

Abstract
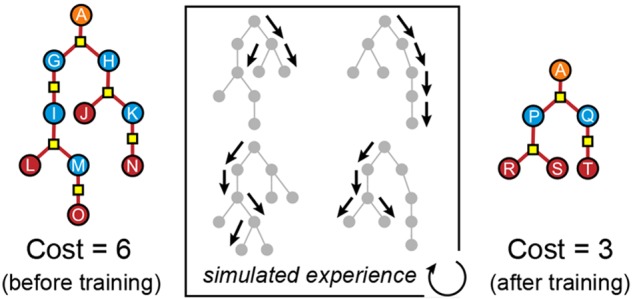
The problem of retrosynthetic planning can be framed as a one-player game, in which the chemist (or a computer program) works backward from a molecular target to simpler starting materials through a series of choices regarding which reactions to perform. This game is challenging as the combinatorial space of possible choices is astronomical, and the value of each choice remains uncertain until the synthesis plan is completed and its cost evaluated. Here, we address this search problem using deep reinforcement learning to identify policies that make (near) optimal reaction choices during each step of retrosynthetic planning according to a user-defined cost metric. Using a simulated experience, we train a neural network to estimate the expected synthesis cost or value of any given molecule based on a representation of its molecular structure. We show that learned policies based on this value network can outperform a heuristic approach that favors symmetric disconnections when synthesizing unfamiliar molecules from available starting materials using the fewest number of reactions. We discuss how the learned policies described here can be incorporated into existing synthesis planning tools and how they can be adapted to changes in the synthesis cost objective or material availability.
Short abstract
Deep reinforcement learning optimizes a retrosynthetic strategy with user-defined pathway cost, finding better pathways than a heuristic scoring function favoring maximally convergent disconnections.
Introduction
The primary goal of computer-aided synthesis planning (CASP) is to help chemists accelerate the synthesis of desired molecules.1−3 Generally, a CASP program takes as input the structure of a target molecule and returns a sequence of feasible reactions linking the target to commercially available starting materials. The number of possible synthesis plans is often astronomical, and it is therefore desirable to identify the plan(s) that minimize some user-specified objective function (e.g., synthesis cost c). The challenge of identifying these optimal syntheses can be framed as a one-player game—the retrosynthesis game—to allow for useful analogies with chess and Go, for which powerful solutions based on deep reinforcement learning now exist.4,5 During play, the chemist starts from the target molecule and identifies a set of candidate reactions by which to make the target in one step (Figure 1). At this point, the chemist must decide which reaction to choose. As in other games such as chess, the benefits of a particular decision may not be immediately obvious. Only when the game is won or lost can one fairly assess the value of decisions that contributed to the outcome. Once a reaction is selected, its reactant or reactants become the new target(s) of successive retrosynthetic analyses. This branching recursive process of identifying candidate reactions and deciding which to use continues until the growing synthesis tree reaches the available substrates (a “win”), or it exceeds a specified number of synthetic steps (a “loss”).
Figure 1.

The objective of the
retrosynthesis game is to synthesize the target
product m0 from available substrates by
way of a synthesis tree that minimizes the cost function. Molecules
and reactions are illustrated by circles and squares, respectively.
Starting from the target, a reaction 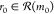 (yellow) is selected according
to a policy
π(r0|m0) that links m0 with precursors m1, m2, m3. The gray squares leading to m0 illustrate the other potential reactions in
(yellow) is selected according
to a policy
π(r0|m0) that links m0 with precursors m1, m2, m3. The gray squares leading to m0 illustrate the other potential reactions in 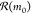 . The game continues one move at a time
reducing intermediate molecules (blue) until there are only substrates
remaining, or until a maximum depth of 10 is reached. Dead-end molecules
(green), for which no reactions are possible, are assigned a cost
penalty of 100, while molecules at maximum depth (purple) are assigned
a cost penalty of 10. Commercially available substrates (red) are
assigned zero cost. The synthesis cost of the product may be computed
according to eq 1 only
on completion of the game. Here, the sampled pathway leading to the
target (red arrows) has a cost of 5.
. The game continues one move at a time
reducing intermediate molecules (blue) until there are only substrates
remaining, or until a maximum depth of 10 is reached. Dead-end molecules
(green), for which no reactions are possible, are assigned a cost
penalty of 100, while molecules at maximum depth (purple) are assigned
a cost penalty of 10. Commercially available substrates (red) are
assigned zero cost. The synthesis cost of the product may be computed
according to eq 1 only
on completion of the game. Here, the sampled pathway leading to the
target (red arrows) has a cost of 5.
Winning outcomes are further distinguished by the cost c of the synthesis pathway identified—the lower the better. This synthesis cost is often ambiguous and difficult to evaluate as it involves a variety of uncertain or unknown quantities. For example, the synthesis cost might include the price of the starting materials, the number of synthetic steps, the yield of each step, the ease of product separation and purification, the amount of chemical waste generated, the safety or environmental hazards associated with the reactions and reagents, etc. It is arguably more challenging to accurately evaluate the cost of a proposed synthesis than it is to generate candidate syntheses. It is therefore common to adopt simple objective functions that make use of the information available (e.g., the number of reactions but not their respective yields).6 We refer to the output of any such function as the cost of the synthesis; optimal synthesis plans correspond to those with minimal cost.
Expert chemists excel at the retrosynthesis game for two reasons: (1) they can identify a large number of feasible reaction candidates at each step, and (2) they can select those candidates most likely to lead to winning syntheses. These abilities derive from the chemists’ prior knowledge and their past experience in making molecules. In contrast to games with a fixed rule set like chess, the identification of feasible reaction candidates (i.e., the possible “moves”) is nontrivial: there may be thousands of possible candidates at each step using known chemistries. To address this challenge, computational approaches have been developed to suggest candidate reactions using libraries of reaction templates prepared by expert chemists7 or derived from literature data.8−10 Armed with these “rules” of synthetic chemistry, a computer can, in principle, search the entire space of possible synthesis pathways and identify the optimal one.
In practice, however, an exhaustive search of possible synthesis trees is not computationally feasible or desirable because of the exponential growth in the number of reactions with distance from the target.6,11 Instead, search algorithms generate a subset of possible synthesis trees, which may or may not contain the optimal pathway(s). For longer syntheses, the subset of pathways identified is an increasingly small fraction of the total available. Thus, it is essential to bias retrosynthetic search algorithms toward those regions of synthesis space most likely to contain the optimal pathway. In the game of retrosynthesis, the player requires a strong guiding model, or policy, for selecting the reaction at each step that leads to the optimal synthetic pathway(s).
Prior reports on retrosynthetic planning have explored a variety of policies for guiding the generation of candidate syntheses.7,12−14 These programs select among possible reactions using heuristic scoring functions,7,15 crowd-sourced accessibility scores,13,16 analogy to precedent reactions,17 or parametric models (e.g., neural networks) trained on literature precedents.18,19 In particular, the Syntaurus software7 allows for user-specified scoring functions that can describe common strategies used by expert chemists20 (e.g., using symmetric disconnections to favor convergent syntheses). By contrast, Segler and Waller used literature reaction data to train a neural network that determines which reaction templates are most likely to be effective on a given molecule.18 The ability to rank order candidate reactions (by any means) allows for guiding network search algorithms (e.g., Monte Carlo tree search19) to generate large numbers of possible synthesis plans. The costs of these candidates can then be evaluated to identify the “best” syntheses, which are provided to the chemist (or perhaps a robotic synthesizer).
Here, we describe a different approach to retrosynthetic planning based on reinforcement learning,21 in which the computer learns to select those candidate reactions that lead ultimately to synthesis plans minimizing a user-specified cost function. Our approach is inspired by the recent success of deep reinforcement learning in mastering combinatorial games such as Go using experience generated by repeated self-play.4,5 In this way, DeepMind’s AlphaGo Zero learned to estimate the value of any possible move from any state in the game, thereby capturing the title of world champion.4,22 Similarly, by repeated plays of the retrosynthesis game, the computer can learn which candidate reactions are most likely to lead from a given molecule to available starting materials in an optimal fashion. Starting from a random policy, the computer explores the synthetic space to generate estimates of the synthesis cost for any molecule. These estimates form the basis for improved policies that guide the discovery of synthesis plans with lower cost. This iterative process of policy improvement converges in time to optimal policies that identify the “best” pathway in a single play of the retrosynthesis game. Importantly, we show that (near) optimal policies trained on the synthesis of ∼100 000 diverse molecules generalize well to the synthesis of unfamiliar molecules.
This approach requires no prior knowledge of synthetic strategy beyond the “rules” governing single-step reactions encoded in a library of reaction templates. The library we use below was extracted algorithmically from the Reaxys database and does not always generate chemically feasible recommendations. However, our approach can be extended to other template libraries, including those curated by human experts7 and/or data-driven filters23,24 to improve the likelihood that proposed reactions are effective and selective. Overall, the goal of this work is to learn the strategy of applying these rules for retrosynthesis, rather than to improve quality of the rules themselves. The learned policies we identify can be incorporated into existing synthesis planning tools and adapted to different cost functions that reflect the changing demands of organic synthesis.
Results and Discussion
The Retrosynthesis Game
We formulate the problem of
retrosynthetic analysis as a game played by a synthetic chemist or
a computer program. At the start of the game, the player is given
a target molecule m to synthesize starting from a
set of buyable molecules denoted 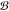 . For any such
molecule, there exist a set
of reactions, denoted
. For any such
molecule, there exist a set
of reactions, denoted 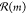 , where each reaction
includes the molecule m as a product. From this set,
the player chooses a particular
reaction
, where each reaction
includes the molecule m as a product. From this set,
the player chooses a particular
reaction 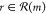 according to a policy
π(r|m), which defines the
probability of selecting
that reaction for use in the synthesis. The cost crxn(r) of performing the chosen reaction—however
defined by the user—is added to a running total, which ultimately
determines the overall synthesis cost. Having completed one step of
the retrosynthesis game, the player considers the reactant(s) of the
chosen reaction in turn. If a reactant m′
is included among the buyable substrates,
according to a policy
π(r|m), which defines the
probability of selecting
that reaction for use in the synthesis. The cost crxn(r) of performing the chosen reaction—however
defined by the user—is added to a running total, which ultimately
determines the overall synthesis cost. Having completed one step of
the retrosynthesis game, the player considers the reactant(s) of the
chosen reaction in turn. If a reactant m′
is included among the buyable substrates, 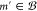 , then the cost of that
molecule csub(m′)
is added to
the running total. Otherwise, the reactant m′
must be synthesized following the same procedure outlined above. This
recursive processes results in a synthesis tree whose root is the
target molecule, and whose leaves are buyable substrates. The total
cost of the resulting synthesis is
, then the cost of that
molecule csub(m′)
is added to
the running total. Otherwise, the reactant m′
must be synthesized following the same procedure outlined above. This
recursive processes results in a synthesis tree whose root is the
target molecule, and whose leaves are buyable substrates. The total
cost of the resulting synthesis is
| 1 |
where the respective sums are evaluated over
all reactions r and all leaf molecules m included in the final synthesis tree. This simple cost function
neglects effects due to interactions between successive reactions
(e.g., costs incurred in switching solvents); however, it has the
useful property that the expected cost vπ(m) of making any molecule m in
one step via reaction r is directly related to the
expected cost of the associated reactants 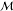
| 2 |
This recursive function terminates at buyable molecules of known cost, for which vπ(m) = csub(m) independent of the policy.
The function vπ(m) denotes the expected cost or “value” of any molecule m under a specified policy π. By repeating the game many times starting from many target molecules, it is possible to estimate the value for each target and its precursors. Such estimates generated from simulated experience can be used to train a parametric representation of the value function, which predicts the expected cost of any molecule. Importantly, knowledge of the value function under a suboptimal policy π enables the creation of new and better policies π′ that reduce the expected cost of synthesizing a molecule (according to the policy improvement theorem21). Using methods of reinforcement learning, such iterative improvement schemes, leads to the identification of optimal policies π∗, which identify synthesis trees of minimal cost. The value of a molecule under such a policy is equal to the expected cost of selecting the “best” reaction at each step such that
| 3 |
From a practical perspective, the optimal value function takes as input a molecule (e.g., a representation of its molecular structure) and outputs a numeric value corresponding to the minimal cost with which it can be synthesized.
Here,
we considered a set of 100 000 target molecules selected
from the Reaxys database on the basis of their structural diversity
(see the Methods section). The set of buyable
substrates 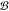 contained
∼300 000 molecules
selected from the Sigma-Aldrich,25 eMolecules,26 and LabNetwork27 catalogs
that have list prices less than $100/g. At each step, the possible
reactions
contained
∼300 000 molecules
selected from the Sigma-Aldrich,25 eMolecules,26 and LabNetwork27 catalogs
that have list prices less than $100/g. At each step, the possible
reactions 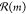 were identified using a set of 60 000
reaction templates derived from more than 12 million single-step reaction
examples reported in the Reaxys database (see the Methods section). Because the application of reaction templates
is computationally expensive, we used a template prioritizer to identify
those templates most relevant to a given molecule.18 On average, this procedure resulted in up to 50 possible
reactions for each molecule encountered during synthesis planning.
We assume the space of molecules and reactions implicit in these transformations
is representative of real organic chemistry while recognizing the
inevitable limitations of templates culled from incomplete and sometimes
inaccurate reaction databases. For simplicity, the cost of each reaction
step was set to one, crxn(r) = 1, and the substrate costs to zero, csub(m) = 0. With these assignments, the cost of making
a molecule is equivalent to the number of reactions in the final synthesis
tree.
were identified using a set of 60 000
reaction templates derived from more than 12 million single-step reaction
examples reported in the Reaxys database (see the Methods section). Because the application of reaction templates
is computationally expensive, we used a template prioritizer to identify
those templates most relevant to a given molecule.18 On average, this procedure resulted in up to 50 possible
reactions for each molecule encountered during synthesis planning.
We assume the space of molecules and reactions implicit in these transformations
is representative of real organic chemistry while recognizing the
inevitable limitations of templates culled from incomplete and sometimes
inaccurate reaction databases. For simplicity, the cost of each reaction
step was set to one, crxn(r) = 1, and the substrate costs to zero, csub(m) = 0. With these assignments, the cost of making
a molecule is equivalent to the number of reactions in the final synthesis
tree.
To prohibit the formation of unreasonably deep synthesis
trees,
we limited our retrosynthetic searches to a maximum depth of dmax = 10. As detailed in the Methods section, the addition of this termination criterion
to the recursive definition of the value function (eq 2) requires some minor modifications
to the retrosynthesis game. In particular, the expected cost of synthesizing
a molecule m depends also on the residual depth, vπ = vπ(m, δ), where δ = dmax – d is the difference between
the maximum depth and the current depth d within
the tree. If a molecule m not included among the
buyable substrates is encountered at a residual depth of zero, it
is assigned a large cost vπ(m, 0) = P1, thereby penalizing
the failed search. Additionally, in the event that no reactions are
identified for a given molecule m (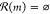 ), we assign
an even larger penalty P2, which encourages
the player to avoid such
dead-end molecules if possible. Below, we use the specific numeric
penalties of P1 = 10 and P2 = 100 for all games.
), we assign
an even larger penalty P2, which encourages
the player to avoid such
dead-end molecules if possible. Below, we use the specific numeric
penalties of P1 = 10 and P2 = 100 for all games.
Heuristic Policies
En route to the development of optimal policies for retrosynthesis, we first consider the performance of some heuristic policies that provide context for the results below. Arguably the simplest policy is one of complete ignorance, in which the player selects a reaction at random at each stage of the synthesis—that is, π(r|m) = constant. We use this “straw man” policy to describe the general process of policy evaluation and provide a baseline from which to measure subsequent improvements, although it is unlikely to be used in practice.
During the evaluation process, the computer plays the retrosynthesis game to the end making random moves at each step of the way. After each game, the cost of each molecule in the resulting synthesis tree is computed. This process is repeated for each of the 100 000 target molecules considered. These data points—each containing a molecule m at residual depth δ with cost c—are used to update the parametric approximation of the value function vπ(m, δ). As detailed in the Methods section, the value function is approximated by a neural network that takes as input an extended-connectivity fingerprint (ECFP) of the molecule m and the residual depth δ and outputs a real valued estimate of the expected cost under the policy π.28 This process is repeated in an iterative manner as the value estimates of the target molecules, vπ(m, dmax), approach their asymptotic values.
Figure 2a shows the total synthesis cost ctot for a single target molecule under the random policy (markers). Each play of the retrosynthesis game has one of three possible outcomes: a “winning” synthesis plan terminating in buyable substrates (blue circles), a “losing” plan that exceeds the maximum depth (green triangles), and a “losing” plan that contains dead-end molecules that cannot be bought or made (black pentagons). After many synthesis attempts, the running average of the fluctuating synthesis cost converges to the expected cost vπ(m, dmax) as approximated by the neural network (red line). Repeating this analysis for the 100 000 target molecules, the random policy results in an average cost of ∼110 per molecule with only a 25% chance of identifying a winning synthesis in each attempt. Clearly, there is room for improvement.
Figure 2.
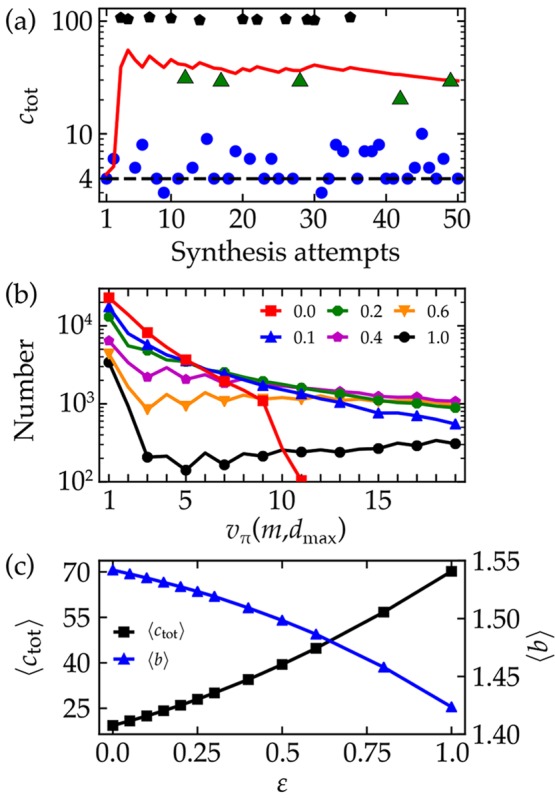
Heuristic policies. (a) Synthesis cost ctot for a single molecule m (N-dibutyl-4-acetylbenzeneacetamide) for successive iterations of the retrosynthesis game under the random policy. Blue circles denote “winning” synthesis plans that trace back to buyable molecules. Green triangles and black pentagons denote “losing” plans that exceed the maximum depth or include unmakeable molecules, respectively. The solid line shows the neural network prediction of the value function vπ(m, dmax) as it converges to the average synthesis cost. The dashed line shows the expected cost under the deterministic “symmetric disconnection” policy with γ = 1.5. (b) Distribution of expected costs vπ(m, dmax) over the set of 100 000 target molecules for different noise levels ε. The red squares and black circles show the performance of the symmetric disconnection policy (ε = 0) and the random policy (ε = 1), respectively. See Figure S1 for the full distribution including higher cost (“losing”) syntheses. (c) The average synthesis cost of the target molecules increases with increasing noise level ε, while the average branching factor decreases. Averages were estimated from 50 plays for each target molecule.
Beyond the random policy, even simple heuristics can be used to improve performance significantly. In one such policy, inspired by Syntaurus,7 the player selects the reaction r that maximizes the quantity
| 4 |
where ns(m) is the length of the canonical smiles
string representing
molecule m, γ is a user-specified exponent,
and the sum is taken over the reactants 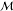 (r) associated with a reaction r. When γ
> 1, the reactions that maximize this function
can be interpreted as those that decompose the product into multiple
parts of roughly equal size. Note that, in contrast to the random
policy, this greedy heuristic is deterministic: each play of the game
results in the same outcome. Figure 2a shows the performance of this “symmetric disconnection”
policy with γ = 1.5 for a single target molecule (dashed line).
Interestingly, while the pathway identified by the greedy policy is
much better on average than those of the random policy (ctot = 4 versus ⟨ctot⟩ = 35.1), repeated application of the latter reveals the
existence of an even better pathway containing only three reactions.
An optimal policy would allow for the identification of that best
synthesis plan during a single play of the retrosynthesis
game.
(r) associated with a reaction r. When γ
> 1, the reactions that maximize this function
can be interpreted as those that decompose the product into multiple
parts of roughly equal size. Note that, in contrast to the random
policy, this greedy heuristic is deterministic: each play of the game
results in the same outcome. Figure 2a shows the performance of this “symmetric disconnection”
policy with γ = 1.5 for a single target molecule (dashed line).
Interestingly, while the pathway identified by the greedy policy is
much better on average than those of the random policy (ctot = 4 versus ⟨ctot⟩ = 35.1), repeated application of the latter reveals the
existence of an even better pathway containing only three reactions.
An optimal policy would allow for the identification of that best
synthesis plan during a single play of the retrosynthesis
game.
The performance of a policy is characterized by the distribution
of expected costs over the set of target molecules. Figure 2b shows the cost distribution
for a series of policies that interpolate between the greedy “symmetric
disconnection” policy and the random policy (see also Figure S1). The intermediate ε-greedy policies
behave greedily with probability 1 – ε, selecting the
reaction that maximizes f(r), but
behave randomly with probability ε, selecting any one of the
possible reactions 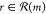 with equal probability. On average, the
addition of such noise is detrimental to policy performance. Noisy
policies are less likely to identify a successful synthesis for a
given target (Figure S2a) and result in
longer syntheses when they do succeed (Figure S2b). Consequently, the average cost ⟨ctot⟩ increases monotonically with increasing noise
as quantified by the parameter ε (Figure 2c).
with equal probability. On average, the
addition of such noise is detrimental to policy performance. Noisy
policies are less likely to identify a successful synthesis for a
given target (Figure S2a) and result in
longer syntheses when they do succeed (Figure S2b). Consequently, the average cost ⟨ctot⟩ increases monotonically with increasing noise
as quantified by the parameter ε (Figure 2c).
The superior performance (lower synthesis costs) of the greedy policy is correlated with the average branching factor ⟨b⟩, which represents the average number of reactants for each reaction in the synthesis tree. Branching is largest for the greedy policy (ε = 0) and decreases monotonically with increasing ε (Figure 2c). On average, synthesis plans with greater branching (i.e., convergent syntheses) require fewer synthetic steps to connect the target molecules to the set of buyable substrates. This observation supports the chemical intuition underlying the symmetric disconnection policy: break apart each “complex” molecule into “simpler” precursors. However, this greedy heuristic can sometimes be short-sighted. An optimal retrosynthetic “move” may increase molecular complexity in the short run to reach simpler precursors more quickly in the longer run (e.g., in protecting group chemistry). An optimal policy would enable the player to identify local moves (i.e., reactions) that lead to synthesis pathways with minimum total cost.
Policy Improvement through Simulated Experience
Knowledge of the value function, vπ, under a given policy π enables the identification of better policies that reduce the expected synthesis cost. To see this, consider a new policy π′ that selects at each step the reaction that minimizes the expected cost under the old policy π
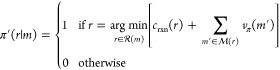 |
5 |
Restated, the new π′ is a deterministic
policy that always selects the reaction r to minimize
the cost of m. It calculates the cost of synthesizing m through reaction r by adding the reaction
cost, crxn(r), and the
costs of the associated precursors, 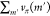 ; the cost of the precursor molecules is
estimated using the value function vπ defined by the old policy π. The new policy function does
not need to be calculated in advance but is used on-the-fly to select
the best reaction from the list of options generated by the template
library.
; the cost of the precursor molecules is
estimated using the value function vπ defined by the old policy π. The new policy function does
not need to be calculated in advance but is used on-the-fly to select
the best reaction from the list of options generated by the template
library.
By the policy improvement theorem,21 this greedy policy π′ is guaranteed to be
as good as or better than the old policy π—that is, 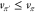 , where equality
holds only for the optimal
policy. This result provides a basis for systematically improving
any policy in an iterative procedure called policy iteration,21 in which the value function vπ leads to an improved policy π′ that
leads to a new value function
, where equality
holds only for the optimal
policy. This result provides a basis for systematically improving
any policy in an iterative procedure called policy iteration,21 in which the value function vπ leads to an improved policy π′ that
leads to a new value function 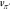 and so on.
and so on.
One of the challenges in using
the greedy policy eq 5 is that it generates only a single
pathway and its associated cost for each of the target molecules.
The limited exposure of these greedy searches can result in poor estimates
of the new value function 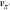 , in particular for molecules that are not
included in the identified pathways. A better estimate of
, in particular for molecules that are not
included in the identified pathways. A better estimate of 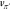 can be achieved by exploring more of the
molecule space in the neighborhood of these greedy pathways. Here,
we encourage exploration by using an ε-greedy policy, which
introduces random choices with probability ε but otherwise follows
the greedy policy eq 5. Iteration of this ε-soft policy is guaranteed to converge
to an optimal policy that minimizes the expected synthesis cost for
a given noise level ε > 0.21 Moreover,
by gradually lowering the noise level, it is possible to approach
the optimal greedy policy in the limit as ε → 0.
can be achieved by exploring more of the
molecule space in the neighborhood of these greedy pathways. Here,
we encourage exploration by using an ε-greedy policy, which
introduces random choices with probability ε but otherwise follows
the greedy policy eq 5. Iteration of this ε-soft policy is guaranteed to converge
to an optimal policy that minimizes the expected synthesis cost for
a given noise level ε > 0.21 Moreover,
by gradually lowering the noise level, it is possible to approach
the optimal greedy policy in the limit as ε → 0.
Training Protocol
Starting from the random policy, we simulated games to learn an improved policy over the course of 1000 iterations, each composed of ∼100 000 retrosynthesis games initiated from the target molecules. During the first iteration, each target molecule was considered in turn using the ε-greedy policy eq 5 with ε = 0.2. Candidate reactions and their associated reactants were identified by application of reaction templates as detailed in the Methods section. Absent an initial model of the value function, the expected costs of molecules encountered during play were selected at random from a uniform distribution on the interval [1, 100]. Following the completion of each game, the costs of molecules in the selected pathway were computed and stored for later use. In subsequent iterations, the values of molecules encountered previously (at a particular depth) were estimated by their average cost. After the first 50 iterations, the value estimates accumulated during play were used to train a neural network, which allowed for estimating the values of new molecules not encountered during the previous games (see the Methods section for details on the network architecture and training). Policy improvement continued in an iterative fashion guided both by the average costs (for molecules previously encountered) and by the neural network (for new molecules), which was updated every 50–100 iterations.
During policy iteration, the noise parameter was reduced from ε = 0.2 to 0 in increments of 0.05 every 200 iterations in an effort to anneal the system toward an optimal policy. Following each change in ε, the saved costs were discarded such that subsequent value estimates were generated at the current noise level ε. The result of this training procedure was a neural network approximation of the (near) optimal value function v∗(m, δ), which estimates the minimum cost of synthesizing any molecule m starting from residual depth δ. In practice, we found that a slightly better value function could be obtained using the cumulative reaction network generated during policy iteration. Following Kowalik et al.,6 we used dynamic programming to compute the minimum synthesis cost for each molecule in the reaction network. These minimum costs were then used to train the final neural network approximation of the value function v∗.
Training Results
Figure 3a shows how the average synthesis cost ⟨ctot⟩ decreased with each iteration over the course of the training process. Initially, the average cost was similar to that of the random policy (⟨ctot⟩ ≈ 70) but improved steadily as the computer learned to identify “winning” reactions that lead quickly to buyable substrates. After 800 iterations, the cost dropped below that of the symmetric disconnection policy (⟨ctot⟩ = 19.3) but showed little further improvement in the absence of exploration (i.e., with ε = 0). The final cost estimate (⟨ctot⟩ = 11.4, cyan square) was generated by identifying the minimum cost pathways present in the cumulative reaction network generated during the training process. The final drop in cost for ε = 0 suggests that further policy improvements are possible using improved annealing schedules. We emphasize that the final near-optimal policy was trained from a state of complete ignorance, as directed by the user-specified objective function to minimize the synthesis cost.
Figure 3.
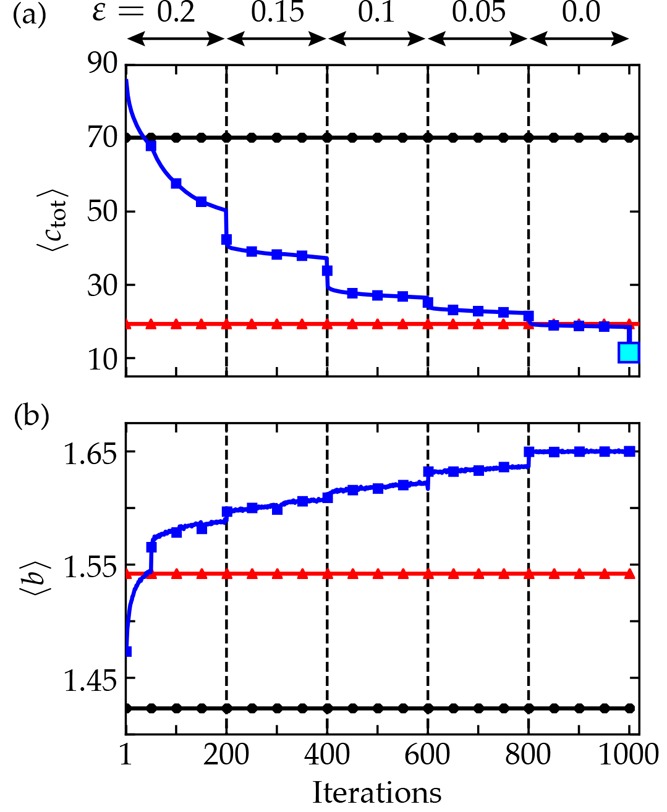
Training results. (a, b) ⟨ctot⟩ and ⟨btot⟩ computed using π∗ are plotted versus policy iterations, respectively (solid blue squares). Solid horizontal lines show these quantities for the heuristic policy πsd (red triangles) and the random policy (black circles). The larger cyan square shows ⟨ctot⟩ after each tree had been searched for the best (lowest) target cost. Dashed vertical lines show points when ε was lowered.
During the training process, the decrease in synthesis cost was guided both by motivation, as prescribed by the cost function, and by opportunity, as dictated by the availability of alternate pathways. Early improvements in the average cost were achieved by avoiding dead-end molecules, which contributed the largest cost penalty, P2 = 100. Of the target molecules, 11% reduced their synthesis cost from ctot > P2 to P2 > ctot > P1 by avoiding such problematic molecules. By contrast, only 2% of targets improved their cost from P2 > ctot > P1 to P1 > ctot. In other words, if a synthesis tree was not found initially at a maximum depth of dmax = 10, it was unlikely to be discovered during the course of training. Perhaps more interesting are those molecules (ca. 10%) for which syntheses were more easily found but subsequently improved (i.e., shortened) during the course of the training process. See Table 1 for a more detailed breakdown of these different groups.
Table 1. Training and Testing Results for the Symmetric Disconnection Policy πsd and the Learned Policy π∗a.
| train (100 000) |
test (25 000) |
|||
|---|---|---|---|---|
| πsd | π∗ | πsd | π∗ | |
| ⟨ctot⟩ | 19.3 | 13.1 | 19.2 | 11.5 |
| ⟨b⟩ | 1.54 | 1.65 | 1.54 | 1.58 |
| ctot < P1 | 64% | 83% | 65% | 73% |
| P1 ≤ ctot < P2 | 25% | 11% | 24% | 22% |
| ctot ≥ P2 | 11% | 6% | 11% | 5% |
Percentages were computed based the sizes of the training set (∼100 000) and the testing set (∼25 000).
Consistent with our observations above, lower-cost pathways were again correlated with the degree of branching b along the synthesis trees (Figure 3b). Interestingly, the average branching factor for synthesis plans identified by the learned policy was significantly larger than that of the symmetric disconnection policy (⟨b⟩ = 1.65 versus 1.54). While the latter favors branching, it does so locally based on limited information—namely, the heuristic score of eq 5. By contrast, the learned policy uses information provided in the molecular fingerprint to select reactions that increase branching across the entire synthesis tree (not just the single step). Furthermore, while the heuristic policy favors branching a priori, the learned policy does so only in the service of reducing the total cost. Changes in the objective function (e.g., in the cost and availability of the buyable substrates) will lead to different learned policies.
Model Validation
Figure 4 compares the performance of the learned policy evaluated on the entire set of ∼100 000 target molecules used for training and on a different set of ∼25 000 target molecules set aside for testing. For the training molecules, the value estimates v∗(m) predicted by the neural network are highly correlated with the actual costs obtained by the final learned policy π∗ (Figure 4a). We used the same near-optimal policy to determine the synthesis cost of the testing molecules, ctot(π∗). As illustrated in Figure 4b, these costs were correlated to the predictions of the value network v∗(m) albeit more weakly than those of the training data (Pearson coefficient of 0.5 for testing versus 0.99 for training). This correlation was stronger for the data in Figure 4b, which focuses on those molecules that could actually be synthesized (Pearson coefficient of 0.7 for the 73% testing molecules with “winning” syntheses).
Figure 4.

Model Validation. A 2D histogram illustrates the relationship between the synthesis cost ctot determined by the learned policy π∗ and that predicted by the value network v∗ for (a) the ∼100 000 training molecules and (b) the ∼25 000 testing molecules. A 2D histogram compares the synthesis cost ctot determined by the symmetric disconnection policy πsd to that of learned policy π∗ for (c) training molecules and (d) testing molecules. The percentage of molecules for which π∗ (πsd) found the cheaper pathway is listed below (above) the red line. In parts a–d, the gray scale intensity is linearly proportional to the number of molecules within a given bin; the red line shows the identity relation. Distributions of synthesis costs ctot determined under policies πsd and π∗ are shown for (e) training molecules and (f) testing molecules.
Figure 4c,d compares the synthesis costs of the symmetric disconnection policy πsd against that of the learned policy π∗ for both the training and testing molecules. The figure shows that the results are highly correlated (Pearson coefficient 0.84 and 0.86 for training and testing, respectively), indicating that the two policies make similar predictions. However, closer inspection reveals that the learned policy is systematically better than the heuristic as made evident by the portion of the histogram below the diagonal (red line). For these molecules (42% and 31% of the training and testing sets, respectively), the learned policy identifies synthesis trees containing fewer reactions than those of the heuristic policy during single deterministic plays of the retrosynthesis game. By contrast, it is rare in both the training and testing molecules (about 4% and 11%, respectively) that the symmetric disconnection policy performs better than the learned policy. Additionally, the learned policy is more likely to succeed in identifying a viable synthesis plan leading to buyable substrates (Figure 4c). Of the ∼25 000 testing molecules, “winning” synthesis plans were identified for 73% using the learn policy as compared to 64% using the heuristic. These results suggest that the lessons gleaned from the training molecules can be used to improve the synthesis of new and unfamiliar molecules.
Figures 5 and 6 show proposed synthesis pathways for two molecules in the test set as identified by the heuristic policy and by the learned policy at different stages of training. We emphasize that what is being learned is a strategy for applying retrosynthetic templates given a fixed template library, not their chemical feasibility. Figure 5 shows a target for which there is a precedent three-component Povarov reaction;29 however, this transformation is not present in the template library and is thus unavailable to the heuristic and trained policies. Instead, the heuristic policy greedily proposes a substantial disconnection followed by a retro-oxidation and the final retrosubstitution. By contrast, the learned policy proposes a retroketone reduction, which does not lead to a large structural simplification of the molecule but rather sets up an elegant three-component Mannich reaction. It is worth noting that the learned policy based on deep neural networks cannot explain why it selects the retroketone reduction, only that “similar” choices led to favorable outcomes in past experience. Early during training, without the benefit of such experience, the learned policy shows virtually no synthetic strategy (Figure 5d). In Figure 6, the first disconnection is shared by the heuristic and the learned policies; however, the latter (Figure 6b) identifies a path to install the phenyl group without requiring additional redox chemistry (Figure 6a). In these representative examples, the learned policy identifies pathways with fewer reaction steps than the heuristic as directed by the chosen cost function.
Figure 5.

Pathways for target 417, COC1C=CC2NC(CC(C3C=CC=CC=3O)C=2C=1)C1C=CC(Cl)=CC=1, obtained (a) using the heuristic policy and using learned policies at different stages of training: (b) the final (near) optimal policy; (c) after 400–800 epochs of training; and (d) after fewer than 400 epochs.
Figure 6.

Pathways for target 2757, C=CC(C)(C)C(=NNC1C=CC(Cl)=CC=1)C1C=CC=CC=1, obtained (a) using the heuristic policy and using learned policies at different stages of training: (b) the final (near) optimal policy; (c) after 400–800 epochs of training; and (d) after fewer than 400 epochs.
Conclusions
We have shown that reinforcement learning can be used to identify effective policies for the computational design of retrosynthetic pathways given a fixed library of retrosynthetic templates defining the “rules”. In this approach, one specifies the global objective function to be minimized (here, the synthesis cost) without the need for ad hoc models or heuristics to guide local decisions during generation of the synthesis plan. Starting from a random policy, repeated plays of the retrosynthesis game are used to systematically improve performance in an iterative process that converges in time to an optimal policy. The learned value function provides a convenient estimate for the synthesis cost of any molecule, while the associated policy allows for rapid identification of the synthesis path.
In practice, synthesis design and pathway optimization are a multiobjective problem that benefits from a detailed consideration of process costs. Ease of purification, estimated yield and purity, chemical availability, presence of genotoxic intermediates or impurities, and overall process mass intensity may all factor into the decision. Importantly, the cost function of eq 2 is readily adapted to accommodate any combination of such costs at the single chemical or single reaction level. Policy iteration using a different cost function will result in a different policy that reflects the newly specified objectives.
The chemical feasibility of synthetic pathways identified by the learned policy is largely determined by the quality of the reaction templates. The present templates are derived algorithmically from reaction precedents reported in the literature; however, an identical approach based on reinforcement learning could be applied using template libraries curated by human experts.7,30 Alternatively, it may be possible to forgo the use of reaction templates altogether in favor of machine learning approaches that suggest reaction precursors by other means.31 Ideally, such predictions should be accompanied by recommendations regarding the desired conditions for performing each reaction in high yield.32−35 There are, however, some challenges in using data-driven models for predicting chemical reactions that limit their predictive accuracy. These include incomplete reporting of reaction stoichiometries, ambiguity in the reported reaction outcomes, and data sparsity when considering rare or under-reported reaction types.
In the present approach, the deterministic policy learned during training is applied only once to suggest one (near) optimal synthesis pathway. Additional pathways are readily generated, for example, using Monte Carlo Tree Search (MCTS) to bias subsequent searches away from previously identified pathways.18 A similar approach is used by Syntaurus, which relies on heuristic scoring functions to guide the generation of many possible synthesis plans, from which the “best” are selected. The main advantage of a strong learned policy is to direct such exploration more effectively toward these best syntheses, thereby reducing the computational cost of exploration.
We note, however, that the computational costs of training the learned policy are significant (ca. several million CPU hours for the training in Figure 3). While the application of reaction templates remains the primary bottleneck (ca. 50%), the additional costs of computing ECFP fingerprints and evaluating the neural network were a close second (ca. 45%). These costs can be greatly reduced by using simple heuristics to generate synthetic pathways, from which stronger policies can be learned. We found that eq 4 performed remarkably well and was much faster to evaluate than the neural network. Such fast heuristics could be used as starting points for iterative policy improvement or as roll-out policies within MCTS-based learning algorithms.21 This approach is conceptually similar to the first iteration of AlphaGo introduced by DeepMind.36 Looking forward, we anticipate that the retrosynthesis game will soon follow the way of chess and Go, in which self-taught algorithms consistently outperform human experts.
Methods
Target Molecules
Training/testing sets of 95 774/23 945 molecules were selected from the Reaxys database on the basis of their structural diversity. Starting from more than 20 million molecules in the database, we excluded (i) those listed in the database of buyable compounds, (ii) those with SMILES strings shorter than 20 or longer than 100, and (iii) those with multiple fragments (i.e., molecules with “.” in the SMILES string). The resulting ∼16 million molecules were then aggregated using the Taylor–Butina (TB) algorithm37 to form ∼1 million clusters, each composed of “similar” molecules. Structural similarity between two molecules i and j was determined by the Tanimoto coefficient
| 6 |
where mi is the ECFP4 fingerprint for molecule i.38 We used fingerprints of length 1024 and radius 3. Two molecules within a common cluster were required to have a Tanimoto coefficient of T > 0.4. The target molecules were chosen as the centroids of the ∼125 000 largest clusters, each containing more than 20 molecules and together representing more than ∼12 million molecules. These target molecules were partitioned at random to form the final sets for training and testing.
Buyable Molecules
A molecule is defined to be a substrate
if it is listed in the commercially available Sigma-Aldrich,25 eMolecules,26 or
LabNetwork catalogs27 and does not cost
more than $100/g. The complete set of molecules in these catalogs
with price per gram ≤ $100 is denoted 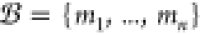 with n ≈ 300 000.
with n ≈ 300 000.
Reaction Templates
Given a molecule m, we used a set of ∼60 000 reaction templates to generate sets of possible precursors m′, which can be used to synthesize m in one step. As detailed previously,39 these templates were extracted automatically from literature precedents and encoded using the SMARTS language. The application of the templates involves two main steps, substructure matching and bond rewiring, which were implemented using RDKit.40 Briefly, we first search the molecule m for a structural pattern specified by the template. For each match, the reaction template further specifies the breaking and making of bonds among the constituent atoms to produce the precursor molecule(s) m′. We used the RDChiral package41 to handle the creation, destruction, and preservation of chiral centers during the reaction. The full code used for retrosynthetic template extraction is available in ref (41).
The application of reaction templates to produce candidate reactions represents a major computational bottleneck in the retrosynthesis game due to the combinatorial complexity of substructure matching. Additionally, even when a template generates a successful match, it may fail to account for the larger molecular context resulting in undesired byproducts during the forward reaction. These two challenges can be partially alleviated by use of a “template prioritizer”,18 which takes as input a representation of the target molecule m and generates a probability distribution over the set of templates based on their likelihood of success. By focusing only on the most probable templates, the prioritizer can serve to improve both quality of the suggested reactions and the speed with which they are generated. In practice, we trained a neural network prioritizer on 5.4 million reaction examples from Reaxys and selected the top 99.5% of templates for each molecule m encountered. This filtering process drastically reduced the total number templates applied from 60 000 to less than 50 for most molecules. The training and validation details as well as the model architecture are available on Github.42
Policy Iteration
As noted in the main text, the depth constraint imposed on synthesis trees generated during the retrosynthesis requires some minor modifications to the value function of eq 2. The expected cost of synthesizing a molecule m now depends on the residual depth δ as
| 7 |
where the first sum is over
candidate reactions with m as product, and the second
is over the reactants 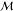 (r) associated with a reaction r. For the present
cost model, the expected cost vπ(m, δ) increases
with decreasing δ due to the increased likelihood of being penalized
(to the extent P1) for reaching the maximum
depth (d = dmax such
that δ = 0). Similarly, the ε-greedy policy used in policy
improvement must also account for the residual depth at which a molecule
is encountered
(r) associated with a reaction r. For the present
cost model, the expected cost vπ(m, δ) increases
with decreasing δ due to the increased likelihood of being penalized
(to the extent P1) for reaching the maximum
depth (d = dmax such
that δ = 0). Similarly, the ε-greedy policy used in policy
improvement must also account for the residual depth at which a molecule
is encountered
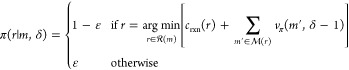 |
8 |
These recursive functions are fully specified
by three terminating conditions introduced in the main text: (1) buyable
molecule encountered, v(m, δ
≠ 0) = csub(m)
for 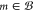 ; (2) maximum depth reached, v(m, 0) = P1; and (3)
unmakeable molecule encountered, v(m, δ ≠ 0) = P2 for
; (2) maximum depth reached, v(m, 0) = P1; and (3)
unmakeable molecule encountered, v(m, δ ≠ 0) = P2 for 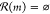 .
.
Neural Network Architecture and Training
We employed
a multilayer neural network illustrated schematically in Figure 7. The 17 million
model parameters were learned using gradient descent on training data
generated by repeated plays of the retrosynthesis game. Training was
performed using Keras with the Theano backend and the Adam optimizer
with an initial learning rate of 0.001, which decayed with the number
of model updates k as 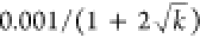 (13 updates were used to compute π∗). During
each update, batches of 128 molecules and
their computed average costs at a fixed ε were selected from
the most recent data and added to a replay buffer. Batches of equivalent
size were randomly selected from the buffer and passed through the
model for up to 100 epochs (1 epoch was taken as the total number
of new data points having passed through the network). The mean-average
error between the averaged (true) and predicted costs was used as
the loss function. The latest model weights were then used as the
policy for the next round of synthesis games. The full code used to
generate the learned policies is available in ref (43).
(13 updates were used to compute π∗). During
each update, batches of 128 molecules and
their computed average costs at a fixed ε were selected from
the most recent data and added to a replay buffer. Batches of equivalent
size were randomly selected from the buffer and passed through the
model for up to 100 epochs (1 epoch was taken as the total number
of new data points having passed through the network). The mean-average
error between the averaged (true) and predicted costs was used as
the loss function. The latest model weights were then used as the
policy for the next round of synthesis games. The full code used to
generate the learned policies is available in ref (43).
Figure 7.
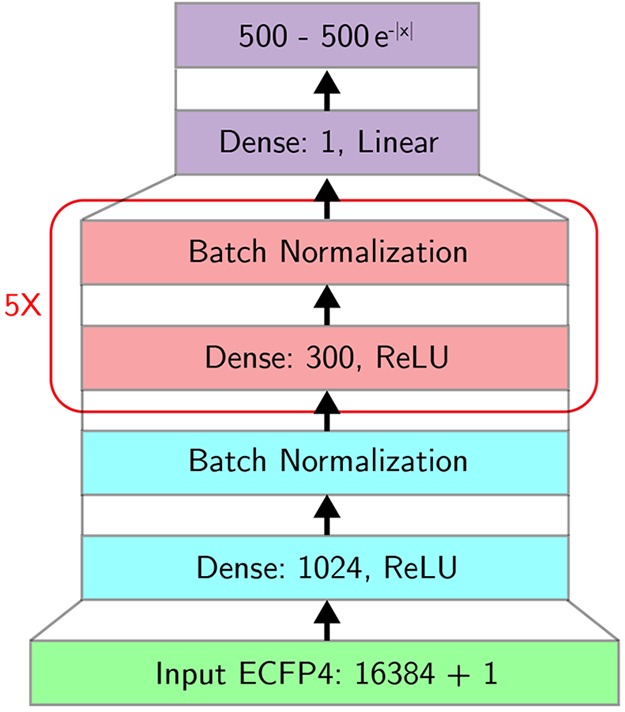
The neural model for the cost of molecules is a feed-forward neural network that accepts as input (green) an ECFP fingerprint of size 16 384 extended to include the residual depth δ of the molecule. The architecture includes one input layer (blue) consisting of 1024 nodes, five hidden layers (red) each containing 300 nodes, and one output layer (purple) of size one plus a filter (also purple) that scales the initial output number to be within the range [0, 500]. We also used batch normalization after each layer. The final output represents the estimated cost.
Acknowledgments
This work was supported by the DARPA Make-It program under Contract ARO W911NF-16-2-0023. We acknowledge computing resources from Columbia University’s Shared Research Computing Facility project, which is supported by NIH Research Facility Improvement Grant 1G20RR030893-01, and associated funds from the New York State Empire State Development, Division of Science Technology and Innovation (NYSTAR) Contract C090171, both awarded April 15, 2010.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.9b00055.
Additional results and figures including a distribution of expected costs, the probability of successfully synthesizing target molecules, normalized probability distributions, and a 2D histogram (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Warr W. A. A short review of chemical reaction database systems, computer-aided synthesis design, reaction prediction and synthetic feasibility. Mol. Inf. 2014, 33, 469–476. 10.1002/minf.201400052. [DOI] [PubMed] [Google Scholar]
- Engkvist O.; Norrby P.-O.; Selmi N.; Lam Y.-h.; Peng Z.; Sherer E. C.; Amberg W.; Erhard T.; Smyth L. A. Computational prediction of chemical reactions: current status and outlook. Drug Discovery Today 2018, 23, 1203–1218. 10.1016/j.drudis.2018.02.014. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Green W. H.; Jensen K. F. Machine learning in computer-aided synthesis planning. Acc. Chem. Res. 2018, 51, 1281–1289. 10.1021/acs.accounts.8b00087. [DOI] [PubMed] [Google Scholar]
- Silver D.; et al. Mastering the game of Go without human knowledge. Nature 2017, 550, 354–359. 10.1038/nature24270. [DOI] [PubMed] [Google Scholar]
- Silver D.; Hubert T.; Schrittwieser J.; Antonoglou I.; Lai M.; Guez A.; Lanctot M.; Sifre L.; Kumaran D.; Graepel T.; Lillicrap T.; Simonyan K.; Hassabis D. A general reinforcement learning algorithm that masters chess, shogi, and Go through self-play. Science 2018, 362, 1140–1144. 10.1126/science.aar6404. [DOI] [PubMed] [Google Scholar]
- Kowalik M.; Gothard C. M.; Drews A. M.; Gothard N. A.; Weckiewicz A.; Fuller P. E.; Grzybowski B. A.; Bishop K. J. Parallel optimization of synthetic pathways within the network of organic chemistry. Angew. Chem., Int. Ed. 2012, 51, 7928–7932. 10.1002/anie.201202209. [DOI] [PubMed] [Google Scholar]
- Szymkuć S.; Gajewska E. P.; Klucznik T.; Molga K.; Dittwald P.; Startek M.; Bajczyk M.; Grzybowski B. A. Computer-assisted synthetic planning: the end of the beginning. Angew. Chem., Int. Ed. 2016, 55, 5904–5937. 10.1002/anie.201506101. [DOI] [PubMed] [Google Scholar]
- Law J.; Zsoldos Z.; Simon A.; Reid D.; Liu Y.; Khew S. Y.; Johnson A. P.; Major S.; Wade R. A.; Ando H. Y. Route designer: a retrosynthetic analysis tool utilizing automated retrosynthetic rule generation. J. Chem. Inf. Model. 2009, 49, 593–602. 10.1021/ci800228y. [DOI] [PubMed] [Google Scholar]
- Christ C. D.; Zentgraf M.; Kriegl J. M. Mining electronic laboratory notebooks: analysis, retrosynthesis, and reaction based enumeration. J. Chem. Inf. Model. 2012, 52, 1745–1756. 10.1021/ci300116p. [DOI] [PubMed] [Google Scholar]
- Bøgevig A.; Federsel H.-J.; Huerta F.; Hutchings M. G.; Kraut H.; Langer T.; Löw P.; Oppawsky C.; Rein T.; Saller H. Route design in the 21st century: The IC SYNTH software tool as an idea generator for synthesis prediction. Org. Process Res. Dev. 2015, 19, 357–368. 10.1021/op500373e. [DOI] [Google Scholar]
- Grzybowski B. A.; Bishop K. J.; Kowalczyk B.; Wilmer C. E. The ‘wired’ universe of organic chemistry. Nat. Chem. 2009, 1, 31–36. 10.1038/nchem.136. [DOI] [PubMed] [Google Scholar]
- Bertz S. H. The first general index of molecular complexity. J. Am. Chem. Soc. 1981, 103, 3599–3601. 10.1021/ja00402a071. [DOI] [Google Scholar]
- Sheridan R. P.; Zorn N.; Sherer E. C.; Campeau L.-C.; Chang C.; Cumming J.; Maddess M. L.; Nantermet P. G.; Sinz C. J.; O’Shea P. D. Modeling a crowdsourced definition of molecular complexity. J. Chem. Inf. Model. 2014, 54, 1604–1616. 10.1021/ci5001778. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Rogers L.; Green W. H.; Jensen K. F. SCScore: Synthetic complexity learned from a reaction corpus. J. Chem. Inf. Model. 2018, 58, 252–261. 10.1021/acs.jcim.7b00622. [DOI] [PubMed] [Google Scholar]
- Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Model. 1988, 28, 31–36. 10.1021/ci00057a005. [DOI] [Google Scholar]
- Ertl P.; Schuffenhauer A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminf. 2009, 1, 8. 10.1186/1758-2946-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley C. W.; Rogers L.; Green W. H.; Jensen K. F. Computer-assisted retrosynthesis based on molecular similarity. ACS Cent. Sci. 2017, 3, 1237–1245. 10.1021/acscentsci.7b00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segler M. H.; Waller M. P. Neural-Symbolic machine learning for retrosynthesis and reaction prediction. Chem. - Eur. J. 2017, 23, 5966–5971. 10.1002/chem.201605499. [DOI] [PubMed] [Google Scholar]
- Segler M. H.; Preuss M.; Waller M. P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. 10.1038/nature25978. [DOI] [PubMed] [Google Scholar]
- Corey E. J.The logic of chemical synthesis; John Wiley & Sons, 1991. [Google Scholar]
- Sutton R.; Barto A.. Reinforcement learning: an introduction, 2nd ed.; MIT Press, 2017. [Google Scholar]
- Mnih V.; et al. Human-level control through deep reinforcement learning. Nature 2015, 518, 529–533. 10.1038/nature14236. [DOI] [PubMed] [Google Scholar]
- Coley C. W.; Jin W.; Rogers L.; Jamison T. F.; Jaakkola T. S.; Green W. H.; Barzilay R.; Jensen K. F. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci. 2019, 10, 370–377. 10.1039/C8SC04228D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller P.; Laino T.; Gaudin T.; Bolgar P.; Bekas C.; Lee A. A.. Molecular Transformer for chemical reaction prediction and uncertainty estimation. 2018, arXiv:1811.02633. arXiv.org e-Print archive. https://arxiv.org/abs/1811.02633.
- Sigma-Aldrich, Inc. https://www.sigmaaldrich.com (accessed Dec 17, 2018).
- E-molecules . https://www.emolecules.com/info/plus/download-database (accessed Dec 17, 2018).
- LabNetwork Collections . https://www.labnetwork.com/frontend-app/p/#!/screening-sets (accessed Dec 17, 2018).
- Rogers D.; Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- Shi F.; Xing G.-J.; Tao Z.-L.; Luo S.-W.; Tu S.-J.; Gong L.-Z. An asymmetric organocatalytic povarov reaction with 2-hydroxystyrenes. J. Org. Chem. 2012, 77, 6970–6979. 10.1021/jo301174g. [DOI] [PubMed] [Google Scholar]
- Klucznik T.; et al. Efficient syntheses of diverse, medicinally relevant targets planned by computer and executed in the laboratory. Chem. 2018, 4, 522–532. 10.1016/j.chempr.2018.02.002. [DOI] [Google Scholar]
- Liu B.; Ramsundar B.; Kawthekar P.; Shi J.; Gomes J.; Luu Nguyen Q.; Ho S.; Sloane J.; Wender P.; Pande V. Retrosynthetic reaction prediction using neural sequence-to-sequence models. ACS Cent. Sci. 2017, 3, 1103–1113. 10.1021/acscentsci.7b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcou G.; Aires de Sousa J.; Latino D. A.; de Luca A.; Horvath D.; Rietsch V.; Varnek A. Expert system for predicting reaction conditions: the Michael reaction case. J. Chem. Inf. Model. 2015, 55, 239–250. 10.1021/ci500698a. [DOI] [PubMed] [Google Scholar]
- Lin A. I.; Madzhidov T. I.; Klimchuk O.; Nugmanov R. I.; Antipin I. S.; Varnek A. Automatized assessment of protective group reactivity: a step toward big reaction data analysis. J. Chem. Inf. Model. 2016, 56, 2140–2148. 10.1021/acs.jcim.6b00319. [DOI] [PubMed] [Google Scholar]
- Segler M. H.; Waller M. P. Modelling chemical reasoning to predict and invent reactions. Chem. - Eur. J. 2017, 23, 6118–6128. 10.1002/chem.201604556. [DOI] [PubMed] [Google Scholar]
- Gao H.; Struble T. J.; Coley C. W.; Wang Y.; Green W. H.; Jensen K. F. Using machine learning to predict suitable conditions for organic reactions. ACS Cent. Sci. 2018, 4, 1465–1476. 10.1021/acscentsci.8b00357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver D.; et al. Mastering the game of Go with deep neural networks and tree search. Nature 2016, 529, 484–489. 10.1038/nature16961. [DOI] [PubMed] [Google Scholar]
- Butina D. Unsupervised data base clustering based on daylight’s fingerprint and Tanimoto similarity: a fast and automated way to cluster small and large data sets. J. Chem. Inf. Comput. Sci. 1999, 39, 747–750. 10.1021/ci9803381. [DOI] [Google Scholar]
- Glen R. C.; Bender A.; Arnby C. H.; Carlsson L.; Boyer S.; Smith J. Circular fingerprints: flexible molecular descriptors with applications from physical chemistry to ADME. IDrugs 2006, 9 (3), 199–204. [PubMed] [Google Scholar]
- Coley C. W.; Barzilay R.; Jaakkola T. S.; Green W. H.; Jensen K. F. Prediction of organic reaction outcomes using machine learning. ACS Cent. Sci. 2017, 3, 434–443. 10.1021/acscentsci.7b00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RDKit : Open-source cheminformatics. http://www.rdkit.org (accessed Dec 17, 2018).
- Coley C. W.RDChiral. https://github.com/connorcoley/rdchiral (accessed July 18, 2018).
- Coley C. W.Retrotemp. https://github.com/connorcoley/retrotemp/tree/master/retrotemp (accessed Dec 17, 2018).
- Schreck J. S.retroRL. https://github.com/jsschreck/retroRL.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.