

Abstract
Objective
To identify genes related to normal-pressure hydrocephalus (NPH) in one Japanese family with several members with NPH.
Methods
We performed whole-exome sequencing (WES) on a Japanese family with multiple individuals with NPH and identified a candidate gene. Then we generated knockout mouse using CRISPR/Cas9 to confirm the effect of the candidate gene on the pathogenesis of hydrocephalus.
Results
In WES, we identified a loss-of-function variant in CFAP43 that segregated with the disease. CFAP43 encoding cilia- and flagella-associated protein is preferentially expressed in the testis. Recent studies have revealed that mutations in this gene cause male infertility owing to morphologic abnormalities of sperm flagella. We knocked out mouse ortholog Cfap43 using CRISPR/Cas9 technology, resulting in Cfap43-deficient mice that exhibited a hydrocephalus phenotype with morphologic abnormality of motile cilia.
Conclusion
Our results strongly suggest that CFAP43 is responsible for morphologic or movement abnormalities of cilia in the brain that result in NPH.
Normal-pressure hydrocephalus (NPH) usually occurs sporadically, but several studies have shown its familial aggregation.1,2 In 2011, a large family featuring individuals with NPH through 3 generations was reported.3 Although family studies have suggested that genetic factors underlie the development of NPH, the genetic risk factors of NPH have remained unclear.
Whole-exome sequencing (WES) is a powerful tool to detect genes causative of single-gene neurodegenerative diseases.4,5 However, to our knowledge, no studies have explored genes causative of NPH using WES. To identify genes related to NPH, we performed WES on one Japanese family with several members with NPH and identified a loss-of-function variant in the CFAP43 gene linked to this disease in this family. Next, we generated Cfap43 knockout mice, and the model showed clear dilation of the lateral and third ventricles of the brain similar to human NPH. We show that mutation in CFAP43 elicits abnormalities in cilia and leads to NPH.
Methods
Clinical presentation of a Japanese family with multiple patients with NPH
The proband was a 55-year-old woman (II-1, figure 1A). She was hospitalized owing to major symptoms including gait impairment (decreased step height, length, and cadence; Timed Up and Go test [TUG]: 33 seconds), cognitive impairment (Mini-Mental State Examination [MMSE]: 24/30, Frontal Assessment Battery [FAB] at bedside: 3/18), and urinary incontinence. Her CSF opening pressure was 150 mm H2O, CSF total protein 38 mg/dL, CSF glucose 64 mg/dL, and CSF chlorine 128 mEq/L. Her symptoms improved temporarily after a tap test (72 hours after tap test: TUG 25 seconds, MMSE 25/30, FAB 9/18). MRI of her brain showed significant dilation of the third, lateral ventricle and Sylvian fissure and high-convexity tightness (figure 1, C–E). She was diagnosed with NPH, in accordance with the reported criteria.6 She also had recurrent respiratory tract infection and chronic sinusitis but did not show bronchiectasis or visceral inversions. She died of pneumonia at 60 years of age. In addition, 2 of her brothers (II-4 and II-7) also showed 3 major symptoms of NPH. II-7 began to show gait impairment, urinary incontinence, and cognitive impairment (MMSE 24/30) at age 44. His brain MRI showed significant dilation of the third, lateral ventricle and Sylvian fissure and high-convexity tightness (figure 1E), and he also had recurrent respiratory tract infection and chronic sinusitis. He was also diagnosed with NPH. Their mother developed hydrocephalus at age 50 and died at 62 years of age (I-7, figure 1A). None of the patients in this family had a history of diseases causing secondary NPH, including head trauma, intracerebral haemorrhage, or meningitis. The patients in this family (II-1, II-4, and II-7) were unmarried, and there was no information about infertility. Other siblings (II-2, II-3, II-5, II-6, and II-8) did not show any symptoms of NPH or chronic infection.
Figure 1. Pedigree, mutation, and MRI.

(A) Japanese family with multiple individuals with normal-pressure hydrocephalus (NPH). Black symbol represents individuals (I-7, II-1, II-4, and II-7) diagnosed with NPH with recurrent respiratory and sinus infection. (B) The nonsense mutation (chr10:105893468C>T on hg19, p.W1502X in ENST00000357060.3) in CFAP43 segregated with the hydrocephalus phenotype. (C–E) MRI demonstrated remarkable dilation of the lateral ventricle and Sylvian fissure in affected individuals (II-1, II-7). (C) Coronal MRI of II-1. Dilation of lateral ventricle (green arrow), Sylvian fissure (red arrows), and interpeduncular cistern (white arrow). In addition, high-convexity tightness (yellow arrows) was observed. (D) Sagittal MRI of II-1. The stenotic site in cerebral aqueduct was not observed. (E) Axial MRI of family members. MRI demonstrated dilation of the third ventricle (blue arrows), lateral ventricle (green arrows), and Sylvian fissure (red arrows) and high-convexity tightness (yellow arrows) in affected individuals (II-1, II-7), but not in unaffected individuals (II-2, II-6).
Samples and DNA extraction
We recruited 5 members of the family for WES and Sanger sequencing, which comprised 2 affected and 3 unaffected individuals (II-1, II-2, II-6, II-7, and II-8). A total of 10 mL of peripheral blood was collected from each individual after obtaining written informed consent, and genomic DNA was extracted from blood lymphocytes using QIAamp DNA Midi Kit (Qiagen, Hilden, Germany). Experimental procedures were approved by the Committee for Ethical Issues on Human Genome and Gene Analysis at Nagasaki University.
Whole-exome sequencing
We performed WES on 3 individuals in this family (II-1, II-6, and II-8), including 1 individual with NPH and 2 unaffected individuals, using the SureSelect Human All Exon V4+UTRs kit (Agilent Technologies, Santa Clara, CA). We also performed massively parallel sequencing using the 5500 SOLiD system (Thermo Fisher Scientific, Waltham, MA).
Sequencing data analysis
NovoAlignCS software (Novocraft, Selangor, Malaysia) version 1.02.03 was used to align SOLiD reads on the hg19/GRCh37 reference genome sequence. Aligned reads were sorted using Novosort software (Novocraft) version 1.00.01. PCR and optical duplications were removed using MarkDuplicates in the Picard tools package (broadinstitute.github.io/picard/) version 1.84. Genome Analysis Toolkit (GATK)7 version 3.4–46 was used to perform local realignment (IndelRealigner), variant call (HaplotypeCaller), multiple sample joint call (GenotypeGVCFs), and depth measurement (DepthOfCoverage). Raw single-nucleotide variants (SNVs) and small insertions/deletions (indels) were hard-filtered in accordance with GATK best practice.8 Passed variants were annotated using the ANNOVAR software.9 This procedure was executed using an in-house workflow management tool.10 The average mean depth of all samples was 57.07 (II-1: 59.01, II-6: 51.93, II-8: 60.28) and the average coverage rate of >×10 depth regions was 94.13% (II-1: 94.7%, II-6: 93.3%, II-8: 94.4%).
Selection of familial pathogenic substitutions
To detect the familial pathogenic mutation from WES data, deleterious variants corresponding to the following criteria were considered as pathogenic candidates: (1) variants: lead stop gain, stop loss, nonsynonymous, or splice site mutations, in accordance with GENCODE basic version 19 downloaded from the UCSC genome browser; (2) alternative allele frequencies at variant loci ≤0.5% in the following databases: 1000 Genomes Project data on all populations released October 2014,11 NIH National Heart, Lung, and Blood Institute6515 exome data (evs.gs.washington.edu/EVS/), Exome Aggregation Consortium (ExAC) exome data,12 Human Genomic Variation Database (HGVD) exome data of 1,208 individuals collected in Japan,13 and SNV allele frequency collected from whole-genome sequencing data of a population of 2,049 healthy Japanese (2KJPN) (ijgvd.megabank.tohoku.ac.jp); (3) mutations not included in the UCSC segmental duplication region; and (4) rare SNVs defined as damaging by functional annotation programs (Polyhen_2,14 SIFT,15 PROVEN,16 or MutationTaster17). After this filtration, 13 substitutions remained as potential candidates, all data with latest annotation and allele frequencies of which are available from Dryad (supplemental data, doi.org/10.5061/dryad.d82n3f8).
Sanger sequencing
We performed Sanger sequencing of 13 substitutions exclusively found in this family that emerged from WES. Chr7:149489295;G>T in the SSPO gene and chr10:105727538;T>C in the SLK gene were not sequenced because of technical difficulties. All primers were designed using Primer3.18 Genomic DNA (5 ng) was amplified using KOD FX Neo polymerase (Toyobo, Osaka, Japan) by PCR in 20-μL reactions. Sequence reactions were performed using the BigDye Terminator Cycle Sequencing Kit v3.1 (Applied Biosystems, Foster City, CA), in accordance with the manufacturer's instructions. After purification using CleanSEQ (Agen Court, Sugar Land, TX), the products were separated on an ABI Genetic Analyzer 3130 (Applied Biosystems) and the electropherograms were visually evaluated using 4Peaks software.19
Generation of CFAP43 knockout mouse using CRISPR-Cas9 technology
We designed guide RNA onto exon 35 of mouse ortholog Cfap43, corresponding to the mutation position in the family, to knock out the gene. Single-guide RNA was designed using the ATUM gRNA Design Tool (atum.bio/eCommerce/cas9/input) as follows: 5′-GATGGAGGACCTAAACCAGA-3′. Single-guide RNA, crRNA, and tracrRNA (FASMAC, Kanagawa, Japan) were injected into wild-type (WT) zygotes using a microinjector system: Olympus IX70 Fluorescence Microscope Cutaway (Olympus, Tokyo, Japan), TransferMan NK2 (Eppendorf, Hamburg, Germany), FemtoJet 4i (Eppendorf), and Cell Tram Oil (Eppendorf). After RNA injection, zygotes were cultured until the blastocyst stage in vitro and then transferred to the uteri of recipient pseudopregnant females. To verify the genotype of the mice, PCR amplification was performed using KOD FX Neo polymerase from mouse DNA and products were cloned into the T-vector (Takara-bio, Kusatsu, Japan) for sequencing. PCR primers were as follows: forward, 5′-GGGAAATAAGAAAAGAGTCATGGA-3′; reverse, 5′-CACATGCTTTATCCAGGAGGA-3′. Experimental procedures were approved by the Ethics Review Committee for Animal Experimentation at Nagasaki University.
Quantitative RT-PCR
Total RNA was extracted from mouse brain and tracheal tissues using NucleoSpin RNA Plus (MACHERY-NAGEL, Düren, Germany). One microgram of total RNA was converted into cDNAs with ReverTra Ace qPCR RT Master Mix with gDNA remover (Toyobo). Fifty nanograms of cDNA was used as a template for amplification using THUNDERBIRD SYBR qPCR MiX (Toyobo) to quantify the expression on LightCycler 480 System II (Roche, Basel, Switzerland) in 10-μL reactions. Mouse Gapdh (forward, 5′-CTGCACCACCAACTGCTTAG-3′; reverse, 5′-TTCAGCTCAGGGATGACCTT-3′) was used as an internal control and mouse Cfap43 q-PCR primers were as follows (forward, 5′-AGGACAAAAGAAAGTTGCTAGCA-3; reverse, 5′-TCCAATCTGAATGGCAATCACA-3′). The presence of a single PCR amplicon was confirmed by melting curve analysis. mRNA expression was quantified using the 2−∆∆Ct method and Student t test was applied for 2-sample significance tests.
Histologic analysis of mouse tissues
Fresh mouse brain and tracheal tissues were fixed in 10% formalin for 72 hours, embedded in paraffin, and then sectioned at 5-μm intervals for tissue slides. After deparaffinization, slides were stained with hematoxylin & eosin (H&E) by standard methods for the visualization of structures. We viewed and photographed sections using BZ-9000 All-in-one Fluorescence Microscope (Keyence, Osaka, Japan).
For immunohistochemical staining, tissue slides were deparaffinized, rehydrated, and rinsed in Tris-buffered saline. For antigen retrieval, tissue sections were microwaved for 20 minutes in 10 mM citrate buffer using an MI-77 microwave processor (Azumaya Corporation, Tokyo, Japan). Then, sections were blocked in DAKO Protein Block Serum-Free (Dako, Carpinteria, CA) for 1 hour at room temperature, followed by incubation for 1 hour at room temperature with primary antibodies.
Immunofluorescent staining was also performed using the following primary antibodies: monoclonal anti-acetylated tubulin antibody, polyclonal anti-SPEF2 antibody, and anti-RSPH4A antibody (all from Sigma-Aldrich, St. Louis, MO). Secondary antibodies were as follows: goat anti-mouse immunoglobulin G (IgG) H&L (Alexa Fluor 488), ab150113; and goat anti-rabbit IgG H&L (Alexa Fluor 555), ab150078 (Abcam, Cambridge, UK). Primary and secondary antibodies were diluted using Antibody Diluent with Background Reducing Components (Dako). Sections were mounted with Vectashield Mounting medium for fluorescence analysis with DAPI (Vector Laboratories, Burlingame, CA). Fluorescent images were taken using a BZ-9000 All-in-one Fluorescence Microscope.
Transmission electron microscopy (TEM)
Samples were fixed with 2% glutaraldehyde (Nacalai Tesque, Kyoto, Japan) in 0.1 M sodium cacodylate buffer containing 1 mM CaCl2 and 1 mM MgCl2 (cacodylate buffer, pH 7.4) at 4°C for 60 minutes. The samples were rinsed with cacodylate buffer, and then postfixed with 1% OsO4 (Nacalai Tesque) in cacodylate buffer at 4°C for 60 minutes. Next, they were washed with cacodylate buffer, dehydrated in a graded series of ethanol and acetone, and embedded in Quetol 651 epoxy resin (Nisshin EM, Tokyo, Japan). The resin-embedded samples were trimmed and sectioned using a diamond knife on an ultramicrotome (Reichert-Jung, Vienna, Austria). Ultrathin sections were collected on grids and stained with uranyl acetate and lead citrate. The samples were examined at 80 kV under a transmission electron microscope (JEM-1230; JEOL, Tokyo, Japan).
Data availability statement
Study data for the primary analyses presented in this report are available upon reasonable request of other investigators.
Results
Mutation in the family
To identify potential pathogenic changes, we performed WES on 3 members of this family (figure 1A) and identified 72 variants found only in affected individuals. To identify compelling candidates, we first filtered out simple missense variants. After filtration, 13 loci remained as candidates (supplemental data, doi.org/10.5061/dryad.d82n3f8). After Sanger sequencing of those candidates in 5 members (affected, II-1 and II-7; unaffected, II-2, II-6, and II-8), we found one variant (chr12:66838468T>C in GRIP1) and one nonsense mutation (chr10:105893468C>T in CFAP43) that segregated with the disease. A homozygous mutation in glutamate receptor-interacting protein 1 (GRIP1) has already been reported to be a gene causative of Fraser syndrome, a rare disorder characterized by cryptophthalmos, cutaneous syndactyly, and genitourinary anomalies.20 Because none of the patients in this family showed any symptoms of Fraser syndrome, DRIP1 was ruled out as a causative candidate. Alternatively, mutation in the gene encoding cilia- and flagella-associated protein 43 (CFAP43) has been reported to be responsible for male infertility with multiple morphologic abnormalities of flagella.21,22 Because recurrent infection and hydrocephalus are frequently observed in primary cilia dyskinesia (PCD),23 the ciliary dysfunction in affected individuals in this family was the most likely factor behind the pathogenesis. Since the nonsense mutation in this gene resulted in truncation of the protein, CFAP43 is suggested to be relevant to the pathogenesis of PCD (figure 2A).
Figure 2. Position of the familial nonsense mutation in exon 35 of the CFAP43 gene and ciliary ultrastructure of nasal mucosa.

(A) The position of the familial nonsense mutation in exon 35 of the CFAP43 gene, which is located within the SMC-N (N-terminus of structural maintenance of chromosomes proteins) domain that is highly conserved among multiple species. (B) Ciliary ultrastructure of nasal mucosa in a patient (II-1). Blue box: normal 9 + 2 axoneme. Orange box: abnormal compound cilia, involving 2 axonemes in one ciliary shaft.
To confirm the ciliary ultrastructure in a patient, we analyzed the nasal mucosa by TEM and found a number of abnormal compound cilia (figure 2B). Considering both our genetic findings and the TEM observations, it is strongly suggested that nonsense mutation in CFAP43 is involved in the pathogenesis of NPH in this family.
Phenotype of Cfap43−/− mice
To confirm the effect of truncation of CFAP43 on the pathogenesis of hydrocephalus, we introduced a 14-bp deletion in exon 35 of mouse ortholog CFAP43 using CRISPR/Cas9 gene editing technology (figure 3A). This deletion generates a premature stop codon, resulting in truncation of the Cfap43 protein (figure 3B). The expression of frameshifted Cfap43 mRNA in the brain and trachea of mutant mice was confirmed by RT-PCR (figure 3C). The low expression level of mutated Cfap43 mRNA was confirmed by quantitative RT-PCR, suggesting that nonsense-mediated mRNA decay reduced the level of mRNA harboring the premature termination codon (figure 3D). Moreover, the lower number of Cfap43−/− offspring from interbreeding mice indicated that embryonic lethality or development difficulties had occurred in some homozygous mutant offspring (table).
Figure 3. Electropherograms, the 14-bp deletion, and quantitative RT-PCR.

(A) Electropherograms of the 14-bp deletion in exon 35 of mouse Cfap43 produced using CRISPR/Cas9 gene editing technology. (B) The 14-bp deletion generates a premature stop codon and is predicted to produce a truncated nonfunctional protein. (C) Electropherograms of brain and tracheal cDNA. The 14-bp deletion mutation was confirmed by RT-PCR using cDNA templates from wild-type (WT) and Cfap43−/− mouse tissues. (D) Quantitative RT-PCR. Cfap43 was detected at a very low expression level in the brain and trachea of homozygous mutant mice.
Table.
Non-Mendelian ratios from interbreeding of Cfap43+/− mice

The brain of Cfap43−/− mice showed dilation of the third and lateral ventricles in H&E staining. Epithelial cells of the lateral ventricle, choroid plexus, and trachea of Cfap43−/− mice showed no obvious difference from those in WT mice on H&E staining (figure 4, A and B). In testis and epididymal tissue, distorted sperm flagella and excess cytoplasm were observed, which correspond to the findings in previous reports21,22 (figure 4C). Cfap43+/− mice showed no obvious differences from WT mice in terms of H&E staining in testis and epididymis.
Figure 4. Mouse histology.

(A) Cfap43−/− mouse brain showed dilation of third and lateral ventricles on hematoxylin & eosin staining. Epithelial cells of the lateral ventricle and choroid plexus of Cfap43−/− mice did not show obvious abnormalities compared with those of wild-type (WT) mice under a conventional optical microscope. CP = choroid plexus; LV = lateral ventricle. Black scale bar = 50 μm. White scale bar = 20 μm. (B) Tracheal epithelial cells of Cfap43−/− mice did not show remarkable change on hematoxylin & eosin staining. Black scale bar = 200 μm. White scale bar = 20 μm. (C) In testis and epididymal tissue, distorted sperm flagella and excess cytoplasm in Cfap43−/− mice were found. Black scale bar = 50 μm. White scale bar = 20 μm.
To better characterize the molecular defect of Cfap43−/− mice, we performed immunofluorescent staining (IF) using an antibody targeting ciliary proteins, since the loss of several proteins comprising the axoneme in sperm was previously reported in Cfap43-deficient mice.22 In IF, decrease in number of acetylated tubulin (as a marker of cilium mortality) was found in the choroid plexus of Cfap43−/− mice (figure 5A). Moreover, some of the epithelial cells of the lateral ventricle and trachea of Cfap43−/− mice showed defect of Spef2 (as a marker of the axoneme central pair) and Rsph4a (as a marker of radial spokes) (figure 5, B and C). This IF result indicates the abnormal protein composition in the cilia of Cfap43-deficient mice.22
Figure 5. Immunofluorescent staining of ciliary protein.
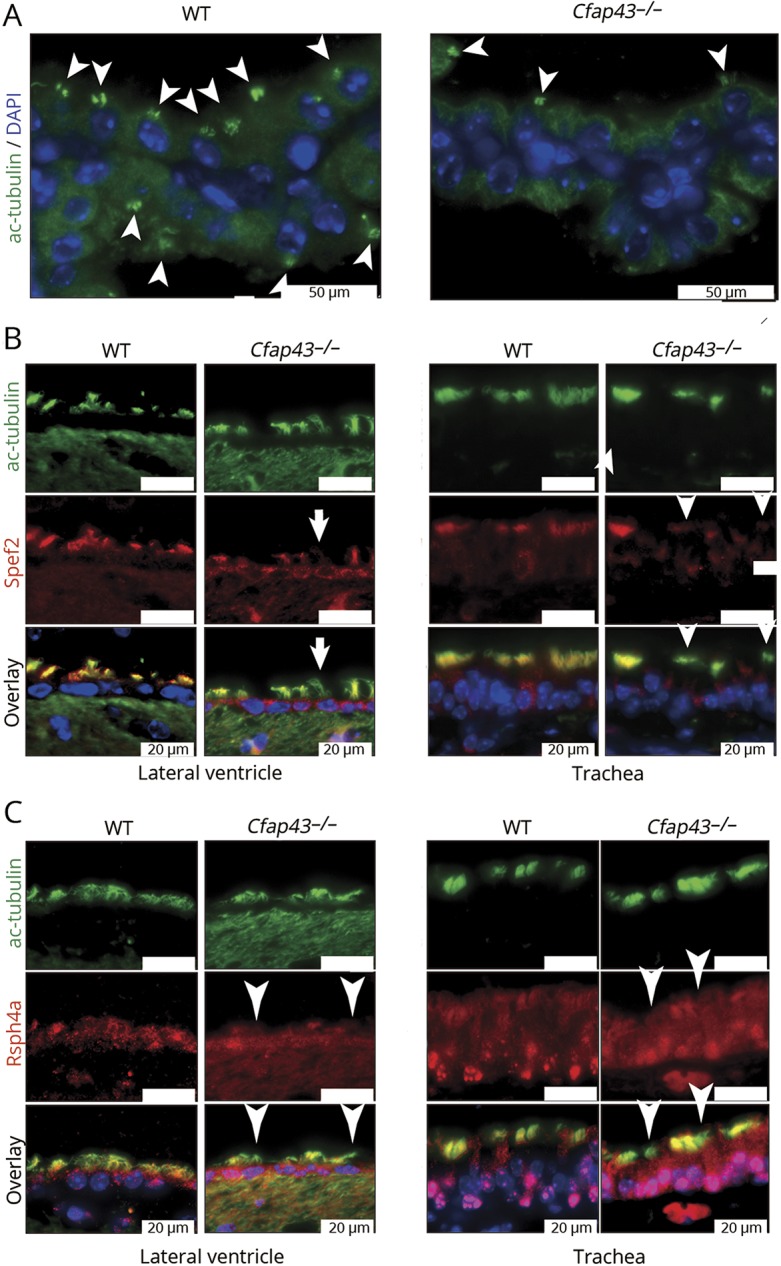
(A) Decrease in number of acetylated tubulin (white arrowheads) was found in the choroid plexus of Cfap43−/− mice. Green: ac-tubulin, marker of cilia. Blue: DAPI. White scale bar = 50 μm. (B) Some epithelial cells of the lateral ventricle and trachea of Cfap43−/− mice showed defect of Spef2 protein. Green: ac-tubulin, marker of cilia. Red: Spef2, a marker of the axoneme central pair. Blue: DAPI. White scale bar = 20 μm. (C) Some epithelial cells of the lateral ventricle and trachea of Cfap43−/− mice showed a defect of Rsph4a protein expression. Green: ac-tubulin, a marker of cilia. Red: Rsph4a, a marker of radial spokes. Blue: DAPI. White scale bar = 20 μm. (C) Some epithelial cells of the lateral ventricle and trachea of Cfap43−/− mice showed a defect of Rsph4a protein expression. Green: ac-tubulin, a marker of cilia. Red: Rsph4a, a marker of radial spokes. Blue: DAPI. White scale bar = 20 μm. WT = wild-type.
To better characterize the ciliary ultrastructure of Cfap43-deficient mice, we performed TEM analysis. TEM revealed a normal 9 + 2 axoneme in the cross-section of WT tracheal cilia (figure 6, A–C) but abnormal 8 or 10 + 2 peripheral microtubules in Cfap43−/−(figure 6, D–F). In addition, compound cilia were observed only in Cfap43−/− mice (figure 6, G–J). This result is consistent with the observation in nasal epithelia in a patient with familial NPH (figure 2B).
Figure 6. Ciliary ultrastructure of tracheal epithelium.

(A–C) Wild-type (WT) tracheal cilia. Blue box: The normal 9 + 2 axoneme was observed in the cross-section of WT cilia. Scale bars: a = 1 μm; b, c = 100 nm. (D–F) Cfap43−/− tracheal cilia. Green box: Abnormal 10 + 2 peripheral microtubules were observed. Orange box: Abnormal 8 + 2 peripheral microtubules were observed. Scale bars: D = 1 μm; E, F = 100 nm. (G, H) WT tracheal cilia. No compound cilia were observed. Scale bars: G = 1 μm, H = 200 nm. (I, J) Cfap43−/− tracheal cilia. Compound cilia were observed. Scale bars: I = 1 μm, J = 200 nm.
Discussion
In this study, using WES, we found a nonsense mutation in CFAP43 in a Japanese family including multiple individuals with NPH. Subsequently, we generated Cfap43-deficient mice using CRISPR/Cas9, which showed the hydrocephalus phenotype with the morphologic abnormality of motile cilia mimicking human NPH. These results strongly suggest that CFAP43 is responsible for NPH. Recent studies have reported that biallelic mutations in CFAP43 cause male infertility with multiple morphologic abnormalities of the sperm flagella and suggested that this gene is strongly associated with the function and morphology of flagella.21,22 Our results obtained from patients and model mice show that ciliary dysfunction due to CFAP43 affects not only the testis but also other tissues including the choroid plexus and epithelial cells of the ventricles of the brain and trachea. Considering both our genetic and pathologic findings, it is highly likely that the NPH phenotype observed in this family is one of the PCD-associated symptoms caused by the haploinsufficiency of CFAP43 in humans.
Intriguingly, the mutation identified in the family was heterozygous. PCD is usually inherited in an autosomal recessive manner,23–25 and previous genetic studies of PCD showed that its symptoms were clearly observable only in cases with biallelic mutations. A recent investigation has demonstrated the N-terminal domain and C-terminal domain of Fap43p have different intracellular localization signals, and only the C-terminal domain of Fap43p localizes to the cilia.26 This suggests the importance of the C-terminal domain of CFAP43 for ciliary function in specific tissues, without redundancy to other ciliary proteins in humans. This raises the possibility that mutation in the C-terminal domain could have induced NPH even in members of the family heterozygous for this mutation. NPH could be a heterozygous phenotype and homozygous or biallelic mutations may result in an additional phenotype such as male infertility in humans. In this study, Cfap43−/− mice showed a considerably lower birth rate than Cfap43+/+ and Cfap43+/− mice (table). The phenotypes found in humans and mice are similar to each other but are expressed at different zygotic states. This clearly suggests a species difference in the effects of the gene mutation. The molecular function and localization in the cilia of CFAP43 are unclear; further functional studies such as confirming the interaction and ultrastructural localization in cilia are required to reveal the pathogenicity in this NPH-affected family.
NPH often occurs in elderly people and its cause remains unclear in many patients; therefore, most patients with NPH are diagnosed with idiopathic NPH. Since it would be difficult to assume that late-onset hydrocephalus is one of the PCD-associated symptoms based only on clinical symptoms and signs, mutation screening in CFAPs should be useful for precise categorization leading to elucidation of the pathogenesis of NPH and the development of appropriate treatments.
Acknowledgment
The authors thank the patients and their family members for participating in this study; Chisa Hayashida, Yukie Takahashi, Hiromu Yano, and Takahiro Motoyama for technical support in the analysis; and Edanz (edanzediting.co.jp) for editing the English text of a draft of this manuscript.
Glossary
- FAB
Frontal Assessment Battery
- GATK
Genome Analysis Toolkit
- H&E
hematoxylin & eosin
- IF
immunofluorescent staining
- IgG
immunoglobulin G
- MMSE
Mini-Mental State Examination
- NPH
normal-pressure hydrocephalus
- PCD
primary cilia dyskinesia
- SNV
single-nucleotide variant
- TEM
transmission electron microscopy
- TUG
Timed Up and Go test
- WES
whole-exome sequencing
- WT
wild-type
Appendix. Authors
Footnotes
Editorial, page 933
Study funding
No targeted funding reported.
Disclosure
Y. Morimoto, S. Yoshida, A. Kinoshita, C. Satoh, H. Mishima, N. Yamaguchi, K. Matsuda, M. Sakaguchi, T. Tanaka, Y. Komohara, A. Imamura, H. Ozawa, and M. Nakashima report no disclosures relevant to the manuscript. N. Kurotaki has received research support from a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of Science and a grant from SENSHIN Medical Research Foundation. T. Kishino reports no disclosures relevant to the manuscript. K. Yoshiura has received research support from a Grant-in-Aid for Scientific Research (B) (16H05159). S. Ono was supported by The Japan Spina Bifida & Hydrocephalus Research Foundation. Go to Neurology.org/N for full disclosures.
References
- 1.McGirr A, Cusimano MD. Familial aggregation of idiopathic normal pressure hydrocephalus: novel familial case and a family study of the NPH triad in an iNPH patient cohort. J Neurol Sci 2012;321:82–88. [DOI] [PubMed] [Google Scholar]
- 2.Huovinen J, Kastinen S, Komulainen S, et al. Familial idiopathic normal pressure hydrocephalus. J Neurol Sci 2016;368:11–18. [DOI] [PubMed] [Google Scholar]
- 3.Takahashi Y, Kawanami T, Nagasawa H, Iseki C, Hanyu H, Kato T. Familial normal pressure hydrocephalus (NPH) with an autosomal-dominant inheritance: a novel subgroup of NPH. J Neurol Sci 2011;308:149–151. [DOI] [PubMed] [Google Scholar]
- 4.Funayama M, Ohe K, Amo T, et al. CHCHD2 mutations in autosomal dominant late-onset Parkinson's disease: a genome-wide linkage and sequencing study. Lancet Neurol 2015;14:274–282. [DOI] [PubMed] [Google Scholar]
- 5.Kim HJ, Kim NC, Wang YD, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013;495:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Relkin N, Marmarou A, Klinge P, Bergsneider M, Black PM. Diagnosing idiopathic normal-pressure hydrocephalus. Neurosurgery 2005;57:S4–S16. [DOI] [PubMed] [Google Scholar]
- 7.McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van der Auwera GA, Carneiro M, Hartl C, et al. From FastQ data to high-confidence variant calls: the genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics 2013;43:11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from next-generation sequencing data. Nucleic Acids Res 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mishima H, Sasaki K, Tanaka M, Tatebe O, Yoshiura K. Agile parallel bioinformatics workflow management using Pwrake. BMC Res Notes 2011;4:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Higasa K, Miyake N, Yoshimura J, et al. Human genetic variation database, a reference database of genetic variations in Japanese population. J Hum Genet 2016;61:547–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 2009;4:1073–1081. [DOI] [PubMed] [Google Scholar]
- 16.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One 2012;7:e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361–362. [DOI] [PubMed] [Google Scholar]
- 18.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 2000;132:365–386. [DOI] [PubMed] [Google Scholar]
- 19.4Peaks software [computer program]. Version 1.8. Aalsmeer, The Netherlands: Nucleobytes; 2017. Available at: https://nucleobytes.com/4peaks/index.html. Accessed January 20, 2016. [Google Scholar]
- 20.Tessier A, Sarreau M, Pelluard F, et al. Fraser syndrome: features suggestive of prenatal diagnosis in a review of 38 cases. Prenat Diagn 2016;36:1270–1275. [DOI] [PubMed] [Google Scholar]
- 21.Tang S, Wang X, Li W, et al. Biallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagella. Am J Hum Genet 2017;100:854–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coutton C, Vargas AS, Amiri-Yekta A, et al. Mutations in CFAP43 and CFAP44 cause male infertility and flagellum defects in trypanosoma and human. Nat Commun 2018;9:686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee L. Mechanisms of mammalian ciliary motility: insights from primary ciliary dyskinesia genetics. Gene 2011;473:57–66. [DOI] [PubMed] [Google Scholar]
- 24.Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia: recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med 2013;188:913–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horani A, Brody SL, Ferkol TW. Picking up speed: advances in the genetics of primary ciliary dyskinesia. Pediatr Res 2014;75:158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urbanska P, Joachimiak E, Bazan R, et al. Ciliary proteins Fap43 and Fap44 interact with each other and are essential for proper cilia and flagella beating. Cell Mol Life Sci 2018;75:4479–4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Study data for the primary analyses presented in this report are available upon reasonable request of other investigators.