

Abstract
The Barker Hypothesis states change to the maternal environment may have significant impacts on fetal development, setting the stage for adult disease to occur. The development of the maternofetal vasculature during implantation and maintenance during pregnancy is extremely precise, yet dynamic. Delays or dysfunction in the orchestration of anatomical remodeling, maintenance of blood pressure, or responsiveness to metabolic demand may have severe consequences to the developing fetus. While these intermissions may not be fatal to the developing fetus, an interruption, reduction, or an inability to meet fetal demand of blood flow during crucial stages of development may predispose young to disease later in life. Maternal inability to meet fetal demand can be attributed to improper placental development and vascular support through morphological change or physiological function will significantly limit nutrient delivery and waste exchange to the developing fetus. Therefore, we present an overview of the uteroplacental vascular network, maternal cardiovascular adaptations that occur during pregnancy, placental blood flow, and common maternal comorbidities and/or exposures that may perturb maternal homeostasis and affect fetal development. Overall, we examine uterine microvasculature pathophysiology contributing to a hostile gestational environment and fetal predisposition to disease as it relates to the Barker Hypothesis.
Abbreviations
- AGEs
advanced glycation end products
- ENM
engineered nanomaterials
- eNOS
endothelial nitric oxide synthase
- GDM
gestational diabetes mellitus
- IUGR
intrauterine growth restriction
- LPS
Lipopolysaccharide (Endotoxin)
- MetS
metabolic syndrome
- NADPH
nicotinamide adenine dinucleotide phosphate hydrogen
- ROS
reactive oxygen species
- TNF
tumor necrosis factor
- uNK
uterine natural killer
- VEGF
Vascular endothelial growth factor
- VEGFR
Vascular Endothelial Growth Factor Receptors
1. INTRODUCTION
Developmental Origins of Health and Disease is a concept proposed by Dr. David Barker that suggests change to the maternal environment and fetal milieu during gestation has significant impacts on health of the progeny in adulthood, later referred to as the Barker Hypothesis.1, 2 Many studies, including those conducted by Dr. David Barker, have associated small for development age at birth with the development of cardiovascular and/or metabolic disease later in life. Given cardiovascular disease is the leading cause of death worldwide, identification of all risk factors is vital. The mechanisms connecting the in utero environment and maternal pathophysiology to the cardiovascular health of the next generation remain elusive.
One common development in a hostile gestational environment is the development of IUGR, in this case, typically due to a reduction in placental and fetal perfusion; in these cases, babies are born small for gestational age. In response to poor health and nutrition in utero, the fetus may be reprogrammed to have a “thrifty phenotype” adapted to a perceived maternal low‐nutrient environment.3, 4 Offspring with IUGR tend to have altered glucose metabolism and insulin signaling despite a normal diet later in life.5 Faced with ample available calories, these individuals develop obesity and other presentations of MetS as adults due to alterations in homeostatic mechanisms that occurred during development. There is growing evidence that maternal health status can amend the fetal genome and imprint gene expression in subsequent developmental stages.
In this review, we will provide an overview of non‐pregnant uterine vascular function and subsequent changes associated with fetal implantation and placentation, highlighting the importance of the macro‐ and microvasculature with each state. Special attention will be given to the role of uterine endothelial function in adequate uteroplacental perfusion and how dysfunction contributes to maternal and fetal disease. Further we discuss the maternal and fetal implications associated with changes to the maternal environment. These include disease (hypertension, obesity, diabetes), systemic inflammation, external stressors (noise pollution and sleep alterations), and environmental exposure (smoking, heavy metals, air pollution, and nanomaterials). Each of these variations to the maternal environment has fetal implications, which may act as a risk factor for the development of cardiovascular disease in adulthood. However, the mechanisms associated with the developmental onset of disease have not been elucidated.
1.1. Overview of uterine circulation before pregnancy
From the outset, uterine arteries are extended bifurcation of the aorta shared with the ovary and distally from the internal iliac artery; this redundancy is in place to prevent ischemia downstream in cases of occlusion.6 Further, the uterine arteries branch to form arcuate networks, providing a consistent quality of oxygenated blood throughout the uterus in preparation to support implantation. These extend as smaller radial arteries penetrating the myometrium. The radial arteries then divide into daughter branches, identified as straight (basal) arterioles which feed the uterine muscle or coiled (spiral) arterioles spanning deeper across the myoendometrial junction also described as preplacental arterioles (Figure 1). In the non‐pregnant uterus, the lineage of arteriolar vessels ends here. Capillaries meet and venules then drain and merge throughout the uterine layers ultimately meeting the inferior vena cava.
Figure 1.
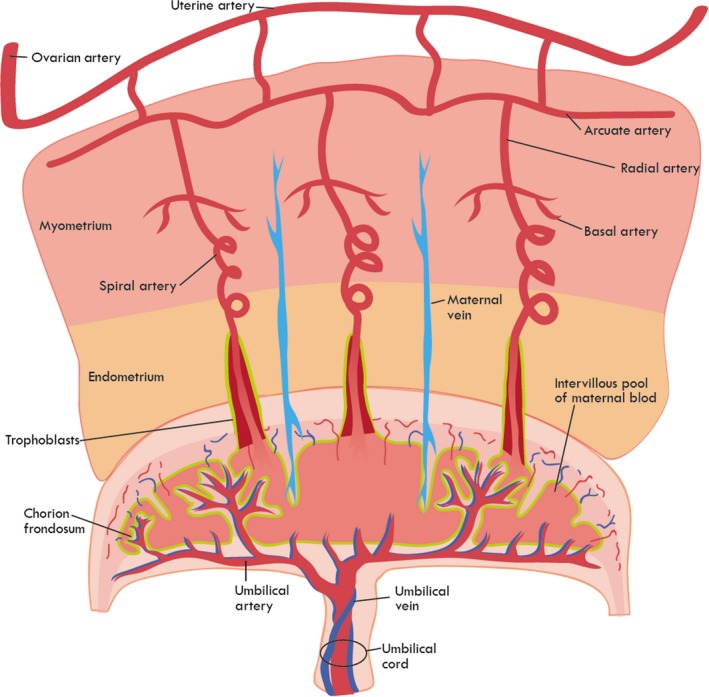
Schematic of uteroplacental vasculature. Development and responsiveness of the maternal‐fetal macro‐ and microvasculature are crucial for proper placental perfusion, fetal development, and successful gestation. Perturbations to vascular maturity, trophoblast invasion, spiral arteriolar remodeling, or arteriolar (radial, basal, or spiral) reactivity, during pregnancy, may limit placental perfusion and have dire consequences for the fetus
The rodent, commonly used as an animal model for reproductive studies, has a duplex or dual‐horn uterus to maximize surface area for litter yielding, where the main uterine arteries run widely spaced from and alongside each horn. Where humans possess penetrating arcuate networks, rodents have secondary vessels that loop arcuate arteries back to the uterine artery to support a horn full of pups lengthwise and tertiary vessels extending those loops down into the myometrium.6
The uterus is one of the few adult organs that undergoes routine vascular expansion and reduction, dictated by hormonal cycling of namely estradiol and progesterone.7 Spiral arterioles are the most hormone‐responsive vessels, whereupon stimulation during the estrogen‐driven proliferative phase undergoes significant growth and coiling.8 Estradiol, VEGF/VEGFR, and angiogenic factors collectively control arteriole proliferation.9 The spiral arterioles are destined to become the uteroplacental arteries, as they are the main supply and terminal extension to the endometrium.
During the first trimester of pregnancy, the mammalian uterus undergoes extensive decidualization, a process initiated during the luteal phase of the reproductive cycle independent of an implanted fetus to improve receptivity of the uterine endometrium. Decidualization involves morphological and physiological changes including infiltration of local immune cell populations and spiral arteriole remodeling. Ramified spiral arterioles from the proliferative phase are surrounded by uNK cells, along with the presence of other immune cells, that appear to aid in the spiral arteriole remodeling and immunotolerance of invading fetal trophoblasts upon implantation.10 In the absence of a conceptus, the richly vascularized bed facing the lumen is shed in response to falling levels of progesterone.
1.2. Implantation
The success of implantation and maintenance through the first 4 weeks of human pregnancy (pre‐placentation) is highly dependent upon the uterine preparation and overall health status of a female. Irregular cardiovascular, endocrine, and/or immune function threatens this process, with an emphasis on the requirement of reactive and responsive microvasculature for uterine priming. Implantation is a two‐way interaction between the blastocyst and endometrial stromal cell types. This cross talk is accomplished by molecular cues such as paracrine factors regulated by estrogen and progesterone.
Decidualization involves the critical process of remodeling the spiral arterioles, the microvessels of the endometrium. The spiral arterioles from the proliferative phase of the reproductive cycle are now poised for invasion of fetal cells to establish the uteroplacental vessels, which will ultimately control maternal blood flow into the placenta. uNK cells and fetal extravillous trophoblasts target the extracellular matrix and smooth muscle of the spiral arterioles, promoting vasodilation to enhance nutrient delivery.11 The trophoblasts further invade the arterioles and supplant the endothelium, assuming a phenotype as a pseudoendothelium.12 Due to the hemochorial placentation of both humans and rodents, uterine microvasculature must also adapt to bring maternal blood into direct contact with fetal villous trophoblast during pregnancy13; therefore, the terminal ends of the spiral arterioles are held open, emptying into the lumen, which is remodeled to become the intervillous space of the placenta where maternal blood bathes the villous trophoblasts.14 The remodeling of the uterine microvessels and thus the establishment of the uteroplacental vasculature will initiate placentation and act as a conduit for nutrition for the developing fetus.
As the interface between the invasive trophoblasts and maternal spiral arterioles is responsible for placental perfusion, inadequate microvascular remodeling, perfusion, and/or reactivity have been associated with reduced placental blood flow and reproductive disorders, such as IUGR and preeclampsia.15 Abnormal fat deposition, nutrient balance, oxidative stress, and energy state have also been associated with higher rates of implantation failure and defective implantation leading to adverse consequences for pregnancy.16, 17
1.3. Changes/Development throughout pregnancy
Throughout pregnancy, uterine circulation undergoes significant restructuring to accommodate fetal demand for nutrients. Critical transformation events include the following: (a) luminal dilation; (b) trophoblast invasion of vessel media and endothelium; and (c) deposition of fibrinoid material in place of vascular smooth muscle.18 Altogether, these changes achieve maximal delivery to intervillous space by increasing vessel diameter and decreasing reactivity to vasoconstricting agents.
During the first trimester of human pregnancy, invasive extravillous trophoblasts travel retrograde through spiral arterioles to serve as intraluminal plugs at the myometrial junction and permit slow release of plasma, but no true perfusion.19 At week 18, the low‐pressure uteroplacental vessels are established by conversion of luminal plugs into circumferential stents.20 About one third of spiral arterioles will be transformed into uteroplacental arterial openings into the intervillous space (Figure 1). Spiral arterioles at the center of the placenta are more likely to be transformed into uteroplacental vessels over those located at the periphery.21
As pregnancy progresses, local changes initiate adaptive responses in the uterine vessels; adaptations of the uterine vessels occur by lengthening (axial) to accommodate the growing uterus and fetus and increase in diameter (circumferential) to, according to Poiseuille's law, exponentially increase flow. Experimentally, myometrial stretch alone is sufficient to induce vessel remodeling in the non‐pregnant rat.22 Further, shear stress‐induced activation of eNOS becomes more sensitive.23, 24 Precise mechanisms by which these phenomena occur are still unclear.
In addition to local uterine vascular changes, systemic hemodynamic accommodations are made by the mother during pregnancy. There is a significant increase in blood volume, increased in cardiac output, elevated resting heart rate, decreased peripheral resistance, and decreased blood pressure.25 Collectively, the physiological alterations made at the systemic level as well as the microcirculation level at the placenta dynamically meet the increasing need of blood to supply the fetus as pregnancy progresses. Readers interested in further information are referred to the following reviews.26, 27
2. PLACENTAL BLOOD FLOW
The placenta is an incredibly unique, transient organ, utilized only during fetal gestation to transfer fresh nutrients to the fetus and waste to the mother. Therefore, the placenta has two circulations, receiving blood from the maternal vasculature (maternal‐placental [uteroplacental]) and fetal systems (fetal‐placental [fetoplacental]). The counter‐current pressure between the two systems drives perfusion and nutrient‐waste exchange. Pressure in the uteroplacental system has been measured at 80‐100 mm Hg, 70 mm Hg in spiral arterioles, and 10 mm Hg in the intervillous space.28 This pressure gradient in combination with the low resistance arterioles permits efficient perfusion of intervillous space to ensure exchange of nutrients for waste. The efficient exchange of blood gases and nutrients takes place due to maternal blood in the intervillous space bathing the placental villi at a low pressure vs driving force. The reduction of maternal mean arterial pressures to the low flows required for proper placental function highlights the importance of the resistance vasculature within the uteroplacental system. Therefore, impairments to uterine microcirculation function and/or reactivity are a significant risk factor for adverse pregnancy outcomes.
Normal uteroplacental blood flow is achieved by the coordinated maternal hemodynamic accommodations, uterine vascular adaptations, and invasion of trophoblasts. The doubling of the uterine artery diameter (circumferentially) enables an increase in blood flow to the uterus, directed at the placenta during fetal development (10 to 30 mL/min compared to ½ L/min).6, 29 During the first trimester, spiral arterioles are luminally obstructed by trophoblastic plugs, which then loosen and permit maternal blood flow into the intervillous space during the second and third trimesters.6, 30 The venous system also undergoes enlargement in diameter, increased distensibility, and reduced elastin content which should enhance venous capacity.27
The umbilical cord arises from embryonic cells and is the conduit between the fetus and placenta. It extends from the fetal umbilicus to the fetal‐facing aspect of the placenta. In humans, the cord contains two arteries (umbilical arteries) and one vein (umbilical vein). As the umbilical vessels are of embryological/fetal origin, the anatomical terminology is similar to that of the pulmonary system, where the umbilical vein supplies the fetus with oxygenated, nutrient‐rich blood from the placenta, and the umbilical arteries return blood that is oxygen and nutrient‐depleted. At the intersection of umbilical cord and placenta, each umbilical artery branches into at least eight stem arteries with average diameters of 1.5 mm.28 These first‐order branches divide into four to eight horizontal vessels of the secondary order that have an average diameter of 1 mm. These arteries span at varying lengths and curve toward the center of the placenta as they begin branching into terminal third‐order 0.1‐0.6 mm villous branches that form capillary beds. Villous capillaries fill an encasement created by another fetal trophoblast cell type and bring fetal blood into close proximity to maternal blood without direct intermingling. Gases and nutrients therefore have a two‐cell layer barrier to traverse between blood circulations: the fetal villous trophoblast and the fetal endothelium. The blood pressure in the umbilical arteries leaving the fetal heart is about 50 mm Hg, which falls to about 30 mm Hg in the villous capillaries, whereas umbilical vein pressure is about 20 mm Hg.28
3. UTEROPLACENTAL MICROVASCULAR FUNCTION
One of the defining characteristics of the microcirculation is to discuss the arterioles, capillaries, and venules within a tissue or organ of interest. Experimentally, this vasculature may then be challenged, cellular function evaluated, and reactivity assessed to identify impairments in blood flow to the tissue which may impact organ function. Physiological studies to identify the mechanisms associated with normal microvascular control, and by default microvasculopathy, continue. Microvascular perturbations that may affect organ perfusion (eg, impaired remodeling or endothelial dysfunction) can limit blood flow within the tissue and impair the physiological function. The heart is a classical case, wherein reduced blood flow by either physical blockage or reduced arteriolar dilation may culminate in localized ischemia, reduced oxygenation and nutrient availability, and symptomatic angina. For simplicity within this review, we will discuss hemodynamic control in yes or no terms, either the uterine microvasculature is capable of placental perfusion to meet fetal demand or it is not; in some cases, this may be associated with poor trophoblast invasion, inadequate spiral arteriolar remodeling, blunted arteriolar (radial, basal, or spiral) vasodilation responsiveness to chemical/mechanical stimuli, or heightened sensitivity to vasoconstrictive signaling molecules (Figure 1). Unfortunately, impaired uterine arteriolar function, implantation, placental perfusion, and the delivery of nutrients to the fetal compartment are much more complex, traditionally asymptomatic in the short term, and may propagate by affecting fetal cardiovascular health.
4. PERTURBATIONS
4.1. Hypertension/Preeclampsia
In general, hypertension is an abnormally high blood pressure which may be identified in an acute state after a physiological stressor (eg, fight or flight) or in a chronic condition attributed to many lifestyle choices and family history. With respect to the microcirculation, chronic hypertension can result in systemic damage to the luminal endothelial lining and smooth muscle cells of blood vessels.31 Histological modifications lead to the weakening of the vessel wall and narrowing of the vessel lumen.31 These changes culminate in microvascular dysfunction and contribute to peripheral resistance, further propagating blood pressure dysregulation and inadequate blood delivery downstream. In the non‐pregnant state, consequences of hypertension include myocardial infarction, strokes, and aneurysms.32
Patients with hypertension prior to becoming pregnant tend to remain on their antihypertensive medications and are closely monitored by their physicians during pregnancy to limit maternal and fetal morbidity and mortality. During pregnancy, major cardiovascular adaptations, including uterine microvascular remodeling, occur to increase uteroplacental blood flow. A hypertensive state during pregnancy is referred to as preeclampsia and is a consequence of failed uterine microvascular remodeling. This stoppage causes an increase in peripheral resistance sourcing from the uteroplacental vessels and, thus, a raise in maternal blood pressure. Along with preeclampsia is the release of placental hypoxic and oxidative stress factors that send additional signaling for a compensatory rise in maternal blood pressure. Moreover, the resulting local oxidative/nitrosative stress generated with poor perfusion has been shown to cause endothelial dysfunction of the microvessels providing blood to the pregnancy, lending to additional vessel narrowing and fetal risk.33
The mechanisms associated with inappropriate uterine remodeling, culminating in poor uteroplacental perfusion, and preeclampsia are currently being investigated34; however, maternal inflammation, insulin resistance, metabolic syndromes, and genetic predispositions participate independently or in association with increased risk.35 Each of these risk factors has been identified within the literature to impact microvascular function in a variety of tissue beds, thereby limiting blood flow to downstream tissues36; perfusion of the placenta is not uniquely impacted in this case; however, tissue ischemia of the placenta can impair fetal development. Implications of preeclampsia involve maternal and fetal hazards including IUGR, placental abruption, and maternal and fetal death.
Poor uteroplacental blood flow, as seen with preeclampsia, may cause growth restriction to the fetus. Hypoperfusion of the placenta ablates nutrient and waste exchange, impeding maximal growth potential for the developing fetus which manifests by low birthweight. IUGR is associated with perinatal adverse events (prematurity, cerebral palsy, stillbirth, neonatal death) and a “preprograming” for adult disease (obesity, hypertension, diabetes mellitus type 2).37
4.2. MetS
The substantial global prevalence of disorders involving perturbed energy utilization is increasing patient morbidity and mortality with cardiovascular disease. MetS is diagnosed as a collection of at least three of the following conditions: obesity, hypertension, hyperglycemia, and dyslipidemia.38 It is postulated that the mal utilization and storage of energy constituting these disorders are predisposing patients to microvascular disease by inducing endothelial dysfunction.38 This is believed to be caused by increased endothelial‐derived NO quenching from elevated ROS, adipose distribution and adipokine production, and endogenous corticosteroid production.39 Various cytokines (TNF‐α, IL‐6, IL‐10) that are elevated in MetS indirectly contribute to endothelial dysfunction by increasing endothelin‐1 secretion and inhibiting eNOS dysfunction.39 Adiponectin, having a vasoprotective activity, is reduced in obesity,40 while increased levels of leptin, deleterious to the cardiovascular system, lend to vascular calcification, vascular smooth muscle proliferation limiting expandability of vessel walls.41 Excess circulating corticosteroids may also contribute to vasoconstriction.42
MetS conditions in women who become pregnant are at higher risk to develop GDM.43 Maternal insulin resistance is a normal process of human pregnancy and is critical to maintain maternal blood glucose to support the growing fetus, especially in the third trimester. Women with obesity enter pregnancy with preexisting insulin resistance that intensifies as pregnancy progresses.44 The placenta is most impacted by GDM through multiple mechanisms of reduced oxygen supply to the fetus and modifications in placental transport.45 Interestingly, when assessed during pregnancy, one study found no differences in microvascular reactivity in women with or without GDM.46 Further, obesity has an effect on uteroplacental remodeling. Increased levels of AGEs are significantly found within obese versus lean uterine cavities, which activated cellular stress pathways resulting in impaired decidualization, embryo implantation, and inhibited trophoblast invasion.47 Uteroplacental micro‐ and macrovascular endothelial dysfunction has also been associated with GDM and obesity during pregnancy. Several studies have identified abnormal eNOS expression and activity of the endothelial L‐arginine/nitric oxide signaling pathway.48
Exposure to adverse nutritional environment in utero permanently alters fetal metabolism and physiology with long‐term health consequences. Offspring of GDM are often born macrosomic as a result of the steeper maternal‐to‐fetal concentration gradient of glucose across the placenta, a risk to the mother during delivery and to the fetus as glucose level plummet after birth. Fetal β‐cell development is permanently altered during critical periods of development where organogenesis aims to adapt to maternal environment.4, 45 Children who are born large for gestational age and were developmentally exposed to maternal diabetes or obesity are at increased risk to develop the constellation of conditions constituting MetS themselves.49
4.3. Inflammation
Acute and chronic inflammation may result in altered microvascular form and function. Unstressed endothelial cells serve as a nonreactive interface between circulation and tissue. Under inflammatory conditions, the endothelium of the microcirculation serves as a major contributor to a local response. Endothelial cells normally respond to neural, humoral, and endothelial‐derived signals; therefore, in a pro‐inflammatory environment, endothelial cells activate by thrusting selectins and integrins to the surface to signal the local recruitment of acute and chronic pro‐inflammatory cells. Increased venular permeability is also a feature of the inflammatory process, under normal circumstances to allow for the immune cells and components to extravasate to the inflamed tissue.50 Molecularly, a pro‐inflammatory environment many usurp the activity of eNOS to produce superoxide and thereby reducing the bioavailability of the endothelium‐dependent vasodilator nitric oxide.51 Oxidative stress from inflammation may cause endothelial cells to increase production of the vasoconstrictive protein endothelin‐1.52 Further, NO may be consumed as it reacts with oxidants, thereby limiting the bioavailability for vascular function.53 Decreased endothelial‐derived vasodilatory signals can inappropriately diminish perfusion to a specific tissue.
In healthy pregnancy, immune mediator cells prevent excessive maternal inflammation and subsequent rejection of the invading fetal tissue.54 However, in some cases interactions between the transformed decidual cell populations the fetal extravillous trophoblasts lend to failed spiral arteriole remodeling. Putative mechanisms include immune cell activation and subsequent secretion of cytokines and placental factors impeding trophoblast invasion, angiogenesis, and, in turn, placentation. Maternal inflammation (as demonstrated by LPS administration) has been associated with deficient trophoblast invasion, uteroplacental hemodynamic alterations, placental nitrosative stress, and spiral arteriole remodeling, possibly associated with an increased activated macrophage population identified at spiral arteries.55 An in vitro study confirmed T‐cell activated macrophage inhibits trophoblast motility and migration by secreting TNF.56, 57 Dams with lipopolysaccharide‐induced inflammation had a reversal of these effects with the administration of nitric oxide mimetic glyceryl trinitrate, indicating the role of inflammatory quenching of nitric oxide in perturbed uteroplacental circulation.55 Conclusively, maternal inflammation causes macrophage‐derived TNF inhibited spiral arteriole remodeling and may thereby lend to the pathological processes of growth restriction sequelae. Further, cytokine‐induced disassembly of cell‐to‐cell junctions at trophoblastic and endothelial cells in inflamed placental tissues may put the fetus at risk through an inability to maintain proper barrier function.58
Fetal development may become compromised with abnormal maternal inflammation. Impaired placentation, uteroplacental perfusion, and homeostasis result and are shared characteristics of preeclampsia, IUGR, and fetal demise. Indeed, it has been shown that women who have history of miscarriage also have elevated levels of inflammation compared to non‐pregnant women who went on to have healthy pregnancies.59 Maternal inflammatory mediators (IL‐6 and IL‐8) have been associated with preterm delivery, low birthweight, and respiratory insufficiency increasing fetal risk of long‐term cardiac, metabolic, and pulmonary deficiencies.60 Additionally, maternal inflammation in mid‐pregnancy changes fetal neurodevelopment resulting in long‐term behavioral deficits may persist into adulthood.61
4.4. Maternal homeostasis (Noise pollution and circadian rhythm disruption)
Homeostasis may become challenged under psychological and/or physiological stressors, where vessels need to undergo changes to restore equilibrium. The microvasculature plays a significant role in buffering fluctuations in blood pressure and blood flow to maintain a dynamic equilibrium. It has been hypothesized that stressors of ambient noise and sleep deprivation have adverse effects on human vascular health.62, 63 Noise pollution can cause high‐stress levels, sleep disturbances, among other harmful effects. Nighttime noise in particular has been observed to increase levels of stress hormones and vascular oxidative stress, thereby contributing to hypertension and diabetes.63 Interestingly, one study reported impaired endothelial function in patients with or at risk of cardiovascular disease that appeared to be independent from annoyance or attitude toward nighttime aircraft noise.64 Noise‐induced vascular damage appears to be mediated by activation of NADPH oxidase, which uncouples eNOS and activates inflammatory cells.63 Sleep disorders in addition are associated with metabolic dysfunction and increased inflammation, which also contribute to endothelial cell dysfunction.65 One group has identified that while sexually mature female mice are more susceptible to chronic stress, they exhibit a vasoprotective phenotype, wherein arteriolar reactivity is blunted, but not as severely as the male counterparts.66 Unfortunately, there is a severe lack of research in this area; however, available studies do implicate these environmental stressors in microvascular dysfunction and a reduced ability to achieve and maintain homeostasis.
Within the context of pregnancy, there is epidemiological evidence showing an association with noise pollution and gestational hypertension, low birthweight, and congenital malformations.67, 68 One mouse study evaluating circadian rhythm disruption on pregnancy outcomes found no significant impact on gestation length, litter size, and birthweight, suggesting there may be maternal compensatory mechanisms to maintain fetal growth during sleep disturbances.69 Further, non‐pregnant female mice following an 8‐week unpredictable chronic mild stress protocol demonstrate impaired arteriolar reactivity66; while the estrous status of these mice is unknown, arteriolar dysfunction at crucial stages may affect trophoblast invasion and adequate placentation. Given the paucity of research evaluating noise, sleep disturbance, and uteroplacental function, it is plausible that the increase in oxidative stress as a result of inflammation during pregnancy lends to uteroplacental microvascular dysfunction. In turn, it is plausible that these perturbations could increase susceptibility to the development of preeclampsia, IUGR, and other adverse fetal outcomes to ensue as a result. Further microvascular research in this space is urgently needed.
4.5. Environmental exposure
Accumulating evidence implicates environmental exposures to microvascular dysfunction and disease. To limit the overwhelming possibilities in this field, we will limit the scope in this review to nicotine, heavy metals as found in polluted drinking water, and air pollution/particulate matter. Nicotine is well known to have deleterious effects such as vasoconstriction and increased blood pressure, overall perturbing microvascular patency.70 Heavy metals such as cadmium alter microvascular permeability by disturbing junctional complexes that join adjacent endothelial cells.71 Additionally, the particulate matter fraction of air pollution (of a diameter of 2.5 μm and smaller; PM2.5) has been shown to escape the lung and reach distal organs through blood circulation.72 Proposed mechanisms for exposure to PM2.5 include high oxidative stress, hypercoagulability, and inflammation that lend to cardiovascular outcomes.73
Smoking has also been linked to vasoconstriction of uteroplacental arteries, thereby reducing maternal blood flow into the intervillous space.74 Further, nicotine increases free radicals within the placenta and exposes maternal and fetal systems to genetic and cellular oxidative damage.75 There are scarce data describing the effects other heavy metals on uteroplacental vasculature. However, the unifying factor is the ability for these metals to generate reactive oxygen and nitrogen species. With evidence that these metals occupy and traverse the placenta, local oxidative stress may cause damage to trophoblastic and maternal endothelial cells, overall contributing to reduced placental function.76 Exposure to air pollution during gestation changes the morphology of multiple placental compartments including maternal spiral arteries, fetal capillaries, and the interface of exchange. Moreover, these changes are found in association with low birthweights for exposed fetuses.77 Engineered nanomaterials have been primarily used as a surrogate for air pollution; however, more recently given an increase in manufacturing, biomedical applications, and nanotechnology, there is risk of exposure to the pregnant population.78, 79, 80 Maternal inhalation of ENM led to increased uterine vascular tone, smaller active diameter, and inappropriate response to an increase in luminal shear stress.81 Even a single exposure, 24 hours prior to assessment, caused significant impairment to endothelium‐dependent dilation during late‐stage pregnancy, indicating a reduced ability to react to vasodilatory mediators and limit maternal‐fetal exchange.82
Environmental contaminants have the potential to directly and indirectly affect the placenta and fetus. Limited data show that fetal systems may be susceptible to maternal inhalation of particulate matter from direct translocation, as well as secondary inflammatory processes. Maternal smoking and nicotine lead to impaired fertility, type 2 diabetes, obesity, hypertension, neurobehavioral deficits, and respiratory dysfunction in the offspring.83 A large prospective study observed as smoking increased, placentas enlarged, and developed microscopic lesions typical of under‐perfusion from the uterine vasculature, which leads to low birthweight after maternal exposure.83 Heavy metal exposure leads to a myriad of fetal effects, from low birthweight to death due to toxicity.85 Heavy metals (mercury, lead, cadmium) are also well known to cross the placental barrier and cause both direct and indirect damage to the fetus and cause IUGR and perturbed neurodevelopment.86, 87, 88 Maternal exposure to air pollution results in abolished fetal endothelium‐dependent reactivity which may predispose offspring to a myriad of adult diseases.77, 81 Further, in our model, cardiac dysfunction is evident in surviving offspring including reduced coronary function, epicardial microvascular dysfunction, diminished cardiomyocyte contractility, and decreased mitochondrial energetics culminating in impaired metabolic function of the heart in adult offspring.89, 90, 91 This hypoperfusion along with toxicant accumulation in the placenta showed long‐lasting neurodevelopmental impairments and low birthweight for the offspring. A robust understanding of fetal outcomes after various gestational exposures cause uteroplacental microvessel dysfunction has yet to be established.
5. CONCLUSION
Placental trophoblast invasion, remodeling of preplacental spiral arterioles, and fetal microvascular endothelial function are key for a successful pregnancy. The joining of maternal and fetal circulations is one of the most acutely demanding and complex processes set up to meet fetal nutritional needs. Uterine spiral arterioles are remodeled by embryonic cells to establish large vessels with low resistance. The importance of successful uterine microvascular remodeling is underscored by its failure resulting in pathologies which impact fetal development and impair fetal growth.
The effects of disease and environmental conditions on cardiovascular form and function have become increasingly understood and emphasized. Uterine microvasculature exhibits a rare example of adult cyclic angiogenesis where extensive remodeling prepares for optimal invasion and uteroplacental perfusion. This complex process is a critical set up for normal pregnancy and fetal development, with major pathological consequences if failed. The “Barker Hypothesis” suggests that fetal development within a hostile environment may underpin susceptibility to adult disease. While the available amount of literature is modest, internal and external variables such as maternal hypertension, obesity, diabetes, inflammation, or disruption of homeostasis derived from stressors or environmental exposure may impede the ability to distribute blood flow to the fetal compartment. Further research in this area is urgently required to better understand the mechanisms of uterine microvascular disease creating a hostile gestational environment and the adult health risks involved for the offspring.
CONFLICT OF INTEREST
The authors report no conflict of interest.
ACKNOWLEDGMENTS
We thank Jane Salmon for her assistance in the preparation of Figure 1 and Dr. Sara Fournier for her critical review of the manuscript. This work was supported by the National Institute of Environmental Health Sciences (R00‐ES024783), Rutgers Center for Environmental Exposures and Disease (P30‐ES005022), and Rutgers Joint Graduate Program in Toxicology (T32‐ES007148).
D'Errico JN, Stapleton PA. Developmental onset of cardiovascular disease—Could the proof be in the placenta?. Microcirculation. 2019;26:e12526 10.1111/micc.12526
REFERENCES
- 1. Barker DJ. The fetal and infant origins of adult disease. BMJ. 1990;301:1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barker DJ. Fetal origins of cardiovascular disease. Ann Med. 1999;31(Suppl 1):3‐6. [PubMed] [Google Scholar]
- 3. Barker DJ, Eriksson JG, Forsen T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235‐1239. [DOI] [PubMed] [Google Scholar]
- 4. Simmons RA. Developmental origins of adult disease. Pediatr Clin North Am. 2009;56:449‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thamotharan M, Garg M, Oak S, et al. Transgenerational inheritance of the insulin‐resistant phenotype in embryo‐transferred intrauterine growth‐restricted adult female rat offspring. Am J Physiol Endocrinol Metab. 2007;292:E1270‐1279. [DOI] [PubMed] [Google Scholar]
- 6. Osol G, Moore LG. Maternal uterine vascular remodeling during pregnancy. Microcirculation. 2014;21:38‐47. [DOI] [PubMed] [Google Scholar]
- 7. Demir R, Yaba A, Huppertz B. Vasculogenesis and angiogenesis in the endometrium during menstrual cycle and implantation. Acta Histochem. 2010;112:203‐214. [DOI] [PubMed] [Google Scholar]
- 8. Losordo DW, Isner JM. Estrogen and angiogenesis: a review. Arterioscler Thromb Vasc Biol. 2001;21:6‐12. [DOI] [PubMed] [Google Scholar]
- 9. Bonagura TW, Pepe GJ, Enders AC, Albrecht ED. Suppression of extravillous trophoblast vascular endothelial growth factor expression and uterine spiral artery invasion by estrogen during early baboon pregnancy. Endocrinology. 2008;149:5078‐5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tessier DR, Yockell‐Lelievre J, Gruslin A. Uterine spiral artery remodeling: the role of uterine natural killer cells and extravillous trophoblasts in normal and high‐risk human pregnancies. Am J Reprod Immunol. 2015;74:1‐11. [DOI] [PubMed] [Google Scholar]
- 11. Wallace AE, Fraser R, Cartwright JE. Extravillous trophoblast and decidual natural killer cells: a remodelling partnership. Hum Reprod Update. 2012;18:458‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Damsky CH, Fisher SJ. Trophoblast pseudo‐vasculogenesis: faking it with endothelial adhesion receptors. Curr Opin Cell Biol. 1998;10:660‐666. [DOI] [PubMed] [Google Scholar]
- 13. Robertson WB. Uteroplacental vasculature. J Clin Pathol. 1976;10:9‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pijnenborg R, Vercruysse L, Brosens I. Deep placentation. Best Pract Res Clin Obstet Gynaecol. 2011;25:273‐285. [DOI] [PubMed] [Google Scholar]
- 15. Furuya M, Ishida J, Aoki I, Fukamizu A. Pathophysiology of placentation abnormalities in pregnancy‐induced hypertension. Vasc Health Risk Manag. 2008;4:1301‐1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mathew H, Castracane VD, Mantzoros C. Adipose tissue and reproductive health. Metabolism. 2018;86:18‐32. [DOI] [PubMed] [Google Scholar]
- 17. Mori M, Bogdan A, Balassa T, Csabai T, Szekeres‐Bartho J. The decidua—the maternal bed embracing the embryo—maintains the pregnancy. Semin Immunopathol. 2016;38:635‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Espinoza J, Romero R, Mee Kim Y, et al. Normal and abnormal transformation of the spiral arteries during pregnancy. J Perinat Med. 2006;34:447‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Browne VA, Julian CG, Toledo‐Jaldin L, Cioffi‐Ragan D, Vargas E, Moore LG. Uterine artery blood flow, fetal hypoxia and fetal growth. Philos Trans R Soc Lond B Biol Sci. 2015;370:20140068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brosens JJ, Pijnenborg R, Brosens IA. The myometrial junctional zone spiral arteries in normal and abnormal pregnancies: a review of the literature. Am J Obstet Gynecol. 2002;187:1416‐1423. [DOI] [PubMed] [Google Scholar]
- 21. Brosens I, Robertson WB, Dixon HG. The physiological response of the vessels of the placental bed to normal pregnancy. J Pathol Bacteriol. 1967;93:569‐579. [DOI] [PubMed] [Google Scholar]
- 22. Osol G, Barron C, Mandala M. Uterine distension differentially affects remodelling and distensibility of the uterine vasculature in non‐pregnant rats. Reprod Fertil Dev. 2012;24:835‐842. [DOI] [PubMed] [Google Scholar]
- 23. Ko NL, John L, Gelinne A, Mandala M, Osol G. Venoarterial communication mediates arterial wall shear stress‐induced maternal uterine vascular remodeling during pregnancy. Am J Physiol Heart Circ Physiol. 2018;315(3):H709‐H717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Osol G, Barron C, Gokina N, Mandala M. Inhibition of nitric oxide synthases abrogates pregnancy‐induced uterine vascular expansive remodeling. J Vasc Res. 2009;46:478‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fu Q. Hemodynamic and electrocardiographic aspects of uncomplicated singleton pregnancy. Adv Exp Med Biol. 2018;1065:413‐431. [DOI] [PubMed] [Google Scholar]
- 26. Mandala M, Osol G. Physiological remodelling of the maternal uterine circulation during pregnancy. Basic Clin Pharmacol Toxicol. 2012;110:12‐18. [DOI] [PubMed] [Google Scholar]
- 27. Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology (Bethesda, Md). 2009;24:58‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Y, Zhao S. Vascular Biology of the Placentaedn. San Rafael, CA: Morcan and Claypool Life Sciences; 2010. [PubMed] [Google Scholar]
- 29. Bulletti C, Jasonni VM, Lubicz S, Flamigni C, Gurpide E. Extracorporeal perfusion of the human uterus. Am J Obstet Gynecol. 1986;154:683‐688. [DOI] [PubMed] [Google Scholar]
- 30. Weiss G, Sundl M, Glasner A, Huppertz B, Moser G. The trophoblast plug during early pregnancy: a deeper insight. Histochem Cell Biol. 2016;146:749‐756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leitschuh M, Chobanian A. Vascular changes in hypertension. Med Clin North Am. 1987;71:827‐841. [DOI] [PubMed] [Google Scholar]
- 32. Kocemba J, Kawecka‐Jaszcz K, Gryglewska B, Grodzicki T. Isolated systolic hypertension: pathophysiology, consequences and therapeutic benefits. J Hum Hypertens. 1998;12:621‐626. [DOI] [PubMed] [Google Scholar]
- 33. Sánchez‐Aranguren LC, Prada CE, Riaño‐Medina CE, Lopez M. Endothelial dysfunction and preeclampsia: role of oxidative stress. Front Physiol. 2014;5:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fisher SJ. Why is placentation abnormal in preeclampsia? Am J Obstet Gynecol. 2015;213:S115‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eskenazi B, Fenster L, Sidney S. A multivariate analysis of risk factors for preeclampsia. JAMA. 1991;266:237‐241. [PubMed] [Google Scholar]
- 36. Romeo GR, Lee J, Shoelson SE. Metabolic syndrome, insulin resistance, and roles of inflammation–mechanisms and therapeutic targets. Arterioscler. Thromb. Vasc. Biol. 2012;32:1771‐1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Albu AR, Anca AF, Horhoianu VV, Horhoianu IA. Predictive factors for intrauterine growth restriction. J Med Life. 2014;7:165‐171. [PMC free article] [PubMed] [Google Scholar]
- 38. Aguilar M, Bhuket T, Torres S, Liu B, Wong RJ. Prevalence of the metabolic syndrome in the United States, 2003‐2012. JAMA. 2015;313:1973‐1974. [DOI] [PubMed] [Google Scholar]
- 39. Vykoukal D, Davies MG. Vascular biology of metabolic syndrome. J Vasc Surg. 2011;54:819‐831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goldstein BJ, Scalia R. Adiponectin: a novel adipokine linking adipocytes and vascular function. J Clin Endocrinol Metabol. 2004;89:2563‐2568. [DOI] [PubMed] [Google Scholar]
- 41. Knudson JD, Dick GM, Tune JD. Adipokines and coronary vasomotor dysfunction. Exp Biol Med. 2007;232:727‐736. [PubMed] [Google Scholar]
- 42. Ullian ME. The role of corticosteroids in the regulation of vascular tone. Cardiovasc Res. 1999;41:55‐64. [DOI] [PubMed] [Google Scholar]
- 43. Chu SY, Callaghan WM, Kim SY, et al. Maternal obesity and risk of gestational diabetes mellitus. Diabetes Care. 2007;30:2070‐2076. [DOI] [PubMed] [Google Scholar]
- 44. Friedman JE. Obesity and gestational diabetes mellitus pathways for programming in mouse, monkey, and man—where do we go next? The 2014 Norbert Freinkel Award Lecture. Diabetes Care. 2015;38:1402‐1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vambergue A, Fajardy I. Consequences of gestational and pregestational diabetes on placental function and birth weight. World J Diabetes. 2011;2:196‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pontes IEA, Afra KF, Silva JR, Borges PSN, Clough GF, Alves JGB. Microvascular reactivity in women with gestational diabetes mellitus studied during pregnancy. Diabetol Metab Syndr. 2015;7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Antoniotti GS, Coughlan M, Salamonsen LA, Evans J. Obesity associated advanced glycation end products within the human uterine cavity adversely impact endometrial function and embryo implantation competence. Hum Reprod. 2018;33:654‐665. [DOI] [PubMed] [Google Scholar]
- 48. Leiva A, Pardo F, Ramírez MA, Farías M, Casanello P, Sobrevia L. Fetoplacental vascular endothelial dysfunction as an early phenomenon in the programming of human adult diseases in subjects born from gestational diabetes mellitus or obesity in pregnancy. Exp Diabetes Res. 2011;2011:349286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115:e290‐296. [DOI] [PubMed] [Google Scholar]
- 50. Pober JS, Sessa WC. Inflammation and the blood microvascular system. Cold Spring Harbor Perspect Biol. 2015;7:a016345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829‐837, 837a‐d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marsden PA, Brenner BM. Transcriptional regulation of the endothelin‐1 gene by TNF‐alpha. Am J Physiol. 1992;262:C854‐861. [DOI] [PubMed] [Google Scholar]
- 53. Incalza MA, D'Oria R, Natalicchio A, Perrini S, Laviola L, Giorgino F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul Pharmacol. 2018;100:1‐19. [DOI] [PubMed] [Google Scholar]
- 54. PrabhuDas M, Bonney E, Caron K, et al. Immune mechanisms at the maternal‐fetal interface: perspectives and challenges. Nature Immunol. 2015;16:328‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cotechini T, Komisarenko M, Sperou A, Macdonald‐Goodfellow S, Adams MA, Graham CH. Inflammation in rat pregnancy inhibits spiral artery remodeling leading to fetal growth restriction and features of preeclampsia. J Exp Med. 2014;211:165‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Renaud SJ, Postovit LM, Macdonald‐Goodfellow SK, McDonald GT, Caldwell JD, Graham CH. Activated macrophages inhibit human cytotrophoblast invasiveness in vitro. Biol Reprod. 2005;73:237‐243. [DOI] [PubMed] [Google Scholar]
- 57. Todt JC, Yang Y, Lei J, et al. Effects of tumor necrosis factor‐alpha on human trophoblast cell adhesion and motility. Am J Reprod Immunol. 1996;36:65‐71. [DOI] [PubMed] [Google Scholar]
- 58. Tossetta G, Paolinelli F, Avellini C, et al. IL‐1beta and TGF‐beta weaken the placental barrier through destruction of tight junctions: an in vivo and in vitro study. Placenta. 2014;35:509‐516. [DOI] [PubMed] [Google Scholar]
- 59. Wilson R, Jenkins C, Miller H, et al. Abnormal cytokine levels in non‐pregnant women with a history of recurrent miscarriage. Eur J Obstet Gynecol Reprod Biol. 2004;115:51‐54. [DOI] [PubMed] [Google Scholar]
- 60. Velten M, Britt RD, Heyob KM, et al. Prenatal inflammation exacerbates hyperoxia‐induced functional and structural changes in adult mice. Am J Physiol Regul Integr Comp Physiol. 2012;303:R279‐R290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Goeden N, Velasquez J, Arnold KA, et al. Maternal inflammation disrupts fetal neurodevelopment via increased placental output of serotonin to the fetal brain. J Neurosci. 2016;36:6041‐6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abrams RM. Sleep deprivation. Obstet Gynecol Clin North Am. 2015;42:493‐506. [DOI] [PubMed] [Google Scholar]
- 63. Munzel T, Schmidt FP, Steven S, Herzog J, Daiber A, Sorensen M. Environmental noise and the cardiovascular system. J Am Coll Cardiol. 2018;71:688‐697. [DOI] [PubMed] [Google Scholar]
- 64. Schmidt F, Kolle K, Kreuder K, et al. Nighttime aircraft noise impairs endothelial function and increases blood pressure in patients with or at high risk for coronary artery disease. Clin Res Cardiol. 2015;104:23‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Qiu C, Sanchez SE, Gelaye B, Enquobahrie DA, Ananth CV, Williams MA. Maternal sleep duration and complaints of vital exhaustion during pregnancy is associated with placental abruption. J Matern Fetal Neonatal Med. 2015;28:350‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stanley SC, Brooks SD, Butcher JT, d'Audiffret AC, Frisbee SJ, Frisbee JC. Protective effect of sex on chronic stress‐ and depressive behavior‐induced vascular dysfunction in BALB/cJ mice. J Appl Physiol. 2014;117:959‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dzhambov AM, Dimitrova DD, Dimitrakova ED. Noise exposure during pregnancy, birth outcomes and fetal development: meta‐analyses using quality effects model. Folia Med. 2014;56:204‐214. [DOI] [PubMed] [Google Scholar]
- 68. Yinon D, Lowenstein L, Suraya S, et al. Pre‐eclampsia is associated with sleep‐disordered breathing and endothelial dysfunction. Eur Respir J. 2006;27:328‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Varcoe TJ, Voultsios A, Gatford KL, Kennaway DJ. The impact of prenatal circadian rhythm disruption on pregnancy outcomes and long‐term metabolic health of mice progeny. Chronobiol Int. 2016;33:1171‐1181. [DOI] [PubMed] [Google Scholar]
- 70. Benowitz NL, Burbank AD. Cardiovascular toxicity of nicotine: implications for electronic cigarette use. Trends Cardiovasc Med. 2016;26:515‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Prozialeck WC, Edwards JR, Nebert DW, Woods JM, Barchowsky A, Atchison WD. The vascular system as a target of metal toxicity. Toxicol Sci. 2008;102:207‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brook RD, Rajagopalan S, Pope CA 3rd, et al. Particulate matter air pollution and cardiovascular disease: an update to the scientific statement from the American Heart Association. Circulation. 2010;121:2331‐2378. [DOI] [PubMed] [Google Scholar]
- 73. Brook Robert D. Cardiovascular effects of air pollution. Clin Sci. 2008;115:175. [DOI] [PubMed] [Google Scholar]
- 74. Philipp K, Pateisky N, Endler M. Effects of smoking on uteroplacental blood flow. Gynecol Obstet Invest. 1984;17:179‐182. [DOI] [PubMed] [Google Scholar]
- 75. Mund M, Louwen F, Klingelhoefer D, Gerber A. Smoking and pregnancy — a review on the first major environmental risk factor of the unborn. Int J Environ Res Public Health. 2013;10:6485‐6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr Med Chem. 2005;12:1161‐1208. [DOI] [PubMed] [Google Scholar]
- 77. Backes CH, Nelin T, Gorr MW, Wold LE. Early life exposure to air pollution: how bad is it? Toxicol Lett. 2013;216:47‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fournier SB, D'Errico JN, Stapleton PA. Engineered nanomaterial applications in perinatal therapeutics. Pharmacol Res. 2018;130:36‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stapleton PA. Gestational nanomaterial exposures: microvascular implications during pregnancy, fetal development and adulthood. J Physiol. 2016;594:2161‐2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Stapleton PA, Nurkiewicz TR. Maternal nanomaterial exposure: a double threat to maternal uterine health and fetal development? Nanomedicine (Lond). 2014;9:929‐931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Stapleton PA, Minarchick VC, Yi J, Engels K, McBride CR, Nurkiewicz TR. Maternal engineered nanomaterial exposure and fetal microvascular function: does the Barker hypothesis apply? Am J Obstet Gynecol. 2013;209(227):e221‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Stapleton PA, McBride CR, Yi J, Abukabda AB, Nurkiewicz TR. Estrous cycle‐dependent modulation of in vivo microvascular dysfunction after nanomaterial inhalation. Reprod Toxicol. 2018;78:20‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bruin JE, Gerstein HC, Holloway AC. Long‐term consequences of fetal and neonatal nicotine exposure: a critical review. Toxicol Sci. 2010;116:364‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Naeye RL. Effects of maternal cigarette smoking on the fetus and placenta. Br J Obstet Gynaecol. 1978;85:732‐737. [DOI] [PubMed] [Google Scholar]
- 85. Wai KM, Mar O, Kosaka S, Umemura M, Watanabe C. Prenatal heavy metal exposure and adverse birth outcomes in myanmar: a birth‐cohort study. Int J Environ Res Public Health. 2017;14:1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bose‐O'Reilly S, McCarty KM, Steckling N, Lettmeier B. Mercury exposure and children's health. Curr Probl Pediatr Adolesc Health Care. 2010;40:186‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Potula V, Kaye W. Report from the CDC. Is lead exposure a risk factor for bone loss? J Women Health. 2005;14:461‐464. [DOI] [PubMed] [Google Scholar]
- 88. Stasenko S, Bradford EM, Piasek M, et al. Metals in human placenta: focus on the effects of cadmium on steroid hormones and leptin. J Appl Toxicol. 2010;30:242‐253. [DOI] [PubMed] [Google Scholar]
- 89. Hathaway QA, Nichols CE, Shepherd DL, et al. Maternal‐engineered nanomaterial exposure disrupts progeny cardiac function and bioenergetics. Am J Physiol Heart Circ Physiol. 2017;312:H446‐H458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Stapleton PA, Hathaway QA, Nichols CE, et al. Maternal engineered nanomaterial inhalation during gestation alters the fetal transcriptome. Part Fibre Toxicol. 2018;15:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Stapleton PA, Nichols CE, Yi J, et al. Microvascular and mitochondrial dysfunction in the female F1 generation after gestational TiO2 nanoparticle exposure. Nanotoxicology. 2015;9:941‐951. [DOI] [PMC free article] [PubMed] [Google Scholar]