

Abstract
Background
NADPH oxidase 4 (NOX4) catalyzes the formation of hydrogen peroxide (H2O2). NOX4 is highly expressed in the kidney, but its role in renal injury is unclear and may depend on its specific tissue localization.
Methods
We performed immunostaining with a specific anti-NOX4 antibody and measured NOX4 mRNA expression in human renal biopsies encompassing diverse renal diseases. We generated transgenic mice specifically overexpressing mouse Nox4 in renal tubular cells and subjected the animals to the unilateral ureteral obstruction (UUO) model of fibrosis.
Results
In normal human kidney, NOX4 protein expression was at its highest on the basolateral side of proximal tubular cells. NOX4 expression increased in mesangial cells and podocytes in proliferative diabetic nephropathy. In tubular cells, NOX4 protein expression decreased in all types of chronic renal disease studied. This finding was substantiated by decreased NOX4 mRNA expression in the tubulo-interstitial compartment in a repository of 175 human renal biopsies. Overexpression of tubular NOX4 in mice resulted in enhanced renal production of H2O2, increased NRF2 protein expression and decreased glomerular filtration, likely via stimulation of the tubulo-glomerular feedback. Tubular NOX4 overexpression had no obvious impact on kidney morphology, apoptosis, or fibrosis at baseline. Under acute and chronic tubular injury induced by UUO, overexpression of NOX4 in tubular cells did not modify the course of the disease.
Conclusions
NOX4 expression was decreased in tubular cells in all types of CKD tested. Tubular NOX4 overexpression did not induce injury in the kidney, and neither modified microvascularization, nor kidney structural lesions in fibrosis.
Keywords: NOX4, Tubular cells, Kidney fibrosis, Renal biopsy, Diabetes, IgA nephropathy, Chronic kidney disease
Graphical abstract
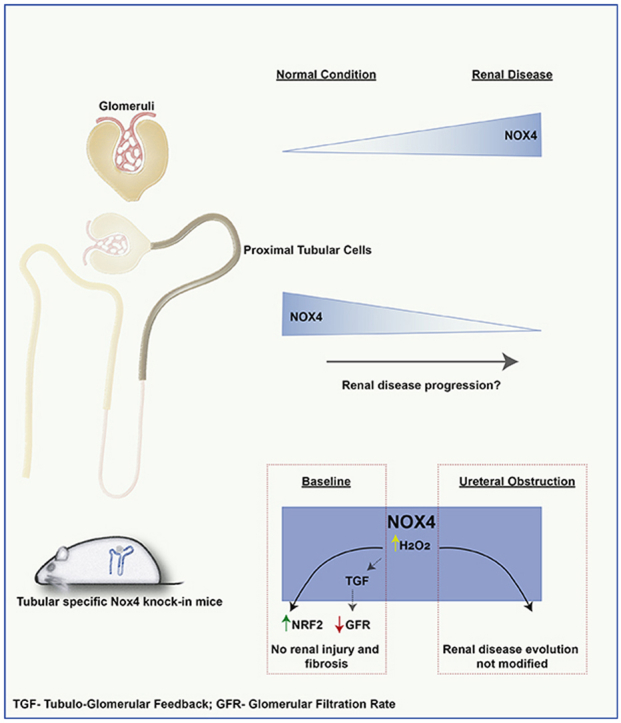
1. Introduction
Chronic Kidney Disease (CKD) is a leading cause of morbidity and mortality worldwide. Excess production of reactive oxygen species (ROS) is considered as a contributor to CKD progression and approaches to mitigate ROS may have therapeutic potential [1]. Even though excessive ROS production may have harmful effects, low levels of ROS are recognized as essential signaling mediators for normal cellular function [[2], [3], [4], [5]].
In cells, ROS are produced by mitochondria, NADPH oxidases and peroxisomes among other sources. Seven NADPH oxidases have been described, with specific tissue distribution and distinct physiological roles [6]. NADPH oxidase type 4 (NOX4) isoform generates mostly H2O2 [7] and is highly expressed in the kidney at baseline, mainly in proximal tubular cells in rodents as detected by in situ hybridization [8]. Because of the unavailability of specific antibodies, the pattern of NOX4 expression in human kidney is unclear [9].
The potential role of NOX4 in diseases is debated. H2O2 generated by NOX4 was proposed to promote aging. Nox4 knock-out mice however do not show significant enhanced lifespan [10]. In fibrotic lung disease, NOX4 expression is increased and plays a deleterious role by reducing fibroblast apoptosis, leading to fibroblast accumulation and fibrosis progression [11]. In the vascular system, Nox4 deletion contributes to atherosclerosis progression and NOX4 is therefore believed to be physiologically protective [[12], [13], [14], [15]]. In the kidney, the physiological role of the very high endogenous expression of NOX4 in the proximal tubular cells is unknown. Germline Nox4 knock-out mice do not display any major renal phenotype at baseline, but NOX4 may modulate tubular cell survival in conditions of injury [9,[16], [17], [18]]. ROS production by NOX4 in the macula densa may also participate in the tubulo-glomerular feedback (TGF) [19]. Nox4 deletion in mice leads to enhanced renal tubular apoptosis during urinary obstruction, acute ischemic nephropathy and tunicamycin-induced acute kidney injury (AKI) [9,[16], [17], [18]]. This anti-apoptotic effect appears to be partly mediated through regulation of NRF2, BCL-2 and elF2 stress signaling pathways [17,18]. In contrast, NOX4 expression increases in podocytes and mesangial cells, where it participates to renal injury by enhancing podocyte apoptosis and mesangial cell proliferation in a diabetic milieu [[20], [21], [22]]. In fact, complete or podocyte specific deletion of Nox4 rescued diabetic nephropathy in several models [9,23,24].
Cell type specific regulation of NOX4 expression during experimental kidney disease is unclear. One study showed decreased NOX4 expression in diabetic nephropathy in the kidney lysate using well-characterized antibodies, whereas increased NOX4 expression was detected in mesangial cells in diabetic nephropathy by others [9,25].
In this study, we took advantage of a recently developed antibody against NOX4 [[17], [26]]. This antibody recognizes both human and murine NOX4 protein in immunoblotting and immunohistochemistry [27]. We first determined the evolution of NOX4 expression in the glomerular versus tubular compartment in normal human kidney and in common kidney diseases. We further analyzed NOX4 mRNA expression according to CKD stage in a bank of human kidney biopsies. We finally studied the role of Nox4 overexpression in kidney tubular cells at baseline and under acute and chronic tubular injury using a mouse model overexpressing Nox4 in a tubular cell specific manner.
2. Methods
2.1. NOX4 antibody validation
To validate the NOX4 antibody for immunohistochemistry analysis on the human renal sections, we made use of a cell pellet of tetracycline inducible human NOX4 overexpressing T-Rex TM cells [28]. Briefly, NOX4 expression was induced by addition of tetracycline (1 μg/ml) or vehicle (PBS). After 24 h of tetracycline treatment, the cells were rinsed with PBS, followed by trypsinization for 5 min. Detached cells were washed with phosphate buffered saline (PBS) containing 10% fetal bovine serum (FBS) and centrifuged at 1500 rpm for 5 min at room temperature. The cell pellet was fixed by adding 4% paraformaldehyde (PFA) for 24 h at 4 °C. The cell pellet was centrifuged again at 1500 rpm for 5 min and the PFA was removed. Ethanol (70%) was added to the pellet for 24 h at 4 °C. Warm 0.8% agarose (1 ml) was added to the pellet and the mixture was transferred to a pre-prepared tube with 250 μl of cold agarose. Once cold, the agarose was then removed from the tube and processed for immunohistochemistry [16,17].
2.2. NOX4 expression in human biopsies
Human renal tissues fixed in formaldehyde and embedded in paraffin were selected from the tissue bank of the Service of Pathology, University Hospital of Geneva. The following samples were included for this study: 5 normal renal tissues (control) obtained from patients who underwent nephrectomy performed for neoplasia diagnosis, and 42 biopsy specimens obtained from patients with renal diseases including 14 diabetic nephropathy, 15 IgA nephropathy, 12 nephroangiosclerosis, and 1 primary FSGS. Among the various pathologies, 19 specimens presented secondary FSGS lesions within the biopsy. For all biopsy specimens, standard analyses were performed. The clinical parameters for the patients enrolled in this study are provided in the Supplementary Table 1. Each patient gave informed consent before enrollment in the study. The institutional ethical committee board approved the clinical protocol (CEREH number 03–081). The research was performed according to the Helsinki's declaration principles.
Immunohistochemistry was performed as previously described [29]. Briefly, sections (3 μm thick) of each biopsy specimen were incubated with a monoclonal rabbit anti- NOX4 antibody at 1:100 dilution [17]. For colocalization experiments, double staining was performed on serial sections using NOX4 and the following two antibodies: monoclonal mouse anti-Na,K-ATPase [30] at 1:200 dilution and mouse monoclonal anti-cytokeratin 19 (DakoCytomation) at 1:40 dilution.
2.3. RNA isolation, RNA preparation, and microarray data analyses of microdissected human kidney biopsies
Human kidney biopsies for Affymetrix microarray expression data were obtained within the scope of the European Renal cDNA Bank Kröner-Fresenius Biopsy Bank [31,32]. Biopsies were received from patients after an informed written consent, approved by the local ethical committees. Following renal biopsy, the tissue was transferred to RNase inhibitor reagent and microdissected into glomerular and tubulo-interstitial fragments. Total RNA was isolated from microdissected glomeruli and tubulointerstitium, reverse transcribed, and linearly amplified according to a protocol previously reported [31,32]. Affymetrix Gene Chip Human Genome U133A and U133 Plus2.0 Arrays were used in this study. The microarray expression data came from individual patients with different chronic kidney diseases (cadaveric donor (CD), tumor nephrectomy (TN), diabetic nephropathy (DN), thin basement disease (TMD), minimal change disease (MCD), hypertensive nephropathy (HTN), IgA nephropathy (IgA), focal segmental glomerulosclerosis (FSGS), membranous nephropathy (MGN), lupus nephritis (LN) and ANCA-vasculitis (ANCA)). Pre-transplantation kidney biopsies from living donors were used as controls. Fragmentation, hybridization, staining, and imaging were performed according to the Affymetrix Expression Analysis Technical Manual (Affymetrix, Santa Clara, CA). The raw data was normalized using Robust Multichip Algorithm (RMA) and annotated by Human Entrez Gene custom CDF annotation version 18 (http://brainarray.mbni.med.umich.edu/Brainarray/default.asp). The log transformed dataset was corrected for batch effect using ComBat from the Gene Pattern pipeline (http://www.broadinstitute.org/cancer/software/genepattern/) [31].
For the differential gene expression analysis in the tubulo-interstitium, patients were grouped into different CKD stages (CKD1-5) according to their estimated GFR calculated by the CKD-EPI equation [32] (Tub: CKD1: n = 56; CKD2: n = 46; CKD3: n = 37; CKD4: n = 26; CKD5: n = 10). Pre-transplantation kidney biopsies from living donors (Tub: LD n = 42) served as the control group. To identify differentially expressed genes the SAM (Significance analysis of Microarrays) method was applied using TiGR (MeV, Version 4.8.1) [33]. A q-value below 5% was considered to be statistically significant.
2.4. Generation of the Nox4 knock-in mouse
Conditional Nox4 knock-in mice were generated by targeted integration of a mNox4 cDNA (NM_015760.5) into mRosa26 locus by Cyagen Biosciences Inc, USA. In brief, the LOX-P-Stop-LoxP-mNox4 cDNA cassette was placed downstream of a CAG promoter in the targeting vector. The Neo cassette was flanked by FrT site, and diphtheria toxin A (DTA) was used for negative selection. The homology arm was obtained by PCR using BAC clones from the C57BL/6 library. C57BL/6 ES cells were used for gene targeting and implanted into C57BL/6 mice. The scheme illustrating the strategy is provided in Supplementary Fig. 2. The conditional Nox4 knock-in mice were then bred with Pax8-Cre mice [34] in C57BL/6 background to obtain a renal tubular specific overexpression of Nox4. Genomic DNA was obtained from ear biopsies. The following genotyping primers were used: FW 5′-ACAGTTGGAGTTTTCTGCTGTGGAC-3′, RV: 5′-AAGACTCCCGCCCATCTTCTAGAT-3′ and the strategic location of primers are indicated in the scheme. Nox4 knock-in animals were identified by a band at 373bp. We refer to these mice displaying Nox4 overexpression in kidney tubular cells under the Pax8 promoter as Nox4KI in the text and figures. Nox4wt/CKI. Pax8 CreNeg and/or Nox4wt/wt. Pax8 CrePos and/or Nox4wt/wt. Pax8 Creneg animals were used as controls.
All the animals had free access to food and water. For all experiments, littermate males were used at 8–10 weeks of age. All animal experiments were approved by the Institutional Ethical Committee of Animal Care in Geneva and Cantonal authorities. All animal experiments were conducted in accordance with Swiss legislation on animal welfare under the animal authorization number GE/11/16 and 181/18.
2.5. Unilateral ureteral obstruction
Male mice (8–10 weeks) were anesthetized using isoflurane gas and unilateral ureteral obstruction (UUO) procedure was performed [16]. The animals were sacrificed 3 days or 10 days after surgery. Both the obstructed and non-obstructed contralateral kidneys were harvested for performing Real-time PCR, Western blotting and immunohistological analysis.
2.6. Hydrogen peroxide (H2O2) production in primary cultured cells and renal organoids
Primary cultures from kidney were obtained by digesting cortical bands with 1% collagenase type 1 and the cells were cultured in DMEM/F12 complemented with 10% FBS. After 3 days of culture, H2O2 measurement was performed using the Amplex Red (Invitrogen)-Horseradish peroxidase (HRP) (Sigma Aldrich) assay. Briefly, cells were incubated in PBS containing Amplex Red (25 μM) and HRP (0.005U/ml) and the resulting fluorescence was measured using a plate reader with excitation at 544 nm and emission at 590 nm. Fluorescence measurement was acquired every 30 s for 90 min with shaking between each reading [16,17].
For measuring H2O2 production in the kidneys, Wt and Nox4KI mice were anesthetized with isoflurane gas and intra-cardiac perfusion with ice cold PBS was performed to remove the blood from the kidneys. A small piece of kidney including both the cortex and medulla was chopped into small pieces and incubated with Amplex Red (25 μM) and HRP (0.005U/ml). After 5 min, image of the reaction tubes was acquired with a camera, and the end point fluorescence was measured. H2O2 production was expressed as fold increase of fluorescence per miligram of the renal tissue utilized for the analysis.
2.7. RNA extraction and Real-time PCR
Total RNA extraction from the renal cortex and medulla was performed using NucleoSpin RNA II RNA extraction kit (Macherey-Nagel). RNA was reversed transcribed using qScript cDNA Super Mix (Quanta Biosciences).10 ng of cDNA was used for the PCR reaction with SYBR Green Master Mix (Applied Biosystems) and the Real-time PCR reaction was performed in duplicates using the StepOne Plus Real-Time PCR System (AB Applied Biosystems) [16,17]. Primers used for gene expression analysis are documented in Supplementary Table 3. The data were normalized to the reference gene, Ribosomal Protein Lateral Stalk Subunit P0 (Rplp0) and results were expressed as fold change in mRNA expression compared to the Wt control samples.
2.8. Western blotting
Protein extraction from kidney tissue was performed as described [17]. 20–30 μg of proteins were loaded after quantification of proteins using the bicinchoninic acid protein assay (Thermo Scientific, Pierce) and Coomassie Blue gel staining (Thermo Scientific GelCode blue stain reagent). After SDS-PAGE, proteins were transferred to either PVDF (Immobilon-P, Millipore) or nitrocellulose (Amersham Protran Premium 0.45 NC) membranes. Membranes were blocked with 5% milk-TBST and incubated with the primary antibodies overnight at 4 °C. The following primary antibodies were used: NOX4 (rabbit monoclonal, 1:500) [[27], [35]], BCL-2 (Rabbit, 1:500, sc-492), NRF-2 (Rabbit, 1:500, sc-722), α-SMA (Mouse, 1:2000, kind gift of Dr. Marie-Luce Bochaton-Piallat, Department of Immunology, University of Geneva), Collagen1α (Rabbit, 1:500, Rockland 600-401-103S), Fibronectin (Rabbit, 1:500, ab-2413), HIF1α (Rabbit, 1:500, NB100-479). Goat anti-mouse HRP (1:2000, BD 554002) and goat anti-rabbit HRP (1:2000, BD 554021) secondary antibodies were used. For collagen1α blots, protein samples were not denatured. ImageJ software (NIH) was used to quantify the band density. The band density for each protein was normalized either to Coomassie blue staining on an independent gel run in parallel or to the Ponceau S staining on the membrane after the transfer. Results were presented as fold change in protein expression compared to the Wt control samples.
2.9. Immunohistochemistry on mouse sections
Kidneys harvested from mice were fixed in 4% paraformaldehyde for 2 h at room temperature, dehydrated, and embedded in paraffin. 4–5 μm sections were used for Sirius red (SR), Hematoxylin and Eosin (H&E) staining and immunohistochemistry. Blocking was performed with 5% bovine serum albumin (BSA). For immunohistochemistry, the following primary antibodies and antigen retrieval methods were used. Anti-F4/80 (Rat, 1:100, ab-6640, Tris 50 mM/EDTA 1 mM), Endomucin (Rat, 1:200, ab-106100, Citrate 10 mM), Ki-67 (Rabbit, 1:5000, ab-15580, Citrate 10 mM) and active Caspase-3 (Rabbit, 1:100, CS -9661, EDTA 10 mM). Goat anti-rat HRP (1:200, Invitrogen 629520) and donkey anti-rabbit HRP (1:200, Jackson 711-035-152) secondary antibodies were used.
2.10. Quantification of histology
The slides were scanned with Axioscan image scanner (Zeiss) at 20x magnification and the quantification of Sirius red and immunostainings were performed using Definiens Tissue Phenomics® software. Quantification was performed on the cortex area. Results were expressed as percentage of positive cells for cleaved caspase-3 and Ki-67 staining. For Sirius red, Endomucin, and F4/80 stainings, results were expressed as percentage of positively stained area.
2.11. Electrolytes measurement
Animals were acclimatized in the metabolic cages under normal water and diet and 24 h urine samples were collected on the last experimental day. Plasma and urine parameters were measured using the UniCel® DxC 800 analyser (Beckman Coulter) and advanced Osmometer 2020 (Advanced Instruments) at the Zurich Integrative Rodent Physiology (ZIRP) platform, University of Zurich.
2.12. Glomerular filtration rate (GFR) measurement
Transcutaneous glomerular filtration rate measurement by Sinistrin-Fluorescein isothiocyanate (sinistrin-FITC) excretion rate were performed on the live animals [36]. Briefly, the skin on the back of the mice was shaved and the camera (NIC-Kidney device, Mannheim Pharma & Diagnostics GmbH) was fixed on the dorsal side close to where the kidney is located using an adhesive tape and the background reading was recorded when mice were anesthetized with isoflurane. Then, sinistrin-FITC was injected via tail vein at the dose of 7.5 mg/100 g body weight (BW) and the animals were housed separately. After 1 h of recording, the camera was removed and t1/2 of sinistrin-FITC was established. GFR was calculated as reported [36].
2.13. Statistics
GraphPad Prism 7 software was used to perform unpaired two tailed t-test (comparison of two experimental groups) and One-way ANOVA analysis with Sidak's multiple comparisons test (comparison of four experimental groups). Data are shown as Mean ± SD.
3. Results
3.1. NOX4 is predominantly expressed in kidney proximal tubular cells
A rabbit monoclonal anti-NOX4 antibody was previously validated in mouse kidney using Nox4 knock-out mice [17]. To verify the specificity of this antibody for human NOX4, we tested the antibody in a cellular model of human NOX4 overexpression. In these cells [28], we show that the rabbit monoclonal antibody detects two bands at around 35 kDa and 70 kDa only in human NOX4 expressing cells using western blotting (Fig. 1, A). Immunohistochemistry of the pelleted T-Rex cells shows an intense staining in NOX4 expressing cells (Fig. 1B and C). Immunohistochemistry using this antibody on normal human kidney sections detected a cytoplasmic expression of NOX4 in proximal tubular cells, whereas expression was much lower in distal segments of the nephron (Fig. 2, A). NOX4 co-localized with the basolateral Na+,K+-ATPase pump in proximal but not in distal tubular segments (Fig. 2, B). In normal glomeruli, NOX4 expression was low and significant expression was restricted to parietal glomerular epithelial cells (Fig. 1, D upper and lower panel). Distinct expression of NOX4 was visible in endothelial cells of large blood vessels (Supplementary Figs. 1 and A), but not in peritubular capillaries or glomerular endothelial cells (Fig. 1, D, Fig. 2, B and Supplementary Figs. 1 and A).
Fig. 1.

NOX4 Antibody validation, NOX4 expression pattern in the normal human glomerular compartment, and in renal nephropathies.
Representative Western blot analysis of T-Rex cells overexpressing (right lane) or not (left lane) the human NOX4 protein (A). Representative images of NOX4 immunohistochemical analysis of T-REX cells overexpressing (tetracycline inducible) (C) or not (B), the human NOX4 protein. D–F: Representative images of NOX4 immunohistochemical analysis of normal human kidney, diabetic and IgA nephropathies. In normal glomeruli, NOX4 expression is restricted to parietal glomerular epithelial cells (D, upper and lower panel). NOX4 expression is observed in the glomerular mesangium of diabetic nephropathy (E, upper and lower panel) and IgA nephropathy (F, upper and lower panel).
Fig. 2.

NOX4 expression in normal renal tubular cells and in tubulo-interstitial injury.
A-B: Representative images of NOX4 immunohistochemical analysis of normal human kidney. A marked cytoplasmic NOX4 immunostaining (brown) is observed in proximal tubules, whereas expression is much lower in distal segments of the nephron (lower magnification of the image shown in Fig. 1D) (A). NOX4 and Na, K-ATPase (red) double immunostaining. NOX4 (in brown) appears predominant at the basolateral side of proximal tubular cells and colocalizes with basolateral sodium-potassium pump Na, K-ATPase (B). C–D: Representative images of NOX4 immunohistochemical analysis in chronic tubulo-interstitial injury. NOX4 expression is decreased and almost absent in atrophic tubules in diabetes nephropathy (C). Periodic acid Schiff staining (D) showing area of fibrosis. Analysis of NOX4, GSTA1, NOX2 and TGFβ1 gene expression in the Affymetrix microarray expression dataset generated by the European Renal cDNA Kröner-Fresenius Biopsy bank demonstrating a marked decrease in NOX4 mRNA expression correlating with the different stages of CKD (E). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.2. NOX4 expression increases in mesangial cells during the course of diabetic and IgA nephropathies
We then assessed the pattern of NOX4 expression in various types of renal disease. Clinical parameters of the patients enrolled in this study are provided in Supplementary Table 1. In diabetic nephropathy (14 cases), strong NOX4 staining was detected in the glomerular mesangium consistent with previous observations (Fig. 1, E upper and lower panel). Moderate podocyte staining was also observed. In advanced stages of diabetic nephropathy, we observed no NOX4 expression in Kimmestiel Wilson nodules that are characterized by nodular mesangial matrix accumulation without mesangial cells (Supplementary Figs. 1 and F). IgA nephropathy is another frequent renal disease where mesangial proliferation plays an important pathogenic role. In IgA nephropathy (n = 15), NOX4 expression was also clearly detected in mesangial cells, but not in podocytes (Fig. 1, F upper and lower panel). NOX4 expression did not increase in hypertensive vascular nephropathy and decreased in areas of fibrosis (Supplementary Figs. 1 and G). NOX4 expression was present in FSGS lesions (Supplementary Fig. 1, B-E).
3.3. Decreased tubular NOX4 protein and mRNA expression in human CKD
In chronic tubulo-interstitial injury and fibrosis, independent of the origin of the disease (IgA, diabetic or hypertensive), proximal tubular cell NOX4 protein expression decreased in areas of fibrosis and capillary rarefaction and was almost absent in dedifferentiated tubules (Fig. 2C and D). To confirm our observations, using the Affymetrix microarray expression data generated by the European Renal cDNA Bank Kröner-Fresenius Biopsy Bank, we observed that NOX4 mRNA decreases in the tubulo-interstitial compartment proportionally to the stage of chronic kidney disease (Fig. 2, E). This decrease was seen in parallel with a decrease in GSTA1 expression, whereas the markers of inflammation (NOX2) and fibrosis (TGFβ1) were increased (Fig. 2, E).
3.4. Increased H2O2 production in NOX4KI mice does not affect kidney structure
NOX4 protein expression also clearly decreases in an experimental model of renal injury and fibrosis induced by UUO in mice, similar to human CKD (Fig. 3, A). In order to determine whether the decrease in NOX4 expression plays a role in the course of fibrosis progression in CKD, we generated a mouse model constitutively overexpressing Nox4 and obtained a specific kidney tubular cell overexpression using Cre recombinase driven by Pax8 promoter [34] (Supplementary Fig. 2). Nox4 overexpression was observed in the kidney by mRNA and protein expression in the cortex and medulla of the Nox4KI animals (Fig. 3B and C). Similar to the T-REX cells expressing human NOX4, the rabbit monoclonal antibody detected two bands on Western blot. H2O2 production levels was increased in primary renal tubular epithelial cell cultures derived from the kidney cortex of Nox4KI mice compared to Wt (Fig. 3D and E). H2O2 production was enhanced in freshly isolated renal organoids (Fig. 3, F). Nox4KI mice display a normal renal structure (Fig. 3, G and Supplementary Figs. 3 and B). Glomerular filtration rate was lower in Nox4KI than in Wt mice, as assessed through transcutaneous measurement of sinistrin-FITC excretion and by endogenous creatinine clearance (Fig. 3H and I). Plasma and urinary electrolytes were unchanged (Supplementary Table 2). There was neither fibrosis, nor evidence of tubular injury in the Nox4KI mice (Fig. 3, G and Supplementary Figs. 3 and B). There was no tubular apoptosis either, as assessed by cleaved caspase-3 immunostaining (Supplementary Figs. 3 and C). NRF2 and HIF1α protein expression was enhanced in the renal cortex of Nox4KI at baseline (Fig. 4, B). At the level of mRNA only a modest increase in Nrf2 and Nqo1, an NRF2 target gene, was observed in the cortex (Fig. 4, A). mRNA expression of Renin and Epo was not different in both renal cortex and medulla of Wt and Nox4KI animals (Supplementary Figs. 3 and A).
Fig. 3.

Generation and validation of tubular cell specific Nox4 knock-in mice.
NOX4 protein expression in renal cortex harvested from wild-type (Wt) mice and mice subjected to 10 days of Unilateral Ureteral Obstruction (A). B–C: Gene expression and Western blot analysis of renal cortex and medulla dissected from Wt and tubular cell specific Nox4KI kidneys. Nox4 mRNA expression (B) and NOX4 protein levels (C) are increased in both cortex and medulla of Nox4KI kidneys. HE-higher exposure of the blot. Quantification of high (70 kDa) and low molecular weight (35 kDa) NOX4 protein bands are shown. D–F: Detection of H2O2 production in the primary epithelial cells and renal organoids isolated from Wt and Nox4KI kidneys. Fold increase in Amplex red fluorescence in the primary epithelial cells is shown in D. Expression levels of Nox4 mRNA in the cultured primary epithelial cells (E). Panel showing the visualization of increased H2O2 generation in renal organoids isolated from Nox4KI kidney by the production of intense florescent pink color and the fold increase in H2O2 production (F). Representative Sirius red images of Wt and Nox4KI kidney sections demonstrating that the overall kidney structure was not modified (G). Transcutaneous glomerular filtration rate measurement by sinistrin-FITC excretion rate on Wt and Nox4KI mice (H). Estimation of glomerular filtration rate by endogenous creatinine clearance (I). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Fig. 4.

Pro-survival, antioxidant and angiogenesis pathways are activated in Nox4KI mice at baseline.
Gene expression analysis of pro-survival (Nrf2, Bcl2) and antioxidant pathway (Gstα, Nqo1) genes at baseline. The gene expression level for each gene is normalized to the housekeeping gene Rplp0 (A). Representative Western blot images of proteins involved in the cell survival, angiogenesis and fibrosis processes. Quantitation of Western blot experiments are shown in (B).
3.5. Tubular NOX4 overexpression does not affect fibrosis after moderate tubular injury
To test whether the decrease in tubular NOX4 expression in CKD contributes to the disease, we subjected Wt or NOX4KI mice to mild tubular injury by performing UUO for 3 days and severe tubular injury for 10 days. Renal function was slightly decreased after UUO compared to normal animals, but with no differences between WT and NOX4KI mice (Supplementary Table 2).
Upon 3 days of UUO, in the renal cortex, mRNA levels of several markers of cellular stress (cMyc), inflammation (Nox2, Ccl5) and fibrosis (Tgfβ1) were induced confirming the efficacy of the UUO procedure (Fig. 5, A and Supplementary Figs. 4 and A). The expression of antioxidant response genes Gstα and Nqo1 expression was stable in both Wt and NOX4KI mice without differences. In UUO model, Nox4KI mice displayed a higher expression of Nox4 due to the transgene and Nox2, cMyc and Il1β, suggesting increased inflammatory response. Western blot experiments confirmed decrease in NOX4 protein expression following UUO in the cortex of Wt (significant by unpaired two-tailed t-test) but not Nox4KI mice (Fig. 6, A). Additional markers of fibrosis were tested: as expected, protein levels of fibronectin and α-SMA were increased following UUO, but no difference between Wt and Nox4KI was observed following UUO. Altogether, this suggests that, after 3 days of UUO, compensatory NOX4 expression in tubular cells has no or little impact on fibrotic markers.
Fig. 5.

Gene expression changes in Wt and Nox4KI animals subjected to 3 days and 10 days of UUO.
A-B: Gene expression analysis on Wt and Nox4KI kidneys harvested 3 days (A) and 10 days (B) after subjecting the animals to UUO. The non-obstructed kidneys of the same animals were used as controls. Gene expression levels of Nox4, Nox2, pro-survival pathway genes (Bcl2, Nrf2) and antioxidant genes (Gstα, Nqo1) are shown. The gene expression level for each gene is normalized to the housekeeping gene Rplp0.
Fig. 6.

Renal NOX4 protein expression is decreased in a severe fibrotic renal disease mouse model but not in NOX4KI mice.
A–B: Western blot analysis on Wt and Nox4KI kidneys harvested 3 days (A) and 10 days (B) after subjecting to UUO. The non-obstructed kidneys were used as controls. Protein expression levels of NOX4, and proteins implicated in cell survival pathways (NRF2, BCL2) and fibrosis (Collagen 1α, Fibronectin and α-SMA) and the quantification are shown.
We further studied tubular cell apoptosis by measuring active caspase- 3, cell proliferation by measuring Ki-67, and extracellular fibrotic deposition by unpolarized Sirius red staining. Fibrosis, tubular proliferation and apoptosis were increased upon UUO but were similar between Wt and Nox4KI mice in the control state and following UUO (Fig. 7, A and E and Supplementary Fig. 5, A-C). Assessment of macrophage infiltration by F4/80 staining and vascularization by endomucin staining was performed on the kidney sections from Wt and Nox4KI mice. Immunostaining for endomucin and F4/80 did not show any differences between groups (Fig. 7, C and F and Supplementary Figs. 6 and A).
Fig. 7.

Constitutive Nox4 expression in renal tubular cells does not prevent fibrotic disease progression.
A, B, E: Representative images of Sirius red staining and quantification on the sections of Wt and Nox4KI kidneys harvested 3 days (A, E) and 10 days (B, E) after subjecting to UUO. Endomucin immunostaining and quantification on the sections of Wt and Nox4KI kidneys harvested 3 days (C, F) and 10 days (D, F) after subjecting to UUO are presented. The non-obstructed kidneys were used as controls. . (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.6. NOX4 overexpression in tubular cells does not modify fibrosis progression in severe tubular injury
Upon 10 days of UUO, Nox4 mRNA and protein decreased in Wt animals (significant by unpaired two-tailed t-test), but not in Nox4KI mice (Fig. 5, B, Fig. 6, B). The kidneys subjected to UUO displayed enhanced inflammation, immune cells infiltration and fibrosis (Fig. 7 B and E, Supplementary Figs. 4 and B and Supplementary Figs. 6 and B). Nox4KI kidneys undergoing chronic UUO surgery did not significantly differ from Wt kidneys. In particular, fibronectin, α-SMA and extracellular matrix deposition as detected by Sirius red staining were unaltered, and capillary density was not modified by Nox4 overexpression notably with a persistent decrease in Vegf (Supplementary Figs. 4 and B). Inflammation was very prominent at 10 days of UUO but overall similar between Wt and Nox4KI, as demonstrated by similar expression levels of pro-inflammatory cytokines or Nox2 mRNA expression in the renal cortex (Fig. 5, B, Supplementary Figs. 4 and B and Supplementary Figs. 6 and B).
4. Discussion
In this study, we show for the first time specific differences in cellular compartmentalization of NOX4 expression in healthy and pathological human kidney samples. We show an upregulation of NOX4 expression in glomeruli of diabetic and IgA kidney disease and a downregulation of tubular NOX4 mRNA and protein expression in all types of studied CKD. The decrease of NOX4 expression was more pronounced according to CKD stage. We further demonstrate that overexpression of NOX4 in tubular cells increases H2O2 production but does not result in pronounced tubular cell injury or inflammation. In contrast, a mild induction of the NRF2 and HIF1α protein was observed. We further show that overexpressing NOX4 in tubular cells in a model of early or late urinary tract obstruction does not significantly modify the course of kidney disease at the level of fibrosis and microvascularization.
Oxidative stress is a key component of chronic kidney disease pathologies, and is associated with endothelial dysfunction, inflammation and fibrosis [37]. NOX4 is highly expressed in the kidney where it generates H2O2 constitutively and is induced in fibroblasts following TGF-β treatment. Thus, NOX4 represents a potential therapeutic target of interest for treating the fibrotic component of CKD. Small molecules targeting NOX4 are currently in clinical trials for diabetic nephropathy. However, the role of NOX4 as a therapeutic target in CKD is debated as NOX4-derived H2O2 is increasingly recognized as a signaling molecule which may be rather protective than deleterious in tubular cells [5]. In addition, the cellular localization of NOX4 in the kidney is still debatable due to the lack of sufficiently validated commercial antibodies. We used a rabbit anti-NOX4 antibody to test the localization of this enzyme in human kidney biopsies. We first tested it by using a tetracycline inducible system to show specificity in both Western blot experiments and immunostaining. In Western blots, the antibody recognizes a long (70 kDa) and a short (35 kDa) form. A splice variant of 28 kDa containing the C-terminus of NOX4 was previously described [38]. However, the small band most likely corresponds to a degradation product corresponding to 35 kDa of the C terminus of NOX4 as in Nox4KI mice containing the mouse Nox4 cDNA (splicing not possible), the small form increases concomitantly with the 70 kDa form. Once validated, the novel antibody for NOX4 was used to detect the localization of the protein in healthy human kidney. We observed a predominant localization of NOX4 in proximal tubular cells compared to other nephron segments, which is in line with the initial description of NOX4 expression in rodents by in situ hybridization [8], and in contrast with a subsequent human study documenting high NOX4 expression in distal tubular cells, and low expression in proximal tubules [39]. No expression of NOX4 was observed in peritubular capillaries, and only endothelial cells of middle size arteries displayed NOX4 expression. Finally, podocytes and mesangial cells did not show relevant NOX4 expression, in contrast to parietal glomerular cells.
In conditions of disease, we observed an important increase in NOX4 protein expression in mesangial cells of diabetic and IgA nephropathies, two common renal pathologies displaying mesangial proliferation. NOX4 overexpression in mesangial cells may enhance ROS formation and profibrotic pathways leading to extracellular matrix deposition, as previously described [20,40]. NOX4 was recently described as an anti-apoptotic protein in various cells types [11,16,17], prompting us to speculate that increased NOX4 expression observed in mesangial cells may also decrease apoptosis and promote mesangial cell accumulation [24]. In contrast, in all chronic human kidney diseases studied, independently of the primary disease, we observed a decrease in tubular NOX4 expression at both the mRNA and protein levels, in parallel to loss of tubular cell differentiation and fibrosis. This decreased expression was highly reproducible between disease types and indicate a compartment specific regulation and role of NOX4 in CKD. The cause for this downregulation is unknown since TGF-β, a major regulator of NOX4 expression, is rather increased during CKD. NOX4 is however considered as a good marker of differentiated proximal tubular cells, and its expression is rapidly lost in cell culture (personal observation). We hypothesize that the decrease in NOX4 expression may be related to dedifferentiation of tubular cells in the diseased tubulo-interstitial compartment. This observation may explain why global NOX4 expression was described to decrease in experimental diabetic kidney disease [9,25]. Altogether this decrease in tubular NOX4 expression is an important marker of CKD that should be further investigated as a prognostic tool in future studies.
The role of NOX4 in proximal tubular cells is still unknown. We and others have previously shown a protective role of NOX4 in the kidney as a complete deficiency of Nox4 is detrimental in conditions of tubular injury in the kidney [9,[16], [17], [18]]. Here, we observe a decrease in tubular NOX4 expression in both human biopsies of CKD and an animal model of UUO. Loss of NOX4 expression was seen in parallel to capillary rarefaction and decreased GSTA1 expression. NOX4 is known to modulate angiogenesis and regulate NRF2 in the heart and vessels [41,42]. On the other hand, NOX4 is believed to be an important source of ROS and oxidative stress in the kidney [1]. Therefore, it is conceivable that the decrease in tubular NOX4 expression may either be a defense against oxidative stress during injury, or in opposition, may play a role in capillary rarefaction or apoptosis in CKD.
In order to address the cell-specific role of NOX4, we generated mice overexpressing Nox4 specifically in tubular cells under the control of mouse Pax8 promoter. In these transgenic animals, no apoptosis, tubular injury, fibrosis or change in vascularization was observed. Kidney size and structure were preserved. This is in contrast with the effect of NOX4 overexpression in podocytes. The NRF2 and HIF pathway were mildly induced in NOX4 overexpressing animals in line with the role of NOX4 in the cardiovascular system [12,14,15,43]. GFR was decreased in mice overexpressing tubular NOX4, despite a normal kidney structure and the absence of tubular injury, inflammation or fibrosis. ROS are involved in the regulation of the tubulo-glomerular feedback (TGF) [44]. By overexpressing NOX4 in the tubular compartment, including the macula densa, it is likely that the TGF is activated, thus leading to decreased GFR at baseline. The hemodynamic aspect of this regulation is supported by the absence of difference in structural parameters at baseline or upon UUO. Furthermore, renal clearance is similar following UUO, a situation where the TGF may likely be abrogated because of the necessary hyperfiltration of the remaining kidney. Electrolyte handling, renin expression levels and urine concentration mechanisms were unchanged, as expected with activation of the TGF.
In conditions of fibrotic disease induced by urinary tract obstruction, a classical model of tubular injury leading to fibrosis, endogenous Nox4 expression decreased in Wt animals, whereas transgenic Nox4 overexpression was not affected. Despite sustained Nox4 overexpression in tubular cells, no major difference was observed between Wt and Nox4KI animals in terms of renal function, tubular apoptosis or fibrosis in mild and severe tubular injury. There was a modest transient increase in inflammatory markers in Nox4KI mice in early disease. Interestingly, circumventing decrease in NOX4 expression did not modify fibrosis and microvascularization, suggesting that loss of Nox4 may not be a major player in fibrosis or capillary rarefaction of CKD. NRF2 increase observed in Nox4KI mice at baseline was not sufficient to attenuate the course of the disease in contrast to what has been observed in the cardiovascular system, possibly because of the very high NOX4 and NRF2 endogenous expression in the kidney cortex of Wt animals. Taken together with our previous data, a complete absence of NOX4 in tubular cells is detrimental in injury but rescuing the partial decrease of NOX4 expression in CKD does not modify the course of the disease. Expression of NOX4 in tubular cells is therefore rather protective and does not induce injury. Decreased NOX4 expression in kidney tubular cells in CKD may rather be a marker of the evolving disease than a key player in its evolution.
In summary, NOX4 expression and function are regulated in a cell compartment-specific manner in the context of CKD. We clearly demonstrate that although glomerular NOX4 expression may be enhanced in some nephropathies, loss of tubular NOX4 is a hallmark of all types of CKD. Overexpression of NOX4 in the tubular compartment did not induce injury or fibrosis and rather induced the NRF2 pathway mildly, further demonstrating that NOX4-derived ROS are not harmful in tubular cells, in contrast to what is known in podocytes. NOX4 overexpression did not modify the course of a fibrotic disease such as UUO. While NOX4 emerges as a potential therapeutic target, its functions appear to be cell compartment specific and caution should be taken with regard to systemic NOX4 inhibition. The role of tubular NOX4 as a marker of the severity and prognosis of kidney disease should be studied further.
Acknowledgements
Prof. Sophie de Seigneux is supported by a grant from Swiss National Science Foundation (PP00P3 157454). The authors thank Nicolas Liaudet, Bioimaging Core Facility, CMU, University of Geneva for the technical assistance provided for the quantification of the histological analysis. We also thank all participating centers of the European Renal cDNA Bank - Kröner-Fresenius biopsy bank (ERCB-KFB) and their patients for their cooperation. For active members at the time of the study refer to Shved et al. [45]. The ERCB-KFB is supported by the Else Kröner-Fresenius Foundation.
Abbreviations:
- αSMA
Alpha smooth muscle actin
- BCL2
B-cell lymphoma 2
- elF2
Eukaryotic Initiation Factor 2
- Ccl5
C–C Motif Chemokine Ligand 5 or Rantes
- CKI
Conditional knock-in
- cMyc
Avian myelocytomatosis virus oncogene cellular homolog
- Epo
Erythropoietin
- FSGS
Focal segmental glomerulosclerosis
- Gstα
Glutathione S-Transferase Alpha
- HIF1α
Hypoxia Inducible Factor 1 alpha
- IgA nephropathy
Immunoglobulin A (IgA) nephropathy
- Il18
Interleukin 18
- Il1β
Interleukin 1 Beta
- kDa
Kilo Dalton
- Na,K-ATPase
sodium-potassium adenosine triphosphatase
- NOX2
NADPH oxidase 2
- Nqo1
NAD(P)H quinone oxidoreductase 1
- NRF2
Nuclear factor (erythroid-derived 2)-like 2
- Pax8
Paired box gene 8
- PTC
Proximal Tubular cells
- Tgfβ1
Transforming growth factor beta 1
- Vegf
Vascular endothelial growth factor
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101234.
The results presented in this paper have not been published previously in whole or part, except in abstract format.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Gorin Y. The kidney: an organ in the front line of oxidative stress-associated pathologies. Antioxidants Redox Signal. 2016;25:639–641. doi: 10.1089/ars.2016.6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diebold L., Chandel N.S. Mitochondrial ROS regulation of proliferating cells. Free Radic. Biol. Med. 2016;100:86–93. doi: 10.1016/j.freeradbiomed.2016.04.198. [DOI] [PubMed] [Google Scholar]
- 3.Shadel G.S., Horvath T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163:560–569. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garcia-Redondo A.B., Aguado A., Briones A.M., Salaices M. NADPH oxidases and vascular remodeling in cardiovascular diseases. Pharmacol. Res. 2016;114:110–120. doi: 10.1016/j.phrs.2016.10.015. [DOI] [PubMed] [Google Scholar]
- 5.Rajaram R.D., Dissard R., Jaquet V., de Seigneux S. Potential benefits and harms of NADPH oxidase type 4 in the kidneys and cardiovascular system. Nephrol. Dial. Transplant. 2018;34(4):567–576. doi: 10.1093/ndt/gfy161. [DOI] [PubMed] [Google Scholar]
- 6.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 7.Takac I. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011;286:13304–13313. doi: 10.1074/jbc.M110.192138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geiszt M., Kopp J.B., Varnai P., Leto T.L. Identification of renox, an NAD(P)H oxidase in kidney. Proc. Natl. Acad. Sci. U. S. A. 2000;97:8010–8014. doi: 10.1073/pnas.130135897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Babelova A. Role of Nox4 in murine models of kidney disease. Free Radic. Biol. Med. 2012;53:842–853. doi: 10.1016/j.freeradbiomed.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 10.Rezende F. Knock out of the NADPH oxidase Nox4 has no impact on life span in mice. Redox biology. 2017;11:312–314. doi: 10.1016/j.redox.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hecker L. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014;6:231ra247. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brandes R.P., Takac I., Schroder K. No superoxide--no stress?: Nox4, the good NADPH oxidase! Arterioscler. Thromb. Vasc. Biol. 2011;31:1255–1257. doi: 10.1161/ATVBAHA.111.226894. [DOI] [PubMed] [Google Scholar]
- 13.Schroder K., Wandzioch K., Helmcke I., Brandes R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler. Thromb. Vasc. Biol. 2009;29:239–245. doi: 10.1161/ATVBAHA.108.174219. [DOI] [PubMed] [Google Scholar]
- 14.Schroder K. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012;110:1217–1225. doi: 10.1161/CIRCRESAHA.112.267054. [DOI] [PubMed] [Google Scholar]
- 15.Zhang M. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 2010;107:18121–18126. doi: 10.1073/pnas.1009700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nlandu Khodo S. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J. Am. Soc. Nephrol. 2012;23(12):1967–1976. doi: 10.1681/ASN.2012040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nlandu-Khodo S. NADPH oxidase 4 deficiency increases tubular cell death during acute ischemic reperfusion injury. Sci. Rep. 2016;6:38598. doi: 10.1038/srep38598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santos C.X. Targeted redox inhibition of protein phosphatase 1 by Nox4 regulates eIF2alpha-mediated stress signaling. EMBO J. 2016;35:319–334. doi: 10.15252/embj.201592394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang J. Enhanced expression and activity of Nox2 and Nox4 in the macula densa in ANG II-induced hypertensive mice. Am. J. Physiol. Renal. Physiol. 2014;306:F344–F350. doi: 10.1152/ajprenal.00515.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Block K. Nox4 NAD(P)H oxidase mediates Src-dependent tyrosine phosphorylation of PDK-1 in response to angiotensin II: role in mesangial cell hypertrophy and fibronectin expression. J. Biol. Chem. 2008;283:24061–24076. doi: 10.1074/jbc.M803964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xia L. Mesangial cell NADPH oxidase upregulation in high glucose is protein kinase C dependent and required for collagen IV expression. Am. J. Physiol. Renal. Physiol. 2006;290:F345–F356. doi: 10.1152/ajprenal.00119.2005. [DOI] [PubMed] [Google Scholar]
- 22.Eid A.A. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes. 2009;58:1201–1211. doi: 10.2337/db08-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jha J.C. Genetic targeting or pharmacologic inhibition of NADPH oxidase Nox4 provides renoprotection in long-term diabetic nephropathy. J. Am. Soc. Nephrol. 2014;25(6):1237–1254. doi: 10.1681/ASN.2013070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jha J.C. Podocyte-specific Nox4 deletion affords renoprotection in a mouse model of diabetic nephropathy. Diabetologia. 2016;59:379–389. doi: 10.1007/s00125-015-3796-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Block K., Gorin Y., Abboud H.E. Subcellular localization of Nox4 and regulation in diabetes. Proc. Natl. Acad. Sci. U. S. A. 2009;106:14385–14390. doi: 10.1073/pnas.0906805106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holl M. ROS signaling by NADPH oxidase 5 modulates the proliferation and survival of prostate carcinoma cells. Mol. Carcinog. 2016;55:27–39. doi: 10.1002/mc.22255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meitzler J.L. Decoding NADPH oxidase 4 expression in human tumors. Redox biology. 2017;13:182–195. doi: 10.1016/j.redox.2017.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Serrander L. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 2007;406:105–114. doi: 10.1042/BJ20061903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prunotto M. Tubular cytoplasmic expression of zinc finger protein SNAI1 in renal transplant biopsies: a sign of diseased epithelial phenotype? Am. J. Pathol. 2017;187:55–69. doi: 10.1016/j.ajpath.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 30.Carranza M.L., Feraille E., Favre H. Protein kinase C-dependent phosphorylation of Na(+)-K(+)-ATPase alpha-subunit in rat kidney cortical tubules. Am. J. Physiol. 1996;271:C136–C143. doi: 10.1152/ajpcell.1996.271.1.C136. [DOI] [PubMed] [Google Scholar]
- 31.Cohen C.D., Frach K., Schlondorff D., Kretzler M. Quantitative gene expression analysis in renal biopsies: a novel protocol for a high-throughput multicenter application. Kidney Int. 2002;61:133–140. doi: 10.1046/j.1523-1755.2002.00113.x. [DOI] [PubMed] [Google Scholar]
- 32.Cohen C.D. Comparative promoter analysis allows de novo identification of specialized cell junction-associated proteins. Proc. Natl. Acad. Sci. U. S. A. 2006;103:5682–5687. doi: 10.1073/pnas.0511257103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson W.E., Li C., Rabinovic A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 2007;8:118–127. doi: 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- 34.Bouchard M., Souabni A., Busslinger M. Tissue-specific expression of cre recombinase from the Pax8 locus. Genesis. 2004;38:105–109. doi: 10.1002/gene.20008. [DOI] [PubMed] [Google Scholar]
- 35.Koziel R. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem. J. 2013;452:231–239. doi: 10.1042/BJ20121778. [DOI] [PubMed] [Google Scholar]
- 36.Schreiber A. Transcutaneous measurement of renal function in conscious mice. Am. J. Physiol. Renal. Physiol. 2012;303:F783–F788. doi: 10.1152/ajprenal.00279.2012. [DOI] [PubMed] [Google Scholar]
- 37.Jha J.C., Banal C., Chow B.S., Cooper M.E., Jandeleit-Dahm K. Diabetes and kidney disease: role of oxidative stress. Antioxidants Redox Signal. 2016;25:657–684. doi: 10.1089/ars.2016.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anilkumar N. A 28-kDa splice variant of NADPH oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arterioscler. Thromb. Vasc. Biol. 2013;33:e104–112. doi: 10.1161/ATVBAHA.112.300956. [DOI] [PubMed] [Google Scholar]
- 39.Shiose A. A novel superoxide-producing NAD(P)H oxidase in kidney. J. Biol. Chem. 2001;276:1417–1423. doi: 10.1074/jbc.M007597200. [DOI] [PubMed] [Google Scholar]
- 40.Gorin Y. Nox4 NAD(P)H oxidase mediates hypertrophy and fibronectin expression in the diabetic kidney. J. Biol. Chem. 2005;280:39616–39626. doi: 10.1074/jbc.M502412200. [DOI] [PubMed] [Google Scholar]
- 41.Chen C., Li L., Zhou H.J., Min W. The role of NOX4 and TRX2 in angiogenesis and their potential cross-talk. Antioxidants. 2017;6 doi: 10.3390/antiox6020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craige S.M. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. 2011;124:731–740. doi: 10.1161/CIRCULATIONAHA.111.030775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schurmann C. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur. Heart J. 2015;36:3447–3456. doi: 10.1093/eurheartj/ehv460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sedeek M., Nasrallah R., Touyz R.M., Hebert R.L. NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J. Am. Soc. Nephrol. 2013;24:1512–1518. doi: 10.1681/ASN.2012111112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shved N. Transcriptome-based network analysis reveals renal cell type-specific dysregulation of hypoxia-associated transcripts. Sci. Rep. 2017;7:8576. doi: 10.1038/s41598-017-08492-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.