

Abstract
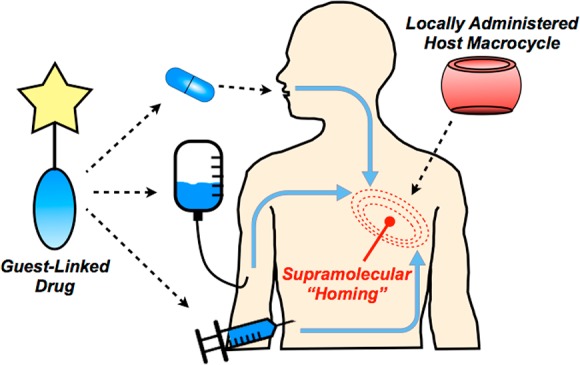
Ensuring effective drug concentration specifically at sites of need, while limiting systemic side effects, remains a challenge in the discovery and use of new drug molecules. Carriers targeted through biological affinity (e.g., antibodies) afford a common means of drug localization, yet often deliver considerably less than 1% of an administered drug to a desired site in the body. We report on an alternative targeting paradigm using pendant guest motifs to direct molecules to sites distinguished by a hydrogel bearing a high density of a complementary cucurbituril supramolecular host. Host–guest affinity (Keq) of 1012 M–1 serves to spatially localize ∼4% of a model small molecule within hours of its administration in mice. These high-affinity interactions furthermore ensure long-lasting retention of the model compound at the site of interest, and the site can be serially targeted upon repeated dosing. This supramolecular homing axis extends the localization of small molecule payloads beyond injectable hydrogels, enabling targeting of modified biomaterials. This approach also has promising therapeutic utility, improving efficacy of a guest-modified chemotherapeutic agent in a tumor model.
Short abstract
A new strategy is shown for site-specific accumulation of guest-linked drug molecules on the basis of supramolecular affinity for a complementary host locally applied using a temperature-responsive injectable hydrogel.
Introduction
The discovery of new drugs, often validated using in vitro screening, has been supported by advances in the fields of chemical biology, medicinal chemistry, and diversity-oriented synthesis to realize new classes of compounds acting against a variety of diseases.1,2 Yet, many compounds with promising in vitro function exhibit dose-limiting toxicity and off-target activity when administered in vivo.3 These challenges can result in drug candidates with desirable activity stalling in development. To overcome this challenge, active drug targeting methods seek to increase the therapeutic index (LD50/EC50) by enhancing regional drug concentration and limiting systemic activity. The field of drug delivery, which includes the packaging of drugs in nanoscale carriers, has been explored to localize drugs to sites of need.4,5 Technologies often rely on biological recognition by antibodies or other biomolecules to direct drugs to specific sites.6−8 Unfortunately, nanoparticles targeted using a gold-standard antibody (Herceptin) show local accumulation of <1%, with only 14 of every 1 million diseased cells (0.0014%) successfully targeted.9 The emerging class of antibody–drug conjugates, wherein a drug is directly tethered to a targeting antibody, has led to a number of clinically approved therapies with many others in the final stages of clinical evaluation.10,11 However, these carriers circulate for days or more in reaching their target, leading to toxicity from release of free drug systemically by linker rupture as well as a toxic bystander effect.12 In addition, only 0.001–0.01% of the total antibody administered finds its way to sites of disease.13,14 As such, and in spite of affinities of ∼108–1012 M–1 and biological target specificity, antibodies and related biomolecules do not dramatically enhance localization of therapeutic payloads. Strategies to improve delivery of a drug to its target while limiting concomitant side effects remain in need of further discovery. One route being explored to overcome the limitations of traditional targeting entails “pre-targeting” a desired site to enable subsequent homing of an administered agent, with approaches using antibodies or reagents for bio-orthogonal “click” chemistry to facilitate recognition and accumulation at the pretargeted site.15−19
Host–guest supramolecular chemistry is classified by the formation of a noncovalent complex of a guest molecule in the portal of a macrocyclic cavitand.20 Applications of this chemistry in drug delivery have primarily used hydrophilic macrocycles as formulation excipients to solubilize or stabilize hydrophobic drugs.21 Yet, there may be opportunities afforded by high-affinity host–guest interactions to promote recognition in complex environments, thus mimicking biological recognition motifs such as biotin/avidin.22 Cucurbit[n]uril (CB[n]) macrocycles are prepared from acid-catalyzed polymerization of glycoluril and formaldehyde to form cavitands with a discrete number [n] of monomers.23 One member of this family, CB[7], forms complexes with certain guests that have monovalent affinities (Keq) of ∼1015 M–1 in buffer.24 For sake of comparison, the most explored family of macrocycles in pharmaceutical practice, cyclodextrins, do not typically exceed Keq of 105 M–1 for binding to any guest.25 CB[7] is water-soluble and has demonstrated low toxicity, with a reported LD50 of 250 mg/kg when administered intravenously in mice.26
We sought in this work to exploit a new paradigm in targeted drug delivery, using supramolecular recognition by CB[7] as an affinity axis in targeting and locally retaining guest-appended small molecules (Figure 1). By first pretargeting a desired site with the localized supramolecular “homing” cue, such as a CB[7]-rich injectable hydrogel, affinity between host and guest would then be used to facilitate drug localization. Guest modification of the drug may further be viewed as a strategy to attenuate systemic activity in the context of prodrug methodology.27 Contrasting with methods that have used antibodies and related large biomolecules, this approach instead uses small molecules which offer improved tissue distribution and more rapid clearance from circulation. In addition, small molecule approaches should circumvent risks of immunogenicity and expensive large-scale production that may limit the use of antibodies.28 Host–guest complexes are typically diffusion-governed (kon ≈ 108 M–1 s–1),29 affording a key advantage over pretargeting methods based on in situ “click” chemistry reactions that are more kinetically limited (kon ≈ 100–104 M–1 s–1).30−32 Furthermore, supramolecular affinity does not permanently consume a targeted site, and in principle the same host site may be subsequently retargeted. In spite of the affinity offered by CB[7]-guest interactions, leveraging its recognition as an axis for therapeutic targeting has not been studied extensively. Such supramolecular recognition becomes increasingly interesting and complicated in the contaminated and dilute milieu of a living animal, necessitating high-affinity motifs.
Figure 1.
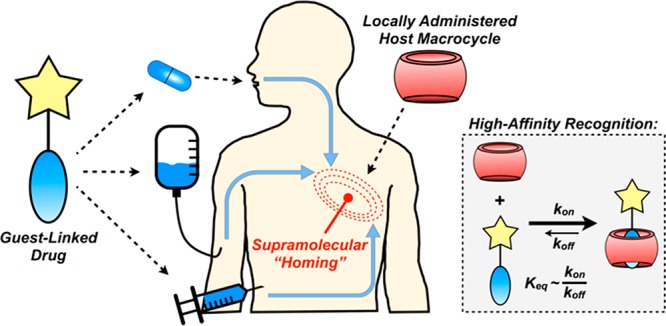
Schematic of approach for supramolecular homing of guest-appended small molecules on the basis of affinity for locally applied host macrocycles.
Results and Discussion
To afford a high concentration of locally applied CB[7] for use as a supramolecular “homing” device, a hydrogel platform was explored with inspiration from the class of injectable biomaterials realized using host–guest supramolecular cross-linking.33,34 Leveraging synthetic precedent to create azide-modified monofunctional CB[7],35 Pluronic F-127 (F127, 12.6 kDa) was end-modified with CB[7] by azide–alkyne copper “click” chemistry (F127-CB[7], Figure 2a, Figures S1–S2). F127 is an FDA-approved poly(ethylene oxide)/poly(propylene oxide)/poly(ethylene oxide) triblock copolymer that undergoes thermally triggered micelle formation near physiologic temperatures.36,37 CB[7] modification was quantitative, resulting in F127 end-modified with two macrocycles. Temperature-dependent 1H NMR, following reported methods,38 revealed limited impact on F127 micelle formation resulting from CB[7] appendage (Figure 2b, Figure S3), with a critical micelle temperature of ∼19 °C and full maturation of micelles once temperatures reached ∼30 °C. The number of surface-presented CB[7] groups on these micelles may be estimated from the aggregation number of F127, reported to be in the range of 35–54,39,40 yielding ∼70–100 CB[7] macrocycles per micelle (two CB[7] per F127).
Figure 2.

Design of a thermoresponsive supramolecular hydrogel for localized drug homing. (a) Pluronic F127 end-modified with cucurbit[7]uril (CB[7]) through copper-catalyzed click chemistry. (b) Variable temperature 1H NMR of F127-CB[7], with PPO-specific signal (purple-shaded region) integrated to quantify micelle formation compared to unmodified F127. (c) A strong ferrocene guest attached to eight-arm polyethylene glycol (PEG8-Fc) to cross-link F127 micelles and form a percolated network. (d) The guest molecule presented on PEG8-Fc was determined by competition 1H NMR to bind CB[7] with an affinity of 3.5 × 1012 M–1. (e) Variable temperature oscillatory rheology to determine the critical gelation temperature for F127-CB[7] and PEG8-Fc at 10 wt % solids, mixed at a CB[7]/Fc ratio of 3:1. Critical gelation temperature was defined by the crossover between G′ and G′′ (tan δ = 1). The solid line corresponds to sample heating from 20 to 45 °C while the dotted line corresponds to cooling over the same range. (f) Shear-thinning and self-healing demonstrated for the same hydrogels at 37 °C, alternating between 2% and 200% strain. (g) Evidence for instant gelation upon injection of a sol of F127-CB[7]:PEG8-Fc into a 37 °C bath, compared to (h) injection of the same sol into a 23 °C bath. (i) Proposed mechanism for thermoresponsive gelation entailing a “loop-rich” precursor with limited cross-linking at ambient temperatures, which transitions to a percolated hydrogel upon cross-linking of F127 micelles at physiologic temperatures.
To cross-link F127-CB[7] micelles and form a percolated hydrogel network, eight-arm polyethylene glycol macromers (20 kDa) were end-functionalized with a ferrocene (Fc) guest for CB[7] (PEG8-Fc, Figure 2c, Figures S4–S5). The binding of CB[7] to a model compound of this ferrocene guest (Figure S6) was measured at 3.5 × 1012 M–1 by competition 1H NMR (Figure 2d, Figure S7). This technique was performed following a published method to determine binding constants in high-affinity regimes.24 By mixing F127-CB[7] with PEG8-Fc at a CB[7]/Fc molar ratio of 3:1 and a concentration of 10 wt % total solids, a thermally reversible hydrogel formed with a critical gelation temperature (G′ = G′′, tan δ = 1) between 31 and 32 °C (Figure 2e). Hydrogels prepared from alternate ratios of CB[7]/Fc (2:1 and 1:1) with a concentration maintained at 10 wt % total solids showed similar thermally reversible hydrogel formation, while F127-CB[7] alone at 10 wt % did not form a hydrogel (Figure S8). The formed network from 3:1 CB[7]/Fc exhibited instantaneous self-healing properties under step-strain perturbation at physiologic temperature (Figure 2f). Thermally induced gelation was effectively instant when the sol was injected into a 37 °C solution (Figure 2g, Movie S1). By comparison, the hydrogel did not form when the viscous sol was injected into a 23 °C bath (Figure 2h, Movie S2).
From these rheology studies, hydrogel network formation depended on host–guest cross-linking between CB[7] and the Fc guests appended from the eight-arm PEG macromer. This is supported by a lack of gel formation in F127-CB[7] alone, in spite of its formation of micelles upon heating. It is noted that F127 forms hydrogels alone at concentrations of ≥20 wt %.41 The interaction between CB[7] and PEG-appended Fc guests should be essentially independent of temperature, and at these concentrations an affinity of ∼1012 M–1 predicts complete guest inclusion in CB[7] portals. When then assessing these findings in the context of Flory–Stockmayer theory,42 it would be expected that F127-CB[7] (f = 2) and PEG8-Fc (f = 8) would form a hydrogel network regardless of temperature. Yet this is not observed in the data presented. Temperature-dependent hydrogel formation is furthermore not a result of excess CB[7] contributing to extensive network defects, as hydrogels prepared from a 1:1 ratio of CB[7]/Fc also showed temperature-dependent network formation. As such, the temperature dependence observed here is likely the result of extensive “loop” formation in the low temperature sol, wherein macrocycles on the bifunctional F127-CB[7] interact primarily with Fc guests on the same PEG8-Fc macromer (Figure 2i). This can be explained according to principles of avidity; once the first CB[7] binds to an Fc guest, the other CB[7] is more likely to bind an Fc guest on the same macromer. Hydrogelation thus arises upon thermally induced aggregation of PPO segments, with host–guest supramolecular interactions then serving to cross-link these micelles to form a network. This mechanism was further supported by an observation of thermoresponsive hydrogel formation upon mixing F127-CB[7] with linear (PEG2-Fc, f = 2) and four-arm (PEG4-Fc, f = 4) guest macromers of molecular weight affording similar arm lengths as PEG8-Fc (Figure S9). In addition, the rapid self-healing of these hydrogels following cessation of high shear supports an assembly mechanism in which PPO aggregation, rather than high-affinity host–guest complexation, governs hydrogel formation.43
Instant thermally induced gelation, coupled with shear-thinning and self-healing character, are desirable traits for injectable biomaterials as it would allow a low viscosity sol to be administered using a syringe and subsequently gel at the site of injection.44 This function was demonstrated for F127-CB[7]:PEG8-Fc hydrogels, which were easily injected as a sol subcutaneously into mice using a 26G syringe and showed clear evidence by palpation of immediate hydrogel formation. Visual inspection of the hydrogel by gross necropsy and tissue histology (H&E) performed at 3, 7, 14, 30, 45, and 60 days revealed a very mild inflammatory response to the injected material which consisted in the acute phase of infiltrating neutrophils and gave rise to macrophages at later times (Figure S10). The gel volume had an apparent decrease with time implanted, until 60 days when hydrogels were no longer visible by necropsy or recoverable for histology.
At 10 wt %, hydrogels with a CB[7]/Fc ratio of 3:1 afford 7.8 mM free CB[7], assuming 100% CB[7]/Fc complexation. Thus, there remains a significant concentration of free CB[7] within the formed hydrogel to enable its use in the spatially defined capture of systemically administered guest-linked small molecules envisioned here. These CB[7]-rich hydrogels were thus well-suited to serve as an injectable hydrogel “homing beacon” and facilitate drug localization on the basis of supramolecular affinity. The monovalent affinity required to home a systemically administered small molecule to the site of the hydrogel in the complex physiologic milieu was next evaluated using a model set of small molecules offering a range of affinity for CB[7]. From methods to afford sulfonated cyanine dyes,45 disulfo-Cy5 with a single pendant carboxylate was synthesized (Figure S11). Sulfonate groups enhance solubility and were included to facilitate rapid clearance in the body, while the near-infrared fluorescence of the dye was intended to enable in vivo imaging with moderate tissue penetration and limited background interference.46 From this dye, different guests for CB[7] were attached to create model “prodrugs” with expected affinities informed by literature precedent.47 The first compound, Fc-O-Cy5, termed here as the “weak” guest, bound CB[7] with a measured Keq of 9.5 × 108 M–1 (Figure 3a, Figures S12–S16), determined using competition 1H NMR.24 In spite of nomenclature, this “weak” guest still bound CB[7] with an affinity roughly 3 orders of magnitude higher than cyclodextrin binds virtually any guest. Next, an amide-linked adamantyl (Ad) variant, Ad-Am-Cy5, was synthesized as a “medium” guest with a measured Keq of 2.1 × 1010 M–1 in binding to CB[7] (Figure 3b, Figures S17–S21). Finally, Fc-N-Cy5, termed the “strong” guest, was prepared and found to bind CB[7] with Keq of 1.5 × 1012 M–1 (Figure 3c, Figures S22–S26).
Figure 3.

Determination of host–guest affinity required for complex formation in the body after systemic administration. (a) Structure of model prodrug from a ferrocene guest conjugated to a near-infrared cyanine dye (Cy5), termed the “Weak” guest (Fc-O-Cy5), with a measured affinity for CB[7] of 9.5 × 108 M–1. (b) Structure of model prodrug from an adamantyl guest conjugated to Cy5, termed the “Medium” guest (Ad-Am-Cy5), with a measured affinity for CB[7] of 2.1 × 1010 M–1. (c) Structure of model prodrug from a ferrocene guest conjugated to Cy5, termed the “Strong” guest (Fc-N-Cy5), with a measured affinity for CB[7] of 1.5 × 1012 M–1. (d) F127-CB[7]:PEG8-Fc hydrogels injected subcutaneously, with subsequent administration of the three model dye-linked guests and representative in vivo fluorescence imaging to quantify dye homing to the site of the hydrogel. (e) Quantification of the average intensity in the hydrogel region of interest over time following administration of the three model dye-linked guests (n = 4). (f) Results from dye quantification following explantation of 100 μL and 200 μL hydrogels, dye extraction, and quantification. (g) Studies evaluating repeat loading of subcutaneous hydrogels with nine consecutive doses of Fc-N-Cy5 administered with 12-h spacing and imaging conducted immediately prior to administration of the next dose, as displayed in the study timeline. The average signal intensity arising from the dye at the hydrogel site was quantified and plotted (n = 4).
Dorsal subcutaneous injection in mice of F127-CB[7]:PEG8-Fc hydrogels with a CB[7]/Fc ratio of 3:1 was followed 48 h later by systemic intraperitoneal administration of the three guest-linked model agents at equal dose. Within only 30 min of administration, there was already a dramatic affinity-dependent difference in dye accumulation at the site of hydrogel implantation, as observed by in vivo imaging (Figure 3d–e). The “strong” conjugate, Fc-N-Cy5, showed rapid accumulation and retention at the site of the hydrogel (Movie S3), with the hydrogel even adopting a blue color that was apparent through the skin (Figure S27). By comparison, and on the basis of fluorescence, 3-fold less of the “medium” conjugate was retained at the site of the hydrogel, while the “weak” conjugate showed no hydrogel accumulation by imaging. A small increase in signal was observed in groups receiving Fc-N-Cy5 and Ad-Am-Cy5 between days 3 and 5, which is attributed to the observation of some hydrogel swelling in the early stages following injection. Swelling would reduce self-quenching from nearby cyanine dyes and increase the size of the gel within the region of interest quantified in the course of image analysis.
Subsequent studies explanted hydrogels 24 h after systemic administration of the “strong” Fc-N-Cy5 conjugate. Extracting the dye from these hydrogels revealed 2.2% (±1.1%) of the total administered dye was retained within a 100 μL hydrogel, while 4.2% (±1.2%) was retained within a 200 μL hydrogel (Figure 3f). This level of homing is exciting when compared to the typical accumulation seen in previously mentioned reports using antibody-based targeting. Though there was correlation between hydrogel volume and the percent of administered agent which homed to the site, these hydrogels were far from saturated upon a single injection of the Fc-N-Cy5 conjugate. This was evident by studying repeat dosing in mice bearing a 100 μL hydrogel (Figure 3g). Dosing nine consecutive times, with 12 h spacing between doses and follow-up imaging, resulted in a linear increase in signal at the hydrogel site up to the limit where the detector of the imaging instrument was saturated.
With kon roughly diffusion-limited (∼108 M–1 s–1) for host–guest interactions of small molecules, koff for the “strong” Fc-N-Cy5 conjugate is ∼10–4 s–1. Accordingly, there should be limited release of dye once bound to CB[7], and the hydrogel serves to promote local retention of the small molecule once it has bound. The signal reduction observed over 45 days furthermore corresponded to observations for gel clearance made in the course of necropsy and histology, where the gel volume showed a marked decrease by 45 days and was completely cleared by 60 days. Taken together, this supports a mechanism wherein dye clearance occurs primarily in the course of material erosion and clearance rather than dye releasing from its CB[7]-bound state.
The homing of Fc-N-Cy5 to the site of the CB[7]-rich hydrogel was remarkably efficient, especially in light of the rapid clearance of these small molecules. If the CB[7]-rich hydrogel is not present, within hours the fluorescent signal in mice returned to the preinjected baseline (Figure S28). This imaging-based clearance study, enabled by near-infrared dyes,46 provides information similar to more conventional pharmacokinetic studies. The majority of dye cleared quickly by renal excretion, as evidenced by fluorescent signal from the kidneys in early imaging times as well as a distinct blue color of urine and bedding evident within only 30 min of dye administration. Rapid renal clearance suggests that only a fraction of administered dose is even exposed to the hydrogel. As such, the finding that 2–4% of Fc-N-Cy5 homes to the site of the hydrogel is all the more impressive. In the case of Fc-N-Cy5, affinity of ∼1012 M–1 ensured effective retention at the site for any agent that was exposed to the hydrogel in the course of its distribution in the body. Comparatively weaker-binding guests would have more rapid exchange (koff) in their binding to CB[7], impacting their retention at the site even when their distribution afforded exposure to the hydrogel. The equilibrium state of weaker-binding agents is similarly more highly impacted by dilution in the body as well as competition from native physiologic binders of CB[7]; among the best-binding competitors present in the body include N-terminal aromatic amino acids on proteins (Keq ≈ 106 M–1).48 These findings also highlight a key limitation of the present technology related to rapid clearance of the guest-linked small molecule and point to the possibility to further increase drug accumulation by extending circulation half-life.
Supramolecular affinity should also be a versatile approach to facilitate spatial localization of small molecules to other desired sites. As an example, biomedical devices are plagued by a number of interface-mediated modes of failure.49,50 Supramolecular homing was thus explored for its ability to enable retention of model small molecules at the interface of an implanted device. Solid glass and polystyrene beads were surface-modified with CB[7]. Following implantation of the beads, administered Fc-N-Cy5 dye localized to the site of these CB[7]-modified biomaterials (Figure S29). This example points to a possible broader use for this approach in the context of biomedical device coatings.
To demonstrate preliminary proof of function in using supramolecular affinity for drug homing, doxorubicin was modified with the same N-linked ferrocene used to enable homing in the “strong” Fc-N-Cy5 conjugate. Given the expected slow off-rate of the guest once bound to CB[7], a labile hydrazone linker was included between drug and guest, yielding a prodrug referred to here as Fc-Hdz-Dox (Figure 4a, Figures S30–S31). Hydrazones are common linkers used in the modification of drugs, even including some doxorubicin variants evaluated clinically.51 Hydrazones are fairly stable at neutral pH but rupture more quickly under acidic conditions, making these useful linkers for controlled drug delivery in treating cancer.52 Studies performed with 1H NMR on Fc-Hdz-Dox samples in neutral and acidic D2O confirmed pH-dependent rupture of the hydrazone linker (Figure S32). To further validate pH-responsive release of the Fc-linked prodrug upon loading, F127-CB[7]:PEG8-Fc hydrogels with a 3:1 ratio of CB[7]/Fc were loaded with Fc-Hdz-Dox, and the release of free doxorubicin was monitored over time at pH 5.5 and 7.4 (Figure S33). Drug release progressed over the course of 100 h at pH 5.5, but only a small percentage of drug was released at pH 7.4. Release from the hydrogel environment, where the prodrug is also bound to CB[7], demonstrated a reduced rate of hydrazone rupture compared to free drug studies performed by 1H NMR; such an observation is common for labile bonds when confined within a material. Modification of doxorubicin by a hydrazone linkage is known to result in attenuation of its potency. Indeed, Fc-Hdz-Dox was found to have an IC50 of 6.8 μM for human breast cancer cells (MDA-MB-231) in culture (Figure 4b); this modified variant is thus an order of magnitude less potent than unmodified doxorubicin (IC50 = 0.40 μM) in vitro. Fluorescence imaging on cultured cells at serial time points following administration confirmed that both doxorubicin and Fc-Hdz-Dox have comparable uptake by cells (Figure S34). However, this data also pointed to greater nuclear overlap—the site at which doxorubicin acts—for the unmodified drug relative to the hydrazone-modified prodrug especially at earlier times following treatment.
Figure 4.

Supramolecular homing applied to therapeutic design. (a) Doxorubicin modified with a strong ferrocene guest through a hydrazone linkage (Fc-Hdz-Dox) as well as a control of doxorubicin modified with a hydrazone but lacking a CB[7]-binding guest (Me-Hdz-Dox). (b) Potency of doxorubicin and hydrazone conjugates in vitro in MDA-MB-231 cancer cells fit to a variable slope inhibitor vs response curve with least-squares fitting (R2 > 0.97 for all drugs). (c) Tumor model to evaluate supramolecular homing of Fc-Hdz-Dox compared to Me-Hdz-Dox following application of F127-CB[7]:PEG8-Fc hydrogel adjacent to a tumor prepared from MDA-MB-231 (luc+) cells. Tumors were induced and 14 days following induction the hydrogel was injected (day 0). Animal weights were tracked as was bioluminescent signal measured by imaging following administration of d-luciferin. Each line on the tumor signal plot represents a single mouse, with group averages noted by bold lines (*P < 0.05, **P < 0.01). All mice survived until the day 28 time point.
In order to demonstrate a role for supramolecular homing in therapy, an orthotopic xenograft tumor model was deployed in immunocompromised mice. Once tumors had formed, the F127-CB[7]:PEG8-Fc hydrogel was applied adjacent to the tumor, and animals were dosed with 3 mg/kg/day of unmodified doxorubicin or a doxorubicin-equivalent dose of Fc-Hdz-Dox for three consecutive days (Figure S35). While this dose of doxorubicin is below its reported LD50 in mice (11.2 mg/kg i.p. from RTECS database), three consecutive doses proved detrimental to health of these mice and resulted in weight loss and poor survival outcomes. Fc-Hdz-Dox was well-tolerated by these same measures. Furthermore, Fc-Hdz-Dox slowed the rate of tumor growth relative to doxorubicin treatment alone, for which tumors continued to grow in spite of treatment. It is noted that standard doxorubicin administration for therapeutic evaluation has often dosed the drug one time per week over the course of several weeks; for example, dosing up to 8 mg/kg once weekly for 6 weeks has been reported.53 However, the objective of these studies was to determine impact of supramolecular homing and subsequently sustained peritumoral drug retention and availability, and accordingly the chosen dosing strategy to assess this effect deviates from common therapeutic uses of doxorubicin.
To probe supramolecular homing to tumors while limiting drug toxicity, another hydrazone-modified doxorubicin variant was synthesized, termed Me-Hdz-Dox (Figure 4a, Figures S36–S37). It was reasoned that prodrug modification of doxorubicin with a hydrazone linker that did not bind CB[7] would have similar attenuated potency to Fc-Hdz-Dox, yet not home to the hydrogel. When evaluated for toxicity in MDA-MB-231 cells in culture, this new variant had an IC50 of 4.7 μM (Figure 4b) and as such was comparable to the in vitro potency of Fc-Hdz-Dox. In addition, this new appending group did not facilitate measurable binding to CB[7]. This compound was thus explored for the explicit purpose of evaluating supramolecular homing of doxorubicin for treating cancer (Figure 4c). Fc-Hdz-Dox and Me-Hdz-Dox were evaluated on the basis of their ability to control the growth of tumors adjacent to F127-CB[7]:PEG8-Fc hydrogels, dosing at 6 mg/kg/day doxorubicin-equivalence for three consecutive days. This dose was chosen following pilot studies to determine the maximum Fc-Hdz-Dox dose without eliciting outward morbidity in these mice (∼8–10 mg/kg/day). Indeed, animal weight remained stable throughout the study, suggesting both compounds were well-tolerated. Excitingly, mice treated with Fc-Hdz-Dox showed a significant and sustained reduction in the rate of tumor growth that lasted well beyond the initial period where compounds were dosed (Figure 4c). All animals survived for the duration of the study, which was terminated at day 28 due to excessive tumor burden in many of the Me-Hdz-Dox mice. On the other hand, mice treated with Fc-Hdz-Dox had body conditions which were notably better compared to the cohort treated with Me-Hdz-Dox. These findings suggest a mechanism entailing initial homing on the basis of supramolecular affinity, drug retention within the hydrogel, and prolonged drug availability near the tumor as hydrazone linkers slowly rupture. Though some have postulated the peritumoral environment to be of somewhat acidic pH,54 release studies suggest the rupture rate of the hydrazone may yet be too slow and as such might limit free drug concentrations reaching the level needed for complete tumor regression. Studies using model fluorescent compounds point to rapid clearance, suggesting a limited role for the ferrocene guest in extending circulation half-life. More detailed pharmacokinetic studies exploring the concentration of both Fc-Hdz-Dox and released free doxorubicin in blood and local tissue will aid in understanding and redesigning the present approach to further improve therapeutic efficacy. Future work will also extend this approach to the study of other labile linkers, new drugs, and drug combinations, buoyed by the promising demonstration of a role for supramolecular affinity as a homing cue to direct therapies in the body.
The targeting and retention afforded by high-affinity host–guest recognition have a variety of benefits and possible applications. As mentioned, methods for bio-orthogonal “click” chemistry have been explored in a similar context. Yet, these reactions are often kinetically limited compared to the typical kon for host–guest motifs. Evidence from the field of flow chemistry suggests an inherent benefit for reactions that occur quickly as a means to combat short residence times;55 there are many scenarios in the body, such as vascular applications, where flow and short residence time may dictate a need for faster kinetics of association than are presently offered by “click” chemistry. In the context of the application described here to treat solid tumors, the present approach offers an alternative to intratumoral administration of chemotherapy either directly or provided through controlled release technologies.56,57 Infusion requires repeated access, while controlled release depots have an exhaustible drug supply. As such, the current approach may have a number of benefits, particularly in directing drugs to disease sites which require more invasive access. Provided unoccupied CB[7] sites remain, the present technology could be periodically loaded remotely to ensure sustained local drug presence at the site. This technology also affords greater ease in temporal control and adjustment of the therapeutic identity and dose, two features that are predetermined at the time when a controlled release device is implanted. Thus, therapeutics may be cycled, or adjustments may be made to a therapeutic regimen in response to clinical measures of disease, without reaccessing the site. Fully demonstrating these features of the system offers an exciting new direction in the use of host–guest recognition for spatiotemporal control of therapeutics.
Safety
See safety statement in the Supporting Information.
Conclusions
High-affinity supramolecular interactions, such as those afforded by CB[7] in binding to certain guests, offer a new strategy for targeting drugs in the body. In the work shown here, pretargeting with an injectable CB[7]-rich hydrogel served to spatially define the desired site of drug action by localizing and retaining a systemically administered guest-linked small molecule on the basis of supramolecular affinity. Provided the site of desired drug activity is known, there are several advantages to this approach. Small molecules, such as the guest-modified variants shown here, should have more extensive tissue distribution than larger antibodies or even larger nanoscale carriers. At the same time, if they do not find their desired site of action, these small molecules should clear rapidly, as opposed to alternative strategies that have challenges arising from toxic drug molecules that are shed from a carrier in the course of prolonged circulation. Synthetic modification of a drug with a guest motif may also attenuate its potency and enable higher dosing without concomitant issues from off-site toxicity, as was demonstrated here when modifying doxorubicin. Host–guest complexes associate at the diffusion limit, contrasting with many in situ chemical ligation strategies which can be kinetically limited. Though the host–guest interactions used here have a very slow off-rate, there remain opportunities with this approach to explore the “regeneration” of host macrocycle sites so that these may be subsequently retargeted to increase the longevity of a device. This is a possibility not afforded by common methods for in situ chemical ligation. Accordingly, we have great excitement for the future exploration of drug targeting on the basis of host–guest supramolecular affinity.
Acknowledgments
M.J.W. acknowledges funding support from the Harper Cancer Research Institute American Cancer Society Institutional Research Grant (IRG-14-195-01) and the University of Notre Dame “Advancing our Vision” initiative. The authors are grateful to the ND Energy Materials Characterization Facility for use of the rheometer and to the Notre Dame Integrated Imaging Facility for access to in vivo imaging and histology services.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00195.
The authors declare no competing financial interest.
Supplementary Material
References
- Schreiber S. L. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 2000, 287 (5460), 1964–1969. 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- Schenone M.; Dančík V.; Wagner B. K.; Clemons P. A. Target Identification and Mechanism of Action in Chemical Biology and Drug Discovery. Nat. Chem. Biol. 2013, 9 (4), 232–240. 10.1038/nchembio.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin W. G. Jr. The Concept of Synthetic Lethality in the Context of Anticancer Therapy. Nat. Rev. Cancer 2005, 5 (9), 689–698. 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Langer R. Drug Delivery and Targeting. Nature 1998, 392 (6679 Suppl), 5–10. [PubMed] [Google Scholar]
- Allen T. M.; Cullis P. R. Drug Delivery Systems: Entering the Mainstream. Science 2004, 303 (5665), 1818–1822. 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- Arap W. Cancer Treatment by Targeted Drug Delivery to Tumor Vasculature in a Mouse Model. Science 1998, 279 (5349), 377–380. 10.1126/science.279.5349.377. [DOI] [PubMed] [Google Scholar]
- Allen T. M. Ligand-Targeted Therapeutics in Anticancer Therapy. Nat. Rev. Cancer 2002, 2 (10), 750–763. 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- Farokhzad O. C.; Karp J. M.; Langer R. Nanoparticle-Aptamer Bioconjugates for Cancer Targeting. Expert Opin. Drug Delivery 2006, 3 (3), 311–324. 10.1517/17425247.3.3.311. [DOI] [PubMed] [Google Scholar]
- Dai Q.; Wilhelm S.; Ding D.; Syed A. M.; Sindhwani S.; Zhang Y.; Chen Y. Y.; MacMillan P.; Chan W. C. W. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano 2018, 12 (8), 8423–8435. 10.1021/acsnano.8b03900. [DOI] [PubMed] [Google Scholar]
- Schrama D.; Reisfeld R. A.; Becker J. C. Antibody Targeted Drugs as Cancer Therapeutics. Nat. Rev. Drug Discovery 2006, 5 (2), 147–159. 10.1038/nrd1957. [DOI] [PubMed] [Google Scholar]
- Doronina S. O.; Toki B. E.; Torgov M. Y.; Mendelsohn B. A.; Cerveny C. G.; Chace D. F.; DeBlanc R. L.; Gearing R. P.; Bovee T. D.; Siegall C. B.; et al. Development of Potent Monoclonal Antibody Auristatin Conjugates for Cancer Therapy. Nat. Biotechnol. 2003, 21 (7), 778–784. 10.1038/nbt832. [DOI] [PubMed] [Google Scholar]
- Ducry L.; Stump B. Antibody-Drug Conjugates: Linking Cytotoxic Payloads to Monoclonal Antibodies. Bioconjugate Chem. 2010, 21 (1), 5–13. 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- Epenetos A. A.; Snook D.; Durbin H.; Johnson P. M.; Taylor-Papadimitriou J. Limitations of Radiolabeled Monoclonal Antibodies for Localization of Human Neoplasms. Cancer Res. 1986, 46 (6), 3183–3191. [PubMed] [Google Scholar]
- Bornstein G. G. Antibody Drug Conjugates: Preclinical Considerations. AAPS J. 2015, 17 (3), 525–534. 10.1208/s12248-015-9738-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stéen E. J. L.; Edem P. E.; Nørregaard K.; Jørgensen J. T.; Shalgunov V.; Kjaer A.; Herth M. M. Pretargeting in Nuclear Imaging and Radionuclide Therapy: Improving Efficacy of Theranostics and Nanomedicines. Biomaterials 2018, 179, 209–245. 10.1016/j.biomaterials.2018.06.021. [DOI] [PubMed] [Google Scholar]
- Brudno Y.; Pezone M. J.; Snyder T. K.; Uzun O.; Moody C. T.; Aizenberg M.; Mooney D. J. Replenishable Drug Depot to Combat Post-Resection Cancer Recurrence. Biomaterials 2018, 178, 373–382. 10.1016/j.biomaterials.2018.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czuban M.; Srinivasan S.; Yee N. A.; Agustin E.; Koliszak A.; Miller E.; Khan I.; Quinones I.; Noory H.; Motola C.; et al. Bio-Orthogonal Chemistry and Reloadable Biomaterial Enable Local Activation of Antibiotic Prodrugs and Enhance Treatments against Staphylococcus Aureus Infections. ACS Cent. Sci. 2018, 4, 1624–1632. 10.1021/acscentsci.8b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia Oneto J. M.; Khan I.; Seebald L.; Royzen M. In Vivo Bioorthogonal Chemistry Enables Local Hydrogel and Systemic Pro-Drug To Treat Soft Tissue Sarcoma. ACS Cent. Sci. 2016, 2 (7), 476–482. 10.1021/acscentsci.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang P. V.; Prescher J. A.; Sletten E. M.; Baskin J. M.; Miller I. A.; Agard N. J.; Lo A.; Bertozzi C. R. Copper-Free Click Chemistry in Living Animals. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (5), 1821–1826. 10.1073/pnas.0911116107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehn J.-M. Supramolecular Chemistry—Scope and Perspectives Molecules, Supermolecules, and Molecular Devices(Nobel Lecture). Angew. Chem., Int. Ed. Engl. 1988, 27 (1), 89–112. 10.1002/anie.198800891. [DOI] [Google Scholar]
- Webber M. J.; Langer R. Drug Delivery by Supramolecular Design. Chem. Soc. Rev. 2017, 46 (21), 6600–6620. 10.1039/C7CS00391A. [DOI] [PubMed] [Google Scholar]
- Liu W.; Samanta S. K.; Smith B. D.; Isaacs L. Synthetic Mimics of Biotin/(strept)avidin. Chem. Soc. Rev. 2017, 46 (9), 2391–2403. 10.1039/C7CS00011A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Jung I.-S.; Kim S.-Y.; Lee E.; Kang J.-K.; Sakamoto S.; Yamaguchi K.; Kim K. New Cucurbituril Homologues: Syntheses, Isolation, Characterization, and X-Ray Crystal Structures of Cucurbit[n]uril (n= 5, 7, and 8). J. Am. Chem. Soc. 2000, 122 (3), 540–541. 10.1021/ja993376p. [DOI] [Google Scholar]
- Cao L.; Śekutor M.; Zavalij P. Y.; Mlinarić-Majerski K.; Glaser R.; Isaacs L. Cucurbit[7]uril·guest Pair with an Attomolar Dissociation Constant. Angew. Chem., Int. Ed. 2014, 53 (4), 988–993. 10.1002/anie.201309635. [DOI] [PubMed] [Google Scholar]
- Chen G.; Jiang M. Cyclodextrin-Based Inclusion Complexation Bridging Supramolecular Chemistry and Macromolecular Self-Assembly. Chem. Soc. Rev. 2011, 40 (5), 2254–2266. 10.1039/c0cs00153h. [DOI] [PubMed] [Google Scholar]
- Uzunova V. D.; Cullinane C.; Brix K.; Nau W. M.; Day A. I. Toxicity of cucurbit[7]uril and cucurbit[8]uril: An Exploratory in Vitro and in Vivo Study. Org. Biomol. Chem. 2010, 8 (9), 2037–2042. 10.1039/b925555a. [DOI] [PubMed] [Google Scholar]
- Rautio J.; Kumpulainen H.; Heimbach T.; Oliyai R.; Oh D.; Järvinen T.; Savolainen J. Prodrugs: Design and Clinical Applications. Nat. Rev. Drug Discovery 2008, 7 (3), 255–270. 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- Beck A.; Wurch T.; Bailly C.; Corvaia N. Strategies and Challenges for the next Generation of Therapeutic Antibodies. Nat. Rev. Immunol. 2010, 10 (5), 345–352. 10.1038/nri2747. [DOI] [PubMed] [Google Scholar]
- Tang H.; Fuentealba D.; Ko Y. H.; Selvapalam N.; Kim K.; Bohne C. Guest Binding Dynamics with Cucurbit[7]uril in the Presence of Cations. J. Am. Chem. Soc. 2011, 133 (50), 20623–20633. 10.1021/ja209266x. [DOI] [PubMed] [Google Scholar]
- Taylor M. T.; Blackman M. L.; Dmitrenko O.; Fox J. M. Design and Synthesis of Highly Reactive Dienophiles for the Tetrazine-Trans-Cyclooctene Ligation. J. Am. Chem. Soc. 2011, 133 (25), 9646–9649. 10.1021/ja201844c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning X.; Guo J.; Wolfert M. A.; Boons G.-J. Visualizing Metabolically Labeled Glycoconjugates of Living Cells by Copper-Free and Fast Huisgen Cycloadditions. Angew. Chem., Int. Ed. 2008, 47 (12), 2253–2255. 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liong M.; Fernandez-Suarez M.; Issadore D.; Min C.; Tassa C.; Reiner T.; Fortune S. M.; Toner M.; Lee H.; Weissleder R. Specific Pathogen Detection Using Bioorthogonal Chemistry and Diagnostic Magnetic Resonance. Bioconjugate Chem. 2011, 22 (12), 2390–2394. 10.1021/bc200490r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber M. J.; Appel E. A.; Meijer E. W.; Langer R. Supramolecular Biomaterials. Nat. Mater. 2016, 15 (1), 13–26. 10.1038/nmat4474. [DOI] [PubMed] [Google Scholar]
- Mantooth S. M.; Munoz-Robles B. G.; Webber M. J. Dynamic Hydrogels from Host-Guest Supramolecular Interactions. Macromol. Biosci. 2019, 19, 1800281 10.1002/mabi.201800281. [DOI] [PubMed] [Google Scholar]
- Vinciguerra B.; Cao L.; Cannon J. R.; Zavalij P. Y.; Fenselau C.; Isaacs L. Synthesis and Self-Assembly Processes of Monofunctionalized Cucurbit[7]uril. J. Am. Chem. Soc. 2012, 134 (31), 13133–13140. 10.1021/ja3058502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohorquez M.; Koch C.; Trygstad T.; Pandit N. A Study of the Temperature-Dependent Micellization of Pluronic F127. J. Colloid Interface Sci. 1999, 216 (1), 34–40. 10.1006/jcis.1999.6273. [DOI] [PubMed] [Google Scholar]
- Alexandridis P.; Holzwarth J. F.; Hatton T. A. Micellization of Poly(ethylene Oxide)-Poly(propylene Oxide)-Poly(ethylene Oxide) Triblock Copolymers in Aqueous Solutions: Thermodynamics of Copolymer Association. Macromolecules 1994, 27 (9), 2414–2425. 10.1021/ma00087a009. [DOI] [Google Scholar]
- Ma J.-H.; Guo C.; Tang Y.-L.; Liu H.-Z. 1H NMR Spectroscopic Investigations on the Micellization and Gelation of PEO–PPO–PEO Block Copolymers in Aqueous Solutions. Langmuir 2007, 23 (19), 9596–9605. 10.1021/la701221f. [DOI] [PubMed] [Google Scholar]
- Wanka G.; Hoffmann H.; Ulbricht W. Phase Diagrams and Aggregation Behavior of Poly(oxyethylene)-Poly(oxypropylene)-Poly(oxyethylene) Triblock Copolymers in Aqueous Solutions. Macromolecules 1994, 27 (15), 4145–4159. 10.1021/ma00093a016. [DOI] [Google Scholar]
- Nagarajan R. Solubilization of Hydrocarbons and Resulting Aggregate Shape Transitions in Aqueous Solutions of Pluronic® (PEO–PPO–PEO) Block Copolymers. Colloids Surf., B 1999, 16 (1–4), 55–72. 10.1016/S0927-7765(99)00061-2. [DOI] [Google Scholar]
- Malmsten M.; Lindman B. Self-Assembly in Aqueous Block Copolymer Solutions. Macromolecules 1992, 25 (20), 5440–5445. 10.1021/ma00046a049. [DOI] [Google Scholar]
- Flory P. J. Molecular Size Distribution in Three Dimensional Polymers. I. Gelation1. J. Am. Chem. Soc. 1941, 63 (11), 3083–3090. 10.1021/ja01856a061. [DOI] [Google Scholar]
- Zou L.; Braegelman A. S.; Webber M. J. Dynamic Supramolecular Hydrogels Spanning an Unprecedented Range of Host-Guest Affinity. ACS Appl. Mater. Interfaces 2019, 11 (6), 5695–5700. 10.1021/acsami.8b22151. [DOI] [PubMed] [Google Scholar]
- Sahoo J. K.; VandenBerg M. A.; Webber M. J. Injectable Network Biomaterials via Molecular or Colloidal Self-Assembly. Adv. Drug Delivery Rev. 2018, 127, 185–207. 10.1016/j.addr.2017.11.005. [DOI] [PubMed] [Google Scholar]
- Mujumdar R. B.; Ernst L. A.; Mujumdar S. R.; Lewis C. J.; Waggoner A. S. Cyanine Dye Labeling Reagents: Sulfoindocyanine Succinimidyl Esters. Bioconjugate Chem. 1993, 4 (2), 105–111. 10.1021/bc00020a001. [DOI] [PubMed] [Google Scholar]
- Hilderbrand S. A.; Weissleder R. Near-Infrared Fluorescence: Application to in Vivo Molecular Imaging. Curr. Opin. Chem. Biol. 2010, 14 (1), 71–79. 10.1016/j.cbpa.2009.09.029. [DOI] [PubMed] [Google Scholar]
- Barrow S. J.; Kasera S.; Rowland M. J.; Del Barrio J.; Scherman O. A. Correction to Cucurbituril-Based Molecular Recognition. Chem. Rev. 2016, 116 (19), 12651–12652. 10.1021/acs.chemrev.6b00577. [DOI] [PubMed] [Google Scholar]
- Li W.; Bockus A. T.; Vinciguerra B.; Isaacs L.; Urbach A. R. Predictive Recognition of Native Proteins by cucurbit[7]uril in a Complex Mixture. Chem. Commun. 2016, 52 (55), 8537–8540. 10.1039/C6CC03193E. [DOI] [PubMed] [Google Scholar]
- Costerton J. W. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 1999, 284 (5418), 1318–1322. 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- Anderson J. M.; Rodriguez A.; Chang D. T. Foreign Body Reaction to Biomaterials. Semin. Immunol. 2008, 20 (2), 86–100. 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratz F. DOXO-EMCH (INNO-206): The First Albumin-Binding Prodrug of Doxorubicin to Enter Clinical Trials. Expert Opin. Invest. Drugs 2007, 16 (6), 855–866. 10.1517/13543784.16.6.855. [DOI] [PubMed] [Google Scholar]
- Ulbrich K.; Subr V. Polymeric Anticancer Drugs with pH-Controlled Activation. Adv. Drug Delivery Rev. 2004, 56 (7), 1023–1050. 10.1016/j.addr.2003.10.040. [DOI] [PubMed] [Google Scholar]
- Ottewell P. D.; Monkkonen H.; Jones M.; Lefley D. V.; Coleman R. E.; Holen I. Antitumor Effects of Doxorubicin Followed by Zoledronic Acid in a Mouse Model of Breast Cancer. JNCI Journal of the National Cancer Institute 2008, 100, 1167–1178. 10.1093/jnci/djn240. [DOI] [PubMed] [Google Scholar]
- Danhier F.; Feron O.; Préat V. To Exploit the Tumor Microenvironment: Passive and Active Tumor Targeting of Nanocarriers for Anti-Cancer Drug Delivery. J. Controlled Release 2010, 148 (2), 135–146. 10.1016/j.jconrel.2010.08.027. [DOI] [PubMed] [Google Scholar]
- Plutschack M. B.; Pieber B.; Gilmore K.; Seeberger P. H. The Hitchhiker’s Guide to Flow Chemistry. Chem. Rev. 2017, 117, 11796–11893. 10.1021/acs.chemrev.7b00183. [DOI] [PubMed] [Google Scholar]
- Walter K. A.; Tamargo R. J.; Olivi A.; Burger P. C.; Brem H. Intratumoral Chemotherapy. Neurosurgery 1995, 37 (6), 1129–1145. 10.1097/00006123-199512000-00013. [DOI] [PubMed] [Google Scholar]
- Ruel-Gariépy E.; Shive M.; Bichara A.; Berrada M.; Le Garrec D.; Chenite A.; Leroux J.-C. A Thermosensitive Chitosan-Based Hydrogel for the Local Delivery of Paclitaxel. Eur. J. Pharm. Biopharm. 2004, 57 (1), 53–63. 10.1016/S0939-6411(03)00095-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.