

Abstract
CNS small vessel disease (CSVD) causes 25% of strokes and contributes to 45% of dementia cases. Prevalence increases with age, affecting about 5% of people aged 50 years to almost 100% of people older than 90 years. Known causes and risk factors include age, hypertension, branch atheromatous disease, cerebral amyloid angiopathy, radiation exposure, immune-mediated vasculitides, certain infections, and several genetic diseases. CSVD can be asymptomatic; however, depending on location, lesions can cause mild cognitive dysfunction, dementia, mood disorders, motor and gait dysfunction, and urinary incontinence. CSVD is diagnosed on the basis of brain imaging biomarkers, including recent small subcortical infarcts, white matter hyperintensities, lacunes, cerebral microbleeds, enlarged perivascular spaces, and cerebral atrophy. Advanced imaging modalities can detect signs of disease even earlier than current standard imaging techniques. Diffusion tensor imaging can identify altered white matter connectivity, and blood oxygenation level-dependent imaging can identify decreased vascular reactivity. Pathogenesis is thought to begin with an etiologically specific insult, with or without genetic predisposition, which results in dysfunction of the neurovascular unit. Uncertainties regarding pathogenesis have delayed development of effective treatment. The most widely accepted approach to treatment is to intensively control well-established vascular risk factors, of which hypertension is the most important. With better understanding of pathogenesis, specific therapies may emerge. Early identification of pathologic characteristics with advanced imaging provides an opportunity to forestall progression before emergence of symptoms.
CNS small vessel disease (CSVD) is one of the most prevalent pathologic processes encountered by neurologists in clinical practice. The increase in life expectancy worldwide has increased CSVD prevalence, affecting almost everyone older than 90 years. In addition, the use of MRI has increased CSVD detection rates. CSVD is the attributable cause of 25% of strokes and more than doubles the odds of recurrent stroke1; furthermore, it contributes to 45% of dementia cases2 and to global functional decline.3 The purpose of this review is to provide a clinical update of CSVD, including its epidemiologic characteristics, risk factors, theories on pathogenesis, clinical presentation, diagnosis, biomarkers, prevention, and treatment. In addition, we propose future directions for advancing against a disease process in need of effective therapies.
Methods
We queried PubMed by using the following keywords and Medical Subject Headings (MeSH) terms: “cerebral small vessel disease,” “cerebral SVD,” “CSVD,” “leukoaraiosis,” “white matter hyperintensities,” “white matter lesions,” “lacunar infarctions,” “lacunes,” “microbleeds,” “cerebral amyloid angiopathy,” and “CADASIL.” Subcategory queries were framed with MeSH terms, including “etiologies,” “pathology,” “pathogenesis,” “gait disorders,” and “treatment.” The range of dates queried was January 1, 1982, to July 31, 2018, and no language restrictions were applied. When selecting articles to review, we gave preference to recently published, clinically focused randomized controlled trials (RCTs), systematic reviews, and meta-analyses. In addition, the literature cited within the articles identified by the initial PubMed queries were reviewed and included if appropriate (data availability: this manuscript will not share individual deidentified participant data).
Discussion
Background
In the 1960s, Fisher4 performed postmortem examinations of patients with lacunar stroke and described the pathologic characteristics of CSVD. The small vessels examined and implicated in CSVD included penetrating arterioles, capillaries, and venules, which are typically <1 mm in diameter.5 The small vessel networks begin as penetrating arterioles branching off the large cerebral arteries and pial arterioles, course through the parenchyma, flow into capillary beds, and end as venules flowing into veins. Small blood vessels play a role in regulating cerebral blood flow (CBF). In a study of brain tissue specimens from hypertensive and normotensive individuals, a negative relationship was observed between tunica media-to-lumen diameter ratios in small resistance arteries and CBF.6 The vascular tree of the brain differs from that of other organs because it is embedded in the neurovascular unit (NVU), a term coined at the 2001 Stroke Progress Review Group meeting of the National Institute of Neurological Disorders and Stroke.7
The NVU consists of neurons, astrocytes, endothelial cells, pericytes, and vascular smooth muscle cells (SMC).7 The specific NVU architecture differs in each vascular segment. How the architectural differences affect function is poorly understood and is a major focus of neuroscience research, as it is hypothesized to be involved in not only cerebrovascular disease but also neurodegenerative diseases. Basic functions of the NVU include regulating entry of pathogens and substances from the blood into the parenchyma by means of the blood–brain barrier (BBB); coupling neural activity with CBF for increased delivery of oxygen and nutrients; and clearing metabolic by-products including proteins and heat that pose a threat to normal cellular function. The biochemical processes and structures thought to be responsible for normal NVU function are currently based on animal models. How well this translates to humans is unknown, but there is a need for further cross-validation studies.
Fisher's4 postmortem pathologic descriptions of lacunar stroke were critical to defining CSVD; however, the modern definition is being molded by the field of neuroradiology. Brain imaging provides a premortem and noninvasive means of identifying and monitoring CSVD. Unlike the large vessels of the brain, small vessels are difficult to image directly; therefore, lesions seen with MRI have been adopted as biomarkers of CSVD. These lesions include recent small subcortical infarct, white matter hyperintensity (WMH), lacune, cerebral microbleed (CMB), enlarged perivascular space (PVS), and cerebral atrophy.8
Epidemiologic characteristics, associations, and risk factors
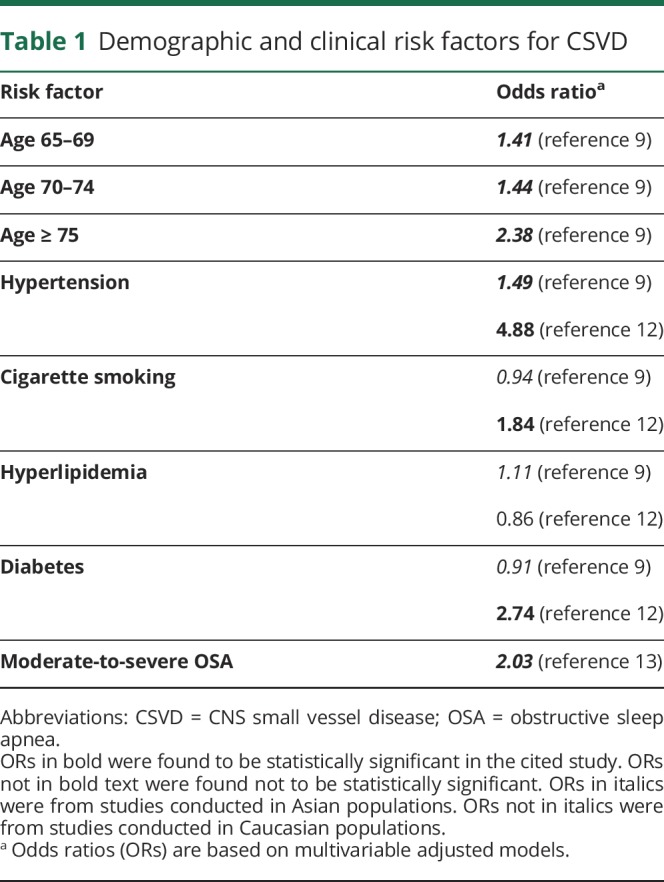
The prevalence of CSVD increases with age, with no significant sex differences9 and no currently known differences across racial-ethnic groups or geography. Specifically, the prevalence of WMH increases from about 5% for people aged 50 years to nearly 100% for people aged 90 years.10 Similarly, the prevalence of CMB increases from 6.5% for people aged 45–50 years to about 36% for people aged 80–89 years.11 The most important modifiable risk factor is arterial hypertension, defined here as blood pressure greater than 140/90 mm Hg.9 Other risk factors include current and former smoking,12 diabetes mellitus,12 obstructive sleep apnea,13 chronic kidney disease,14 and branch atheromatous disease with associated subcortical stroke15 (table 1). Although hypercholesterolemia is a risk factor for large vessel disease, its effect on the risk of CSVD is difficult to estimate in modern-day populations because of the widespread use of statin medications.16
Table 1.
Demographic and clinical risk factors for CSVD
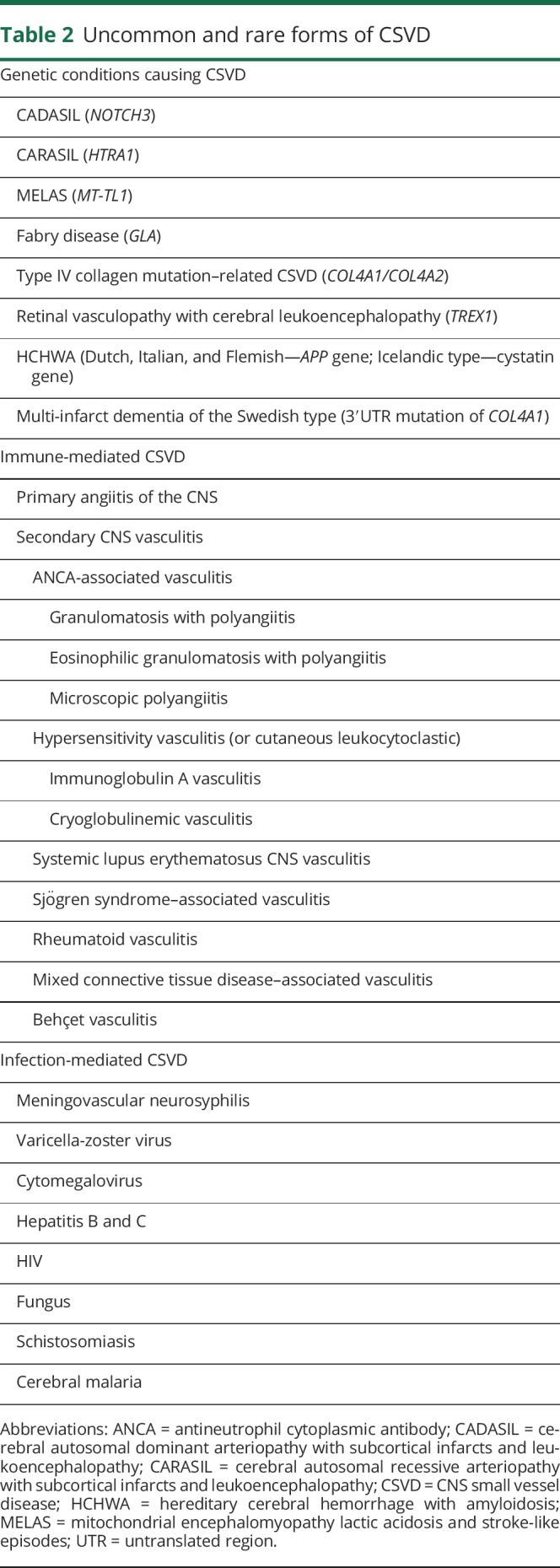
Single-gene disorders are infrequently the cause of CSVD. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL), perhaps the most common inherited cause of CSVD, has a population prevalence among working age adults of about 2–4 per 100,000.17 Several other single-gene and mitochondrial disorders are associated with CSVD but are exceptionally rare. Other uncommon causes, including CSVD associated with immune- and infection-mediated processes, must be considered because the treatments are markedly different. Table 2 summarizes the uncommon and rare causes of CSVD.
Table 2.
Uncommon and rare forms of CSVD
Pathogenesis
The uncertain pathogenesis of CSVD hinders the creation of animal models that might lead to effective therapies. The uncertainty perhaps stems from the complexity of the NVU itself and from the multitude of pathogenic pathways and diseases that exist under the CSVD umbrella. Understanding the sequence of the pathogenesis for each CSVD type is the key to prevention and treatments.
Fisher provided the first pathologic description of the arterial pathology caused by small subcortical infarcts that helped define hypertension-related microangiopathy. The pathologies thought to be secondary to uncontrolled hypertension include hyaline arteriolosclerosis (figure 1A), hyperplastic arteriolosclerosis, segmental arterial disorganization, and microaneurysm (figure 1B).4,5 The increase in media-to-lumen ratio decreases CBF,6 leading to a poorly understood cascade of NVU dysfunction secondary to hypoxia, BBB leakage, inflammation and edema, and oligodendrocyte dysfunction.16,18,19 The resultant loss of myelin and gliosis manifests on MRI as WMH.16,18,19 Severe ischemia of the small vessel territory results in small subcortical infarction. Leakage of blood products out of a microaneurysm results in CMB; rupture results in hypertensive cerebral hemorrhage, typically in the subcortical white matter and deep gray nuclei.
Figure 1. The most common pathologic characteristics of CNS small vessel disease.

(A, B) Hypertensive CSVD (H&E stain, original magnification ×400), as evidenced by hyaline arteriolar sclerosis (A) and a microaneurysm (B). (C, D) CAA (H&E stain, original magnification ×200), as evidenced by amyloid-β-laden vessels in the subarachnoid space, with the double-barreled appearance (C) and capillaries with calcium mineralization (D) (H&E stain, original magnification ×400). (E) Leptomeningeal CAA (thioflavin S, original magnification ×200). (F) Parenchymal CAA (thioflavin S, original magnification ×400). (G) CADASIL showing loss of smooth muscle cells (H&E stain, original magnification ×400). (H) CADASIL showing collagen deposition and loss of smooth muscle cells (trichrome stain, original magnification ×400). (I) Ubiquitinated proteins in granular osmiophilic material (ubiquitin immunohistochemistry, original magnification ×400). (J) Smooth muscle cell degradation due to CADASIL (smooth muscle actin immunohistochemistry, original magnification ×400). CAA = cerebral amyloid angiopathy; CADASIL = cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CSVD = CNS small vessel disease; H&E = hematoxylin-eosin.
The pathogenesis of cerebral amyloid angiopathy (CAA) differs from other types of CSVD in anatomical location and mechanism. Small to medium cortical and leptomeningeal arterioles and arteries are primarily affected. Amyloid-β peptide (Aβ) is deposited in the walls of these vessels, with greater concentrations in the perivascular basement membrane surrounding the SMC in the tunica media (figure 1C).5 Sequelae of CAA include loss of vessel compliance; decreased cerebral vascular reactivity; and increased susceptibility to cortical CMB, convexal subarachnoid hemorrhage (cSAH), cortical superficial siderosis (cSS), greater volume of WMH (posterior predominance), altered structural network connectivity, and cortical atrophy.20
Like CAA, CADASIL causes CSVD by aggregation and deposition of abnormal protein.18 CADASIL is an autosomal dominant genetic disease caused by a mutation in NOTCH3, which encodes a transmembrane receptor found almost exclusively in vascular SMCs and pericytes18; it functions in the regulation of cell-fate determination with other membrane-bound ligands. The mutation typically results in an altered number of cysteine residues in the extracellular domain of NOTCH3, which accumulates, binds to the tissue inhibitor of metalloproteinase 3 and vitronectin, and forms granular osmiophilic material that deposits extracellularly (figure 1I).18 Degeneration of vascular SMCs ensues (figure 1J).18 Other less-common pathologic conditions can result in rare forms of CSVD (figure 2).
Figure 2. Various pathologic characteristics of CNS small vessel disease.

Left column, venous collagenosis. H&E stain of a periventricular venule in the white matter with thickened walls and collagen deposition. Hypersensitivity vasculitis. (Top left) H&E stain showing features of leukoctyoclastic vasculitis; (top right) CD68 immunohistochemistry for macrophages; (bottom left) CD20 showing sparse B lymphocytes; (bottom right) CD3 showing rare T lymphocytes. Schistosomiasis-induced CSVD vasculitis. (Left and right) H&E stains showing granulomatous inflammation. Middle column, postradiation CSVD. (Top left) H&E stain showing fibrinoid necrosis; (top right) H&E stain showing capillary ectasia and atypical nuclear changes; (bottom left) trichrome stain showing collagenosis (blue) and fibrinoid material; (bottom right) H&E stain showing obliterative collagenosis of small vessels with nuclear atypia. Center image, H&E stain showing a normal subcortical white matter arteriole. Immune-mediated CSVD vasculitis. (Top left) H&E stain showing cellular infiltrates in vessel wall; (top right) HLA-DR immunohistochemistry showing macrophage infiltrates; (bottom left) CD20 immunohistochemistry showing B-lymphocytic infiltrates; (bottom right) CD3 showing T-lymphocytic infiltrates. Right column, hypertensive CSVD. (Left) H&E stain showing hyaline arteriolar sclerosis; (right) H&E stain showing a microaneurysm. Cerebral Amyloid Angiopathy. (Top left) H&E stain showing amyloid-β-laden vessels in the subarachnoid spacewith the doublebarreled appearance; (top right) capillaries with calcium mineralization; (bottom left) leptomeningeal CAA on thioflavin S fluorescent microscopy; (bottom right) parenchymal CAA on thioflavin S fluorescent microscopy. Genetic CAA/novel transthyretin mutation. (Left) H&E stain showing leptomeningeal arteriole involvement; (right) transthyretin immunohistochemistry. CADASIL. (Top left) H&E stain showing loss of smooth muscle cells; (top right) trichrome stain showing collagen deposition (blue) in wall of affected arteriole; (bottom left) ubiquitin immunohistochemistry showing granular deposits in arterial wall; (bottom right) smooth muscle actin immunohistochemistry showing fragmentation and loss of smooth muscle cells in affected arteriole. Abbreviations: CAA = cerebral amyloid angiopathy; CADASIL = cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy; CSVD = central nervous systemsmall vessel disease. H&E = hematoxylin-eosin (Figure used with permission of Mayo Foundation for Medical Education and Research).
Diagnosis and biomarkers
CSVD is often incidentally diagnosed with standard MRI sequences obtained by using 1.5- to 3-Tesla (T) scanners. Imaging biomarkers include recent small subcortical infarct, WMH, lacune, CMB, enlarged PVS, and cerebral atrophy.9 A lacune typically is a lesion ≤20 mm in diameter that affects the subcortical white matter and the deep gray matter of the brain and brainstem.9 It is a fluid-filled, gliotic cavity thought to be secondary to prior small subcortical infarcts (figure 3, B.a, B.b).4 WMH, also known as leukoaraiosis, is by definition punctate, patchy, or confluent T2-weighted hyperintensity seen on MRI (figure 3A.a). An enlarged PVS is a dilated space filled with CSF that surrounds perforating arterioles and venules, as they course from the subarachnoid space through the brain parenchyma (figure 3, C.a and C.b).21
Figure 3. MRI findings of CNS small vessel disease.

Patient 1, images A.a, A.b, B.a, B.b. (A.a) Axial T2 FLAIR image shows a typical WMH (arrow) with increased FLAIR signal. (A.b) Corresponding intermediate-signal intensity (arrow) on T1-weighted MRI. (B.a) By contrast, an axial T2 FLAIR image taken slightly more superior in location shows a lacune with marginal increased FLAIR hyperintensity reflecting gliosis with central hypointensity (arrow) representing cavitation. (B.b) The corresponding axial T1-weighted image shows marginal intermediate signal (arrow) corresponding to areas of gliosis with central hypointensity indicating cavitation. Patient 2, images C.a and C.b. (C.a) Coronal T2 FLAIR image shows a left-sided FLAIR hyperintensity (arrowhead) that represents incomplete fluid suppression within a PVS, which is a common diagnostic pitfall and may be erroneously interpreted as a WMH. (C.b) Corresponding coronal T1-weighted image better illustrates the linear branching configuration (arrowhead) typical of a PVS. FLAIR = fluid-attenuated inversion recovery; PVS = perivascular space; WMH = white matter hyperintensity.
Distinguishing among WMH, lacune, and enlarged PVS can occasionally be challenging by imaging. The T2-weighted fluid-attenuated inversion recovery (FLAIR) sequence is commonly used to identify lacunes, WMH, and enlarged PVS; however, FLAIR must be interpreted with caution. MRI signal within lacunes varies by numerous factors. The rates and degrees of cavitation and hence fates of subcortical lesions are highly variable. One study of consecutive acute stroke patients (mean age, 60.7 years) found that about 20% of patients with recent small subcortical strokes failed to show cavitation of their lesion on MRI by 3 months.22 When cavitation does occur, it is more likely to be partial than complete.23 There are currently no studies correlating MRI lesion signals with the contents of the small subcortical infarct or resultant cavitation in necropsy. Another variable is the imaging sequence. FLAIR often fails to suppress the fluid signal within a lacune, thereby misidentifying it as WMH.24 A similar lack of fluid suppression can be seen with enlarged PVS (figure 3, C.a and C.b). Because both lacunes and enlarged PVS can have variable fluid contents, the inversion time may be altered, resulting in a lack of suppression of fluid on the FLAIR sequences. As such, hypointensity seen with T1-weighted imaging or fluid signal intensity on T2-weighted imaging routinely outperforms FLAIR for differentiating lacunes, WMH, and enlarged PVS.24 Another common pitfall is the traditional teaching that a marginally hyperintense rim seen with FLAIR is suggestive of a lacune instead of enlarged PVS. However, up to 50% of enlarged PVS identified in white matter are associated with marginal FLAIR hyperintensity that may reflect microperivascular spaces, gliosis, or both. The typical anatomical location of enlarged PVS may be the best differentiating feature.
CMB is a small area of focal hemosiderin deposition that indicates the previous extravasation of blood from damaged small vessels (figure 4B). Susceptibility-weighted imaging (SWI), gradient-recalled echo (GRE), and T2*-weighted sequences detect small distortions in the magnetic field that can be induced by heavy metals, such as iron. These distortions are shown as a hypointense signal with “blooming artifact,” which is useful for identification of CMB (figure 4B). SWI is preferred for assessing CMB because it is considerably more sensitive than GRE and T2*-weighted sequences.25 The sensitivity of CMB detection is also dependent on the MRI field strength, with 7-T MRI showing greater detection than 3-T MRI, while both have been shown to be superior to 1.5-T MRI.26
Figure 4. MRI findings characteristic of cerebral amyloid angiopathy.

(A) Axial postcontrast vessel wall image showing enhancement of the posterior vessel walls (arrows) with multiple chronic perivascular microhemorrhages (arrowheads). (B) Susceptibility-weighted image showing numerous chronic peripheral CMBs (arrow) and cSS (arrowhead) in a patient with CAA. The microhemorrhages show “blooming artifact” in which the lesions appear larger than their actual sizes because of a magnetic susceptibility effect. (C) T2-weighted FLAIR sequence showing a confluent hyperintensity that is consistent with vasogenic edema from CAA–related inflammation. CAA = cerebral amyloid angiopathy; CMB = cerebral microbleed; cSS = cortical superficial siderosis; FLAIR = fluid-attenuated inversion recovery.
Vessel wall imaging (VWI) is an emerging MRI technique that may help diagnose CSVD. VWI may better depict many types of CSVD compared with luminal imaging techniques.27 Although data are limited regarding the use of VWI for evaluation of CSVD, perhaps as many as 50% of patients with CAA have vessel wall enhancement (figure 4A).28 The presence of enhancement may portend a poor prognosis for neurovascular events.28 VWI may also benefit from ultra-high-field MRI, such as 7-T MRI. Further studies are needed to better understand the role of VWI in detecting and managing CSVD.
More advanced imaging modalities, such as 7-T MRI and diffusion tensor imaging (DTI), can detect microscopic tissue damage earlier than standard MRI sequences.8 One of these early biomarkers seen on 7-T MRI is a microinfarct, which is a 0.2–2.9-mm sharply demarcated microscopic area of parenchymal necrosis due to ischemia.29 Although disturbances have been identified with traditional tensor metrics, such as mean diffusivity and fractional anisotropy, these metrics are nonspecific and may not accurately distinguish between normal white matter and axonal damage.30 More recently, additional metrics, determined by using diffusion imaging with free-water analysis, better predicted CSVD-induced damage than traditional DTI measures.31 Increased extracellular fluid may be a result of increased permeability of the BBB as a sequela of small vessel injury. In the future, these metrics may serve as additional biomarkers for early CSVD.
Assessment of brain perfusion with MRI may become a beneficial tool for diagnosis and prognostication of CSVD.32 Technical improvements in perfusion measures by MRI arterial spin labeling have led to increased interest in this technique. Similarly, fMRI with blood oxygen level-dependent scanning may be a useful tool and has been shown to be abnormal in some presymptomatic patients.33 Thus far, data are conflicting regarding the roles of both CBF and cerebrovascular reactivity in CSVD. Future longitudinal studies in larger patient groups are needed to better understand the role of perfusion imaging in CSVD.
There are several MRI scoring systems that can be easily applied by clinicians to characterize CSVD severity, many of which can predict clinical outcomes. One commonly used scale is the Fazekas scale, which is used to evaluate WMH on T2-weighted FLAIR sequence. The scale grades the severity from 0 to 3—grade 0 represents occasional or nonpunctate WMH; grade 1, multiple punctate WMHs; grade 2, bridging of punctate WMHs leading to confluent lesions; and grade 3, widespread confluent WMH. Fazekas grades 2 and 3 are associated with disability at 90 days and 1 year after ischemic stroke.34
Clinical presentation
Many patients found to have CSVD on MRI incidentally may have mild signs or symptoms of neurocognitive dysfunction previously attributed to normal aging. Up to 20% of asymptomatic elderly persons have evidence of incidental lacunes on MRI. These incidental lacunes more than double the risk of subsequent stroke and dementia.1 Patient presentations can include acute stroke syndromes, various subjective cognitive impairments, mild cognitive impairment, dementia, mood or behavioral disturbances, gait dysfunction, movement disorders, and a general decline in function.
Fisher4 described 5 classic lacunar syndromes in his autopsy series. Syndromes with anatomical localizations include hemisensory loss and hemiparesis (thalamocapsular), pure hemisensory loss (thalamus), pure motor hemiparesis (internal capsule, corona radiata, or basis pontis), dysarthria–clumsy hand syndrome (genu of the internal capsule and basis pontis), and ataxic hemiparesis (pons, midbrain, internal capsule, or parietal white matter). The classic lacunar syndromes have a positive predictive value of 87% overall for detecting lacunes on imaging, with pure hemisensory loss (100%) and ataxic hemiparesis (95%) being the most predictive.35
CAA commonly presents with transient focal neurologic episodes, also known as amyloid spells, which typically include recurrent stereotyped focal weakness or numbness (or both). Some evidence suggests that these spells may be caused by cortical spreading depression from acute blood during cSAH. The recurrent stereotyped events may be from subsequent hemosiderin deposition in the region forming cSS or recurrent cSAH.36 Although transient, the spells are not benign because of the increased risk of subsequent intracerebral hemorrhage (ICH). A recent systematic review showed that patients with cSAH attributed to CAA had a 19% annual risk of ICH.37 Ultimately, CAA is implicated in 37%–74% of nontraumatic ICH cases and associated with considerable morbidity and mortality.9
Another common presentation of CAA is cognitive impairment, which presents as a spectrum of symptoms from subjective concerns to severe dementia. The cognitive changes may be secondary to the ischemic damage caused by CAA (WMH, microinfarcts, and microstructural tissue changes seen on DTI).38 In addition, the presence of Aβ has been associated with Alzheimer disease, and capillary-type CAA is thought to contribute to dementia among patients with Alzheimer disease. A bidirectional relationship between CAA and Alzheimer disease has been proposed.39 A rarer entity among patients with CAA is an inflammatory response to Aβ, termed CAA-related inflammation (figure 4C). These patients have altered mental status, headaches, focal neurologic deficits, and seizures. This is important to recognize because it can be treated with steroids.
WMH and lacunes were independently associated with general cognitive function and strongly predictive of rapid global functional decline in a sample of independently living older persons (mean age, 74.1 years).3 To date, prognosticating which patients with CSVD will progress to dementia has proven difficult; however, newly developed MRI biomarkers show promise. For example, free-water measures determined with diffusion-weighted MRI had greater correlation with cognitive impairment than structural imaging or standard diffusion tensor metrics.31 In addition, the Radboud University Nijmegen Diffusion Tensor and Magnetic Imaging Cohort (RUN DMC) study determined that greater Fazekas grade (i.e., 2 or 3) indicates greater pathologic burden; corresponds to the lower baseline Mini-Mental State Examination score; and indicates a steeper decline in the Mini-Mental State Examination score.40 Differentiating cognitive decline from mood disorder can pose a clinical challenge. Furthermore, both entities can be a part of the clinical presentation of CSVD. Mood changes can be prominent, depression being the most common. The RUN DMC study showed that frontal subcortical white matter disease is associated with depressive symptoms. This finding is likely secondary to disruption of the neural circuitry involved in mood regulation.41
Gait dysfunction is another clinical consequence of CSVD. WMH is the most important predictor of gait dysfunction, with more severe WMH-associated deficits located in the internal capsule, centrum semiovale, periventricular frontal lobes, and genu of the corpus callosum.42 Normal-appearing white matter observed with FLAIR sequence does not exclude CSVD-related gait dysfunction because these patients can have disrupted white matter integrity in the genu of the corpus callosum detectable on DTI.42 The clinical presentation can be similar to that of a patient with a lacunar lesion in the anterior corpus callosum and frontal gait dysfunction, which is characterized by slower velocity, wider base, and shorter stride.42
The typical gait dysfunction seen in CSVD should not be confused with vascular parkinsonian gait. Both entities are characterized by bradykinesia and short stride but can be differentiated. Vascular parkinsonism is a sudden-onset movement disorder with later onset and shorter disease course than Parkinson disease. Clinical signs include a syndrome with lower-body parkinsonism (bilateral lower-extremity bradykinesia and rigidity), urinary incontinence, pyramidal signs, freezing gait, postural instability, falls, dementia, absence of tremor, and poor responsiveness to levodopa. In addition, patients with vascular parkinsonism have large-volume WMH and multifocal lacunes on imaging. Damage in the caudate, putamen, and globus pallidus externa shows variable correlations with vascular parkinsonism.
Prevention and treatment
Incomplete understanding of the pathogenesis of CSVD limits prevention and treatment efforts. However, predictors of progression are rational therapeutic targets. Currently, the treatment approach is individualized on the basis of the risk factor profile, type and severity of biomarkers, and severity of clinical sequelae.
Reducing blood pressure
Blood pressure is the most important modifiable risk factor for CSVD. A meta-analysis of 4 trials on the effect of antihypertensive medication on CSVD showed that patients in the intensive antihypertensive medication groups had significantly less progression of WMH.43 The trials did not study the progression of lacunes, CMBs, enlarged PVS, or acute small subcortical infarcts. In addition, the effects of intensive antihypertensive medication on brain atrophy have been conflicting (studied in ACCORD-MIND and SCOPE).43
The effect of lowering blood pressure in secondary stroke prevention for patients with small subcortical strokes was studied in one multicenter trial, the Secondary Prevention of Small Subcortical Strokes (SPS3) trial. A total of 3,020 patients from multiple centers with recent symptomatic lacunar stroke were randomly assigned to a target systolic blood pressure <130 mm Hg or 130–149 mm Hg. The primary outcome, reduction of all recurrent strokes, was not significant; however, the group with a target systolic blood pressure <130 mm Hg had a significantly reduced rate of hemorrhagic stroke.44
Antiplatelet therapy
Pooled analysis of randomized trials has shown aspirin monotherapy after acute subcortical infarction reduces the risk of recurrent stroke by 30%.45 Aspirin monotherapy was compared with dual antiplatelet therapy (DAPT) in SPS3. Specifically, 325-mg aspirin was compared with 325-mg aspirin plus 75-mg clopidogrel daily. No significant difference was noted for the primary outcome, reduction of all strokes. Importantly, DAPT doubled the annual risk of major hemorrhages.46,47 Criticisms of this trial include the following: (1) the high dose of aspirin used in the DAPT arm, which likely increased the risk of hemorrhage, and (2) the patients were randomized 2 weeks to 6 months after the index subcortical stroke, rendering the results nongeneralizable to the acute poststroke period (the period of highest risk). The following year, another multicenter, randomized, double-blind study, The Clopidogrel in High-risk patients with Acute Nondisabling Cerebrovascular Events trial, concluded that 21 days of DAPT within 24 hours of stroke onset reduced recurrent 90-day strokes when compared with aspirin monotherapy.48 The generalizability of the study has been questioned because it included only Chinese patients who tend to have higher rates of large vessel strokes and polymorphisms affecting clopidogrel metabolism.49
Finally, a recently published international multicenter, randomized, double-blind, placebo-controlled trial provided support for DAPT in the United States and Europe. The Platelet-Oriented Inhibition in New TIA and Minor Ischemic Stroke (POINT) trial randomized patients within 12 hours of symptom onset with high-risk TIA (ABCD2 score ≥4) or minor ischemic stroke (National Institute of Health Stroke Scale ≤ 3) to either aspirin 50–325 mg or DAPT (aspirin 50–325 mg and clopidogrel loading dose of 600 mg with 75 mg daily thereafter) for 90 days. DAPT reduced the 90-day incidence of stroke but increased the incidence of hemorrhagic adverse events.50 Secondary analysis showed the benefit of DAPT was significant in the first 7–30 days, whereas the major hemorrhages occurred more often from day 8 to 90.51 Criticisms of this trial include the following: high loading dose of clopidogrel; long (90-day) duration of DAPT (both of which may have contributed to the higher rates of hemorrhagic adverse events); and the lack of capturing stroke etiology. In addition, patients with CMB, history of ICH, and history of systemic bleeding were excluded from the POINT trial and, therefore, may not be good candidates for DAPT.51
Thrombolysis
IV recombinant tissue plasminogen activator (IV r-tPA) is standard of care for patients suspected of acute subcortical stroke presenting within 4.5 hours from symptom onset. A study of patients with acute small subcortical stroke showed that those who received IV r-tPA had better neurologic outcome than patients who received a placebo.52 However, the presence of CMB and WMH (seen with pretreatment brain MRI) increased the risk of symptomatic ICH by >50% and severe WMH increased the risk of symptomatic ICH by >2.5-fold.53 Therefore, caution is warranted when administering IV r-tPA to patients with MRI findings of CMB and severe WMH,52 especially if presenting with a nondisabling stroke.54 The recent PRISMS trial showed no significant difference in the modified Rankin Scale 0–1 at 90 days in patients presenting with a mild nondisabling stroke treated with IV r-tPA vs aspirin.54 However, this study was underpowered due to early termination. These results should be interpreted with caution; perhaps, the information can support withholding IV r-tPA in favor of aspirin in nondisabling stroke patients with moderate to severe CSVD on prior imaging.
Statins
Statin therapy is another evidence-based treatment for cerebrovascular disease. Statins have lipid-lowering, anti-inflammatory, and endothelial protective properties. Administration of statins to patients with WMH was shown to decrease the risk of stroke, WMH progression, and cognitive decline.55 The Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) trial reported that the use of 80-mg atorvastatin daily was similarly efficacious for preventing ischemic stroke in the small vessel disease and large vessel disease groups.56 In addition, Zhang et al.16 postulated that statins improved endothelial function and stabilized BBB. Despite the benefits for patients with ischemic stroke, conflicting data exist regarding the use of statins for the treatment of ICH and CAA because of the risk of ICH. The SPARCL trial reported an increased risk of ICH recurrence among patients who received high-dose atorvastatin, especially those with CSVD.56 The Heart Protection Study also reported that statin use protected against ischemic stroke; however, statins increased the risk of ICH.57 The link between statin use and ICH appeared greater among patients with lobar hemorrhage, as noted by the recent Multicenter Study on Cerebral Hemorrhage in Italy.58 In addition, other observational data suggest that patients with CAA may be even more susceptible to ICH while receiving statins. Therefore, caution is advised when prescribing statins to patients with CAA.36
Treatment specifics in cerebral amyloid angiopathy
Prevention of ICH is the main goal when treating patients with CAA due to the significant morbidity and mortality associated with ICH. Subsidiary analysis of Perindopril Protection Against Recurrent Stroke Study showed that reducing the blood pressure of patients with CAA decreased ICH by 77% over a mean follow-up of 3.9 years, regardless of preexisting hypertension.59 While tight blood pressure control is an absolute necessity in CAA, controversy surrounds the use of antiplatelet and anticoagulant medications for the reduction of ischemic stroke in CAA due to the increased bleed risk. Despite the strong link between CAA and hemorrhage, CAA is also associated with ischemic lesions. Furthermore, patients with concomitant atherosclerotic disease and atrial fibrillation have even greater risk of ischemic stroke. This clinical dilemma is common because the prevalence of CAA and atrial fibrillation increases with age. The problem is compounded by the lack of RCTs evaluating these medications in CAA patients. The RCTs evaluating anticoagulation for stroke prevention among patients with atrial fibrillation did not screen for CMB, cSS, or cSAH. Patients with prior ICH were excluded from these trials. Despite the limited data on patients with CAA, the high baseline risk of ICH in CAA with features of prior ICH, cSAH, or cSS has led some experts to recommend avoiding anticoagulation.36 Antiplatelets also increase the risk of ICH and should be avoided if possible. A risk–benefit conversation with this population is warranted when on antiplatelet therapy for prior vascular stents.
The most controversial population in the thrombosis-bleeding dilemma is patients with CMBs, especially the CAA population with solely CMBs. A recent prospective observational cohort study, Clinical Relevance of Microbleeds in Stroke trial (CROMIS-2), looked at patients presenting with TIA/ischemic stroke and nonvalvular atrial fibrillation who were candidates for oral anticoaguation.60 CMBs and cSS were screened for with a baseline MRI. The primary outcome was symptomatic ICH during 24 months of follow-up. The study found baseline CMBs independently increased the risk of ICH in a dose-dependent manner. In addition, the ICH group had significantly higher rates of diabetes and the use of vitamin K antagonists. The CMB threshold where the risk of ICH outweighs the benefit of ischemic stroke reduction was not determined. Furthermore, the absolute rate of recurrent ischemic stroke was much higher than the absolute rate of ICH in all patients, including the CMB populations. The key question remains, “When does the risk of ICH outweigh the benefit of anticoagulation therapy for ischemic stroke prevention?” RCTs are needed to help guide antithrombotic management of patients with CMBs, especially CAA with solely CMBs on imaging.
Conclusion and future directions
CSVD affects most elderly individuals worldwide. Some risk factors and causes are well established, but many questions remain regarding how these relate to pathogenesis. Furthermore, disease biomarkers are evolving. With the increasing availability of DTI, blood oxygen level-dependent scanning, VWI, and ultra-high-field MRI, CSVD can be diagnosed earlier, opening the possibility for a reversal in the earliest stages. Another promising area of research is serum biomarkers, which have shown preliminary associations with CSVD. The NIH has launched a consortium, MarkVCID, to further investigate putative biomarkers of CSVD because of the strong association between CSVD and dementia. As the understanding of pathogenesis and biomarkers evolves, the long-term goal will be to establish additional specific pharmacologic and nonpharmacologic treatments.
Glossary
- Aβ
amyloid-β peptide
- BBB
blood–brain barrier
- CAA
cerebral amyloid angiopathy
- CADASIL
cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy
- CBF
cerebral blood flow
- CMB
cerebral microbleed
- CROMIS-2
Clinical Relevance of Microbleeds in Stroke
- cSAH
convexal subarachnoid hemorrhage
- cSS
cortical superficial siderosis
- CSVD
CNS small vessel disease
- DAPT
dual antiplatelet therapy
- DTI
diffusion tensor imaging
- FLAIR
fluid-attenuated inversion recovery
- GRE
gradient-recalled echo
- ICH
intracerebral hemorrhage
- IV r-tPA
IV recombinant tissue plasminogen activator
- MeSH
Medical Subject Headings
- NVU
neurovascular unit
- POINT
Platelet-Oriented Inhibition in New TIA and Minor Ischemic Stroke Trial
- PVS
perivascular space
- RCT
randomized controlled trial
- RUN DMC
Radboud University Nijmegen Diffusion Tensor and Magnetic Imaging Cohort
- SMC
smooth muscle cell
- SPARCL
Stroke Prevention by Aggressive Reduction in Cholesterol Levels
- SPS3
Secondary Prevention of Small Subcortical Strokes
- SWI
susceptibility-weighted imaging
- VWI
vessel wall imaging
- WMH
white matter hyperintensity
Appendix. Authors
Study funding
No targeted funding reported.
Disclosure
Dr. Meschia receives support from the Earl & Nyda Swanson Neurosciences Research Fund and the Harley N. and Rebecca N. Hotchkiss Endowed Fund in Neuroscience Research, Honoring Ken and Marietta. Go to Neurology.org/N for full disclosures.
References
- 1.Nam KW, Kwon HM, Lim JS, Han MK, Nam H, Lee YS. The presence and severity of cerebral small vessel disease increases the frequency of stroke in a cohort of patients with large artery occlusive disease. PLoS One 2017;12:e0184944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke 2011;42:2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Inzitari D, Pracucci G, Poggesi A, et al. Changes in white matter as determinant of global functional decline in older independent outpatients: three year follow-up of LADIS (leukoaraiosis and disability) study cohort. BMJ 2009;339:b2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher CM. Lacunar strokes and infarcts: a review. Neurology 1982;32:871–876. [DOI] [PubMed] [Google Scholar]
- 5.Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol 2010;9:689–701. [DOI] [PubMed] [Google Scholar]
- 6.De Ciuceis C, Cornali C, Porteri E, et al. Cerebral small-resistance artery structure and cerebral blood flow in normotensive subjects and hypertensive patients. Neuroradiology 2014;56:1103–1111. [DOI] [PubMed] [Google Scholar]
- 7.Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron 2017;96:17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12:822–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hilal S, Mok V, Youn YC, Wong A, Ikram MK, Chen CL. Prevalence, risk factors and consequences of cerebral small vessel diseases: data from three Asian countries. J Neurol Neurosurg Psychiatry 2017;88:669–674. [DOI] [PubMed] [Google Scholar]
- 10.de Leeuw FE, de Groot JC, Achten E, et al. Prevalence of cerebral white matter lesions in elderly people: a population based magnetic resonance imaging study. The Rotterdam Scan Study. J Neurol Neurosurg Psychiatry 2001;70:9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Poels MM, Vernooij MW, Ikram MA, et al. Prevalence and risk factors of cerebral microbleeds: an update of the Rotterdam scan study. Stroke 2010;41:S103–S106. [DOI] [PubMed] [Google Scholar]
- 12.Khan U, Porteous L, Hassan A, Markus HS. Risk factor profile of cerebral small vessel disease and its subtypes. J Neurol Neurosurg Psychiatry 2007;78:702–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim H, Yun CH, Thomas RJ, et al. Obstructive sleep apnea as a risk factor for cerebral white matter change in a middle-aged and older general population. Sleep 2013;36:709–715B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu B, Lau KK, Li L, et al. Age-specific associations of renal impairment with magnetic resonance imaging markers of cerebral small vessel disease in transient ischemic attack and stroke. Stroke 2018;49:899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caplan LR. Lacunar infarction and small vessel disease: pathology and pathophysiology. J Stroke 2015;17:2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang CE, Wong SM, van de Haar HJ, et al. Blood-brain barrier leakage is more widespread in patients with cerebral small vessel disease. Neurology 2017;88:426–432. [DOI] [PubMed] [Google Scholar]
- 17.Narayan SK, Gorman G, Kalaria RN, Ford GA, Chinnery PF. The minimum prevalence of CADASIL in Northeast England. Neurology 2012;78:1025–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mestre H, Kostrikov S, Mehta RI, Nedergaard M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin Sci 2017;131:2257–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wardlaw JM, Smith C, Dichgans M. Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol 2013;12:483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenberg SM, William M. Feinberg award for excellence in clinical stroke: big pictures and small vessels. Stroke 2017;48:2628–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Potter GM, Doubal FN, Jackson CA, et al. Enlarged perivascular spaces and cerebral small vessel disease. Int J Stroke 2015;10:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinter D, Gattringer T, Enzinger C, et al. Longitudinal MRI dynamics of recent small subcortical infarcts and possible predictors. J Cereb Blood Flow Metab Epub 2018 May 8. [DOI] [PMC free article] [PubMed]
- 23.Loos CM, Makin SDJ, Staal J, et al. Long-term morphological changes of symptomatic lacunar infarcts and surrounding white matter on structural magnetic resonance imaging. Stroke 2018;49:1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moreau F, Patel S, Lauzon ML, et al. Cavitation after acute symptomatic lacunar stroke depends on time, location, and MRI sequence. Stroke 2012;43:1837–1842. [DOI] [PubMed] [Google Scholar]
- 25.Cheng AL, Batool S, McCreary CR, et al. Susceptibility-weighted imaging is more reliable than T2*-weighted gradient-recalled echo MRI for detecting microbleeds. Stroke 2013;44:2782–2786. [DOI] [PubMed] [Google Scholar]
- 26.Benjamin P, Viessmann O, MacKinnon AD, Jezzard P, Markus HS. 7 Tesla MRI in cerebral small vessel disease. Int J Stroke 2015;10:659–664. [DOI] [PubMed] [Google Scholar]
- 27.Mossa-Basha M, Alexander M, Gaddikeri S, Yuan C, Gandhi D. Vessel wall imaging for intracranial vascular disease evaluation. J Neurointerv Surg 2016;8:1154–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Havenon A, Lobo R, Eisenmenger L, et al. MRI detection of vessel wall inflammation and contrast leakage in cerebral amyloid angiopathy [abstract]. Stroke 2017;48:AWP138. [Google Scholar]
- 29.Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol 2012;11:272–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alba-Ferrara LM, de Erausquin GA. What does anisotropy measure? Insights from increased and decreased anisotropy in selective fiber tracts in schizophrenia. Front Integr Neurosci 2013;7:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ji F, Pasternak O, Liu S, et al. Distinct white matter microstructural abnormalities and extracellular water increases relate to cognitive impairment in Alzheimer's disease with and without cerebrovascular disease. Alzheimers Res Ther 2017;9:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bahrani AA, Powell DK, Yu G, Johnson ES, Jicha GA, Smith CD. White matter hyperintensity associations with cerebral blood flow in elderly subjects stratified by cerebrovascular risk. J Stroke Cerebrovasc Dis 2017;26:779–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Opstal AM, van Rooden S, van Harten T, et al. Cerebrovascular function in presymptomatic and symptomatic individuals with hereditary cerebral amyloid angiopathy: a case-control study. Lancet Neurol 2017;16:115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zerna C, Yu AYX, Modi J, et al. Association of white matter hyperintensities with short-term outcomes in patients with minor cerebrovascular events. Stroke 2018;49:919–923. [DOI] [PubMed] [Google Scholar]
- 35.Gan R, Sacco RL, Kargman DE, Roberts JK, Boden-Albala B, Gu Q. Testing the validity of the lacunar hypothesis: the Northern Manhattan Stroke Study experience. Neurology 1997;48:1204–1211. [DOI] [PubMed] [Google Scholar]
- 36.Banerjee G, Carare R, Cordonnier C, et al. The increasing impact of cerebral amyloid angiopathy: essential new insights for clinical practice. J Neurol Neurosurg Psychiatry 2017;88:982–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson D, Hostettler IC, Ambler G, et al. Convexity subarachnoid haemorrhage has a high risk of intracerebral haemorrhage in suspected cerebral amyloid angiopathy. J Neurol 2017;264:664–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reijmer YD, van Veluw SJ, Greenberg SM. Ischemic brain injury in cerebral amyloid angiopathy. J Cereb Blood Flow Metab 2016;36:40–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thal DR, Griffin WS, de Vos RA, Ghebremedhin E. Cerebral amyloid angiopathy and its relationship to Alzheimer's disease. Acta Neuropathol 2008;115:599–609. [DOI] [PubMed] [Google Scholar]
- 40.van Leijsen EMC, van Uden IWM, Ghafoorian M, et al. Nonlinear temporal dynamics of cerebral small vessel disease: the RUN DMC study. Neurology 2017;89:1569–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van Uden IW, van der Holst HM, Schaapsmeerders P, et al. Baseline white matter microstructural integrity is not related to cognitive decline after 5 years: the RUN DMC study. BBA Clin 2015;4:108–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Laat KF, Tuladhar AM, van Norden AG, Norris DG, Zwiers MP, de Leeuw FE. Loss of white matter integrity is associated with gait disorders in cerebral small vessel disease. Brain 2011;134:73–83. [DOI] [PubMed] [Google Scholar]
- 43.van Middelaar T, Argillander TE, Schreuder FHBM, Deinum J, Richard E, Klijn CJM. Effect of antihypertensive medication on cerebral small vessel disease: a systematic review and meta-analysis. Stroke 2018;49:1531–1533. [DOI] [PubMed] [Google Scholar]
- 44.Benavente OR, Coffey CS, Conwit R, et al. Blood-pressure targets in patients with recent lacunar stroke: the SPS3 randomised trial. Lancet 2013;382:507–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwok CS, Shoamanesh A, Copley HC, Myint PK, Loke YK, Benavente OR. Efficacy of antiplatelet therapy in secondary prevention following lacunar stroke: pooled analysis of randomized trials. Stroke 2015;46:1014–1023. [DOI] [PubMed] [Google Scholar]
- 46.Nakamura T, Tsuruta S, Uchiyama S. Cilostazol combined with aspirin prevents early neurological deterioration in patients with acute ischemic stroke: a pilot study. J Neurol Sci 2012;313:22–26. [DOI] [PubMed] [Google Scholar]
- 47.Benavente OR, Hart RG, McClure LA, et al. Effects of clopidogrel added to aspirin in patients with recent lacunar stroke. N Engl J Med 2012;367:817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang Y, Wang Y, Zhao X, et al. Clopidogrel with aspirin in acute minor stroke or transient ischemic attack. N Engl J Med 2013;369:11–19. [DOI] [PubMed] [Google Scholar]
- 49.Hankey GJ. Dual antiplatelet therapy in acute transient ischemic attack and minor stroke. N Engl J Med 2013;369:82–83. [DOI] [PubMed] [Google Scholar]
- 50.Johnston SC, Easton JD, Farrant M, et al. Clopidogrel and aspirin in acute ischemic stroke and high-risk TIA. N Engl J Med 2018;379:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grotta JC. Antiplatelet therapy after ischemic stroke or TIA. N Engl J Med 2018;379:291–292. [DOI] [PubMed] [Google Scholar]
- 52.Mok V, Kim JS. Prevention and management of cerebral small vessel disease. J Stroke 2015;17:111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Charidimou A, Pasi M, Fiorelli M, et al. Leukoaraiosis, cerebral hemorrhage, and outcome after intravenous thrombolysis for acute ischemic stroke: a meta-analysis (v1). Stroke 2016;47:2364–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khatri P, Kleindorfer DO, Devlin T, et al. Effect of alteplase vs aspirin on functional outcome for patients with acute ischemic stroke and minor nondisabling neurologic deficits: the PRISMS randomized clinical trial. JAMA 2018;320:156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xiong Y, Wong A, Cavalieri M, et al. Prestroke statins, progression of white matter hyperintensities, and cognitive decline in stroke patients with confluent white matter hyperintensities. Neurotherapeutics 2014;11:606–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amarenco P, Benavente O, Goldstein LB, et al. Results of the stroke prevention by aggressive reduction in cholesterol levels (SPARCL) trial by stroke subtypes. Stroke 2009;40:1405–1409. [DOI] [PubMed] [Google Scholar]
- 57.Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002;360:7–22.12114036 [Google Scholar]
- 58.Pezzini A, Grassi M, Iacoviello L, et al. Serum cholesterol levels, HMG-CoA reductase inhibitors and the risk of intracerebral hemorrhage: the multicenter study on cerebral hemorrhage in Italy (MUCH-Italy). J Neurol Neurosurg Psychiatry 2016;87:924–929. [DOI] [PubMed] [Google Scholar]
- 59.Arima H, Tzourio C, Anderson C, et al. Effects of perindopril-based lowering of blood pressure on intracerebral hemorrhage related to amyloid angiopathy: the PROGRESS trial. Stroke 2010;41:394–396. [DOI] [PubMed] [Google Scholar]
- 60.Wilson D, Ambler G, Shakeshaft C, et al. Cerebral microbleeds and intracranial haemorrhage risk in patients anticoagulated for atrial fibrillation after acute ischemic stroke or transient ischemic attack (CROMIS-2): a multicenter observational cohort study. Lancet Neurol 2018;17:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]