

Abstract
Methamphetamine (METH) is a powerfully addictive psychostimulant that has a pronounced effect on the central nervous system (CNS). The present study aimed to assess METH toxicity in differentiated C6 astroglia-like cells through biochemical and toxicity markers with acute (1 h) and chronic (48 h) treatments. In the absence of external stimulants, cellular differentiation of neuronal morphology was achieved through reduced serum (2.5%) in the medium. The cells displayed branched neurite-like processes with extensive intercellular connections. Results indicated that acute METH treatment neither altered the cell morphology nor killed the cells, which echoed with lack of consequence on reactive oxygen species (ROS), nitric oxide (NO) or inhibition of any cell cycle phases except induction of cytoplasmic vacuoles. On the other hand, chronic treatment at 1 mM or above destroyed the neurite-like processors and decreased the cell viability that paralleled with increased levels of ROS, lipid peroxidation and lactate, depletion in glutathione (GSH) level and inhibition at G0/G1 phase of cell cycle, leading to apoptosis. Pre-treatment of cells with N-acetyl cysteine (NAC, 2.5 mM for 1 h) followed by METH co-treatment for 48 h rescued the cells completely from toxicity by decreasing ROS through increased GSH. Our results provide evidence that increased ROS and GSH depletion underlie the cytotoxic effects of METH in the cells. Since loss in neurite connections and intracellular changes can lead to psychiatric illnesses in drug users, the evidence that we show in our study suggests that these are also contributing factors for psychiatric-illnesses in METH addicts.
Subject terms: Cellular neuroscience, Cell death
Introduction
METH, an amphetamine derivative, is one of the popular synthetic illegal psycho-stimulants abused not only in the US1 but also worldwide2,3. At present, there are about 10 million METH users in the US, and about 35 million worldwide4–6. Unfortunately, its use is gaining popularity again in the US7,8.
Crystal meth, ice and speed are some popular club names for METH, and it is portrayed as a poor man’s cocaine and the drug of choice for economically poor addicts. Though both METH and cocaine work as CNS stimulants, METH triggers much stronger pharmacological effect9,10 than cocaine because the latter is metabolized most rapidly and disposed of the body in contrast to the former9. Due to a different mechanism of action3,9, METH releases more dopamine in the brain as opposed to cocaine. For these reasons, METH is considered dangerously addictive.
While increased alertness, decreased appetite, hyperthermia, cerebrovascular accidents, and diarrhea11 are some of the acute effects of METH, weight loss, depression, agitation & insomnia12, neurotoxicity13 and cognitive sequelae14–17 are a few chronic psychiatric symptoms. The acute and chronic complications may be related to the toxic effects induced by METH at the cellular level in the CNS. In order to understand the mechanism of toxicity of METH, in vitro studies were conducted using various neuronal cell types due to METH interaction with neurons in the CNS18–25. However, not many studies have addressed the METH-induced toxic effect in astrocytes. Since astrocytes are considerably more abundant than neurons in many regions of mammalian brain26,27, it is possible that events of METH toxicity could manifest in these cells long before they die. It is not yet known what toxic markers METH induces in astrocytes. Therefore, identifying various toxic markers in astrocytes is imperative so that safe therapeutic strategies can be developed against the neurodegeneration associated with chronic use of METH.
The primary aim of our study is to discern the cytotoxic markers for METH using rat C6 astroglia-like cells. We tested these cells at acute (1 h) and chronic (48 h) time points. These cells behave like astrocytes in terms of expression of GFAP28, a marker protein in differentiated matured astrocytes29,30, and exhibit similarities to humans in terms of gene expression31 and enzymes32. The cytotoxic markers we focused on include vacuolation, viability, ROS, NO release, morphology, lipid peroxidation, lactate release, GSH level and apoptosis at acute and chronic treatments. Furthermore, the inhibitory role of METH on cell cycle phases was also assessed.
Results
Lack of acute METH effect on cells
Acute treatment for 1 h was chosen based on an earlier report28. Initial treatment of the cells for 1 h at METH concentrations lower than 500 μM did not result in any cell death (data not shown). As reported on various cell types24,33–37, we used concentrations of 0.5, 1, 2, and 3 mM METH in our studies. METH did cause an induction of cytoplasmic vacuoles (n = 12, F = 4.5, P < 0.05), which is an indication of cell injury, in the treated cells compared to the control (Fig. 1); however, METH did not trigger significant alteration in any cellular parameters, such as cell viability (Fig. 2A) or ROS production (Fig. 2B) or NO release (Fig. 2C) or cell cycle progression (Fig. 2D).
Figure 1.
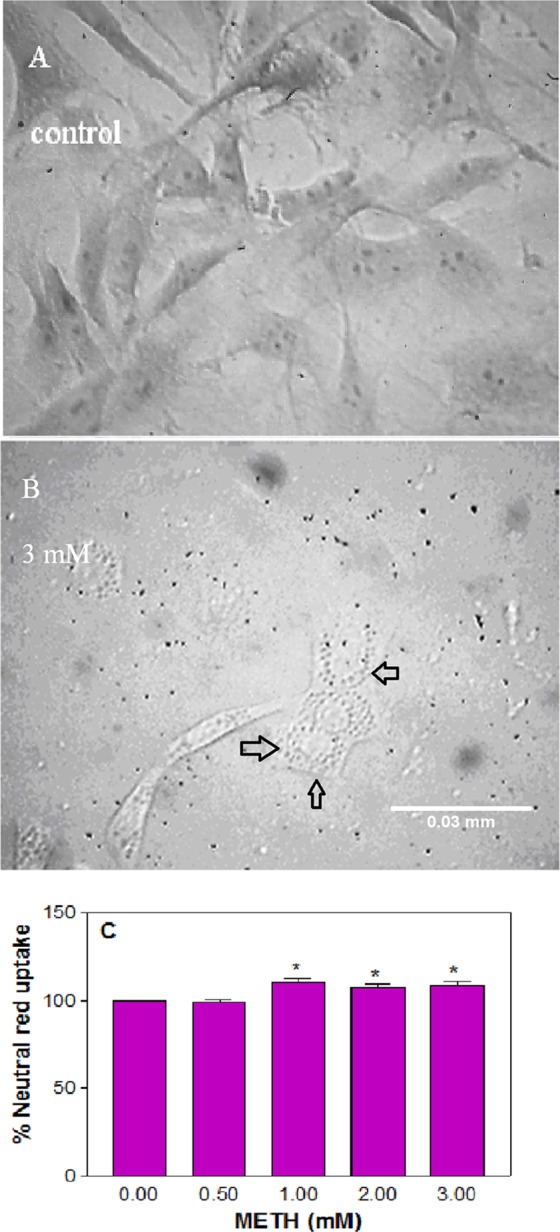
METH-induced vacuolation. C6 astroglia-like cells were treated with equal volume of vehicle (PBS control) or 0.5 to 3 mM METH for 1 h. Images of unstained cells were taken using an inverted phase contrast IX-70 Olympus microscope with a 40x objective. No vacuoles were seen in the control cells (A) but seen in 3 mM treated cells (B). Scale bar: 0.03 mm. For the sake of quantification of vacuoles (C), the cells were stained with neutral red for 10 min and quantified in a plate reader. Data were represented as mean ± SEM (n = 12), *P < 0.05, significant compared to control, one-way ANOVA, Dunnett’s multiple comparison test.
Figure 2.

Effect of acute METH in C6 astroglia-like cells on viability (A), ROS generation (B), nitric oxide release (C) and cell cycle phases (D). Cells were treated with an equal volume of vehicle (PBS control) or 0.5 to 3 mM METH for 1 h. Cell viability was evaluated by a Celltiter 96 Aqueous one solution kit (n = 16). ROS generation was measured by staining the cells with an H2DCFDA dye (20 μM, 30 min), followed by measurement of DCF in a micro plate fluorometer with excitation and emission filters set at 485 and 530 nm respectively (n = 8–12). Nitric oxide release was assayed by Griess reagent (n = 12), and the cell cycle phases were analyzed by flow cytometry (n = 2). Data were represented as mean ± SEM, P > 0.05, insignificant compared to corresponding controls, one-way ANOVA, Dunnett’s multiple comparison test.
Alteration in cell morphology with chronic METH treatment
Chronic treatment for 48 h was selected based on a previous study38. We initially focused on the chronic effect of METH on cell morphology because alteration in cell shape is considered an index of toxicity. After treating the cells with METH at 0.5, 1 and 2 mM for 48 h, the cells were stained with crystal violet39 and observed under the microscope. It was found that in the absence of external stimulants, the cells in 2.5% FBS in medium exhibited a high degree of differentiation. For instance, the control cells (Fig. 3A) exhibited neuronal morphology ranging from a polygonal to stellate shapes and displayed high branching of bi- or tri-polar neurite-like processes40 with extensive intercellular connections (Fig. 3A, black arrows). The morphology of cells treated with 0.5 mM METH remained the same as the control with extensive branching and intact intercellular joints (Fig. 3B); however, treatment with 1 and 2 mM METH gradually destroyed such associations (Fig. 3C,D, red arrows), causing most cells to become rounded. In addition, the cells treated with 2 mM METH lost the anchorage property, and the intercellular gaps expanded. Vacuoles were not observed in the treated cells at any concentration.
Figure 3.

Effect of chronic METH on cell morphology. C6 astroglia-like cells were treated with equal volume of vehicle (PBS control, A) or 0.5 (B) or 1 (C) or 2 mM METH (D) for 48 h. Morphological images of crystal violet dye stained cells were taken using an inverted phase contrast IX-70 Olympus microscope with a 40x objective lens. Inter-cellular connections in control cells (A) are shown by black arrows, while their loss in 2 mM treated cells (D) is shown with red arrows. Scale bar: 20 μm.
Decreased cell viability
METH concentrations below 500 μM did not show significant cell death even after 48 h treatment (data not shown); however, at higher concentrations like 0.5, 1 and 2 mM, there was a significant (n = 6, F = 10.8, P < 0.01) dose-dependent decrease in cell viability (Fig. 4A). The average cell viability was 89.5 ± 1.6, 70.2 ± 7.8 and 53.0 ± 9.7% of the control (100%) at 0.5, 1 and 2 mM, respectively.
Figure 4.

Effect of chronic METH in C6 astroglia-like cells on viability (A) and ROS generation (B). Cells were treated with an equal volume of vehicle (PBS control) or 0.5 to 2 mM METH for 48 h. Cell viability was evaluated by crystal violet dye (n = 6). ROS generation was measured by staining the cells with an H2DCFDA dye (20 μM, 30 min), followed by measurement of DCF in a micro plate fluorometer with excitation and emission filters set at 485 and 530 nm respectively (n = 11). Data were represented as mean ± SEM, *P < 0.05 or **P < 0.01, significant compared to corresponding controls, one-way ANOVA, Dunnett’s multiple comparison test.
Increased ROS production
It was speculated that the dose-dependent decrease in cell viability (Fig. 4A) with METH treatment was related to ROS production. In order to confirm it, the cells were treated with METH at 0.5, 1, and 2 mM for 48 h and assessed for ROS release by dichlorodihydrofluorescein (DCF) measurement. It was found that there was a significant (n = 11, F = 17.9, P < 0.01) dose-dependent increase in ROS level compared to the control (Fig. 4B). The average increase in ROS at 1 and 2 mM METH was (±SEM) 111.9 ± 3.2 and 118.0 ± 1.9%, respectively compared to the control (100%).
Increased lipid peroxidation
Since the dose-dependent increase in ROS can compromise the cell membrane integrity, we next measured the lipid peroxidation in the treated cells. There was a significant (n = 10, F = 13.1, P < 0.01) increase in the lipid peroxidation due to METH treatment compared to the control (Fig. 5A). The average peroxidation at 1 and 2 mM METH was ( ± SEM) 107.9 ± 2.6 and 137.4 ± 10.2%, respectively compared to the control.
Figure 5.

Effect of chronic METH on cells. C6 astroglia-like cells were treated with METH for 48 h. Lipid peroxidation (A, n = 6), lactate release (B, n = 9–12) and glutathione level (C, n = 6) were evaluated calorimetrically in a plate reader. For cell cycle analysis (D, n = 3), the cells were harvested and stained by PI staining solution for 1 h in the dark and analyzed by flow cytometry. Data were represented as mean ± SEM, *P < 0.05 or **P < 0.01, significant compared to control, one-way ANOVA, Dunnett’s multiple comparison test.
Increased lactate level
Treatment of cells with METH at 0.5, 1 and 2 mM for 48 h significantly (n = 12, F = 122.3, P < 0.01) and dose-dependently increased lactate release into the medium compared to the control (Fig. 5B). The average increase (±SEM) was 139.1 ± 2.8 and 193.7 ± 6.4% compared to the control (100%) at 1 and 2 mM METH, respectively.
Decreased GSH level
Glutathione is one of the most available antioxidants in cells. In order to explore the potential role of METH on glutathione level, the cells were treated with METH at 0.5, 1 and 2 mM for 48 h. It was found that METH treatment caused a significant (n = 6, F = 18.5, P < 0.05) decrease in GSH level compared to the control (Fig. 5C). The GSH levels were (±SEM) 83.8 ± 19.5, 77.8 ± 5.5 and 73.6 ± 9.3% of the control (100%) at 0.5, 1 and 2 mM METH, respectively.
METH inhibition at G0/G1 phase
It is widely known that proliferation is an extremely controlled event in the cells. In order to study the effect of METH on various phases of the cell cycle, cells were treated with METH at 1, 1.5 and 2 mM for 48 h and analyzed by flow cytometry. The data indicated that METH treatment caused a significant (n = 3, F = 4.9, P < 0.05) arrest in the G0/G1 phase (Fig. 5D). The percent cells accumulated at G0/G1 progression was significant at 1.5 mM (69.17 ± 2.2%) and 2 mM (75.4 ± 2.4%) METH compared to the control (62.4 ± 3.7%). This accumulation of cells correlated with a subsequent decrease in S phased cells at these concentrations. The cells at G2/M remained nearly the same at all METH treatments. METH-induced G0/G1 inhibition was reported earlier in dentate gyrus culture41.
METH induces apoptosis
METH treatment gradually increased the early apoptotic cells in a significant (n = 3, F = 29.2, P < 0.05) and dose-dependent manner, which subsequently reflected in the gradual decrease in a number of live cells (Fig. 6). The average early apoptotic cells at 0.5 and 1 mM METH were (±SEM) 24.3 ± 2.1 and 35.9 ± 1.2%, respectively.
Figure 6.
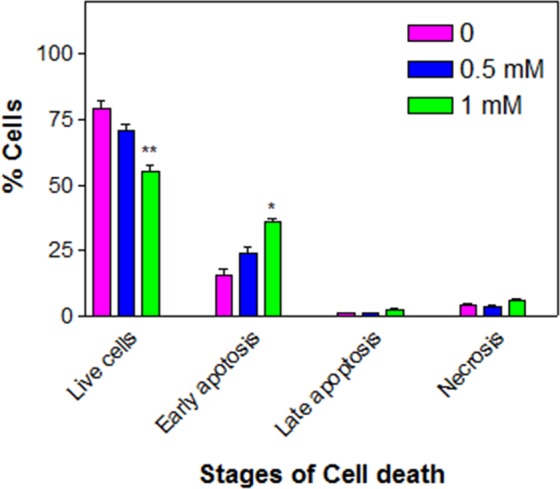
METH-induced apoptosis. C6 astroglia-like cells were treated with equal volume of vehicle (PBS control) or 0.5 or 1 mM METH for 48 h. For apoptosis assay (n = 3), the cells were harvested and stained by annexin-V-FITC and PI in the dark and analyzed by flow cytometry. Data were represented as mean ± SEM, *P < 0.05 or **P < 0.01, significant compared to control, one-way ANOVA, Dunnett’s multiple comparison test.
NAC pre-treatment prevents METH-induced cytotoxicity
Rescue against METH-Induced cytotoxicity by NAC was evaluated for viability, ROS and GSH in the cells treated with METH for 48 h. Based on our previous studies28, we selected 2.5 mM NAC for 48 h exposure. Data indicated that NAC pre-treatment alone did not compromise cell viability, while it fully prevented the cells (n = 12, F = 17, P < 0.01) from METH-induced toxicity compared to METH alone treated cells (Fig. 7A). It was further observed that NAC pre-treatment decreased (n = 7, F = 35.9, P < 0.01) the METH-induced ROS generation (Fig. 7B) and increased (n = 5, F = 18.5, P < 0.01) the GSH level (Fig. 7C) compared to the corresponding METH alone treated cells. These data clearly indicate that NAC pre-treatment prevent METH-induced cytotoxicity.
Figure 7.
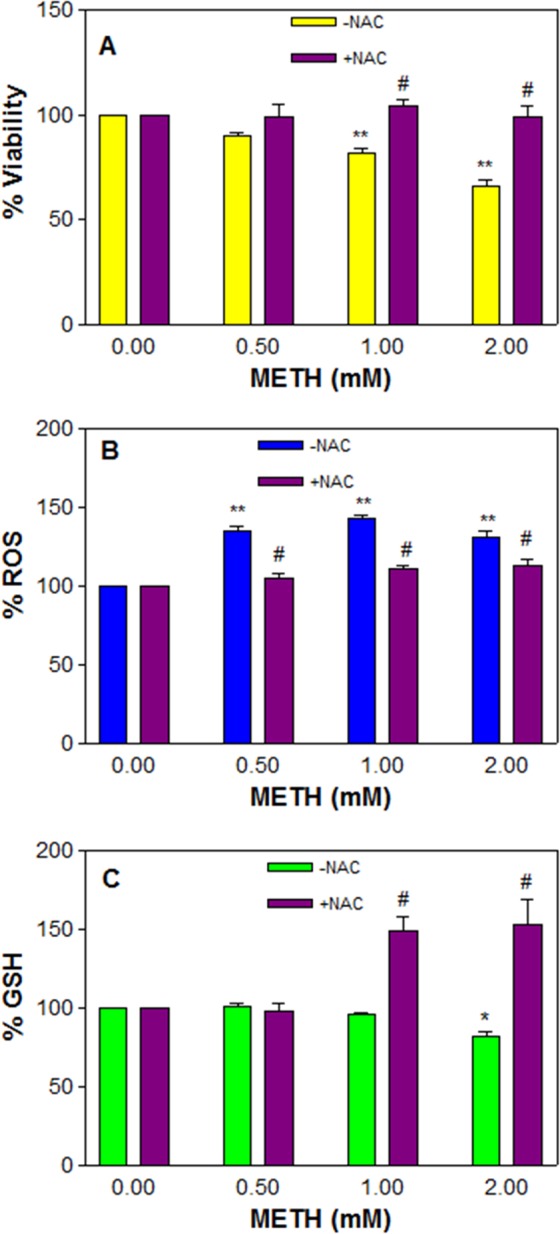
NAC rescues against METH-induced toxicity. C6 astroglia-like cells were pre-treated with 2.5 mM NAC or equal volume of vehicle (PBS control) for 1 h followed by co-treatment with 0.5, 1 and 2 mM METH for 48 h. Cell viability (n = 12) was evaluated by crystal violet dye (A), ROS generation (B, n = 7) by H2DCFDA (20 μM, 30 min) and glutathione level (C, n = 5) by Ellman’s reagent in a plate reader. Data were represented as mean ± SEM, *P < 0.05 or **P < 0.01, significant compared to control, or #P < 0.01, significant compared to corresponding METH treatments, one-way ANOVA, Bonferroni’s multiple comparison test.
Discussion
Studies have demonstrated that substances of drug abuse cause psychological disorders by altering neuronal structure42–44 or mitochondrial function45 in the CNS. Given that these changes could last life-long46, they may have the propensity to manifest in several neurodegenerative diseases including Parkinson’s disease47, Schizophrenia, and Alzheimer disease. Since astrocytes insinuate between the neurons, interact in signal transmission, release trophic factors required for the growth48 and differentiation of neurons, METH-induced astrocytic death could potentially contribute to neuronal dysfunction and thereby lead to above neurodegenerative diseases. Several in vivo49–52 and in vitro53 studies showed that METH treatment induced astrogliosis, a sign of CNS injury, leading to modulation of neuronal activity54. Studies in human astrocytes and neurons further showed that METH toxicity was through glucose energy deprivation55. So far, there is no clear understanding of the mechanism of astrocyte death in vivo with METH exposure.
Direct assessment of METH toxic effect under in vivo is impeded due to body complexity. Employing primary cultures is not practical on account of restricted growth potential, finite life span and lack of cell homogeneity; thus, we employed C6 astroglia-like cells under in vitro conditions to gain insights on toxicity underlying cell death. These cells represent a good model system for astrocytes due to various merits outlined earlier28–32. These cells undergo differentiation and are shown to propagate calcium ion waves, called astrocyte excitability56, in the brain as well as under in vitro conditions57,58. Treatment with dibutyryl cAMP59,60 or taxol54 enabled these cells to differentiate, giving typical neuronal morphology. In our study, we found that C6 cells grown in reduced FBS (2.5%) without external growth factors induced a high level of differentiation, exhibiting neuronal morphology with extensive neurite-like processors and intercellular connections (Fig. 3A arrows). This observation is comparable with dibutyryl cAMP-induced differentiation in C6 cells60 but appears greater (Fig. 3A) than taxol-induced differentiation in the same cell line61.
The concentration of METH in plasma depends on several factors -like amount of drug intake, its frequency, drug tolerance, drug hydrolysis by blood esterases62,63, gender, genetics, age and time gap between drug intake & assessment. For example, METH level in serum after 3 h of intake was found to be 1.94 mg/L64, which is equal to 10.4 μM; (METH MW: 185.69), while the level was 6 μM after 22 h. It is important to know that these micro molar levels do not indicate consumption of METH in micro quantities by addicts. At the time of METH intake, its concentration in blood would be in milli molar range. For example, neurotoxic studies in rats were conducted65 at a maximum of 80 mg/kg METH as a binge dose (20 mg/kg, 4 times a day). In another study, these authors tested at 20 mg/kg/day METH for 10 days as a chronic dose in rats. Testing at 80 or 20 mg/kg in rats would translate to 8.11 or 2 mM METH respectively in the blood, taking a total volume of 12.8 ml/240 grams of rat weight (64 ml/kg). Similarly, in humans, a well adopted addict can use 1 g66 or more METH62 per day. On an average, 1 g translates to 1.07 mM METH in the blood assuming a total of 5 L. Since blood contains both plasma and corpuscles roughly in 1:1 ratio, the volume of plasma alone would now be equal to 2.5 L. Under this situation, METH concentration in plasma would be 2.14 mM. A comparison of highest METH concentration (2 mM) at chronic treatment in our study with a well tolerated human dose (1 g/day/2.5 L plasma = 2.14 mM) would reveal that the doses tested in our study (0.5, 1 and 2 mM) are within the range found in animals and humans.
Acute METH treatment did not induce cell death (Fig. 2A). Lack of ROS production or NO generation or cell cycle inhibition with acute METH treatment (Fig. 2B–D) corroborated with the lack of cell death (Fig. 2A). No studies were reported with acute METH effects for the induction of toxic markers in these cells except reports on activation of cells treated with METH67; similarly, the protective role of melatonin against METH-induced endoplasmic reticulum stress was reported37.
Lack of cell death (Fig. 2A) cannot be construed as METH’s inability to enter the cells, because METH treatment resulted in vacuolation (Fig. 1B), demonstrating its intracellular access. Vacuolation in our study is in agreement with earlier reports on different cell-types68,69. Despite vacuolation (Fig. 1B), lack of concomitant METH toxicity to cells (Fig. 2A) indicates that the formation of vacuoles in these cells was a secondary effect of toxicity. Although vacuolation is commonly associated with substances of drug abuse28,39,70–73, it was not noticed with chronic cocaine treatment in PC12 cells38 or with chronic METH treatment in C6 astroglia-like cells (Fig. 4B). On the other hand, vacuolation in mesencephalic cultures treated with chronic METH68 may suggest that this phenomenon is cell-type specific.
Chronic METH treatment in our study caused an alteration in cell morphology, destroyed the neurite-like processors (Fig. 3B, red arrows) and resulted in a decrease in viability (Fig. 4A), which corresponded with increased ROS production (Fig. 4B), an observation consistent with previous studies that showed METH-induced ROS generation in cultured astrocytes74. Furthermore, lactate release indicated that the treated cells were in the state of hypoxia (Fig. 5B), an alternate way for cell survival under mitochondrial dysfunction as shown earlier75. Increased ROS and dysfunctional mitochondria in our study are consistent with previous studies on long term METH exposure76. GSH is one of the most available antioxidants meant to cope with oxidative stress in cells. Consistent with the concept of the inverse relationship between increased ROS and decreased antioxidant levels in mammalian cells77, we found that the increased ROS level (Fig. 4B) with chronic METH treatment correlated with depletion of GSH level (Fig. 5C). This decrease was further paralleled with the decreased viability (Fig. 4A) and increased lipid peroxidation (Fig. 5A), suggesting that GSH depletion was one of the causes of cell death.
Increased ROS is associated with DNA damage78,79. Cells under such condition cease to proliferate in an attempt to repair DNA damage; however, if the damage is beyond repair, such cells undergo apoptotic death. Based on the observation of METH-induced cell death (Fig. 4A), increased ROS level (Fig. 4B), and inhibition of G0/G1 cell cycle phase (Fig. 5D), we suspected that the observed cell death could be apoptotic; further investigation proved that METH treatment indeed caused apoptosis in cells (Fig. 6), which is consistent with earlier reports24,25,33. Interestingly, NAC pre-treatment protected the cells completely (Fig. 7A) by decreasing the ROS (Fig. 7B) through increased GSH level (Fig. 7C), thus further supporting the results that increased ROS and consequent depletion of GSH were the main reasons of cell death in C6 cells treated with METH.
Conclusion
Drug addicts often suffer from psychiatric illnesses such as depression80, anger, aggressiveness, and paranoia81. Damage to neuronal or astrocyte structures could be some of the contributing factors for these illnesses. In this study, we have shown that METH damages neurite-like processors (Fig. 3D). These data suggest that this damage could be a contributing factor to these illnesses.
It is disheartening to learn that METH is the main substance of drug abuse among pregnant women82. As per some estimations, its prevalence of use during pregnancy is ranged between 0.7–5.2%83, which is considered very high. Detrimental effects of METH on the developing fetus were reported earlier84–88. Thus, the impact of METH usage by pregnant women requires special national attention because there is a 3-fold increase in admissions of these women for METH treatment82, making this problem an economical burden to the society. The depletion in GSH89 due to increased oxidative stress90 is one of the causes of several psychiatric illnesses in METH addicts; therefore, compounds -such as NAC, which support GSH synthesis, could play a vital role in reducing the METH toxicity to neurons and alleviate from psychiatric-illnesses.
Materials and Methods
Materials
RPMI 1640, fetal bovine serum (FBS), penicillin/streptomycin sulfate, amphotericin B, phosphate-buffered saline (PBS) and L-glutamine were purchased from Media Tech (Herndon, VA). Crystal violet, L glutaraldehyde, trypan blue, methamphetamine (METH) hydrochloride (MW: 185.69), 2′,7′-dichlorofluorescin diacetate (H2DCFDA), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), and EDTA were supplied by Sigma-Aldrich Company (St. Louis, MO). All other routine chemicals were of analytical grade.
Cell culture maintenance
The CNS-derived rat C6 astroglia-like cell line (CCL-107) was purchased from American Type Culture Collection (Rockville, MD) and maintained as an adherent monolayer culture as described earlier28 in a humidified atmosphere containing 5% CO2 in air at 37 °C in an incubator. Cells were sub-cultured twice a week.
METH and N-acetyl cysteine (NAC) treatments
The cells were seeded in 96-well microtiter plates at a starting density of 1 × 104 cells per well in a total volume of 195 μl growth medium supplemented with 2.5% FBS. The cells were allowed to adhere to the wells in the incubator for 18–24 h before drug exposure. Stocks (1 M) and working stocks (20 to 120 mM) of METH were always prepared fresh in PBS. The cells, typically about 60–70% confluent, were treated with increasing concentrations of METH (0.5, 1, 2 and 3 mM for 1 h or 0.5, 1, and 2 mM for 48 h) in a final volume of 5 μl to prevent pH alteration of cell medium. Cells in medium alone or PBS in medium containing cells served as controls. Both controls and the treated samples were always present in the same culture plate. These plates were incubated for 1 or 48 h continuously without further renewal of growth media in a 5% CO2 at 37 °C incubator with plates capped in a normal fashion. For rescue experiments, the cells in 96-well plates were pretreated with 2.5 mM NAC for 1 h prior to METH co-treatment for 48 h. Cell viability, ROS and GSH levels were assayed at the end of the using methods outlined below.
Microscopy
Morphology of crystal violet stained cells was evaluated as per the method described earlier28 using an inverted phase contrast IX70 Olympus microscope (Olympus, Ontario, NY) with a 40x objective. Photomicrographs were taken by a CCD camera (DP70, Olympus) with the image-acquisition system (DP-Controller, Olympus).
Cell vacuolation
Cells were seeded in 96-well culture plates as described above. After treating with various concentrations of METH (0.5, 1, 2 and 3 mM) for 1 h, the cytoplasmic vacuoles in unstained cells were observed using an inverted phase contrast IX-70 Olympus microscope (Olympus) with a 40x objective. Photomicrographs were taken by an ocular video-camera system (MD35 Electronic eyepiece, Zhejiang Jincheng Scientific & Technology Co., Ltd., Hangzhou, China) using C-Imaging System Software (Compix Inc. Cranberry Township, PA). Vacuoles were quantified by neutral red dye uptake method91. This dye selectively deposits in the vacuoles besides leaving the cytoplasm unstained. The incorporated dye was extracted with 70% ethanol and 0.37% HCl, and the absorbance at 540 nm was taken in a micro plate reader.
Viability assay
For acute treatment, the cell viability was evaluated using a Celltiter 96 Aqueous one solution kit (MTS, 10 μl, Promega, Madison, WI) as per the instructions provided by the manufacturer. After 30 min incubation at 37 °C, absorbance was taken in a micro plate reader at 490 nm. Because of false positive result due to interaction between NAC and MTS (unpublished observation), cell viability for chronic treatment was evaluated by crystal violet dye-uptake method as described previously92. Absorbance was taken in a micro plate reader at 540 nm.
Measurement of intracellular ROS
Cells in 96-well plates in media devoid of phenol red with 2.5% FBS were treated with METH at 0.5, 1, 2 and 3 mM for 1 h or 0.5, 1 and 2 mM for 48 h followed by staining with the cell permeable dye H2DCFDA (20 μM final) for 30 min. After gentle washing and air drying of the cells, PBS (100 μl/well) was added. The fluorescent DCF was measured in an automatic fluorometer micro plate reader with the excitation filter set at 485 nm and the emission filter at 530 nm respectively (Synergy HTX multimode micro plate reader, BioTek Instruments, Winooski, VT).
NO assay
Cells (2 × 104 cells/well) were seeded in 96-well titer plates in media devoid of phenol red with 2.5% FBS. Next day, the cells were treated with METH at various concentrations (0.5, 1, 2 and 3 mM) for 1 h. At the end of incubation, 50 μl of media was transferred into a new plate and mixed with an equal volume of Griess reagent (1% sulfanilamide/0.1% N-naphthyl-ethylenediamine in 5% phosphoric acid) followed by a 10 min incubation in the dark. The absorbance at 546 nm was measured in a microtiter culture plate reader.
Lipid peroxidation assay
Cells were seeded at a starting density of 0.5 × 106 cells per well in 6-well culture plates in media devoid of phenol red with 2.5% FBS. After METH treatment at 0.5, 1 and 2 mM for 48 h, the cells were harvested and centrifuged at 13,000 rpm on a table top micro centrifuge for 6 min. All cell pellets were sonicated in PBS on ice for 3 sec and transferred into glass tubes. The lysates were further processed as per the earlier report75 and absorbance at 535 nm was measured in a micro plate reader. Clear medium without cells was used as a blank.
Lactate assay
After treating the cells with METH at different concentrations (0.5, 1 and 2 mM) for 48 h in 96-well plates in media containing 2.5% FBS, lactate release was determined calorimetrically as per the study reported earlier38. Absorbance was measured at 490 nm in a microtiter culture plate reader.
GSH level
After treating with METH at 0.5, 1 and 2 mM for 48 h in 96-well microtiter plates, the cells were fixed with 0.25% glutaraldehyde for 30 min, followed by gentle washing and air drying. Total cellular GSH was assayed with Ellman’s reagent (DTNB) as per earlier study93. The absorbance was measured at 412 nm in a micro plate reader.
Cell cycle analysis
Treatments with METH at 1, 1.5, 2, 2.5 and 3 mM for 1 h or 1, 1.5 and 2 mM for 48 h were performed in 100 mm plastic culture dishes. The cells were harvested and fixed in 95% ice-cold ethanol for at least 24 h at 4 °C. The next day, the tubes were centrifuged at 1217 g for 7 min and re-suspended in 100 μl ethanol. Cells were stained as described elsewhere94. The proportion of cells in each stage of the cell cycle was determined within 2 h by using FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). First the instrument was aligned with Calibrite beads (BD Biosciences). Then, prior to cell cycle analysis, the linearity of the fluorescence pulse detector was checked using chicken erythrocyte nuclei (BD Biosciences) on a linear scale with Doublet Discrimination Module at a flow rate of 12 μl/min as per the protocol supplied by the manufacturer. A total of 10,000 individual gated events were analyzed separately. CellQuest Software was used for the acquisition of the data, and the results were displayed as histograms (FL2-A vs counts). The percentage of cells in each phase was determined by using ModFit LT 3.0 (Verity Software House, Topsham, ME).
Apoptosis assay
Annexin V-FITC Apoptosis Detection Kit (BD Biosciences Pharmingen, San Diego, CA) was utilized to measure the percent of apoptosis. In brief, after treatment with 0.5 and 1 mM METH in triplicate wells in 6-well plates for 48 h, the cells were harvested by trypsinization, washed in PBS and stained with annexin V/propidium iodide (PI) as per the protocol supplied by the manufacturer except for the steps of fixation and RNase reported earlier95. Samples were analyzed within 1 h by using FACSCalibur flow cytometer (BD Biosciences). CellQuest Pro software was used for the acquisition of the data at a flow rate of 12 μl/min, and the results were displayed as quadrant dot plots in the log mode; the X-axis indicates the fluorescence of annexin-V (green), while Y-axis indicates PI (red). Unstained cells (double negative), annexin-V-FITC single stained cells, and PI single stained cells were used for quadrant settings (compensation). In this study, untreated (control) and treated samples were stained with both dyes. A total of 10,000 individual gated events were analyzed separately for each sample. Quadrant statistics were used to quantify the cell population in the quadrants.
Statistical analysis
The experimental results were presented as mean ± SEM. Since there is only one variable in each experiment, the data were analyzed for significance by one-way ANOVA and compared by Dunnett’s or Bonferroni’s multiple comparison tests using GraphPad Prism Software, version 3.00 (San Diego, CA). The test values of P < 0.05 and P < 0.01 were considered significant and highly significant, respectively.
Acknowledgements
The authors thank Cheryl A. Fitch-Pye for reading the manuscript and for suggestions. This study was supported by the National Institute on Minority Health and Health Disparities of the National Institutes of Health under award number G12MD007582, P20 MD006738 and the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R25GM107777. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health.
Author Contributions
R.B.B., C.B.G., L.M.L. and K.F.A.S. conceived and designed research. R.B.B., C.W., S.F.D., K.R. and M.A. performed research, R.B.B., C.B.G. and C.W. analyzed the data, R.B.B. wrote the paper. All authors gave the final approval and agreed to be accountable for all aspects of the work.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Roehr B. Half a million Americans use methamphetamine every week. BMJ. 2005;331(7515):476. doi: 10.1136/bmj.331.7515.476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rawson RA, Gonzales R, Brethen P. Treatment of Methamphetamine use disorders: an update. J Subst Abuse Treat. 2002;23:145–150. doi: 10.1016/S0740-5472(02)00256-8. [DOI] [PubMed] [Google Scholar]
- 3.Barr AM, et al. The need for speed: an update on methamphetamine addiction. J Psychiatry Neurosci. 2006;31:301–313. [PMC free article] [PubMed] [Google Scholar]
- 4.Schifano F, et al. Smokable (“ice”, “crystal meth”) and non smokable amphetamine- type stimulants: clinical pharmacological and epidemiological issues, with special reference to the UK. Ann Ist Super Sanita. 2007;43:110–115. [PubMed] [Google Scholar]
- 5.Vearrier D, et al. Methamphetamine: history, pathophysi-ology, adverse health effects, current trends, and hazards associated with the clandestine manufacture of metham-phetamine. Dis Mon. 2012;58:38–89. doi: 10.1016/j.disamonth.2011.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Kirby T, Thornber-Dunwell M. High-risk drug practices tighten grip on London gay scene. Lancet. 2013;381:101–102. doi: 10.1016/S0140-6736(13)60032-X. [DOI] [PubMed] [Google Scholar]
- 7.Maxwell J, Brecht ML. Methamphetamine: here we go again? Addict Behav. 2011;36:1168–1173. doi: 10.1016/j.addbeh.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carnevale, J. The Current Status of the Methamphetamine Epidemic (Policy Brief). Gaithersburg, MD: Carnevale Associates (2013).
- 9.NIDA. Methamphetamine: Abuse and Addiction. Rockville, MD (Sep. 2007).
- 10.CDC. Methamphetamine Use and Risk for HIV/AIDS. Atlanta, GA (Jan. 2007).
- 11.Hart CL, et al. Acute physiological and behavioral effects of intranasal methamphetamine in humans. Neuropsychopharmacology. 2008;33(8):1847–1855. doi: 10.1038/sj.npp.1301578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Newton TF, et al. Methamphetamine abstinence syndrome: preliminary findings. Am J Addict. 2004;13(3):248–55. doi: 10.1080/10550490490459915. [DOI] [PubMed] [Google Scholar]
- 13.Sekine Y, et al. Methamphetamine causes microglial activation in the brains of human abusers. J Neurosci. 2008;28(22):5756–61. doi: 10.1523/JNEUROSCI.1179-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rawson RA, Condon TP. Why do we need an Addiction supplement focused on methamphetamine? Addiction. 2007;102(Suppl. 1):1–4. doi: 10.1111/j.1360-0443.2006.01781.x. [DOI] [PubMed] [Google Scholar]
- 15.Grant KM, et al. Methamphetamine-associated psychosis. J Neuroimmune Pharmacol. 2012;7(1):133–39. doi: 10.1007/s11481-011-9288-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Forray A, Sofuoglu M. Future pharmacological treatments for substance use disorders. Br J Clin Pharmacol. 2014;77:382–400. doi: 10.1111/j.1365-2125.2012.04474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh JH, Stein DJ, Howells FM. The neurobiology of methamphetamine induced psychosis. Front Hum Neurosci. 2014;8:537. doi: 10.3389/fnhum.2014.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricaurte GA, et al. Dopamine nerve terminal degeneration produced by high doses of methamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- 19.Gibb, J. W., Hanson, G. R. & Johnson, M. Neurochemical mechanisms of toxicity. In: Amphetamine and its analogs (Cho, A. K. & Segal D. S., eds), pp 269–295, Los Angeles: Academic (1994).
- 20.Pu C, Broening HW, Vorhees CV. Effect of methamphetamine on glutamate-positive neurons in the adult and developing rat somatosensory cortex. Synapse. 1996;23:328–334. doi: 10.1002/(SICI)1098-2396(199608)23:4<328::AID-SYN11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 21.Lew, R. et al. Evidence for mechanism of action of neurotoxicity of amphetamine related compounds. In: Highly selective neurotoxins: basic and clinical applications (Kostrzewa, R. M. ed.), pp 235–268, Totowa, N. J.: Humana (1998).
- 22.Xie T, et al. Effect of temperature on dopamine transporter function and intracellular accumulation of methamphetamine: Implications for methamphetamine-induced dopaminergic neurotoxicity. J Neurosci. 2000;20(20):7838–7845. doi: 10.1523/JNEUROSCI.20-20-07838.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stumm G, et al. Amphetamines induce apoptosis and regulation of bcl-x splice variants in neocortical neurons. FASEB J. 1999;13:1065–1072. doi: 10.1096/fasebj.13.9.1065. [DOI] [PubMed] [Google Scholar]
- 24.Kanthasamy A, et al. Methamphetamine induces autophagy and apoptosis in a mesencephalic dopaminergic neuronal culture model role of cathepsin-D in methamphetamine-induced apoptotic cell death. Ann N Y Acad Sci. 2006;1074:234–244. doi: 10.1196/annals.1369.022. [DOI] [PubMed] [Google Scholar]
- 25.Tian C, Murrin LC, Zheng JC. Mitochondrial fragmentation is involved in methamphetamine-induced cell death in rat hippocampal neural progenitor cells. PLoS One. 2009;4(5):e5546. doi: 10.1371/journal.pone.0005546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Candel, E. R., Schwartz, J. H. & Jessel, T. M. Principles of Neural Science. Norwalk, CT: Appleton and Lange. 20 p (1991).
- 27.Tsacopoulos M, Magistretti PJ. Metabolic coupling between glia and neurons. J Neurosci. 1996;16:877–885. doi: 10.1523/JNEUROSCI.16-03-00877.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Badisa Ramesh B., Kumar Sanjay S., Mazzio Elizabeth, Haughbrook Rasheda D., Allen John R., Davidson Michael W., Fitch-Pye Cheryl A., Goodman Carl B. N-Acetyl Cysteine Mitigates the Acute Effects of Cocaine-Induced Toxicity in Astroglia-Like Cells. PLOS ONE. 2015;10(1):e0114285. doi: 10.1371/journal.pone.0114285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dahl D. The vimentinGFA protein transition in rat neuroglia cytoskeleton occurs at the time of myelination. J. Neurosci. Res. 1981;6:741–748. doi: 10.1002/jnr.490060608. [DOI] [PubMed] [Google Scholar]
- 30.Dahl D, et al. Vimentin, the 57,000 molecular weight protein of fibroblast filaments is the major cytoskeletal component in immature glia. Eur. J. Cell Biol. 1981;24(5):13–522. [PubMed] [Google Scholar]
- 31.Sibenaller ZA, et al. Genetic characterization of commonly used glioma cell lines in the rat animal model system. Neurosurg Focus. 2005;19:1–9. doi: 10.3171/foc.2005.19.4.2. [DOI] [PubMed] [Google Scholar]
- 32.Zimmer DB, Van Eldik LJ. Analysis of the calcium-modulated proteins S100 and calmodulin, and their target proteins during C6 glioma cell differentiation. J Cell Biol. 1989;108:141–151. doi: 10.1083/jcb.108.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cadet JL, Ordonez S. Serum withdrawal potentiates the toxic effects of methamphetamine in vitro. Ann. N.Y. Acad. Sci. 2000;914:82–91. doi: 10.1111/j.1749-6632.2000.tb05186.x. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X, et al. N-Acetylcysteine amide protects against methamphetamine-induced oxidative stress and neurotoxicity in immortalized human brain endothelial cells. Brain Res. 2009;1275(87–9):5. doi: 10.1016/j.brainres.2009.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El Ayadi A, Zigmond MJ. Low concentrations of methamphetamine can protect dopaminergic cells against a larger oxidative stress injury: Mechanistic study. PLoS One. 2011;6(10):e24722. doi: 10.1371/journal.pone.0024722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Y, et al. Taurine attenuates methamphetamine-induced autophagy and apoptosis in PC12 cells through mTOR signaling pathway. Toxicol Lett. 2012;215:1–7. doi: 10.1016/j.toxlet.2012.09.019. [DOI] [PubMed] [Google Scholar]
- 37.Tungkum W, et al. Melatonin suppresses methamphetamine-triggered endoplasmic reticulum stress in C6 cells glioma cell lines. J Toxicol Sci. 2017;42(1):63–71. doi: 10.2131/jts.42.63. [DOI] [PubMed] [Google Scholar]
- 38.Badisa RB, et al. Identification of biochemical and cytotoxic markers in cocaine treated PC12 cells. Sci. Rep. 2018;8(1):2710. doi: 10.1038/s41598-018-21182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Badisa RB, Darling-Reed SF, Goodman CB. Cocaine induces alterations in mitochondrial membrane potential and dual cell cycle arrest in rat C6 astroglioma cells. Neurochem Res. 2010;35:288–297. doi: 10.1007/s11064-009-0053-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henschlet D, et al. The inhibitory effect of neuropathic organophosphate esters on neurite outgrowth in cell cultures: a basis for screening for delayed neurotoxicity. Toxicol In Vitro. 1992;6:327–335. doi: 10.1016/0887-2333(92)90022-J. [DOI] [PubMed] [Google Scholar]
- 41.Baptista S, et al. Methamphetamine decreases dentate gyrus stem cell self-renewal and shifts the differentiation towards neuronal fate. Stem Cell Research. 2014;13(2):329–341. doi: 10.1016/j.scr.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 42.Sapolsky RM. Why stress is bad for your brain. Science. 1996;273(5276):749–750. doi: 10.1126/science.273.5276.749. [DOI] [PubMed] [Google Scholar]
- 43.Gould E, et al. Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation. J. Neurosci. 1997;17:2492–2498. doi: 10.1523/JNEUROSCI.17-07-02492.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moeller S, et al. Irreversible brain damage caused by methamphetamine persisting structural brain lesions. Alcoholism and Drug. Addiction. 2016;29:39–41. [Google Scholar]
- 45.Karabatsiakis A, et al. Mitochondrial respiration in peripheral blood mononuclear cells correlates with depressive subsymptoms and severity of major depression. Transl Psychiatry. 2014;4:e397. doi: 10.1038/tp.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Su TP, Hayashi T. Cocaine affects the dynamics of cytoskeletal proteins via sigma(1) receptors. Trends Pharmacol Sci. 2001;22:456–458. doi: 10.1016/S0165-6147(00)01740-5. [DOI] [PubMed] [Google Scholar]
- 47.Lloyd SA, Faherty CJ, Smeyne RJ. Adult and in utero exposure to cocaine alters sensitivity to the Parkinsonian toxin 1- methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. Neuroscience. 2006;137:905–913. doi: 10.1016/j.neuroscience.2005.09.035. [DOI] [PubMed] [Google Scholar]
- 48.Sobkowicz HM, Waclawik AJ, August BK. The astroglial cell that guides nerve fibers from growth cone to synapse in organotypic cultures of the fetal mouse spinal cord. Synapse. 2006;59:183–200. doi: 10.1002/syn.20222. [DOI] [PubMed] [Google Scholar]
- 49.Pu C, Vorhees CV. Developmental dissociation of methamphetamine-induced depletion of dopaminergic terminals and astrocytes reaction in rat striatum. Brain Res Dev Brain Res. 1993;72:325–328. doi: 10.1016/0165-3806(93)90201-K. [DOI] [PubMed] [Google Scholar]
- 50.Pu C, et al. The effects of amfonelic acid, a dopamine uptake inhibitor, on methamphetamine-induced dopaminergic terminal degeneration and astrocytic response in rat striatum. Brain Res. 1994;649:217–224. doi: 10.1016/0006-8993(94)91067-7. [DOI] [PubMed] [Google Scholar]
- 51.Fukumura M, et al. A single dose model of methamphetamine-induced neurotoxicity in rats: effects on neostriatal monoamines and glial fibrillary acidic protein. Brain Res. 1998;806:1–7. doi: 10.1016/S0006-8993(98)00656-8. [DOI] [PubMed] [Google Scholar]
- 52.Hebert MA, O’Callaghan JP. Protein phosphorylation cascades associated with methamphetamine-induced glial activation. Ann. N.Y. Acad. Sci. 2000;914:238–262. doi: 10.1111/j.1749-6632.2000.tb05200.x. [DOI] [PubMed] [Google Scholar]
- 53.Stadlin A, Lau JW, Szeto YK. A selective regional response of cultured astrocytes to methamphetamine. Ann. N. Y. Acad. Sci. 1998;844:108–121. doi: 10.1111/j.1749-6632.1998.tb08226.x. [DOI] [PubMed] [Google Scholar]
- 54.Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat. Rev. Neurosci. 2005;6(8):626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- 55.Abdul Muneer PM, et al. Methamphetamine inhibits the glucose uptake by human neurons and astrocytes: stabilization by acetyl-L-carnitine. PLoS One. 2011;6(4):e19258. doi: 10.1371/journal.pone.0019258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pivneva T, et al. Store-operated Ca(2+) entry in astrocytes: Different spatial arrangement of endoplasmic reticulum explains functional diversity in vitro and in situ. Cell Calcium. 2007;43:591–601. doi: 10.1016/j.ceca.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Charles AC, et al. Intercellular signaling in glial cells: calcium waves and oscillations in response to mechanical stimulation and glutamate. Neuron. 1991;6:983–992. doi: 10.1016/0896-6273(91)90238-U. [DOI] [PubMed] [Google Scholar]
- 58.Cornell-Bell AH, Finkbeiner SM. Ca2+ waves in astrocytes. Cell Calcium. 1991;12:185–204. doi: 10.1016/0143-4160(91)90020-F. [DOI] [PubMed] [Google Scholar]
- 59.Messens J, Slegers H. Synthesis of glial fibrillary acidic protein in rat C6 glioma in chemically defined medium: cyclic AMP-dependent transcriptional and translational regulation. J. Neurochem. 1992;58:2071–2080. doi: 10.1111/j.1471-4159.1992.tb10948.x. [DOI] [PubMed] [Google Scholar]
- 60.Hu W, et al. Change of morphology and cytoskeletal protein gene expression during dibutyryl cAMP-induced differentiation in C6 glioma cells. Cell Mol Neurobiol. 2008;28:519–528. doi: 10.1007/s10571-007-9229-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chao C, et al. Induction of neural differentiation in rat C6 glioma cells with taxol. Brain and Behavior. 2015;5(12):e00414. doi: 10.1002/brb3.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Melega WP, et al. Methamphetamine blood concentrations in human abusers: application to pharmacokinetic modeling. Synapse. 2007;61(4):216–220. doi: 10.1002/syn.20365. [DOI] [PubMed] [Google Scholar]
- 63.Kraemer T, Maurer HH. Toxicokinetics of amphetamines: metabolism and toxicokinetic data of designer drugs, amphetamine, methamphetamine, and their N- alkyl derivatives. Ther. Drug Monit. 2002;24(2):277–289. doi: 10.1097/00007691-200204000-00009. [DOI] [PubMed] [Google Scholar]
- 64.Molina NM, Jejurikar SG. Toxicological findings in a fatal ingestion of methamphetamine. J. Anal. Toxicol. 1999;23:67–68. doi: 10.1093/jat/23.1.67. [DOI] [PubMed] [Google Scholar]
- 65.Moszczynska A, Turenne S, Kish SJ. Rat striatal levels of the antioxidant glutathione are decreased following binge administration of methamphetamine. Neurosci Lett. 1998;255(1):49–52. doi: 10.1016/S0304-3940(98)00711-3. [DOI] [PubMed] [Google Scholar]
- 66.Simon SL, et al. A comparison of patterns of methamphetamine and cocaine use. J. Addict. Dis. 2001;21(1):35–44. doi: 10.1300/J069v21n01_04. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, et al. Role of high-mobility group box 1 in methamphetamine-induced activation and migration of astrocytes. J Neuroinflammation. 2015;12:156–168. doi: 10.1186/s12974-015-0374-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bennett BA, et al. Differing neurotoxic potencies of methamphetamine, mazindol, and cocaine in mesencephalic cultures. J Neurochem. 1993;60:1444–1452. doi: 10.1111/j.1471-4159.1993.tb03307.x. [DOI] [PubMed] [Google Scholar]
- 69.Cubells JF, et al. Methamphetamine neurotoxicity involves vacuolation of endocytic organelies and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;74(4):2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vitullo JC, et al. Cocaine-induced small vessel spasm in isolated rat hearts. Am J Pathol. 1989;135:85–91. [PMC free article] [PubMed] [Google Scholar]
- 71.Welder AA. A primary culture system of adult rat heart cells for the evaluation of cocaine toxicity. Toxicology. 1992;72:175–187. doi: 10.1016/0300-483X(92)90111-Q. [DOI] [PubMed] [Google Scholar]
- 72.Repetto G, et al. Morphological, biochemical and molecular effects of cocaine on mouse neuroblastoma cells culture in vitro. Toxicol in Vitro. 1997;11:519–525. doi: 10.1016/S0887-2333(97)00066-0. [DOI] [PubMed] [Google Scholar]
- 73.Yu RC, et al. Characterization of cocaine elicited cell vacuolation: the involvement of calcium/calmodulin in organelle deregulation. J Biomed Sci. 2008;15:215–226. doi: 10.1007/s11373-007-9213-z. [DOI] [PubMed] [Google Scholar]
- 74.Lau JW, Senok S, Stadlin A. Methamphetamine-induced oxidative stress in cultured mouse astrocytes. Ann. N. Y. Acad. Sci. 2000;914:146–156. doi: 10.1111/j.1749-6632.2000.tb05192.x. [DOI] [PubMed] [Google Scholar]
- 75.Badisa, R. B. et al. Cellular and molecular responses to acute cocaine treatment in neuronal-like N2a cells: potential mechanism for its resistance in cell death. Cell Death Discovery, Volume 5, 10.1038/s41420-018-0078-x (2018). [DOI] [PMC free article] [PubMed]
- 76.Quinton MS, Yamamoto BK. Causes and consequences of methamphetamine and MDMA toxicity. AAPS. J. 2006;8:E337–E347. doi: 10.1007/BF02854904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barayuga SM, et al. Methamphetamine decreases levels of glutathione peroxidases 1 and 4 in SH-SY5Y neuronal cells: protective effects of selenium. Neurotoxicology. 2013;37:240–246. doi: 10.1016/j.neuro.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schraufstatter IU, et al. Oxidant injury of cells: DNA strand breaks activate polyadenosine diphosphate_ribose polymerase and lead to depletion of nicotinamide adenine dinucleotide. J Clin Invest. 1986;77:1312–1320. doi: 10.1172/JCI112436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schraufstatter IU, et al. Hydrogen peroxide-induced injury of cells and its prevention by inhibitors of poly(ADP-ribose) polymerase. Proc Natl Sci USA. 1986;83:4908–4912. doi: 10.1073/pnas.83.13.4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rounsaville BJ, et al. Heterogenicity of psychiatric diagnosis in treated opiate addicts. Arch Gen Psychiatry. 1982;39:161–166. doi: 10.1001/archpsyc.1982.04290020027006. [DOI] [PubMed] [Google Scholar]
- 81.Satel SL, Southwick SM, Gawin FH. Clinical features of cocaine-induced paranoia. Am J Psychiatry. 1991;148:495–498. doi: 10.1176/ajp.148.4.495. [DOI] [PubMed] [Google Scholar]
- 82.Terplan M, et al. Methamphetamine use among pregnant women. Obstet Gynecol. 2009;113:1285–1291. doi: 10.1097/AOG.0b013e3181a5ec6f. [DOI] [PubMed] [Google Scholar]
- 83.Derauf C, et al. Demographic and psychosocial characteristics of mothers using methamphetamine during pregnancy: preliminary results of the infant development, environment, and lifestyle study (IDEAL) Am J drug and alcohol abuse. 2007;33(2):281–289. doi: 10.1080/00952990601175029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Robinson TE, Kolb B. Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci. 1997;17:8491–8497. doi: 10.1523/JNEUROSCI.17-21-08491.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Robinson TE, Kolb B. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 1999;11:1598–1604. doi: 10.1046/j.1460-9568.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- 86.Good MM, et al. Methamphetamine use during pregnancy: maternal and neonatal implications. Obstet Gynecol. 2010;116:330–334. doi: 10.1097/AOG.0b013e3181e67094. [DOI] [PubMed] [Google Scholar]
- 87.Leino O, et al. Pollutant concentrations in placenta. Food Chem Toxicol. 2011;54:59–69. doi: 10.1016/j.fct.2011.10.058. [DOI] [PubMed] [Google Scholar]
- 88.Plotka, J. et al. Effects of addictive substances during pregnancy and infancy and their analysis in biological materials. In: Whitacre, D. (eds) Reviews of environmental contamination and toxicology, Volume 227. Reviews of Environmental Contamination and Toxicology (Continuation of Residue Reviews), vol 227. Springer, Cham 227 (2014). [DOI] [PubMed]
- 89.Dean OM, et al. A role for glutathione in the pathophysiology of bipolar disorder and schizophrenia? Animal models and relevance to clinical practice. Curr. Med. Chem. 2009;16(23):2965–2976. doi: 10.2174/092986709788803060. [DOI] [PubMed] [Google Scholar]
- 90.Ng F, et al. Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int. J. Neuropsychoph. 2008;11:851–876. doi: 10.1017/S1461145707008401. [DOI] [PubMed] [Google Scholar]
- 91.Cover TL, et al. Effects of urease on HeLa cell vacuolation induced by Helicobacter pyroli cytotoxin. Infect Immun. 1991;59:1264–1270. doi: 10.1128/iai.59.4.1264-1270.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Badisa RB, et al. Cytotoxic activities of some Greek Labiatae herbs. Phytother. Res. 2003;17:472–476. doi: 10.1002/ptr.1175. [DOI] [PubMed] [Google Scholar]
- 93.Smith IK, Vierheller TL, Thorne CA. Assay of glutathione reductase in crude tissue homogenates using 5, 5′- dithiobis(2-nitrobenzoic acid) Anal. Biochem. 1988;175:408–413. doi: 10.1016/0003-2697(88)90564-7. [DOI] [PubMed] [Google Scholar]
- 94.Badisa RB, et al. Selective cytotoxic activities of two novel synthetic drugs on human breast carcinoma MCF-7 cells. Anticancer Res. 2009;29:2993–2996. [PMC free article] [PubMed] [Google Scholar]
- 95.Rieger AM, et al. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp. 2011;(50):e2597. doi: 10.3791/2597. [DOI] [PMC free article] [PubMed] [Google Scholar]