

Abstract

Mitragyna speciosa, more commonly known as kratom, is a plant native to Southeast Asia, the leaves of which have been used traditionally as a stimulant, analgesic, and treatment for opioid addiction. Recently, growing use of the plant in the United States and concerns that kratom represents an uncontrolled drug with potential abuse liability, have highlighted the need for more careful study of its pharmacological activity. The major active alkaloid found in kratom, mitragynine, has been reported to have opioid agonist and analgesic activity in vitro and in animal models, consistent with the purported effects of kratom leaf in humans. However, preliminary research has provided some evidence that mitragynine and related compounds may act as atypical opioid agonists, inducing therapeutic effects such as analgesia, while limiting the negative side effects typical of classical opioids. Here we report evidence that an active metabolite plays an important role in mediating the analgesic effects of mitragynine. We find that mitragynine is converted in vitro in both mouse and human liver preparations to the much more potent mu-opioid receptor agonist 7-hydroxymitragynine and that this conversion is mediated by cytochrome P450 3A isoforms. Further, we show that 7-hydroxymitragynine is formed from mitragynine in mice and that brain concentrations of this metabolite are sufficient to explain most or all of the opioid-receptor-mediated analgesic activity of mitragynine. At the same time, mitragynine is found in the brains of mice at very high concentrations relative to its opioid receptor binding affinity, suggesting that it does not directly activate opioid receptors. The results presented here provide a metabolism-dependent mechanism for the analgesic effects of mitragynine and clarify the importance of route of administration for determining the activity of this compound. Further, they raise important questions about the interpretation of existing data on mitragynine and highlight critical areas for further research in animals and humans.
Short abstract
Mitragynine is the major active alkaloid of Mitragyna speciosa (kratom). In mice, this compound is converted to an active metabolite, 7-hydroxymitragynine, which mediates its analgesic effect via the mu-opioid receptor.
Introduction
Mitragyna speciosa, often referred to by the common name kratom, is an evergreen tree native to the tropical jungles of Southeast Asia, where it has been used for at least the last century as a traditional medicine. In its native regions, leaves of the kratom tree are typically consumed as a tea or chewed directly, and are purported to induce stimulant and opioid-like analgesic effects in the user in a dose- and time-dependent manner. These effects have traditionally made kratom popular among agricultural workers, who use the plant to aid in long hours of hard labor. Kratom has also been reported to be useful in ameliorating withdrawal symptoms following cessation of opioid use, and it has achieved some popularity for this use in its native regions.1−8
Over the past decade, kratom use has become increasingly popular in the United States (US). Many thousands of US users have reported that consumption of kratom preparations (most often in the form of dried, powdered leaf material) is an efficacious treatment not only for physical pain, but also for mood and anxiety disorders, particularly in cases where other available treatments have either failed or elicited intolerable side effects.9−13 A significant portion of users have also reported use of kratom as a tool to stop or reduce consumption of prescription or illicit opioids, a potential application that is presently of high interest given the ongoing opioid abuse epidemic in the US. Unfortunately, there have been no controlled clinical studies either examining these alleged therapeutic effects or quantifying any potential risks of kratom use. At the same time, kratom is primarily distributed through unregulated Internet sales and used outside medical supervision. Considering these factors, as well as the reported opioid-like effects and adverse health effects of the plant, the US Drug Enforcement Administration (DEA) and the US Food and Drug Administration (FDA) have both raised concerns that kratom represents a potential drug of abuse and public health threat.14−16 In September 2016, the DEA announced its intent to place the active compounds found in kratom into Schedule I of the Controlled Substances Act but ultimately decided not to proceed with this action following a large public outcry from the kratom user community, including over 23 000 comments logged in the federal register docket for this issue.13,17−19 Despite this turn of events, kratom at this time remains in regulatory limbo, with an uncertain future in the US. Accordingly, our laboratories have been working to develop a preclinical profile of kratom’s biological effects, with the hope that such knowledge will prove useful for the development of future therapeutics based on kratom, as well as guide regulatory decision making on the plant itself.
Kratom contains a number of indole alkaloids that are believed to be the primary contributors to its psychoactive effects. Chief among these is mitragynine (Figure 1), which typically constitutes 1–2% of the dry leaf mass and up to approximately two-thirds of the total alkaloid content.1,5 This compound is joined by 7-hydroxymitragynine (7-OH, Figure 1) in much lower concentrations, typically less than 0.05% of the dried leaf mass.1,20 The other predominant alkaloids found in kratom leaf are speciogynine, speciociliatine, and paynantheine (each ∼0.2–0.5% of dry leaf mass), but at this time little is known about their pharmacology.1,5
Figure 1.

Known (Pathway A) and proposed (Pathway B) metabolic transformations of mitragynine.
We have reported that mitragynine and 7-OH are partial agonists of the human μ-opioid receptor (hMOR) in vitro, with 7-OH (EC50 = 34.5 nM; Emax = 47%) being ∼10-fold more potent than mitragynine (EC50 = 339 nM; Emax = 34%).21 Further, both compounds are known to exhibit opioid-receptor-dependent analgesic effects in rodent models, with 7-OH again being significantly more potent.6,22−25 At the same time, we have also found that both mitragynine and 7-OH are G protein-biased agonists of the mu-opioid receptor (MOR).21,25 We have thus classified the kratom alkaloids as “atypical opioids” and have postulated that these compounds may provide a greater therapeutic window between analgesia and classical opioid side effects. For example, other MOR agonists that activate G protein signaling without recruiting the beta-arrestin pathway exhibit attenuated respiratory depression and reduced inhibition of gastrointestinal (GI) transit compared to classical opioids.26−30 In fact, an early study on the pharmacology of mitragynine demonstrated its superiority compared to the classical opioid codeine in this regard, providing preliminary support for this hypothesis.24 Similarly, mitragynine pseudoindoxyl, a chemical rearrangement product of 7-OH, has been found to be both G-protein biased and exhibit an improved therapeutic window in mice.25
Both methanolic and crude alkaloid extracts of kratom have also demonstrated analgesic properties in rodents.1 However, it currently remains unsettled which of kratom’s alkaloids are the most important contributors to the analgesic properties of such preparations or to those of the raw leaf. Despite its higher potency, we previously estimated on the basis of preclinical studies that the quantity of 7-OH contained in typical dry kratom leaf preparations is insufficient to induce opioid-like effects directly at typical doses consumed by human subjects.1 At the same time, examination of the existing literature on the pharmacology of mitragynine revealed a number of unusual observations that complicated our understanding at the outset of the present investigations. First, despite low to moderate oral bioavailability (20–30% in rats), mitragynine has been found in prior investigations to be paradoxically more potent as an analgesic when administered by the oral (p.o.) and intraperitoneal (i.p.) routes compared to the subcutaneous (s.c.) route in rats and mice.1,24,31 These findings suggested to us the involvement of an active metabolite produced via first-pass metabolism in mediating the analgesic activity of mitragynine.1 At the same time, our own prior in vitro studies at the mouse mu-opioid receptor (mMOR) had shown that mitragynine acts as either a competitive antagonist or a partial agonist depending on the assay system used, calling into question the ability of this compound to have a direct agonist effect on MOR in rodents and consistent with a metabolite-driven analgesic effect.21,25 However, mitragynine has also been found to induce analgesic effects when administered intracerebroventricularly (i.c.v), which argued conversely against the involvement of an active metabolite (unless metabolism occurs directly in the brain).23 Therefore, we set out to further elucidate the pharmacological and metabolic mechanism(s) leading to mitragynine’s analgesic effects.
Results
Known Metabolites Do Not Explain Analgesic Activity
Given our working hypothesis that an active metabolite might be involved in mediating the analgesic effects of mitragynine, we were interested in identifying metabolites that might exhibit agonist activity at MOR. The metabolites of mitragynine in rat and human urine had been previously studied, revealing a large number of metabolites modified primarily through demethylation at one or more of mitragynine’s three O-methyl groups (Figure 1, Pathway A), followed by either glucuronidation or sulfation of the exposed nucleophile, or oxidative or reductive transformations of the acrylate moiety.32 Considering previously observed structure–activity relationships (SAR) in the mitragynine scaffold, it was expected that the demethylated metabolites would exhibit similar or lower potency activity at MOR compared to mitragynine.21 This was confirmed by synthesis of these compounds and testing using in vitro functional assays at hMOR (see Supporting Information, Table S1). Similarly, conjugated metabolites were also expected to be both inactive at MOR and have low penetration of the blood–brain barrier (BBB). Accordingly, when we began our investigation, known metabolites were insufficient to explain the opioid-mediated analgesic activity of mitragynine.
Deuteration of Mitragynine Has Little Effect on Metabolism in Vitro
Our above supposition was further strengthened by findings with a deuterated analogue. On the basis of the reported metabolic pathways of mitragynine, we synthesized an analogue of this compound fully deuterated at each of its three O-methyl groups, mitragynine-d9 (see Supporting Information, Scheme S1), expecting that, through kinetic isotope effects, the metabolism of this derivative would be slowed. Surprisingly, when we examined the stability of mitragynine-d9 head-to-head with its undeuterated counterpart in human liver microsomes (HLM) and mouse liver microsomes (MLM), stability was unchanged (Figure 2A). Therefore, we concluded that demethylation is only a minor pathway of hepatic metabolism (demethylated metabolites may accumulate slowly in the urine or be formed extrahepatically) and that the previously reported metabolites are unlikely to explain the hypothesized role of a hepatically formed metabolite in the analgesic activity of mitragynine.
Figure 2.

Decomposition of mitragynine and formation of 7-OH in liver microsomes was quantified by LC-MS/MS. (A) Mitragynine was unstable in both MLM and HLM, while deuteration of all three methyl groups, as in mitragynine-d9, had little effect on microsomal stability. (B) Loss of mitragynine in microsomal preparations was accompanied by formation of 7-OH as a metabolite, and this conversion was more efficient in HLM. There was a strong main effect of species on 7-OH concentration. Two-way ANOVA: F1,30 = 51.66, p < 0.0001. ****p < 0.0001 relative to MLM, **p < 0.01 relative to MLM, *p < 0.05 relative to MLM, ns = p > 0.05 relative to MLM. All data points represent the means of two independent experiments with two incubations per experiment, with error bars representing ± SEM.
Chemistry Suggests a New Site of Metabolism
With known metabolites seemingly unable to explain the observations pointing toward an active metabolite, we began exploring alternative metabolic pathways that might yield such a compound. For inspiration, we turned to known chemical transformations of indole alkaloids, including mitragynine, under oxidative conditions. For example, 2,3-disubstituted indoles are known to undergo functionalization under oxidation/halogenation conditions to afford the corresponding 3-substituted indolenines.33 Specifically, mitragynine is known to be oxidized by [bis(trifluoroacetoxy)iodo]benzene (PIFA) to give 7-OH (Scheme 1).6 During our ongoing synthetic explorations of the mitragynine scaffold, we also found that singlet oxygen21 and potassium peroxymonosulfate (Oxone) were effective oxidants for the conversion of mitragynine into 7-OH (Scheme 1). Therefore, we postulated that the 2–3 indole double bond attacked by these chemical oxidants might also be a site for oxidation by cytochrome P450 enzymes (CYPs) to produce 7-OH as a metabolite (Figure 1, Pathway B). Given the much greater potency of 7-OH as an MOR agonist, we hypothesized that even minor conversion to this product might contribute significantly to mitragynine’s analgesic activity and help to explain the apparent contradictions in the literature.
Scheme 1. Conversion of Mitragynine to 7-OH by Chemical Oxidants.

7-OH is a Mitragynine Metabolite in Vitro
To test our hypothesis, we monitored formation of 7-OH by liquid chromatography-tandem mass spectrometry (LC-MS/MS) during incubation of mitragynine with both HLM and MLM. In both microsome preparations, 7-OH was produced concomitant with disappearance of mitragynine (Figure 2B). Further, 7-OH appeared to be the major metabolite in each case, as the approximate decreases in molar mitragynine concentration from the starting level (2 μM) were accompanied by similar increases in 7-OH concentration. The metabolic conversion was more efficient in HLM, suggesting that an appreciation of interspecies differences is likely to be important for understanding the pharmacology of mitragynine. However, note that our findings conflict with an earlier report, which found that mitragynine was stable in microsomes.34 The reason for this discrepancy remains unclear at this time, but it may relate to variable metabolic activity of microsome preparations from different sources. Further, we confirmed the low stability of mitragynine in HLM and concomitant formation of 7-OH in an independent laboratory (Biotranex, LLC, data not shown).
7-OH is Resistant to Phase I Metabolism
Having found that 7-OH was indeed formed from mitragynine in vitro, we next assessed whether this compound was itself stable in liver preparations. We found that 7-OH was stable in both HLM and MLM, with more than 90% remaining after 40 min of incubation (Figure S1). This finding that 7-OH was stable to oxidative Phase I metabolism was consistent with its accumulation during microsomal incubations of mitragynine.
Mitragynine and 7-OH are Stable in Plasma
Before proceeding further, we also tested the stability of mitragynine and 7-OH in blood plasma. Both mitragynine and 7-OH were highly stable in mouse plasma (Figure S2), indicating that plasma hydrolysis or other plasma metabolism does not contribute significantly to the biotransformation of these compounds.
Conversion of Mitragynine to 7-OH is Mediated by CYP3A Isoforms in Vitro
We next set out to determine which CYP isoform is responsible for conversion of mitragynine to 7-OH. Mitragynine was incubated in vitro with purified recombinant preparations of the five major human CYP isoforms (CYP3A4, 2C19, 2C9, 1A2, 2D6) alongside a reference substrate of each isoform as positive control. Decomposition of mitragynine was nearly complete in the presence of CYP3A4 (2% remaining at 60 min). In contrast, there was little or no decomposition in the incubations with CYP2C19, 2C9, 1A2, and 2D6 (77%, 99%, 96%, and 82% remaining at 60 min, respectively). The activity of each enzyme preparation was confirmed by nearly complete disappearance of the reference substrates (Figure 3A). During these incubations, the formation of 7-OH was also monitored by LC-MS/MS, revealing that formation of 7-OH was most robust in the presence of CYP3A4, whereas little conversion to 7-OH was observed in the incubations with other CYPs (Figure 3B).
Figure 3.

CYP3A4 mediates conversion of mitragynine to 7-OH. (A) Mitragynine was incubated in vitro with recombinant preparations of the five major human CYP isoforms alongside a reference substrate of each isoform as positive control. The relative percent remaining of mitragyine or reference substrate in each incubation was quantified by LC-MS/MS. Disappearance of mitragynine was most rapid in the presence of CYP3A4, whereas incubations with the other isoforms resulted in little or no decomposition. The activity of each preparation was confirmed by the nearly complete disappearance of the corresponding reference substrates. (B) Formation of 7-OH was monitored by LC-MS/MS during incubation of mitragynine with each of the recombinant CYP preparations. CYP3A4 resulted in the most robust conversion to 7-OH. All data points represent the means of two independent experiments with two incubations per experiment, with error bars representing ± SEM.
Having identified CYP3A4 as the predominant metabolic pathway responsible for hepatic metabolism of mitragynine and concurrent formation of 7-OH using purified enzyme preparations, we next sought to confirm these results in liver microsomes, a more complex system with endogenous expression of relevant enzymes. To that end, mitragynine was incubated with HLM alone and in the presence of the CYP3A inhibitor ketoconazole (1 or 10 μM), the CYP2C19 inhibitor ticlopidine (20 μM), or a combination of the two. Ketoconazole robustly inhibited both decomposition of mitragynine (Figure 4A) and formation of 7-OH (Figure 4B), whereas ticlopidine had little effect. Analogous results were found in MLM (Figure S3). In sum, our findings demonstrate that mitragynine is converted to 7-OH in both mouse and human liver preparations and that this conversion is mediated by CYP3A isoforms (including CYP3A4 in humans).
Figure 4.
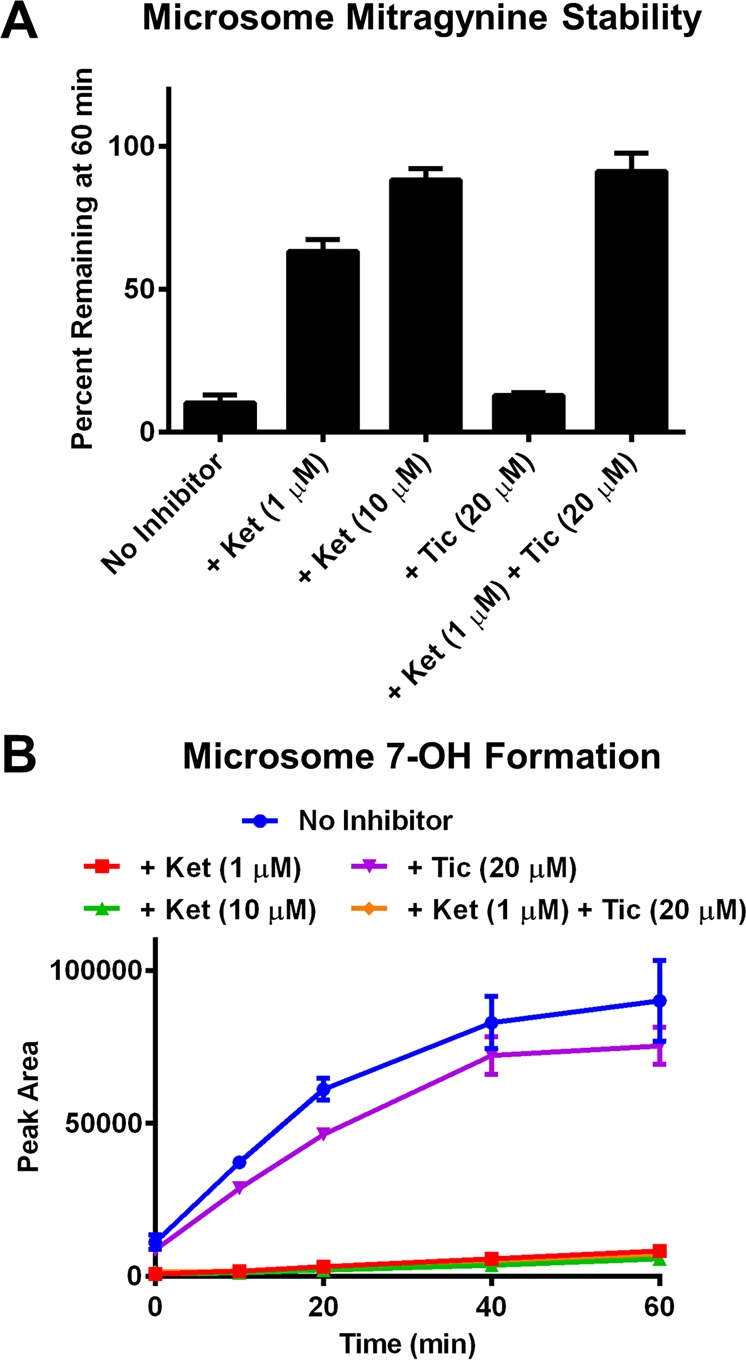
CYP3A-dependence of 7-OH formation was confirmed in HLM. Disappearance of mitragynine and formation of 7-OH was monitored by LC-MS/MS. (A) In HLM, the CYP3A isoform inhibitor ketoconazole (Ket) dose-dependently attenuated metabolic decomposition of mitragynine. In contrast, the CYP2C19 inhibitor ticlopidine (Tic) had little effect. (B) During incubation of mitragynine with HLM, ketoconazole (and to a small degree, ticlopidine) also inhibited formation of 7-OH. All data points represent the means of two independent experiments with two incubations per experiment, with error bars representing ± SEM.
Mitragynine is Analgesic in Mice Through an MOR-Dependent Mechanism
To build on the preceding results, we hoped to examine whether 7-OH is in fact formed as a metabolite of mitragynine in vivo. Further, we sought to test whether 7-OH, as a metabolite, could be a key mediator of the analgesic effects of mitragynine. However, before proceeding with these experiments, we first set out to confirm the analgesic activity of mitragynine in our own hands and to determine whether these effects are dependent on MOR.
For this purpose, we used the 129 mouse strain, which we found to be sensitive to mitragynine-induced analgesia, consistent with the documented high sensitivity of this strain to opioid agonists.35,36 Further, genetic knockouts of opioid receptors are available on this genetic background. Dose–response curves were generated for mitragynine administered by both oral (p.o.) and subcutaneous (s.c.) routes using the tail-flick test, a classic rodent model for analgesic activity (Figure 5A). We found that mitragynine was much more potent when administered p.o. (ED50 = 2.1 mg/kg) than when administered s.c. (ED50 = 106 mg/kg), consistent with earlier literature reports. To establish the opioid receptor dependence of these effects, an efficacious analgesic dose of mitragynine (10 mg/kg, p.o.) was administered to MOR knockout (KO), κ-opioid receptor (KOR) KO, and δ-opioid receptor (DOR) KO mice, and to wild-type (WT) mice following pretreatment with the opioid receptor antagonist naloxone (1 mg/kg, s.c.). Both MOR KO and naloxone pretreatment significantly attenuated the analgesic effect of mitragynine, whereas KOR KO and DOR KO had no effect (Figure 5B). Therefore, the analgesic activity of mitragynine was found to be MOR-dependent, consistent with prior literature demonstrating pharmacological blockade of analgesia with the opioid receptor antagonist naloxone.6,23
Figure 5.

Analgesic activity of mitragynine in 129 mice. (A) Dose–responses of mitragynine and 7-OH in the tail-flick assay at time of maximal analgesic effect (15 min). Mice (n = 5–17 per dose, per treatment; 129S1 strain) were treated with ascending doses of mitragynine or 7-OH by the indicated route of administration in a cumulative dosing procedure, and tail-flick latency was recorded for each animal at 15 min post drug administration (peak analgesic effect). ED50s (95% CI) were as follows: mitragynine, s.c. = 106 (57.4–195) mg/kg; mitragynine, p.o. = 2.05 (1.24–3.38); 7-OH, s.c. = 0.57 (0.19–1.7). The means of each point were calculated as percentage maximal possible effect (%MPE) [(observed latency – baseline latency)/(maximal latency – baseline latency)] × 100. Error bars represent ± SEM. (B) The analgesic effect of mitragynine (10 mg/kg, p.o.) in the tail-flick assay was evaluated 15 min post drug administration in WT, MOR-1 KO, KOR-1 KO, and DOR-1 KO mice, and WT mice following pretreatment with naloxone (1 mg/kg, s.c.), in two independent experiments (n = 5 per group per experiment, n = 10 total per group; 129S6 strain). The analgesic effect of mitragynine was attenuated by naloxone pretreatment and in MOR-1 KO, while the effect was found intact in KOR-1 KO and DOR-1 KO mice. One-way ANOVA: F4,45 = 19.90, p < 0.0001. ****p < 0.0001 relative to WT, ns = p > 0.05 relative to WT. All values are expressed as the mean ± SEM. (C) The analgesic effect of 7-OH (1 mg/kg, s.c.) in the tail-flick assay was evaluated 15 min post drug administration in WT, MOR-1 KO, KOR-1 KO, and DOR-1 KO mice, and WT mice following pretreatment with naloxone (1 mg/kg, s.c.), in two independent experiments (n = 5 per group per experiment, n = 10 total per group; 129S6 strain). The analgesic effect of 7-OH was attenuated by naloxone pretreatment and in MOR-1 KO, while the effect was found intact in KOR-1 KO and DOR-1 KO mice. One-way ANOVA: F4,45 = 101.7, p < 0.0001. ****p < 0.0001 relative to WT, *p < 0.05 relative to WT. All values are expressed as the mean ± SEM.
7-OH is a Potent Analgesic in Mice Acting Through an MOR-Dependent Mechanism
Before further exploring our hypothesis that 7-OH is a key active metabolite of mitragynine, we also wanted to confirm the potent analgesic activity reported for this compound. A dose–response curve was generated for 7-OH in 129 mice using the tail-flick assay (Figure 5A). Consistent with previous reports,22,25 it was found that 7-OH was a highly potent analgesic (ED50 = 0.6 mg/kg, s.c.), ∼fivefold more potent than p.o. mitragynine. This high potency was consistent with our hypothesis that 7-OH formed as a metabolite, even in small quantities, could be responsible for mediating much of mitragynine’s analgesic activity. Both MOR KO and naloxone pretreatment significantly attenuated the analgesic effects of 7-OH, whereas the analgesic effect was retained in KOR KO and DOR KO animals (Figure 5C). Therefore, the analgesic activity induced by 7-OH was also MOR dependent. We note that the reason for the slight enhancement of analgesia in KOR KO and DOR KO animals remains unclear, but some prior literature has suggested that blockade of DOR may enhance MOR-dependent analgesia.37 Further, any potential antagonism of KOR or DOR by mitragynine or 7-OH, as has been suggested by in vitro studies,21 would not be expected to contribute to analgesic effects directly, since antagonists of these receptors are not known to be analgesic on their own. Thus, our findings are consistent with agonism of MOR as the primary mechanism of analgesia for both compounds.
Conversion of Mitragynine to 7-OH also Occurs in Vivo
Having profiled the analgesic effects of mitragynine and 7-OH in mice, we next set out to confirm that the metabolic conversion that we had previously observed in vitro also occurred in living animals. Mice (129S1) were treated with mitragynine (10 mg/kg, s.c.), and plasma and brain samples were collected at 15 and 60 min and analyzed by LC-MS/MS for mitragynine and 7-OH. Both mitragynine (Figure 6A) and 7-OH (Figure 6B) were detected at both time points in both plasma and brain, confirming that 7-OH is indeed formed as a metabolite of mitragynine in vivo and that it enters the brain. These results also permitted a preliminary assessment of the BBB penetration of the two alkaloids by comparison of plasma to whole brain concentrations. By this metric, mitragynine brain penetration was very high (∼1:1 brain/plasma), whereas 7-OH brain penetration was more modest (∼1:5 brain/plasma). However, note that this estimation does not account for tissue binding and, thus, does not necessarily reflect free concentrations of drug.
Figure 6.

Conversion of mitragynine to 7-OH was confirmed in vivo in 129S1 mice using LC-MS/MS. (A) Mitragynine was detected in both the plasma and brains of mice treated with mitragynine (10 mg/kg, s.c.). n = 4 per time point for plasma; n = 8–9 per time point for brain. (B) At the same time, 7-OH was also detected in the plasma and brains of the same animals, but at lower concentrations.
It is interesting to note the differences between our findings in vitro in microsome preparations and those in vivo. In microsomes, we found that 7-OH was a major hepatic metabolite. In contrast, in mice, 7-OH was found to be only a minor metabolite in terms of relative concentrations, with a mitragynine/7-OH ratio in plasma of ∼15:1 or more (dependent on time point). This suggests both inherent differences between the metabolic activity of in vitro and in vivo systems, as well as additional hepatic or extrahepatic routes of mitragynine and/or 7-OH metabolism not well accounted for by in vitro microsome preparations.
7-OH Contributes to the Analgesic Activity of Mitragynine as a Metabolite
Having shown that 7-OH was indeed a metabolite of mitragynine in mice, we last examined whether the concentration of this metabolite formed in vivo might be sufficient to contribute to mitragynine’s opioid-mediated analgesic effects. To demonstrate this, we planned an experiment in which the brain concentration of 7-OH observed as a metabolite following administration of an analgesic dose of mitragynine would be compared to the brain concentration of 7-OH observed following direct administration of an equianalgesic dose of 7-OH. Under these conditions, we hypothesized that 7-OH concentrations would be similar if this metabolite was in fact playing a significant role in mediating the analgesic effects of mitragynine. In testing this hypothesis, we chose to administer both compounds by the s.c. route, as we found that it was difficult to obtain reliable dose–response curves for p.o. 7-OH, likely as a result of variable p.o. absorption of the compound. Thus, s.c. administration was necessary to allow the PK of mitragynine and 7-OH to be compared following the same route of administration, which was desirable from the standpoint of consistency.
Equianalgesic doses of mitragynine (140 mg/kg, s.c.) and 7-OH (0.7 mg/kg, s.c.) were selected based on the previously obtained dose–response curves (∼1.3-fold ED50, Figure 5A). Mice (129S1) were treated with the selected doses, and analgesic activity was confirmed at 15 min in the tail-flick assay. As expected, there was no significant difference in tail-flick latency between the two groups (Figure 7A). Immediately after determination of tail-flick latency, mice were sacrificed, and brain samples were collected for analysis. There was no significant difference in the mean brain concentration of 7-OH found in the mitragynine group (formed as metabolite) compared to that found in the 7-OH group (from direct administration) (Figure 7B), consistent with 7-OH being the primary mediator of central analgesic activity in both cases. At the same time, very brain high concentrations (16.6 ± 2.7 μM; mean ± standard error of the mean (SEM)) of mitragynine were observed in the mitragynine-treated animals, while minimal concentrations (0.097 ± 0.012 μM; mean ± SEM) were observed in the 7-OH-treated animals (Figure 7C). This 170-fold difference in mitragynine brain concentration between the two groups had no significant effect on analgesia (Figure 7A), despite the fact that the mitragynine brain concentration in the mitragynine-treated group was ∼50-fold higher than the compound’s binding affinity for mMOR (0.23 μM21). Accordingly, we conclude that 7-OH formed as a metabolite is sufficient to explain the opioid-mediated analgesic activity of mitragynine and that the parent compound does not make a significant contribution to its own analgesic activity in mice.
Figure 7.

7-OH formed as a metabolite is sufficient to explain mitragynine’s analgesic effect in 129S1 mice. (A) Selected doses of mitragynine (140 mg/kg, s.c.) and 7-OH (0.7 mg/kg, s.c.) were equianalgesic at 15 min in the tail-flick assay and induced ∼50% MPE. n = 10 per treatment. Two-tailed t test, t(18) = 0.66, p = 0.52. ns = p > 0.05. (B) 7-OH was detected in the brains of the same animals at 15 min (sacrifice and sample collection immediately after tail-flick), and there was no difference in mean brain concentration between the two treatments. n = 9 for mitragynine, n = 10 for 7-OH. Two-tailed t test, t(17) = 1.19, p = 0.25. ns = p > 0.05. (C) Mitragynine was detected in the brains of the same animals at 15 min, and there was a significant difference between the two treatments, with a 170-fold higher brain concentration of mitragynine in the animals directly treated with this drug compared to those treated with 7-OH. n = 10 for mitragynine, n = 6 for 7-OH (four values below the lower limit of quantitation, 5 ng/g, were excluded from analysis). Two-tailed t test, t(13) = 4.88, p = 0.0003. ***p < 0.001. All bars represent mean ± SEM.
Although it does not appear to play an important role in mediating analgesia, the above observation of small quantities of mitragynine formed as a metabolite of 7-OH in vivo was surprising to us. We did not expect to observe this reductive metabolic process, which is effectively the reverse of the observed oxidative metabolism of mitragynine to 7-OH. However, prior literature had indeed reported the partial conversion of 7-OH to mitragynine in vitro in liver microsomes and simulated gastric fluid.34 The exact mechanism of this transformation remains unclear at this time.
Discussion
In the present report, we provide evidence that hepatic formation of 7-OH as a metabolite is important in mediating the analgesic activity (and presumably other MOR-mediated effects) of mitragynine, the major active alkaloid of the kratom plant. The analgesic effects of mitragynine and 7-OH each depended on activation of MORs. Further, the analgesia induced by mitragynine appears to depend largely on formation of 7-OH as a metabolite and not on the parent compound. Accordingly, the pharmacological profile of mitragynine may mirror that of 7-OH under certain circumstances, assuming appropriate dose corrections are made to account for the efficiency of conversion to this active metabolite in the system under study. It should be mentioned that during the preparation of this manuscript, other groups separately reported the formation of 7-OH as a metabolite of mitragynine in vitro and in rats, but the mechanistic significance of this finding was not established through pharmacological studies.38,39 Here we confirmed these concurrent research findings and provided the essential in vivo link demonstrating the pharmacological relevance of 7-OH.
Our results highlight the critical need to consider metabolic pathways and interspecies differences when interpreting preclinical data on kratom or attempting translation of such to man. For example, greater or lesser conversion (relative to mice) of mitragynine to 7-OH in a given species would be expected to increase or decrease, respectively, the potency of mitragynine’s opioid activity in that species. At the same time, if similar metabolic conversion is required for the expression of opioid agonist activity in humans, there exists the possibility that metabolic saturation at high doses might provide a built-in ceiling to the opioid effects of kratom, improving the inherent safety of this material. In fact, preliminary evidence of such metabolic saturation can be inferred from the present results, where the 7-OH brain concentration (0.157 μM) at 15 min after a high dose (140 mg/kg, s.c.) of mitragynine was only around sixfold higher than that (0.027 μM) formed from a 14-fold lower dose (10 mg/kg, s.c.). However, full PK profiles at multiple doses will be needed to confirm the existence of such a phenomenon. Likewise, metabolic differences between individuals will need to be considered, and some individuals may be more sensitive to the opioid agonist effects of kratom by virtue of more efficient CYP3A metabolism.
Indeed, here we demonstrated that 7-OH is formed from mitragynine in HLM and that this conversion appears more efficient compared to that in MLM. Accordingly, we expect that 7-OH will also be observed as a metabolite in humans. This hypothesis is supported by a post-mortem toxicological report from a kratom-associated fatality, where mitragynine and 7-OH were quantified in the blood and urine of the decedent.40 Interestingly, the mitragynine/7-OH ratios found in blood and urine were ∼7:1 and ∼1.5:1, respectively. It is our opinion that such high relative concentrations of 7-OH compared to mitragynine would be impossible to achieve in the circulation through direct absorption of the minimal quantities of 7-OH found in kratom leaf preparations and commercial extracts, where the mitragynine/7-OH ratio is typically greater than 50:1. Instead, assuming no adulteration of the ingested material, the high concentrations of 7-OH observed in this case support the hypothesis that 7-OH is also formed as an important metabolite in humans. PK studies will ultimately be required to elucidate the importance of 7-OH as a mitragynine metabolite in man.
It is interesting to note the apparent parallel of mitragynine to codeine, a classical opioid that depends on metabolic conversion to the active metabolite morphine by CYP2D6 for inducement of opioid effects.41 Accordingly, individuals with genetic mutations leading to abnormally high or low activity of CYP2D6 are more or less sensitive to codeine, respectively.41 However, life-threatening respiratory depression with codeine is rare even in cases of intentional overdose, potentially due to metabolic saturation.42 Considering that preclinical studies have found mitragynine to induce less respiratory depression than codeine,24 which is itself relatively safe compared to other classical opioids, we hypothesize that severe respiratory depression or death induced through an opioid mechanism following oral mitragynine (or kratom) consumption is likely to be rare. Consistent with this hypothesis, mitragynine has been found to have low toxicity in mice when administered orally (LD50 > 400 mg/kg).31,43 Further, the active metabolite 7-OH has itself been found to be a partial, G protein-biased agonist at MOR, signaling properties that are hypothesized to lead to improved respiratory safety among MOR agonists.21,25 Overall, these considerations are consistent with the limited reports of kratom overdose deaths (44 in total for all time as of 2017, despite millions of worldwide users)14 in comparison to all opioid overdose deaths (∼50 000 in 2017 alone).44 However, such hypotheses must be confirmed in humans before definitive conclusions are reached regarding the respiratory safety of mitragynine or kratom.
Metabolic processes are also likely to be an important determinant of the abuse liability of kratom and its alkaloids. It has recently been found in two independent studies that mitragynine does not support self-administration (SA) in rats, while 7-OH is self-administered.45,46 We suspect that this apparent contradiction may be explained, at least in part, by the metabolic effects reported here. Mitragynine has been shown to be more potent (in terms of analgesic effects) in rats by the p.o. route24 versus the intravenous (i.v.) route used in the reported self-administration studies, thus complicating appropriate dose selection and challenging the relevance of i.v. SA studies for assessing the abuse liability of p.o. mitragynine (or kratom). Further, the likely necessity of metabolic conversion of mitragynine to 7-OH for the induction of opioid agonist effects might result in a temporal dissociation between drug administration and experience of rewarding effects, further complicating the interpretation of SA studies. Lastly, certain individuals with enhanced or deficient CYP3A4 activity may experience greater or lesser reinforcing effects following mitragynine (or kratom) consumption depending on the extent of conversion to 7-OH. Again, a comparison to codeine is warranted, as the abuse liability of this compound has been found to depend on formation of active metabolites and vary depending on interindividual differences in CYP metabolic efficiency.47 There also remains the possibility that mitragynine exerts a buffering effect on the opioid agonist activity of its 7-OH metabolite, either through direct competitive antagonism at MOR or through other as-yet-unknown pharmacological mechanisms. Ultimately, only controlled clinical trials will be able to definitively resolve these issues for mitragynine.
In addition to interspecies metabolic differences, the in vitro pharmacology of mitragynine at the opioid receptors is also species-dependent and complicates the translation of the present results to humans. For example, we have previously shown in an in vitro assay that mitragynine acts as a competitive antagonist at the mMOR but as a weak partial agonist at the hMOR.21 Accordingly, in humans, mitragynine itself might contribute to its own analgesic effects directly via the parent compound, in contrast to the present mouse study, where most of the analgesic effects appeared to be induced through the 7-OH metabolite. The interplay between mitragynine and 7-OH in mediating kratom’s purported analgesic effects will thus require further exploration in humans.
Lastly, it is important to mention that extracts of the kratom plant have been found to both inhibit and induce various CYP enzymes.48,49 Accordingly, it remains a possibility that the mixture of compounds contained in the plant or its extracts may present a distinct opioid pharmacology compared to isolated mitragynine, by virtue of interference of other chemical species with the hepatic conversion of mitragynine to 7-OH. Thus, additional study of metabolic drug–drug interactions among kratom compounds will also be necessary.
Conclusion
We have found that the opioid pharmacology of mitragynine is complicated by the important role of an active metabolite, 7-OH, in mediating its activity. This apparent requirement for metabolic activation is consistent with the higher analgesic potency of mitragynine administered orally versus parenterally in animals. Further, it suggests a possible explanation for the seemingly improved safety profile of mitragynine compared to classical opioid agonists. However, the critical involvement of hepatic metabolism also complicates our understanding of mitragynine’s pharmacology and introduces the possibility of interindividual variability in the compound’s potential therapeutic effects and side effects. We believe mitragynine and related compounds have great potential as future therapeutics, but metabolic processes must be carefully considered as the field continues to advance.
Acknowledgments
This work was generously supported by the National Institute on Drug Abuse (R01DA046487 and R21DA045884), the American Kratom Association, and the Hope for Depression Research Foundation. S.M. also received support from the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program (W81XWH-17-1-0256) and acknowledges National Cancer Institute Cancer Center Support Grant No. P30 CA008748. We also thank Dr. B. Fowler (Dept. of Chemistry, Columbia Univ.) for HRMS analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00141.
Opioid functional activity of desmethyl metabolites, additional microsome and plasma stability experiments, formation of 7-OH in mouse microsomes, synthetic scheme and procedures for preparation of deuterated analogues, biological procedures (PDF)
Author Contributions
◆ A.C.K. and R.U. contributed equally to this work.
The authors declare the following competing financial interest(s): A.C.K., D.S., J.A.J., S.M., and G.W.P. are named as inventors on patent applications related to mitragynine analogs, which may lead to royalties or other licensing revenues from future commercial products.
Supplementary Material
References
- Kruegel A. C.; Grundmann O. The Medicinal Chemistry and Neuropharmacology of Kratom: A Preliminary Discussion of a Promising Medicinal Plant and Analysis of Its Potential for Abuse. Neuropharmacology 2018, 134, 108–120. 10.1016/j.neuropharm.2017.08.026. [DOI] [PubMed] [Google Scholar]
- Adkins J. E.; Boyer E. W.; McCurdy C. R. Mitragyna Speciosa, a Psychoactive Tree from Southeast Asia with Opioid Activity. Curr. Top. Med. Chem. 2011, 11 (9), 1165–1175. 10.2174/156802611795371305. [DOI] [PubMed] [Google Scholar]
- Prozialeck W. C.; Jivan J. K.; Andurkar S. V. Pharmacology of Kratom: An Emerging Botanical Agent with Stimulant, Analgesic and Opioid-like Effects. J. Am. Osteopath. Assoc. 2012, 112 (12), 792–799. [PubMed] [Google Scholar]
- Singh D.; Narayanan S.; Vicknasingam B. Traditional and Non-Traditional Uses of Mitragynine (Kratom): A Survey of the Literature. Brain Res. Bull. 2016, 126, 41–46. 10.1016/j.brainresbull.2016.05.004. [DOI] [PubMed] [Google Scholar]
- Takayama H. Chemistry and Pharmacology of Analgesic Indole Alkaloids from the Rubiaceous Plant, Mitragyna Speciosa. Chem. Pharm. Bull. 2004, 52 (8), 916–928. 10.1248/cpb.52.916. [DOI] [PubMed] [Google Scholar]
- Takayama H.; Ishikawa H.; Kurihara M.; Kitajima M.; Aimi N.; Ponglux D.; Koyama F.; Matsumoto K.; Moriyama T.; Yamamoto L. T.; Watanabe K.; Murayama T.; Horie S. Studies on the Synthesis and Opioid Agonistic Activities of Mitragynine-Related Indole Alkaloids: Discovery of Opioid Agonists Structurally Different from Other Opioid Ligands. J. Med. Chem. 2002, 45 (9), 1949–1956. 10.1021/jm010576e. [DOI] [PubMed] [Google Scholar]
- Raffa R. B.; Beckett J. R.; Brahmbhatt V. N.; Ebinger T. M.; Fabian C. A.; Nixon J. R.; Orlando S. T.; Rana C. A.; Tejani A. H.; Tomazic R. J. Orally Active Opioid Compounds from a Non-Poppy Source. J. Med. Chem. 2013, 56 (12), 4840–4848. 10.1021/jm400143z. [DOI] [PubMed] [Google Scholar]
- Suhaimi F. W.; Yusoff N. H. M.; Hassan R.; Mansor S. M.; Navaratnam V.; Müller C. P.; Hassan Z. Neurobiology of Kratom and Its Main Alkaloid Mitragynine. Brain Res. Bull. 2016, 126, 29–40. 10.1016/j.brainresbull.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Grundmann O. Patterns of Kratom Use and Health Impact in the US—Results from an Online Survey. Drug Alcohol Depend. 2017, 176, 63–70. 10.1016/j.drugalcdep.2017.03.007. [DOI] [PubMed] [Google Scholar]
- Swogger M. T.; Hart E.; Erowid F.; Erowid E.; Trabold N.; Yee K.; Parkhurst K. A.; Priddy B. M.; Walsh Z. Experiences of Kratom Users: A Qualitative Analysis. J. Psychoact. Drugs 2015, 47 (5), 360–367. 10.1080/02791072.2015.1096434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pain News Network . KRATOM SURVEY—Pain News Network https://www.painnewsnetwork.org/kratom-survey/ (accessed Dec 19, 2018).
- Smith K. E.; Lawson T. Prevalence and Motivations for Kratom Use in a Sample of Substance Users Enrolled in a Residential Treatment Program. Drug Alcohol Depend. 2017, 180, 340–348. 10.1016/j.drugalcdep.2017.08.034. [DOI] [PubMed] [Google Scholar]
- Regulations.gov –Docket Folder Summary https://www.regulations.gov/docket?D=DEA-2016-0015 (accessed Dec 19, 2018).
- FDA Office of the Commissioner . Press Announcements–Statement from FDA Commissioner Scott Gottlieb, M.D., on the agency’s 2019 policy and regulatory agenda for continued action to forcefully address the tragic epidemic of opioid abuse https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-agencys-scientific-evidence-presence-opioid-compounds (accessed Dec 19, 2018).
- Drug Enforcement Administration . Kratom https://www.dea.gov/factsheets/kratom (accessed Dec 19, 2018).
- Swetlitz I.HHS recommended that the DEA ban kratom, documents show–STAT https://www.statnews.com/2018/11/09/hhs-recommended-dea-ban-kratom-documents-show/ (accessed Dec 19, 2018).
- Regulations.gov –Proposed Rule Document https://www.regulations.gov/document?D=DEA-2016-0015-0003 (accessed Dec 19, 2018).
- Regulations.gov –Proposed Rule Document https://www.regulations.gov/document?D=DEA-2016-0015-0006 (accessed Dec 19, 2018).
- DEA opts against ban on herbal supplement kratom https://www.statnews.com/2016/10/12/kratom-dea-ban-opioids/ (accessed Dec 19, 2018).
- Ponglux D.; Wongseripipatana S.; Takayama H.; Kikuchi M.; Kurihara M.; Kitajima M.; Aimi N.; Sakai S. A New Indole Alkaloid, 7α-Hydroxy-7H-Mitragynine, from Mitragyna Speciosa in Thailand. Planta Med. 1994, 60 (6), 580–581. 10.1055/s-2006-959578. [DOI] [PubMed] [Google Scholar]
- Kruegel A. C.; Gassaway M. M.; Kapoor A.; Váradi A.; Majumdar S.; Filizola M.; Javitch J. A.; Sames D. Synthetic and Receptor Signaling Explorations of the Mitragyna Alkaloids: Mitragynine as an Atypical Molecular Framework for Opioid Receptor Modulators. J. Am. Chem. Soc. 2016, 138 (21), 6754–6764. 10.1021/jacs.6b00360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K.; Horie S.; Ishikawa H.; Takayama H.; Aimi N.; Ponglux D.; Watanabe K. Antinociceptive Effect of 7-Hydroxymitragynine in Mice: Discovery of an Orally Active Opioid Analgesic from the Thai Medicinal Herb Mitragyna Speciosa. Life Sci. 2004, 74 (17), 2143–2155. 10.1016/j.lfs.2003.09.054. [DOI] [PubMed] [Google Scholar]
- Matsumoto K.; Mizowaki M.; Suchitra T.; Takayama H.; Sakai S.; Aimi N.; Watanabe H. Antinociceptive Action of Mitragynine in Mice: Evidence for the Involvement of Supraspinal Opioid Receptors. Life Sci. 1996, 59 (14), 1149–1155. 10.1016/0024-3205(96)00432-8. [DOI] [PubMed] [Google Scholar]
- Macko E.; Weisbach J. A.; Douglas B. Some Observations on the Pharmacology of Mitragynine. Arch. Int. Pharmacodyn. thérapie 1972, 198 (1), 145–161. [PubMed] [Google Scholar]
- Váradi A.; Marrone G. F.; Palmer T. C.; Narayan A.; Szabó M. R.; Le Rouzic V.; Grinnell S. G.; Subrath J. J.; Warner E.; Kalra S.; Hunkele A.; Pagirsky J.; Eans S. O.; Medina J. M.; Xu J.; Pan Y.-X.; Borics A.; Pasternak G. W.; McLaughlin J. P.; Majumdar S. Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit β-Arrestin-2. J. Med. Chem. 2016, 59 (18), 8381–8397. 10.1021/acs.jmedchem.6b00748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid C. L.; Kennedy N. M.; Ross N. C.; Lovell K. M.; Yue Z.; Morgenweck J.; Cameron M. D.; Bannister T. D.; Bohn L. M. Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171 (5), 1165–1175e13. 10.1016/j.cell.2017.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soergel D. G.; Subach R. A.; Burnham N.; Lark M. W.; James I. E.; Sadler B. M.; Skobieranda F.; Violin J. D.; Webster L. R. Biased Agonism of the μ-Opioid Receptor by TRV130 Increases Analgesia and Reduces on-Target Adverse Effects versus Morphine: A Randomized, Double-Blind, Placebo-Controlled, Crossover Study in Healthy Volunteers. Pain 2014, 155 (9), 1829–1835. 10.1016/j.pain.2014.06.011. [DOI] [PubMed] [Google Scholar]
- Raehal K. M.; Schmid C. L.; Groer C. E.; Bohn L. M. Functional Selectivity at the μ-Opioid Receptor: Implications for Understanding Opioid Analgesia and Tolerance. Pharmacol. Rev. 2011, 63 (4), 1001–1019. 10.1124/pr.111.004598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly E. Efficacy and Ligand Bias at the μ-Opioid Receptor. Br. J. Pharmacol. 2013, 169 (7), 1430–1446. 10.1111/bph.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar S.; Devi L. A. Strategy for Making Safer Opioids Bolstered. Nature 2018, 553 (7688), 286–288. 10.1038/d41586-018-00045-1. [DOI] [PubMed] [Google Scholar]
- Sabetghadam A.; Navaratnam V.; Mansor S. M. Dose-Response Relationship, Acute Toxicity, and Therapeutic Index between the Alkaloid Extract of Mitragyna Speciosa and Its Main Active Compound Mitragynine in Mice. Drug Dev. Res. 2013, 74 (1), 23–30. 10.1002/ddr.21052. [DOI] [Google Scholar]
- Philipp A. A.; Wissenbach D. K.; Zoerntlein S. W.; Klein O. N.; Kanogsunthornrat J.; Maurer H. H. Studies on the Metabolism of Mitragynine, the Main Alkaloid of the Herbal Drug Kratom, in Rat and Human Urine Using Liquid Chromatography-Linear Ion Trap Mass Spectrometry. J. Mass Spectrom. 2009, 44 (8), 1249–1261. 10.1002/jms.1607. [DOI] [PubMed] [Google Scholar]
- Finch N.; Gemenden C. W.; Hsu I. H. C.; Kerr A.; Sim G. A.; Taylor W. I. Oxidative Transformations of Indole Alkaloids. III. Pseudoindoxyls from Yohimbinoid Alkaloids and Their Conversion to “Invert” Alkaloids1,2. J. Am. Chem. Soc. 1965, 87 (10), 2229–2235. 10.1021/ja01088a024. [DOI] [PubMed] [Google Scholar]
- Manda V.; Avula B.; Ali Z.; Khan I.; Walker L.; Khan S. Evaluation of In Vitro Absorption, Distribution, Metabolism, and Excretion (ADME) Properties of Mitragynine, 7-Hydroxymitragynine, and Mitraphylline. Planta Med. 2014, 80 (07), 568–576. 10.1055/s-0034-1368444. [DOI] [PubMed] [Google Scholar]
- Semenova S.; Kuzmin A.; Zvartau E. Strain Differences in the Analgesic and Reinforcing Action of Morphine in Mice. Pharmacol., Biochem. Behav. 1995, 50 (1), 17–21. 10.1016/0091-3057(94)00221-4. [DOI] [PubMed] [Google Scholar]
- Mogil J. S.Opioid Analgesia, Strain Differences. In Encyclopedia of Pain ;Springer Berlin Heidelberg: Berlin, Germany, 2007; pp 1528–1531. [Google Scholar]
- Gomes I.; Gupta A.; Filipovska J.; Szeto H. H.; Pintar J. E.; Devi L. A. A Role for Heterodimerization of Mu and Delta Opiate Receptors in Enhancing Morphine Analgesia. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (14), 5135–5139. 10.1073/pnas.0307601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusof S. R.; Mohd Uzid M.; Teh E.-H.; Hanapi N. A.; Mohideen M.; Mohamad Arshad A. S.; Mordi M. N.; Loryan I.; Hammarlund-Udenaes M. Rate and Extent of Mitragynine and 7-Hydroxymitragynine Blood-Brain Barrier Transport and Their Intra-Brain Distribution: The Missing Link in Pharmacodynamic Studies. Addict. Biol. 2018, 10.1111/adb.12661. [DOI] [PubMed] [Google Scholar]
- Kamble S. H.; Sharma A.; King T. I.; León F.; McCurdy C. R.; Avery B. A. Metabolite Profiling and Identification of Enzymes Responsible for the Metabolism of Mitragynine, the Major Alkaloid of Mitragyna Speciosa (Kratom). Xenobiotica 2018, 1–31. 10.1080/00498254.2018.1552819. [DOI] [PubMed] [Google Scholar]
- Karinen R.; Fosen J. T.; Rogde S.; Vindenes V. An Accidental Poisoning with Mitragynine. Forensic Sci. Int. 2014, 245, e29–e32. 10.1016/j.forsciint.2014.10.025. [DOI] [PubMed] [Google Scholar]
- Dean L.Codeine Therapy and CYP2D6 Genotype. Medical Genetics Summaries ;National Center for Biotechnology Information (US), 2012. [PubMed] [Google Scholar]
- Heppell S. P. E.; Isbister G. K. Lack of Respiratory Depression in Paracetamol-Codeine Combination Overdoses. Br. J. Clin. Pharmacol. 2017, 83 (6), 1273–1278. 10.1111/bcp.13224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith L. C.; Lin L.; Hwang C. S.; Zhou B.; Kubitz D. M.; Wang H.; Janda K. D. Lateral Flow Assessment and Unanticipated Toxicity of Kratom. Chem. Res. Toxicol. 2019, 32, 113. 10.1021/acs.chemrestox.8b00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute on Drug Abuse . Overdose Death Rates. National Institute on Drug Abuse (NIDA) https://www.drugabuse.gov/related-topics/trends-statistics/overdose-death-rates (accessed Dec 26, 2018).
- Hemby S. E.; McIntosh S.; Leon F.; Cutler S. J.; McCurdy C. R. Abuse Liability and Therapeutic Potential of the Mitragyna Speciosa (Kratom) Alkaloids Mitragynine and 7-Hydroxymitragynine. Addict. Biol. 2018, 10.1111/adb.12639. [DOI] [PubMed] [Google Scholar]
- Yue K.; Kopajtic T. A.; Katz J. L. Abuse Liability of Mitragynine Assessed with a Self-Administration Procedure in Rats. Psychopharmacology (Berl). 2018, 235 (10), 2823–2829. 10.1007/s00213-018-4974-9. [DOI] [PubMed] [Google Scholar]
- Kathiramalainathan K.; Kaplan H. L.; Romach M. K.; Busto U. E.; Li N. Y.; Säwe J.; Tyndale R. F.; Sellers E. M. Inhibition of Cytochrome P450 2D6Modifies Codeine Abuse Liability. J. Clin. Psychopharmacol. 2000, 20 (4), 435–444. 10.1097/00004714-200008000-00008. [DOI] [PubMed] [Google Scholar]
- Kong W. M.; Chik Z.; Ramachandra M.; Subramaniam U.; Aziddin R. E. R.; Mohamed Z. Evaluation of the Effects of Mitragyna Speciosa Alkaloid Extract on Cytochrome P450 Enzymes Using a High Throughput Assay. Molecules 2011, 16 (12), 7344–7356. 10.3390/molecules16097344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manda V. K.; Avula B.; Dale O. R.; Ali Z.; Khan I. A.; Walker L. A.; Khan S. I. PXR Mediated Induction of CYP3A4, CYP1A2, and P-Gp by Mitragyna Speciosa and Its Alkaloids. Phytother. Res. 2017, 31 (12), 1935–1945. 10.1002/ptr.5942. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.