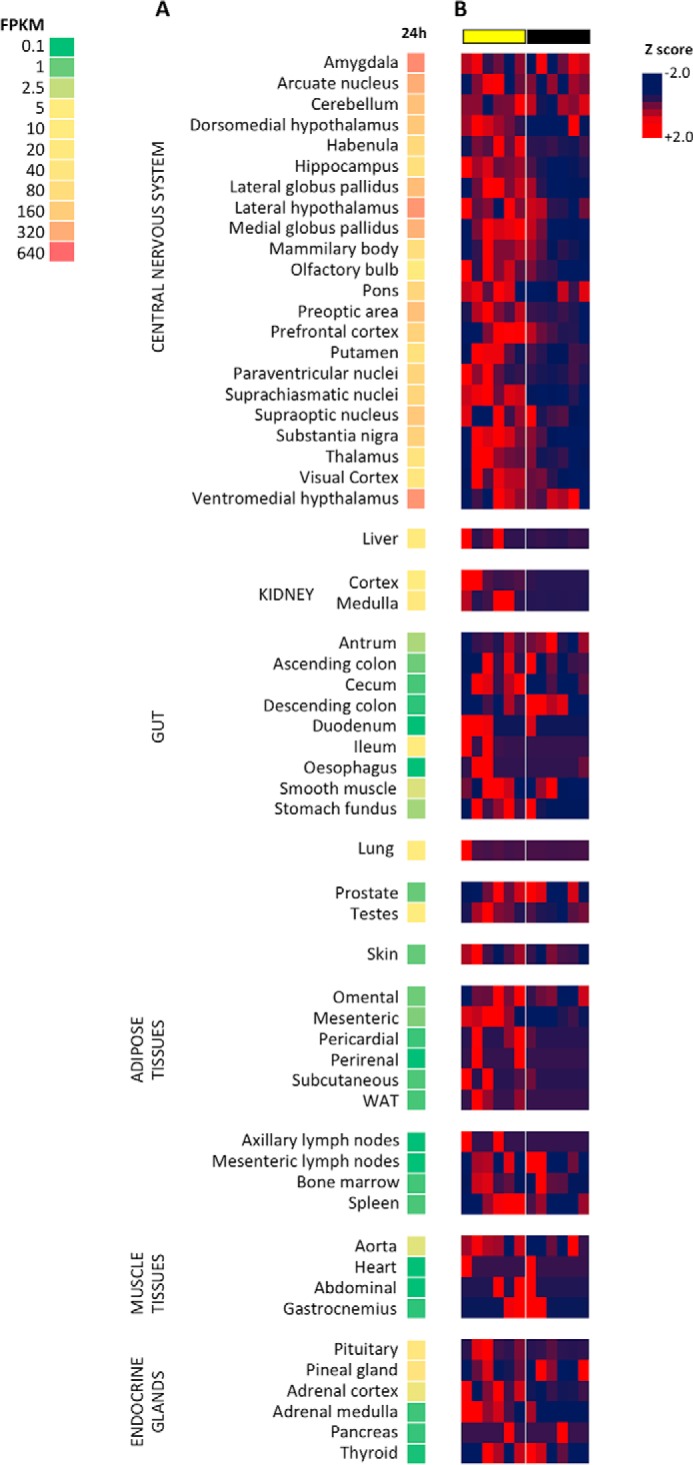
Relative ENHO expression in baboon tissues (A) and diurnal expression profile within tissues (B). The heat map in A is a single column that shows relative expression between tissues and is read from top to bottom. These data are derived from FPKM averaged over 24 h within each tissue. The heat maps in B show relative expression as a deviation from the 24-h mean (z-score) during a 24-h period within each tissue and should be read left (ZT0) to right (ZT24). The heat maps illustrate the diurnal profile of ENHO expression. Peak ENHO expression (red) is observed in the daytime in most tissues. Lights-on (day time) and lights-off (night time) are indicated by the yellow and black bars, respectively.
Studies in mice indicate liver adropin expression is nutritionally regulated (1, 8). In baboon liver, ENHO expression is limited to zeitgeber time 0 (ZT0, transition between dark and light phases; the first meal was provided at ZT3) and ZT6 (3 h before the second meal at ZT9) (Fig. 2A). A matrix of genes ranked by their Pearson coefficient compared with ENHO indicates over 1200 genes share this profile, using a correlation coefficient (r) of >0.7 as selection criteria (Fig. 2B). Hepatic ENHO expression also appears to be mutually exclusive (r < −0.7) for a similar number of genes (Fig. 2B). Global analysis identified “MIKKELSEN_MCV6_HCP_WITH_H3K27ME3” and “MIKKELSEN_MEF_HCP_WITH_H3K27ME3” as the top two gene sets enriched in the population of ENHO co-regulated genes in liver (Table S1). The promoter regions of these genes contain markers of high CpG density and epigenetically silent histone H3K27 trimethylation (H3K27me3). These genes may thus share a common regulatory pathway involving CpG-coupled epigenetic mechanisms, possibly acting in a concerted fashion. A broad CpG island domain found 5′ of the ENHO gene spans the entire promoter and exon 1 regions. This CpG island sequence is highly conserved across 46 different species, indicated by high phastCon and SiPhy scores (data not shown). DNA methylation and ChIP-Seq results from HepG2 cells published by the ENCODE Consortium (36) indicated heavy methylation of the ENHO gene promoter at the CpG island region. Histone H3 was also strongly methylated at H3K27, generating extensive H3K27me3 repressive markers surrounding the entire ENHO promoter region. The active histone marker (acetyl-H3K27 (H3K27ac)) is absent at the ENHO promoter (data not shown).
Figure 2.
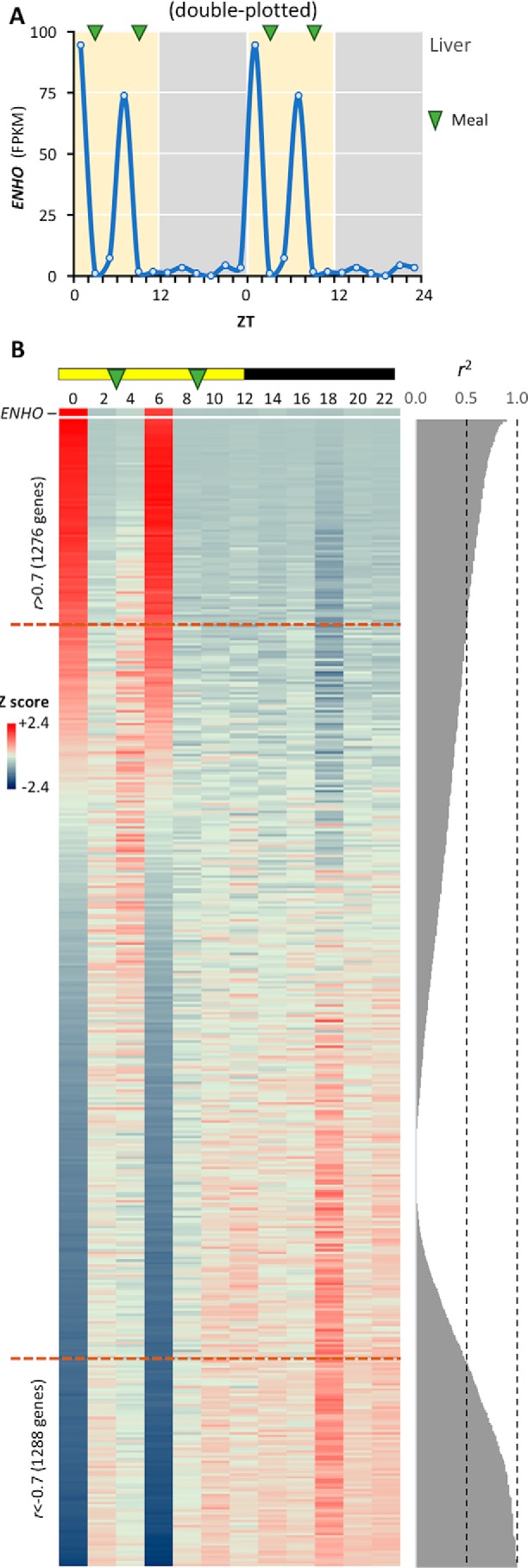
Hepatic ENHO expression shares a dynamic expression profile with >1200 genes. A, diurnal profile of hepatic ENHO expression shown as a line graph. Meal times are indicated by green arrows. B, heat map comparing profile of ENHO expression (top) with genes ranked by their correlation coefficient (r, high to low when read from the top down) with ENHO. The coefficient of determination (R2) is shown to the right. Lights-on (day time) and lights-off (night time) are indicated by the yellow and black bar at the top of the figure. There are 1276 genes showing a marked positive association and whose expression is mutually inclusive (r < −0.7) with ENHO. For a similar number of genes, ENHO expression appears be mutually exclusive (ENHO is highly expressed as indicated by red coloring and their expression is repressed as indicated by blue coloring).
Studies in rodents have linked adropin to glucose and lipid metabolism (1, 3, 9, 10, 12). In the baboon liver, genes involved in glucose metabolism (for example hexokinase-1, phosphofructokinase, and GLUT1), and in G-protein–coupled receptor and receptor tyrosine kinase signaling exhibit expression profiles similar to ENHO (Fig. S1). Lipid and lipoprotein gene sets/pathways are also enriched in genes that are negatively correlated with ENHO expression (e.g. “REACTOME_LIPOPROTEIN_METABOLISM”). To confirm this, we extracted the list of genes contributing to the negative association (NES = −2.16, padj = 6.1e-03) reported by GSEA. All of these genes exhibit an expression profile that is negatively correlated with ENHO (Table S2).
Other nonneural tissues with relatively robust levels of diurnal expression (Fig. 1A) also exhibit dynamic profiles of expression with discrete peaks (Fig. S2A). An unbiased analysis examining gene clusters correlating with ENHO expression in all tissues indicates strong relationships between liver, kidney medulla, lung, and ileum (Fig. S3A). Using r >0.9 as selection criteria in the correlation matrix, 426 genes exhibit an expression profile common in these tissues (Fig. S3B and Table S3). As observed in liver, this cluster is enriched for epigenetic silencing by histone H3K27me3 (p = 4.9 × 10−46). However, this cluster is also enriched for genes linked to neural function and neurodegenerative diseases (supporting Results, GO_426_geneset). Of note, this cluster includes genes encoding secreted peptides' cholecystokinin (CCK) involved in appetite control (37) and growth differentiation factor 11 (GDF11) implicated in development and aging (38). Diurnal ENHO expression in the ileum also correlates strongly with expression of CCK and apolipoproteins (ApoA4 and ApoE) (Fig. S2B).
Low plasma adropin concentrations signal metabolic dysregulation in rhesus macaques
We next tested the hypothesis that low plasma adropin concentrations signal increased risk for metabolic dysregulation. Fasting plasma adropin concentrations were measured in rhesus macaques at baseline and when challenged with a high-sugar (fructose) beverage diet for 3 months. Biometric and metabolic parameters from the animals used for this study have been published (39). In brief, dietary fructose supplementation rapidly induces weight gain, increases of fasting plasma insulin, leptin, triglycerides (TG), ApoC3, and ApoE, and decreases plasma adiponectin concentrations (39).
At baseline, simple modeling identified correlations between fasting plasma concentrations of adropin and leptin (ρ = −0.476, p < 0.001, Fig. 3A), glucose (ρ = −0.523, p < 0.001, Fig. 3B), and ApoA1 (ρ = −0.352, p < 0.01). Fasting glucose concentrations >100 mg/dl are used as diagnostic criteria for type 2 diabetes in rhesus macaques (40–43). In this study, animals with type 2 diabetes all have low plasma adropin concentrations (Fig. 3B). There is, however, no consistent correlation between fasting plasma concentrations of adropin and insulin at baseline (Fig. 3C). Elevated circulating leptin concentrations in animals with low plasma adropin concentrations suggest greater body adiposity. However, body weight does not correlate with plasma adropin concentrations (Fig. S4, A and B).
Figure 3.

Relationships between plasma adropin concentrations and fasting plasma concentrations of leptin (A), glucose (B), insulin (C), and ApoC3 (D) in male rhesus macaques (n = 59). Scatterplots present the relationships at baseline (chow-fed) and after 1 and 3 months of consuming fructose-sweetened beverages (300 kcal/day). Fasting plasma glucose concentrations exceeding 100 mg/dl (indicated with red symbols, orange shading in B) have been used as a diagnostic criteria for type 2 diabetes in rhesus macaques (38–41). Of the seven animals with fasting glucose >100 mg/dl after 3 months of fructose, four had glucose levels >100 mg/dl at baseline (>50%). The fasting glucose levels of the other three animal at baseline were higher (85, 83, and 92 mg/dl) relative to the group average (mean 80 mg/dl, S.D. 15 mg/dl).
This observation could indicate a correlation between plasma adropin concentration and partitioning of nutrients between fat and lean tissues. A nutrient partitioning phenotype is observed in male B6 adropin transgenic mice. Adropin knockout B6 mice exhibit increased fat mass while having normal body weight (11). Comparison of body composition in adropin transgenic (AdrTG) B6 mice and wildtype (WT) littermate controls (mean age, 16.6 weeks; S.D., 3.4 weeks; range 11.4–20.4 weeks) further supports this finding. Body weight is not significantly different (age-adjusted weight for AdrTG, 31.2 ± 0.7 g; WT, 30.8 ± 0.7 g) (Fig. S4C). However, relative to controls AdrTG exhibit reduced fat mass (3.9 ± 0.4 versus 5.6 ± 0.4 g, age- and weight-adjusted, p < 0.01) and increased fat-free mass (19.1 ± 0.2 versus 17.9 ± 0.2 g, age- and weight-adjusted, p < 0.005) (Fig. S4, C–E).
During fructose consumption, animals with low adropin concentrations exhibited more pronounced increases of plasma leptin (Fig. 3A). Fasting hyperglycemia appears more severe in animals with type 2 diabetes, and again is only observed in animals with low plasma adropin concentration (Fig. 3B).
We employed multiple linear regression to determine correlates of plasma adropin concentrations based on age, body weight, and indices of glucose homeostasis and lipid metabolism at each time point. In general terms, indices of adiposity and lipoprotein metabolism are strong predictors of plasma adropin concentration (R2 = 0.74, 0.71, and 0.74 at baseline and after 1 or 3 months of fructose, all p < 0.001) (Table 1). Leptin, HDL-C, ApoA1 and ApoC3 are highly significant predictors of plasma adropin concentrations, irrespective of diet (Table 1). After 3 months of fructose consumption and dyslipidemia, TG and ApoB are also significant predictors of plasma adropin concentrations. Plasma adropin and ApoC3 concentrations exhibit an inverse correlation that is enhanced during fructose consumption (Fig. 3D). However, relationships between plasma concentrations of adropin, HDL-C, and ApoA1 appear less robust when assessed individually (Fig. S5).
Table 1.
Calculation of plasma adropin concentration using multiple linear regression in male rhesus macaques on chow, and after 1 or 3 months of consuming a fructose beverage (300 kcal/d) (n = 59)
Log-transformed data were used for the analysis. The 1st column presents the independent variables, and the next 3 columns present results from the analysis of data from animals collected at specific time points in the study: baseline (chow) and after 1 or 3 months of fructose consumption. R2 and p values are shown for each calculation in the 1st row. At each time point in the study, modeling was successful in calculating plasma adropin concentration using the variables described in the 1st column, explaining 70–74% of the variation in plasma adropin concentration between individual animals. B is the coefficient, representing the independent contributions of the independent variables (age, body weight, glucose, insulin, etc.) to the prediction of the dependent variable (adropin). The t statistic is the coefficient divided by its standard error.

Relationships between baseline plasma adropin concentration and fructose-induced weight gain, insulin resistance, and dyslipidemia were explored by separating the animals into three groups (tertiles) (low adropin, n = 20; medium adropin, n = 19; and high adropin, n = 20) (Table 2, 3). Animals in the low-adropin group had significantly higher fasting glucose concentrations compared with the high-adropin group, irrespective of diet (Table 2). Differences in fasting insulin between adropin groups was significant (p < 0.05) when two outliers from the low-adropin group with insulin values >600 milliunits/ml were excluded (>1.5 times the interquartile range). However, it is notable that these extreme insulin values after 3 months on the fructose beverage diet were limited to animals in the low-adropin group (Fig. 3C). Fructose-induced increases of plasma leptin concentrations (Δleptin) adjusted for age and baseline values are also significantly higher in animals in the low-adropin group (Table 2). Plasma ApoC3 concentrations exhibit marked (50%) and significant differences between adropin groups (Table 3), consistent with an inverse association (Fig. 3D). ApoC3 regulates lipoprotein metabolism to increase TG (44, 45). Animals with low baseline plasma adropin levels appear to have a greater propensity for developing hypertriglyceridemia after 3 months of fructose consumption (Fig. S6). However, there are no significant correlations between adropin and plasma concentrations of IDL or VLDL particles (Fig. S6).
Table 2.
Indices of glucose control in rhesus macaques grouped by low, medium, or high baseline plasma adropin concentration
Age is reported as mean ± S.D. Measurements at baseline, 1, and 3 months are shown as estimated marginal means ± S.E.; covariates are indicated in the table subheading. Changes relative to baseline (Δ) after 3 months on fructose are estimated marginal means ± S.E. adjusted for age and baseline values. The increase in plasma leptin concentrations indicated in the final row (Δ) is after 3 months of fructose consumption. * indicates significantly different from medium (p < 0.05) and high (p < 0.01) adropin groups. ** indicates significantly different from medium and high adropin groups (p < 0.005). #, ## indicates significantly different from low adropin group (#, p < 0.05; ##, p < 0.01).

a Differences between all groups are highly significant, p < 0.001.
Table 3.
Indices of lipid metabolism
Data shown are estimate marginal means ± S.E. adjusted for age and body weight. Data are significantly different from low- and medium-adropin groups, *, p < 0.05; data are significantly different from medium- and high-adropin groups, **, p < 0.05 versus medium = 0.001 versus low; and data are significantly different from low-adropin group, #, p < 0.005.

Animals in the high-adropin group appear to have consistently higher LDL-C levels during the study, although the difference is significant only at the 3-month time point (Table 3). Fasting plasma adropin concentrations also exhibit significant positive relationships with LDL particles at baseline (LDL-4C, ρ = 0.386; LDL-4B, ρ = 0.485; LDL-4A, ρ = 0.377; p < 0.005; LDL3B, ρ = 0.308, p < 0.05; see Table S4 for plasma lipoprotein particle concentrations at baseline) but not during fructose consumption (data not shown).
Correlations between fructose-induced changes of plasma adropin and ApoA1 concentrations
Overall, a small but highly significant increase of plasma adropin concentrations is observed after 1 month of fructose supplementation (diet effect, p < 0.001 by Friedman's ANOVA; mean ± S.D. for plasma adropin concentration in nanograms/ml at baseline, 1.69 ± 0.92; 1 month of fructose, 1.91 ± 0.99; 3 months of fructose, 1.68 ± 0.90; 1 month versus pre- and 3 months of fructose, p < 0.001, n = 59). In humans, a “responder/nonresponder” situation is observed with the effect of consuming fructose beverages as 25% of daily energy requirements on plasma adropin concentrations (29). The impact of fructose consumption on plasma adropin concentrations also varies markedly between animals (Fig. S7, A and B; Δadropin range from −0.7 to +1.3 ng/ml). Multiple linear regression identified ΔApoA1 as the strongest predictor of Δadropin (t = 2.846, p = 0.005). Comparing ΔApoA1 and Δadropin confirmed a modest association (Fig. 4, A and B). Although highly significant (p = 0.001), the model explains only ∼31% of the variation in Δadropin (R2 = 0.306). Independent variables included in the model were Δbody weight, age, and Δ pooled from the 1-month- and 3-month–sampling for fasting glucose, plasma concentrations of hormones involved in glucose and lipid homeostasis, apolipoproteins, total lipids, and lipoprotein–cholesterol levels.
Figure 4.

Relationships between fructose-induced changes in ApoA1 (A and B), TG (C and D), and ApoC3 (E and F). The data shown are for fructose-induced changes (Δ) after 1 month (A, C, and E) and 3 months (B, D, and F). The proportion of variance dependent on the variable (coefficient of determination, R2 with p value) is shown in each graph.
Declining adropin signals increase risk for fructose-induced atherogenic dyslipidemia
Of note, animals exhibiting decreases of plasma adropin concentration have larger increases of plasma concentrations of TG (Fig. 4, C and D) and ApoC3 concentrations (Fig. 4, E and F). The physiological significance of fructose-induced changes of plasma adropin in individual animals was further investigated by ranking animals by Δadropin after 1 month of fructose consumption. The animals were separated into three groups that exhibit a robust but transient increase of plasma adropin (“high-responders,” n = 20), an intermediate but persistent increase (“mid-responders,” n = 19), or decreases of adropin (“low-responders,” n = 20) (Fig. 5, A and B).
Figure 5.
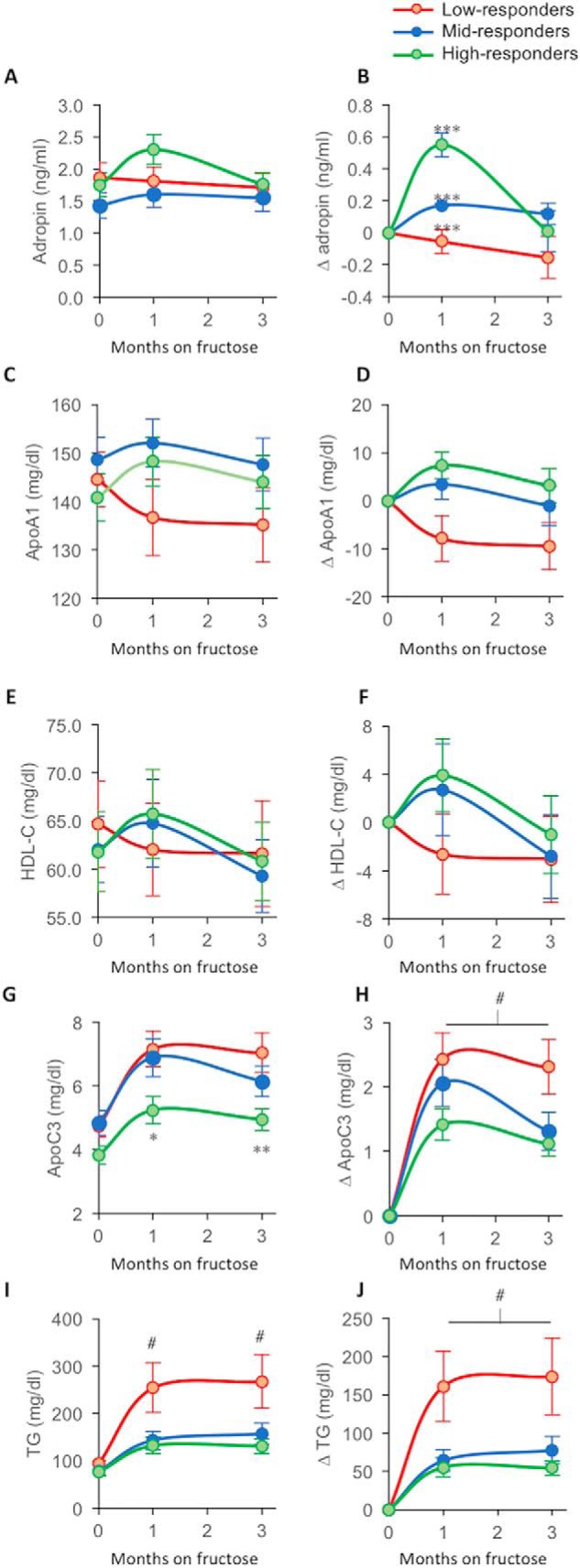
Lipid parameters in subpopulations of rhesus macaques exhibiting low, medium, or high responses of plasma adropin concentrations after 1 month of fructose consumption. Animals were ranked by changes of plasma adropin concentrations after 1 month of fructose consumption. They were then subdivided into three groups: high-responders, n = 20, green lines/symbols; mid-responders, n = 19, blue lines/symbols; and low-responders, n = 20, red lines/symbols. Shown are the averages for each time point based on the ranking at the 1-month time point. Plasma concentrations of the variable defined in the y axis label are shown in A, C, E, G, and H. Fructose-induced Δs of plasma concentrations of the variable defined in the y axis label (1- and 3-month values subtracted from baseline) are shown in B, D, F, H, and J. “High-responders” exhibit a marked increase in plasma adropin concentration at the 1-month time point (B). ***, p < 0.001 between all groups; #, p < 0.05 versus mid- and high-responders; *, p < 0.05; **, p < 0.01 versus low-responders (within time points). #, low- versus medium-responder, p < 0.05, versus high-responder, p < 0.01 (ANOVA with repeated measures).
Fructose-induced changes of adropin, ApoA1, and HDL-C (Apoa1 is a major apoliprotein in HDL) were similar between groups (Fig. 5, C–F). However, the differences for ApoA1 and HDL-C were not statistically significant. For plasma concentrations of ApoC3 and TG, a different pattern was evident. Animals exhibiting the largest increases of plasma adropin concentrations at 1 month have lower ApoC3 (estimated marginal means of fasting ApoC3 for low-responders, 6.3 ± 0.4 mg/dl; mid-responders, 6.0 ± 0.4; high-responders, 4.7 ± 0.4 mg/dl; low versus high, p < 0.01). They also exhibit less-pronounced increases of ApoC3 during fructose consumption (Fig. 5, G and H). A similar pattern was evident for TG (estimated marginal means of fasting TG for responders, 205 ± 25 mg/dl; mid-responders, 126 ± 25; high-responders, 113 ± 25 mg/dl; low versus medium and high, p < 0.05). Low-adropin responders have larger increases of plasma TG in response to fructose consumption (Fig. 5, I and J). For both ApoC3 and TG, repeated measures ANOVA indicated a significant impact of time on the fructose diet (p < 0.001) and a significant interaction between diet and Δadropin-group (low-, medium-, or high-responder; p < 0.05). Analysis of lipoprotein particles fractionated by size also indicated correlations with risk for atherogenic dyslipidemia during fructose consumption with low-responders exhibiting more pronounced increases of small, dense LDL (LDL4) (Fig. S8).
Discussion
There are two major findings from these studies. First, data from the baboon study are indicative of a diurnal pattern of ENHO expression in most tissues examined. These studies suggest co-regulation of ENHO expression and genes involved in hepatic carbohydrate and lipoprotein metabolism. Second, data from the rhesus macaque study indicate low plasma adropin concentrations are associated with indices of lipoprotein metabolism and features of the metabolic syndrome (fasting hyperglycemia, dyslipidemia, and hyperleptinemia). The relationship between plasma concentrations of adropin and leptin are independent of body weight. This observation could indicate a correlation between circulating adropin and nutrient partitioning between fat and lean tissues, with low plasma adropin concentrations related to increases of body adiposity.
Overall, the expression profiling results in a nonhuman primate are consistent with previous data from mice indicating regulation by the biological clock (8). In male B6 mice, liver ENHO expression peaks late in the dark cycle when nocturnal mice are most active. In diurnal baboons, ENHO expression peaks during the daytime in the liver, and in most other tissues examined with significant levels of ENHO expression. The expression profiling data are also consistent with elevated plasma adropin concentrations around meal times during the daytime, and a steady decline at night-time, in rhesus macaques (8). The finding that liver ENHO expression peaks 3 h prior to both meals may be coincidental. Another caveat is the small sample size. It is nevertheless consistent with a gene linking metabolism with nutrient intake. Further studies examining the link between meal timing and hepatic adropin expression are clearly needed.
The finding that ENHO expression shares a dynamic expression profile with a cluster of genes regulated by histone H3K27 trimethylation is novel. H3K27me3 is an evolutionarily conserved pathway for repressing gene expression (46, 47). Regulation of gene expression by H3K27me3 has classically been linked to control of gene expression during development and in tumorigenesis. Interesting hypotheses raised by this observation include links between epigenetic silencing of transcription and variation in ENHO expression between tissues, and with plasma adropin concentrations that vary markedly in humans (8, 48).
ENHO expression in the periphery also clusters with genes linked to neural functions. The significance of this finding is unclear. One possible interpretation is evidence of interactions between adropin signaling and the activity of the somatic and autonomic nervous systems in peripheral tissues.
Carbohydrate and lipid metabolism in the liver are controlled by elements of the biological clock (49, 50). In this study, peak hepatic ENHO expression during the daytime appeared to coincide with phases of increased receptivity to endocrine and autonomic inputs. Expression of enzymes involved in signal transduction pathways emanating from G-protein–coupled receptors and receptor tyrosine kinases peaks exhibits similarly dynamic profiles with peaks in expression coinciding with those for ENHO. Expression profiling data also indicate the potential for co-regulation of the expression of adropin and genes involved in carbohydrate and lipid metabolism. This observation may also explain the associations between plasma adropin concentrations and apolipoproteins observed in rhesus macaques.
Another interesting observation is the tight correlation between ENHO expression and gut-secreted peptides linked to the regulation of satiety and intestinal motility (CCK and ApoA4) and lipid absorption (ApoA4 and ApoE) in the ileum (37, 51). A caveat to interpreting these data are that we do not know whether differences in ENHO expression translates to the level of protein. However, diurnal rhythms in ApoA4 and ApoE mRNA and protein have been reported in rat tissues (52–54).
The second study presented here examined the relationships between plasma adropin concentrations and circulating parameters of carbohydrate and lipid metabolism. We investigated whether plasma adropin concentrations are predictive of metabolic dysregulation and sensitivity in response to a high-sugar diet. Experiments using B6 mice indicate that even modest suppression of adropin is related to insulin resistance and more pronounced glucose intolerance with diet-induced obesity (11, 15).
Rhesus macaques weigh less and have fasting glucose levels that are typically ∼20–30 mg/dl lower than humans (40–43). Type 2 diabetes occurs spontaneously, particularly in animals with elevated body fat mass (43). There is an inverse relationship between plasma adropin concentrations and risk of dysregulation of glucose metabolism (i.e. fasting hyperglycemia/type 2 diabetes) in rhesus macaques. Animals with low adropin exhibit increased incidence of overt type 2 diabetes and elevated plasma leptin, indicative of increased body adiposity. In these animals, fructose consumption may be considered a “risk-amplifier.” Fructose consumption induces further weight gain, increases adiposity, and accelerates metabolic dysregulation. Animals with low plasma adropin concentrations may thus exhibit a more rapid progression of pre-existing metabolic conditions toward a more overt disease state (type 2 diabetes and adverse changes of apolipoprotein profiles associated with atherosclerosis in humans). Although the relationship with insulin is less clear, the development of more severe hyperinsulinemia with fructose consumption is limited to animals with low plasma adropin concentrations. Low plasma adropin is also closely related to elevations of plasma ApoC3, indicating increased risk for hypertriglyceridemia resulting from the effects of ApoC3 to augment hepatic lipogenesis and impair TG clearance (45, 55–57).
High circulating leptin/adiponectin levels in animals with low adropin despite normal body weight suggest an increased adipose–to–lean mass ratio. A “TOFI” (thin-outside–fat-inside) phenotype would also be consistent with insulin resistance (58). High plasma adropin concentrations are associated with a leaner phenotype in young men; however, this association is not observed later in life (8, 59). Whether low plasma adropin levels signal increased visceral fat accumulation in rhesus macaques, or in young adult humans, will require further study.
Plasma adropin concentrations at baseline are positively correlated with age (ρ = 0.373, p < 0.005). Animals in the low plasma adropin tertile were actually significantly younger (∼2 years) compared with the mid- and upper tertiles. Indeed, animals in the low-adropin group had yet to enter middle age (mean age, 10.4 years, range 7.8–14.8 years; rhesus macaque life expectancy is ∼30 years). Aging per se is therefore not a factor in the relationships between low circulating adropin levels and increased risk for metabolic dysregulation. However, this study suggests increases of obesity-related co-morbidities that could reduce life expectancy in animals with low circulating adropin.
The positive correlation between plasma adropin concentration and age in this study differs from previous results from humans and rodents showing a decline with aging (8, 48, 60, 61). There are two potential explanations for the discrepancy that also serve as a precaution when comparing results between studies. First, type 2 diabetes is often used as an exclusion criterion in studies involving human subjects, introducing potential bias. Plasma adropin concentrations have been reported to be lower in people with type 2 diabetes (15, 23–25). Aging and metabolic dysregulation associated with type 2 diabetes may act through independent and common mechanisms to suppress adropin expression. The relative mixture of young or aged and healthy or diabetic subjects could thus influence the observed associations. Second, another bias is introduced because the animals used in rodent studies are almost always highly inbred (e.g. B6 mice) and are maintained in a more controlled environment. This would reduce the contribution of genetic and environmental variability to the effects of aging on adropin expression and circulating adropin levels.
Studies using mice and cultured rat atrial cells indicate adropin rapidly improves insulin action and enhances glycolysis (3, 9, 10). Enhanced oxidation of glucose in skeletal muscle provides a plausible mechanism underlying the lean phenotype suggested by the leptin/adiponectin data and observed in this study using adropin transgenic mice. Enhanced oxidative glucose disposal in muscle could also improve lipid metabolism through shuttling carbohydrates away from lipogenesis. Further studies exploring the relationship between plasma adropin concentrations and glucose metabolism are clearly needed. In addition, further experiments examining the acute response of nonhuman primates with low adropin to the administration of exogenous adropin34–76 are warranted based on these results.
It is also important to consider the potential impact of a combination of risk factors, including elevated leptin and ApoC3 levels, when interpreting the phenotype of animals with low plasma adropin concentrations. Relationships between low circulating adropin concentrations and the heightened risk for metabolic dysfunction cannot be interpreted as a “direct” consequence of altered adropin action. For example, ApoC3 is positively associated with atherogenic dyslipidemia and the risk of cardiovascular disease (62–64). Accordingly, low adropin levels may be secondary to metabolic dysregulation driven by independent stochastic variables. Indeed, a modest decline of plasma adropin concentrations associated with more severe dyslipidemia. However, adropin-deficient mice exhibit insulin resistance and exaggerated impaired glucose tolerance with diet-induced obesity (11), indicating low adropin increases risk factors for metabolic disease. Clearly, further studies using a primate model are needed to ascertain causality between differences in plasma adropin concentrations and risk of metabolic dysregulation. These would include interventions to determine the physiological impact of increasing plasma adropin concentration in animals with low plasma adropin concentrations.
Plasma adropin concentrations are correlated with self-reported and observed macronutrient selection in humans (26, 28). These studies suggest a relationship between plasma adropin concentrations and the ratio of fat to carbohydrate in the diet. Further studies are needed to determine whether diet affects adropin expression in primates or adropin expression correlates with macronutrient preferences. The latter finding would not be without precedent. For example, fibroblast growth factor-21 (FGF21) regulates consumption of protein and sugars (65, 66), whereas melanocortin-4 receptor (MC4R) signaling in the central nervous system regulates fat and sucrose preferences (67).
In summary, these results strongly indicate that low adropin expression is associated with metabolic dysregulation in a nonhuman primate model of sugar (fructose) diet-induced metabolic syndrome. A strength of the study is that the data were derived from experiments conducted in nonhuman primates, suggesting a higher probability of clinical relevance (30–32).The diurnal profile of ENHO transcript indicates that peak adropin expression and signaling are temporally linked to food intake and occur when feeding behavior is maximal in primates. Peaks in ENHO expression coincide with those of genes linked to signaling from cell-surface G-protein–coupled receptors and receptor tyrosine kinases. If gene expression data are indicative of protein levels, then the peak hepatic ENHO expression appears to coincide with enhanced responsiveness of the liver to neural and hormonal signals of metabolic status. Low plasma adropin concentrations are predictive of a pre-diabetic phenotype at an earlier age in rhesus macaques and may also be related to increased body adiposity. These results provide a strong rationale for further studies investigating relationships between rhythms in adropin signaling and metabolic homeostasis, particularly the metabolic effects of adropin administration/replacement in nonhuman primate models.
Experimental procedures
In silico analysis in a nonhuman primate diurnal transcriptome atlas
Animal studies used to generate the atlas (GSE98965) were reported elsewhere by the laboratories responsible for conducting the experiments (34). Briefly, animals were acclimated to housing in semi-natural conditions under 12-h light/12-h dark cycles for 1 month; meals (fruits and primate meal pellets) were provided at ZT3 and ZT9. The dataset contains 29,202 genes from 108 tissues of P. anubis collected at 12 time points over 24 h (ZT00, ZT02, ZT04, ZT06, ZT08, ZT10, ZT12, ZT14, ZT16, ZT18, ZT20, and ZT22, ZT0 = start of the light cycle and ZT12 = start of the dark cycle).
Processed RPKM values were downloaded from the National Institutes of Health GEO website (GEO accession number GSE98965), decompressed, and analyzed in R and Bioconductor. Gene symbols were updated by querying the BioMart “panubis_gene_ensembl” dataset and matching Ensembl IDs using the R “biomaRt” package (68).
ENHO expression exhibits a dynamic profile in baboon tissues. To explore whether the profile is unique or shared by a cluster of genes regulated by the common regulatory pathway, we screened for genes exhibiting similar profiles during the light and dark periods. The dataset was consolidated by removing genes with zero counts at all time points. A Pearson correlation coefficient matrix was calculated between all gene pairings. Correlation coefficients were then extracted between the expression profile of all genes and ENHO. This matrix was ranked by correlation coefficient from the highest (+1, ENHO:ENHO) to lowest (−0.9471788, ENHO:ABCB7). An example correlation matrix is shown in Table S1. GSEA was then used to identify globally enriched or depleted gene sets/pathways in the Molecular Signatures Database (MSigDB Version 5.2, http://software.broadinstitute.org/gsea/msigdb/index.jsp)5 (69) in genes showing high- or low-correlation coefficients. We used R values as selection criteria (R >0.7 or < −0.7 for liver and >0.9 or < −0.9 for the unbiased screen of all genes in all tissues). To perform the GSEA, we examined specific gene sets/pathways, enrichment, or depletion among the ENHO-coexpressed genes using the fgsea package (http://bioconductor.org/packages/release/bioc/html/fgsea.html)5 in R (69, 70, 76). Enriched or depleted genes within significant gene sets/pathways were extracted using the “fgsea” function provided in the package.
For comparing the pattern of correlations between ENHO expression and gene clusters between tissues, we computed Pearson correlation coefficients between ENHO and all other genes in the dataset based on expression levels across the different time points, producing a column vector for each tissue. We then computed the distance between each pair of the tissues by obtaining a sample-wise correlation matrix using the column vectors obtained above. Finally, the correlation matrix was subjected to unsupervised hierarchical clustering to identify clusters of tissues with similar ENHO regulatory pathways (i.e. time-dependent expression of ENHO-coregulated genes).
ENHO promoter features were analyzed using Integrative Genome Browser (Version 2.4). Various tracks, including “CpG islands,” “SiPhy” score using 10-mer (sequence conservation score, higher scores indicate stronger conservation) (71), and “phastCons” (conservation score based on phylogenetic hidden Markov model) (72), were used to analyze CpG density and sequence conservation of the ENHO promoter region across 46 vertebrate species. ENCODE datasets (https://www.encodeproject.org)5 (36) revealed DNA methylation and histone methylation status at the ENHO promoter region in HepG2 cells.
Rhesus macaque model of diet-induced obesity, insulin resistance, and dyslipidemia
Plasma adropin concentrations were measured in samples from a previously published group of rhesus macaques (31, 35, 39). In brief, 59 adult male rhesus macaques maintained by the California National Primate Research Center (CNPRC, University of California, Davis) were provided ad libitum access to a high-protein diet (LabDiet 5047: 30.4% kcal/protein, 13.2% kcal/fat, and 56.4% kcal/carbohydrates) and water. Dietary carbohydrates are primarily starch; fats are primarily linoleic acid and monounsaturated and saturated fatty acids. After collection of baseline biometric and plasma samples, animals were provided 500 ml/day of a flavored 15% fructose solution providing 75 g or ∼300 kcal/day. Fasting plasma samples and body weight measurements were collected after 1 and 3 months. The research protocols were approved both by the Institutional Animal Care and Use Committees of the University of California, Davis, and conducted in accordance with the Department of Agriculture Animal Welfare Act and National Institutes of Health Guidelines for the Care and Use of Animals.
Blood chemistries
Plasma insulin, leptin, and adiponectin concentrations were measured by RIA (EMD Millipore, Billerica, MA). Plasma adropin concentrations were measured by ELISA (Peninsula Laboratories, San Carlos, CA). Plasma glucose concentrations were measured with a YSI Glucose Analyzer (YSI Life Sciences). Plasma total cholesterol, HDL cholesterol (HDL-C), direct LDL cholesterol (LDL-C), TG, ApoA1, ApoB, ApoC3, and ApoE concentrations were determined by using a Polychem Chemistry Analyzer (PolyMedCo). VLDL-C was calculated by subtracting HDL-C and LDL-C from total cholesterol.
Measurement of lipoprotein particle size
Particle concentrations of VLDL, IDL, LDL, and HDL subfractions were analyzed in specific particle-size intervals using ion mobility (IM), which uniquely allows for direct particle quantification as a function of particle diameter (73), following a procedure to remove other plasma proteins (74). The IM instrument utilizes an electrospray to create an aerosol of particles, which then pass through a differential mobility analyzer coupled to a particle counter.
Particle concentrations (nmol/liter) were determined for subfractions defined by the following size intervals (nm): VLDL: large (42.40–54.70), medium (33.50–42.39), and small (29.60–33.49); IDL: large (25.00–29.59) and small (23.33–24.99); LDL: large (22.0–23.32), medium (21.41–21.99), small (20.82–21.40), and very small (18.0–20.81); and HDL: large (10.50–14.50) and small (7.65–10.49). Peak LDL diameter (nm) was determined as described previously (73).
Adropin transgenic mice
Adropin transgenic mice expressing a synthetic gene driven by the human β-actin promoter have been described previously (1, 8, 10). Fat mass, fat-free mass, and free H2O measured by NMR (Bruker Minispec; Bruker, Billerica, MA) and analyzed using regression was described previously (75). Studies using adropin transgenic mice were reviewed and approved by the Saint Louis University Institutional Animal Care and Use Committee.
Statistical analysis
Statistical analysis used IBM SPSS Statistics (Version 24). Relationships between adropin and indices of body weight and metabolic homeostasis were assessed using log-transformed data in simple modeling and multiple regression. Animals were also separated into groups ranked by baseline plasma adropin concentration (low, n = 20; medium, n = 19; or high, n = 20). Group differences were initially assessed for significance using parametric or, if required, nonparametric tests (Kruskal-Wallis test). Interactions between diet and adropin groups were ranked by baseline or Δs using repeated measures ANOVA. Variables exhibiting significant differences between adropin groups were further explored using univariate analysis (independent variable: adropin group) with post-hoc comparisons. Potentially confounding variables (age, body weight) were controlled for as required by inclusion as covariates in the analysis. Determinants of plasma adropin concentrations were calculated using linear regression with log-transformed data.
Author contributions
A. A. Butler, A. A. Bremer, and P. J. H. conceptualization; A. A. Butler, C. A. P., J. R. S., J. L. G., S. K., and R. M. K. data curation; A. A. Butler, J. Z., C. A. P., J. R. S., J. L. G., K. L. S., and P. J. H. formal analysis; A. A. Butler, J. Z., J. R. S., and J. L. G. investigation; A. A. Butler visualization; A. A. Butler writing-original draft; J. Z., C. A. P., J. R. S., J. L. G., K. L. S., R. M. K., A. A. Bremer, and P. J. H. writing-review and editing; J. L. G., S. K., R. M. K., and P. J. H. methodology; K. L. S. and R. M. K. supervision; P. J. H. resources; P. J. H. funding acquisition; P. J. H. project administration.
Acknowledgments
We thank Marinelle Nunez, Guoxia Chen, Vanessa Bakula, Ross Allen, Marinelle Nunez, Sarah Davis, Jenny Short, and the staff and administration of the California National Primate Research Center for their technical and logistical contributions and support for this study. We thank Professor Joel Eissenberg (Edward A. Doisy Dept. of Biochemistry and Molecular Biology, Saint Louis University School of Medicine) for comments during manuscript preparation. We also acknowledge the important contributions of the ENCODE consortium, and the laboratories that contributed data to the consortium and to the current project.
This work was supported in part by National Institutes of Health Grants R21NS108138, AT250099, and AT003645, the American Diabetes Association, California National Primate Research Center Base Grant OD-0111107, and UC Davis Clinical and Translational Science Center Grant UL1 RR024146. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Figs. S1–S8, Tables S1–S4, and supporting Results.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- DIO
- diet-induced obese
- ANOVA
- analysis of variance
- ROR
- retinoic acid–related orphan receptor
- LDL
- low density lipoprotein
- HDL
- high density lipoprotein
- IDL
- intermediate-density lipoprotein
- VLDL
- very-low density lipoprotein
- VMH
- ventromedial hypothalamus
- FPKM
- fragments per kb of transcript per million
- TG
- triglyceride
- AdrTG
- adropin transgenic
- IM
- ion mobility
- CCK
- cholecystokinin
- GSEA
- Gene Set Enrichment Analysis
- CpG
- cytosine-phosphate-guanine
- -C
- -cholesterol.
References
- 1. Kumar K. G., Trevaskis J. L., Lam D. D., Sutton G. M., Koza R. A., Chouljenko V. N., Kousoulas K. G., Rogers P. M., Kesterson R. A., Thearle M., Ferrante A. W. Jr., Mynatt R. L., Burris T. P., Dong J. Z., Halem H. A., et al. (2008) Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab. 8, 468–481 10.1016/j.cmet.2008.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 3. Thapa D., Stoner M. W., Zhang M., Xie B., Manning J. R., Guimaraes D., Shiva S., Jurczak M. J., and Scott I. (2018) Adropin regulates pyruvate dehydrogenase in cardiac cells via a novel GPCR–MAPK–PDK4 signaling pathway. Redox Biol. 18, 25–32 10.1016/j.redox.2018.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sato K., Yamashita T., Shirai R., Shibata K., Okano T., Yamaguchi M., Mori Y., Hirano T., and Watanabe T. (2018) Adropin contributes to anti-atherosclerosis by suppressing monocyte-endothelial cell adhesion and smooth muscle cell proliferation. Int. J. Mol. Sci. 19, E1293 10.3390/ijms19051293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yang C., DeMars K. M., Hawkins K. E., and Candelario-Jalil E. (2016) Adropin reduces paracellular permeability of rat brain endothelial cells exposed to ischemia-like conditions. Peptides 81, 29–37 10.1016/j.peptides.2016.03.009 [DOI] [PubMed] [Google Scholar]
- 6. Akcilar R., Kocak F. E., Simsek H., Akcilar A., Bayat Z., Ece E., and Kokdasgil H. (2016) Antidiabetic and hypolipidemic effects of adropinin streptozotocin-induced type 2 diabetic rats. Bratisl. Lek. Listy. 117, 100–105 [DOI] [PubMed] [Google Scholar]
- 7. Akcıilar R., Emel Koçak F., Şimşek H., Akcıilar A., Bayat Z., Ece E., and Kökdaşgil H. (2016) The effect of adropin on lipid and glucose metabolism in rats with hyperlipidemia. Iran J. Basic Med. Sci. 19, 245–251 [PMC free article] [PubMed] [Google Scholar]
- 8. Ghoshal S., Stevens J. R., Billon C., Girardet C., Sitaula S., Leon A. S., Rao D. C., Skinner J. S., Rankinen T., Bouchard C., Nuñez M. V., Stanhope K. L., Howatt D. A., Daugherty A., Zhang J., et al. (2018) Adropin: an endocrine link between the biological clock and cholesterol homeostasis. Mol. Metab. 8, 51–64 10.1016/j.molmet.2017.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gao S., McMillan R. P., Zhu Q., Lopaschuk G. D., Hulver M. W., and Butler A. A. (2015) Therapeutic effects of adropin on glucose tolerance and substrate utilization in diet-induced obese mice with insulin resistance. Mol. Metab. 4, 310–324 10.1016/j.molmet.2015.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gao S., McMillan R. P., Jacas J., Zhu Q., Li X., Kumar G. K., Casals N., Hegardt F. G., Robbins P. D., Lopaschuk G. D., Hulver M. W., and Butler A. A. (2014) Regulation of substrate oxidation preferences in muscle by the peptide hormone adropin. Diabetes 63, 3242–3252 10.2337/db14-0388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ganesh Kumar K., Zhang J., Gao S., Rossi J., McGuinness O. P., Halem H. H., Culler M. D., Mynatt R. L., and Butler A. A. (2012) Adropin deficiency is associated with increased adiposity and insulin resistance. Obesity 20, 1394–1402 10.1038/oby.2012.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thapa D., Xie B., Zhang M., Stoner M. W., Manning J. R., Huckestein B. R., Edmunds L. R., Mullett S. J., McTiernan C. F., Wendell S. G., Jurczak M. J., and Scott I. (2019) Adropin treatment restores cardiac glucose oxidation in pre-diabetic obese mice. J. Mol. Cell. Cardiol. 129, 174–178 10.1016/j.yjmcc.2019.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bremer A. A., Stanhope K. L., Graham J. L., Cummings B. P., Ampah S. B., Saville B. R., and Havel P. J. (2014) Fish oil supplementation ameliorates fructose-induced hypertriglyceridemia and insulin resistance in adult male rhesus macaques. J. Nutr. 144, 5–11 10.3945/jn.113.178061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wong C. M., Wang Y., Lee J. T., Huang Z., Wu D., Xu A., and Lam K. S. (2014) Adropin is a brain membrane-bound protein regulating physical activity via the NB-3/Notch signaling pathway in mice. J. Biol. Chem. 289, 25976–25986 10.1074/jbc.M114.576058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen S., Zeng K., Liu Q. C., Guo Z., Zhang S., Chen X. R., Lin J. H., Wen J. P., Zhao C. F., Lin X. H., and Gao F. (2017) Adropin deficiency worsens HFD-induced metabolic defects. Cell Death Dis. 8, e3008 10.1038/cddis.2017.362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuhla A., Hahn S., Butschkau A., Lange S., Wree A., and Vollmar B. (2014) Lifelong caloric restriction reprograms hepatic fat metabolism in mice. J. Gerontol. A Biol. Sci. Med. Sci. 69, 915–922 10.1093/gerona/glt160 [DOI] [PubMed] [Google Scholar]
- 17. Partridge C. G., Fawcett G. L., Wang B., Semenkovich C. F., and Cheverud J. M. (2014) The effect of dietary fat intake on hepatic gene expression in LG/J AND SM/J mice. BMC Genomics 15, 99 10.1186/1471-2164-15-99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bushkofsky J. R., Maguire M., Larsen M. C., Fong Y. H., and Jefcoate C. R. (2016) Cyp1b1 affects external control of mouse hepatocytes, fatty acid homeostasis and signaling involving HNF4α and PPARα. Arch. Biochem. Biophys. 597, 30–47 10.1016/j.abb.2016.03.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Larsen M. C., Bushkofsky J. R., Gorman T., Adhami V., Mukhtar H., Wang S., Reeder S. B., Sheibani N., and Jefcoate C. R. (2015) Cytochrome P450 1B1: an unexpected modulator of liver fatty acid homeostasis. Arch. Biochem. Biophys. 571, 21–39 10.1016/j.abb.2015.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kojetin D. J., and Burris T. P. (2014) REV-ERB and ROR nuclear receptors as drug targets. Nat. Rev. Drug Discov. 13, 197–216 10.1038/nrd4100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lazar M. A. (2016) in A Time for Metabolism and Hormones (Sassone-Corsi P., and Christen Y., eds) pp. 63–70, Springer, New York: [PubMed] [Google Scholar]
- 22. Bass J. (2016) in A Time for Metabolism and Hormones (Sassone-Corsi P., and Christen Y., eds) pp. 25–32, Springer, New York: [PubMed] [Google Scholar]
- 23. Zang H., Jiang F., Cheng X., Xu H., and Hu X. (2018) Serum adropin levels are decreased in Chinese type 2 diabetic patients and negatively correlated with body mass index. Endocr J. 65, 685–691 10.1507/endocrj.EJ18-0060 [DOI] [PubMed] [Google Scholar]
- 24. Beigi A., Shirzad N., Nikpour F., Nasli Esfahani E., Emamgholipour S., and Bandarian F. (2015) Association between serum adropin levels and gestational diabetes mellitus; a case-control study. Gynecol. Endocrinol. 31, 939–941 10.3109/09513590.2015.1081681 [DOI] [PubMed] [Google Scholar]
- 25. Celik E., Yilmaz E., Celik O., Ulas M., Turkcuoglu I., Karaer A., Simsek Y., Minareci Y., and Aydin S. (2013) Maternal and fetal adropin levels in gestational diabetes mellitus. J. Perinat. Med. 41, 375–380 10.1515/jpm-2012-0227 [DOI] [PubMed] [Google Scholar]
- 26. Stevens J. R., Kearney M. L., St-Onge M. P., Stanhope K. L., Havel P. J., Kanaley J. A., Thyfault J. P., Weiss E. P., and Butler A. A. (2016) Inverse association between carbohydrate consumption and plasma adropin concentrations in humans. Obesity 24, 1731–1740 10.1002/oby.21557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Orsso C. E., Butler A. A., Muehlbauer M. J., Cui H. N., Rubin D. A., Pakseresht M., Butler M. G., Prado C. M., Freemark M., and Haqq A. M. (2019) Obestatin and adropin in Prader-Willi syndrome and nonsyndromic obesity: associations with weight, BMI-z, and HOMA-IR. Pediatr. Obes. 14, e12493 10.1111/ijpo.12493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. St-Onge M. P., Shechter A., Shlisky J., Tam C. S., Gao S., Ravussin E., and Butler A. A. (2014) Fasting plasma adropin concentrations correlate with fat consumption in human females. Obesity 22, 1056–1063 10.1002/oby.20631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Butler A. A., St-Onge M. P., Siebert E. A., Medici V., Stanhope K. L., and Havel P. J. (2015) Differential responses of plasma adropin concentrations to dietary glucose or fructose consumption in humans. Sci. Rep. 5, 14691 10.1038/srep14691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kleinert M., Clemmensen C., Hofmann S. M., Moore M. C., Renner S., Woods S. C., Huypens P., Beckers J., de Angelis M. H., Schürmann A., Bakhti M., Klingenspor M., Heiman M., Cherrington A. D., Ristow M., et al. (2018) Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 14, 140–162 10.1038/nrendo.2017.161 [DOI] [PubMed] [Google Scholar]
- 31. Havel P. J., Kievit P., Comuzzie A. G., and Bremer A. A. (2017) Use and importance of nonhuman primates in metabolic disease research: current state of the field. ILAR J. 58, 251–268 10.1093/ilar/ilx031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cox L. A., Olivier M., Spradling-Reeves K., Karere G. M., Comuzzie A. G., and VandeBerg J. L. (2017) Nonhuman primates and translational research-cardiovascular disease. ILAR J. 58, 235–250 10.1093/ilar/ilx025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rogers J., and Gibbs R. A. (2014) Comparative primate genomics: emerging patterns of genome content and dynamics. Nat. Rev. Genet. 15, 347–359 10.1038/nrg3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mure L. S., Le H. D., Benegiamo G., Chang M. W., Rios L., Jillani N., Ngotho M., Kariuki T., Dkhissi-Benyahya O., Cooper H. M., and Panda S. (2018) Diurnal transcriptome atlas of a primate across major neural and peripheral tissues. Science 359, eaao0318 10.1126/science.aao0318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bremer A. A., Stanhope K. L., Graham J. L., Cummings B. P., Wang W., Saville B. R., and Havel P. J. (2011) Fructose-fed rhesus monkeys: a nonhuman primate model of insulin resistance, metabolic syndrome, and type 2 diabetes. Clin. Transl. Sci. 4, 243–252 10.1111/j.1752-8062.2011.00298.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. ENCODE Project Consortium. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Begg D. P., and Woods S. C. (2013) The endocrinology of food intake. Nat. Rev. Endocrinol. 9, 584–597 10.1038/nrendo.2013.136 [DOI] [PubMed] [Google Scholar]
- 38. Jamaiyar A., Wan W., Janota D. M., Enrick M. K., Chilian W. M., and Yin L. (2017) The versatility and paradox of GDF 11. Pharmacol. Ther. 175, 28–34 10.1016/j.pharmthera.2017.02.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Butler A. A., Price C. A., Graham J. L., Stanhope K. L., King S., Hung Y. H., Sethupathy P., Wong S., Hamilton J., Krauss R. M., Bremer A. A., and Havel P. J. (2019) Fructose-induce hypertriglyceridemia in rhesus macaques: relationships with ApoC3 and attenuation with fish oil or ApoC3 RNAi. J. Lipid Res. 60, 805–818 10.1194/jlr.M089508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wagner J. E., Kavanagh K., Ward G. M., Auerbach B. J., Harwood H. J. Jr., and Kaplan J. R. (2006) Old world nonhuman primate models of type 2 diabetes mellitus. ILAR J. 47, 259–271 10.1093/ilar.47.3.259 [DOI] [PubMed] [Google Scholar]
- 41. Mattison J. A., Colman R. J., Beasley T. M., Allison D. B., Kemnitz J. W., Roth G. S., Ingram D. K., Weindruch R., de Cabo R., and Anderson R. M. (2017) Caloric restriction improves health and survival of rhesus monkeys. Nat. Commun. 8, 14063 10.1038/ncomms14063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bodkin N. L., Alexander T. M., Ortmeyer H. K., Johnson E., and Hansen B. C. (2003) Mortality and morbidity in laboratory-maintained Rhesus monkeys and effects of long-term dietary restriction. J. Gerontol. A Biol. Sci. Med. Sci. 58, 212–219 [DOI] [PubMed] [Google Scholar]
- 43. Hansen B. C., and Bodkin N. L. (1986) Heterogeneity of insulin responses: phases leading to type 2 (non-insulin-dependent) diabetes mellitus in the rhesus monkey. Diabetologia 29, 713–719 10.1007/BF00870281 [DOI] [PubMed] [Google Scholar]
- 44. Ramms B., and Gordts P. (2018) Apolipoprotein C-III in triglyceride-rich lipoprotein metabolism. Curr. Opin. Lipidol. 29, 171–179 10.1097/MOL.0000000000000502 [DOI] [PubMed] [Google Scholar]
- 45. Khetarpal S. A., Qamar A., Millar J. S., and Rader D. J. (2016) Targeting ApoC-III to reduce coronary disease risk. Curr. Atheroscler. Rep. 18, 54 10.1007/s11883-016-0609-y [DOI] [PubMed] [Google Scholar]
- 46. Holoch D., and Margueron R. (2017) Mechanisms regulating PRC2 recruitment and enzymatic activity. Trends Biochem. Sci. 42, 531–542 10.1016/j.tibs.2017.04.003 [DOI] [PubMed] [Google Scholar]
- 47. Conway E., Healy E., and Bracken A. P. (2015) PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr. Opin. Cell Biol. 37, 42–48 10.1016/j.ceb.2015.10.003 [DOI] [PubMed] [Google Scholar]
- 48. Butler A. A., Tam C. S., Stanhope K. L., Wolfe B. M., Ali M. R., O'Keeffe M., St-Onge M. P., Ravussin E., and Havel P. J. (2012) Low circulating adropin concentrations with obesity and aging correlate with risk factors for metabolic disease and increase after gastric bypass surgery in humans. J. Clin. Endocrinol. Metab. 97, 3783–3791 10.1210/jc.2012-2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reinke H., and Asher G. (2016) Circadian clock control of liver metabolic functions. Gastroenterology 150, 574–580 10.1053/j.gastro.2015.11.043 [DOI] [PubMed] [Google Scholar]
- 50. Li S., and Lin J. D. (2015) Transcriptional control of circadian metabolic rhythms in the liver. Diabetes Obes. Metab. 17, Suppl. 1, 33–38 10.1111/dom.12520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang F., Kohan A. B., Lo C. M., Liu M., Howles P., and Tso P. (2015) Apolipoprotein A-IV: a protein intimately involved in metabolism. J. Lipid Res. 56, 1403–1418 10.1194/jlr.R052753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shen L., Ma L. Y., Qin X. F., Jandacek R., Sakai R., and Liu M. (2005) Diurnal changes in intestinal apolipoprotein A-IV and its relation to food intake and corticosterone in rats. Am. J. Physiol. Gastrointest. Liver Physiol. 288, G48–G53 10.1152/ajpgi.00064.2004 [DOI] [PubMed] [Google Scholar]
- 53. Liu M., Shen L., Liu Y., Tajima D., Sakai R., Woods S. C., and Tso P. (2004) Diurnal rhythm of apolipoprotein A-IV in rat hypothalamus and its relation to food intake and corticosterone. Endocrinology 145, 3232–3238 10.1210/en.2003-1554 [DOI] [PubMed] [Google Scholar]
- 54. Shen L., Carey K., Wang D. Q., Woods S. C., and Liu M. (2009) Food-entrained rhythmic expression of apolipoprotein E expression in the hypothalamus of rats. Brain Res. 1273, 66–71 10.1016/j.brainres.2009.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nordestgaard B. G., Nicholls S. J., Langsted A., Ray K. K., and Tybjærg-Hansen A. (2018) Advances in lipid-lowering therapy through gene-silencing technologies. Nat. Rev. Cardiol. 15, 261–272 10.1038/nrcardio.2018.3 [DOI] [PubMed] [Google Scholar]
- 56. Matikainen N., Adiels M., Söderlund S., Stennabb S., Ahola T., Hakkarainen A., Borén J., and Taskinen M. R. (2014) Hepatic lipogenesis and a marker of hepatic lipid oxidation, predict postprandial responses of triglyceride-rich lipoproteins. Obesity 22, 1854–1859 10.1002/oby.20781 [DOI] [PubMed] [Google Scholar]
- 57. Sacks F. M. (2015) The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia. Curr. Opin. Lipidol. 26, 56–63 10.1097/MOL.0000000000000146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Thomas E. L., Parkinson J. R., Frost G. S., Goldstone A. P., Doré C. J., McCarthy J. P., Collins A. L., Fitzpatrick J. A., Durighel G., Taylor-Robinson S. D., and Bell J. D. (2012) The missing risk: MRI and MRS phenotyping of abdominal adiposity and ectopic fat. Obesity 20, 76–87 10.1038/oby.2011.142 [DOI] [PubMed] [Google Scholar]
- 59. Chang J. B., Chu N. F., Lin F. H., Hsu J. T., and Chen P. Y. (2018) Relationship between plasma adropin levels and body composition and lipid characteristics amongst young adolescents in Taiwan. Obes. Res. Clin. Pract. 12, 101–107 10.1016/j.orcp.2017.03.001 [DOI] [PubMed] [Google Scholar]
- 60. Yang C., DeMars K. M., and Candelario-Jalil E. (2018) Age-dependent decrease in adropin is associated with reduced levels of endothelial nitric oxide synthase and increased oxidative stress in the rat brain. Aging Dis. 9, 322–330 10.14336/AD.2017.0523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tuna B. G., Atalay P. B., Altunbek M., Kalkan B. M., and Dogan S. (2017) Effects of chronic and intermittent calorie restriction on adropin levels in breast cancer. Nutr. Cancer 69, 1003–1010 10.1080/01635581.2017.1359314 [DOI] [PubMed] [Google Scholar]
- 62. Saleheen D., Natarajan P., Armean I. M., Zhao W., Rasheed A., Khetarpal S. A., Won H. H., Karczewski K. J., O'Donnell-Luria A. H., Samocha K. E., Weisburd B., Gupta N., Zaidi M., Samuel M., Imran A., et al. (2017) Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 544, 235–239 10.1038/nature22034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pollin T. I., Damcott C. M., Shen H., Ott S. H., Shelton J., Horenstein R. B., Post W., McLenithan J. C., Bielak L. F., Peyser P. A., Mitchell B. D., Miller M., O'Connell J. R., and Shuldiner A. R. (2008) A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science 322, 1702–1705 10.1126/science.1161524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ito Y., Azrolan N., O'Connell A., Walsh A., and Breslow J. L. (1990) Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science 249, 790–793 10.1126/science.2167514 [DOI] [PubMed] [Google Scholar]
- 65. Morrison C. D., and Laeger T. (2015) Protein-dependent regulation of feeding and metabolism. Trends Endocrinol. Metab. 26, 256–262 10.1016/j.tem.2015.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Adams A. C., and Gimeno R. E. (2016) The sweetest thing: regulation of macronutrient preference by FGF21. Cell Metab. 23, 227–228 10.1016/j.cmet.2016.01.013 [DOI] [PubMed] [Google Scholar]
- 67. van der Klaauw A. A., Keogh J. M., Henning E., Stephenson C., Kelway S., Trowse V. M., Subramanian N., O'Rahilly S., Fletcher P. C., and Farooqi I. S. (2016) Divergent effects of central melanocortin signalling on fat and sucrose preference in humans. Nat. Commun. 7, 13055 10.1038/ncomms13055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Durinck S., Spellman P. T., Birney E., and Huber W. (2009) Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 4, 1184–1191 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sergushichev A. A. (2016) An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 10.1101/060012 [DOI] [Google Scholar]
- 71. Garber M., Guttman M., Clamp M., Zody M. C., Friedman N., and Xie X. (2009) Identifying novel constrained elements by exploiting biased substitution patterns. Bioinformatics 25, i54–62 10.1093/bioinformatics/btp190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Siepel A., Bejerano G., Pedersen J. S., Hinrichs A. S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L. W., Richards S., Weinstock G. M., Wilson R. K., Gibbs R. A., Kent W. J., Miller W., and Haussler D. (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050 10.1101/gr.3715005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Caulfield M. P., Li S., Lee G., Blanche P. J., Salameh W. A., Benner W. H., Reitz R. E., and Krauss R. M. (2008) Direct determination of lipoprotein particle sizes and concentrations by ion mobility analysis. Clin. Chem. 54, 1307–1316 10.1373/clinchem.2007.100586 [DOI] [PubMed] [Google Scholar]
- 74. Mora S., Caulfield M. P., Wohlgemuth J., Chen Z., Superko H. R., Rowland C. M., Glynn R. J., Ridker P. M., and Krauss R. M. (2015) Atherogenic lipoprotein subfractions determined by ion mobility and first cardiovascular events after random allocation to high-intensity statin or placebo: the justification for the use of statins in prevention: an intervention trial evaluating rosuvastatin (JUPITER) trial. Circulation 132, 2220–2229 10.1161/CIRCULATIONAHA.115.016857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Butler A. A., Girardet C., Mavrikaki M., Trevaskis J. L., Macarthur H., Marks D. L., and Farr S. A. (2017) A life without hunger: the ups (and downs) to modulating melanocortin-3 receptor signaling. Front. Neurosci. 11, 128 10.3389/fnins.2017.00128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sergushichev A. A. (2016) An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 10.1101/060012 [DOI] [Google Scholar]