

Abstract
A new series of 5,5-diphenylhydantoin derivatives containing benzylidene or isatin (4–19) was synthesized. Their anticancer activity against HeLa, a cervical cancer cell line, A549, a lung cancer cell line, and MDA-MB-231, a breast cancer cell line, was evaluated. Compounds 13, 16, 17 and 18 exhibited potent anticancer activity with average IC50 values against the tested cell lines of 109, 59, 81 and 113 μM, respectively. Compound 16 showed potent EGFR and VEGFR2 inhibitory activity with IC50 values of 6.17 and 0.09 μM, respectively. In addition, compound 16 induced caspase-dependent apoptosis and reactive oxygen species (ROS) production at 5 and 10 μM. Moreover, a molecular docking simulation was performed for compound 16 and sunitinib to predict the protein-ligand interactions with the active site of VEGFR2.
Keywords: 5,5-Diphenylhydantoin; Isatin; Apoptosis; EGFR; VEGFR2; Molecular docking
1. Introduction
Cancer is one of the most critical public health issues and a leading cause of human death worldwide (Senwar et al., 2016, Weir et al., 2016). Many of currently available anticancer agents lack sufficient selectivity towards cancer cells, causing a wide range of toxic side effects. Therefore, the development of more effective anticancer agents with improved pharmacokinetic properties is an urgent medical need.
Imidazolidine-2,4-dione (hydantoin) is a versatile scaffold that exerts a wide range of pharmacological effects such as anticancer activity and inhibition of COX-1/2 and carbonic anhydrase (Abdel-Aziz et al., 2016, Abdel-Aziz et al., 2015, Alanazi et al., 2013, Zuliani et al., 2009, Zhang et al., 2017, Azizmohammadi et al., 2013, Mostafa et al., 2016, Hmuda et al., 2014). A 5,5-diphenylimidazolidine-2,4-dione derivative (I) displayed a promising growth inhibitory activity against several distinct cancer cell lines at 10 μM (Fig. 1) (Alanazi et al., 2013). The proposed molecular mechanism of anticancer activity was mediated by EGFR kinase inhibition (Alanazi et al., 2013). Another imidazolidine-2,4-dione derivative (II) showed good antiproliferative activity against the lung cancer cell line A549 (Fig. 1) (Zuliani et al., 2009) and inhibited autophosphorylation of epidermal growth factor receptor (EGFR) in the same cell line at an IC50 of 19 μM (Zuliani et al., 2009).
Fig. 1.

Reported and proposed 5,5-diphenylimidazolidine-2,4-dione–isatin conjugates with anticancer and protein kinase inhibitor activity.
Isatin-containing compounds and their Schiff’s bases are known to possess anticancer activity (Senwar et al., 2016, Teng et al., 2016, Havrylyuk et al., 2011, Evdokimov et al., 2016, Ali et al., 2014, Karthikeyan et al., 2013, Eldehna et al., 2015, Solomon et al., 2010, Solomon et al., 2009, Arun et al., 2013). Sunitinib (III) is an isatin-containing drug approved by the FDA for the treatment of renal cell carcinoma (RCC) and gastrointestinal stromal tumor (GIST) (Fig. 1) (Motzer et al., 2006, Prenen et al., 2006). Sunitinib is a receptor tyrosine kinase (RTK) inhibitor that blocks the vascular endothelial growth factor receptor 2 (VEGFR2) with an IC50 value of 80 nM (Sun et al., 2003). Another isatin derivative (IV) is a potent growth inhibitor of colorectal and breast cancer cell lines at an average IC50 of 10 μM that induces apoptosis at the same concentration (Fig. 1) (El-Azab et al., 2017). Moreover, this compound showed good inhibitory activity against cellular EGFR at 10 μM (El-Azab et al., 2017).
In this study, we sought to develop effective cytotoxic agents by synthesizing novel hybrid derivatives 14–19 of 5,5-diphenylimidazolidine-2,4-dione conjugated to 5-substituted isatin that were evaluated for their anticancer activity. Moreover, to identify the most potent compound among the new derivatives, they were tested for their activity as EGFR and VEGFR2 kinase inhibitors and as apoptosis and caspase inducers. Molecular docking studies were performed to predict the structural determinants for the kinase inhibitor activity toward the target molecules.
2. Results and discussion
2.1. Chemistry
The synthetic route for target compounds 4–19 is presented in Scheme 1. The structures of target compounds 4–19 were confirmed by IR and NMR spectroscopy as well as mass spectrometry. To generate ethyl (5,5-diphenylhydantoin-3-yl)acetate (2) as reported previously, 5,5-diphenylhydantoin (1) and ethyl bromoacetate were heated in acetone containing anhydrous K2CO3 (Handzlik et al., 2012, Abdel-Aziz et al., 2016). The resulting ester was combined with hydrazine in refluxing ethanol to yield a hydrazide derivative (3) (Botros et al., 2012, Abdel-Aziz et al., 2016) that was reacted with either an appropriate benzaldehyde derivative or 3-formylindole to produce Schiff’s bases 4–12 or 13, respectively, according to the method reported previously (Abdel-Aziz et al., 2016). Similarly, Schiff’s bases 14–19 were prepared by reacting 3 with a suitable isatin derivative.
Scheme 1.

Synthesis of 5,5-diphenylimidazolidine-2,4-dione conjugated to isatin, benzaldehyde or 3-formylindole. Reagents and conditions: (a) ethyl bromoacetate, K2CO3, acetone, reflux, 24 h; (b) N2H4, EtOH, rt, 24 h; (c) appropriate benzaldehyde or isatin, AcOH, MeOH, reflux, 6 h.
2.2. Biological activity
2.2.1. Cytotoxicity assay
The Alamar Blue assay was performed in vitro to assess the cytotoxic effect of 2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetamide derivatives along with the reference compound docetaxel on HeLa, human lung adenocarcinoma (A549), and human breast adenocarcinoma (MDA-MB-231) cell. After treating the cancer cells for 24 h, most compounds exhibited a promising antiproliferative activity (Table 1).
Table 1.
Structures of compounds 2–19 and their cytotoxic activity (IC50) against human cancer cell lines.
 | ||||
|---|---|---|---|---|
| Compound | R | 24 h IC50 (μM) |
||
| HeLa | A549 | MDA-MB-231 | ||
| 2 | – | 241 ± 1.3 | 108 ± 3.7 | 263 ± 3.4 |
| 3 | – | 236 ± 3.5 | 278 ± 7.5 | 278 ± 2 |
| 4 | 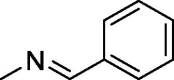 |
170 ± 1.3 | 350 ± 5.2 | 240 ± 0.9 |
| 5 | 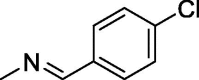 |
123 ± 2.6 | 157 ± 1.4 | 196 ± 1.7 |
| 6 | 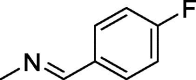 |
168 ± 1.2 | 347 ± 7.2 | 173 ± 2 |
| 7 | 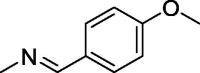 |
123 ± 1.6 | >350 | 150 ± 2.4 |
| 8 | 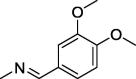 |
60 ± 3.3 | 162 ± 2.3 | 154 ± 1.1 |
| 9 | 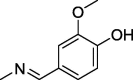 |
50 ± 1 | 175 ± 4.3 | 136 ± 3.9 |
| 10 | 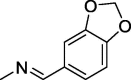 |
113 ± 0.9 | 267 ± 6 | 200 ± 4.1 |
| 11 | 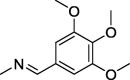 |
53 ± 2.1 | 170 ± 4.2 | 133 ± 0.7 |
| 12 | 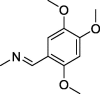 |
39 ± 3.1 | 116 ± 1.3 | >350 |
| 13 | 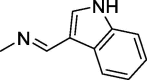 |
10 ± 2.1 | 160 ± 0.8 | 156 ± 3.1 |
| 14 | 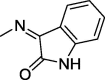 |
30 ± 2.6 | 116 ± 1.6 | 123 ± 0.8 |
| 15 | 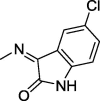 |
53 ± 2.2 | 72 ± 3 | 311 ± 5.5 |
| 16 | 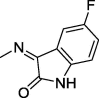 |
18.5 ± 1.6 | 95 ± 1 | 64 ± 0.6 |
| 17 | 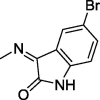 |
29 ± 1.7 | 77 ± 1.5 | 138 ± 2.6 |
| 18 | 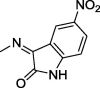 |
30 ± 1.2 | 229 ± 5.2 | 80 ± 0.7 |
| 19 | 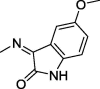 |
118 ± 1.3 | 170 ± 2.2 | 174 ± 2.9 |
| Docetaxel | – | 100 ± 3.8 | 50 ± 1.3 | 100 ± 2.2 |
Compounds 8, 9 and 11–18 showed high cytotoxic activity against HeLa cells (IC50: 10–60 μM); the comparative IC50 value for docetaxel was 100 μM. On the other hand, compounds 2–7, 10 and 19 were less active than docetaxel with IC50 values 114–241 μM. A549 cells, however, were significantly less sensitive to all of the tested compounds (IC50: 72–350 μM); the comparative IC50 value for docetaxel was 100 μM. Finally, only compounds 16 and 18 showed more potent cytotoxic activity against MDA-MB-231 cells than docetaxel with IC50 values of 64, 80 and 100 μM, respectively. Overall, the results indicated that compound 16 is the most potent compound with average IC50 against the tested cell lines of 59 μM whereas the average IC50 of docetaxel is 83 μM.
Generally, the vast majority of the tested compounds showed promising antiproliferative activity with HeLa cells being most sensitive cell line (average IC50 = 93 μM). The cytotoxic activity was improved upon introducing indole or isatin at R. In contrast, the derivatives with phenyl groups exerted a cytotoxic activity comparable to docetaxel only against HeLa cells whereas their IC50 values were significantly higher than docetaxel against A549 and MDA-MB-231. Substitution on the phenyl ring (compounds 5–12) improved the anticancer activity against HeLa cells (IC50: 39–168 μM) when compared with the unsubstituted derivative 4 (IC50: 170 μM). Moreover, compounds 8–12 with di- and trisubstituion showed further improvement in the anticancer activity against the same cells (IC50: 39–113 μM).
Schiff’s bases of indole and isatins 13–19 exhibited significant improvement in anticancer activity against HeLa cells when compared to compounds 4–12 with compound 13 being the most cytotoxic agent. Introducing electron-donating group to 5-position of isatin as in 19 abolished the cytotoxic activity (IC50: 118 μM) when compared to compounds 14–18 with electron-withdrawing groups or no substitution at the same position (IC50: 18.5–53 μM).
The results of the cytotoxicity test were promising for an investigation of the possible mechanism of the anticancer activity exerted by compounds 16 and 18.
2.2.2. Kinase inhibitory activity and molecular docking studies
EGFR and VEGFR2 inhibition assays were used to assess the RTK inhibitory activity of compounds 13, 16 and 18 (Table 2). Compounds 13 and 18 exhibited a potent inhibitory activity against EGFR with IC50 values of 0.10 and 0.37 μM. The substitution of the nitro group at position 5 in compound 18 by a fluorine group in compound 16 significantly reduced the inhibitory activity against EGFR. Interestingly, compound 16 exhibited a promising inhibitory activity against VEGFR2 with an IC50 of 0.09 μM whereas compounds 13 and 18 inhibited the same receptor in the micromolar ranges.
Table 2.
Summary of in vitro evaluation of compounds 13, 16 and 18.
 | ||||
|---|---|---|---|---|
| Compound | R | Kinase inhibition IC50 (μM) |
HeLa IC50 (μM) | |
| EGFR | VEGFR2 | |||
| 13 | – | 0.10 ± 0.02 | 0.47 ± 0.09 | 10 ± 2.1 |
| 16 | F | 6.17 ± 0.22 | 0.09 ± 0.01 | 18.5 ± 1.6 |
| 18 | NO2 | 0.37 ± 0.01 | 2.52 ± 0.09 | 30 ± 1.2 |
| Erlotinib | – | 0.10 ± 0.01 | 0.004 ± 0.11 | NT |
| Sorafenib | – | 0.06 ± 0.004 | 0.005 ± 0.001 | NT |
The anticancer activity along with the inhibition of VEGFR2 and EGFR by compound 16 and compound 18, respectively, prompted us to investigate the possible binding interactions of compound 16 and compound 18 with the active site of VEGFR2 and EGFR, respectively, by performing molecular docking simulation using Autodock vina.
We assumed that compound 16 exerts its anticancer activity by inhibiting VEGFR2. The crystal structure of VEGFR2 in complex with sunitinib (PDB code: 4AGD) was obtained from the protein data bank. The potential interactions along with the alignment of compound 16 and the original inhibitor sunitinib were investigated by docking compound 16 into the active site of VEGFR2 (Fig. 2A). The binding energies of compound 16 and sunitinib docked into the active site of VEGFR2 were −9.8 and −9.7 kcal/mol, respectively. The docking simulation predicts that the isatin group of compound 16 is overlaying the isatin group of sunitinib in the hydrophobic binding pocket of VEGFR2. Furthermore, like the isatin group of sunitinib, the amide substituent on the isatin group of compound 16 is making two hydrogen bonds with GLU 917 and CYS 919. The hydantoin moiety in compound 16 is making two hydrogen bonds with LEU 840 and ASN 923, indicating that the hydantoin moiety has a different orientation than the other parts of sunitinib. Fig. 2B presents the pose and binding mode of compound 16 at the active site of VEGVR2 that supports the hypothesis that compound 16 may inhibit the same target enzyme as sunitinib.
Fig. 2.

A. 3D alignment of compound 16 (blue) and sunitinib (green) in the active site of VEGFR2. B. 2D presentation of the interactions between compound 16 and the active site of VEGFR2. Hydrogen bonds are indicated as green dotted lines.
To predict the protein-ligand binding properties of compound 18, the crystal structure of EGFR in complex with gefitinib (PDB code: 2ITY) was obtained from the protein data bank. The potential interactions along with the alignment of compound 18 and the original inhibitor gefitinib were simulated by docking compound 18 into the active site of EGFR (Fig. 3A). Based on the results of the anticancer activity tests and the EGFR assay, we assumed that compound 18 exerts its anticancer activity by inhibiting EGFR through binding. The binding energies of compound 18 and gefitinib docked into the active site of EGFR were −9.6 and −7.8 kcal/mol, respectively. Fig. 3B shows that the hydantoin moiety of compound 18 is occupying the hydrophobic pocket of EGFR by making hydrophobic interactions with LEU 714, LEU 844, ALA 743, and VAL 726, whereas the isatin moiety forms hydrogen bonds with SER 720, GLY 721, GLY 719, and LYS 745. It appears that the nitro group on position 5 in compound 18 reduces the preference of the isatin moiety for the hydrophobic pocket. Hence, these docking simulations predict why compound 18 is selective for EGFR and compound 16 is selective for VEGFR2.
Fig. 3.

A. 3D alignment of compound 18 (blue) and gefitinib (violet) in the active site of EGFR. B. 2D presentation of the interactions between compound 18 and the active site of EGFR. Hydrogen bonds are indicated as green dotted lines.
Molecular docking of compound 4, the least active cytotoxic agent, into the active site of EGFR and VEGFR2 was performed to investigate the potential differences in orientation and type of interactions in comparison to compound 16 and compound 18 on VEGFR2 and EGFR, respectively. The molecular docking results showed that compound 4 is making only one hydrogen bond with VEGFR2 (Fig. 4A and B) while no hydrogen bond was found with EGFR (Fig. 4C and D). This can probably explain the low potency of compound 4 on the three cell lines in comparison to compounds 16 and 18.
Fig. 4.

A. 3D alignment of compound 4 (blue) and sunitinib (green) in the active site of VEGFR2. B. 2D presentation of the interactions between compound 4 and the active site of VEGFR2. C. 3D alignment of compound 4 (blue) and gefitinib (violet) in the active site of EGFR. D. 2D presentation of the interactions between compound 4 and the active site of EGFR. Hydrogen bonds are indicated as green dotted lines.
2.2.3. Induction of apoptosis and caspase-3/7, -8, and -9
Cell death induced by anticancer agents can occur by either apoptosis or by necrosis. This was assessed for compound 16 using the annexin V/PI assay after treating HeLa cells with compound 16 for 24 h. Fig. 5 shows that the percentage of apoptotic cells after treatment with 5 and 10 µM of compound 16 were 48.6% and 63.4%, respectively.
Fig. 5.

The percentage of HeLa cells undergoing apoptosis after a 24 h treatment without compound 16 (Control, upper panel) or with 5 and 10 μM of compound 16 (lower right and left panel, respectively).
The activity of caspases -3/7, -8, and -9 in HeLa cells was assayed after 24 h of treatment with 0, 5, and 10 µM of compound 16 (Fig. 6). There were no significant changes in the activity of caspase-8 and -9. However, for the executioner caspase activity (caspase-3/7), the results show that the 24 h treatment of HeLa cells with 10 µM of compound 16 caspase-3/7 activity by approximately 40%.
Fig. 6.

Levels of caspase -3/7, -8, and -9 in HeLa cells after treatment with 5 and 10 μM of compound 16 for 24 h.
2.2.4. Reactive oxygen species (ROS) generation
Reactive oxygen species (ROS) play a significant role in cell regulation, activation of signaling cascades, and apoptosis. Changes in ROS levels were measured after a 24 h incubation of cells with 0, 10, and 40 µM of compound 16. The principle behind the determination of HeLa cells undergoing oxidative stress is based on the intracellular detection of superoxide radicals. After the 24 h incubation, we detected an increased level of ROS production after treatment with 10 µM and 40 µM of 16 in 68.8% and 37.7% of cells, respectively (Fig. 7).
Fig. 7.

Measurement of ROS level in response to 16. Level of ROS in control (A), 10 µM (B), and 40 µM (C).
3. Conclusion
A series of fifteen derivatives (4–19) was designed, synthesized, and evaluated in vitro for anticancer activity and EGFR and VEGFR2 inhibitory activity. Compound 16 showed potent VEGFR2 inhibitory activity (IC50 = 0.09 μM), whereas compounds 13 and 18 were more potent against EGFR (IC50 = 0.10 and 0.37 μM, respectively) than compound 16. Compounds 13, 16, 17 and 18 showed strong cytotoxic activity against HeLa cells (IC50 values = 10, 18.5, 29 and 30 μM, respectively) in comparison to that of docetaxel (IC50 = 100 μM). Compound 16 induced caspase 3/7-dependent apoptosis and ROS production in HeLa cells.
4. Experimental
4.1. Chemistry
Melting points (uncorrected) and IR spectra were recorded on a Barnstead 9100 Electrothermal melting apparatus and an FT-IR Perkin-Elmer spectrometer, respectively. 1H and 13C NMR spectra were recorded in deuterated dimethyl sulfoxide (DMSO‑d6) on a Bruker 500 and a 125 MHz NMR spectrometer, respectively, using tetramethylsilane (TMS) as an internal standard (chemical shifts in ppm). Agilent 6320 Ion Trap mass spectrometer was used to record mass spectra. Compounds 2, 3, 4, 6, and 7 were prepared as reported previously (Abdel-Aziz et al., 2016).
4.1.1. General procedure for the synthesis of compounds 4–13
A solution of 2-(2,4-dioxo-5,5-diphenylimidazolidin-3-yl)acetohydrazide (3) (1.0 mmol, 0.324 g) and an appropriate benzaldehyde (1.1 mmol) in dry methanol (10 mL) was refluxed for 6 h. The reaction mixture was cooled and the formed precipitate recovered by filtration, dried, and recrystallized from an appropriate solvent.
4.1.1.1. N'-(4-Chlorobenzylidene)-2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetohydrazide (5)
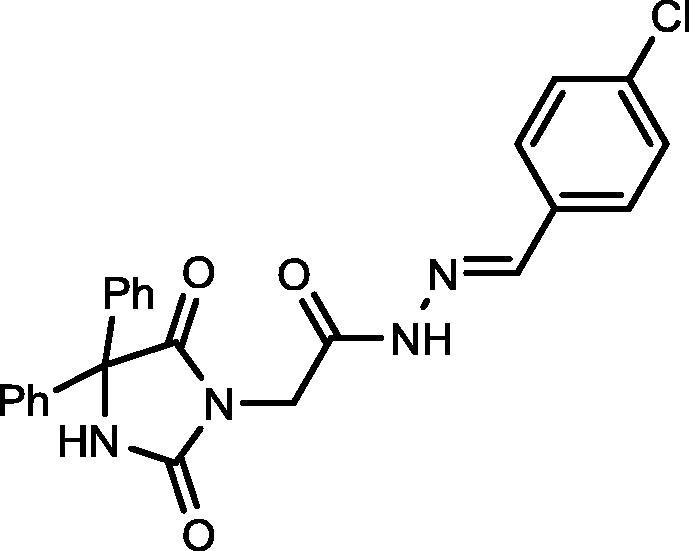
Yield, 96%; mp: >350 °C; IR (KBr) νmax/cm−1: 3412, 3229 (NH), 1773, 1713 (C O); 1H NMR (500 MHz, DMSO‑d6): δ 11.75 (s, 1H), 9.71 (s, 1H), 8.19 (s, 0.27H), 8.03 (s, 0.73H), 7.74 (d, 2H, J = 8.5 Hz), 7.52–7.37 (m, 12H), 4.62 (s, 1.49H), 4.25 (s, 0.51H); 13C NMR (DMSO‑d6): δ 69.58, 126.93, 128.19, 128.44, 128.58, 128.74, 128.83, 128.88, 132.77, 132.90, 134.49, 134.67, 139.49, 139.56, 143.17, 145.99, 154.89, 155.03, 162.76, 167.38, 173.45; C24H19ClN4O3: m/z (446).
4.1.1.2. N′-(3,4-Dimethoxybenzylidene)-2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetohydrazide (8)
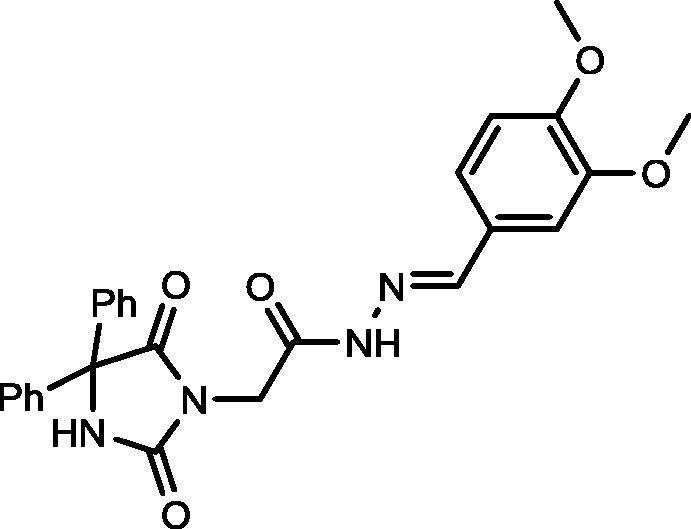
Yield, 91%; mp: 310-312 °C; IR (KBr) νmax/cm−1: 3414, 3230 (NH), 1778, 1732 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.61 (s, 0.22H), 11.50 (s, 0.78H), 9.58 (s, 1H), 8.11 (s, 0.22H), 7.94 (s, 0.78H), 7.49 (d, 2H, J = 7.5 Hz), 7.47 (d, 2H, J = 7.0 Hz), 7.39–7.31 (m, 7H), 7.12 (d, 0.22H, J = 8.5), 7.11 (d, 0.78H, J = 8.5 Hz), 6.93 (dd, 1H, J = 8.0 & 7.5 Hz), 4.65 (s, 1.56H), 4.25 (s, 0.44H), 3.82 (dd, 6H, J = 1.5 Hz); 13C NMR (DMSO‑d6-CDCl3): δ 55.38, 55.44, 69.59, 108.04, 111.02, 121.51, 126.57, 126.70, 127.90, 128.13, 139.51, 139.57, 144.33, 148.99, 150.56, 155.12, 166.93, 173.42, 173.51; C26H24N4O5: m/z (472).
4.1.1.3. 2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(4-hydroxy-3-methoxybenzylidene)acetohydrazide (9)
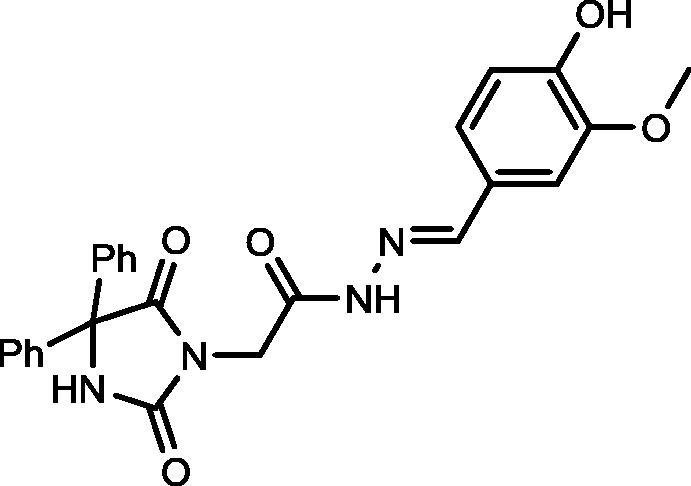
Yield, 85%; mp: 348-350 °C; IR (KBr) νmax/cm−1: 3362, 3176 (NH), 1780, 1715 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.59 (s, 0.25H), 11.46 (s, 0.75H), 9.61 (s, 1H), 9.41 (s, 1H), 8.07 (s, 0.25H), 7.92 (s, 0.75H), 7.46–7.30 (m, 11H), 7.07–7.02 (m, 1H), 6.84–6.81 (m, 1H), 4.62 (s, 1.75H), 4.23 (s, 0.25H), 3.83 (s, 3H); 13C NMR (DMSO‑d6-CDCl3): δ 48.67, 55.46, 69.55, 108.83, 115.23, 121.69, 122.19, 125.33, 126.56, 127.00, 127.96, 128.20, 139.59, 144.71, 147.72, 147.90, 148.82, 149.04, 154.92, 155.09, 162.23, 166.84, 173.41, 173.50; C25H22N4O5: m/z (458).
4.1.1.4. N′-(Benzo[d][1,3]dioxol-5-ylmethylene)-2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetohydrazide (10)
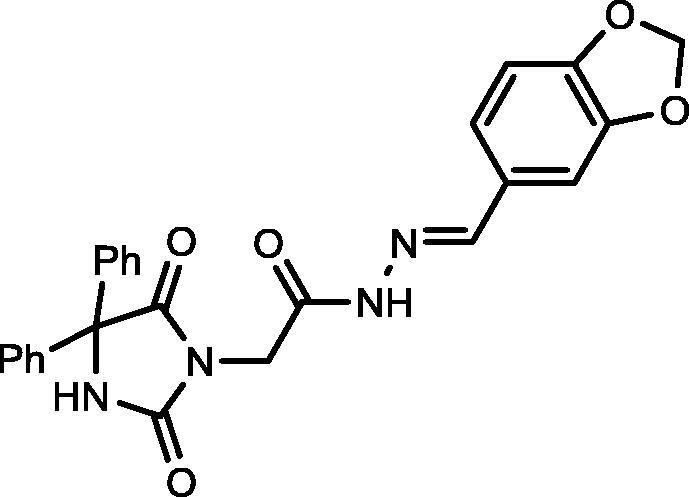
Yield, 87%; mp: >350 °C; IR (KBr) νmax/cm−1: 3413, 3243 (NH), 1775, 1721 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.77 (s, 0.22H), 11.65 (s, 0.78H), 9.62 (s, 1H), 8.19 (d, 0.22H, J = 8.5 Hz), 8.02 (s, 0.78H), 7.73 (s, 2H), 7.45–7.36 (m, 11H), 7.18 (d, 2H, J = 8.5 Hz), 4.61 (s, 1.40H), 4.23 (s, 0.60H); 13C NMR (DMSO‑d6-CDCl3): δ 69.55, 115.51, 115.68, 126.97, 127.94, 128.20, 128.87, 128.93, 129.19, 130.39, 139.58, 143.12, 146.05, 154.86, 155.02, 162.04, 162.58, 164.01, 167.14, 173.43; C25H20N4O5: m/z (456).
4.1.1.5. 2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(3,4,5-trimethoxybenzylidene)acetohydrazide (11)
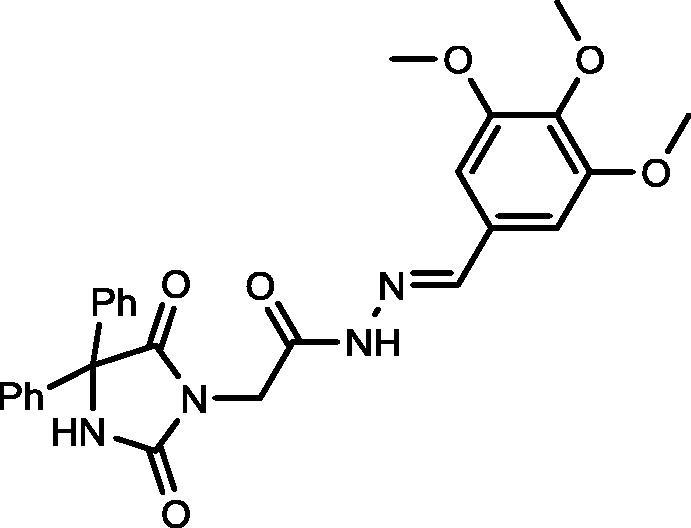
Yield, 95%; mp: 346-348 °C; IR (KBr) νmax/cm−1: 3414, 3310 (NH), 1772, 1717, 1700 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.51 (s, 0.2H), 11.60 (s, 0.8H), 9.58 (s, 1H), 8.10 (s, 0.2H), 7.935 (s, 0.8H), 7.45 (d, 4H, J = 7.5 Hz), 7.37–7.31 (m, 6H), 6.99 (s, 0.4H), 6.95 (s, 1.6H), 4.64 (s, 1.6H), 4.25 (s, 0.4H), 3.82 (s, 6H), 3.74 (d, 3H, J = 4.0 Hz); 13C NMR (DMSO‑d6-CDCl3): δ 55.77, 60.02, 69.59, 104.01, 104.35, 126.55, 126.99, 127.89, 128.12, 129.313, 139.14, 139.34, 139.48, 144.18, 147.28, 153.03, 155.10, 167.09, 173.39, 173.48; C27H26N4O6: m/z (502).
4.1.1.6. 2-(2,5-]ioxo-4,4-diphenylimidazolidin-1-yl)-N′-(2,4,5-trimethoxybenzylidene)acetohydrazide (12)
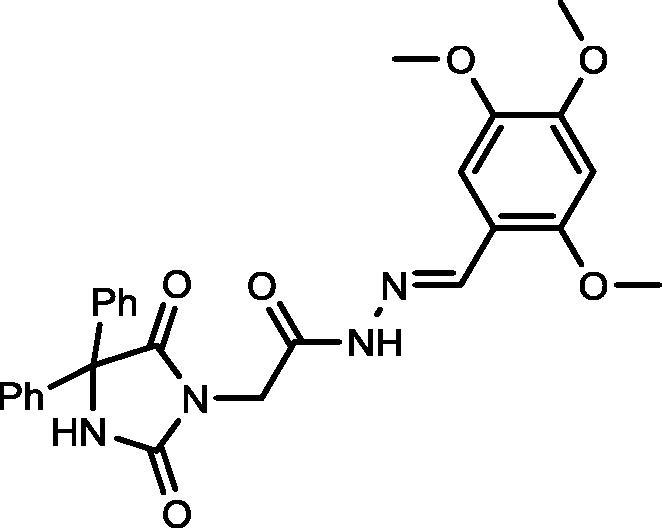
Yield, 93%; mp: >350 °C; IR (KBr) νmax/cm−1: 3545, 3214 (NH), 1772, 1700 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.57 (s, 0.25H), 11.356 (s, 0.75H), 9.53 (s, 0.25H), 9.51 (s, 0.75H), 8.44 (s, 0.25H), 8.28 (s, 0.75H), 7.45–7.43 (m, 3H), 7.36–7.29 (m, 8H), 6.60 (d, 1H, J = 5 Hz), 4.60 (s, 1.5H), 4.19 (s, 0.50H), 3.85 (d, 3H, J = 3.0 Hz), 3.82 (d, 3H, J = 6.5 Hz), 3.75 (d, 3H, J = 4.0 Hz); 13C NMR (DMSO‑d6-CDCl3): δ 55.62, 55.88, 56.19, 69.5779.14, 107.83, 108.03, 113.27, 113.46, 127.01, 127.82, 128.04, 139.44, 139.50, 140.09, 143.10, 151.97, 153.10, 155.15, 162.08, 166.69, 173.52; C27H26N4O6: m/z (502).
4.1.1.7. N′-((1H-indol-3-yl)methylene)-2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetohydrazide (13)
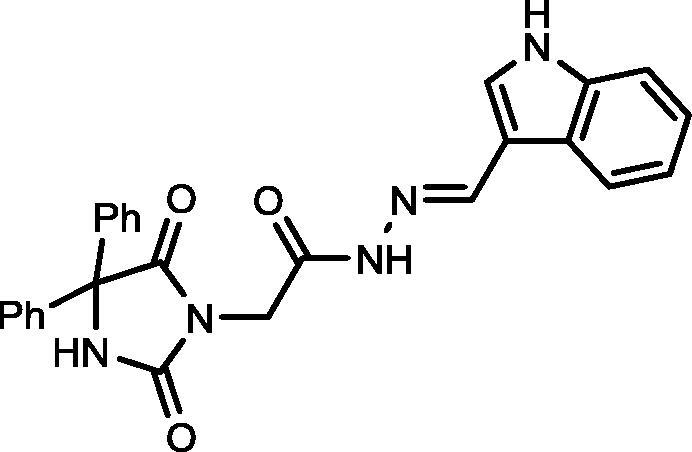
Yield, 84%; mp: >350 °C; IR (KBr) νmax/cm−1: 3364, 3253 (NH), 1774, 1701 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.77 (s, 1H), 10.75 (s, 1H), 9.53 (s, 1H), 8.26 (s, 1H), 7.44–7.27 (m, 14H), 6.85–6.79 (m, 1H), 4.64 (s, 2H); 13C NMR (DMSO‑d6-CDCl3): δ 69.69, 110.96, 112.28, 116.12, 126.33, 126.97, 127.48, 127.61, 127.88, 128.09, 128.52, 128.76, 139.41, 142.45, 154.88, 164.23, 173.40; C26H21N5O3: m/z (451).
4.1.2. General procedure for the synthesis of compounds 14–19
A solution of 2-(2,4-dioxo-5,5-diphenylimidazolidin-3-yl)acetohydrazide (3) (1.0 mmol) and an appropriate isatin (1.1 mmol) in dry methanol (10 mL) and glacial acetic acid (1 mL) was refluxed for 12 h. The resulting mixture was cooled and the formed precipitate was filtered, dried, and recrystallized from an appropriate solvent.
4.1.2.1. 2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(2-oxoindolin-3-ylidene)acetohydrazide (14)
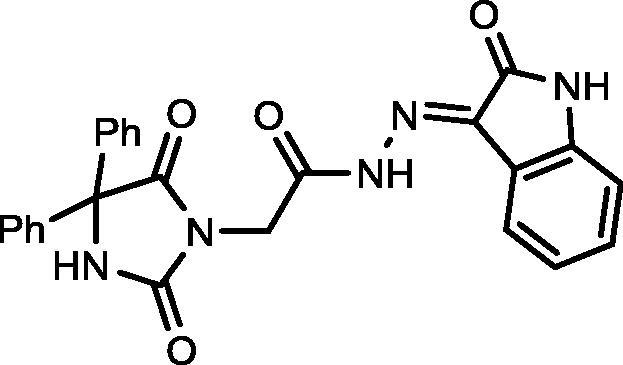
Yield, 91%; mp: 340-342 °C; IR (KBr) νmax/cm−1: 3413, 3263 (NH), 1769, 1732, 1715 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.77 (s, 1H), 10.85 (s, 1H), 9.62 (s, 1H), 8.43 (s, 1H), 7.44–6.82 (m, 13H), 4.69 (s, 2H); 13C NMR (DMSO‑d6-CDCl3): δ 69.69, 112.20, 112.87, 113.58, 116.56, 126.98, 127.94, 128.15, 128.35, 133.71, 134.59, 139.45, 142.95, 154.86, 164.00, 173.40; C25H19N5O4: m/z (453).
4.1.2.2. N′-(5-Chloro-2-oxoindolin-3-ylidene)-2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetohydrazide (15)
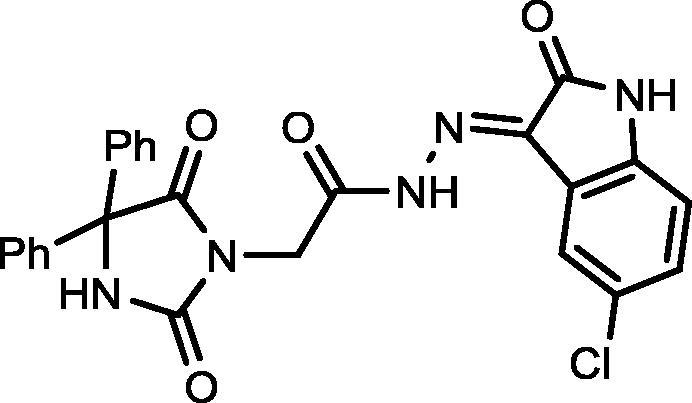
Yield, 88%; mp: 325-327 °C; IR (KBr) νmax/cm−1: 3414, 3254 (NH), 1773, 1731 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.71 (s, 0.25H), 11.59 (s, 0.75H), 9.56 (s, 1H), 8.17 (s, 0.25H), 8.01 (s, 0.75H), 7.68–7.64 (m, 2H), 7.45 (d, 4H, J = 6.5 Hz), 7.363–7.346 (m, 7H), 4.62 (s, 1.5H), 4.23 (s, 0.5H); 13C NMR (DMSO‑d6-CDCl3): δ 69.59, 126.55, 126.70, 126.97, 127.86, 128.10, 128.44, 129.68, 129.89, 133.80, 139.54, 144.23, 147.21, 154.89, 155.07, 162.56, 167.09, 173.45; C25H18ClN5O4: m/z (487, 489).
4.1.2.3. 2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(5-fluoro-2-oxoindolin-3-ylidene)acetohydrazide (16)
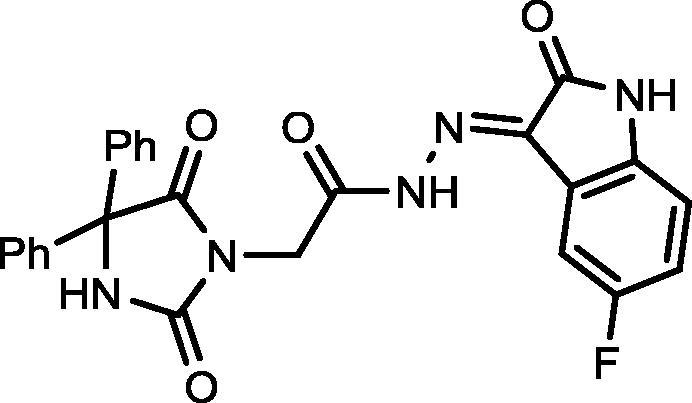
Yield, 84%; mp: 315-317 °C; IR (KBr) νmax/cm−1: 3466, 3198 (NH), 1776, 1715 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.52 (s, 1H), 10.69 (s, 1H), 9.57 (s, 1H), 7.40–7.27 (m, 10H) 7.22–7.15 (m, 1H), 7.12–7.07 (m, 1H), 6.83–6.79 (m, 1H), 4.73 (s, 2H); C25H18FN5O4: m/z (471).
4.1.2.4. N′-(5-Bromo-2-oxoindolin-3-ylidene)-2-(2,5-dioxo-4,4-diphenylimidazolidin-1-yl)acetohydrazide (17)
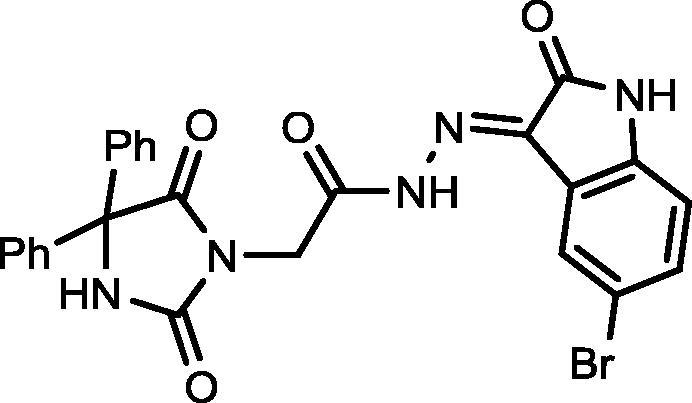
Yield, 90%; mp: 294-296 °C; IR (KBr) νmax/cm−1: 3414, 3264 (NH), 1771, 1732, 1716 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.51 (s, 1H), 10.76 (s, 1H), 9.65 (s, 1H), 8.11 (d, 1H, J = 7.5 Hz), 7.52–7.32 (m, 10H), 7.00 (s, 1H), 6.88 (d, 1H, J = 7.5 Hz), 4.71 (s, 2H); 13C NMR (DMSO‑d6-CDCl3): δ 69.69, 110.56,111.07, 115.06, 120.68, 121.53, 122.39, 126.09, 126.93, 126.99, 127.97, 128.19, 131.60, 132.50, 139.39, 139.47, 142.61, 143.96, 154.88, 164.36, 173.43; C25H18BrN5O4: m/z (532).
4.1.2.5. 2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(5-nitro-2-oxoindolin-3-ylidene)acetohydrazide (18)
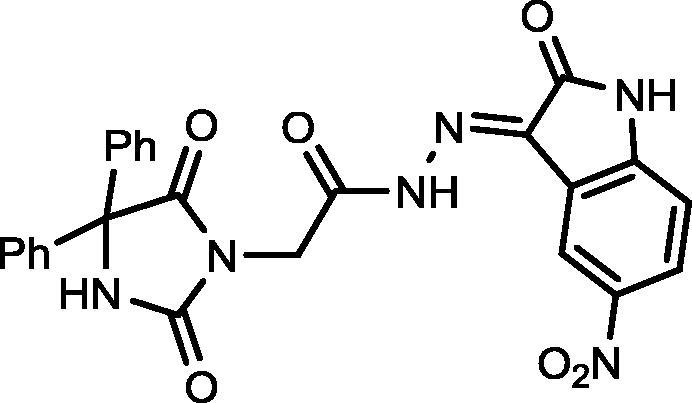
Yield, 81%; mp: 267-269 °C; IR (KBr) νmax/cm−1: 3413, 3234 (NH), 1792, 1773, 1717 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 12.51 (s, 1H), 11.43 (s, 1H), 11.83 (s, 1H), 9.68 (s, 0.5H), 9.65 (s, 0.5H), 9.14 (s, 0.5H), 8.32 (s, 0.5H), 8.27–8.23 (m, 1H), 7.46–7.31 (m, 4H), 7.39–7.31 (m, 5H), 7.10 (d, 0.5H, J = 8.5 Hz), 7.04 (d, 0.5H, J = 8.5 Hz), 4.84 (s, 1H), 4.71 (s, 1H); 13C NMR (DMSO‑d6-CDCl3): δ 110.47, 111.25, 114.83, 116.08, 121.69, 126.90, 126.97, 127.28, 127.97, 128.18, 139.45, 142.15, 142.85, 147.58, 149.27, 154.70, 164.59, 171.88, 173.23, 173.39; C25H18N6O6: m/z (498).
4.1.2.6. 2-(2,5-Dioxo-4,4-diphenylimidazolidin-1-yl)-N′-(5-methoxy-2-oxoindolin-3-ylidene)acetohydrazide (19)
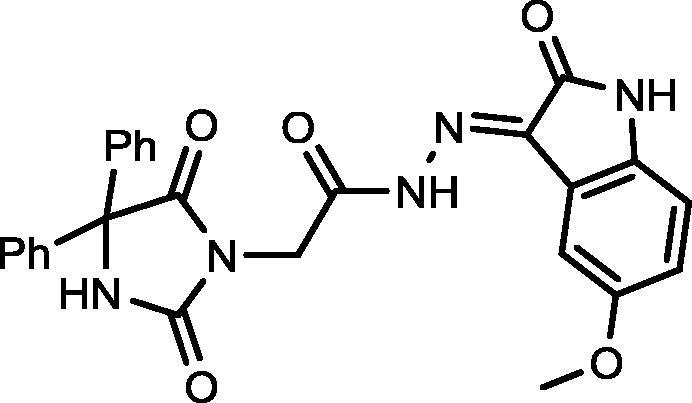
Yield, 82%; mp: 349-351 °C; IR (KBr) νmax/cm−1: 3545, 3214 (NH), 1772, 1700 (C O); 1H NMR (500 MHz, DMSO‑d6-CDCl3): δ 11.62 (s, 0.2H), 11.51 (s, 0.8H), 9.61 (s, 1H), 8.12 (s, 0.2H), 7.97 (s, 0.8), 7.65–7.61 (m, 2H), 7.45 (d, 4H, J = 6.5 Hz), 7.40–7.34 (m, 5H), 6.97–6.93 (m, 2H), 4.60 (s, 1.6H), 4.22 (s, 0.4H), 3.36 (d, 3H, J = 4.0 Hz); 13C NMR (DMSO‑d6-CDCl3): δ 55.11, 69.56, 114.05, 126.45, 126.99, 127.94, 128.20, 128.35, 128.63, 129.82, 139.54, 139.61, 144.15, 147.05, 154.90, 155.07, 160.70, 160.86, 162.30, 166.88, 173.47; C26H21N5O5: m/z (483).
4.2. Biological activity
4.2.1. Cytotoxicity assay
The cancer cell lines were maintained in DMEM supplemented with L-glutamine (GIBCO), 10% FBS (GIBCO), and 1% penicillin–streptomycin (GIBCO). Cells were cultured at 37 °C in a 5% CO2 incubator. Cells were recovered from cultures and seeded in growth medium into 96-flat well microtiter plates (1 × 104 cells/well) for a 24 h incubation at 37 °C in a 5% CO2 incubator. The test compounds were adjusted to test concentrations of 10, 25, 50, and 1000 μM/ml by dilution with the growth medium. Cells on microtiter plates were incubated for 24 h, medium was removed, and the test compound dilutions were added to the wells. Control wells with cells did not contain any test compound. After 24 h of treatment with test compounds, 10 µL of Alamar Blue reagent (AbD Serotec, UK) was added to each well (final concentration, 10 μg/ml). Then, the plates were incubated at 37 °C for 2–4 h. After incubation, plates were read using Spectra Max M5 plate reader (Molecular Devices, Inc.) at 560 nm emissions and 590 nm excitation for recording the fluorescence intensity. Cell viability was calculated using the formula:
4.2.1.1. Kinase inhibition assay
EGFR kinase inhibitory activity was determined according to the manufacturer's instructions (EGFR Kinase Assay Kit Catalog # ab126419 of ABCAM, Cambridge, MA) as reported previously (El-Husseiny et al., 2018). VEGFR2 kinase inhibitory activity was determined according to the manufacturer's instructions (VEGFR2 Kinase Assay Kit Catalog # 40352 of BPS Biosciences, San Diego, CA).
4.2.1.2. Caspase-3/7, -8, and -9 activity assay
Prior to treatment with 16, cells were seeded in 12-well plates (1 × 105/well) in duplicates and incubated for 24 h. Then, cells were then treated with 16 at concentrations of 1, 5, 10 and 20 µM and control cells were incubated at 0.1% of DMSO (v/v) without test compound. After a 24 h incubation, the caspase-3/7, -8, and -9 activities were measured using the Caspase Glo® 3/7, Caspase Glo® 8 and Caspase Glo® 9 assay kits (Promega, USA), respectively, according to manufacturer’s instruction.
4.2.1.3. Measurement of ROS production
Quantitative ROS measurements in HeLa cells undergoing oxidative stress after treatment with varying concentrations of 16 (10 and 40 µM) was performed using the Muse Oxidative Stress Kit (Merck Millipore) according to manufacturer’s procedure. Control cells were incubated with 0.1% DMSO (v/v). After a 24 h incubation, cells were analyzed on the Muse Cell Analyzer.
4.3. Molecular docking
The protein crystal structure data was obtained from the RCSB Protein Databank. The 3D-crystal structures used for docking are 4AGD (VEGFR2 crystal structure data of juxtamembrane and kinase domain in complex with sunitinib) and 2ITY (EGFR crystal structure data of kinase domain in complex with gefitinib). Software programs used for docking simulations were Discovery Studio, AutoDock Tools, Vina, and PyRx (The Scripps Research Institute, La Jolla, CA). First, using Discovery Studio, the protein crystal structure data was processed by eliminating all extra molecules such as water, ligand, and sulfate. The processed data was saved as in PDB file format. Second, polar hydrogens were introduced using AtuoDock Tools and the file was saved in PDBQT format. Third, the co-crystallized ligands were isolated and saved in PDB format using Discovery Studio. Compounds 16 and 18 were also saved in PDB file format. Grid box was used to suppress non-specific binding predictions, reduce processing time, and pinpoint the docking size and dimension. Finally, docking simulations were performed using PyRx; the lowest energy poses of the compounds were and compared with the co-crystallized ligand poses.
Footnotes
Peer review under responsibility of King Saud University.
References
- Abdel-Aziz A.A., El-Azab A.S., Abou-Zeid L.A., Eltahir K.E., Abdel-Aziz N.I., Ayyad R.R., Al-Obaid A.M. Synthesis, anti-inflammatory, analgesic and COX-1/2 inhibition activities of anilides based on 5,5-diphenylimidazolidine-2,4-dione scaffold: molecular docking studies. Eur. J. Med. Chem. 2016;115:121–131. doi: 10.1016/j.ejmech.2016.03.011. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz A.A., El-Azab A.S., Ekinci D., Senturk M., Supuran C.T. Investigation of arenesulfonyl-2-imidazolidinones as potent carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2015;30:81–84. doi: 10.3109/14756366.2014.880696. [DOI] [PubMed] [Google Scholar]
- Alanazi A.M., El-Azab A.S., Al-Swaidan I.A., Maarouf A.R., El-Bendary E.R., Abu El-Enin M.A., Abdel-Aziz A.A.M. Synthesis, single-crystal, in vitro antitumor evaluation and molecular docking of 3-substitued 5,5-diphenylimidazolidine-2,4-dione derivatives. Med. Chem. Res. 2013;22:6129–6142. [Google Scholar]
- Ali A.Q., Teoh S.G., Salhin A., Eltayeb N.E., Ahamed M.B.K., Majid A.M.S.A. Synthesis of platinum(II) complexes of isatin thiosemicarbazones derivatives: in vitro anti-cancer and deoxyribose nucleic acid binding activities. Inorg. Chim. Acta. 2014;416:235–244. [Google Scholar]
- Arun Y., Bhaskar G., Balachandran C., Ignacimuthu S., Perumal P.T. Facile one-pot synthesis of novel dispirooxindole-pyrrolidine derivatives and their antimicrobial and anticancer activity against A549 human lung adenocarcinoma cancer cell line. Bioorg. Med. Chem. Lett. 2013;23:1839–1845. doi: 10.1016/j.bmcl.2013.01.023. [DOI] [PubMed] [Google Scholar]
- Azizmohammadi M., Khoobi M., Ramazani A., Emami S., Zarrin A., Firuzi O., Miri R., Shafiee A. 2H-chromene derivatives bearing thiazolidine-2,4-dione, rhodanine or hydantoin moieties as potential anticancer agents. Eur. J. Med. Chem. 2013;59:15–22. doi: 10.1016/j.ejmech.2012.10.044. [DOI] [PubMed] [Google Scholar]
- Botros S., Khalil N.A., Naguib B.H., El-Dash Y. Phenytoin-based bivalent ligands: design, synthesis and anticonvulsant activity. Arch. Pharm. Res. 2012;35:2105–2116. doi: 10.1007/s12272-012-1207-3. [DOI] [PubMed] [Google Scholar]
- El-Azab A.S., Al-Dhfyan A., Abdel-Aziz A.A., Abou-Zeid L.A., Alkahtani H.M., Al-Obaid A.M., Al-Gendy M.A. Synthesis, anticancer and apoptosis-inducing activities of quinazoline-isatin conjugates: epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies. J. Enzyme Inhib. Med. Chem. 2017;32:935–944. doi: 10.1080/14756366.2017.1344981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Husseiny W.M., El-Sayed M.A., Abdel-Aziz N.I., El-Azab A.S., Ahmed E.R., Abdel-Aziz A.A. Synthesis, antitumour and antioxidant activities of novel alpha, beta-unsaturated ketones and related heterocyclic analogues: EGFR inhibition and molecular modelling study. J. Enzyme Inhib. Med. Chem. 2018;33:507–518. doi: 10.1080/14756366.2018.1434519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldehna W.M., Altoukhy A., Mahrous H., Abdel-Aziz H.A. Design, synthesis and QSAR study of certain isatin-pyridine hybrids as potential anti-proliferative agents. Eur. J. Med. Chem. 2015;90:684–694. doi: 10.1016/j.ejmech.2014.12.010. [DOI] [PubMed] [Google Scholar]
- Evdokimov N.M., Magedov I.V., Mcbrayer D., Kornienko A. Isatin derivatives with activity against apoptosis-resistant cancer cells. Bioorg. Med. Chem. Lett. 2016;26:1558–1560. doi: 10.1016/j.bmcl.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handzlik J., Bajda M., Zygmunt M., Maciag D., Dybala M., Bednarski M., Filipek B., Malawska B., Kiec-Kononowicz K. Antiarrhythmic properties of phenylpiperazine derivatives of phenytoin with alpha(1)-adrenoceptor affinities. Bioorg Med Chem. 2012;20:2290–2303. doi: 10.1016/j.bmc.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Havrylyuk D., Kovach N., Zimenkovsky B., Vasylenko O., Lesyk R. Synthesis and anticancer activity of isatin-based pyrazolines and thiazolidines conjugates. Arch. Pharm. (Weinheim) 2011;344:514–522. doi: 10.1002/ardp.201100055. [DOI] [PubMed] [Google Scholar]
- Hmuda S., Trisovic N., Rogan J., Poleti D., Vitnik Z., Vitnik V., Valentic N., Bozic B., Uscumlic G. New derivatives of hydantoin as potential antiproliferative agents: biological and structural characterization in combination with quantum chemical calculations. Monatsh. Chem. 2014;145:821–833. [Google Scholar]
- Karthikeyan C., Solomon V.R., Lee H., Trivedi P. Design, synthesis and biological evaluation of some isatin-linked chalcones as novel anti-breast cancer agents: a molecular hybridization approach. Biomed. Prev. Nutr. 2013;3:325–330. [Google Scholar]
- Mostafa A.A., Al-Rahmah A.N., Kumar R.S., Manilal A., Idhayadhulla A. biological evaluation of some imidazolidine-2,4-dione and 2-thioxoimidazolidin-4-one derivatives as anticoagulant agents and inhibition of MCF-7 breast cancer cell line. Int. J. Pharmacol. 2016;12:290–303. [Google Scholar]
- Motzer R.J., Michaelson M.D., Redman B.G., Hudes G.R., Wilding G., Figlin R.A., Ginsberg M.S., Kim S.T., Baum C.M., Deprimo S.E., Li J.Z., Bello C.L., Theuer C.P., George D.J., Rini B.I. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J. Clin. Oncol. 2006;24:16–24. doi: 10.1200/JCO.2005.02.2574. [DOI] [PubMed] [Google Scholar]
- Prenen H., Cools J., Mentens N., Folens C., Sciot R., Schoffski P., Van Oosterom A., Marynen P., Debiec-Rychter M. Efficacy of the kinase inhibitor SU11248 against gastrointestinal stromal tumor mutants refractory to imatinib mesylate. Clin. Cancer Res. 2006;12:2622–2627. doi: 10.1158/1078-0432.CCR-05-2275. [DOI] [PubMed] [Google Scholar]
- Senwar K.R., Reddy T.S., Thummuri D., Sharma P., Naidu V.G., Srinivasulu G., Shankaraiah N. Design, synthesis and apoptosis inducing effect of novel (Z)-3-(3′-methoxy-4′-(2-amino-2-oxoethoxy)-benzylidene)indolin-2-ones as potential antitumour agents. Eur. J. Med. Chem. 2016;118:34–46. doi: 10.1016/j.ejmech.2016.04.025. [DOI] [PubMed] [Google Scholar]
- Solomon V.R., Hu C., Lee H. Hybrid pharmacophore design and synthesis of isatin-benzothiazole analogs for their anti-breast cancer activity. Bioorg. Med. Chem. 2009;17:7585–7592. doi: 10.1016/j.bmc.2009.08.068. [DOI] [PubMed] [Google Scholar]
- Solomon V.R., Hu C., Lee H. Design and synthesis of anti-breast cancer agents from 4-piperazinylquinoline: a hybrid pharmacophore approach. Bioorg. Med. Chem. 2010;18:1563–1572. doi: 10.1016/j.bmc.2010.01.001. [DOI] [PubMed] [Google Scholar]
- Sun L., Liang C., Shirazian S., Zhou Y., Miller T., Cui J., Fukuda J.Y., Chu J.Y., Nematalla A., Wang X., Chen H., Sistla A., Luu T.C., Tang F., Wei J., Tang C. Discovery of 5-[5-fluoro-2-oxo-1,2- dihydroindol-(3Z)-ylidenemethyl]-2,4- dimethyl-1H-pyrrole-3-carboxylic acid (2-diethylaminoethyl)amide, a novel tyrosine kinase inhibitor targeting vascular endothelial and platelet-derived growth factor receptor tyrosine kinase. J. Med. Chem. 2003;46:1116–1119. doi: 10.1021/jm0204183. [DOI] [PubMed] [Google Scholar]
- Teng Y.O., Zhao H.Y., Wang J., Liu H., Gao M.L., Zhou Y., Han K.L., Fan Z.C., Zhang Y.M., Sun H., Yu P. Synthesis and anti-cancer activity evaluation of 5-(2-carboxyethenyl)-isatin derivatives. Eur. J. Med. Chem. 2016;112:145–156. doi: 10.1016/j.ejmech.2015.12.050. [DOI] [PubMed] [Google Scholar]
- Weir H.K., Anderson R.N., Coleman King S.M., Soman A., Thompson T.D., Hong Y., Moller B., Leadbetter S. Heart Disease and cancer deaths – trends and projections in the United States, 1969–2020. Prev. Chronic Dis. 2016;13:E157. doi: 10.5888/pcd13.160211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M., Liang Y.R., Li H., Liu M.M., Wang Y. Design, synthesis, and biological evaluation of hydantoin bridged analogues of combretastatin A-4 as potential anticancer agents. Bioorg. Med. Chem. 2017;25:6623–6634. doi: 10.1016/j.bmc.2017.10.045. [DOI] [PubMed] [Google Scholar]
- Zuliani V., Carmi C., Rivara M., Fantini M., Lodola A., Vacondio F., Bordi F., Plazzi P.V., Cavazzoni A., Galetti M., Alfieri R.R., Petronini P.G., Mor M. 5-Benzylidene-hydantoins: synthesis and antiproliferative activity on A549 lung cancer cell line. Eur. J. Med. Chem. 2009;44:3471–3479. doi: 10.1016/j.ejmech.2009.01.035. [DOI] [PubMed] [Google Scholar]