

Abstract
Gut-brain signaling controls feeding behavior and energy homeostasis; however, the underlying molecular mechanisms and impact of diet-induced obesity (DIO) on these pathways are poorly defined. We tested the hypothesis that elevated endocannabinoid activity at cannabinoid CB1 receptor (CB1Rs) in the gut of mice rendered DIO by chronic access to a high fat and sucrose diet for 60 days inhibits nutrient-induced release of satiation peptides and promotes overeating. Immunoreactivity for CB1Rs was present in enteroendocrine cells in the mouse’s upper small-intestinal epithelium that produce and secrete the satiation peptide, cholecystokinin (CCK), and expression of mRNA for CB1Rs was greater in these cells when compared to non-CCK producing cells. Oral gavage of corn oil increased levels of bioactive CCK (CCK-8) in plasma from mice fed a low fat no-sucrose diet. Pretreatment with the cannabinoid receptor agonist, WIN55,212-2, blocked this response, which was reversed by co-administration with the peripherally-restricted CB1R neutral antagonist, AM6545. Furthermore, monoacylglycerol metabolic enzyme function was dysregulated in the upper small-intestinal epithelium from DIO mice, which was met with increased levels of a variety of monoacylglycerols including the endocannabinoid, 2-arachidonoyl-sn-glycerol. Corn oil failed to affect levels of CCK in DIO mouse plasma; however, pretreatment with AM6545 restored the ability for corn oil to stimulate increases in levels of CCK, which suggests that elevated endocannabinoid signaling at small intestinal CB1Rs in DIO mice inhibits nutrient-induced CCK release. Moreover, the hypophagic effect of AM6545 in DIO mice was reversed by co-administration with the CCKA receptor antagonist, devazepide. Collectively, these results provide evidence that hyperphagia associated with DIO is driven by a mechanism that includes CB1R-mediated inhibition of gut-brain satiation signaling.
Keywords: CB1R, cholecystokinin, enteroendocrine cell, gut-brain, obesity, satiation
Introduction
Food intake and energy homeostasis are controlled by a dynamic interplay of gut-brain signaling pathways that are not well-defined but are thought to become dysregulated in obesity (Steinert et al., 2017). Recent studies in humans and rodents suggest a critical role for the endocannabinoid (eCB) system in these processes (DiPatrizio and Piomelli, 2012, 2015; DiPatrizio, 2016). The eCB system is located in cells throughout the body and is comprised of the eCBs, 2-arachidonoyl-sn-glycerol (2-AG) and anandamide (AEA), their biosynthetic and degradative enzymes, and the cannabinoid receptor subtypes 1 and 2 [CB1R and CB2R, respectively (Piomelli, 2003; Pertwee, 2015)]. CB1Rs in the brain control food intake and energy homeostasis (DiPatrizio and Piomelli, 2012; Simon and Cota, 2017); however, targeting central CB1Rs with antagonists for the treatment of human obesity led to psychiatric side-effects that preclude their use as safe and effective anti-obesity therapeutics (Christensen et al., 2007). In contrast, CB1Rs antagonists that are restricted to the periphery and do not readily cross the blood-brain barrier are associated with improvements in a variety of metabolic parameters in rodents, and may be an effective anti-obesity strategy that is devoid of psychiatric side-effects inherent to brain-permeable drugs (LoVerme et al., 2009; Randall et al., 2010; Cluny et al., 2011; DiPatrizio et al., 2011, 2013, 2015; Tam et al., 2012, 2017; Maccarrone et al., 2015). Nonetheless, peripheral mechanisms influence brain function [e.g., signals from the gut microbiome (Cani and Knauf, 2016)]; thus, the impact of disrupting endocannabinoid signaling at peripheral CB1Rs on these functions is largely unknown and warrants future investigation.
Studies from our lab and others suggest key roles for the peripheral eCB system in controlling feeding behavior and energy homeostasis (DiPatrizio, 2016; Hillard, 2017; Simon and Cota, 2017). Indeed, eCB levels are increased in the small intestinal epithelium of rodents (1) during a fast (Gomez et al., 2002; Izzo et al., 2009; DiPatrizio et al., 2015; Argueta and DiPatrizio, 2017), (2) after oral exposure to dietary fats (DiPatrizio et al., 2011, 2013), and (3) in a mouse model of western diet-induced obesity (DIO) (Argueta and DiPatrizio, 2017). Pharmacological inhibition of elevated eCB signaling at small-intestinal CB1Rs with peripherally-restricted CB1R antagonists blocks (1) re-feeding after a fast (DiPatrizio et al., 2015), (2) intake of dietary fats based on their orosensory properties (DiPatrizio et al., 2011, 2013), and (3) overeating (i.e., increased meal size and caloric intake) associated with DIO, (Argueta and DiPatrizio, 2017). These studies suggest that the eCB system in the small intestinal epithelium plays a key role in feeding behavior and energy balance, and becomes dysregulated in DIO.
The mechanism(s) underlying eCB control of gut-brain signaling and its dysregulation in DIO is largely unknown. Nonetheless, CB1Rs are expressed on the afferent vagus nerve and suggested to control feeding behavior and energy balance by directly modifying gut-brain vagal signaling important for food intake (Burdyga et al., 2004, 2006). For example, expression of CB1Rs in the rat nodose ganglion is upregulated after fasting, and refeeding or administration of the gut-derived satiation peptide, cholecystokinin (CCK), reversed this effect (Burdyga et al., 2004, 2010). Moreover, both, fasting-induced increases in CB1R expression in the nodose ganglion and the ability for CCK to decrease this response were blunted in rats fed a high-fat diet (Cluny et al., 2013). Vianna and colleagues, however, reported that select deletion of CB1Rs on the afferent and efferent vagus nerve had no effect on food intake or body weight in mice fed a standard rodent chow or high-fat diet (Vianna et al., 2012). These findings suggest that CB1Rs expressed on the vagus nerve may be dispensable for feeding behavior and maintenance of body weight.
Dietary fats and other macronutrients are sensed by enteroendocrine cells in the small intestinal epithelium and stimulate release of satiation peptides including CCK (McLaughlin et al., 1998, 1999; Raybould et al., 2006; Steinert et al., 2017), which controls meal size and satiation by activating CCKA receptors on the afferent vagus nerve (Smith et al., 1981, 1985; Schwartz and Moran, 1994; Raybould, 2007; Dockray, 2013; Kaelberer et al., 2018; Schwartz, 2018) and possibly in the brain (Reidelberger et al., 2004; Ripken et al., 2015). Furthermore, CCK-containing I-cells in the upper small intestinal epithelium of mice express genes for CB1Rs (Sykaras et al., 2012). Thus, CB1Rs in the small intestinal epithelium may control feeding behavior by an indirect mechanism that includes controlling release of gut-derived satiation peptides. We investigated this possibility by testing the hypothesis that elevated endocannabinoid activity at CB1Rs in the gut of mice rendered DIO by chronic access to a high-fat and sucrose diet inhibits nutrient-induced release of satiation peptides, which in turn, leads to overeating by delaying satiation.
Materials and Methods
Animals
Eight-week old C57BL/6 mice (Taconic, Oxnard, CA, USA) were group-housed with ad libitum food and water access and maintained on a 12 h dark/light cycle. C57BL/6-Tg (Cck-EGFP)2Mirn/J mice with enhanced green fluorescent protein on the promoter for cholecystokinin were used for immunohistochemistry and fluorescence-activated cell sorting (FACS) of small intestinal CCK-containing cells (Jackson Laboratories, Bar Harbor, ME, USA). Test diets included Teklad 2020x soy-purified Standard Rodent Chow (SD; Envigo, Huntingdon, UK) or Western-style diet (WD; Research Diets D12709B, New Brunswick, NJ, USA; 40% kcal as fat, 43% kcal as carbohydrates, mainly sucrose). Body weights were recorded every other day at noon. To assess feeding behaviors, mice were single-housed in behavior chambers (TSE Systems, Chesterfield, MO, USA). All procedures met the U.S. National Institute of Health guidelines for care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of the University of California, Riverside.
Feeding Behaviors
Animals were placed into feeding chambers 5 days prior to recording for acclimation, and testing began at 60 days after being placed on their respective experimental diets. Feeding behaviors were assessed starting 1 h prior to dark cycle (1,700 h) over a 24 h period for acclimation and for 12 h following drug administrations. Behavioral parameters include total caloric intake, average meal size, average rate of intake, average number of meals, first meal size, average meal duration, and average post meal interval. Data were processed using TSE Phenomaster software.
Chemicals and Administration Schedule
AM6545, a peripherally-restricted CB1R neutral antagonist, was given by IP injection at 10 mg per kg (Northeastern University Center for Drug Discovery, Boston, MA, USA). Devazepide (Tocris, Bristol, UK), a CCKA receptor antagonist, was given IP at 0.3 mg per kg. Both drugs were dissolved in vehicle consisting of 7.5% DMSO, 7.5% Tween80, and 85% sterile saline, and warmed in a water bath to ensure solubility. All control conditions were matched, using vehicle in place of drugs and injections occurred 1 h prior to behavior recording (1,600 h). A 72-h washout period was allowed between drug treatments. JZL184 (Tocris, Bristol, UK), a potent inhibitor of monoacylglycerol lipase (MGL), was used to prevent monoacylglycerol hydrolysis in the diacylglycerol lipase (DGL) assay and to validate our MGL assay (described below). Tetrahydrolipstatin (Tocris, Bristol, UK), a lipase inhibitor used routinely to study DGL activity (Gregg et al., 2012; Jung et al., 2012), was used to validate our DGL assay.
Measurement of Intestinal Lipids
Tissue Harvest and Lipid Extraction
Animals were anesthetized with isoflurane at time of tissue harvest (1,500–1,700 h) following ad libitum food and water access. Blood was collected by cardiac puncture and deposited into vacutainers containing EDTA; plasma was collected as supernatant following 10 min centrifugation at 1,500 g (kept at 4°C). Jejunum was quickly removed and washed in phosphate-buffered saline (PBS), opened longitudinally on a stainless steel tray on ice, and contents were removed. Jejunum mucosa was isolated using glass slides to scrape the epithelial layer and was snap-frozen in liquid N2. Samples were stored at −80°C pending analysis. Frozen tissues were weighed and then homogenized in 1 ml methanol solution containing 500 pmol [2H5]-2-AG (Cayman Chemicals, Ann Arbor, MI) as an internal standard. Lipids were extracted as previously described (Argueta and DiPatrizio, 2017) and resuspended in 0.1 ml methanol:chloroform (9:1) and analyzed via ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS).
LCMS Detection of 2-Arachidonoyl-sn-Glycerol and Other Monoacylglycerols
Data were acquired using an Acquity I Class UPLC with direct connection to a Xevo TQ-S Micro Mass Spectrometer (Waters Corporation, Milford, MA, USA) with electrospray ionization (ESI) sample delivery. Lipids were separated using an Acquity UPLC BEH C18 column (2.1 mm × 50 mm i.d., 1.7 μm, Waters Corporation) and inline Acquity guard column (UPLC BEH C18 VanGuard PreColumn; 2.1 mm × 5 mm i.d.; 1.7 μm, Waters Corporation), and eluted by a gradient of water and methanol (containing 0.25% acetic acid, 5 mM ammonium acetate) at a flow rate of 0.4 ml per min and gradient: 80% methanol 0.5 min, 80 to 100% methanol 0.5–2.5 min, 100% methanol 2.5–4.5 min, 100 to 80% methanol 4.5–4.6 min, and 80% methanol 4.6–5.5 min. The column was maintained at 40°C, and samples were kept at 10°C in accompanying sample manager. MS/MS detection was in positive ion mode with capillary voltage maintained at 1.10 kV, and argon (99.998%) was used as collision gas. Cone voltages and collision energies for respective analytes: 2-AG (20:4) = 30v, 12v; 2-DG (22:6) = 34v, 14v; 2-PG (16:0) = 18v, 10v; 2-OG (18:1) = 42v, 10v; 2-LG (18:2) = 30v, 10v; monononadecadienoin (19:2 monoacylglycerol; product of DGL assay, see below in “DGL Activity Assay”) = 18v, 10v; and [2H5]-2-AG = 25v, 44v. Lipids were quantified using a stable isotope dilution method detecting H+ or Na+ adducts of the molecular ions [M + H/Na]+ in multiple reaction monitoring (MRM) mode. Acyl migration occurs in monoacylglycerols; thus, the sum of 2-AG and 1-AG is presented. Tissue processing and LCMS analyses for experiments occurred independently of other experiments. Extracted ion chromatograms for MRM transitions were used to quantify analytes: 2-AG (m/z = 379.3 > 287.3), 2-DG (m/z = 403.3 > 311.1), 2-PG (m/z = 331.3 > 239.3), 2-OG (m/z = 357.4 > 265.2), 2-LG (m/z = 355.3 > 263.3), 19:2 monoacylglycerol (m/z = 386.4 > 277.2), and [2H5]-2-AG (m/z = 384.3 > 93.4), which was used as an internal standard for quantitation of monoacylglycerols.
ELISA Analysis of CCK-8 Octapeptide
Mice were fasted for 12 h in order to ensure an empty stomach. Mice were pretreated with CB1R ligands, then administered corn oil (0.5 ml) by oral gavage 30 min later. Levels of CCK-8 were assessed in blood plasma 30 min following gavage. Blood was placed in BD vacutainer lavender-top EDTA blood collection tubes on ice and plasma obtained by centrifugation of tubes at 1,500 g for 10 min at 4°C by a sensitive and selective CCK-8 ELISA (Cloud Clone Corp; Katy, TX, USA). Mice were maintained for 60 days on standard diet (SD) and given IP injection of vehicle or the general cannabinoid receptor agonist, WIN55,212-2 (3 mg per kg), or WIN 55,212-2 in combination with the peripherally-restricted CB1R antagonist, AM6545 (10 mg per kg). In addition, mice maintained for 60 days on Western diet (WD) were given IP injection of vehicle or AM6545 (10 mg per kg). ELISA reaction was measured using iMark microplate reader (BioRad, Hercules, CA, USA).
Immunohistochemistry
Intact proximal small intestine was removed, and contents were flushed with ice-cold 4% paraformaldehyde (PFA)/PBS, then fixed in 4% PFA for 4 h at 4°C. Samples were transferred to 20% sucrose/PBS and incubated for 1 day at 4°C for cryopreservation. Cross sections of upper small intestine were cut and frozen in OCT (Fisher Healthcare, Chino, CA, USA) on dry ice. About 16 μm sections were taken on a cryostat (Leica) and mounted onto charged glass slides. Sections were permeabilized with 0.5% Tween-20/PBS and then blocked with 0.1% Tween in casein solution (Thermo Fisher). Primary antibodies from rabbit for Cholecystokinin (CCK; ABcam, Cambridge, UK) and Cannabinoid Receptor 1 (Generously provided by Dr. Ken Mackie, Indiana University) were diluted 1:500 in blocking buffer and separately added to slides. Slides were washed three times with 0.1% Tween/PBS solution before being incubated with AlexaFluor 647 (Donkey anti-rabbit, Thermo Fisher). Tissue was washed again and mounted with Prolong Gold Antifade reagent with DAPI (Thermo Fisher) for nuclear counterstaining. Images were obtained at room temperature using an Axio Observer Z1 Inverted Microscope (Zeiss, Oberkochen, Germany) at 63× magnification with a CSU-X1 Confocal Scanner Unit (Yokogawa, Tokyo, Japan), and images were captured using a Prime 95B Scientific CMOS Camera (Photometrics, Huntington Beach, CA, USA). Micro-Manager open source software was used for image capture, and final images were optimized using ImageJ 1.51n (NIH, Bethesda, MD, USA).
Fluorescence-Activated Cell Sorting
Isolation of Intestinal Epithelial Cells
Approximately, 4 cm of proximal small intestine was inverted and mechanically disrupted with frosted glass slides into ice-cold buffer containing 5% BSA, 0.6 mM dithiothreitol (DTT) and 1 mM EDTA in PBS to disrupt mucosal cell layer. Live cells were counted following trypan blue staining and 20 × 106 cells were pelleted at 200 g for 5 min and resuspended in 1 ml of 3% BSA containing 1 mM EDTA in PBS. Cell suspension was filtered through 30-micron mesh and subsequently processed by fluorescence-activated cell sorting (FACS).
FACS Sorting of eGFP (+) and eGFP (−) Cells
Isolated cells were sorted and analyzed on a MoFlo Astrios (Beckman Coulter, Brea, CA, USA). Debris was detected and excluded using forward and side scatter profiles generated with a 488 nm laser. eGFP positive (+) cells were detected by fluorescence intensity, using excitation and emission spectra of 488 and 513/26, respectively. A wild-type mouse from C57Bl/6 J background was used to establish autofluorescence, and gating for eGFP was used for final sorting (See Figures 1D,E). Samples were sorted into fresh resuspension buffer prior to qPCR analysis of gene expression. Mice were fasted for 10 h prior to acquisition of cells.
Figure 1.

CB1Rs co-localize with CCK-containing cells in the upper small-intestinal epithelium. Immunohistochemical detection of eGFP [CCK-eGFP (B, F, J)] and CB1Rs [CB1R-ab (C, G, K)] reveals co-localization [merge (A, E, I)] in villi of intestinal epithelium. Arrows indicate separate enteroendocrine cells that contain immunoreactivity for CB1Rs that co-localize with CCK-eGFP cells. Representative images from three CCK-eGFP mice [DAPI stain (D,H,L)].
Enzyme Activity Assays
Tissue Preparation
Intestinal epithelium was collected as described above (2.3.1) and approximately 100 mg of frozen tissue was homogenized in 2 ml of ice-cold 50 mM Tris-HCl, 320 mM sucrose (pH 7.5) buffer. Homogenates were centrifuged at 800 g for 10 min at 4°C and supernatant was collected. Protein supernatants were sonicated twice for 10 s and then freeze-thawed in liquid nitrogen twice. Samples were spun again, and supernatant protein content was quantified using BCA assay and diluted to working concentration with Tris-HCl/sucrose buffer.
DGL Activity Assay
Small-intestinal epithelial tissue homogenates (25 μg, room temperature) were incubated with the MGL inhibitor, JZL184 (0.3 μM), for 10 min in order to block MGL activity during the assay. Homogenates were then incubated in 0.2 ml solution of Tris-HCL with 0.2% Triton X-100 (pH 7.0) containing 20 nmol dinonadecadienoin (19:2 DAG) at 37°C for 30 min. Reactions were stopped by adding 1 ml of ice-cold MeOH containing 25 pmol [2H5]-2-AG as internal standard. Lipids were extracted and the product of the reaction, monononadecadienoin (19:2 monoacylglycerol), was analyzed via UPLC/MS/MS as described above for 19:2 monoacylglycerol (See “LCMS Detection of 2-Arachidonoyl-sn-Glycerol and Other Monoacylglycerols”).
MGL Activity Assay
Small-intestinal epithelial tissue (10 μg) was incubated with 0.4 ml solution of Tris-HCL with 0.1% BSA (pH 8.0) containing 50 nmol nonadecadienoin (19:2 monoacylglycerol; Nu-Chek Prep, Waterville, MN, USA; final volume 0.5 ml per reaction) at 37°C for 10 min. Reactions were stopped by adding 1 ml of MeOH containing 10 nmol heptadecanoic acid (17:1 FFA; Nu-Chek Prep) as internal standard. Lipids were extracted and the product of the reaction (19:2 free fatty acid) was analyzed via UPLC/MS/MS according to the following protocol. Data were acquired using equipment described above (See “LCMS Detection of 2-Arachidonoyl-sn-Glycerol and Other Monoacylglycerols”) and eluted by a gradient of water and methanol (containing 0.25% acetic acid, 5 mM ammonium acetate) at a flow rate of 0.4 ml per min and gradient: 90% methanol 0.1 min, 90–100% methanol 0.1–2.0 min, 100% methanol 2.0–2.1 min, 100 to 90% methanol 2.1–2.2 min, and 90% methanol 2.2–2.5 min. Column was maintained at 40°C and samples were kept at 10°C in sample manager. MS detection was in negative ion mode with capillary voltage maintained at 3.00 kV. Cone voltages for nonadecadienoic acid (19:2 FFA) = 48v and heptadecanoic acid (17:1 FFA) = 64v. Lipids were quantified using a stable isotope dilution method of proton adducts of the molecular ions [M–H]− in selected ion recording (SIR) mode. Tissue processing and LCMS analyses for experiments occurred independently of other experiments. Extracted ion chromatograms for SIR masses were used to quantify analytes: 19:2 FFA (m/z = 293.2) product of MGL enzyme assay and 17:1 FFA (m/z = 267.2) as internal standard.
Gastric Emptying
To evaluate drug or endogenous endocannabinoid effects on gastric emptying, corn oil was spiked with 1.0 nmol 19:2 FFA and administered by oral gavage (500 μl), then quantities of 19:2 FFA remaining in the stomach were evaluated at the time of blood collection 30 min after gavage. The stomach was removed and immediately placed into methanol containing 17:1 FFA as internal standard. Lipids were extracted and 19:2 FFA was detected and quantified as above (See “MGL Activity Assay”).
Gene Expression Analysis
RNA Isolation From Intestinal Epithelium
Total RNA was extracted from intestinal epithelium using RNeasy kit (Qiagen, Valencia, CA, USA) method, and first-strand complementary DNA was generated using M-MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA). All surfaces for tissue collection and processing were sanitized using 70% ethanol and then treated with an RNAse inhibitor (RNAse out, G-Biosciences, St. Louis, MO, USA) to maintain integrity of isolated RNA. Reverse transcription of total RNA (1 μg epithelium) was performed as previously described (Argueta and DiPatrizio, 2017).
RNA Isolation From Sorted Cells
Sorted cell suspensions were pelleted at 3,000 g for 10 min and resuspended in 0.5 ml of Qiazol (Qiagen, Valencia, CA) and subsequently processed using RNeasy kit to isolate total RNA. Reverse transcription was performed as described above using 50 ng total RNA.
Quantitative Polymerase Chain Reaction Analysis
RT-qPCR was carried out using PrimePCR Sybr Green Assays (Biorad, Hercules, CA, USA) with the following primers for mouse genes: CB1R (Cnr1), CB2R (Cnr2), cholecystokinin (Cck), fatty-acid amide hydrolase (Faah), n-acyl phosphatidyl ethanolamine-specific phospholipase D (Napepld), diacylglycerol lipase alpha (Dagla) and beta (Daglb), monoacylglycerol lipase (Mgll), alpha-beta-hydrolyzing domain 6 (Abhd6) with Hprt and Actb as housekeeping genes for epithelium and sorted cells, respectively. Values are expressed as relative mRNA expression based on widely used methods [i.e., delta-delta cq; see (Livak and Schmittgen, 2001)]. Reactions were run in triplicate for each animal.
Statistical Analysis
Values are expressed as means ± SEM. Unpaired Student’s two-tailed t-test was used to compare data for standard diet- and western diet-fed groups. Repeated measures two-way ANOVA was used for groups measured over time. Additionally, regular one-way and two-way ANOVA were used to determine differences in multiple groups with post-hoc Sidak’s multiple comparisons tests or Newman-Keul’s, as appropriate. Data were analyzed using GraphPad Prism6 software. Significance was determined as p < 0.05. Statistical outliers were determined using Grubb’s test in all datasets.
Results
CB1Rs Are Expressed in CCK-Containing Cells in the Upper Small-Intestinal Epithelium
We reported that eCB levels are increased in the upper small-intestinal epithelium from mice maintained on a Western Diet (WD; high-fat and sucrose diet) for 60 days when compared to lean controls maintained on a low-fat and low-sugar diet, and inhibiting peripheral CB1Rs blocked overeating associated with consumption of WD (i.e., increased meal size, rate of food intake, and total caloric intake) (Argueta and DiPatrizio, 2017). To identify the molecular underpinnings of gut-brain eCB signaling important for feeding behavior and its dysregulation in DIO, we first evaluated whether CB1Rs are expressed in cells that produce and secrete the satiation peptide, CCK. CCK controls meal size and induces satiation by activating CCKA receptors on the afferent vagus nerve (Smith et al., 1981, 1985; Schwartz and Moran, 1994; Reidelberger et al., 2004; Raybould, 2007; Dockray, 2013; Ripken et al., 2015). CB1R immunoreactivity was found in CCK-eGFP-positive cells from the upper small intestinal epithelium (Figure 1) in a mouse line that expresses eGFP selectively in CCK-expressing cells [C57BL/6-Tg(Cck-EGFP)2Mirn/J] (Schmidt et al., 2014). Furthermore, immunoreactivity for CCK was co-localized with eGFP in the upper small-intestinal epithelium, which confirms expression of CCK in eGFP-containing cells from this mouse line (Supplementary Figure S1). We, next isolated eGFP-positive and eGFP-negative cells from the upper small intestinal epithelium by fluorescence-activated cell sorting (FACS). Messenger RNA (mRNA) for CB1Rs (Cnr1) was enriched in CCK-eGFP-positive cells when compared to CCK-eGFP-negative cells (Figure 2A; eGFP-positive = 1.00 ± 0.24, eGFP-negative = 0.04 ± 0.04; p = 0.016; data from three mice). Moreover, mRNA for CCK was present in CCK-eGFP-positive cells isolated by FACS but was not present in CCK-eGFP-negative cells, which highlights the specificity of our FACS gating strategy for isolating CCK-eGFP cells and further confirms expression of CCK in these cells (Figure 2B). Our gating strategy was optimized for sorting of eGFP-positive and eGFP-negative events from cells isolated from the upper small-intestinal epithelium of CCK-eGFP mice (see Figure 2C). Cells from wild-type mice (see Figure 2D) show minimal fluorescence at less than 10% of levels found in CCK-eGFP cells: eGFP-positive cells comprise 0.63% of total cells analyzed from CCK-eGFP mice, and wild-type show 0.06%, likely due to autofluorescence (see Supplementary Figure S2 for detailed FACS report). These results suggest that CCK-containing I-cells in the mouse upper small-intestinal epithelium are enriched in expression of CB1Rs.
Figure 2.

CB1R mRNA expression is enriched in CCK-containing cells in the upper small-intestinal epithelium. Fluorescence-activated cell sorting (FACS) of eGFP-CCK-positive (+) and eGFP-CCK-negative (−) cells from the upper small-intestinal epithelium reveals enhanced Cnr1 expression in eGFP-CCK-positive cells (A). Expression of mRNA for CCK is found in eGFP-CCK-positive cells but not in eGFP-CCK-negative cells (B). Gating strategy shown for sorting of eGFP-positive and eGFP-negative events, with eGFP-positive cells highlighted in green and demarked by thin line (C) and compared to upper small-intestinal epithelial cells from a wild-type (WT) mouse (D). Data expressed as mean ± S.E.M. Analyzed using Student’s t-test, two-tailed (C); n = 3 per group; *p < 0.05.
Peripheral CB1Rs Control Fat-Induced CCK Secretion
The arrival of fat and other macronutrients into the duodenum stimulates release of a variety of signaling molecules that include CCK, which is produced and secreted by enteroendocrine I-cells lining the upper small-intestinal epithelium (McLaughlin et al., 1999; Raybould, 1999; Raybould et al., 2006; Cvijanovic et al., 2017; Steinert et al., 2017). We next tested the hypothesis that CB1Rs control nutrient-induced release of CCK from the upper small-intestinal epithelium. Oral gavage of corn oil (CO) in lean mice maintained on a standard rodent diet (SD; low-fat no-sucrose chow) increased plasma levels of bioactive CCK, CCK-8 (octapeptide), when compared to control mice that received oral gavage of saline [Figure 3A; CO = 0.69 ± 0.11 ng per ml, saline control = 0.28 ± 0.02 ng per ml; p < 0.05 CO versus saline control, n = 3–5). Peripheral administration of the general cannabinoid receptor agonist, WIN55,212-2 (WIN, 3 mg per kg), blocked CO-induced secretion of CCK-8 (Figure 3A; CO + WIN = 0.36 ± 0.04 ng per ml; p < 0.05 CO + WIN versus CO alone, n = 5). Furthermore, the effect of WIN administration on CO-induced secretion of CCK-8 was reversed by co-treatment with the peripherally-restricted neutral CB1R-selective antagonist, AM6545 (Figure 3A; CO + WIN+AM6545 = 0.75 ± 0.14 ng per ml; p < 0.05 CO + WIN versus CO + WIN+AM, n = 5; AM6545 10 mg per kg). These results suggest that exogenous activation of CB1Rs inhibits nutrient-induced CCK release from the upper gut.
Figure 3.
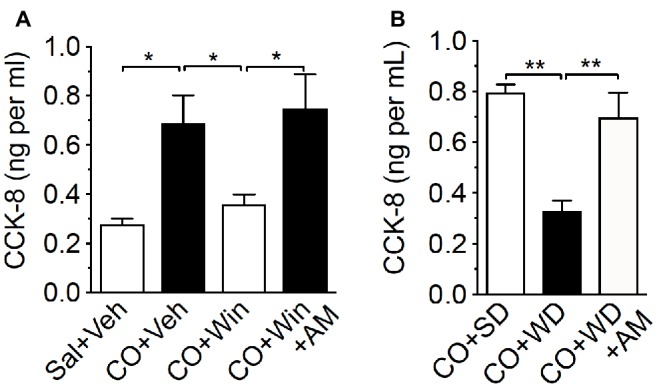
Exogenous or endogenous activation of peripheral CB1Rs inhibits fat-induced CCK release. Compared to control [0.5 ml saline (Sal) by oral gavage and vehicle (Veh) by IP injection], corn oil (CO; 0.5 ml by oral gavage) increased levels of CCK-8 in plasma of lean mice fed a low-fat no-sugar standard diet (SD), an effect blocked by the CB1R agonist, WIN 55,212-2 (WIN, IP 3 mg per kg 30 min before CO) (A). The effects of WIN were inhibited by co-administration with the peripherally-restricted CB1R antagonist, AM6545 (AM; 10 mg per kg 30 min before CO). When compared to control SD mice (CO + SD), CO failed to elicit changes in levels of CCK-8 in plasma in mice fed western diet (WD) for 60 days, and inhibition of peripheral CB1Rs with AM6545 normalized levels of CCK-8 to those found in SD CO controls (B). Data expressed as means ± S.E.M. Analyzed by one-way ANOVA with post hoc Newman-Keuls multiple comparison test. n = 3–5 per condition, * p < 0.05, ** p < 0.01.
We next tested the hypothesis that elevated endogenous activity (e.g., increased 2-AG levels) at upper small-intestinal CB1Rs in mice maintained on Western Diet (WD; high-fat and sucrose diet) for 60 days inhibits CO-induced increases in circulating levels of CCK-8. We first confirmed that levels of 2-AG—among other monoacylglycerols—were increased in the upper small intestinal epithelium of WD mice, when compared to lean mice fed SD for 60 days (see Table 1). Next, we tested the ability for oral gavage of CO to increase CCK-8 levels in plasma of WD mice. CO failed to affect levels of CCK-8 in WD mice when compared to mice fed a standard diet (SD) that is low in fat and absent of sucrose (Figure 3B; CO + WD = 0.33 ± 0.04 ng per ml, CO + SD = 0.8 ± 0.03 ng per ml; p < 0.01, n = 5). Furthermore, AM6545 treatment in WD mice that received oral gavage of CO increased levels of CCK-8 to those comparable to SD mice under the same conditions (Figure 3B; CO + WD + AM = 0.7 ± 0.1 ng per ml; p < 0.01 CO + WD versus CO + WD + AM, n = 6). Collectively, these results suggest that exogenous or endogenous activation of CB1Rs in the upper small intestinal epithelium inhibits nutrient-induced CCK secretion.
Table 1.
Impact of diet on monoacylglycerols in mouse small-intestinal epithelium.
| MAG | 20:4 (2-AG) (nmol g−1) |
18:1 (nmol g−1) |
18:2 (nmol g−1) |
16:0 (nmol g−1) |
22:6 (nmol g−1) |
Total (nmol g−1) |
|---|---|---|---|---|---|---|
| SD | 80.23 ± 8.542 | 49.57 ± 9.804 | 217.7 ± 52.09 | 33.99 ± 4.125 | 9.413 ± 1.996 | 390.9 ± 72.24 |
| WD | 132.5 ± 22.20 | 109.0 ± 22.03 | 415.3 ± 83.26 | 150.1 ± 21.53 | 22.63 ± 3.703 | 829.4 ± 144.2 |
| p | 0.0353 | 0.0206 | 0.0554 | <0.0001 | 0.0049 | 0.0122 |
MAG, Monoacylglycerol represented as fatty acid chain; SD, Standard diet n = 10; WD, Western diet n = 9. Mean values are shown as ± S.E.M. Bold values are significantly different determined by two-tailed unpaired t-test.
All levels of CCK-8 in these experiments fell within the range of the standard curve for CCK-8 quantitation by a sensitive and selective CCK-8 ELISA, which shows no cross-reactivity for gastrin (see Supplementary Figure S3), another gut-derived peptide that shares some common molecular features with CCK-8 (Walsh et al., 1982; Eysselein et al., 1984; Wolfe et al., 1985; Shulkes and Baldwin, 1997). Furthermore, the range of CCK-8 levels in our studies (from 0.27 ± 0.02 to 0.8 ± 0.03 ng per ml or 0.23 ± 0.02 to 0.7 ± 0.03 nM) aligns with reported Ki and EC50 values of sulfated CCK-8 in several binding and in vitro bioassays (e.g., amylase release from pancreatic acini and ileum contractions in guinea pig) (Charpentier et al., 1988).
CB1R activation is reported to decrease gastric emptying, an effect also found in mice fed a high-fat diet for 14 weeks (Pertwee, 2001; Di Marzo et al., 2008). To identify if altered gastric emptying occurs under our conditions and may contribute in part to inhibited corn oil-induced CCK release, we developed a novel UPLC/MS/MS-based method to evaluate if CB1R activation with WIN 55,212-2 or exposure to WD for 60 days impacts gastric emptying following oral gavage of corn oil in SD and WD mice, respectively. Thirty minutes after administration of drugs, we administered by oral gavage corn oil (500 μl) that contained 19:2 free-fatty acid (1 nmol) as a tracer and measured by UPLC/MS/MS the remaining quantities of 19:2 free-fatty acid in the stomach 30 min after gavage. WIN 55,212-2 (3 mg per kg) alone or in combination with AM6545 (10 mg per kg) had no effect on gastric emptying of corn oil in SD mice (see Supplementary Figure S4A). Similarly, WD mice displayed no changes in gastric emptying of corn oil when compared to SD mice (see Supplementary Figure S4B). These data suggest that exogenous activation (WIN in SD mice) or endogenous activation (elevated small intestinal epithelial eCB levels in WD mice) of CB1Rs does not affect gastric emptying of corn oil under our conditions, and does not likely impact CCK release by a mechanism that includes alterations in gastric emptying in mice.
CB1Rs in pancreatic beta cells control insulin release and glucose homeostasis (Juan-Pico et al., 2006; De Petrocellis et al., 2007; Bermudez-Silva et al., 2008; Nakata and Yada, 2008; Li et al., 2010; Gonzalez-Mariscal et al., 2018). Thus, we tested if drug treatment impacted glucose levels in response to corn oil gavage in SD mice, which in turn, could affect gastric emptying, motility, or enteroendocrine hormones from small intestinal enteroendocrine cells. Glucose levels in blood were collected from tail vein and monitored via hand-held glucose monitor at (1) time of drug administration, (2) 30 min later just prior to corn oil gavage, and (3) 30 min later at time of kill (See Supplementary Figure S5). Drug treatment had no significant impact on blood glucose levels at any time point prior to or after gavage of corn oil (See Supplementary Figure S5). These data suggest that, under our conditions, activating CB1Rs does not impact blood glucose levels following oral gavage of corn oil in mice.
Activity of Enzymes Responsible for Metabolism of 2-AG and Other Monoacylglycerols Is Dysregulated in the Upper Small-Intestinal Epithelium in Mice Chronically Fed WD
We next aimed to identify the mechanism(s) of increased 2-AG and related monoacylglycerol levels (see Table 1) in WD mice by analyzing activity of their biosynthetic [diacylglycerol lipase (DGL)] and degradative enzymes [monoacylglycerol lipase (MGL)] using our lab’s UPLC/MS/MS-based functional enzyme assay methods (see Supplementary Figure S6 for validation of enzyme assays). When compared to SD mice, WD mice displayed an increase in activity of DGL (Figure 4A; SD = 0.12 ± 0.02, WD = 0.22 ± 0.03 nmol per mg protein per min; p = 0.016, reactions from six mice per diet group), and MGL (Figure 4B; SD = 36.32 ± 3.82, WD = 51.60 ± 4.95 nmol per mg protein per min; p = 0.035, reactions from six mice per diet group) in isolated tissue from the upper small intestinal epithelium. Congruent with data in Table 1 and (Argueta and DiPatrizio, 2017), these effects were met with increased levels of 2-AG in upper small intestinal epithelium of separate mice (Figure 4C; SD = 45.71 ± 6.93, WD = 92.57 ± 16.41 nmol per g; p = 0.014, n = 9–10 per diet group). See Figure 4D for diagram of the 2-AG metabolic pathways. Together, these results suggest that monoacylglycerol metabolic pathways are dysregulated after chronic exposure to WD, which leads to a net increase in monoacylglycerols, including 2-AG, in the upper small-intestinal epithelium.
Figure 4.

2-AG biosynthesis and degradation are upregulated in small intestine during obesity. Hydrolytic activity of DGL (A) and MGL (B) are increased in mice maintained on western diet (WD) when compared to controls fed a standard diet (SD). Levels of the endocannabinoid, 2-AG, are increased in jejunum mucosa of WD mice, when compared to SD mice (C). 2-AG is formed by the hydrolysis of its 1,stearoyl,2-arachidonoyl-sn-glycerol precursor by DGL and is subsequently degraded by MGL into arachidonic acid and glycerol (D). Data expressed as mean ± S.E.M. Analyzed using Student’s two-tailed t-test. n = 6 per condition, *p < 0.05.
Expression of Select eCB System Components in the Upper Small-Intestinal Epithelium Is Dysregulated in Mice Chronically Fed WD and Partially Conserved in CCK-Positive Cells
Relative expression of mRNA for intestinal CCK, CB1Rs, and CB2Rs (Cnr2) was unchanged in whole upper small intestinal epithelial scrapings from mice fed WD versus SD mice (Figure 5A; CCK, SD = 1.00 ± 0.76, WD = 0.56 ± 0.45, p = 0.64; Cnr1, SD = 1.00 ± 0.36, WD = 0.79 ± 0.31, p = 0.67; Cnr2, SD = 1.00 ± 0.31, WD = 0.83 ± 0.19, p = 0.65; data from four mice per diet group). Expression of mRNA for the alpha isoform of DGL (Dagla) was also unaffected by diet (Figure 5A; SD = 1.00 ± 0.25, WD = 0.90 ± 0.29, p = 0.80); however, expression of mRNA for the beta isoform of DGL (Daglb) was reduced in WD versus SD mice (Figure 5A; SD = 1.00 ± 0.15, WD = 0.35 ± 0.03, p = 0.005), while mRNA for MGL (Mgll) and the serine hydrolase alpha/beta hydrolase domain 6 (Abhd6) were increased in small intestinal epithelium under the same conditions (Figure 5A; Mgll, SD = 1.00 ± 0.17, WD = 2.71 ± 0.46, p = 0.013; Abhd6, SD = 1.00 ± 0.16, WD = 1.54 ± 0.05, p = 0.02). No changes were found for the fatty acid ethanolamide biosynthetic enzyme, NAPE-PLD, or the fatty acid ethanolamide degradative enzyme, FAAH (Figure 5A; NAPE-PLD, SD = 1.00 ± 0.18, WD = 0.89 ± 0.08, p = 0.6; FAAH, SD = 1.00 ± 0.17, WD = 1.00 ± 0.07, p = 0.99). Furthermore, the upper small-intestinal epithelium is enriched in expression of mRNA for Daglb when compared to Dagla (Figure 5A inset; Dagla = 1.00 ± 0.19, Daglb = 29.73 ± 4.3; p = 0.001; data from four mice fed SD).
Figure 5.

Expression of select components of the eCB system is dysregulated in the upper small intestine of DIO mice and partially conserved in CCK-eGFP+ cells. Expression of mRNA for cholecystokinin (Cck), cannabinoid receptor subtype 1 (Cnr1) and 2 (Cnr2), and other components of the eCB system in upper small-intestinal mucosal scrapings are not impacted by western diet (WD) exposure when compared to controls fed a standard diet (SD) (A). Expression of diacylglycerol lipase beta (Daglb) is decreased, and expression of the degradative enzymes monoacylglycerol lipase (Mgll) and alpha-beta hydrolyzing domain 6 (Abhd6) are increased in WD mice. Expression of mRNA for CCK or components of the eCB system were not significantly affected by diet in eGFP (+) sorted cells (B). Expression of mRNA for diacylglycerol lipase alpha (Dagla), Mgll, and N-acyl phosphatidylethanolamine specific phospholipase D (Napepld) was not detected (ND) (B). Data expressed as mean ± S.E.M. Analyzed using Student’s two-tailed t-test. n = 3 per group in triplicate and *p < 0.05, **p < 0.01, ***p < 0.001 (A); n = 3 per group in triplicate, p > 0.05 (B).
It is important to note, in contrast to our previous report that included analysis of eCB system expression in the upper small intestinal epithelium of mice maintained on WD and SD [Lab Diet 5001 used in (Argueta and DiPatrizio, 2017)], in this study we used a soy protein-free Teklad 2020x as a control SD in order to eliminate any potential effects of phytoestrogen-containing soy protein on eCB metabolism or behavior [see (McFarland et al., 2004; Thors et al., 2007, 2008; Peroni et al., 2012)]. We found two differences in results when comparing use of the two control diets versus WD. We reported no changes in expression of mRNA for the beta isoform of DGL and increases in expression of mRNA for FAAH in WD mice when compared to control SD mice (Argueta and DiPatrizio, 2017); however, in this study, we found decreased expression of mRNA for the beta isoform of DGL and no changes in expression of mRNA for FAAH in WD mice when compared to SD mice. These differences highlight possible effects of diets that utilize soy protein on expression of eCB metabolic enzymes and eCB metabolic function. A direct comparison of the impact of specific control diets on expression of eCB system components, however, remains to be evaluated.
CCK-eGFP-positive cells isolated from mice fed SD or WD mice displayed no differences between diet condition in expression of mRNA for CCK and components of the eCB system that include Cnr1, Cnr2, Daglb, Abhd6, and FAAH (Figure 5B; p > 0.05 not significant, data from three mice per diet group). Dagla, Mgll, and Napepld mRNA were below detectable levels, which suggest a lack of expression of these eCB system components in CCK-containing cells.
Collectively, these results identify select eCB system gene transcripts in CCK-containing cells, and changes in expression of biosynthetic and degradative enzyme gene transcripts in whole epithelium of WD mice that do not fully correspond to changes in activity of their proteins, including DGL and MGL (see Figure 4). The latter suggests possible post-transcriptional and/or post-translational changes in expression of these enzymes in the upper small-intestinal epithelium in WD mice when compared to lean SD mice, although this hypothesis remains to be directly tested. Furthermore, a lack of expression of the fatty acid ethanolamide (FAE) biosynthetic enzyme, NAPE-PLD, in CCK-containing cells suggests that FAEs including anandamide—which is also found in small intestinal epithelium of rodents (Izzo et al., 2009; DiPatrizio et al., 2011, 2013, 2015; Argueta and DiPatrizio, 2017; Perez and DiPatrizio, 2018)—is generated in neighboring cells and therefore may act in a paracrine manner with I-cells that contain CB1Rs. In contrast, expression of mRNA for the beta isoform of the monoacylglycerol biosynthetic enzyme, DGL, is abundantly expressed in CCK-containing cells, which suggests that 2-AG may signal at CB1Rs in an autocrine manner at these cells. Expression of the primary 2-AG degradative enzyme, MGL, is absent in CCK-containing I-cells, which suggests that 2-AG is degraded at adjacent cells and therefore may additionally signal CB1Rs on adjacent cells in a paracrine manner. A comprehensive analysis of eCB system architecture and its cell-specific expression in the upper small-intestinal epithelium of mice remains for future studies.
Western Diet Exposure for 60 Days Is Associated With Obesity and Hyperphagia in Male Mice
Consistent with our previous studies (Argueta and DiPatrizio, 2017), exposure to WD for 60 days, when compared to lean mice fed SD for 60 days, was associated with (1) a rapid and sustained increase in body mass when compared to control mice fed SD for 60 days, (2) increased 24 h meal size, (3) rate of food intake, and (4) total 24 h caloric intake (see Supplementary Figure S7 and Table 2 for details and data). No significant changes were found for other feeding behaviors including (1) first meal size, (2) meal frequency, (3) meal duration, and (4) post-meal interval. As discussed above in “Expression of Select eCB System Components in the Upper Small Intestinal Epithelium is Dysregulated in Mice Chronically Fed WD and Partially Conserved in CCK-Positive Cells,” in contrast to our previous study (Argueta and DiPatrizio, 2017), in this study we used a soy-protein-free lab chow. Irrespective of control diet, however, WD intake was consistently associated with increased 2-AG levels (Table 1) and hyperphagia across relevant parameters in both studies (Table 2, Figure 6; Argueta and DiPatrizio, 2017). Together, these data suggest that exposure to a WD rapidly induces body weight gain that is met with increased meal size, rate of intake, and daily caloric intake, when compared to lean controls.
Table 2.
Consumption of WD is associated with hyperphagia.
| Δ Body mass (g) | Meal size (kcal) | Intake rate (kcal min−1) | 24 h Intake (kcal) | First meal (kcal) | Frequency (meals day−1) | Duration (Min) | PMI (Min) | |
|---|---|---|---|---|---|---|---|---|
| SD | 8.90 ± 0.31 | 0.69 ± 0.04 | 0.33 ± 0.02 | 8.10 ± 0.61 | 0.72 ± 0.11 | 11.67 ± 0.45 | 8.61 ± 1.16 | 114.7 ± 8.4 |
| WD | 18.14 ± 0.46 | 1.29 ± 0.10 | 0.71 ± 0.08 | 13.28 ± 0.81 | 1.99 ± 0.66 | 9.83 ± 1.42 | 6.85 ± 0.98 | 129.3 ± 14.3 |
| p | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.07 | 0.23 | 0.26 | 0.31 |
PMI, Post meal interval. Mean values are shown as ± S.E.M. n = 10. Bold values are significantly different determined by two-tailed unpaired t-test.
Figure 6.

Peripheral eCB signaling drives hyperphagia in mice maintained on a WD via a CCK-dependent mechanism. Caloric intake (A), meal size (B), and rate of intake (C) of a western diet (closed bars) are significantly reduced during a 12 h test following inhibition of peripheral CB1Rs with AM6545 (AM, 10 mg per kg), an effect absent in low-fat chow fed mice (open bars) and that is blocked by co-administration with the CCKA receptor antagonist, devazepide (Dev; 0.1 mg per kg). Diet and drug had no effect on meal frequency (D), meal duration (E), post meal interval (F), or first meal size (G). All data represented as means ± SEM. Analyzed using regular 2-Way ANOVA with post hoc Newman-Keuls multiple comparison’s test. n = 11–12 per condition, *p < 0.05, **p < 0.01.
Pharmacological Inhibition of CCKA Receptors Blocks the Anorexic Effect of AM6545 in Mice Chronically Fed a WD
We next tested the hypothesis that peripheral CB1Rs control feeding behavior by a mechanism that includes control of CCK-mediated satiation signaling. When compared to vehicle treatment in mice fed WD for 60 days, AM6545 treatment (10 mg per kg) in WD mice reduced meal size (Figure 6A; vehicle = 1.47 ± 0.15 kcal, AM6545 = 1.13 ± 0.67 kcal; p < 0.05, n = 12), rate of intake (Figure 6B; vehicle = 0.76 ± 0.12 kcal per min, AM6545 = 0.46 ± 0.05 kcal per min; p < 0.01), and total caloric intake (Figure 6C; vehicle = 9.11 ± 0.67 kcal per min, AM6545 = 6.62 ± 0.69 kcal per min; p < 0.01) during a 12 h test, which is consistent with our previous findings (Argueta and DiPatrizio, 2017). Furthermore, AM6545 treatment in mice fed SD for 60 days did not affect meal size (Figure 6A; vehicle = 0.74 ± 0.05 kcal, AM6545 = 0.71 ± 0.04 kcal; p > 0.05, n = 12), rate of intake (Figure 6B; vehicle = 0.30 ± 0.03 kcal per min, AM6545 = 0.32 ± 0.03 kcal per min; p > 0.05), and total caloric intake (Figure 6C; vehicle = 5.51 ± 0.42 kcal per min, AM6545 = 5.45 ± 0.28 kcal per min; p > 0.05) during a 12 h test. Importantly, co-administration of a low dose of the CCKA receptor antagonist, devazepide (Dev; 0.1 mg per kg), in WD mice blocked the effects of AM6545 on reducing meal size (Figure 6A; vehicle = 1.47 ± 0.15 kcal, AM6545 + devazepide = 1.49 ± 0.16 kcal; p > 0.05), rate of intake (Figure 6B; vehicle = 0.76 ± 0.12 kcal per min, AM6545 + devazepide = 0.62 ± 0.06 kcal per min; p > 0.05), and total caloric intake (Figure 6C; vehicle = 9.11 ± 0.67 kcal per min, AM6545 + devazepide = 8.98 ± 0.67 kcal per min; p > 0.05). Administration of devazepide alone affected only total 12-h caloric intake in SD mice (Figure 6C; vehicle = 5.51 ± 0.42 kcal per min, devazepide = 7.61 ± 0.33 kcal per min; p < 0.05). Neither AM6545 nor devazepide affected other meal parameters including meal frequency (Figure 6D), meal duration, (Figure 6E), post-meal interval (Figure 6F), or first-meal size (Figure 6G) in SD or WD mice. These data suggest that the acute anorexic effects of AM6545 in WD mice are dependent on a mechanism that includes activation of CCKA receptors and inhibition of gut-brain satiation signaling.
Discussion
The molecular underpinnings of gut-brain signaling and their dysregulation in DIO are poorly defined. Our studies suggest that eCB activity at CB1Rs in the upper small intestinal epithelium is upregulated in mice chronically fed a WD, which in turn, promotes overeating by a mechanism that includes inhibiting nutrient-induced gut-brain satiation signaling (see Figure 7 for model). Six primary findings support this conclusion: (1) CB1Rs are enriched in CCK-containing cells in the mouse upper small intestinal epithelium; (2) oral gavage of corn oil increased circulating levels of CCK-8 in lean mice, and pharmacological activation of CB1Rs blocked this effect, which was reversed by inhibition of peripheral CB1Rs with a peripherally-restricted CB1R neutral antagonist; (3) levels of 2-AG and other monoacylglycerols were increased in the upper-small intestinal epithelium of WD mice when compared to lean mice, and this effect was associated with dysregulated monoacylglycerol metabolism; (4) oral gavage of corn oil failed to affect circulating levels of CCK-8 in WD mice, and inhibition of peripheral CB1Rs in WD mice restored the ability for corn oil to increase CCK levels; (5) pharmacological inhibition of peripheral CB1Rs in WD mice blocked overeating associated with increased meal size, rate of feeding, and total caloric intake; and (6) the hypophagic effects of peripheral CB1R antagonism in WD mice were reversed by pretreatment with a low-dose CCKA receptor antagonist. Collectively, our studies identify a previously unknown role for the eCB system at the interface of nutrient-sensing and gut-brain satiation signaling that becomes dysregulated in DIO and promotes overeating by delaying satiation.
Figure 7.

Model of CB1R control of nutrient-induced CCK release. Our studies suggest that cannabinoid CB1Rs in the upper small-intestinal epithelium control nutrient-induced satiation signaling, and their signaling is increased in diet-induced obesity, which drives overeating by delaying satiation. The upper small-intestinal epithelium contains enteroendocrine I-cells, which are a subpopulation of enterocytes that secrete cholecystokinin (CCK) when nutrients – including dietary fats – enter the lumen (Rehfeld, 1998; Tanaka et al., 2008; Wang et al., 2011; Liou et al., 2011a; Brennan et al., 2012). Dietary fats (e.g., corn oil), in the form of triacylglycerols, are hydrolyzed in the lumen into mostly monoacylglycerols and free-fatty acids (FFAs) that are sensed by free-fatty acid receptors (FFARs) located on enteroendocrine cells in the small-intestinal epithelium. Activation of FFARs stimulates secretion of CCK by a mechanism that requires calcium (Ca2+) influx and/or intracellular (i.e., endoplasmic reticulum, ER) mobilization (McLaughlin et al., 1998; Hira et al., 2004; Tanaka et al., 2008). CCK activates CCKA receptors located on adjacent afferent sensory vagal fibers, which in turn, communicate with the brain and control meal size and satiation (Smith et al., 1981, 1985; Schwartz and Moran, 1994; Kaelberer et al., 2018). Consumption of a Western diet (WD) is associated with increased levels of the endocannabinoids (eCBs) and their activity at CB1Rs in the upper small-intestinal epithelium, which we propose inhibits CCK release and satiation signaling. The molecular mechanism(s) mediating CB1R control of CCK release is unknown, but may include inhibition of Ca2+-mediated CCK release. A future test of this hypothesis is warranted.
Our studies suggest that the eCB system in the small intestinal epithelium controls feeding behavior by a mechanism that includes inhibiting nutrient-induced release of the gut-derived satiation peptide, CCK, which in turn increases meal size and caloric intake. CCK is secreted from enteroendocrine I-cells in the upper small intestinal epithelium after nutrients arrive in the lumen (McLaughlin et al., 1999; Raybould et al., 2006; Steinert et al., 2017; Kaelberer et al., 2018) and controls meal size and induces satiation by activating CCKA receptors on afferent vagal fibers (Smith et al., 1981, 1985; Schwartz and Moran, 1994; Raybould, 2007; Kaelberer et al., 2018; Schwartz, 2018) and possibly the brain (Reidelberger et al., 2004; Ripken et al., 2015). Indeed, polymorphisms in CCKA receptor genes in humans are associated with increased meal size and food intake, and obesity (Miller et al., 1995; Marchal-Victorion et al., 2002; de Krom et al., 2007). Furthermore, CCK in a stabilized form resistant to degradation in the GI tract is effective at reducing food intake and body weight in DIO rodents (Pierson et al., 1997; Irwin et al., 2012, 2013b), and activating CCKA receptors enhances the anti-obesity properties of GLP-1 agonists, amylin, and leptin (Trevaskis et al., 2010, 2015; Irwin et al., 2013a, 2015).
Gene transcripts and immunoreactivity for CB1Rs were found in CCK-containing I-cells in the upper small-intestinal epithelium of mice (see Figures 1, 2; Sykaras et al., 2012). Furthermore, the hypophagic effects of AM6545 were blocked by co-administration of the CCKA receptor antagonist, devazepide. These results suggest that when eCB activity is elevated at local CB1Rs in the upper small-intestinal epithelium in DIO, increased CB1R activation may inhibit nutrient-induced release of satiation peptides from small-intestinal enteroendocrine cells and lead to increased meal size and caloric intake. In support of this hypothesis, oral gavage of corn oil—which potently increases circulating levels of bioactive CCK-8 in lean mice that have low levels of small-intestinal eCB levels—failed to affect circulating levels of CCK-8 in mice chronically fed WD that have elevated eCB levels in the small-intestinal epithelium. Moreover, inhibiting elevated eCB signaling at peripheral CB1Rs with AM6545 in WD mice—at a dose that blocked overeating—restored the ability for corn oil to increase circulating levels of CCK-8.
The mechanisms of CB1R control of nutrient-induced release of CCK from enteroendocrine I-cells in the upper small-intestinal epithelium are unknown. Nonetheless, a primary mechanism by which CB1Rs block neurotransmitter release is by inhibiting calcium influx or mobilization (Howlett et al., 2010; Pertwee, 2015), and nutrient-induced CCK release is calcium-dependent (McLaughlin et al., 1998; Gevrey et al., 2002; Hira et al., 2004; Liou et al., 2011b; Nakajima et al., 2012). Thus, CB1R activity may inhibit release of gut peptides by a mechanism that includes inhibiting calcium influx or mobilization; however, a direct test of this hypothesis remains to be performed (see Figure 7 for proposed mechanism).
It is controversial if obesity impacts CCK secretion [see for review, (Steinert et al., 2017)]. In line with our present findings in mice, several studies suggest that CCK secretion is reduced in obese humans: fasting CCK levels were lower than non-obese (Baranowska et al., 2000) and a trend toward lower CCK release after intra-duodenal infusions of oleic acid in overweight or obese subjects (Stewart et al., 2011). Fat-induced CCK secretion and satiation induced by CCK administration were also reduced in rats fed a high-fat diet (Duca et al., 2013). Other studies, however, reported no differences in CCK levels between obese or lean humans following a meal (Brennan et al., 2012), and increases in CCK after a high-fat meal (French et al., 1993). Furthermore, several preclinical studies in rodents suggest that sensitivity of vagal afferent neurons to the satiating effects of CCK may be decreased in DIO (Covasa et al., 2000; Daly et al., 2011; Duca et al., 2013; de Lartigue, 2016). This phenomenon may be due, in part, to changes in membrane properties of neurons in the nodose ganglion. The satiating actions of a physiological dose of CCK, however, were equally effective in suppressing food intake in obese and lean human subjects (Lieverse et al., 1995). Moreover, a variety of studies conducted over the past several decades show that CCK-induced satiation is mediated by the vagus nerve (Smith et al., 1981, 1985; Schwartz and Moran, 1994; Raybould, 2007; Dockray, 2013; Kaelberer et al., 2018; Schwartz, 2018); however, selected studies show that gut-derived CCK may additionally interact with CCK-A receptors in the brain (Reidelberger et al., 2004; Ripken et al., 2015). We used the brain-penetrant CCK-A receptor antagonist, devazepide, in our studies; therefore, we cannot rule out the possibility that CCK-A receptors in the brain participate in the appetite-suppressing effects of CCK release following inhibition of peripheral CB1Rs. Thus, given discrepancies in the literature regarding the underlying mechanisms of gut-brain signaling and its dysregulation in DIO, it is critical to examine the impact of diet and obesity on gut-brain satiation signaling using reliable and reproducible model systems.
It is plausible that CB1R control of nutrient-induced CCK release is one of several mechanisms by which peripheral CB1Rs impact gut-brain signaling pathways (Burdyga et al., 2004, 2006; Cluny et al., 2013). For example, administration of ghrelin – which is produced in the stomach and upper small intestinal epithelium and increases feeding [see for review, (Steinert et al., 2017; Kaelberer and Bohorquez, 2018)] – blocked downregulation of CB1Rs in the nodose ganglion after, both, re-feeding and CCK administration in fasted rats (Burdyga et al., 2006). Moreover, pharmacological inhibition of CB1Rs blocked fasting-induced ghrelin production in rats (Cani et al., 2004; Al-Massadi et al., 2011; Senin et al., 2013), which suggests that CB1Rs in the upper GI tract may control ghrelin signaling. Furthermore, Kunos and colleagues reported that a peripherally-restricted CB1R inverse agonist improved a host of metabolic parameters as well as reducing food intake in DIO mice by a mechanism that may include reversing hyperleptinemia and leptin resistance associated with DIO (Tam et al., 2012) and restoring anorexic melanocortin signaling in the arcuate nucleus of the hypothalamus (Tam et al., 2017). Bellocchio and colleagues reported that the hypophagic effects of CB1R inhibition with the CB1R inverse agonist, rimonabant, is blocked by pharmacological inhibition of peripheral beta-adrenergic neurotransmission (Bellocchio et al., 2013), which suggests that CB1Rs may additionally control feeding behavior via interactions with the peripheral sympathetic nervous system. This study also showed that intact afferent vagal signaling was required for the hypophagic effects of rimonabant, and CB1Rs in the brain were not required for its pharmacological actions. Nonetheless, circulating levels of the eCBs increase in human and rodent models of obesity (Engeli et al., 2005; Bluher et al., 2006; Cote et al., 2007; Di Marzo et al., 2009; Matias et al., 2012; Argueta and DiPatrizio, 2017; Hillard, 2017; Simon and Cota, 2017; Little et al., 2018), which may directly interact with CB1Rs in the brain and control feeding behavior and energy homeostasis. A comprehensive analysis of this possibility remains to be performed. In addition to I-cells in the small intestinal epithelium (see Figures 1, 2; Sykaras et al., 2012), CB1Rs are also expressed in K-cells that produce and secrete glucose-dependent insulinotropic peptide [GIP (Moss et al., 2012; Reimann and Gribble, 2016)]. Pharmacological activation of CB1Rs inhibits GIP release in rodents, which suggests that local CB1Rs may impact glucose homeostasis via a mechanism that includes controlling nutrient-induced incretin release. Lastly, enteroendocrine cells in the intestinal lining form functional synapses with afferent vagal fibers (Kaelberer et al., 2018). Termed “neuropods” by Bohorquez and colleagues, these cells sense nutrients and release glutamate and CCK in a coordinated manner that interact with corresponding receptors on local afferent vagal fibers, which in turn, communicate with the brain. Our data suggest that CB1Rs may be at the interface of this signaling. It is unknown, however, if CB1Rs control glutamate signaling at these synapses in the small intestine as they do in the brain (Jung et al., 2007). Collectively, these studies—in combination with the present report—describe key roles for peripheral CB1Rs in feeding behavior and energy homeostasis.
In summary, our results provide evidence of a previously unknown mechanism of CB1R-mediated inhibition of gut-brain satiation signaling in DIO that promotes overeating. Pharmacological manipulation of these pathways in the periphery may provide a therapeutic advantage for the treatment of obesity and related metabolic disorders when compared to anti-obesity drugs that interact with the brain and display psychiatric side-effects (Christensen et al., 2007; Khera et al., 2016). Despite the peripherally-restricted properties of these CB1R antagonists, however, their impact on cognition and brain function by altering gut microbe activity is unknown and remains to be reported.
Data Availability
The datasets generated for this study are available on request to the corresponding author.
Ethics Statement
All procedures met the U.S. National Institute of Health guidelines for care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee of the University of California, Riverside.
Author Contributions
DA and PP contributed to experimental design, performed experiments, collected data, processed data, and contributed to writing of the manuscript. AM provided the pharmacological compound, AM6545, and reviewed the manuscript. ND orchestrated the project, designed experiments, processed data, and wrote the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the contributions of the following for technical support and expertise: Dr. David D. Lo (microscopy and FACS), Dr. Declan McCole, Dr. Ken Mackie, Dr. Ali Shawki, Marisol Arellano; the graduate students Mark Wiley and Andrea Dillon; and the research volunteers, Arshia Aghasharif, Erica Burnham, Victoria Dinh, Vishal K. Gupta, Kevin Mortazavi, Rishi K. Nanda, Crystal Nguyen, Jasmin D. Sanchez, Doris Xie, and Mellonie Zhang.
Glossary
Abbreviations
- 2-AG
2-Arachidonoyl-sn-glycerol
- Abhd6
alpha-beta-hydrolyzing domain 6
- AEA
Anandamide
- AM
AM6545
- CB1R
Cannabinoid receptor subtype 1
- CB2R
Cannabinoid receptor subtype 2
- CCK
Cholecystokinin
- CO
Corn oil
- DAG
Diacylglycerol
- Dev
Devazepide
- DGL
Diacylglycerol lipase
- DIO
Diet-induced obesity
- eCB
Endocannabinoid
- eGFP
Enhanced green fluorescent protein
- FAAH
Fatty acid amide hydrolase
- FACS
Fluorescence activated cell sorting
- FAE
Fatty acid ethanolamide
- MAG
Monoacylglycerol
- MGL
Monoacylglycerol lipase
- NAPE-PLD
N-acyl phosphatidylethanolamine-specific phospholipase D
- SD
Standard diet
- WD
Western diet
- WIN
WIN 55,212-2.
Footnotes
Funding. This study was funded by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases grants DK119498 and DK114978 to ND, and National Institute on Drug Abuse grant DA034009 to ND. In addition, we thank the Ford Foundation Dissertation Fellowship to DA.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/article/10.3389/fphys.2019.00704/full#supplementary-material
References
- Al-Massadi O., Gabellieri E., Trujillo M. L., Senaris R., Pagotto U., Pasquali R., et al. (2011). Peripheral endocannabinoid system-mediated actions of rimonabant on growth hormone secretion are ghrelin-dependent. J. Neuroendocrinol. 22, 1127–1136. 10.1111/j.1365-2826.2010.02065.x [DOI] [PubMed] [Google Scholar]
- Argueta D. A., Dipatrizio N. V. (2017). Peripheral endocannabinoid signaling controls hyperphagia in western diet-induced obesity. Physiol. Behav. 171, 32–39. 10.1016/j.physbeh.2016.12.044, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranowska B., Radzikowska M., Wasilewska-Dziubinska E., Roguski K., Borowiec M. (2000). Disturbed release of gastrointestinal peptides in anorexia nervosa and in obesity. Diabetes Obes. Metab. 2, 99–103. 10.1046/j.1463-1326.2000.00070.x, PMID: [DOI] [PubMed] [Google Scholar]
- Bellocchio L., Soria-Gomez E., Quarta C., Metna-Laurent M., Cardinal P., Binder E., et al. (2013). Activation of the sympathetic nervous system mediates hypophagic and anxiety-like effects of CB1 receptor blockade. Proc. Natl. Acad. Sci. USA 110, 4786–4791. 10.1073/pnas.1218573110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bermudez-Silva F. J., Suarez J., Baixeras E., Cobo N., Bautista D., Cuesta-Munoz A. L., et al. (2008). Presence of functional cannabinoid receptors in human endocrine pancreas. Diabetologia 51, 476–487. 10.1007/s00125-007-0890-y, PMID: [DOI] [PubMed] [Google Scholar]
- Bluher M., Engeli S., Kloting N., Berndt J., Fasshauer M., Batkai S., et al. (2006). Dysregulation of the peripheral and adipose tissue endocannabinoid system in human abdominal obesity. Diabetes 55, 3053–3060. 10.2337/db06-0812, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan I. M., Luscombe-Marsh N. D., Seimon R. V., Otto B., Horowitz M., Wishart J. M., et al. (2012). Effects of fat, protein, and carbohydrate and protein load on appetite, plasma cholecystokinin, peptide YY, and ghrelin, and energy intake in lean and obese men. Am. J. Physiol. Gastrointest. Liver Physiol. 303, G129–G140. 10.1152/ajpgi.00478.2011, PMID: [DOI] [PubMed] [Google Scholar]
- Burdyga G., Lal S., Varro A., Dimaline R., Thompson D. G., Dockray G. J. (2004). Expression of cannabinoid CB1 receptors by vagal afferent neurons is inhibited by cholecystokinin. J. Neurosci. 24, 2708–2715. 10.1523/JNEUROSCI.5404-03.2004, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdyga G., Varro A., Dimaline R., Thompson D. G., Dockray G. J. (2006). Ghrelin receptors in rat and human nodose ganglia: putative role in regulating CB-1 and MCH receptor abundance. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G1289–G1297. 10.1152/ajpgi.00543.2005, PMID: [DOI] [PubMed] [Google Scholar]
- Burdyga G., Varro A., Dimaline R., Thompson D. G., Dockray G. J. (2010). Expression of cannabinoid CB1 receptors by vagal afferent neurons: kinetics and role in influencing neurochemical phenotype. Am. J. Physiol. Gastrointest. Liver Physiol. 299, G63–G69. 10.1152/ajpgi.00059.2010, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani P. D., Knauf C. (2016). How gut microbes talk to organs: the role of endocrine and nervous routes. Mol. Metab. 5, 743–752. 10.1016/j.molmet.2016.05.011, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani P. D., Montoya M. L., Neyrinck A. M., Delzenne N. M., Lambert D. M. (2004). Potential modulation of plasma ghrelin and glucagon-like peptide-1 by anorexigenic cannabinoid compounds, SR141716A (rimonabant) and oleoylethanolamide. Br. J. Nutr. 92, 757–761. 10.1079/BJN20041256, PMID: [DOI] [PubMed] [Google Scholar]
- Charpentier B., Pelaprat D., Durieux C., Dor A., Reibaud M., Blanchard J. C., et al. (1988). Cyclic cholecystokinin analogues with high selectivity for central receptors. Proc. Natl. Acad. Sci. USA 85, 1968–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen R., Kristensen P. K., Bartels E. M., Bliddal H., Astrup A. (2007). Efficacy and safety of the weight-loss drug rimonabant: a meta-analysis of randomised trials. Lancet 370, 1706–1713. 10.1016/S0140-6736(07)61721-8, PMID: [DOI] [PubMed] [Google Scholar]
- Cluny N. L., Baraboi E. D., Mackie K., Burdyga G., Richard D., Dockray G. J., et al. (2013). High fat diet and body weight have different effects on cannabinoid CB(1) receptor expression in rat nodose ganglia. Auton. Neurosci. 179, 122–130. 10.1016/j.autneu.2013.09.015, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cluny N. L., Vemuri V. K., Chambers A. P., Limebeer C. L., Bedard H., Wood J. T., et al. (2011). A novel peripherally restricted cannabinoid receptor antagonist, AM6545, reduces food intake and body weight, but does not cause malaise, in rodents. Br. J. Pharmacol. 161, 629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cote M., Matias I., Lemieux I., Petrosino S., Almeras N., Despres J. P., et al. (2007). Circulating endocannabinoid levels, abdominal adiposity and related cardiometabolic risk factors in obese men. Int. J. Obes. 31, 692–699. 10.1038/sj.ijo.0803539, PMID: [DOI] [PubMed] [Google Scholar]
- Covasa M., Grahn J., Ritter R. C. (2000). High fat maintenance diet attenuates hindbrain neuronal response to CCK. Regul. Pept. 86, 83–88. 10.1016/S0167-0115(99)00084-1, PMID: [DOI] [PubMed] [Google Scholar]
- Cvijanovic N., Isaacs N. J., Rayner C. K., Feinle-Bisset C., Young R. L., Little T. J. (2017). Duodenal fatty acid sensor and transporter expression following acute fat exposure in healthy lean humans. Clin. Nutr. 36, 564–569. 10.1016/j.clnu.2016.02.005, PMID: [DOI] [PubMed] [Google Scholar]
- Daly D. M., Park S. J., Valinsky W. C., Beyak M. J. (2011). Impaired intestinal afferent nerve satiety signalling and vagal afferent excitability in diet induced obesity in the mouse. J. Physiol. 589, 2857–2870. 10.1113/jphysiol.2010.204594, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Krom M., Van Der Schouw Y. T., Hendriks J., Ophoff R. A., Van Gils C. H., Stolk R. P., et al. (2007). Common genetic variations in CCK, leptin, and leptin receptor genes are associated with specific human eating patterns. Diabetes 56, 276–280. 10.2337/db06-0473 [DOI] [PubMed] [Google Scholar]
- de Lartigue G. (2016). Role of the vagus nerve in the development and treatment of diet-induced obesity. J. Physiol. 594, 5791–5815. 10.1113/JP271538, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L., Marini P., Matias I., Moriello A. S., Starowicz K., Cristino L., et al. (2007). Mechanisms for the coupling of cannabinoid receptors to intracellular calcium mobilization in rat insulinoma beta-cells. Exp. Cell Res. 313, 2993–3004. 10.1016/j.yexcr.2007.05.012, PMID: [DOI] [PubMed] [Google Scholar]
- Di Marzo V., Capasso R., Matias I., Aviello G., Petrosino S., Borrelli F., et al. (2008). The role of endocannabinoids in the regulation of gastric emptying: alterations in mice fed a high-fat diet. Br. J. Pharmacol. 153, 1272–1280. 10.1038/sj.bjp.0707682, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V., Cote M., Matias I., Lemieux I., Arsenault B. J., Cartier A., et al. (2009). Changes in plasma endocannabinoid levels in viscerally obese men following a 1 year lifestyle modification programme and waist circumference reduction: associations with changes in metabolic risk factors. Diabetologia 52, 213–217. 10.1007/s00125-008-1178-6, PMID: [DOI] [PubMed] [Google Scholar]
- DiPatrizio N. V. (2016). Endocannabinoids in the gut. Cannabis Cannabinoid Res. 1, 67–77. 10.1089/can.2016.0001, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPatrizio N. V., Astarita G., Schwartz G., Li X., Piomelli D. (2011). Endocannabinoid signal in the gut controls dietary fat intake. Proc. Natl. Acad. Sci. USA 108, 12904–12908. 10.1073/pnas.1104675108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPatrizio N. V., Igarashi M., Narayanaswami V., Murray C., Gancayco J., Russell A., et al. (2015). Fasting stimulates 2-AG biosynthesis in the small intestine: role of cholinergic pathways. Am. J. Phys. Regul. Integr. Comp. Phys. 309, R805–R813. 10.1152/ajpregu.00239.2015, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPatrizio N. V., Joslin A., Jung K. M., Piomelli D. (2013). Endocannabinoid signaling in the gut mediates preference for dietary unsaturated fats. FASEB J. 27, 2513–2520. 10.1096/fj.13-227587, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPatrizio N. V., Piomelli D. (2012). The thrifty lipids: endocannabinoids and the neural control of energy conservation. Trends Neurosci. 35, 403–411. 10.1016/j.tins.2012.04.006, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPatrizio N. V., Piomelli D. (2015). Intestinal lipid-derived signals that sense dietary fat. J. Clin. Invest. 125, 891–898. 10.1172/JCI76302, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockray G. J. (2013). Enteroendocrine cell signalling via the vagus nerve. Curr. Opin. Pharmacol. 13, 954–958. 10.1016/j.coph.2013.09.007, PMID: [DOI] [PubMed] [Google Scholar]
- Duca F. A., Zhong L., Covasa M. (2013). Reduced CCK signaling in obese-prone rats fed a high fat diet. Horm. Behav. 64, 812–817. 10.1016/j.yhbeh.2013.09.004, PMID: [DOI] [PubMed] [Google Scholar]
- Engeli S., Bohnke J., Feldpausch M., Gorzelniak K., Janke J., Batkai S., et al. (2005). Activation of the peripheral endocannabinoid system in human obesity. Diabetes 54, 2838–2843. 10.2337/diabetes.54.10.2838, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eysselein V. E., Reeve J. R. Jr., Shively J. E., Miller C., Walsh J. H. (1984). Isolation of a large cholecystokinin precursor from canine brain. Proc. Natl. Acad. Sci. USA 81, 6565–6568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French S. J., Murray B., Rumsey R. D., Sepple C. P., Read N. W. (1993). Preliminary studies on the gastrointestinal responses to fatty meals in obese people. Int. J. Obes. Relat. Metab. Disord. 17, 295–300. PMID: [PubMed] [Google Scholar]
- Gevrey J.-C., Cordier-Bussat M., Némoz-Gaillard E., Chayvialle J.-A., Abello J. (2002). Co-requirement of cyclic AMP- and calcium-dependent protein kinases for transcriptional activation of cholecystokinin gene by protein hydrolysates. J. Biol. Chem. 277, 22407–22413. 10.1074/jbc.M201624200, PMID: [DOI] [PubMed] [Google Scholar]
- Gomez R., Navarro M., Ferrer B., Trigo J. M., Bilbao A., Del Arco I., et al. (2002). A peripheral mechanism for CB1 cannabinoid receptor-dependent modulation of feeding. J. Neurosci. 22, 9612–9617. 10.1523/JNEUROSCI.22-21-09612.2002, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Mariscal I., Montoro R. A., Doyle M. E., Liu Q. R., Rouse M., O’connell J. F., et al. (2018). Absence of cannabinoid 1 receptor in beta cells protects against high-fat/high-sugar diet-induced beta cell dysfunction and inflammation in murine islets. Diabetologia 61, 1470–1483. 10.1007/s00125-018-4576-4, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg L. C., Jung K. M., Spradley J. M., Nyilas R., Suplita R. L. 2nd, Zimmer A., et al. (2012). Activation of type 5 metabotropic glutamate receptors and diacylglycerol lipase-alpha initiates 2-arachidonoylglycerol formation and endocannabinoid-mediated analgesia. J. Neurosci. 32, 9457–9468. 10.1523/JNEUROSCI.0013-12.2012, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillard C. J. (2017). Circulating endocannabinoids: from whence do they come and where are they going? Neuropsychopharmacology 43, 155–172. 10.1038/npp.2017.130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hira T., Elliott A. C., Thompson D. G., Case R. M., McLaughlin J. T. (2004). Multiple fatty acid sensing mechanisms operate in enteroendocrine cells: novel evidence for direct mobilization of stored calcium by cytosolic fatty acid. J. Biol. Chem. 279, 26082–26089. 10.1074/jbc.M400098200, PMID: [DOI] [PubMed] [Google Scholar]
- Howlett A. C., Blume L. C., Dalton G. D. (2010). CB(1) cannabinoid receptors and their associated proteins. Curr. Med. Chem. 17, 1382–1393. 10.2174/092986710790980023, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin N., Frizelle P., Montgomery I. A., Moffett R. C., O’harte F. P. M., Flatt P. R. (2012). Beneficial effects of the novel cholecystokinin agonist (pGlu-Gln)-CCK-8 in mouse models of obesity/diabetes. Diabetologia 55, 2747–2758. 10.1007/s00125-012-2654-6, PMID: [DOI] [PubMed] [Google Scholar]
- Irwin N., Montgomery I. A., Flatt P. R. (2013a). Comparison of the metabolic effects of sustained CCK1 receptor activation alone and in combination with upregulated leptin signalling in high-fat-fed mice. Diabetologia 56, 1425–1435. 10.1007/s00125-013-2878-0 [DOI] [PubMed] [Google Scholar]
- Irwin N., Montgomery I. A., O'harte F. P., Frizelle P., Flatt P. R. (2013b). Comparison of the independent and combined metabolic effects of subchronic modulation of CCK and GIP receptor action in obesity-related diabetes. Int. J. Obes. 37, 1058–1063. 10.1038/ijo.2012.179 [DOI] [PubMed] [Google Scholar]
- Irwin N., Pathak V., Flatt P. R. (2015). A novel CCK-8/GLP-1 hybrid peptide exhibiting prominent insulinotropic, glucose-lowering, and satiety actions with significant therapeutic potential in high-fat-fed mice. Diabetes 64, 2996–3009. 10.2337/db15-0220, PMID: [DOI] [PubMed] [Google Scholar]
- Izzo A. A., Piscitelli F., Capasso R., Aviello G., Romano B., Borrelli F., et al. (2009). Peripheral endocannabinoid dysregulation in obesity: relation to intestinal motility and energy processing induced by food deprivation and re-feeding. Br. J. Pharmacol. 158, 451–461. 10.1111/j.1476-5381.2009.00183.x, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan-Pico P., Fuentes E., Bermudez-Silva F. J., Javier Diaz-Molina F., Ripoll C., Rodriguez De Fonseca F., et al. (2006). Cannabinoid receptors regulate Ca(2+) signals and insulin secretion in pancreatic beta-cell. Cell Calcium 39, 155–162. 10.1016/j.ceca.2005.10.005, PMID: [DOI] [PubMed] [Google Scholar]
- Jung K. M., Astarita G., Zhu C., Wallace M., Mackie K., Piomelli D. (2007). A key role for diacylglycerol lipase-alpha in metabotropic glutamate receptor-dependent endocannabinoid mobilization. Mol. Pharmacol. 72, 612–621. 10.1124/mol.107.037796, PMID: [DOI] [PubMed] [Google Scholar]
- Jung K. M., Sepers M., Henstridge C. M., Lassalle O., Neuhofer D., Martin H., et al. (2012). Uncoupling of the endocannabinoid signalling complex in a mouse model of fragile X syndrome. Nat. Commun. 3:1080. 10.1038/ncomms2045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelberer M. M., Bohorquez D. V. (2018). The now and then of gut-brain signaling. Brain Res. 1693, 192–196. 10.1016/j.brainres.2018.03.027, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelberer M. M., Buchanan K. L., Klein M. E., Barth B. B., Montoya M. M., Shen X., et al. (2018). A gut-brain neural circuit for nutrient sensory transduction. Science 361:eaat5236. 10.1126/science.aat5236, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera R., Murad M. H., Chandar A. K., Dulai P. S., Wang Z., Prokop L. J., et al. (2016). Association of pharmacological treatments for obesity with weight loss and adverse events: a systematic review and meta-analysis. JAMA 315, 2424–2434. 10.1001/jama.2016.7602, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Bowe J. E., Jones P. M., Persaud S. J. (2010). Expression and function of cannabinoid receptors in mouse islets. Islets 2, 293–302. 10.4161/isl.2.5.12729 [DOI] [PubMed] [Google Scholar]
- Lieverse R. J., Jansen J. B., Masclee A. A., Lamers C. B. (1995). Satiety effects of a physiological dose of cholecystokinin in humans. Gut 36, 176–179. 10.1136/gut.36.2.176, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou A. P., Lu X., Sei Y., Zhao X., Pechhold S., Carrero R. J., et al. (2011a). The G-protein-coupled receptor GPR40 directly mediates long-chain fatty acid-induced secretion of cholecystokinin. Gastroenterology 140, 903–912. 10.1053/j.gastro.2010.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou A. P., Sei Y., Zhao X., Feng J., Lu X., Thomas C., et al. (2011b). The extracellular calcium-sensing receptor is required for cholecystokinin secretion in response to l-phenylalanine in acutely isolated intestinal I cells. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G538–G546. 10.1152/ajpgi.00342.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little T. J., Cvijanovic N., Dipatrizio N. V., Argueta D. A., Rayner C. K., Feinle-Bisset C., et al. (2018). Plasma endocannabinoid levels in lean, overweight and obese humans: relationships with intestinal permeability markers, inflammation and incretin secretion. Am. J. Physiol. Endocrinol. Metab. 315, E489–E495. 10.1152/ajpendo.00355.2017, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K. J., Schmittgen T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods 25, 402–408. 10.1006/meth.2001.1262, PMID: [DOI] [PubMed] [Google Scholar]
- LoVerme J., Duranti A., Tontini A., Spadoni G., Mor M., Rivara S., et al. (2009). Synthesis and characterization of a peripherally restricted CB1 cannabinoid antagonist, URB447, that reduces feeding and body-weight gain in mice. Bioorg. Med. Chem. Lett. 19, 639–643. 10.1016/j.bmcl.2008.12.059, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccarrone M., Bab I., Biro T., Cabral G. A., Dey S. K., Di Marzo V., et al. (2015). Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 36, 277–296. 10.1016/j.tips.2015.02.008, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal-Victorion S., Vionnet N., Escrieut C., Dematos F., Dina C., Dufresne M., et al. (2002). Genetic, pharmacological and functional analysis of cholecystokinin-1 and cholecystokinin-2 receptor polymorphism in type 2 diabetes and obese patients. Pharmacogenetics 12, 23–30. 10.1097/00008571-200201000-00004, PMID: [DOI] [PubMed] [Google Scholar]
- Matias I., Gatta-Cherifi B., Tabarin A., Clark S., Leste-Lasserre T., Marsicano G., et al. (2012). Endocannabinoids measurement in human saliva as potential biomarker of obesity. PLoS One 7:e42399. 10.1371/journal.pone.0042399, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland M. J., Porter A. C., Rakhshan F. R., Rawat D. S., Gibbs R. A., Barker E. L. (2004). A role for caveolae/lipid rafts in the uptake and recycling of the endogenous cannabinoid anandamide. J. Biol. Chem. 279, 41991–41997. 10.1074/jbc.M407250200, PMID: [DOI] [PubMed] [Google Scholar]
- McLaughlin J., Grazia Luca M., Jones M. N., D'amato M., Dockray G. J., Thompson D. G. (1999). Fatty acid chain length determines cholecystokinin secretion and effect on human gastric motility. Gastroenterology 116, 46–53. 10.1016/S0016-5085(99)70227-1, PMID: [DOI] [PubMed] [Google Scholar]
- McLaughlin J. T., Lomax R. B., Hall L., Dockray G. J., Thompson D. G., Warhurst G. (1998). Fatty acids stimulate cholecystokinin secretion via an acyl chain length-specific, Ca2+−dependent mechanism in the enteroendocrine cell line STC-1. J. Physiol. 513, 11–18. 10.1111/j.1469-7793.1998.011by.x, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller L. J., Holicky E. L., Ulrich C. D., Wieben E. D. (1995). Abnormal processing of the human cholecystokinin receptor gene in association with gallstones and obesity. Gastroenterology 109, 1375–1380. 10.1016/0016-5085(95)90601-0, PMID: [DOI] [PubMed] [Google Scholar]
- Moss C. E., Marsh W. J., Parker H. E., Ogunnowo-Bada E., Riches C. H., Habib A. M., et al. (2012). Somatostatin receptor 5 and cannabinoid receptor 1 activation inhibit secretion of glucose-dependent insulinotropic polypeptide from intestinal K cells in rodents. Diabetologia 55, 3094–3103. 10.1007/s00125-012-2663-5, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima S., Hira T., Hara H. (2012). Calcium-sensing receptor mediates dietary peptide-induced CCK secretion in enteroendocrine STC-1 cells. Mol. Nutr. Food Res. 56, 753–760. 10.1002/mnfr.201100666, PMID: [DOI] [PubMed] [Google Scholar]
- Nakata M., Yada T. (2008). Cannabinoids inhibit insulin secretion and cytosolic Ca2+ oscillation in islet beta-cells via CB1 receptors. Regul. Pept. 145, 49–53. 10.1016/j.regpep.2007.08.009, PMID: [DOI] [PubMed] [Google Scholar]
- Perez P. A., Dipatrizio N. V. (2018). Impact of maternal western diet-induced obesity on offspring mortality and peripheral endocannabinoid system in mice. PLoS One 13:e0205021. 10.1371/journal.pone.0205021, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peroni R. N., Abramoff T., Neuman I., Podesta E. J., Adler-Graschinsky E. (2012). Phytoestrogens enhance the vascular actions of the endocannabinoid anandamide in mesenteric beds of female rats. Int. J. Hypertens. 2012:647856. 10.1155/2012/647856, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R. G. (2001). Cannabinoids and the gastrointestinal tract. Gut 48, 859–867. 10.1136/gut.48.6.859, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee R. G. (2015). Endocannabinoids and their pharmacological actions. Handb. Exp. Pharmacol. 231, 1–37. 10.1007/978-3-319-20825-1_1 [DOI] [PubMed] [Google Scholar]
- Pierson M. E., Comstock J. M., Simmons R. D., Kaiser F., Julien R., Zongrone J., et al. (1997). Synthesis and biological evaluation of potent, selective, hexapeptide CCK-A agonist anorectic agents. J. Med. Chem. 40, 4302–4307. 10.1021/jm970477u, PMID: [DOI] [PubMed] [Google Scholar]
- Piomelli D. (2003). The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 4, 873–884. 10.1038/nrn1247, PMID: [DOI] [PubMed] [Google Scholar]
- Randall P. A., Vemuri V. K., Segovia K. N., Torres E. F., Hosmer S., Nunes E. J., et al. (2010). The novel cannabinoid CB1 antagonist AM6545 suppresses food intake and food-reinforced behavior. Pharmacol. Biochem. Behav. 97, 179–184. 10.1016/j.pbb.2010.07.021, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould H. E. (1999). Nutrient tasting and signaling mechanisms in the gut. I. Sensing of lipid by the intestinal mucosa. Am. J. Phys. 277, G751–G755. [DOI] [PubMed] [Google Scholar]
- Raybould H. E. (2007). Mechanisms of CCK signaling from gut to brain. Curr. Opin. Pharmacol. 7, 570–574. 10.1016/j.coph.2007.09.006, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybould H. E., Glatzle J., Freeman S. L., Whited K., Darcel N., Liou A., et al. (2006). Detection of macronutrients in the intestinal wall. Auton. Neurosci. 125, 28–33. 10.1016/j.autneu.2006.01.016, PMID: [DOI] [PubMed] [Google Scholar]
- Rehfeld J. F. (1998). Accurate measurement of cholecystokinin in plasma. Clin. Chem. 44, 991–1001. PMID: [PubMed] [Google Scholar]
- Reidelberger R. D., Hernandez J., Fritzsch B., Hulce M. (2004). Abdominal vagal mediation of the satiety effects of CCK in rats. Am. J. Phys. Regul. Integr. Comp. Phys. 286, R1005–R1012. 10.1152/ajpregu.00646.2003, PMID: [DOI] [PubMed] [Google Scholar]
- Reimann F., Gribble F. M. (2016). Mechanisms underlying glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 secretion. J. Diabetes Investig. 7(Suppl. 1), 13–19. 10.1111/jdi.12478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripken D., Van Der Wielen N., Van Der Meulen J., Schuurman T., Witkamp R. F., Hendriks H. F., et al. (2015). Cholecystokinin regulates satiation independently of the abdominal vagal nerve in a pig model of total subdiaphragmatic vagotomy. Physiol. Behav. 139, 167–176. 10.1016/j.physbeh.2014.11.031, PMID: [DOI] [PubMed] [Google Scholar]
- Schmidt M. J., Horvath S., Ebert P., Norris J. L., Seeley E. H., Brown J., et al. (2014). Modulation of behavioral networks by selective interneuronal inactivation. Mol. Psychiatry 19, 580–587. 10.1038/mp.2013.167, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz G. J. (2018). Roles for gut vagal sensory signals in determining energy availability and energy expenditure. Brain Res. 1693, 151–153. 10.1016/j.brainres.2018.04.004, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz G. J., Moran T. H. (1994). CCK elicits and modulates vagal afferent activity arising from gastric and duodenal sites. Ann. N. Y. Acad. Sci. 713, 121–128. 10.1111/j.1749-6632.1994.tb44058.x, PMID: [DOI] [PubMed] [Google Scholar]
- Senin L. L., Al-Massadi O., Folguiera C., Pardo M., Barja-Fernandez S., Roca-Rivada A., et al. (2013). The gasric CB1 receptor modulates ghrelin production through the mTOR pathway to regulate food intake. PLoS One 8:e80339. 10.1371/journal.pone.0080339, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulkes A., Baldwin G. S. (1997). Biology of gut cholecystokinin and gastrin receptors. Clin. Exp. Pharmacol. Physiol. 24, 209–216. 10.1111/j.1440-1681.1997.tb01809.x, PMID: [DOI] [PubMed] [Google Scholar]
- Simon V., Cota D. (2017). Mechanisms in endocrinology: endocannabinoids and metabolism: past, present and future. Eur. J. Endocrinol. 176, R309–R324. 10.1530/EJE-16-1044, PMID: [DOI] [PubMed] [Google Scholar]
- Smith G. P., Jerome C., Cushin B. J., Eterno R., Simansky K. J. (1981). Abdominal vagotomy blocks the satiety effect of cholecystokinin in the rat. Science 213, 1036–1037. 10.1126/science.7268408, PMID: [DOI] [PubMed] [Google Scholar]
- Smith G. P., Jerome C., Norgren R. (1985). Afferent axons in abdominal vagus mediate satiety effect of cholecystokinin in rats. Am. J. Phys. 249, R638–R641. [DOI] [PubMed] [Google Scholar]
- Steinert R. E., Feinle-Bisset C., Asarian L., Horowitz M., Beglinger C., Geary N. (2017). Ghrelin, CCK, GLP-1, and PYY(3-36): secretory controls and physiological roles in eating and glycemia in health, obesity, and after RYGB. Physiol. Rev. 97, 411–463. 10.1152/physrev.00031.2014, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. E., Seimon R. V., Otto B., Keast R. S., Clifton P. M., Feinle-Bisset C. (2011). Marked differences in gustatory and gastrointestinal sensitivity to oleic acid between lean and obese men. Am. J. Clin. Nutr. 93, 703–711. 10.3945/ajcn.110.007583, PMID: [DOI] [PubMed] [Google Scholar]
- Sykaras A. G., Demenis C., Case R. M., McLaughlin J. T., Smith C. P. (2012). Duodenal enteroendocrine I-cells contain mRNA transcripts encoding key endocannabinoid and fatty acid receptors. PLoS One 7:e42373. 10.1371/journal.pone.0042373, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J., Cinar R., Liu J., Godlewski G., Wesley D., Jourdan T., et al. (2012). Peripheral cannabinoid-1 receptor inverse agonism reduces obesity by reversing leptin resistance. Cell Metab. 16, 167–179. 10.1016/j.cmet.2012.07.002, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam J., Szanda G., Drori A., Liu Z., Cinar R., Kashiwaya Y., et al. (2017). Peripheral cannabinoid-1 receptor blockade restores hypothalamic leptin signaling. Mol. Metab. 6, 1113–1125. 10.1016/j.molmet.2017.06.010, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Katsuma S., Adachi T., Koshimizu T. A., Hirasawa A., Tsujimoto G. (2008). Free fatty acids induce cholecystokinin secretion through GPR120. Naunyn Schmiedeberg's Arch. Pharmacol. 377, 523–527. 10.1007/s00210-007-0200-8, PMID: [DOI] [PubMed] [Google Scholar]
- Thors L., Belghiti M., Fowler C. J. (2008). Inhibition of fatty acid amide hydrolase by kaempferol and related naturally occurring flavonoids. Br. J. Pharmacol. 155, 244–252. 10.1038/bjp.2008.237, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thors L., Eriksson J., Fowler C. J. (2007). Inhibition of the cellular uptake of anandamide by genistein and its analogue daidzein in cells with different levels of fatty acid amide hydrolase-driven uptake. Br. J. Pharmacol. 152, 744–750. 10.1038/sj.bjp.0707401, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevaskis J. L., Sun C., Athanacio J., D'souza L., Samant M., Tatarkiewicz K., et al. (2015). Synergistic metabolic benefits of an exenatide analogue and cholecystokinin in diet-induced obese and leptin-deficient rodents. Diabetes Obes. Metab. 17, 61–73. 10.1111/dom.12390, PMID: [DOI] [PubMed] [Google Scholar]
- Trevaskis J. L., Turek V. F., Griffin P. S., Wittmer C., Parkes D. G., Roth J. D. (2010). Multi-hormonal weight loss combinations in diet-induced obese rats: therapeutic potential of cholecystokinin? Physiol. Behav. 100, 187–195. 10.1016/j.physbeh.2010.02.023, PMID: [DOI] [PubMed] [Google Scholar]
- Vianna C. R., Donato J. Jr., Rossi J., Scott M., Economides K., Gautron L., et al. (2012). Cannabinoid receptor 1 in the vagus nerve is dispensable for body weight homeostasis but required for normal gastrointestinal motility. J. Neurosci. 32, 10331–10337. 10.1523/JNEUROSCI.4507-11.2012, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh J. H., Lamers C. B., Valenzuela J. E. (1982). Cholecystokinin-octapeptidelike immunoreactivity in human plasma. Gastroenterology 82, 438–444. 10.1016/S0016-5085(82)80391-0, PMID: [DOI] [PubMed] [Google Scholar]
- Wang Y., Chandra R., Samsa L. A., Gooch B., Fee B. E., Cook J. M., et al. (2011). Amino acids stimulate cholecystokinin release through the Ca2+−sensing receptor. Am. J. Physiol. Gastrointest. Liver Physiol. 300, G528–G537. 10.1152/ajpgi.00387.2010, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe M. M., Paquet R. J., Reel G. M. (1985). Specificity of commercially available antibodies used for gastrin measurement. J. Lab. Clin. Med. 105, 417–421. PMID: [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.