

Abstract
T cell receptor (TCR) binding to agonist peptide major histocompatibility complex (pMHC) triggers signaling events that initiate T cell responses. This system is remarkably sensitive, requiring only a few binding events to successfully activate a cellular response. On average, activating pMHC ligands exhibit mean dwell times of at least a few seconds when bound to the TCR. However, a T cell accumulates pMHC-TCR interactions as a stochastic series of discrete, single-molecule binding events whose individual dwell times are broadly distributed. With activation occurring in response to only a handful of such binding events, individual cells are unlikely to experience the average binding time. Here, we mapped the ensemble of pMHC-TCR binding events in space and time while simultaneously monitoring cellular activation. Our findings revealed that T cell activation hinges on rare, long-dwell time binding events that are an order of magnitude longer than the average agonist pMHC- TCR dwell time. Furthermore, we observed that short pMHC-TCR binding events that were spatially correlated and temporally sequential led to cellular activation. These observations indicate that T cell antigen discrimination likely occurs by sensing the tail end of the pMHC-TCR binding dwell time distribution rather than its average properties.
INTRODUCTION
Antigen discrimination by T cells is the front line of the adaptive immune response. During surveillance, T cell receptors (TCRs) discriminate agonist peptide major histocompatibility complex (pMHC) ligands from self pMHCs on antigen-presenting cells (APCs) to mount an immune response against foreign pathogens while avoiding autoimmunity. T cells are capable of distinguishing between ligands with subtly different binding kinetics (1, 2) and, remarkably, do this with nearly single-molecule sensitivity (3, 4). The biochemical pathways involved in T cell activation have been extensively characterized (5, 6). However, essentially, all current understanding about the TCR signaling system is based on population-averaged information.
For example, the hallmark difference between activating and nonactivating pMHC ligands is the average binding dwell time between pMHC and TCR (2, 7). However, this conclusion comes from experiments that correlate population measurements of pMHC-TCR binding kinetics to cellular activity readouts, such as intracellular calcium flux or cytokine production, on populations of cells (1). The connection between the stochastic sequence of individual pMHC-TCR binding events that each cell experiences and the specific molecular response of that cell is lost in such population-level measurements. This is especially notable in the case of T cell antigen recognition, because only a handful of individual pMHC-TCR binding events lead to each cellular decision (3, 4, 8). Even under identical conditions, each cell will experience a different sequence of binding events, and the sample average from this small set can differ markedly from the overall average for all pMHC-TCR binding events. How a single T cell responds to individual molecular binding events and how these are integrated into the decision to activate are not understood.
In this study, we used an assay in which the series of pMHC-TCR binding events on an individual T cell were mapped in space and time while simultaneously monitoring the cellular decision to activate. The experimental platform was built off a method of directly imaging the binding events between pMHC and TCR on live T cells activated on a supported membrane (9–12). Key to this strategy is the unambiguous resolution of pMHC-TCR binding events themselves, rather than the mere presence of a ligand (3, 4), which is only loosely related to actual binding events due to stochastic variation and active modulation of the T cell-APC interface (10). Here, we used this platform to simultaneously visualize the activation state of individual T cells using the transcription factor NFAT (nuclear factor of activated T cells), which undergoes nuclear translocation in response to early activation of calcium signaling (13). NFAT translocation provides a rapid and easily resolved readout of the decision-making outcome that can be monitored in parallel with single-molecule pMHC-TCR imaging (10). We here refer to this mapping between the sequence of individual pMHC-TCR binding events and NFAT translocation as a molecular impulse-response function, in analogy to electronic signal processing (14–16).
We performed a series of experiments on primary mouse T cells (AND TCR transgenic) at various pMHC ligand densities and TCR affinities (e.g., different mean pMHC-TCR binding dwell times: <τoff> = 1/koff). The results indicated that T cells neither count the number of pMHC-TCR binding events nor integrate the cumulative pMHC-TCR binding duration, or dwell time. Instead, T cells responded disproportionately to rare, long-dwelling binding events that can be an order of magnitude longer than the mean. We also observed that rare cells that activated in response to low densities of weak agonist pMHC experienced an unusual spatiotemporal correlation of short pMHC-TCR binding events. Short-lived binding events that formed a temporal sequence within a limited spatial neighborhood were apparently summed by the cell and produced a response resembling a single, long-dwelling event. The extended sequence in time was important, because a burst of simultaneous binding events in the same location failed to activate cells. Overall, these results revealed that TCR signaling was tuned to respond to the tail end of the pMHC-TCR binding dwell time distribution, rather than its average properties. Thus, although at the population level, T cell activation may be correlated with mean pMHC-TCR binding dwell times of a few seconds, this does not imply that TCR signaling is tuned to the same set point of binding duration. This realization has consequences both in the context of natural antigen discrimination and in the design of synthetic T cell activators.
RESULTS
Mapping individual molecular binding events to the cellular decision to activate
Here, we used single-molecule imaging to track the individual pMHC- TCR binding events experienced by a T cell and simultaneously monitored cellular activation (9,10). In these experiments, peripheral CD4 T cells that expressed a transgenic TCR (AND) were transduced with an NFAT-fluorescent protein (FP) reporter. T cells were then exposed to supported lipid bilayers functionalized with histidine-tagged intercellular adhesion molecule-1 (ICAM-1) and MHC class II (IEk) loaded with peptide (pMHC), which were linked to the membrane through interactions with Ni2+-chelating lipids (17). Agonist peptides were site-specifically labeled with a single organic dye by a maleimide- thiol linkage, which was confirmed by high-performance liquid chromatography. Although most of the measurements were made using peptides conjugated with Atto647N, the behavior of peptides labeled with alternate fluorophores was identical (fig. S1, A and B). These data confirmed that there were no detectable dye-specific interactions between the lig-and and the supported membrane. The pMHC class II molecules used here diffused freely and remained monomeric on the supported membrane, which was experimentally verified by single-step photobleaching observations (9). Although MHC class I clusters on APCs (18–22), whether multiple pMHC class II loaded with the same antigen cluster on APCs remains controversial (23–25). In these studies, we focused on unclustered pMHC class II.
After exposure of live cells to the functionalized supported membranes, the interaction of leukocyte function-associated antigen 1 (LFA-1) and ICAM-1 initiated T cell spreading. These interactions facilitated formation of an essentially planar junction between individual T cells and the supported membrane (Fig. 1A), which functioned as a synthetic APC (Fig. 1B). Initial spreading was followed by pMHC binding to TCRs at the T cell-APC interface. All experiments were performed at 37°C.
Fig. 1. Detection of single pMHC-TCR binding events that initiate T cell signaling.
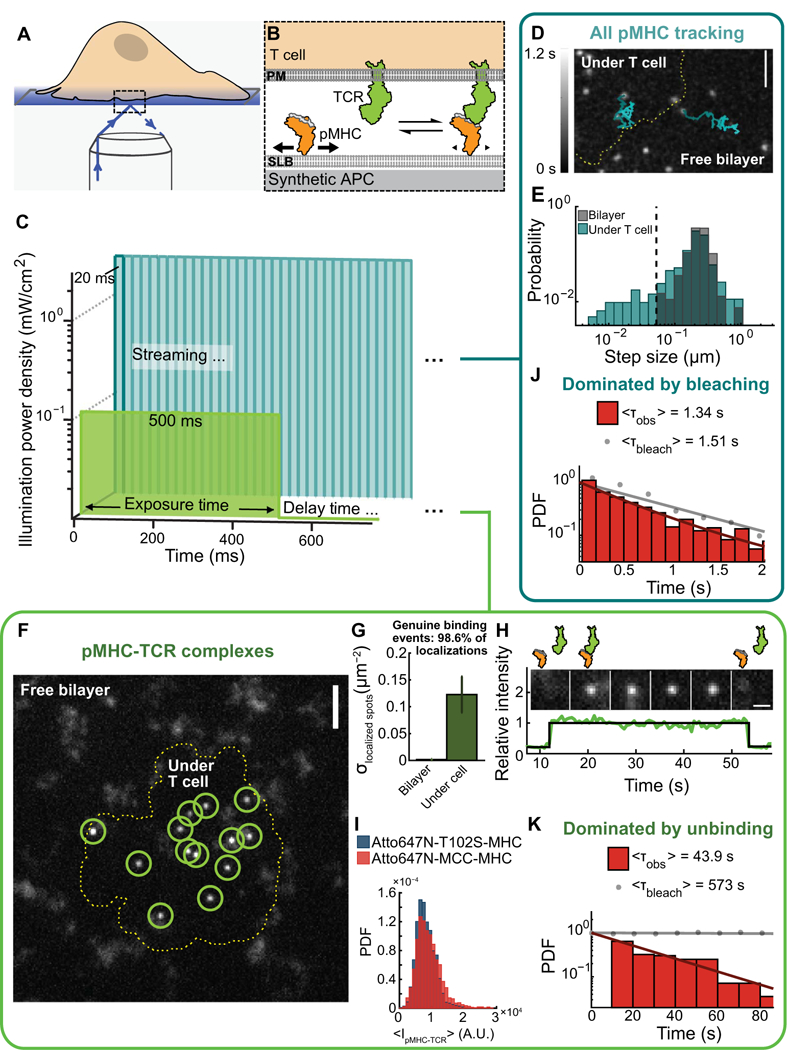
(A and B) Outline of TIRF microscopy experiments in which live, primary T cells form interfaces with its plasma membrane (PM) and a supported lipid bilayer (SLB) and the intermembrane junction is selectively imaged (A). Within the lipid bilayer, freely mobile pMHC (bold arrows) experience rapid diffusion, whereas TCR-ligated pMHC (small arrows) have reduced mobility (B). (C) pMHC-TCR binding events were detected either using short exposure times and high-power intensity by streaming acquisition (no delay time between frame) (cyan) or using long exposure times and low-power intensity at matched energy input after a delay time (green). (D)Sin-gle pMHC trajectories observed using short exposure times and high-power intensity microscopy imaging of the T cell contact site (dashed line) and supported membrane. The trajectories (n > 100) are representative of at least three independent experiments. Scale bar, 3 μm. (E) Step-size distribution of single MCC pMHC molecules shows bimodal mobility under a T cell (cyan) and unimodal mobility on the free supported membrane (gray). Step sizes were calculated for all steps in >4000 trajectories from three independent experiments. (F) Localization of single pMHC-TCR complexes using long exposure times and low-power intensity imaging in the free bilayer or under a T cell (dashed line). Images are representative of at least 20 independent experiments. Scale bar, 3 mm. (G) Density of localized particles on free bilayers and at the T cell contact site. Data are means ± SEM of three independent measurements. (H) Single pMHC-TCR binding and unbinding over time determined by microscopy. Images (top) and intensity traces (bottom) are representative of 20 independent experiments. (I) Single pMHC-TCR complexflu- orescence intensity distributions determined by microscopy. Probability density function (PDF) plot of the mean intensity of the 2×2 pixels around a localized centroid for all steps in all trajectories is from three independent experiments. (J) Dwell time distribution of MCC-Atto647N-MHC (red) using short expo sures and high-power intensity imaging with-out a time delay and corrected for fluorophore photobleaching (gray). Data are from the analysis of >2300 trajectories from five cells in three independent experiments. (K) Dwell time distribution of MCC-Atto647N-MHC using long exposure and low-power intensity imaging after a 10-s time lapse and corrected for fluorophore photobleaching (gray). Data are from the analysis of >1100 trajectories from nine cells in three independent experiments. A.U., arbitrary units.
Individual pMHC molecules were visualized by total internal reflection fluorescence (TIRF) microscopy (Fig. 1, A and B). All pMHC molecules could be resolved using short exposure times (20 ms) and high laser illumination intensity (2.5 mW/cm2, Fig. 1C). The motion of individual molecules, under the cell and on free bilayers, was tracked using streaming acquisitions. In the absence of a T cell, pMHCs on the bilayer underwent simple Brownian motion, which was well described by a unimodal step-size distribution (Fig. 1, D and E). After T cell landing, some pMHC molecules bound TCR to form pMHC-TCR complexes. These bound complexes were detectable in the step-size distribution, which became bimodal with a fast-moving population (corresponding to free pMHC ligands) and another population with slowed mobility (corresponding to pMHC-TCR complexes; Fig. 1, D and E) (9).
The bound pMHC-TCR complexes were conveniently discriminated from free pMHC ligands using long exposure times (500 ms) and low laser illumination intensity (0.1 mW/cm2), maintaining an equivalent energy dosage per exposure (Fig. 1, C and F, and fig. S1, A to E). Under these imaging conditions, fast-moving, free pMHC were blurred out, and only the TCR-bound pMHC-TCR complexes were detected as discrete puncta. Artifact signals would be produced where pMHC molecules were immobilized in defects in the supported bilayer. We measured the density of these artifacts in each experiment by examining bilayer regions outside the cell-contact zone where pMHC-TCR binding could not occur. Typical artifact densities in these experiments were 0.0017 ± 0.0014 mm−2, which compared with measured pMHC-TCR binding event densities of 0.12 ± 0.033 μm−2, which indicated that ~98% of observed events were genuine pMHC- TCR complexes (Fig. 1G). We note that this high precision in bound complex detection required extreme attention to detail in the formation of the supported membrane (9, 17). Further confirmation that the slowed complexes were indeed pMHC bound to TCR on the T cell was provided by control experiments using a null pMHC, which was incapable of binding TCR and did not exhibit slowed pMHC-TCR complexes (fig. S1E).
The pMHC-TCR binding dwell time, as well as any spatial transport of the complex, could be resolved using the low power, long exposure strategy (Fig. 1F). We confirmed that these pMHC-TCR complexes are single molecules by single-step intensity loss observations and molecular brightness (Fig. 1, H and I) (9). We found that the molecular binding kinetics of the pMHC-TCR interactions examined here were long (mean dwell times in seconds to tens of seconds, depending on the peptide). These dwell times were too long to be resolved by streaming imaging because the mean time to photobleach under those conditions was less than 2 s (Fig. 1J). The long-exposure method discriminated bound pMHC-TCR in each image and thus allowed imaging exposures to be spaced as needed to accommodate the molecular binding kinetics (Fig. 1, C and K). Using a delay time between 500-ms exposures of 10 s, the mean time to photobleach was 573 ± 187 s. For most of the experiments described here, individual pMHC-TCR complexes were typically spaced micrometers apart within the T cell-supported membrane interface; thus, complexes could be unambiguously tracked from frame to frame. We note that pMHC-TCR complexes moved under directed motion [by coupling to the retrograde flow of the cytoskeleton (26–28)] at the average velocity of ~0.2 μm/s, and this movement was tracked as well. Ligand densities of 0.1 to 2 molecules/μm2 (see Materials and Methods for density estimate calculation; fig. S2) were near the physiological agonist pMHC densities on APCs (29–33). This density range was at least an order of magnitude below the regime where stable pMHC-TCR microclusters were formed; instead, single pMHC-TCR molecular complexes predominated (28, 34–42). Overall, the combination of long bleach time and spatially isolated pMHC-TCR complexes enabled accurate measurement of pMHC- TCR binding dwell times for all pMHC studied here (Fig. 1K).
The biochemical signaling pathways downstream of triggered TCRs are well understood (5, 6). Although many issues concerning the quantitative features of TCR signaling remain unresolved, the basic sequence of events is known (Fig. 2A). In the standard view, TCR recognition of pMHC stimulates Lck-mediated phosphorylation, which initiates the recruitment and phosphorylation of the kinase ZAP-70 (zeta chain-associated protein kinase 70). Subsequently, photobleaching (gray). Data are from the analysis of >1100 trajectories from nine cells in three independent experiments. A.U., arbitrary units. ZAP-70 phosphorylates the scaffold protein, linker for the activation of T cells (LAT) (43, 44), required for the formation of large-scale assemblies with adaptor protein Grb2 (growth factor receptor-bound protein 2) and guanine nucleotide exchange factor SOS (45–48). These LAT:Grb2:SOS assemblies, which have also been identified as phase-separated protein condensates (46–50), promote activation of the guanosine triphosphatase Ras and MAPK (mitogen-activated protein kinase) signaling (51, 52). LAT assemblies also recruit the enzyme phospholipase C-γ to generate inositol 1,4,5-triphosphate and in response to TCR ligation (Fig. 2C and movie S1). In this study, we focused on the relationship between pMHC-TCR binding event sequences and NFAT translocation, which we here refer to as the molecular impulse-response function.
Fig. 2. A single-cell assay measures both T cell input accumulation and activation.

(A) TCR recognition of pMHC initiates an intracellular signaling cascade in T cells that culminates in nuclear translocation of the transcription factor NFAT and IL-2 production. PLC-γ, phospholipase C-γ; PIP2, phosphatidylinositol 4,5-bisphosphate;IP3, inositol 1,4,5-triphosphate. (B) Experimental schematic of the single-cell pMHC-TCR binding input and the NFAT nuclear translocation response assay. (C) Images of NFAT-mCherry transduced T cell landing and spreading (green, dashed lines), binding pMHC (green circles), and NFAT nuclear translocation (purple bar) after interaction with a bilayer containing MCC-Atto647N-MHC. Cell landing was determined by a dark signature in RICM (yellow arrow), and activation time (t = 0) was defined as the first observation of NFAT translocation (purple dotted line). Images are representative of at least 30 independent experiments. Scale bar, 3 μm. (D) Analysis of NFAT activation determined by the ratio of mean nuclear intensity to mean cytosolic intensity. Epifluorescent images of an NFAT localization in the nucleus (purple line) or cytosol (cyan line) of an activating (left) and nonactivating (right) cell are representative of at least 30 independent experiments. (E) The distribution of single-cell NFAT-green fluorescent protein (GFP) abundance in T cells stimulated on an MCC-MHC bilayer as a function of activation state. Single-cell NFAT-FP intensity measurements at the time of initial T cell landing are pooled from 12 independent experiments.
In these experiments, the binding of pMHC and TCR was measured beginning from the initial contact between the T cell and the functionalized supported bilayer. T cell landing and the formation diacylglycerol, which initiates calcium flux and the sequential activation of calmodulin and calcineurin (53). Calcium signaling is an early T cell response that results in the translocation of the transcription factor NFAT to the nucleus, which coordinates with the AP1 (activating protein 1) transcription factor complex to drive interleukin-2 (IL-2) production (13,54, 55). To monitor single-cell decision-making in response to pMHC-TCR binding event sequences (Fig. 2B), we retrovirally transduced the primary T cells with a truncated, fluorescent protein-tagged NFAT sensor (54), which undergoes nuclear translocation of a large interfacial contact were detected over time using reflection interference contrast microscopy (RICM) (Fig. 2C; movie S1; and fig. S3, A and B). Single-molecule pMHC-TCR TIRF images were acquired at the same time as epifluor-escence images of NFAT sensor distribution (Fig. 2C and movie S1), as described (54). At a focal depth of 3 to 6 μm above the supported membrane-live cell interface, we could clearly visualize both the cytoplasmic and nuclear NFAT pools (Fig. 2, C and D, and fig. S4). We found that T cells that received activating signals initiated nuclear translocation of NFAT within 3 to 5 min, independent of reporter abundance (Fig. 2, C and E, and fig. S4). NFAT translocation preceded spatial segregation of pMHC-TCR and ICAM-1-LFA-1 at the intermembrane junction (fig. S3A), as expected (56). For the purposes of this study, activation was defined as the time of the first detection of NFAT nuclear localization, which was accomplished here with a 30-s temporal resolution (Fig. 2C). T cells continued to accumulate pMHC- TCR binding events after NFAT trans-location (Fig. 2C), and these additional binding events may be required to sustain calcium signaling (57). For each cell, the molecular impulse-response assay recorded pMHC-TCR binding events leading up to and beyond NFAT translocation. In the following analyses, we restricted our focus to the initial translocation of NFAT.
Although photobleaching was minimized with the staggered, long-exposure imaging methods, it could not be ignored. Some pMHC would be photobleached before binding TCR. On the basis of measured bleaching rates, we calculated the number of such unseen events to be no more than 40% in any of the experiments, and often much lower than that. Because photobleaching events accumulated over time, these bleaching limits represented the maximum number of bleached pMHC at the end of the experiment. During the primary course of signaling early on, far fewer binding events were missed. We performed a detailed analysis of photo-bleaching probabilities as a function of time for the different experimental conditions (fig. S5).
Constructing the pMHC-TCR binding event sequence
As a first step, we mapped the sequence of pMHC-TCR binding events and their dwell times leading up to NFAT translocation. The initial time and location of each pMHC-TCR binding event was detected, and the bound complexes were tracked through time (Fig. 3A). In parallel, NFAT nuclear translocation was monitored to detect the time at which activation occurred (marked by the arrow in Fig. 3A). These data could be distilled into the sequence and duration of binding events (Fig. 3B).
Fig. 3. Space, time, and kinetics of pMHC-TCR molecular binding events contribute to activation.

(A) The location, time, and duration of a subset of the pMHC-TCR binding events (color tracks) experienced by a cell (white outline) relative to NFAT activation. Scale bar, 3 μm. (B) Dwell time of all pMHC binding inputs experienced by a single T cell from initial contact (t = 0) until end of acquisition. Each line represents a single pMHC-TCR trajectory. Time of NFAT activation indicated by a purple dashed line. Data in (A) and (B) are representative of >20 independent experiments. (C) Dwell time of T102S-Atto647N-MHC (blue) and MCC-Atto647N-MHC (red) using long exposure and low-power intensity microscopy after 3- and 10-s time lapses, respectively. The mean dwell time (<τoff>) was determined after data fitting (colored lines) and corrected for fluorophore photobleaching (gray). The probability distributions were determined from the analysis of >3000 trajectories from at least 25 cells. (D) Density-dependent change in NFAT activation for a population of AND TCR T cells on T102S pMHC (blue) and MCC (red) bilayers. Peptide densities that approach a half-maximal NFAT response were designated as threshold values [gray dashed lines, σ(MCC) = 0.12 μm−2, σ(T102S) = 0.90 μm−2]. Data are from >30 cells per condition from five independent experiments.
Experiments were performed with a panel of AND TCR-specific agonist peptide ligands of varying TCR affinities and correspondingly different potency with respect to IL-2 secretion (1, 10, 58). For all pMHC-TCR combinations, the distributions of observed in situ dwell times were reasonably well described by a single exponential (Fig. 3, C and D, and fig. S6), indicative of first-order dissociation kinetics. The mean molecular binding dwell times (<τoff>) were calculated using rates extracted from a mathematical fit of the experimental data and calibrated for photobleaching and ranged from 9.6 to 47.5 s. These data are generally consistent with the known in vitro pMHC-TCR binding kinetics of these peptide ligands (1). However, because the imaging methods used here established a temporal detection window that excludes the shortest dwell events (τoff < 500 ms), data fit analysis was essential since the average of measurements will not reflect the true average. Peptide strength was further characterized based on the NFAT activation threshold from the average pMHC density that resulted in activation of half of the cells in the population after 20 min of exposure to the supported membrane (Fig. 3D). The data confirmed that NFAT activation thresholds correlated inversely with themean pMHCdwell times (Fig. 3, C and D), as expected (10).
The mean binding dwell time between MCC [Moth cytochrome C 88–103 peptide (MCC88–103)] pMHC and AND TCR (47.5 s) is more than an order of magnitude longer than the mean dwell time correlated with activation (2,7). Thus, MCC pMHC is an exceptionally strong ligand that enabled exploration of the response of the TCR signaling system to extremely long dwell times. The binding kinetics of the T102S pMHC used here (<τoff> = 9.6 s), may be more representative of physiological ligands (2, 7). Specifically, the naturally originated 5c.c7 recombinant TCR binding to its MCC pMHC ligand (<τoff> = 7.2 s) is quite similar to AND TCR binding to T102S pMHC (9, 10). We note that the only difference between AND and 5c.c7 TCRs are modifications in the hypervariable pMHC binding site, leading to the altered kinetics to MCC pMHC (59, 60). In the subsequent sections, the behaviors of the strong agonist MCC pMHC and the weak agonist T102S pMHC were compared, covering a range of fivefold difference in affinity. Results for the moderate agonist K3 pMHC are provided in the Supplementary Materials.
Separating activating and nonactivating binding event sequences
For a population of T cells, all interacting with the same pMHC/ICAM-1 membrane, each cell experienced a unique sequence of pMHC-TCR binding events (fig. S7, A and B). At pMHC densities equal to or below the threshold activation density, some cells could experience binding event sequences leading to activation, whereas the others failed to activate. Here, we compared activating and nonactivating binding event sequences to examine on what basis the cellular decision to activate was made (Fig. 4, A and B, and fig. S8, A to D).
Fig. 4. Dwell time accumulation does not set the activation threshold.

(A and B) The number of pMHC-TCR binding events (A) and their total dwell time (B) that T cells accumulated at the time of initial NFAT activation. Cells were exposed to distinct input densities with different NFAT activation probabilities: 32% at threshold T102S pMHC density (blue), 41% at threshold MCC density (red), and 5% at low T102S density (dark blue). Scatterplot of single-cell values after correction for events that are not directly observed with population means (black) ± SD (gray) and estimated population distributions (right) from 11 cells from five experiments (Threshold MCC), 15 cells from seven experiments (Threshold T102S), or 7 cells from three experiments (Low T102S).
The simplest activation models suggest that T cells either count the number of pMHC-TCR binding events or integrate their dwell times (61, 62). We tested these hypotheses in the following experiments by comparing cell behavior under three different conditions: MCC pMHC at threshold activation density, T102S pMHC at threshold density, and T102S at low density. Under each of the conditions used, strict control over pMHC presentation in supported membranes, monitored by direct single-molecule pMHC counting, confirmed that cells were exposed to the specified average ligand density (fig. S8A). Individual cells sampled random variations in density, such as those expected for pMHC class II on the surface of an APC (29–31). Cells at low T102S pMHC density took longer to translocate NFAT than those at threshold densities of either MCC or T102S pMHCs (fig. S8D).
The number of pMHC-TCR binding events before NFAT translocation for activating binding sequences was compiled (Fig. 4A and fig. S8B). At their threshold densities, more T102S pMHC binding events were observed in activating cells than MCC binding events. This result was expected because the threshold density of T102S pMHC was higher than that of MCC pMHC (Fig. 3D and fig. S2), and the kinetic on-rate (kon) for different pMHCs were similar (10). At low T102S density, ~5% of the total number of T cells were still able to activate. These rare cells, by random chance, may have experienced more pMHC-TCR binding events than average and thus resemble cells exposed to the higher threshold T102S pMHC density. However, the data indicated that, at low T102S pMHC density, T cells that activated NFAT accumulated fewer pMHC-TCR binding events than those at threshold T102S pMHC density (Fig. 4A).
Analysis of the total integrated pMHC binding dwell time indicated that, at threshold densities of MCC and T102S pMHC, T cell activation required a total binding time of ~1100 s, irrespective of peptide affinity (Fig. 4B and fig. S8C). Although these data may suggest that total pMHC dwell time may be the defining parameter on which cellular activation is based, T cell activation at low T102S pMHC density indicated otherwise. We found that, when T cells activate NFAT in response to low T102S density, they exhibit nearly half the cumulative dwell time (648 ± 110 s) as T cells stimulated with threshold density peptide (Fig. 4B). Furthermore, cells that failed to activate at threshold densities were capable of reaching similar integrated dwell times (fig. S8C). In addition, the rate of dwell time accumulation for T cells that activated NFAT at low T102S density was indistinguishable from that of cells that did not activate (fig. S9). Thus, it was clear that neither integrated binding time nor rate of binding time accumulation was the activation set point.
The integrated total number of binding events and dwell time values were independent of fluorescent labels for the peptide ligand and NFAT (fig. S10). Signal integration from binding inputs was diminished by inhibiting Lck, resulting in cells that accumulated signals at a slower rate and activated with a lower probability (fig. S11, A to E). In the presence of the costimulatory ligand CD80, cells that activated NFAT experienced a decrease in the time to activation, accumulated binding events, and cumulative dwell time (fig. S12). Because neither the cumulative binding events nor integral dwell time correlated with T cell activation, the rare, activating cells were either tuned differently than the average T cells in these populations, or there were other features in the sequences of binding events that determine activation.
Correlated binding events activate T cells stimulated with weak agonist
We hypothesized that T cell activation, particularly for rare, activating cells at low T102S pMHC density, may result from coordination of pMHC-TCR inputs in both space and time. In terms of a molecular mechanism, this amounted to multiple short pMHC-TCR binding events that were sufficiently close in space forming a temporal sequence, contributing to the same localized intracellular signaling reaction, thus resembling a longer binding event. Spatial manipulation of pMHC-TCR binding events can alter phosphorylation levels of membrane proximal proteins and downstream calcium responses (27, 63). In addition, temporal modulation of binding events by mechanically controlling the contact between pMHC and TCR in a micropipette assay demonstrates that input frequency may influence calcium flux (64,65). As a first approach to test this hypothesis, we measured the nearest-neighbor distances between binding events in the single-cell activating sequences.
We analyzed the spatial pair distribution of pMHC-TCR binding events that occurred within 30 s of each other on T cells that did or did not activate NFAT (Fig. 5, A to H). At threshold densities of pMHC, we found that the spatial correlation of pMHC binding was grossly similar regardless of NFAT activation status (Fig. 5, C, D, F, and G). However, for the rare T cells that activated NFAT in response to low T102S pMHC density, we found that 90% of the binding events occurred within 2 mm and 30 s of another binding event (Fig. 5H). In contrast, for binding events on T cells that did not activate NFAT in response to low T102S pMHC density, there was a lower frequency (68%) of nearest-neighbor pMHC binding events within the same space and time (Fig. 5H). This spatiotemporal correlation was not found when binding events occurred simultaneously, suggesting that coupling inputs must occur over time (Fig. 5, C to E, and fig. S13). In other words, short pMHC-TCR binding events must occur sequentially to be summed by the cell. Simultaneous binding events occurring near to each other in space appeared not to be added.
Fig. 5. Rare, activating cells exposed to weak agonist exhibit correlated binding events.

(A to B) Representative nearest-neighbor distance analysis for the proximity between pMHC-TCR binding events on T cells stimulated with MCC pMHC (A) or T102S (B) at comparable densities. For a given pMHC-TCR binding event (orange circle), the distance to its nearest neighbor (yellow line) at the same time point (left) or within a time window (right) was determined from analysis of the data in Fig. 4. Scale bars, 3 μm. (C to H) Nearest-neighbor distance distribution of pMHC-TCR binding events that occurred concurrently (C to E) or within a 30-s time window (F to H) in response to threshold MCC pMHC density (C and F), threshold T102S density (D and G), and low T102S density (E and H) stimulation. Gray areas indicate the fraction of nearest-neighbor separations up to 2 mm in cells that activated NFAT (Act.; filled bars) and those that did not (Nonact.; empty bars) from the analysis of the data from Fig. 4.
We explored the spatiotemporal correlation of binding events more closely by examining how multiple short binding events may be stitched into one, apparently long-lived event. In this ad hoc correlation analysis, individual pMHC-TCR binding events were linked into an effective correlated track if a new binding event occurred within both the spatial neighborhood and the temporal window of an existing binding event (Fig. 6A). We defined the spatial neighborhood as a region within a specified radial distance around the location of the pMHC-TCR complex and the temporal window as a time interval from the current position of the pMHC-TCR complex to some later time in the input accumulation period. Uncorrelated tracks were unaffected by this filtering. A 30-s temporal window was selected on the basis of the observation that 30 s was the shortest time window in which spatiotemporal coupling was detected (Fig. 5H and fig. S13).
Fig. 6. T cells group spatiotemporally correlated pMHC-TCR binding events to create long-dwelling inputs.
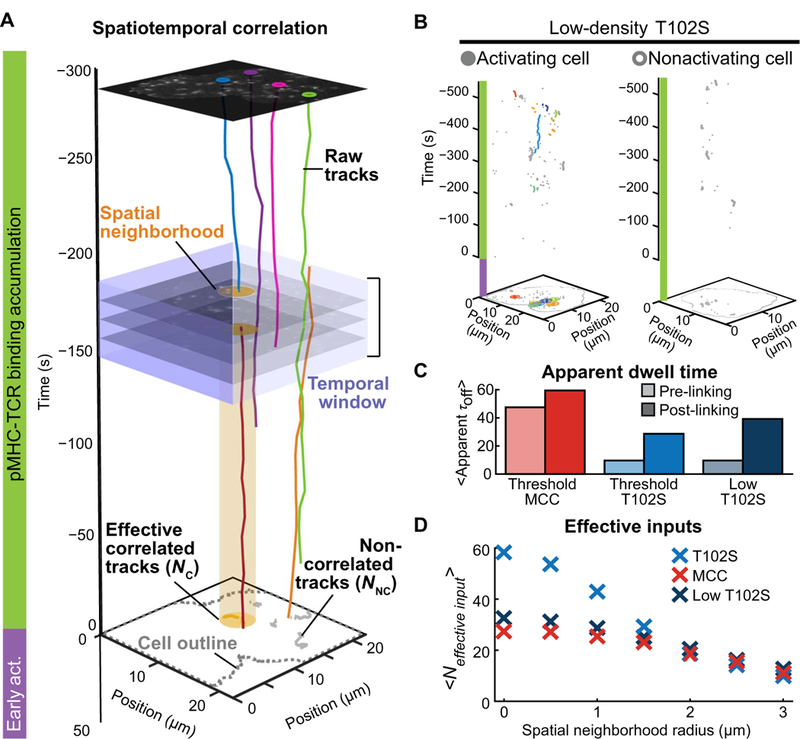
(A)Schematic of the spatiotemporal correlation analysis. Raw data are shown with selected single-molecule pMHC-TCR trajectories that occurred while the cell accumulated impulses (green bar) before activating NFAT (purple bar). Correlated tracks were coupled given coincidence within a neighborhood (yellow disc) and a 30-s time window (purple square region) and require one trajectory to be initiated during that window. Trajectories satisfying these criteria were combined into an effective correlated track (NC) and displayed within a cluster on the two-dimensional projection of accumulated tracks (yellow). Trajectories that did not satisfy the criteria were considered noncorrelated tracks (NNC) (gray). (B) Representative spatiotemporal correlation of a cell that translocated NFAT (left) and one that did not (right) at low T102S pMHC density. Raw correlated tracks (colored) and noncorrelated (gray) pMHC-TCR trajectories were from the analysis of the data in Fig. 4. (C) The mean dwell time of cells that activated NFAT on threshold MCC pMHC density (red), threshold T102S density (blue), and low T102S pMHC density (dark blue) before and after spatiotemporal correlation (light and dark shades, respectively) is from the analysis of the data in Fig. 4. (D) Number of effective inputs after spatiotemporal correlation analysis of the data from Fig. 4 as a function of neighborhood radius.
At low T102S pMHC density, when T cells activated NFAT, we found that new binding events exhibited a high probability of occurring close within space and time to an existing event (Fig. 6B). If these events indeed feed into the same localized signaling reaction, then the result could resemble long-dwelling MCC pMHC binding inputs in terms of mean dwell time (Fig. 6C and fig. S14, A to C). Furthermore, our analysis indicated that, in response to low T102S pMHC density, T cells that activated NFAT accumulated between two and nine correlated pMHC tracks, whereas T cells that did not activate NFAT experienced zero to three correlated pMHC tracks (figs. S15 and S16A). At threshold conditions, T cells activating NFAT had more correlated tracks than T cells that did not activate, and all cells exposed to T102S pMHC had more spatiotemporally correlated pMHC tracks than those exposed to MCC (figs. S15 and S16B). Further analysis found that, after linking pMHC tracks within 2 μm and 30 s, all activating cells collected the same number of total effective inputs, irrespective of pMHC affinity (Fig. 6D). Together, our data indicated that sustained input, consisting of either a single long-dwelling binding event or a sequence of spatially correlated events, was important for T cell activation (fig. S17).
DISCUSSION
The experiments reported here constitute an effort to map the specific sequence of individual pMHC binding events experienced by a T cell to that cell’s decision to activate. Our data revealed that the molecular threshold for activating a T cell was apparently set to a longer average pMHC-TCR binding dwell time than predicted by population-averaged experiments. Effectively, T cells seemed to respond to the tail end of long-lived pMHC-TCR binding interactions [Fig. 6C and fig. S14 (A to C)], exhibiting a disproportionate sensitivity to binding events lasting at least an order of magnitude longer than the population average activation threshold of 3 s (2, 7). In addition, we observed that a sequence of short pMHC-TCR binding events that were sufficiently close in space and time mimicked long-dwelling events in terms of the cellular response. For a physiological agonist pMHC with a mean dwell time typically of a few seconds, the probability that an individual pMHC-TCR binding event lasted for tens of seconds was vanishingly low (Fig. 2, C and D, and fig. S6, see Materials and Methods for detailed calculation). Thus, the spatiotemporal correlation between a series of short binding events could have a prominent influence on T cell activation under both physiological and disease conditions. The fact that no such summation was observed for simultaneous binding events occurring near each other in space further underscores the importance of sustained pMHC-TCR signal in the cellular decision to activate.
There are possible alternate interpretations of the results that we describe here. For example, it may be that the rate of pMHC-TCR binding event accumulation determines T cell activation. However, we found that the rate of pMHC-TCR dwell time accumulation for cells that activate NFAT in response to low T102S pMHC density was indistinguishable from cells that did not active NFAT at threshold densities (fig. S9), which suggests that the rate of binding time accumulation was not a defining characteristic of activating cells. It is also possible that short pMHC-TCR binding events (τoff « 3 s), which were not detected here, provide an invisible contribution to T cell behavior. Single-molecule tracking (11) and fluorescence resonance energy transfer (FRET) (12) studies suggest that T cell activation may correlate with accelerated pMHC-TCR unbinding kinetics in a similar system. Although this superficially seems to contrast the long dwell times reported here, as acknowledged in the tracking study, the fast photobleaching rates used may prevent observation of long-lived pMHC-TCR complexes. Similarly, in the FRET study, it remains unclear whether the loss of FRET signal between pMHC and TCR represents complete unbinding or whether this may correspond to an alternative structural configuration of the bound complex. Such a complex might be predicted on the basis of recent studies of mechanically stimulated TCR activation (66). Nonetheless, it is possible that there is a rapidly unbinding pMHC-TCR species, not observed in the present study, that participates in signaling. However, the short-lived ER60 pMHC ligand fails to activate T cells at any density (10, 67). Thus, longer dwelling events are at least a requirement. For the long binding MCC pMHC (<τoff> = 47.5 s), a substantial fraction of the all pMHC under the T cell are bound as long-lived species, which suggests that there cannot be many unseen short binding events. Ultimate resolution of this issue will require more advanced imaging strategies to establish a broader temporal window to capture a more complete picture of all pMHC-TCR binding events experienced by the cell.
Signal integration from multiple ligands may promote the assembly of the LAT signalosome. LAT is phosphorylated on multiple tyrosine sites by ZAP-70, which is activated by ligated TCR (43, 44). Phosphorylated tyrosine sites on LAT allow Grb2 binding, which in turn recruits SOS, which can lead to a networked assembly of LAT:Grb2: SOS by a gelation or liquid-phase transition mechanism (46, 51). These LAT assemblies elongate Grb2 and SOS interaction times, and it has been speculated that this dynamic modulation could increase the probability of full SOS activation and provide a type of kinetic proofreading mechanism governing downstream activation of Ras by SOS (47). The LAT signalosome assembly process resembles a phase transition and exhibits an extreme non-linearity with respect to the degree of LAT phosphorylation. As a consequence, LAT assembly may be sensitive to spatio-temporally correlated pMHC-TCR binding events.
The distinct pMHC-TCR binding events observed in these experiments almost certainly involve different TCRs, in light of the vast excess of TCR on the T cell surface over pMHC within the supported membranes. Similarly, it is likely that different pMHCs are involved as well because, once unbound, pMHC rapidly diffuse away. This suggests that integration of multiple binding events by the same TCR, such as the input accumulation sequence predicted by the serial rebinding model (62) and other related models (68, 69), is unlikely to occur in these experiments. Some physical confinement of both TCR and pMHC would be required for such repeated engagements to occur. Although such confinement is conceivable under some physiological conditions, the findings that we report here suggest that confinement is not necessary. Even a pMHC-TCR pair with particularly fast kon, which would be more prone to serial engagements of TCR in a spatially correlated manner, is still likely to bind different TCR due to the excess of TCRs in our system. We found that the binding events between different TCR and pMHC ligands could be integrated within the cell when these events form a temporal sequence close in space. We suggest that the molecular memory of spatiotemporally correlated pMHC binding events may be recorded in the phosphorylation state of local LAT molecules and assemblies.
There is a practical need to understand detailed molecular mechanisms underlying T cell activation. Directing the T cell response for tumor clearance is a core element of cancer immunotherapy, which presents great therapeutic promise as well as challenges (70, 71). The insights we have described here may be informative in the development of therapeutic agents to mobilize T cell activation. Current bispecific antibodies such as ertumaxomab, blinatumomab, and EGFRBi induce responses by cross-linking a TCR and a ligand on an apposed cell, such as the surface receptors HER2, CD19, or endothelial growth factor receptor, respectively (72–74). Antibody-based agents typically bind targets with nanomolar affinity, which is an order of magnitude stronger than that of the physiological pMHC-TCR interactions (1, 75). The results presented here suggest that weaker bispecific antibodies may be more effective T cell activators if they are present at a high enough density to enable correlated binding event sequences. Alternatively, extremely strong binders might be active at exceptionally low densities. The methods that we have described here offer a new spectrum of possibilities to examine the individual molecular events underlying signaling in living cells.
MATERIALS AND METHODS
Protein purification
Histidine-tagged MHC class III-EK and ICAM-1 were expressed and purified as previously described (17). MHC II with C-terminal hexahis- tidine tags on both α and β chains were expressed using a baculovirus expression system in S2 cells and purified using a Ni-nitrilotriacetic acid (NTA) agarose column (Qiagen). The histidine-tagged MHC bacmid (76, 77)was a gift of L. Teyton (Scripps Research Institute) and M. Davis (Stanford University). The bacmid for ICAM-1 with a C-terminal dec-ahistidine was synthesized, and it was similarly expressed and purified in High Five cells (Invitrogen) (17).
DNA constructs
A plasmid containing a GFP fusion to the regulatory domain of the murine NFAT1 (NFATc2) protein in a murine stem cell virus vector (54) was a gift of F. Marangoni (Harvard Medical School). This truncated form of NFAT1 [pMSCV-NFAT1(1–460)-GFP] contains the regulatory domain that controls the nucleocytoplasmic shuttling of NFAT but lacks the DNA binding domain. A second version of the plasmid was generated to replace the GFP coding sequence with mCherry.
Peptide synthesis and labeling
Moth cytochrome C 88–103 peptide (MCC88–103; abbreviated as MCC) and previously characterized variants (12, 78) were synthesized and lyophilized on campus (D. King, Howard Hughes Medical Institute Mass Spectrometry Laboratory at University of California, Berkeley) or commercially (Elim Biopharmaceuticals, Hayward, CA). A short flexible linker of three amino acids and terminal cysteine was added to the C terminus for fluorophore labeling. The sequences are as follows: MCC (ANERADLIAYLKQATK), MCC(C) (ANERADLIAYLKQATKGGSC), K3 (ANERADLIAYPKAATKF), K3(C) (ANERADLIAYPK-AATKFGGSC), T102S (ANERADLIAYLKQASK), T102S(C) (ANERA-DLIAYLKQASKGGSC), and Null(C) (ANERAELIAYLTQAAKGGSC).
For dye conjugation, the cysteine-containing peptide sequences were reacted with the maleimide-containing organic fluorophore of interest (Atto647N, Atto565, or Atto488; Atto-Tec GmbH, Siegen, Germany) in phosphate-buffered saline (PBS) with a trace amount of 1-propanol, as previously described (9). The labeled peptides were purified using a H2O/acetonitrile gradient on a C18 reverse-phase column (Grace Vydac, Deerfield, IL) in the AKTA Explorer 100 FPLC system (Amersham Pharmacia Biotech, Piscataway, NJ). Mass spectrometry was used to confirm the peptide identity after purification.
T cell culture and transduction
CD4+ T cells from the lymph nodes and spleens of 6- to 20-week-old AND × B10.BR transgenic mice (Jackson Laboratory) were stimulated with MCC peptide in vitro, as described (9, 36). All animal work was performed with prior approval by Lawrence Berkeley National Laboratory Animal Welfare and Research Committee under the approved protocol 17702.
Activated T cells were retrovirally transduced with NFAT-GFP or NFAT-mCherry using Platinum-Eco cell-derived supernatants. Platinum-Eco cells (Cell Biolabs, San Diego, CA) were transfected with the desired plasmid using linear, polycationic polyethylenimine (Sigma-Aldrich) or Lipofectamine 2000 (Thermo Fisher Scientific). After 24 to 48 hours, retrovirus-containing supernatant was used to spinfect T cells in the presence of polybrene (4 μg/ml) on days 3 and 4 after activation. NFAT-GFP-positive cells were sorted using fluorescence-activated cell sorting based on viability and FP expression. The entire NFAT-GFP-positive population was used for imaging experiments; 0.5 million to 2 million cells were exposed to each bilayer.
Sample and bilayer assembly
Glass-supported lipid bilayer membranes were prepared in imaging chambers and functionalized with proteins using standard protocols (9). At 18 to 24 hours before imaging, MCC and variant peptides were loaded onto MHC III-EK at 37°C in peptide-loading buffer [1% (w/v) bovine serum albumin (BSA) in PBS (pH 4.5) with citric acid]. Just before exposure to bilayers, dye-peptide-MHC complexes were purified using a 10,000-molecular weight cutoff spin concentrator (Vivaspin 500, GE Healthcare, Pittsburgh, PA).
Small unilamellar vesicles (SUVs) are formed using tip sonication with the composition of 98 mole percent (mol %) 1,2-dioleoyl-sn-glycero-3-phosphocholine and 2 mol % 1,2 dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt) (Ni2+-NTA-DOGS) (Avanti Polar Lipids, Alabaster, AL) in Milli-Q water (EMD Millipore, Billerica, MA). In addition, #2 40-mm-diameter round coverslips were ultrasonicated in 1:1 isoporopyl/H2O and etched for 5 min in piranha solution (3:1 H2SO4/H2O2). FCS2 Closed Chamber Systems (flow cells; Bioptechs, Butler, PA) were assembled using etched coverslips and flushed with 1× tris-buffered saline [TBS; 19.98 mM tris and 136 mM NaCl (pH 7.4); Mediatech Inc., Herndon, VA]. SUVs in 2× TBS were introduced into the chambers, and bilayers were allowed to form through vesicle rupture for at least 30 min. After rinsing, the bilayer was activated with 100 mM NiCl2 for 5 min. Samples were exchanged to a live T cell imaging buffer [LCB; 1 mM CaCl2, 2 mM MgCl2, 20 mM Hepes, 137 mM NaCl, 5 mM KCl, 0.7 mM Na2HPO4, 6 mM D-glucose, and 1% (w/v) BSA]. Immediately before imaging, the bilayer was incubated for 35 min with pMHC and ICAM-1 at appropriate concentrations (~100 nM) in LCB. The bilayer was rinsed with imaging buffer after the incubation, and the His-tagged/Ni-NTA- bound proteins were allowed to equilibrate on the bilayer for 35 min. The resulting supported membranes typically display ICAM-1 at 100 to 200 μm−2 and pMHC at 0.1 to 2 μm-2. The chamber was equilibrated to 37°C, and T cells resuspended in T cell imaging buffer were introduced. All imaging was done in 37°C.
Microscopy setup
All imaging experiments were performed on a motorized inverted microscope (Nikon Eclipse Ti-E; Technical Instruments, Burlingame, CA) with a motorized Epi/TIRF illuminator, a motorized Intensilight mercury lamp (Nikon C-HGFIE), and a motorized stage (MS-2000; Applied Scientific Instrumentation, Eugene, OR). A laser launch with 488-, 560-, and 640-nm diode lasers (Coherent OBIS, Santa Clara, CA) was aligned into a custom-built fiber launch (Solamere Technology Group Inc., Salt Lake City, UT). For TIRF imaging, laser illumination was reflected through the appropriate dichroic beam splitter (ZT488/647rpc, Z561rdc with ET575LP) to the objective lens [Nikon (1.47, numerical aperture; 100×), TIRF; Technical Instruments, Burlingame, CA]. RICM and epifluorescent excitation were filtered through a 50/50 beam splitter or band-pass filters (D546/10×, ET470/40×, ET545/30×, and ET620/60×). All emissions were collected through the appropriate emission filters (ET525/50M, ET600/50M, and ET700/75M) and captured on an EM-CCD (iXon 897DU; Andor Inc., South Windsor, CT). All filters were from Chroma Technology Corp. (Bellows Falls, VT). All microscope hardware was controlled using MicroManager (79).
Imaging conditions
Single-molecule TIRF images are taken at the same power dosage, either by high laser illumination intensity (2.5 mW/cm2) and short exposure time (20 ms) or by low laser illumination intensity (0.1 mW/cm2) and long exposure time (500 ms). The illumination intensity for fraction bound measurements is set at 2.5 mW/cm2, and the exposure time is varied.
Initial cell contact was visualized as a small, dark feature in the RICM channel. Single-molecule TIRF images of long exposure time (500 ms) were collected every 3 to 10 s to localize single pMHC-TCR using a laser illumination intensity of 0.1 mW/cm2. Epifluorescence images of 150-ms exposure time were collected from the same cell every 50 s at 3 and 6 μm above the coverslip to monitor NFAT dynamics. Each cell was imaged for 20 to 60 min. For quantification of cell landing, images of bright field and RICM were sequentially acquired over more than 10 fields of view. Cells were hand-counted on the basis of complete detection in the field. Cells in bright field were not required to have contacted the substrate to be scored.
Most of the experiments used an NFAT-mCherry construct, paired with peptide-Atto647N ligands. To verify that the fluorescent protein and organic dye pair were not perturbative, a set of measurements with cells expressing NFAT-GFP and MCC-Atto565 ligand was performed. In this experiment, a supported membrane displaying 0.2 MCC-Atto565- MHC/μm2 is exposed to T cells expressing NFAT-GFP. Image acquisition timing and order are identical to the NFAT-mCherry experiments, but the excitation for TIRF is 561 nm and for Epi is 488 nm.
Costimulation and inhibitor measurements
The murine costimulatory ligand CD80 (B7) protein was designed to be a recombinant, secreted, truncated ectodomain with a flexible linker and decahistidine tag. Cloning began with a synthetic, codon-optimized gene version that was then prepared in a baculovirus vector system (Invitrogen Bac-to-Bac Baculovirus Expression System). An insect cell expression system was used (Gibco High Five), and the recombinant protein-enriched supernatant was purified by affinity chromatography with Ni2+-NTA acid resin (Qiagen, Ni-NTA agarose). CD80 ligand was eluted in an imidazole-stepped gradient, followed by overnight dialysis in tris buffer with 10% glycerol. For experiments, bilayers were incubated with CD80 at concentrations to result in approximate surface densities of 100 to 300 molecules/μm2 (10).
For measurements with Lck inhibitor [4-amino-5-(4-phenoxyphe-nyl)-7H-pyrrolo[3,2-d]pyrimidin-7-yl-cyclopentane; Sigma-Aldrich], cells were incubated with the pharmacological agent (100 nM) for 15 min at 37°C, in complete media. After centrifugation, cells were resus-pended in imaging buffer, in the presence of the drug, and added to an imaging chamber that was equilibrated with the inhibitor for 5 min before imaging.
Estimating APC agonist pMHC density
Quantification of protein processing and MHC class II loading by APC shows that 200 to 80,000 molecules of a particular agonist pMHC are loaded per 106 to 108 molecules of all loaded MHCs (30, 31), and the typical radius of an APC is ~10 μm. Together, each APC is approximated to have 102 to 104 agonist pMHC molecules and an estimated density of 0.08 to 8 agonist pMHC molecules/μm2, assuming that pMHC molecules are monomeric and evenly mixed on the APC surface and that an APC is approximately spherical.
Imaging analysis
Single-molecule fitting and tracking were performed as previously described (9). Briefly, single-molecule puncta of pMHC above a size and intensity threshold were identified using particle detection and tracking algorithm (80) adapted for MATLAB (MathWorks, Natick, MA) as described (81). Single-molecule trajectories were verified using single-step unbinding determined impartially by a Bayesian change point detection algorithm (82). The number and molecular dwell time distributions of the bound pMHC were extracted from the tracks and corrected for photobleaching using data for fluorescent pMHC on a bilayer acquired at the same imaging conditions (illumination power, exposure time, and frame rate).
NFAT translocation was determined by measuring the fluorescence intensity in the cytosol and nucleus. For each cell, the cytosol and nucleus were identified and masked off as different regions using the ISODATA thresholding method in the DIPImage toolbox (83). All images with the masked regions were inspected. The time point at which the nuclear NFAT intensity is first detected to be above the background was set as the time point of initial NFAT translocation. The cumulative binding events is the sum of single-molecule-bound pMHCs that have been detected from cell landing to the point of initial translocation, as explained above, whereas the cumulative dwell time is the sum of the lifetimes of all bound pMHC over the same time period.
Fraction bound
The fraction of localized fluorescent ligands was quantified as previously described (10). Briefly, the number of molecules localized and counted at a given exposure time was normalized to all ligands visualized in the same area using high illumination intensity and short exposure time. Fraction bound under the cell was normalized for cell contact area, detected by RICM.
Analysis of unseen binding events
For a given cell that is observed to accumulate N binding events with T total dwell time from landing to initial NFAT translocation, two types of binding events are not directly visualized: (i) those from pMHC ligands that are bleached before binding (dark events) and (ii) those shorter than the acquisition/time lapse interval (short events). For each cell, we estimate an offset of the mean number of binding events that are not directly detected (〈dN〉) and their integrated dwell time (〈dT〉)
assuming that the mean dwell time 〈dTdark〉 ≪ time to activation. We are determining the term 〈Ndark ∩ long〉 to avoid double counting binding events that are both dark and short.
To account for the dark events (case 1), we consider that bleaching is a Poisson process in time. At illumination exposures of a given energy dose, the probability that a fluorescent pMHC is bleached after n illumination exposures is exponentially distributed
where kbl = 1/<number of exposures to bleach>. The fraction of bleached pMHC grows as a function of cumulative number of exposures
The number of dark binding events accumulated from the frame of landing (nland) to the frame of activation (nact) is the integral of the product of the pMHC-TCR binding probability (on-rate) and the fraction of bleached pMHCs
Some dark binding events are classified as long, on the basis of the criterion that they dwell longer than imaging time lapse interval (t > int). The number of such events depends on the true dwell time distribution (determined from fitting observed data)
The number of short events (case 2) is estimated on the basis of the probability of short events relative to that of the long events. We define short events as events whose lifetimes are from 0.8 s [most probable to contribute to TCR signaling, based on (84)] to the imaging time lapse interval (int)
where the long events can be either observed or dark
For both types of events, the integrated dwell time offset is estimated using the number of each type of events and their corresponding mean dwell time
The offsets in binding number and dwell time from both types of un-seen events are estimated for each cell (fig. S13). After applying the offsets to both cumulative binding events and dwell time to all cell data, we find that overall trends remain similar. Namely, neither cumulative binding events nor integrated dwell time is a convergent property of activating cells (Fig. 4 and fig. S12).
Spatiotemporal analysis
The spatiotemporal correlation analysis was implemented in MATLAB. Trajectories were filtered on the basis of spatial and temporal proximity criteria. Positions of all trajectories were defined as a function of time, where the positions of an existing trajectory and a new trajectory are at time t, respectively. The time to represents the time of the first localization of molecule. For each cell, the trajectories are defined as correlated for a time window tthres and spatial neighborhood radius there exists
In the analysis, the time window is set at 30 s and the neighborhood radius is varied. Multiple trajectories were allowed to form a larger effective correlated track, pC, if individual trajectories are involved in track pairs that met the above criteria. The dwell time of an effective signaling cluster is defined as where m are the individual pMHC trajectories within an effective correlated track pC.
Probability of long-dwelling events for physiological agonist pMHCs
We calculate the probability that a physiological pMHC-TCR binding event reaches the molecular, receptor level threshold of about 40 s, as determined from the spatiotemporal correlation analysis. The T102S pMHC-AND mean dwell time (<τoff> = 9.6 s) is close to the physiological, population activation threshold of 3 s (2, 7). Assuming that pMHC-TCR unbinding follows simple, single exponential kinetics, the probability that a physiological agonist pMHC-TCR binding event dwells for 40 s is 0.02. This is much lower than the probability of 0.43 for long-dwelling MCC pMHC-AND interactions, arguing that single, long dwells are rare.
Supplementary Material
Acknowledgments:
We thank F. Marangoni (Harvard Medical School) for providing the NFAT reporter plasmid. We are grateful for the contribution of G. Graff (ideocraft) in composing the model schematic in the supplementary figures. We thank W.-L. Lo (UCSF) and members of the Groves Laboratory for critical feedback on the manuscript. We thank L. Teyton (Scripps Research Institute) and M. Davis (Stanford University) for providing the MHC bacmids.
Funding: This work was supported by NIH grant P01 AI091580 and by the Novo Nordisk Foundation Challenge Programme as part of the Center for Geometrically Engineered Cellular Systems.
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. The AND TCR mice described in this work may be obtained from J.T.G. through a signed and executed material transfer agreement.
REFERENCES AND NOTES
- 1.Matsui K, Boniface JJ, Steffner P, Reay PA, Davis MM, Kinetics of T-cell receptor binding to peptide/I-Ek complexes: Correlation of the dissociation rate with T-cell responsiveness. Proc. Natl. Acad. Sci. U.SA. 91, 12862–12866 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naeher D, Daniels MA, Hausmann B, Guillaume P, Luescher I, Palmer E, A constant affinity threshold for T cell tolerance. J. Exp. Med. 204, 2553–2559 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Irvine DJ, Purbhoo MA, Krogsgaard M, Davis MM, Direct observation of ligand recognition by T cells. Nature 419, 845–849 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Huang J, Brameshuber M, Zeng X, Xie J, Li Q-J, Chien Y-H, Valitutti S, Davis MM, A single peptide-major histocompatibility complex ligand triggers digital cytokine secretion in CD4+ T cells. Immunity 39, 846–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gorentla BK, Zhong X-P, T cell receptor signal transduction in T lymphocytes. J. Clin. Cell Immunol. 2012, 005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brownlie RJ, Zamoyska R, T cell receptor signalling networks: Branched, diversified and bounded. Nat. Rev. Immunol. 13, 257–269 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Palmer E, Naeher D, Affinity threshold for thymic selection through a T-cell receptor-co-receptor zipper. Nat. Rev. Immunol. 9, 207–213 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Purbhoo MA, Irvine DJ, Huppa JB, Davis MM, T cell killing does not require the formation of a stable mature immunological synapse. Nat. Immunol. 5, 524–530 (2004). [DOI] [PubMed] [Google Scholar]
- 9.O’Donoghue GP, Pielak RM, Smoligovets AA, Lin JJ, Groves JT , Direct single molecule measurement of TCR triggering by agonist pMHC in living primary T cells. eLife 2, e00778 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pielak RM, O’Donoghue GP, Lin JJ, Alfieri KN, Fay NC, Low-Nam ST, Groves JT, Early T cell receptor signals globally modulate ligand:receptor affinities during antigen discrimination. Proc. Natl. Acad. Sci. U.S.A. 114, 12190–12195 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Axmann M, Huppa JB, Davis MM, Schütz GJ, Determination of interaction kinetics between the T cell receptor and peptide-loaded MHC class II via single-molecule diffusion measurements. Biophys. J. 103, L17–L19 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huppa JB, Axmann M, Mörtelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schütz GJ, Davis MM, TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature 463, 963–967 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Podtschaske M, Benary U, Zwinger S, Höfer T, Radbruch A, Baumgrass R, Digital NFATc2 activation per cell transforms graded T cell receptor activation into an all-or-none IL-2 expression. PLOS ONE 2, e935 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morris MK, Saez-Rodriguez J, Sorger PK, Lauffenburger DA, Logic-based models for the analysis of cell signaling networks. Biochemistry 49, 3216–3224 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi H, Kærn M, Araki M, Chung K, Gardner TS, Cantor CR, Collins JJ, Programmable cells: Interfacing natural and engineered gene networks. Proc. Natl. Acad. Sci. U.S.A. 101, 8414–8419 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim WA, Designing customized cell signalling circuits. Nat. Rev. Mol. Cell Biol. 11, 393–403 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nye JA, Groves JT, Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir 24, 4145–4149 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Pentcheva T, Edidin M, Clustering of peptide-loaded MHC class I molecules for endoplasmic reticulum export imaged by fluorescence resonance energy transfer. J. Immunol. 166, 6625–6632 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Edidin M, Class I MHC molecules as probes of membrane patchiness: From biophysical measurements to modulation of immune responses. Immunol. Res. 47, 265–272 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bosch B, Heipertz EL, Drake JR, Roche PA, Major histocompatibility complex (MHC) class II-peptide complexes arrive at the plasma membrane in cholesterol-rich microclusters. J. Biol. Chem. 288, 13236–13242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fooksman DR, Organizing MHC class II presentation. Front. Immunol. 5, 158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu X, Gibbs JS, Hickman HD, David A, Dolan BP, Jin Y, Kranz DM, Bennink JR, Yewdell JW, Varma R, Endogenous viral antigen processing generates peptide-specific MHC class I cell-surface clusters. Proc. Natl. Acad. Sci. U.S.A. 109, 15407–15412 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vrljic M, Nishimura SY, Brasselet S, Moerner WE, McConnell HM, Translational diffusion of individual class II MHC membrane proteins in cells. Biophys. J. 83,2681–2692 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishimura SY, Vrljic M, Klein LO, McConnell HM, Moerner WE, Cholesterol depletion induces solid-like regions in the plasma membrane. Biophys. J. 90, 927–938 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Umemura YM, Vrljic M, Nishimura SY, Fujiwara TK, Suzuki KGN, Kusumi A, Both MHC class II and its GPI-anchored form undergo hop diffusion as observed by single-molecule tracking. Biophys. J. 95, 435–450 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu Y, Smoligovets AA, Groves JT, Modulation of T cell signaling by the actin cytoskeleton. J. Cell Sci. 126, 1049–1058 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Mossman KD, Campi G, Groves JT, Dustin ML, Altered TCR signaling from geometrically repatterned immunological synapses. Science 310, 1191–1193 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Yokosuka T, Sakata-Sogawa K, Kobayashi W, Hiroshima M, Hashimoto-Tane A, Tokunaga M, Dustin ML, Saito T, Newly generated T cell receptor microclusters initiate and sustain T cell activation by recruitment of Zap70 and SLP-76. Nat. Immunol. 6, 1253–1262 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Sykulev Y, Joo M, Vturina I, Tsomides TJ, Eisen HN, Evidence that a single peptide-MHC complex on a target cell can elicit a cytolytic T cell response. Immunity 4, 565–571 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Demotz S, Grey HM, Sette A, The minimal number of class II MHC-antigen complexes needed for T cell activation. Science 249, 1028–1030 (1990). [DOI] [PubMed] [Google Scholar]
- 31.Velazquez C, DiPaolo R, Unanue ER, Quantitation of lysozyme peptides bound to class II MHC molecules indicates very large differences in levels of presentation. J. Immunol. 166, 5488–5494 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Bozzacco L, Yu H, Zebroski HA, Dengjel J, Deng H, Mojsov S, Steinman RM, Mass spectrometry analysis and quantitation of peptides presented on the MHC II molecules of mouse spleen dendritic cells. J. Proteome Res. 10, 5016–5030 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boulanger DSM, Eccleston RC, Phillips A, Coveney PV, Elliott T, Dalchau N, A mechanistic model for predicting cell surface presentation of competing peptides by MHC class I molecules. Front. Immunol. 9, 1538 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campi G, Varma R, Dustin ML, Actin and agonist MHC-peptide complex-dependent T cell receptor microclusters as scaffolds for signaling. J. Exp. Med. 202, 1031–1036 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.DeMond AL, Mossman KD, Starr T, Dustin ML, Groves JT, T cell receptor microcluster transport through molecular mazes reveals mechanism of translocation. Biophys. J. 94, 3286–3292 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu C-H, Wu H-J, Kaizuka Y, Vale RD, Groves JT, Altered actin centripetal retrograde flow in physically restricted immunological synapses. PLOS ONE 5, e11878 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brameshuber M, Kellner F, Rossboth BK, Ta H, Alge K, Sevcsik E, Gohring J, Axmann M, Baumgart F, Gascoigne NRJ, Davis SJ, Stockinger H, Schutz GJ, Huppa JB, Monomeric TCRs drive T cell antigen recognition. Nat. Immunol. 19,487–496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rossboth B, Arnold AM, Ta H, Platzer R, Kellner F, Huppa JB, Brameshuber M, Baumgart F, Schutz GJ, TCRs are randomly distributed on the plasma membrane of resting antigen-experienced T cells. Nat. Immunol. 19, 821–827 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartman NC, Nye JA, Groves JT, Cluster size regulates protein sorting in the immunological synapse. Proc. Natl. Acad. Sci. U.S.A. 106, 12729–12734 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Caculitan NG, Kai H, Liu EY, Fay N, Yu Y, Lohmuller T, O’Donoghue GP, Groves JT, Size-based chromatography of signaling clusters in a living cell membrane. Nano Lett. 14, 2293–2298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dustin ML, Groves JT, Receptor signaling clusters in the immune synapse. Annu. Rev. Biophys. 41, 543–556 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y, Fay NC, Smoligovets AA, Wu H-J, Groves JT, Myosin IIA modulates T cell receptor transport and CasL phosphorylation during early immunological synapse formation. PLOS ONE 7, e30704 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Straus DB, Weiss A, Genetic evidence for the involvement of the lck tyrosine kinase in signal transduction through the T cell antigen receptor. Cell 70, 585–593 (1992). [DOI] [PubMed] [Google Scholar]
- 44.Bunnell SC, Hong DI, Kardon JR, Yamazaki T, McGlade CJ, Barr VA, Samelson LE, T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J. Cell Biol. 158, 1263–1275 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Houtman JCD, Yamaguchi H, Barda-Saad M, Braiman A, Bowden B, Appella E, Schuck P, Samelson LE, Oligomerization of signaling complexes by the multipoint binding of GRB2 to both LAT and SOS1. Nat. Struct. Mol. Biol. 13, 798–805 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, Vale RD, Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 352, 595–599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang WYC, Yan Q, Lin W-C, Chung JK, Hansen SD, Christensen SM, Tu H-L, Kuriyan J, Groves JT, Phosphotyrosine-mediated LAT assembly on membranes drives kinetic bifurcation in recruitment dynamics of the Ras activator SOS. Proc. Natl. Acad. Sci. U.S.A. 113, 8218–8223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang WYC, Chiang H-K, Groves JT, Dynamic scaling analysis of molecular motion within the LAT:Grb2:SOS protein network on membranes. Biophys. J. 113,1807–1813 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nag A, Monine MI, Faeder JR, Goldstein B, Aggregation of membrane proteins by cytosolic cross-linkers: Theory and simulation of the LAT-Grb2-SOS1 system. Biophys. J. 96, 2604–2623 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hyman AA, Weber CA, Julicher F, Liquid-liquid phase separation in biology. Anna. Rev. Cell Dev. Biol. 30, 39–58 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Kortum RL, Balagopalan L, Alexander CP, Garcia J, Pinski JM, Merrill RK, Nguyen PH, Li W, Agarwal I, Akpan IO, Sommers CL, Samelson LE, The ability of Sos1 to oligomerize the adaptor protein LAT is separable from its guanine nucleotide exchange activity in vivo. Sci. Signal. 6, ra99 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Balagopalan L, Kortum RL, Coussens NP, Barr VA, Samelson LE, The linker for activation of T cells (LAT) signaling hub: From signaling complexes to microclusters. J. Biol. Chem. 290, 26422–26429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abraham RT, Weiss A, Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat. Rev. Immunol. 4, 301–308 (2004). [DOI] [PubMed] [Google Scholar]
- 54.Marangoni F, Murooka TT, Manzo T, Kim EY, Carrizosa E, Elpek NM, Mempel TR, The transcription factor NFAT exhibits signal memory during serial T cell interactions with antigen presenting cells. Immunity 38, 237–249 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rao A, Luo C, Hogan PG, Transcription factors of the NFAT family: Regulation and function. Anna. Rev. Immunol. 15, 707–747 (1997). [DOI] [PubMed] [Google Scholar]
- 56.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML, The immunological synapse: A molecular machine controlling T cell activation. Science 285, 221–227 (1999). [DOI] [PubMed] [Google Scholar]
- 57.Varma R, Campi G, Yokosuka T, Saito T, Dustin ML, T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 25, 117–127 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krogsgaard M, Prado N, Adams EJ, He X-L, Chow D-C, Wilson DB, Garcia KC, Davis MM, Evidence that structural rearrangements and/or flexibility during TCR binding can contribute to T cell activation. Mol. Cell 12, 1367–1378 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Hedrick SM, Engel I, McElligott DL, Fink PJ, Hsu ML, Hansburg D, Matis LA, Selection of amino acid sequences in the beta chain of the T cell antigen receptor. Science 239, 1541–1544 (1988). [DOI] [PubMed] [Google Scholar]
- 60.Kaye J, Hsu M-L, Sauron M-E, Jameson SC, Gascoigne NRJ, Hedrick SM, Selective development of CD4+ T cells in transgenic mice expressing a class II MHC-restricted antigen receptor. Nature 341, 746–749 (1989). [DOI] [PubMed] [Google Scholar]
- 61.McKeithan TW, Kinetic proofreading in T-cell receptor signal transduction. Proc. Natl. Acad. Sci. U.S.A. 92, 5042–5046 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Valitutti S, Dessing M, Aktories K, Gallati H, Lanzavecchia A, Sustained signaling leading to T cell activation results from prolonged T cell receptor occupancy. Role of T cell actin cytoskeleton. J. Exp. Med. 181, 577–584 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Manz BN, Groves JT, Spatial organization and signal transduction at intercellular junctions. Nat. Rev. Mol. Cell Biol. 11, 342–352 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Utzny C, Faroudi M, Valitutti S, Frequency encoding of T-cell receptor engagement dynamics in calcium time series. Biophys. J. 88, 1–14 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pryshchep S, Zarnitsyna VI, Hong J, Evavold BD, Zhu C, Accumulation of serial forces on TCR and CD8 frequently applied by agonist antigenic peptides embedded in MHC molecules triggers calcium in T cells. J. Immunol. 193, 68–76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Das DK, Feng Y, Mallis RJ, Li X, Keskin DB, Hussey RE, Brady SK, Wang J-H, Wagner G, Reinherz EL, Lang MJ, Force-dependent transition in the T-cell receptor b-subunit allosterically regulates peptide discrimination and pMHC bond lifetime. Proc. Natl. Acad. Sci. U.S.A. 112, 1517–1522 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ma Z, Sharp KA, Janmey PA, Finkel TH, Surface-anchored monomeric agonist pMHCs alone trigger TCR with high sensitivity. PLOS Biol. 6, e43 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Aleksic M, Dushek O, Zhang H, Shenderov E, Chen JL, Cerundolo V, Coombs D, van der Merwe PA, Dependence of T cell antigen recognition on T cell receptor-peptide MHC confinement time. Immunity 32, 163–174 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Govern CC, Paczosa MK, Chakraborty AK, Huseby ES, Fast on-rates allow short dwell time ligands to activate T cells. Proc. Natl. Acad. Sci. U.SA. 107, 8724–8729 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gross G, Eshhar Z, Therapeutic potential of T cell chimeric antigen receptors (CARs) in cancer treatment: Counteracting off-tumor toxicities for safe CAR T cell therapy. Annu. Rev. Pharmacol. Toxicol. 56, 59–83 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Mei H, Jiang H, Wu Y, Guo T, Xia L, Jin R, Hu Y, Neurological toxicities and coagulation disorders in the cytokine release syndrome during CAR-T therapy. Br. J. Haematol. 181, 689–692 (2018). [DOI] [PubMed] [Google Scholar]
- 72.Reusch U, Sundaram M, Davol PA, Olson SD, Davis JB, Demel K, Nissim J, Rathore R, Liu PY, Lum LG, Anti-CD3 x anti-epidermal growth factor receptor (EGFR) bispecific antibody redirects T-cell cytolytic activity to EGFR-positive cancers in vitro and in an animal model. Clin. Cancer Res. 12, 183–190 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Kiewe P, Hasmüller S, Kahlert S, Heinrigs M, Rack B, Marme A, Korfel A, Jäger M, Lindhofer F, Sommer H, Thiel E, Untch M, Phase I trial of the trifunctional anti-HER2 x anti-CD3 antibody ertumaxomab in metastatic breast cancer. Clin. Cancer Res. 12, 3085–3091 (2006). [DOI] [PubMed] [Google Scholar]
- 74.Löffler A, Kufer P, Lutterbüse R, Zettl F, Daniel PT, Schwenkenbecher JM, Riethmüller H, Dörken B, Bargou RC, A recombinant bispecific single-chain antibody, CD19 x CD3, induces rapid and high lymphoma-directed cytotoxicity by unstimulated T lymphocytes. Blood 95, 2098–2103 (2000). [PubMed] [Google Scholar]
- 75.Corse E, Gottschalk RA, Allison JP, Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J. Immunol. 186, 5039–5045 (2011). [DOI] [PubMed] [Google Scholar]
- 76.Malherbe L, Hausl C, Teyton L, McHeyzer-Williams MG, Clonal selection of helper T cells is determined by an affinity threshold with no further skewing of TCR binding properties. Immunity 21, 669–679 (2004). [DOI] [PubMed] [Google Scholar]
- 77.Sumen C, Dustin ML, Davis MM, T cell receptor antagonism interferes with MHC clustering and integrin patterning during immunological synapse formation. J. Cell Biol. 166, 579–590 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Corse E, Gottschalk RA, Krogsgaard M, Allison JP, Attenuated T cell responses to a high-potency ligand in vivo. PLOS Biol. 8, e1000481 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stuurman N, Edelstein AD, Amodaj N, Hoover KH, Vale RD, Computer control of microscopes using μManager. Curr. Protoc. Mol. Biol. Chapter 14, Unit14.20 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Crocker JC, Grier DG, When like charges attract: The effects of geometrical confinement on long-range colloidal interactions. Phys. Rev. Lett. 77, 1897–1900 (1996). [DOI] [PubMed] [Google Scholar]
- 81.Blair DD, The Matlab Particle Tracking Code Repository (2008). [Google Scholar]
- 82.Ensign DL, Pande VS, Bayesian single-exponential kinetics in single-molecule experiments and simulations. J. Phys. Chem. B 113, 12410–12423 (2009). [DOI] [PubMed] [Google Scholar]
- 83.Luengo Hendriks CL, van Vliet LJ, Rieger B, van Kempen GMP, van Ginkel M, DIPimage: A scientific image processing toolbox for MATLAB. Delft University of Technology (1999). [Google Scholar]
- 84.Stepanek O, Prabhakar AS, Osswald C, King CG, Bulek A, Naeher D, Beaufils-Hugot M, Abanto ML, Galati V, Hausmann B, Lang R, Cole DK, Huseby ES, Sewell AK, Chakraborty AK, Palmer E, Coreceptor scanning by the T-cell receptor provides a mechanism for T cell tolerance. Cell 159, 333–345 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.