

Abstract
The catalytic enantioselective conjunctive coupling of C(sp3) electrophiles can be accomplished with Ni catalysis. The enantioselectivity of the reaction is dependent on reaction mechanism with many substrates able to engage in an asymmetric process with Pybox-Ni complexes, whereas other substrates provide racemic product mixtures. The link between substrate structure and selectivity is addressed.
Graphical Abstract

Chiral alkylboronate esters are important building blocks in contemporary organic chemistry and have been adopted for both academic and industrial asymmetric synthesis.1 New methods for constructing these motifs from untapped classes of starting materials can therefore have a significant impact in a number of venues.2 In this connection, we have been developing transition-metal catalyzed conjunctive cross-coupling reactions as a means to produce alkyl-boronate esters from vinylboron “ate” complexes and organic electrophiles.3,4 Preliminary studies have indicated that conjunctive coupling can be conducted catalytically and enantioselectively with either Ni3d or Pd3a-c complexes and, mechanistically, appears to proceed by way of a metal-induced metallate rearrangement. While these processes operate efficiently and enantioselectively with C(sp2) electrophiles, an important additional dimension in conjunctive coupling would arise by the introduction of reactions that employ C(sp3) electrophiles. In this report, we describe the utility of chiral Ni complexes in efficient catalytic conjunctive coupling reactions with alkyl electrophiles and describe how the reaction path – proceeding either through an enan-tioselective metal-induced metallate rearrangement, or through a non-selective radical-polar cross over manifold as elaborated by Studer5 and Aggarwal6,7 – is intimately linked to substrate structure and therefore controllable by judicious substrate selection.
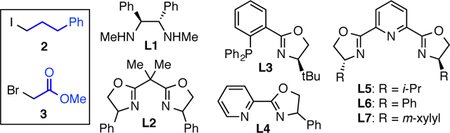
While most research in the field of catalytic cross-coupling8 has focused on the construction of C(sp2)–C(sp2) bonds, recent advances have expanded the scope of cross-coupling methods to include C(sp2)–C(sp3) and C(sp3)–C(sp3) couplings.9 The latter class of C–C bonds are often difficult to forge by cross-coupling due to mechanistic considerations involving challenging oxidative addition, slow transmetallation, and facile b-hydride elimination. In this light, use of alkyl halide electrophiles in conjunctive cross-coupling might appear to be fraught with complication. However, it was considered that the use of Ni complexes10 might ameliorate challenges associated with b-hydrogen elimination from Ni(alkyl) catalytic intermediates. Moreover, barriers to cross-coupling that arise from transmetallation of alkyl-M reagents11 to catalytic centers might be avoided in conjunctive coupling since the transmetallation step is replaced with a metal-induced metallate rearrangement. We therefore began our investigations by subjecting boron “ate” complex 1, generated from PhBpin and vinyl-lithium, to 3-phenyl-iodopropane (2) and either Ni(acac)2 alone, or in combination with diamine L112, the optimal ligand for Ni-catalyzed conjunctive coupling of aryl electro-philes.3d As depicted in Table 1, neither reaction furnished detectable quantities of conjunctive coupling product (4). A survey of other ligands indicated that bis(oxazoline), phosphine-oxazoline, and pyridyl-oxazoline are ineffective ligand scaffolds; however, tri-coordinate Pybox ligands furnished conjunctive coupling products with good levels of asymmetric induction. Modification of the ligand and nickel source revealed L7 and [(methallyl)NiCl]2 as a particularly competent combination, delivering 70% yield of conjunctive coupling product 4 in 96% ee (entry 10). In contrast to the enantioselective reaction with substrate 2, when α-bromo-methyl acetate 3 was employed, the reaction furnished racemic product (entries 11 and 12). Unlike reaction of unactivated substrate 2, reaction of compound 3 is efficient in the absence of ligand and, with addition of NaI, can furnish the racemic product 5 in outstanding yield.
Table 1.
Ni-Catalyzed Conjunctive Cross-Coupling with C(sp3) Electrophiles
 | |||||
|---|---|---|---|---|---|
| entry | E-X | ligand | Ni salt | % yielda | er |
| 1 | 2 | none | Ni(acac)2 | <10% | nd |
| 2 | 2 | L1 | Ni(acac)2 | <10% | nd |
| 3 | 2 | L2 | Ni(acac)2 | <10% | nd |
| 4 | 2 | L3 | Ni(acac)2 | <10 | nd |
| 5 | 2 | L4 | Ni(acac)2 | <5% | nd |
| 6b | 2 | L5 | Ni(acac)2 | 17 | 89:11 |
| 7b | 2 | L6 | Ni(acac)2 | 36 | 99:1 |
| 8b,c | 2 | L6 | Ni(acac)2 | 49 | 98:2 |
| 9b,c | 2 | L6 | [(methallyl)NiCI]2 | 51 | 98:2 |
| 10b,c | 2 | L7 | [(methallyl)NiCI]2 | 70 | 98:2 |
| 11b,c | 3 | L7 | Ni(acac)2 | 34 | 50:50 |
| 12d | 3 | Ni(acac)2 | 92 | nd | |
Determined by NMR versus an internal standard.
Determined by chiral SFC analysis.
Catalyst solution pre-complexed for 1.5 h.
Reaction conducted at 22 °C without ligand and with one equivalent NaI added.
The marked difference in reaction selectivity when using substrate 2 versus 3 suggested that the operative mechanisms for these reactions are likely different. In light of recent studies by Studer5 and Aggarwal6 on radical-polar crossover reactions between organic halides and vinyl boron “ate” complexes, it was suspected that radical processes might intervene in the Ni-catalyzed conjunctive coupling reactions of particular substrates and alter their course. Three mechanistic scenarios were considered as plausible reaction pathways (Scheme 2), with all involving initial halogen atom abstraction13 to convert the C(sp3) halide to a carbon-centered radical and nickel complex A. In one direction, following the precedent of Studer and Aggarwal, reaction of the carbon radical with the olefinic “ate” complex might deliver a-boryl radical B, which undergoes single-electron-transfer with the organohalide substrate thereby regenerating the carbon radical and delivering transient carbocation C; cation C would be expected to undergo metallate shift and necessarily deliver racemic reaction product. Formation of enantiomerically-enriched reaction product could occur by one of two routes: first, recombination of the initially-generated carbon radical with Ni complex A might deliver complex D – a compound that could engage in selective metal-induced metallate shift similar to previously described Pd and Ni complexes. Alternatively, aligned with recent observations by Fu14 on cross-coupling of α-haloboronic esters, α-boryl radical B might reengage Ni complex A to furnish E; subsequent intramolecular transmetallation and reductive elimination would furnish nonracemic product. Following the above-described mechanistic proposals, substrates that would furnish an electrophilic radical would be more prone to react with the electron-rich vinyl boron “ate” complex15 and undergo the radical chain process described by the Stu-der-Aggarwal cycle. Substrates leading to non-stabilized radicals could favor the metallate shift pathway since A would be expected to rapidly recombine with the initially- formed radical, or these substrates might operate the Fu-type cycle since single-electron transfer from B to an alkyl halide might be impeded by the redox potential of the nonactivated electrophile.
Scheme 2.

Prospective Mechanisms for Ni-catalyzed Conjunctive Coupling of C(sp3) Electrophiles.
To disentangle the operative pathways for these conjunctive coupling reactions, a series of mechanistic experiments were undertaken. First, we examined reactions of α-bromo ester 3 in the presence of TEMPO. As depicted in equation 1 (Scheme 3), addition of one equivalent of the radical scavenger is sufficient to inhibit formation of 5, thereby suggesting that radicals are indeed involved in the reaction of this substrate. Consistent with the non-stereospecific nature of the radical-polar crossover mechanism, conjunctive coupling between 3 and isotopically labeled “ate” complex 6 furnished the reaction product as a mixture of diastereomers (eq. 2). Of note, radical intermediates still appear to intervene in the enantioselective reactions of non-activated haloalkanes as indicated by the ring-opening and ring-closing reactions in equations 3 and 4 (Scheme 3)16 as well as the observation that TEMPO inhibits reaction of 1-iodobutane (eq. 5). However, the “Fu-type” cycle is ruled out as a mechanistic possibility by the observation that the enantioselective conjunctive coupling of iodobutane is a stereospecific process (eq. 6).
Scheme 3.

Mechanistic Experiments on Ni-catalyzed Conjunctive Coupling of C(sp3) Electrophiles.
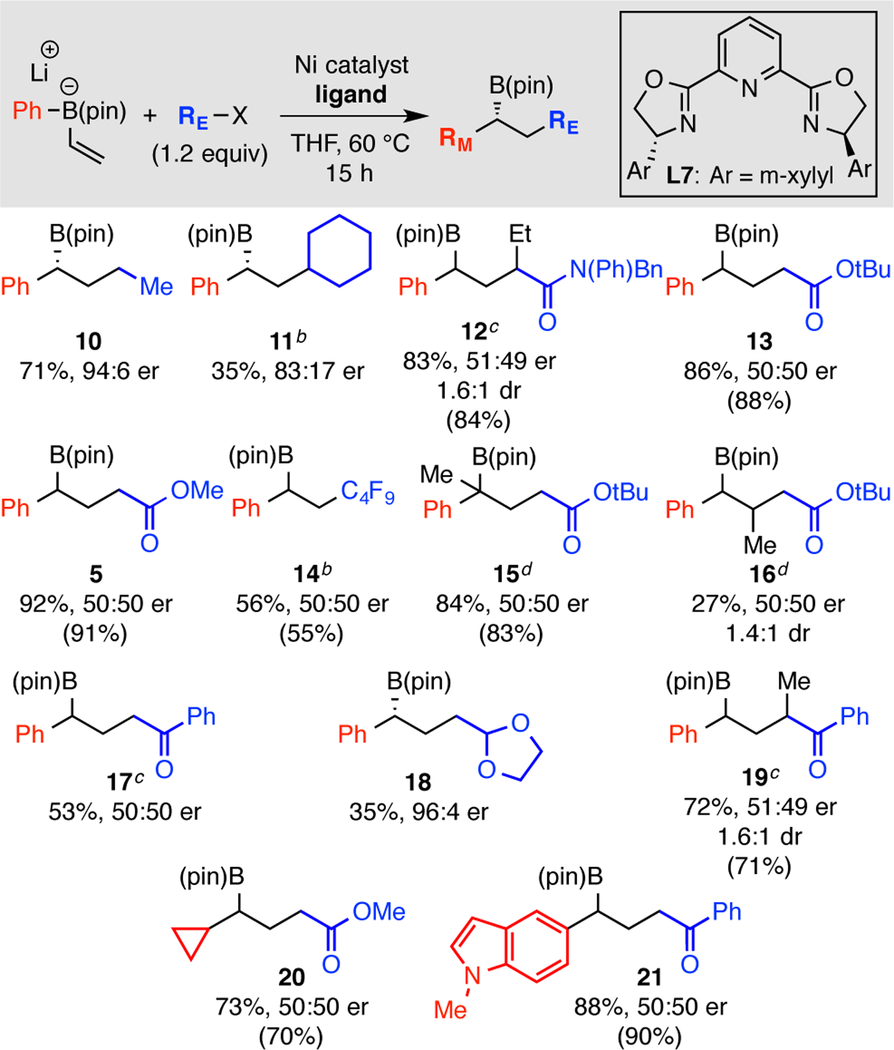
With an appreciation of operative mechanisms that can intervene in conjunctive coupling reactions with Ni-catalysts, we set out to probe classes of C(sp3) electrophiles that might avoid the radical-polar crossover path and instead engage in enantioselective conjunctive coupling by the metallate shift- based pathway (Table 2). Using enantioselectivity in conjunctive coupling as a guide to reaction mechanism, it can be concluded that whereas primary and secondary alkyl halides can engage in conjunctive coupling by the metallate shift pathway (products 10, 11), the presence of a single electron-withdrawing conjugating group is sufficient to render the reaction non-selective. Thus, alkyl halides bearing an adjacent ketone (17, 19), ester (5, 13, 15-16, 20), or amide (12) result in high yielding but non-enantioselective conjunctive coupling, presumably because the intermediate carbon radical is electrophilic enough to engage in rapid reaction with the nucleophilic alkene of the boron “ate” complex. Similar to the observations of Studer5 and Aggarwal6, fluorinated alkyl halides (14) also appear to react by the radical-polar crossover path and provide racemic reaction product. It is worth noting that with activated substrates, the reaction could be conducted equally well with or without the Pybox ligand (Table 2), was generally more effective with Ni(acac)2 as the Ni source, and remained racemic even with very different migrating groups (20, 21). Lastly, whereas substrates with conjugating heteroatoms do not engage in asymmetric couplings, those bearing s-bonded heteroatoms adjacent to the halide follow the metallate-shift based pathway and provide access to products with oxidized hydrocarbon functional groups (18).
Table 2.
Impact of Substrate Functionality on Ni-Catalyzed Conjunctive Coupling with C(sp3) Electrophiles.a
|
Unless otherwise noted, the “ate” complexes were prepared by addition of the alkenyllithium to RB(pin). Data for compounds 10, 11 and 12 from reactions that employed 5% [(methallyl)NiCl]2 and 6% (R,R)-L7, others are for 5% Ni(acac)2 and 6% (R,R)-L7. Yields in parentheses are for reactions that are ligand-free and employ 3% Ni(acac)2 at 22 °C. All yields represent isolated product obtained after chromatographic purification. Products 10 and 11 are derived from the alkyliodide, whereas the others employed the organobromide and one equivalent of NaI.
Product isolated as the derived alcohol after peroxide oxidation.
The reactions were also conducted by addition of PhLi to vinylB(pin) and gave similar results.
Data is for reaction conducted by addition of PhLi to vinylB(pin).
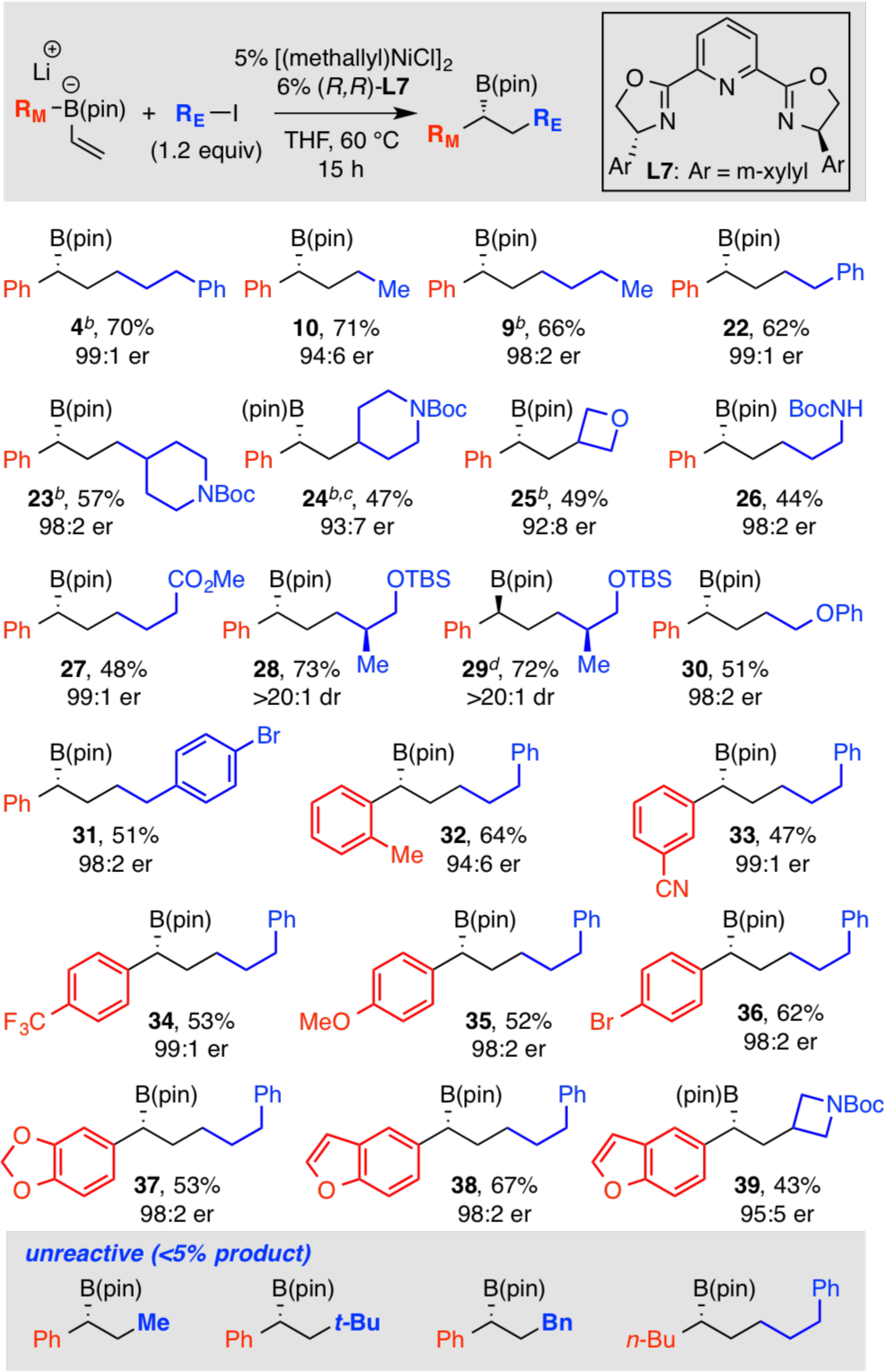
The scope of the asymmetric conjunctive coupling reaction employing unactivated primary and secondary organo-halides was further examined as depicted in Table 3. In addition to 3-phenyliodopropane, iodoethane (product 10) and iodobutane (9) also engage in the reaction suggesting that there is no chelating effect from a tethered functionality that assists in the stereocontrol of this process. β-Branched electrophiles (23, 28, 29) and cyclic secondary electrophiles (24, 25, 39) were also successfully engaged in the reaction though iodocyclohexane (11, Table 2) gave low yield of the conjunctive coupling product. Electron rich (35, 37–39), electron poor (33, 34), sterically encumbered (32), and heterocyclic (37-39) migrating groups can also be successfully employed in the reaction. Lastly, it is worth noting that the reaction appears to be compatible with labile functional groups such as methyl esters (27) and nitriles (33), free car-bamate NH groups (26), and TBS-protected alcohols (28, 29). Also of note, it is possible to selectively transform C(sp3) iodides in the presence of C(sp2) bromides (31, 36). Lastly, for electrophiles with labile b-substituents (30), elimination is not observed. Current challenges to this catalytic cross-coupling include methyl-, t-butyl-, and benzyl-based electrophiles as well as alkyl migrating groups (inset, Table 3).
Table 3.
Ni-Catalyzed Enantioselective Conjunctive Coup-ling with C(sp3) Electrophiles.a
|
Unless otherwise noted, the “ate” complexes were prepared by addition of vinyllithium to RB(pin). All yields represent isolated product obtained after chromatographic purification.
Compounds 4, 9, 23, 24, and 25 were also prepared by addition of PhLi to vinylB(pin) and provide products in 67, 65, 57, 47 and 44% yield, respectively.
Product isolated as the derived alcohol after peroxide oxidation.
(S,S)-L7 employed as ligand.
In conclusion, we have developed a highly enantioselec-tive nickel-catalyzed conjunctive cross-coupling that can engage primary and secondary C(sp3) electrophiles and can tolerate a range of functional groups in the production of chiral alkylboronate ester products. Mechanistic experiments have elucidated the origin of the divergent reactivity between activated and unactivated electrophilic substrates and we anticipate that this knowledge will be useful in the design of new processes.
Supplementary Material
Scheme 1.

Ni-Catalyzed Conjunctive Coupling of C(sp3) Electrophiles.
ACKNOWLEDGEMENTS
This work was supported by the NIH (GM-118641).
Footnotes
Supporting Information. Procedures, characterization and spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Reviews, see:; (a) Brown HS; Singaram B Acc. Chem. Res 1988, 21, 287. [Google Scholar]; (b) Organoboranes for Synthesis, Ramachandran PV; Brown HC, Eds. ACS Symposium Series 783; American Chemical Society: Washington, DC: 2001. [Google Scholar]; (c) Stereodirected Synthesis with Or-ganoboranes Matteson, D. S. Springer: New York, 1995. [Google Scholar]; (d) Leonori D; Aggarwal VK Accts. Chem. Res 2014, 47, 3174. [DOI] [PubMed] [Google Scholar]
- (2).For a recent review:; Collins BSL; Wilson CM; Myers EL; Aggarwal VK Angew. Chem Int. Ed 2017, 56, 11700. [DOI] [PubMed] [Google Scholar]
- (3) (a).Zhang L; Lovinger GJ; Edelstein EK; Szymaniak AA; Chierchia MP; Morken JP Science. 2016, 351, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lovinger GJ; Aparece MD; Morken JP J. Am. Chem. Soc 2017, 139, 3153. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Edelstein EK; Namirembe S; Morken JP J. Am. Chem. Soc 2017, 139, 5027. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chierchia M; Law C; Morken JP Angew. Chem. Int. Ed 2017, 56, 11870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For aligned processes, see:; (a) Ishida N; Shimamoto Y; Mura-kami M Org. Lett 2009, 11, 5434. [DOI] [PubMed] [Google Scholar]; (b) Ishida N; Shinmoto T; Sawano S; Miura T; Murakami M Bull. Chem. Soc. Jpn 2010, 83, 1380. [Google Scholar]; (c) Ishida N; Narumi M; Murakami M Org. Lett 2008, 10, 1279. [DOI] [PubMed] [Google Scholar]; (d) Ishida N; Shimamoto Y; Murakami M Org. Lett 2010, 12, 3179. [DOI] [PubMed] [Google Scholar]; (e) Ishida N; Ikemoto W; Narumi M; Murakami M Org. Lett 2011, 13, 3008. [DOI] [PubMed] [Google Scholar]
- (5).Kischkewitz M; Okamoto K; Mück-Lichtenfeld C; Studer A Science, 2017, 355, 936. [DOI] [PubMed] [Google Scholar]
- (6).Silvi M; Sandford C; Aggarwal VK J. Am. Chem. Soc 2017, 139, 5736. [DOI] [PubMed] [Google Scholar]
- (7).For an overview, see:; Hidasová D; Jahn U Angew. Chem. Int. Ed 2017, 56, 9656. [DOI] [PubMed] [Google Scholar]
- (8).For a comprehensive overview of catalytic cross-coupling, see:; (a) de Meijere A, Diederich F, Eds. Metal-Catalyzed Cross-Coupling Reactions, 2nd Completely Revised and Enlarged ed., Vol. 1; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]; (b) de Meijere A, Diederich F, Eds. Metal-Catalyzed Cross-Coupling Reactions, 2nd Completely Revised and Enlarged ed., Vol. 2; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- (9).For a recent review, see:; Choi J; Fu GC Science, 2017, 356, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Overviews of mechanistic features in cross-coupling with Ni catalysts, see:; (a) Hu X Chem. Soc. Rev 2011, 2, 1867. [Google Scholar]; (b) Tasker SZ; Standley EA; Jamison TF Nature 2014, 509, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Overview:; Jana R; Pathak TP; Sigman MS Chem. Rev 2011, 111, 1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For selected enantioselective Ni-catalyzed cross-coupling with Ni/diamine catalysts, see:; (a) Schmidt J; Choi J; Liu AT; Slusar-czyk M; Fu GC Science, 2016, 354, 1265. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wilsily A; Tramutola F; Owston NA; Fu GC J. Am. Chem. Soc 2012, 134, 5794. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lu Z; Wilsily A; Fu GC J. Am. Chem. Soc 2011, 133, 8154. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Saito B; Fu GC J. Am. Chem. Soc 2007, 129, 9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).For lead references, see:; (a) Fu GC ACS Cent. Sci 2017, 3, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jahn U Top. Curr. Chem 2012, 320, 323. [DOI] [PubMed] [Google Scholar]
- (14).Schmidt J; Choi J; Liu AT; Slusarczyk M; Fu GC Science, 2016, 354, 1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For polar effects in radical addition reactions, see:; Giese B Angew. Chem. Int. Ed. Engl 1983, 22, 753. [Google Scholar]
- (16).For a review of cyclopropylcarbinyl radical chemistry, see:; Nonhebel DC Chem. Soc. Rev 1993, 347. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.