

Abstract
Advances in medical device technology have been dramatic in recent years resulting in both an increased number of medical devices and an increase in the invasiveness and critical function which devices perform. Two new regulations entered into force in Europe in May 2017, the Medical Device Regulation (MDR) and the In Vitro Diagnostic Device Regulation (IVDR). These regulations will replace the current directives over the coming years. These regulations, for the first time introduce requirements relating to registries.
Medical device manufacturers are required to have systematic methods for examining their devices once available on the market, by systematically gathering, recording and analysing data on safety and performance.
Registries can assist public health protection in very practical ways, for example, to help urgently identify patients or devices. Registries can also be powerful tools for collecting and appraising real-world clinical evidence concerning medical devices. Clinical investigations are limited in terms of the sample size and the duration of follow-up which can reasonably be expected. Registries may also be the only available tool to examine rare adverse effects, sub-populations or for time durations which it is not possible or feasible to study in a clinical investigation. By ensuring that a core dataset is collected which can be compared to other registries or trial data, it is possible to pool data to better examine outcomes. There are a range of excellent initiatives which have aimed at ensuring the appropriate regulatory application of registry data.
Cite this article: EFORT Open Rev 2019;4 DOI: 10.1302/2058-5241.4.180061
Keywords: EU regulations, medical device, registry
Introduction
Advances in medical device technology have been dramatic in recent years, resulting in both an increased number of medical devices (estimated to be approximately 500 000 different devices in Europe) and an increase in the invasiveness and critical function which devices perform. Almost everyone will be exposed to a medical device in their lifetime and many more people are being implanted with permanent devices which often cannot be subsequently removed – patients therefore rely on medical devices being safe and performing as intended for their lifetime. The aims of EU policies with respect to public health include measures to set high standards of quality and safety for medical devices, in addition to a range of other areas of co-operation.1 In this context, two new regulations entered into force in Europe in May 2017, the Medical Device Regulation (MDR)2 and the In Vitro Diagnostic Device Regulation (IVDR).3 These regulations will replace the current directives in a phased manner with a three-year transitional period for the MDR and a five-year transition for the IVDR (Fig. 1).
Fig. 1.

Timeline for implementation of the new regulations.
The purpose of the regulators was to establish a modernized and more robust EU legislative framework; therefore these regulations represent a strengthening and reinforcement of a number of elements of the current system. As such they represent a revision, rather than a fundamental redesign of the current system for medical device regulation in Europe. The early failure and other adverse effects associated with certain metal-on-metal (MoM) hip implants were identified as an important weakness of the medical device regulatory system which needed to be addressed.4 In addition to this, the fraudulent use of non-medical-grade silicone in breast implants manufactured by Poly Implant Prothèse (PIP) led the European Commission to establish a joint plan for immediate action in 2012, and one of the five ‘immediate actions’ included support for the development of implant registers.5
Following these two crises regarding MoM hips and PIP, the new regulations were drafted with the explicit aim of ensuring a high level of safety and health whilst supporting innovation (Preamble par.1).2 The MDR also brings a number of new procedures which will be important with respect to the clinical evidence requirements for devices. In this article, we will introduce some of the fundamental elements of medical device regulation in Europe, describe some of the important changes that the new regulations will bring, in addition to providing an update on regulatory developments, with a focus on device registries.
New approach legislation and CE marking of medical devices: a brief introduction
European legislation regarding medical devices was first introduced in the early 1990s, as part of what was known as a ‘new approach’ framework of laws developed in Europe during the 1980s for a range of different products and sectors. Until this time, the trading of goods in Europe under the ‘old approach’ consisted of national authorities creating individual sets of technical legislation, which was often different to that in their neighbouring member states. The new approach, in essence, meant that products could be traded within Europe, as long as they complied with the ‘essential requirements’ of the legislation.6 To comply with these essential requirements, standards were developed, by groups such as the International Organization for Standardization (ISO). These standards became ‘harmonized’ when they were accepted as allowing compliance to the essential requirements of the legislation. A listing of these standards, as they apply to medical devices, is available on the European Commission website.7 It is important to note, however, that the great majority of these standards concern technical (i.e. non-clinical testing) requirements, and as such they do not tend to have any requirements with respect to clinical study design, sample size, selection of endpoints etc.
Depending upon the type of product, conformity was then either claimed directly by the manufacturer, or assessed by third-party certification bodies known as notified bodies. Under the current directive system, there are approximately 55 notified bodies designated for medical devices,8 and the process of designation of notified bodies with respect to the new regulations is underway. Successful conformity assessment by a notified body results in a CE mark, which then allows a product to be marketed without any technical barriers to trade in any of the European member states. Notified bodies must also evaluate devices once they enter the market, by evaluating the post-market data associated with a device and, if needed, the notified body can withdraw, suspend or impose conditions on the CE-marked status of a device. Member states also have competent authorities, who receive incident reports relating to devices and conduct market surveillance and authorities can also take regulatory action against non-conforming devices. This framework still exists today, subject to a number of additional features, with the MDR and the IVDR.
Clinical evidence for medical devices: what is required
To meet the essential requirements, referred to as general safety and performance requirements (GSPR) in the MDR (Annex I),2 a manufacturer must evaluate clinical data, which is defined in the MDR (Article 2 par.48).2 This clinical data may be sourced from the device under evaluation, or from a device which is demonstrated to be ‘equivalent’ to that device. With the MDR, for the first time, the clinical, technical and biological factors which must be taken into account to demonstrate equivalence have been introduced into European law. It is required that the device under evaluation and the claimed equivalent device are similar to the extent that there would be no clinically significant difference in the safety and clinical performance of the device (Annex XIV, Part A, Section 3).2
These clinical data are then evaluated, to determine whether a manufacturer can claim compliance with the GSPR with respect to the intended use a manufacturer ascribes to their device. The clinical evaluation then informs the extent to which the device manufacturer is required to further examine their device once made available on the market, by means of post-market surveillance (PMS) or post-market clinical follow-up (PMCF) (Fig. 2).
Fig. 2.
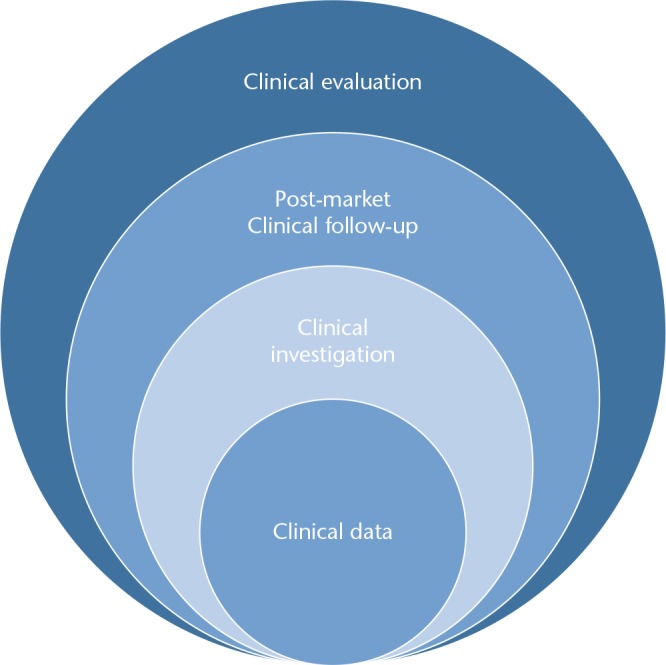
Types of clinical evidence for medical devices.
Clinical evidence and the new regulation
The MDR notes in a number of instances that this clinical data must be ‘sufficient’ (Article 61, par 6a),2 and the clinical evaluation should determine the way in which manufacturers evaluate their device in the post-market phase. Two important new changes in the MDR have the potential to address technical or clinical evidence requirements for medical devices and these are briefly introduced here. Common specifications are rules which can be created for device technologies, which have the potential to introduce harmonized requirements for clinical evidence. The criteria which apply to common specifications are detailed further in the MDR (Article 9).2
A second important procedure has become known as the ‘scrutiny’ procedure, referred to as the ‘clinical evaluation consultation procedure’ in the MDR. This is a mandatory procedure which applies to certain high-risk devices (Article 54),2 and requires that the evidence to support a device, in addition to the assessment of clinical evidence by the notified body, are passed to an expert panel, who may then provide an opinion with respect to the device within a 60-day period.
Registries and the MDR
The MDR mentions registries, for the first time in European legislation for medical devices. Article 108 of the MDR notes that the Commission and member states shall encourage the establishment of registries for specific device technologies, in addition to setting common principles to collect comparable information. Moreover, such registers and databanks shall contribute to the independent evaluation of the long-term safety and performance of devices, or the traceability of implantable devices, or all of such characteristics.
A second important aspect concerning registries and the MDR is the requirement that both manufacturers and notified bodies take registry data into account, as part of their obligations. For manufacturers, taking registry data into account is one of the post-market requirements, to undertake an evaluation of suitable registers or PMCF studies. In particular, for each device, they shall plan, establish, document, implement, maintain and update a post-market surveillance system in a manner that is proportionate to the risk class and appropriate for the type of device. Furthermore, the post-market surveillance system shall be suited to actively and systematically gathering, recording and analysing relevant data on the quality, performance and safety of a device throughout its entire lifetime, to drawing the necessary conclusions and to determining, implementing and monitoring any preventive and corrective actions (Article 83).2 These post-market responsibilities of device manufacturers, and the scope of many device registries, share many commonalities.
For notified bodies, at the time of recertification of a device, manufacturers will be required to submit information regarding changes in medical, scientific and technical knowledge, including data from registries (Annex VII par.4.11).2
In general, the primary aim of implantable device registries is to monitor real-world evidence concerning safety and performance over time; for example, with respect to joint replacements, device performance is primarily examined by measuring the revision rate (a surrogate marker of the rate of failure). To identify devices with lower performances or early failures, it is also important to understand the technical features of each device (characterization) in order to compare it with other similar devices;9 these comparators are what are often referred to as the ‘state of the art’ and these are the benchmarks to which safety and performance metrics are often described. Registries which examine a range of devices have the potential to compare different devices in a standardized way.
Registries and device identification
Registries also serve very practical needs – for example they can support an urgent recall of patients if needed. To do this, traceability of the devices is essential. It is therefore important to know exactly which device was implanted (identification) to facilitate urgent action. The MDR also introduces for the first time, requirements relating to unique device identification (UDI), something which holds much potential for ensuring that data can be tracked to specific device iterations in a more harmonized way. The UDI is defined as a series of numeric or alphanumeric characters that is created through internationally accepted device identification and coding standards and that allows unambiguous identification of specific devices on the market and shall allow the identification and facilitate the traceability of devices. Joint prostheses belong to class III implantable devices. For these devices, the MDR states that health institutions shall store and keep, preferably by electronic means, the UDIs of the devices which they have supplied or with which they have been supplied, and that member states shall encourage, and may require, healthcare professionals to store and keep, preferably by electronic means, the UDIs of the devices with which they have been supplied (Article 27).2 Informatic platforms of registries might support both health institutions and healthcare professionals in fulfilling these requirements. The UDI will also be included on an implant card for implantable devices, so that patients will have this information and other important information regarding their implant made available to them as a matter of routine (Article 18).2
The MDR EUDAMED database
One key aspect in fulfilling the objectives of the new regulations is the creation of a European database on medical devices (EUDAMED) that should integrate different electronic systems to collate and process information regarding devices on the market and the relevant economic operators, certain aspects of conformity assessment, notified bodies, certificates, clinical investigations, vigilance and market surveillance. The objectives of EUDAMED are to enhance overall transparency, including through better access to information for the public and for healthcare professionals, to avoid multiple reporting requirements, to enhance co-ordination between member states and to streamline and facilitate the flow of information between economic operators, notified bodies or sponsors and member states as well as between member states among themselves and with the Commission (Preamble par.44).2 In particular, EUDAMED is organized into the following three electronic systems: (i) on clinical investigations, (ii) on vigilance, and (iii) on market surveillance. The MDR states that EUDAMED’s electronic systems regarding devices on the market, the relevant economic operators and certificates should enable the public to be adequately informed about devices on the European Union market. More specifically, the electronic system on clinical investigations should serve as a tool for the co-operation between member states and for enabling sponsors to submit, on a voluntary basis, a single application for several member states and to report serious adverse events, device deficiencies and related updates. The electronic system on vigilance should enable manufacturers to report serious incidents and other reportable events and to support the co-ordination of the evaluation of such incidents and events by competent authorities. The electronic system regarding market surveillance should be a tool for the exchange of information between competent authorities (Preamble, par.46).2
To facilitate the functioning of EUDAMED, the MDR indicates that an internationally recognized medical device nomenclature should be available free of charge to manufacturers and other natural or legal persons required by this regulation to use it. Furthermore, that nomenclature should also be available, where reasonably practicable, free of charge to other stakeholders (Preamble par.45, Article 26).2 Description of requirements for the future EU medical device nomenclature have recently been published by the Medical Device Coordination Group.10 In particular, the role of registries is recognized in supporting the system/processes that shall be put in place to periodically review the terminology structure and content to incorporate learning from ongoing experience with real-world use of device nomenclature (e.g. EUDAMED, GUDID, registries) as well as from technological innovation.
Improving transparency for medical device clinical evidence
One other notable feature of the new regulations is the summary of safety and clinical performance (SSCP) which will be publicly available on the MDR EUDAMED database (which is currently under development) for high-risk devices (implantable devices and class III devices, other custom-made or investigational devices) which have been granted a CE mark (Article 32).2 This marks an important step for medical device regulation in Europe, as, for the first time, a summary of the clinical evaluation which was used to achieve CE marking will be publicly available. It is hoped that this will help to facilitate decision making between clinicians and patients when considering treatment options, and the need for transparency with respect to medical devices is something which clinical associations such as the European Society of Cardiology have taken a keen interest in.11
International collaboration
There have been a number of collaborations relevant to device registries at the International Medical Device Regulatory Forum (IMDRF), a forum for international collaboration between regulatory authorities. The IMDRF have produced guidance with respect to linking registry data,12 methodological principles13 and tools for assessing the usability of registries to support decision making.14 With respect to device identification, specific nomenclatures for pre-market and post-market adverse events, that will be taken into account in the EUDAMED electronic system on vigilance, are under development in the framework of IMDRF.15 With respect to the medical device registries, the IMDRF developed a definition, in addition to a number of factors which should be considered regarding the impact, value and sustainability of the registry.16
Device regulation and registries
In practice, registries can range from a simple spreadsheet on a ward-based computer to large internationally harmonized registries. Registry design is key to ensuring the best possible value and impact for the data accrued. It is also important to note that medical devices are iterative by nature, with multiple changes often occurring from the time of initial prototype development, to first clinical use and until the device becomes obsolete. This has the potential to introduce variables into datasets and the decision as to whether a change is truly ‘minor’ or ‘significant’ is sometimes one which can be difficult to make when analysing pre-clinical data.
When a trend or safety signal is identified, it is vitally important that this information is shared with the appropriate stakeholders. These ‘governance’ aspects are important to consider well in advance as the data generated by a registry may be of interest to a broad range of stakeholders, including patients, clinicians, authorities, device manufacturers, health technology assessment bodies and other decision makers in a health system. These governance questions go beyond data protection and deciding who has access to data, but should consider who should be the responsible actor, when an issue is identified from emerging registry data.17
Initiatives such as the Beyond Compliance advisory group and the Orthopaedic Data Evaluation Panel (ODEP) in the United Kingdom, have developed methods for working with device manufacturers and other stakeholders to help integrate registry data into a manufacturer’s post-market regulatory obligations. Moreover, there have also been international collaborations of device registries. An example of this is the UK National Joint Registry (NJR), which has recently had its existing component database upgraded and developed, in collaboration with the EndoProthesen Register Deutschland (EPRD). The joint NJR-EPRD component database is a structured database, directly fed by manufacturers that includes both the information necessary to track and identify the implanted device and to describe its technical characteristics, these features being an indispensable requirement to compare the performance of different prostheses. Other registries, such as the Dutch Arthroplasty Register (LROI) and the Italian Arthroplasty registry (RIAP), have also shown interest in participating in this initiative that might be further considered in the MDR framework.
Conclusions
Registries can be powerful tools for collecting and appraising real-world clinical evidence concerning medical devices. Clinical investigations are limited in terms of the sample size and duration of follow-up which can reasonably be expected. Registries may also be the only available tool to examine rare adverse effects, sub-populations or for time durations which it is not possible or feasible to study in a clinical investigation. There is a growing recognition and acceptance of the importance of registries to identify safety issues associated with devices. Orthopaedic registries are some of the best-established device registries available, and the data that are produced by these registries are becoming ever more nuanced.
There are a range of excellent initiatives which have aimed to ensure the appropriate regulatory application of registry data. By ensuring that a core dataset is collected which can be compared to other registries or trial data, it is possible to pool data to better examine the safety of implants in the post-market phase.
Registries can be powerful tools for assessing safety, by helping those responsible for the safety of devices to examine real-world outcomes from large numbers of patients. Registries, however, are not simple or easy undertakings to establish, run, maintain and govern. The MDR, for the first time, introduces requirements for the incorporation of registry data into the data which a manufacturer must evaluate as part of clinical evaluation. It is hoped that this will result in the better integration of registry data in regulatory decision making. To give true effect to registries and the MDR, all interested parties need to work together to achieve the high level of safety that patients expect.
Acknowledgments
We acknowledge Rosaria Boldrini, Head of Vigilance Unit and Annamaria Donato, Head of Medical Devices Regulatory Affairs Unit of the Directorate General of Medical Devices and Pharmaceutical Services, Ministry of Health, Rome (Italy), for their respective advice.
Footnotes
ICMJE Conflict of interest statement: TM is employed by the National Competent Authority for Medical Devices in Ireland.
MT declares travel/accommodations/meeting expenses unrelated to activities listed.
Funding statement
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article. The RIAP project is funded by the Italian Ministry of Health, Medical Devices and Pharmaceutical Service General Directorate. Further information concerning the project is available at www.iss.it/riap.
References
- 1.No authors listed. Consolidated version of the Treaty on the Functioning of the European Union – Part Three: Union Policies And Internal Actions – Title XIV: Public Health – Article 168 (ex Article 152 TEC). Official Journal 09/05/2008;115:122–124. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A12008E168 (date last accessed 18 September 2018).
- 2.No authors listed. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC (Text with EEA relevance). https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32017R0745 (date last accessed 18 September 2018).
- 3.No authors listed. Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and Commission Decision 2010/227/EU (Text with EEA relevance). https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32017R0746 (date last accessed 18 September 2018).
- 4. Scientific Committee on Emerging and Newly Identified Health Risks (SCENIHR). Opinion on the safety of metal-on-metal joint replacements with a particular focus on hip implants, 2014. https://ec.europa.eu/health/sites/health/files/scientific_committees/emerging/docs/scenihr_o_042.pdf (date last accessed 27 August 2018).
- 5. European Commission. Press Release – Medical devices: European Commission calls for immediate actions – tighten controls, increase surveillance, restore confidence, (2012). http://europa.eu/rapid/press-release_IP-12-119_en.htm?locale=en (date last accessed 28 August 2018).
- 6. European Commission. Commission Notice – The ‘Blue Guide’ on the implementation of EU products rules 2016. Official Journal C 272 26/07/2016;59:6–7. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=OJ:C:2016:272:FULL&from=EN (date last accessed 18 September 2018). [Google Scholar]
- 7. European Commission. Summary list of titles and references of harmonised standards under directive 93/42/EEC for medical devices (2018). https://ec.europa.eu/growth/single-market/european-standards/harmonised-standards/medical-devices_en (date last accessed 27 August 2018).
- 8. European Commission. List of bodies notified under directive: 93/42/EEC medical devices, 2018. http://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.pdf&refe_cd=93%2F42%2FEEC&requesttimeout=900 (date last accessed 27 August 2018).
- 9. Torre M, Laricchiuta P, Luzi I, Ceccarelli S, Carrani E, Masciocchi M, Sampaolo L. Italian Arthroplasty Registry Project. Better data quality for better patient safety. Fourth Report 2017 – Addendum. Roma: Il Pensiero Scientifico Editore, 2018. http://www.iss.it/riap (date last accessed 18 September 2018). [Google Scholar]
- 10.No authors listed. MDCG 2018-2 Future EU medical device nomenclature. Description of requirements. https://ec.europa.eu/docsroom/documents/28668 (date last accessed 18 September 2018).
- 11. Fraser AG, Butchart EG, Szymański P, et al. The need for transparency of clinical evidence for medical devices in Europe. Lancet 2018;392:521–530. [DOI] [PubMed] [Google Scholar]
- 12. IMDRF. Principles of international system of registries linked to other data sources and tools. (IMDRF/REGISTRY WG/N33 FINAL:2016) 30/09/2016. http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-160930-principles-system-registries.pdf (date last accessed 18 September 2018).
- 13. IMDRF. Methodological principles in the use of international/medical device registry data. (IMDRF/Registry WG/N42FINAL:2017) 16/03/2017. http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-170316-methodological-principles.pdf (date last accessed 18 September 2018).
- 14. IMDRF. Tools for assessing the usability of registries in support of regulatory decision-making (IMDRF/Registry WG/N46 FINAL:2018) 27/03/2018. http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-180327-usability-tools-n46.pdf (date last accessed 18 September 2018).
- 15. IMDRF. IMDRF terminologies for categorized Adverse Event Reporting (AER): terms, terminology structure and codes (IMDRF/AE WG/N43FINAL:2017 (Edition 2)) 21/09/2017. http://www.imdrf.org/docs/imdrf/final/technical/imdrf-tech-170921-aer-n43-r2.pdf (date last accessed 18 September 2018).
- 16. IMDRF. Patient registry: essential principles, 2/10/2015. http://www.imdrf.org/consultations/cons-essential-principles-151124.asp (date last accessed 28 August 2018).
- 17. Wilkinson J, Crosbie A. A UK medical devices regulator’s perspective on registries. Biomed Tech (Berl) 2016;61:233–237. [DOI] [PubMed] [Google Scholar]