

Molecular simulation and NMR reveal how intrinsically disordered proteins move in synergy with their surrounding environment.
Abstract
Protein and water dynamics have a synergistic relationship, which is particularly important for intrinsically disordered proteins (IDPs), although the details of this coupling remain poorly understood. Here, we combine temperature-dependent molecular dynamics simulations using different water models with extensive nuclear magnetic resonance (NMR) relaxation to examine the importance of distinct modes of solvent and solute motion for the accurate reproduction of site-specific dynamics in IDPs. We find that water dynamics play a key role in motional processes internal to “segments” of IDPs, stretches of primary sequence that share dynamic properties and behave as discrete dynamic units. We identify a relationship between the time scales of intrasegment dynamics and the lifetime of hydrogen bonds in bulk water. Correct description of these motions is essential for accurate reproduction of protein relaxation. Our findings open important perspectives for understanding the role of hydration water on the behavior and function of IDPs in solution.
INTRODUCTION
The activity of proteins is a complex function of their structure and dynamics (1), and understanding the intimate relationship between protein motion and function remains one of the fundamental goals of biophysics. It is becoming increasingly clear that water plays a crucial role in determining the free-energy landscape of proteins and ultimately their structural and dynamic features (2, 3). Folded proteins have been shown to undergo a so-called “dynamic transition,” between 200 and 240 K, below which both motion and function are severely restricted (4–6). It has recently been proposed that dynamic processes associated with different components of the solvated protein, for example, backbone or side-chain fluctuations, are activated at distinct temperatures, because of their specific activation energies, so that, at higher temperatures where proteins function, dynamic modes associated with biological activity are dominant (7). The onset of functional dynamics appears to be coupled with the dynamics of the network formed by protein-water hydrogen bonds (8), which is continuously reshaped by translational diffusion of water molecules (9).
Therefore, it is not surprising that the dynamics of water at the interface with biomolecules has been extensively studied. It was found that the translational diffusion in the hydration layer of protein, whose extent was estimated to concern a single water molecule (10), is, on average, two to three times slower than in the bulk (11, 12). Water dynamics are considerably slowed down (13), while site-resolved solution nuclear magnetic resonance (NMR) studies (14) revealed a heterogeneous distribution of protein-water interactions across the surface of proteins.
While these considerations are general for all proteins, they are particularly relevant for intrinsically disordered proteins (IDPs) that lack a well-defined three-dimensional structure and dynamically access a diverse ensemble of conformations in their functional state and exhibit solvent accessible surfaces several times larger than folded proteins of similar molecular weight. IDPs carry out important functions in living cells, where they are predicted to represent more than 30% of the proteome of eukaryotes and are involved in many signaling and regulatory processes. The lack of a three-dimensional structure implies a predominant role for dynamics in determining the activity of IDPs, which sometimes manifest as folding upon binding to functional partners (15) but more often appear in the formation of dynamic complexes (15–19).
Armstrong et al. (20) used Overhauser dynamic nuclear polarization to propose that water dynamics in the hydration layer of acid-unfolded apomyoglobin is faster and more uniform than that measured in the native state of the protein (but still slower than diffusion in bulk water), suggesting a role of water in determining folding kinetics. Elastic incoherent neutron scattering studies carried out on hydrated powders of folded and unfolded proteins suggested a correlation between water mobility and protein dynamics (21).
Molecular dynamics (MD) simulation is an ideal complement to experimental studies of IDPs (22) that often lack the capability of tracking individual water molecules in time and space. Simulations (23) were used to confirm that disordered proteins bind a significantly larger amount of hydration water than folded proteins of comparable size, as originally indicated by solid-state NMR relaxation (24). In addition, MD simulations show that water is more ordered in the vicinity of disordered peptides, whose hydration layer features slow dynamics and exchange of molecules with bulk solvent (10, 23, 25).
Despite our increasing understanding of the physicochemical properties of water around the surface of IDPs, less is known about the nature of solvent effects on the dynamics of IDPs. Solution-state NMR spectroscopy represents a particularly powerful tool for the study of the dynamics of IDPs at atomic resolution, allowing a detailed mapping of both the conformational space (26) and the associated dynamic time scales (27). In particular, NMR relaxation is exquisitely sensitive to motions that quench the angular correlation functions of NH bond vectors situated throughout the protein and as such can be used to probe time scales that are relevant to the dynamics of both protein and water. MD simulations allow us to investigate the effect of specific molecular parameters on IDP motions occurring on these same time scales, by verifying how variations of force field parameters affect the results of simulations and how they affect the reproduction of relaxation rates.
Because of the very broad conformational space sampled by IDPs in solution, care must be taken to account for the averaging of dynamic parameters. In the limit of fast (τex < 10 μs) exchange between different substates, measured spin relaxation rates report on population-weighted averages of the rates associated with dynamics occurring within each conformational substate on time scales up to the nanosecond. The relaxation rates of each individual state cannot be known a priori, and the populations are difficult to determine. For this reason, we recently developed an ensemble-trajectory approach (28) that reports on fast (<25 ns) motions occurring in an ensemble of conformational states that interconvert on slower (microsecond) time scales. This method [average block selection using relaxation data (ABSURD)] estimates the correct weighting factors for local relaxation times by comparison with experimental data sensitive to motions from tens of picoseconds to tens of nanoseconds. ABSURD thereby actively mimics the true averaging process measured under experimental conditions by allowing the simultaneous observation of relaxation-active dynamic processes over a highly heterogeneous ensemble, significantly improving the reproduction of experimental measurements sensitive to dynamics occurring over at least five orders of magnitude.
We recently collected temperature-dependent, multifield NMR relaxation measurements (up to 57 relaxation rates for each NH site) (29) that were used to classify site-specific IDP dynamics in terms of contributions from fast (~50 ps) local librations, intermediate (~1 ns) conformational sampling of backbone dihedral angles, and slow (up to 20 ns) chain-like segmental motions in a 125–amino acid IDP. The observations were combined with simulation (30) and the ABSURD approach to describe ensemble-averaged correlation functions for each NH bond vector in the protein, allowing us to identify the molecular origin of the different categories of motion. In this study, we further exploit this tool to compare temperature-dependent NMR relaxation with 60 μs of MD simulation to examine the intimate relationship between solvent properties and motions occurring within local segments of the primary chain, demonstrating that NMR spin relaxation is highly sensitive to such motions. We propose a method to explicitly include the segmental nature of IDP dynamics in the analysis of experimental spin relaxation data and analytically describe the nature of these motions and calculate expected relaxation rates from simulation. Comparison of experiment with simulations using different physical descriptions of water not only identifies the most appropriate water model that best reproduces protein motions on a broad range of time scales but also reveals that solvent effects on protein dynamics depend strongly on the properties of bulk water, particularly on the kinetics of hydrogen bond formation. In particular, the time scales of intrasegmental motions appear to be strongly coupled to the dynamics of water. Our results provide previously unidentified insight into the importance of water on the dynamic behavior of IDPs, suggest novel ways to improve the current generation of force fields for IDPs, and allow us to speculate on the possible effect of the crowded cellular environment on both water and protein dynamics.
RESULTS
Temperature dependence of simulated NMR spin relaxation rates depends on the properties of water models
We carried out MD simulations of the 125–amino acid unfolded C-terminal domain of Sendai virus (Ntail) using different combinations of protein force fields and water models, as summarized in table S1, for a total simulated time of 60 μs. Simulation details are given in the Supplementary Materials. Briefly, we started with an ensemble of conformations of Ntail previously validated using first-order NMR observables, such as chemical shifts and residual dipolar couplings (31), which are averaged over all states that interconvert on time scales faster than 10−4 s and therefore effectively cover this conformational space. Randomly selected conformers from the ensemble that are distant on the free-energy landscape are used as starting points of the trajectories that each sample fast (<25 ns) time scale motions. In practice, the goal of individual trajectories is to adequately sample the spin relaxation–active dynamic modes sampled by each conformer; i.e., each trajectory has to be sufficiently long to allow the rotational correlation function of backbone amide NH bond vectors to effectively decay to zero.
We combine the CHARMM36m force field of Huang et al. (32), arguably one of the most reliable force fields currently available for IDPs, with either the CHARMM-modified TIP3P water model (recommended to be used with CHARMM36m, simulation C3P) or the highly optimized TIP4P/2005 water model (simulation C4P) (33). In addition, we combine TIP4P/2005 with a force field that is not optimized for IDPs, Amber ff99SB-ILDN (34), obtaining simulation A4P. The calculations were repeated for different temperatures at which we measured multifield, multitemperature spin relaxation for Ntail (29) between 5° and 25°C, as reported in table S1.
We divide our trajectories into overlapping blocks that are sufficiently long for converged rotational correlation function of backbone amide NH bond vectors to have effectively decayed to zero (in practice, 100 to 300 ns, depending on the temperature, see table S1) so that motions occurring on much faster time scales (<10 to 30 ns) can be adequately sampled. Backbone 15N spin relaxation rates for all blocks are then calculated from the averaged angular correlation function (see the Supplementary Materials). Then, we use the ABSURD approach (28) in combination with experimental data acquired at different temperatures (29) to select ensembles of trajectories that best capture the dynamics of Ntail, as reported by NMR spin relaxation rates. Details of the selection procedure are given in Materials and Methods and table S2.
We validate our simulations in three ways, first, by comparison with experimental observables that report on structural features and, therefore, are orthogonal to the information about dynamics contained in the relaxation data used for selection. 13C chemical shifts (fig. S1), which report on local conformational sampling of backbone dihedral angles, show that in all our simulations, the conformational propensity of residues in Ntail is well reproduced. Second, the distribution of radii of gyration, which are a measure of the effective size of the protein in solution, is realistic in all three simulations (fig. S2), in comparison to data measured (at 298 K) from small-angle scattering (28). The distribution in A4P is closest to that found in the ensemble of conformations used to seed the simulation. Radii of gyration have different temperature dependences in the three simulations: The ensemble seems to contract when temperature is lowered in C3P; the opposite is true for A4P, whereas the temperature dependence is less pronounced in C4P.
Third, to evaluate how accurately the simulations capture the dynamics of Ntail occurring on time scales from tens of picoseconds to tens of nanoseconds, we compare simulated spin relaxation rates with a previously reported (29) set of 4200 experimental relaxation rates (42 rates measured over 100 NH sites) (Fig. 1 and fig. S3). Transverse spin relaxation rates such as R2 and ηxy, which depend on the spectral density function evaluated at zero frequency and have been used, in most cases, in the selection step of the ABSURD procedure (see table S2), are well reproduced at all temperatures and magnetic fields (Fig. 1A and fig. S3). In most cases, C3P reproduces R2 and ηxy slightly more accurately than C4P (figs. S4 to S6). Simulations carried out in TIP4P/2005 clearly capture the temperature dependence of the experimental 15N longitudinal spin relaxation rates (R1, Fig. 1B) and 15N{1H} steady-state nuclear Overhauser effects (NOEs, Fig. 1C) most accurately, although they were not actively used to select the ensemble of trajectories. This is particularly evident in the helical recognition element, whose R1 rates are significantly overestimated in C3P but not in C4P and A4P. Closer inspection reveals that, already at room temperature, TIP4P/2005 simulations reproduce sequence and magnetic field dependence of R1 much more convincingly, not only in the helical element but also in the C-terminal region (residues 500 to 520). These features are summarized in the root mean square deviations (RMSDs) comparing calculated and experimental rates (figs. S4 to S6). In addition, Fig. 1C shows that in C3P at lower temperatures, NOEs are systematically lower than the experimental data, especially between the N terminus and residue 445 and between the C terminus and residue 490, suggesting that either the fastest component of protein dynamics probed by 15N spin relaxation is unphysically fast or the amplitude of picosecond motions is severely overestimated (29).
Fig. 1. Comparison of experimental and simulated NMR relaxation rates at 278 to 298 K.
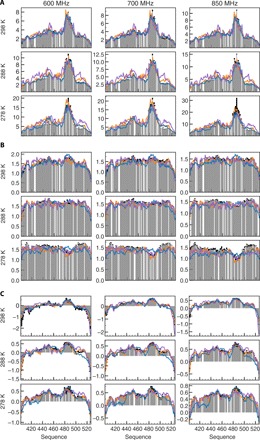
(A) Experimental 15N transverse spin relaxation rates R2 (gray bars) measured on Ntail at different magnetic fields (columns) and temperatures (rows) are compared with the results of simulations C3P (blue line), C4P (orange), and A4P (purple). At all temperatures and fields, simulations in TIP4P/2005 capture the dynamics of Ntail better than simulations in TIP3P water (RMSD given in figs. S4 to S6). All rates are reported in s−1. Rates at 950 MHz are not shown in the interests of space. (B) Experimental 15N longitudinal spin relaxation rates (R1) measured on Ntail at different magnetic fields (columns) and temperatures (rows) [color code as in (A)]. All simulations reproduce, at least qualitatively, the sequence dependence of R2 rates, although simulations are more accurate at room temperature than at lower temperature. All rates are reported in s−1. (C) Experimental 15N{1H} steady-state NOEs measured on Ntail at different magnetic fields (columns) and temperatures (rows) [color code as in (A)]. Simulations in TIP4P/2005 reproduce the experimental values better than C3P, at all temperatures and at all fields (RMSD given in figs. S4 to S6).
In terms of independent cross-validation of experimental rates (table S2), the ABSURD ensemble of trajectories derived using C4P reproduces the entire data set considerably better than C3P, whether considering the entire data set or each independent temperature. Although C4P performs slightly better than A4P, the two models are comparable, suggesting that the TIP4P/2005 water model is the most appropriate for describing picosecond to nanosecond motions occurring in IDPs and that the water model is responsible for the accuracy of the description.
Solvent properties determine protein segmental motions but not backbone dihedral angle dynamics
We investigated which physical mechanism is responsible for the observed water-dependent differences. In our previous work (30), we resolved the ensemble-averaged correlation functions into distinct contributions to 15N spin relaxation. Reorientational dynamics were described in terms of backbone dihedral angle dynamics and long-range segmental motions, revealing that both components display significant sequence dependence. Using the same methods, we have characterized the time scales associated with the dynamics of backbone φ/ψ angles (fig. S7). We find that these motions, occurring on multiple time scales, are progressively restricted as the temperature is lowered. However, we do not find any significant difference between the three sets of simulations, indicating that the presence of these components is relatively independent of the water model.
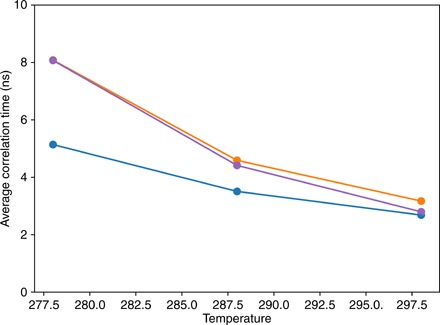
On the contrary, the temperature dependence of segmental dynamics, determined by characterizing the rotational properties of peptide planes (see Materials and Methods) (30), shows clear solvent-dependent features (Fig. 2 and fig. S8). While average time scales are essentially force field independent at room temperature, the use of TIP4P/2005 results in a much steeper temperature dependence of segmental motion time scales than for simulations using TIP3P (Fig. 2). In addition, TIP4P/2005 induces a site-specific variation of time scales that spans a much larger range when simulation temperature is changed (fig. S8). C4P and A4P display segmental dynamics on the same time scales and with the same temperature dependence (Fig. 2 and fig. S8), suggesting that the water model is more important than the protein force field in determining the properties of segmental chain dynamics. Common to all simulations, and similarly to observations reported previously (30), segmental dynamics exhibit two major components: a faster one, between hundreds of picoseconds and 2 ns at room temperature and between 1 ns and several nanoseconds at 5°C, and a slower one, between 2 and 10 ns at room temperature and between 3 and 30 ns at 5°C.
Fig. 2. Average time scales resulted from fitting segmental dynamics correlation functions in C3P (blue), C4P (orange), and A4P (purple).
We use simple correlation metrics (30, 35) to characterize how many consecutive amino acids belong to the same dynamic unit (or segment). In practice, the larger the value of the angular order parameter (fig. S9), the larger the degree of dynamic coupling between two distinct residues. We notice that in all cases, and more clearly when CHARMM36m is used, decreasing the temperature results in an overall increase of coupling. In all simulations, each residue forms a highly correlated core with its two neighbors. However, while in TIP3P, segments generally extend far beyond this three-residue core, the correlation maps A4P and C4P suggest that the effect of few neighboring residues (5 to 7, except for the more rigid helical element) is sufficient to describe the dynamic properties of IDPs probed by NMR relaxation, and this is the case at all temperatures. In particular, while correlations between distinct segments are present in C3P, for instance, between the recognition element and the hydrophobic region centered on residue 450, simulations in TIP4P/2005 display more localized dynamics, in which intersegment correlations are absent at least on the time scales relevant to NMR relaxation. In general, the observation that TIP3P trajectories comprise an overrepresentation of protein-protein interactions substantiates the observation of partial collapse compared to the distribution of radii of gyration present in the initial ensemble that validates experimental small-angle x-ray scattering data.
Intersegment dynamics are not strongly dependent on the water model
To understand the physical source of the observed differences in segmental motions, we make a distinction between two contributions to the motion. The first describes the rotational dynamics of the entire segment with respect to the laboratory frame and, consequently, with respect to other independent segments in the peptide chain—this contribution therefore also reports on intersegment dynamics. A second component reports on intrasegment dynamics, in which the orientation of each NH bond vector is modulated by time-dependent fluctuations described by internal degrees of freedom within each segment (Fig. 3).
Fig. 3. Illustration of inter- and intrasegment dynamics contributing to NMR relaxation.
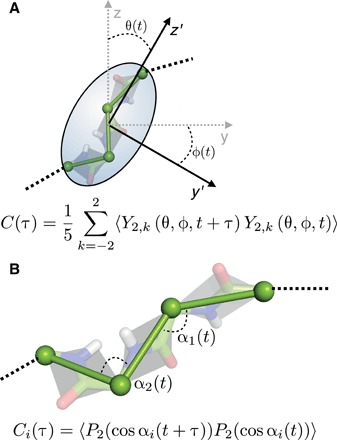
(A) We consider a time-dependent gyration tensor for each segment (here represented by an ellipsoid), as defined in (36). The gyration tensor is diagonalized by a rotation matrix expressed as a function of time-dependent angles θ and φ that are used to compute a correlation function that reports on the time fluctuations of the orientation of the segment in the laboratory frame. (B) In the model presented in (30), all the information regarding segmental motions is encoded in the relative orientation of peptide planes. We label α1, α2 … αn the n = N(N − 1)/2 time-dependent angles identified by two Cα-Cα vectors in a segment of N residues. We compute n correlation functions reporting on intrasegment dynamics.
Segmental rotational dynamics are characterized using the method of Su et al. (36), in which the time-dependent gyration tensor of each segment is calculated and then diagonalized (Fig. 3A). The time-dependent Euler angles derived from the diagonalization of the gyration tensor are used to compute correlation functions that report on the rotational dynamics of the segment. In all our simulations, the decay of such correlation functions is, to a good approximation, monoexponential. Their characteristic time constants are shown in fig. S10. Unexpectedly, we find that the differences in the two water models do not affect segmental rotational dynamics that occur on similar time scales in all simulations.
Figure S10 also shows that, as expected, such dynamics occur on longer time scales at lower temperatures and in the center of the chain compared to the more flexible ends. We find that the time scales of rotational dynamics of segments are largely overlapping with (and therefore apparently account for) the faster of the two dominant components in segmental dynamics (fig. S8).
Intrasegment dynamics are strongly coupled to water properties
Next, we examine the component of motion concerning intrasegment dynamics, characterized by the autocorrelation functions (ACFs) of the N(N − 1)/2 time-dependent angles defined by two Cα-Cα vectors in a segment comprising N residues (Fig. 3B). Notice that this component is not to be confused with the effect of φi/ψi−1 angles on relaxation discussed above: The geometry of peptide planes is such that, to a good approximation, the N-Cα and N-C′ bond vectors are almost collinear with the Cα-Cα vector, whose motion is therefore dominated by internal degrees of freedom in the segment other than φi and ψi−1. Intrasegmental motions as defined here bear some similarity to the so-called “backrub” motions that were used to describe backbone conformational degrees of freedom in loop regions of folded proteins (37, 38).
Figure 4 and figs. S11 and S12 show time scales obtained from the fit of such sets of correlation functions at 298, 288, and 278 K, respectively. Intrasegment dynamics occur on a distribution of time scales, reflecting the presence of multiple and heterogeneous energy barriers to the reorientation of peptide planes with respect to each other. In C3P, this distribution is rather narrow and centered around time scales longer than 10 ns and slower than the segmental motions at all temperatures and for all residues. When the simulation is carried out in TIP4P/2005, however, time scales in intrasegment dynamics are more uniformly distributed from subnanosecond values up to 50 ns. Simulations in A4P and C4P result in a very similar description of intrasegment dynamics, indicating that conformational dynamics within each segment are mostly influenced by solvent properties. This appears to identify intrasegment motions as the most important physical mechanism responsible for the very different dynamic features observed in TIP3P and TIP4P/2005.
Fig. 4. Comparison of intrasegment dynamics and longest relaxation active time scale.
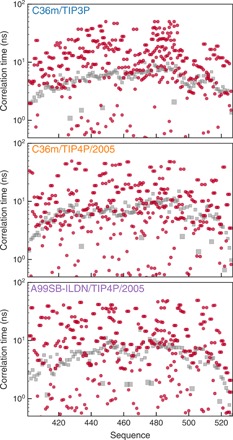
Time scales associated with intrasegment dynamics at 298 K (red circles) in C3P (top), C4P (middle), and A4P (bottom) are compared with the longest time scale resulting from fitting segmental dynamics correlation functions (gray squares, see also fig. S7).
Comparison of intrasegment dynamic time scales with the longest time scale extracted from the global correlation function for segmental dynamics (Fig. 4 and figs. S11 and S12; see also Fig. 2 and fig. S8) reveals an important feature. Because the segmental motions are, in our model (30), the only factors driving the rotational correlation function of NH bond vectors to zero (i.e., they are the only physical mechanisms whose correlation function does not converge to a finite plateau or order parameter), they also define the longest time scale that can be observed using NMR relaxation. In practice, time scales significantly above the gray squares in Fig. 2 and figs. S11 and S12 are associated with processes that are therefore too slow to contribute significantly to measured spin relaxation. Figure 2 shows that this is the case for most of the intrasegmental motional processes in C3P. On the contrary, in A4P and C4P, intrasegment dynamics represent an important contribution to relaxation as they occur within the relaxation-active window of the correlation function (below the gray curve). These observations are confirmed by the simulations at 15° and 5°C (figs. S11 and S12, respectively), reinforcing the suggestion that NMR spin relaxation rates are reproduced significantly better by TIP4P/2005, because this water model allows for more accurate descriptions of intrasegmental protein motions. This underlines the ability of NMR spin relaxation to probe a rich variety of motional processes occurring within local segments with exquisite sensitivity.
The accuracy of the water model defines segmental motions and directly affects picosecond to nanosecond dynamics
To independently test the model of segmental motions, we have calculated expected NMR relaxation rates using the segments identified from the different simulations, imposing that each peptide unit within the segment evolves with the same segmental correlation time. For each segment, internal correlation times were determined for each residue, as well as the relative amplitude of the librational, internal, and segmental components (see the Supplementary Materials). The segments defined using the TIP4P/2005 simulations systematically improve reproduction of NMR relaxation rates reporting on time scales spanning three orders of magnitude (10−11 to 10−8 s) compared to segments derived from TIP3P simulations, and this is the case at all temperatures (Fig. 5). As expected, the improvement is localized in the regions where the segment definitions are significantly different.
Fig. 5. Segmental motional models derived from C4P reproduce overall NMR relaxation rates better than segmental motional models derived from C3P.
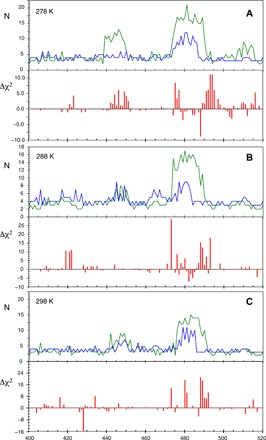
(A) Top: Length and position of segments derived from C3P (green) and C4P (blue) at 278 K. Bottom: Difference in χ2 of the central residue in the segment between C3P- and C4P-derived segmental models (χ2C3P and χ2C4P). (B and C) Similar representation for segments derived from ensembles of trajectories determined from C3P and C4P at 288 K and 298 K.
Temperature dependence of intrasegment dynamics depends on the lifetime of water-water hydrogen bonds
We also investigated whether solvent effects on protein dynamics can be inferred from the physical properties of bulk water. To do so, we carried out simulations of boxes containing TIP4P/2005 or CHARMM-modified TIP3P water (see the Supplementary Materials). It has been suggested (39) that water translation is the main mechanism by which solvent controls protein dynamics. We calculate the self-diffusion coefficients of TIP4P/2005 and TIP3P water at different temperatures (Fig. 6A). Our results are in agreement with data previously published by other groups [e.g., (40)], showing that TIP3P water molecules diffuse much faster than TIP4P/2005, correlating with the observation that fast protein dynamics are excessively accelerated in C3P. Self-diffusion coefficients of TIP4P/2005 are also much closer to the experimental values (40). In both simulations, self-diffusion coefficients increase almost linearly with temperature, with the same slope for both water models (in contrast to the segmental dynamics whose slope varies considerably). This suggests that the different temperature dependence of protein dynamics in the two force fields does not result from translational diffusion. On the contrary, we find that the lifetime of water-water hydrogen bonds decreases much more rapidly with temperature in TIP4P/2005 than in TIP3P (Fig. 6B), indicating that collective modes are much more rapidly decoupled in TIP3P. This supports recent observations (41) that the coupling of water and protein dynamics in TIP3P is weak and insensitive to temperature.
Fig. 6. Comparison of properties of water models.
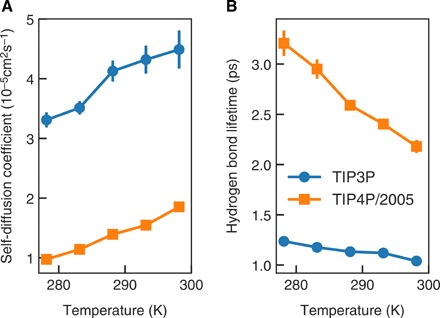
Self-diffusion coefficients (A) and lifetime of hydrogen bonds (B) in TIP3P (blue circles) and TIP4P/2005 (orange squares) water.
DISCUSSION
Protein dynamics are intimately connected to water dynamics (7, 39). In the case of IDPs, for which most of the protein is exposed to solvent, water is expected to have an even more significant impact on dynamics than for folded proteins. This paradigm has been investigated in the past using both experiment (20) and simulation (23); however, the lack of a single, well-defined three-dimensional fold and the impossibility of tracking individual water molecules common to many experimental techniques hinder mechanistic understanding of the effect of water on the structure and dynamics of IDPs.
The dynamics of IDPs are conveniently probed by site-specific NMR relaxation measured throughout the protein, which can only be accurately predicted if the distribution of motions, reporting on modes occurring from tens of picoseconds to tens of nanoseconds, is correctly simulated. In this study, we have combined recent analytical tools developed to interpret NMR relaxation data from IDPs in terms of representative ensembles of time-dependent MD trajectories, high-power computing (60 μs of simulation), and a recently compiled set of self-consistent temperature-dependent experimental NMR relaxation rates to examine water-protein interactions in unique detail. By independently changing the force field parameters describing the disordered peptide and the surrounding water molecules, we are thereby able to identify and analyze the key components controlling the dynamic modes of the protein in solution.
Longitudinal 15N auto- and cross-relaxation rates of the 125–amino acid IDP, Ntail—chosen because it is sufficiently long to probe chain-like behavior and to probe numerous distinct sites—measured at three temperatures and at four magnetic field strengths, are best reproduced when the TIP4P/2005 water model is used in the production of the ensemble of trajectories. We attempted to identify the characteristics of this model that are responsible for the improved predictive behavior for these rates that are sensitive to motions ranging from tens of picoseconds to nanoseconds. By deconvoluting the ensemble-averaged ACF that is responsible for NMR relaxation into its component parts and systematically analyzing their behavior, we show that the motional properties of IDPs are organized in dynamic segments of variable size and identify a clear correlation of one specific component of the dynamics, describing intrasegmental motions, with the physical and, in particular, the dynamic properties of the solvent. These modes are characterized by rotations about vectors connecting Cα-Cα pairs within the segment and include the so-called backrub motions that have been observed from analysis of high-resolution x-ray crystallographic structures and were shown to capture loop motions of folded proteins (37, 38). Such modes have also been observed in β sheets in small folded domains using residual dipolar couplings (42).
Here, we show that the tight coupling between this component of IDP dynamics and the dynamics of the solvent is responsible for the poor reproduction of the motional time scales of the protein if the water model used for simulation is not physically accurate and, concomitantly, an accurate reproduction of relaxation rates if a more correct water model is used. NMR is particularly sensitive to these time scales and modes because motions that are slower than the reorientational time scale of the peptide unit are inefficient in terms of relaxation, while the same motions are highly efficient if they are significantly faster than the overall reorientation of the segment. Combination of NMR and molecular simulation allows these time scales to be compared with exquisite definition. This study therefore provides unique new insight into the coupled dynamics of IDP solutes and surrounding solvent systems.
A number of studies (43, 44) have convincingly identified the imbalance between protein-protein and protein-water forces in currently available force fields as responsible for inaccurate simulations of IDPs, for example, overrepresentation of α-helical elements and collapsed or compact states that disagree with dimensions derived from small-angle scattering. Modified parameters have been introduced to obtain ensembles of structures in better agreement with available dimensions. However, such modifications can also result in unrealistic destabilization of folded domains (28, 45–47) and cannot be easily transferred to model more complex systems, containing both folded and unfolded domains, which would be necessary, for instance, to characterize IDP binding (17).
In this study, we demonstrate the importance of considering the accurate prediction of time scales of molecular motion, in addition to the range of local and long-range conformational sampling. The additional constraint on dynamic time scales implicit in the simulation of spin relaxation contrasts with first-order NMR observables, such as chemical shifts or coupling constants, or small-angle scattering, which are considered to average over an ensemble with little or no restriction on the averaging time scale. In the case of chemical shifts, dynamic interconversion occurring on the fast time scale implies that interconversion between states occurs faster than hundreds of microseconds, which is a relatively weak restraint in such a flat conformational energy surface. In the current study, of which NMR relaxation is the focus, dynamic time scales are crucial to the reproduction of the large volume of experimental data. The additional insight that is gained from considering the dynamics of both protein and solvent provides unique insight into their apparent coupling. Force field assessment and, potentially, development, via such a “holistic” approach, in which parameters describing the free-energy landscape of proteins and water (and possibly nucleic acids) are simultaneously optimized against structural and dynamic properties of both folded and disordered systems, hold great promise for accurate simulation of the behavior of IDPs in solution.
The biological function of IDPs is most often carried out by binding to partner proteins, an event that can lead (15) or not (16, 17, 48) to folding of disordered chains. Our results indicate that water is important in the binding process not only because it determines translational diffusion, and consequently rates of molecular association, but also because it has a fundamental influence on the short-range intrasegment conformational rearrangements required for example for specific binding to folded partners (18). Accurate descriptions of the behavior of water are also essential for understanding entropic and enthalpic contributions to binding free energies involving IDPs. A broad comparison of the physical behavior of available rigid, nonpolarizable water models with experimental observation found that the TIP4P/2005 scored highest over a wide range of properties and thermodynamic states (49). Our study, assessed from the perspective of detailed analysis of dynamic time scales experienced by the solute protein, further supports these conclusions, at least in comparison to TIP3P.
Our results are also important in the context of the interpretation of experimental evidence about dynamics of IDPs in vivo (50), often analyzed in analogy or in contrast to in vitro studies. Several studies (51, 52) used in-cell NMR to demonstrate that IDPs remain disordered in cellular environments, although their dynamics appear considerably slowed down, presumably because of viscosity effects, resulting in faster transverse relaxation and broader spectral lines compared to in vitro studies (50, 53). However, our results suggest that this effect is unlikely to be due to water translation alone, as it has been shown that a significant fraction of cellular water exhibits the same dynamic time scales as pure water (54), thereby excluding significant effects of confinement on mobility. Alternatively, “excluded volume effects,” due to the fact that 10 to 40% of cell volume is occupied by macromolecules, are often evoked to account for the loss of translational and rotational mobility of proteins in cells (55). Our results suggest that a key factor controlling the dynamic properties of IDPs in the cellular environment could be the specific properties of the solvent and in particular the strength of intermolecular hydrogen bonds. It is known that the concentration of osmolytes, macromolecules, and other chemical entities affects the donor/acceptor properties of water molecules (56). In principle, this control may be tuned by the specific chemical composition of cell lines and organelles within cells (57), thereby accounting for differences observed across different cell lines. Further experimental work is required to explore this hypothesis.
MATERIALS AND METHODS
The simulation system consisted of the Ntail polypeptide in a rectangular box with ~100,000 water molecules and a number of Na+ and Cl− ions corresponding to a salt concentration of 0.5 M, with an excess of 20 Na+ ions to neutralize protein charges. Different combinations of protein force fields and water models were used, as summarized in table S1. For each force field/water model combination, 10 to 28 (see table S1) independent simulations were seeded from different protein conformations, randomly selected among the 200 PDBs of an ensemble derived from NMR observables (31).
Trajectories in TIP3P water were calculated using ACEMD Molecular Dynamics version 2016.10.27 (58). Energy minimization with the conjugate gradient minimization algorithm was performed for 500 steps, followed by 100-ps and 1-ns equilibration runs in the NVE and NPT ensembles, respectively. Last, production runs of 200 to 400 ns depending on the simulated temperature (see table S1) were calculated in the NVT ensemble integrating the equation of motions every 2 fs. Holonomic constraints were applied on all hydrogen-heavy atom bond terms to remove fast oscillations. A cutoff of 9 Å was applied to Lennard-Jones and electrostatic interactions, together with smoothing and switching functions to electrostatic and van der Waals forces beyond 7.5 Å. Long-range electrostatic forces were calculated using particle mesh Ewald summation (59) with a grid spacing of 1 Å. During the NPT and NVT runs, the temperature of the system was controlled using a Langevin thermostat with a damping constant of 0.1 ps. In addition, during the NPT equilibration step, the pressure was kept at 1.01325 bar using a Berendsen barostat with a relaxation time of 400 fs.
GROMACS version 5.1.4 (60) was used for the simulations in TIP4P/2005 water, using the same parameters described above with minor differences as follows. Energy minimization of the system was carried out using a steepest descent algorithm for a maximum of 10,000 steps or until the maximum force in the system was <1000 kJ mol−1 nm−1. NVE and NPT equilibrations were carried out for 500 ps and 2 ns, respectively. Temperature control was achieved using a Berendsen thermostat with a relaxation time of 10 ps. A Parrinello-Rahman scheme with a time constant of 2 ps was used to keep the pressure constant.
Calculation of NMR spin relaxation rates
ACFs for the amide bond vector orientations of all trajectory blocks in table S1 were calculated up to a maximum lag time corresponding to half of the length of the block. The procedure used to fit the ACFs to a sum of exponential decays and then compute spin relaxation rates was described in (30).
Average block selection using relaxation data
To minimize force field–dependent inaccuracies, we applied our ABSURD procedure as described in (28, 30). Tentatively, we performed the selection for each simulation using each spin relaxation dataset recorded at the simulated temperature. Then, we considered only the selection that yields the lowest global χ2 value, calculated comparing the selected trajectory segments with all the relaxation rates measured at that temperature. The results of this procedure are summarized in table S2.
Contributions of backbone dihedral angle dynamics to 15N relaxation
The effect of the dynamics of backbone dihedral angles on spin relaxation was calculated as described in (30).
Contributions of segmental dynamics to 15N relaxation
As described in the analytical model presented in (30), segmental dynamics were characterized by calculating the rotational properties of peptide planes in the laboratory frame, approximated by the rotational correlation function of Cα-Cα vectors.
Segment length
We used the definition of angle order parameters of Hyberts et al. (35) in which N is the number of frames in a simulation and α is the time-dependent angle between the Cα-Cα vectors associated with two residues. If , the two residues are considered to belong to the same dynamic segment.
Water simulations
Five Na+ and 5 Cl− ions were added to a cubic simulation box containing 512 TIP3P or TIP4P/2005 water molecules to obtain a salt concentration equal to that used experimentally (500 mM). GROMACS version 5.1.4 (60) was used to simulate the dynamics of the box, using the same protocol described above. For each simulated temperature (5°, 10°, 15°, 20°, and 25°C), we calculated three independent 100-ns trajectories.
Calculation of self-diffusion coefficients and hydrogen bond lifetimes
Analysis of the dynamics of water molecules was carried out using the gmx msd and gmx hbond tools in the GROMACS suite.
Derivation of segmental motional models from experimental spin relaxation rates
The fitted relaxation rates are given by the following functions
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
J(ω) is the spectral density function at frequency ω, and θ is the angle between the N-H dipole-dipole interaction and the principal axis of the chemical shift anisotropy (CSA) tensor (assumed axially symmetric with anisotropy σ∥ − σ⊥= −172 ppm). rNH is the NH internuclear distance (assumed to be 1.02 Å); γH and γN are the gyromagnetic ratio of 1H and 15N nuclei, respectively; μ0 is the permittivity of free space; and ℏ is Planck’s constant.
The spectral density function was modeled by the sum of Lorentzian functions
| (6) |
where ∑kAk = 1.
The model-free analysis optimizes A2, A3, τ1, τ2, τ3, and θ using a nonlinear least-squares fitting approach by minimizing the following function
| (7) |
Segments were analyzed by constraining one of the correlation times that is common to the segment to adopt the same value for each residue, while the other two correlation times and the amplitudes were all optimized simultaneously. The optimization was carried out by summing Eq. 7 over all of the residues in the given segment.
Supplementary Material
Acknowledgments
Funding: We acknowledge financial support from the Swiss National Science Foundation (Advanced Postdoc Mobility Fellowship P300P2_167742 to N.S.), FRISBI (ANR-10-INSB-05-02), GRAL (ANR-10-LABX-49-01), and NanoDisPro (ANR-18-CE29-0003). MD simulations were performed using the HPC resources of CCRT available by GENCI (Grand Equipement National de Calcul Intensif, projects t2016077486, 2017drfr1700, and gen7486/t2016077486). Author contributions: N.S., A.A., and M.B. planned and performed the research, and N.S. and M.B. wrote the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaax2348/DC1
Table S1. Summary of the MD simulations carried out on the C-terminal domain of Sendai virus and discussed in the present work.
Table S2. Summary of the results of the ABSURD procedure.
Fig. S1. Experimental secondary chemical shifts (bars) compared with values calculated using frames extracted from the trajectories every 500 ps as input to SPARTA+.
Fig. S2. Distribution of radii of gyration in the ensemble used to seed the MD simulations (bars) compared with those calculated using frames extracted from the trajectories every 200 ps.
Fig. S3. Experimental 15N chemical shift anisotropy/dipole-dipole cross-correlated cross-relaxation rates (ηxy) measured on Ntail compared with the results of simulations.
Fig. S4. Root-mean-square deviations between experimental and simulated spin relaxation rates at 298K.
Fig. S5. Root-mean-square deviations between experimental and simulated spin relaxation rates at 288K.
Fig. S6. Root-mean-square deviations between experimental and simulated spin relaxation rates at 278K.
Fig. S7. Time scales in the correlation function describing the contribution of backbone dihedral angles dynamics to relaxation of 15N backbone amide nuclei.
Fig. S8. Time scales in the correlation function describing the contribution of segmental motions to relaxation of 15N backbone amide nuclei.
Fig. S9. Fluctuations of the relative orientation of peptide planes measured by the order parameter S2seg.
Fig. S10. Time scales extracted from fits of correlation functions describing the rotational dynamics of segments to mono-exponential decays.
Fig. S11. Time scales associated with intra-segment dynamics at 288K (orange circles) are compared with the longest time scale resulted from fitting segmental dynamics correlation functions (gray squares).
Fig. S12. Time scales associated with intra-segment dynamics at 278K (yellow circles) are compared with the longest time scale resulted from fitting segmental dynamics correlation functions (gray squares).
Reference (61)
REFERENCES AND NOTES
- 1.Karplus M., Kuriyan J., Molecular dynamics and protein function. Proc. Natl. Acad. Sci. U.S.A. 102, 6679–6685 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levy Y., Onuchic J. N., Water mediation in protein folding and molecular recognition. Annu. Rev. Biophys. Biomol. Struct. 35, 389–415 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Laage D., Elsaesser T., Hynes J. T., Water dynamics in the hydration shells of biomolecules. Chem. Rev. 117, 10694–10725 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doster W., Cusack S., Petry W., Dynamical transition of myoglobin revealed by inelastic neutron scattering. Nature 337, 754–756 (1989). [DOI] [PubMed] [Google Scholar]
- 5.Rasmussen B. F., Stock A. M., Ringe D., Petsko G. A., Crystalline ribonuclease A loses function below the dynamical transition at 220 K. Nature 357, 423–424 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Ferrand M., Dianoux A. J., Petry W., Zaccaï G., Thermal motions and function of bacteriorhodopsin in purple membranes: Effects of temperature and hydration studied by neutron scattering. Proc. Natl. Acad. Sci. U.S.A. 90, 9668–9672 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewandowski J. R., Halse M. E., Blackledge M., Emsley L., Direct observation of hierarchical protein dynamics. Science 348, 578–581 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Arcangeli C., Bizzarri A. R., Cannistraro S., Role of interfacial water in the molecular dynamics-simulated dynamical transition of plastocyanin. Chem. Phys. Lett. 291, 7–14 (1998). [Google Scholar]
- 9.Tarek M., Tobias D. J., Role of protein-water hydrogen bond dynamics in the protein dynamical transition. Phys. Rev. Lett. 88, 138101 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Persson F., Söderhjelm P., Halle B., The spatial range of protein hydration. J. Chem. Phys. 148, 215104 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Nilsson L., Halle B., Molecular origin of time-dependent fluorescence shifts in proteins. Proc. Natl. Acad. Sci. U.S.A. 102, 13867–13872 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S.-H., Liu L., Fratini E., Baglioni P., Faraone A., Mamontov E., Observation of fragile-to-strong dynamic crossover in protein hydration water. Proc. Natl. Acad. Sci. U.S.A. 103, 9012–9016 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramakrishnan G., González-Jiménez M., Lapthorn A. J., Wynne K., Spectrum of slow and super-slow (picosecond to nanosecond) water dynamics around organic and biological solutes. J. Phys. Chem. Lett. 8, 2964–2970 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Nucci N. V., Pometun M. S., Wand A. J., Site-resolved measurement of water-protein interactions by solution NMR. Nat. Struct. Mol. Biol. 18, 245–249 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sugase K., Dyson H. J., Wright P. E., Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature 447, 1021–1025 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Kragelj J., Palencia A., Nanao M., Maurin D., Bouvignies G., Blackledge M., Jensen M. R., Structure and dynamics of the MKK7-JNK signaling complex. Proc. Natl. Acad. Sci. U.S.A. 112, 3409–3414 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borgia A., Borgia M., Bugge K., Kissling V., Heidarsson P., Fernandes C., Sottini A., Soranno A., Buholzer K., Nettels D., Kragelund B., Best R., Schuler B., Extreme disorder in an ultrahigh-affinity protein complex. Nature 555, 61–66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schneider R., Maurin D., Communie G., Kragelj J., Hansen D., Ruigrok R. W. H., Jensen M. R., Blackledge M., Visualizing the molecular recognition trajectory of an intrinsically disordered protein using multinuclear relaxation dispersion NMR. J. Am. Chem. Soc. 137, 1220–1229 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Milles S., Jensen M. R., Lazert C., Guseva S., Ivashchenko S., Communie G., Maurin D., Gerlier D., Ruigrok R. W. H., Blackledge M., An ultraweak interaction in the intrinsically disordered replication machinery is essential for measles virus function. Sci. Adv. 4, eaat7778 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armstrong B. D., Choi J., López C., Wesener D., Hubbell W., Cavagnero S., Han S., Site-specific hydration dynamics in the nonpolar core of a molten globule by dynamic nuclear polarization of water. J. Am. Chem. Soc. 133, 5987–5995 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallat F.-X., Laganowsky A., Wood K., Gabel F., van Eijck L., Wuttke J., Moulin M., Härtlein M., Eisenberg D., Colletier J.-P., Zaccai G., Weik M., Dynamical coupling of intrinsically disordered proteins and their hydration water: Comparison with folded soluble and membrane proteins. Biophys. J. 103, 129–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang J., MacKerell A. D. Jr., Force field development and simulations of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 48, 40–48 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rani P., Biswas P., Local structure and dynamics of hydration water in intrinsically disordered proteins. J. Phys. Chem. B 119, 10858–10867 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Bokór M., Csizmok V., Kovács D., Bánki D., Friedrich P., Tompa P., Tompa K., NMR relaxation studies on the hydrate layer of intrinsically unstructured proteins. Biophys. J. 88, 2030–2037 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gavrilov Y., Leuchter J. D., Levy Y., On the coupling between the dynamics of protein and water. Phys. Chem. Chem. Phys. 19, 8243–8257 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Jensen M. R., Zweckstetter M., Huang J., Blackledge M., Exploring free-energy landscapes of intrinsically disordered proteins at atomic resolution using NMR spectroscopy. Chem. Rev. 114, 6632–6660 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Salvi N., Abyzov A., Blackledge M., Atomic resolution conformational dynamics of intrinsically disordered proteins from NMR spin relaxation. Prog. Nucl. Magn. Reson. Spectrosc. 102–103, 43–60 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Salvi N., Abyzov A., Blackledge M., Multi-timescale dynamics in intrinsically disordered proteins from NMR relaxation and molecular simulation. J. Phys. Chem. Lett. 7, 2483–2489 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Abyzov A., Salvi N., Schneider R., Maurin D., Ruigrok R. W. H., Jensen M. R., Blackledge M., Identification of dynamic modes in an intrinsically disordered protein using temperature-dependent NMR relaxation. J. Am. Chem. Soc. 138, 6240–6251 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Salvi N., Abyzov A., Blackledge M., Analytical description of NMR relaxation highlights correlated dynamics in intrinsically disordered proteins. Angew. Chem. Int. Ed. 56, 14020–14024 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Jensen M. R., Salmon L., Nodet G., Blackledge M., Defining conformational ensembles of intrinsically disordered and partially folded proteins directly from chemical shifts. J. Am. Chem. Soc. 132, 1270–1272 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B., Grubmüller H., MacKerell A. Jr., CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Abascal J. L. F., Vega C., A general purpose model for the condensed phases of water: TIP4P/2005. J. Chem. Phys. 123, 234505 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Lindorff-Larsen K., Piani S., Palmo K., Maragakis P., Klepeis J., Dror R., Shaw D., Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hyberts S. G., Goldberg M. S., Havel T. F., Wagner G., The solution structure of eglin c based on measurements of many NOEs and coupling constants and its comparison with X-ray structures. Protein 1, 736–751 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su S. J., Denny M. S., Kovac J., Computer simulation of the dynamics of shape fluctuations in uniform star polymers. Macromolecules 24, 917–923 (1991). [Google Scholar]
- 37.Davis I. W., Arendall W. B. III, Richardson D. C., Richardson J. S., The backrub motion: How protein backbone shrugs when a sidechain dances. Structure 14, 265–274 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Smith C. A., Kortemme T., Backrub-like backbone simulation recapitulates natural protein conformational variability and improves mutant side-chain prediction. J. Mol. Biol. 380, 742–756 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Karplus M., Vitkup D., Ringe D., Petsko G. A., Solvent mobility and the protein ‘glass’ transition. Nat. Struct. Biol. 7, 34–38 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Wang L.-P., Martinez T. J., Pande V. S., Building force fields: An automatic, systematic, and reproducible approach. J. Phys. Chem. Lett. 5, 1885–1891 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haddadian E. J., Zhang H., Freed K. F., Douglas J. F., Comparative study of the collective dynamics of proteins and inorganic nanoparticles. Sci. Rep. 7, 41671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouvignies G., Bernadó P., Meier S., Cho K., Grzesiek S., Bruschweiler R., Blackledge M., Identification of slow correlated motions in proteins using residual dipolar and hydrogen-bond scalar couplings. Proc. Natl. Acad. Sci. U.S.A. 102, 13885–13890 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Best R. B., Zheng W., Mittal J., Balanced protein-water interactions improve properties of disordered proteins and non-specific protein association. J. Chem. Theory Comput. 10, 5113–5124 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Piana S., Donchev A. G., Robustelli P., Shaw D. E., Water dispersion interactions strongly influence simulated structural properties of disordered protein states. J. Phys. Chem. B 119, 5113–5123 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Henriques J., Skepö M., Molecular dynamics simulations of intrinsically disordered proteins: On the accuracy of the TIP4P-D water model and the representativeness of protein disorder models. J. Chem. Theory Comput. 12, 3407–3415 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Mercadante D., Wagner J. A., Aramburu I. V., Lemke E. A., Gräter F., Sampling long- versus short-range interactions defines the ability of force fields to reproduce the dynamics of intrinsically disordered proteins. J. Chem. Theory Comput. 13, 3964–3974 (2017). [DOI] [PubMed] [Google Scholar]
- 47.Robustelli P., Piana S., Shaw D. E., Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U.S.A. 115, E4758–E4766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tompa P., Fuxreiter M., Fuzzy complexes: Polymorphism and structural disorder in protein–protein interactions. Trends Biochem. Sci. 33, 2–8 (2008). [DOI] [PubMed] [Google Scholar]
- 49.Vega C., Abascal J. L. F., Simulating water with rigid non-polarizable models: A general perspective. Phys. Chem. Chem. Phys. 13, 19663–19688 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Theillet F.-X., Binolfi A., Bekei B., Martorana A., Rose H., Stuiver M., Verzini S., Lorenz D., van Rossum M., Goldfarb D., Selenko P., Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530, 45–50 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Ito Y., Mikawa T., Smith B. O., In-cell NMR of intrinsically disordered proteins in prokaryotic cells. Methods Mol. Biol. 895, 19–31 (2012). [DOI] [PubMed] [Google Scholar]
- 52.Luchinat E., Banci L., A unique tool for cellular structural biology: In-cell NMR. J. Biol. Chem. 291, 3776–3784 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Binolfi A., Theillet F.-X., Selenko P., Bacterial in-cell NMR of human α-synuclein: A disordered monomer by nature? Biochem. Soc. Trans. 40, 950–954 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Persson E., Halle B., Cell water dynamics on multiple time scales. Proc. Natl. Acad. Sci. U.S.A. 105, 6266–6271 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Theillet F.-X., Binolfi A., Frembergen-Kesner T., Hingorani K., Sarkar M., Kyne C., Li C., Crowley P., Gierasch L., Pielak G., Elcock A., Gershenson A., Selenko P., Physicochemical properties of cells and their effects on intrinsically disordered proteins (IDPs). Chem. Rev. 114, 6661–6714 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferreira L. A., Breydo L., Reichardt C., Uversky V. N., Zaslavsky B. Y., Effects of osmolytes on solvent features of water in aqueous solutions. J. Biomol. Struct. Dyn. 35, 1055–1068 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Zaslavsky B. Y., Uversky V. N., In Aqua Veritas: The indispensable yet mostly ignored role of water in phase separation and membrane-less organelles. Biochemistry 57, 2437–2451 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Harvey M. J., Giupponi G., De Fabritiis G., ACEMD: Accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 5, 1632–1639 (2009). [DOI] [PubMed] [Google Scholar]
- 59.Essmann U., Perera L., Berkowitz M., A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995). [Google Scholar]
- 60.Abraham M. J., Murtola T., Schulz R., Pail S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 61.Shen Y., Bax A., SPARTA+: A modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J. Biomol. NMR 48, 13–22 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaax2348/DC1
Table S1. Summary of the MD simulations carried out on the C-terminal domain of Sendai virus and discussed in the present work.
Table S2. Summary of the results of the ABSURD procedure.
Fig. S1. Experimental secondary chemical shifts (bars) compared with values calculated using frames extracted from the trajectories every 500 ps as input to SPARTA+.
Fig. S2. Distribution of radii of gyration in the ensemble used to seed the MD simulations (bars) compared with those calculated using frames extracted from the trajectories every 200 ps.
Fig. S3. Experimental 15N chemical shift anisotropy/dipole-dipole cross-correlated cross-relaxation rates (ηxy) measured on Ntail compared with the results of simulations.
Fig. S4. Root-mean-square deviations between experimental and simulated spin relaxation rates at 298K.
Fig. S5. Root-mean-square deviations between experimental and simulated spin relaxation rates at 288K.
Fig. S6. Root-mean-square deviations between experimental and simulated spin relaxation rates at 278K.
Fig. S7. Time scales in the correlation function describing the contribution of backbone dihedral angles dynamics to relaxation of 15N backbone amide nuclei.
Fig. S8. Time scales in the correlation function describing the contribution of segmental motions to relaxation of 15N backbone amide nuclei.
Fig. S9. Fluctuations of the relative orientation of peptide planes measured by the order parameter S2seg.
Fig. S10. Time scales extracted from fits of correlation functions describing the rotational dynamics of segments to mono-exponential decays.
Fig. S11. Time scales associated with intra-segment dynamics at 288K (orange circles) are compared with the longest time scale resulted from fitting segmental dynamics correlation functions (gray squares).
Fig. S12. Time scales associated with intra-segment dynamics at 278K (yellow circles) are compared with the longest time scale resulted from fitting segmental dynamics correlation functions (gray squares).
Reference (61)