

Abstract
Background and purpose
In models of neuropathic pain, inhibition of HCN1 is anti-hyperalgesic. 2,6-di-iso-propyl phenol (propofol) and its non-anesthetic congener, 2,6-di-tert-butyl phenol, inhibit HCN1 channels by stabilizing closed state(s).
Experimental approach
Using in vitro electrophysiology and kinetic modeling, we systematically explore the contribution of ligand architecture to alkylphenol-channel coupling.
Key results
When corrected for changes in hydrophobicity (and propensity for intra-membrane partitioning), the decrease in potency upon 1-position substitution (NCO~OH >> SH >>> F) mirrors the ligands’ H-bond acceptor (NCO > OH > SH >>> F) but not donor profile (OH > SH >>> NCO~F). H-bond elimination (OH to F) corresponds to a ΔΔG of ~4.5 kCal mol−1 loss of potency with little or no disruption of efficacy. Substitution of compact alkyl groups (iso-propyl, tert-butyl) with shorter (ethyl, methyl) or more extended (sec-butyl) adducts disrupts both potency and efficacy. Ring saturation (with the obligate loss of both planarity and π electrons) primarily disrupts efficacy.
Conclusions and implications
A hydrophobicity-independent decrement in potency at higher volumes suggests the alkylbenzene site has a volume of ≥ 800 Å3. Within this, a relatively static (with respect to ligand) H-bond donor contributes to initial binding with little involvement in generation of coupling energy. The influence of π electrons/ring planarity and alkyl adducts on efficacy reveals these aspects of the ligand present towards a face of the channel that undergoes structural changes during opening. The site’s characteristics suggest it is “druggable”; introduction of other adducts on the ring may generate higher potency inverse agonists.
Keywords: HCN channels, inverse agonists, kinetic modeling, Neuropathic pain, anti-hyperalgesia, propofol, alkylphenol, cyclohexanol, phenyl-isocyanate, phenyl-thiol, phenyl-fluorine, 2-fluoro-1, 3-di-isopropylbenzene
Graphical Abstract
1. Introduction
Neuropathic pain is pain caused by a lesion of, or damage to, the somatosensory nervous system [1, 2]. Neuropathic pain is a pathologic feature of numerous conditions, including postherpetic neuralgia, trigeminal neuralgia, sensory (painful) radiculopathy, painful diabetic neuropathy, peripheral nerve injury, stroke, and treatment with antineoplastics [2–4]. Globally, it is estimated that 7– 10% of the population lives with chronic pain with neuropathic features [2–6]. Cardinal symptoms of neuropathic pain are spontaneous pain, abnormal response to non-painful (allodynia) or painful (hyperalgesia) stimuli, dysesthesia (unpleasant/strange sensation – “tingling” “pins and needles”), and enhanced thermal sensitivity; numerous pharmacologic approaches to relieving neuropathic pain have been proposed [3], but their efficacy is limited, resulting in a large unmet need with respect to providing meaningful pain relief [7].
Recent work suggests that neuropathic pain might better be understood at the mechanistic level as a function of sensory profiling rather than disease etiology [8]. Hyperexcitability of primary afferent neurons is thought to be an early causal factor leading to the development and maintenance of peripheral neuropathic pain [1, 9]. Numerous ion channel families contribute to this hyperexcitability, including the hyperpolarization-activated, cyclic nucleotide-gated (HCN) channel family [10]. In animal models of neuropathic pain, non-selective HCN block with either ZD7288 [11–14] or the clinically-available drug ivabradine [15–17] results in significant anti-hyperalgesia. HCN1 expression is upregulated in multiple animal models of neuropathic pain including those wherein the initiating injury was mechanical damage [11–13], chemotherapy administration [16–19], or diabetes [20]. Anti-hyperalgesic activity is retained in HCN1-selective molecules [21–24], suggesting that HCN1-selective blockade has therapeutic potential.
Previously we demonstrated that the intravenous general anesthetic 2,6-di-iso-propyl phenol (propofol, DIP-P) and its closely related, non-anesthetic congener 2,6-di-tert-butyl phenol (DTB-P), were HCN1-selective inhibitors of channel gating [22, 25]. Such inhibition resulted from ligand-mediated stabilization of channel closed state(s), which was solely dependent upon interactions with the membrane embedded channel core [25]. Both DIP-P and DTB-P provided mechanical and thermal anti-hyperalgesia in mouse peripheral nerve injury neuropathic pain models [22, 23].
In silico modelling and crystallography demonstrate alkylphenols dock in cavities in GABAA receptors (GABAA-Rs) [26, 27] where they likely displace either native acyl chains [28] or water [29–31]. Crystallography further reveals acyl chains associate with, and intercalate between, the voltage sensor and pore domains of Kv channels, indicating specific packing at the lipid-channel interface is thermodynamically favored [32]. Given the structural homology between HCN1 and Kv channels [10, 33], we posit that specific acyl (and water) filled cavities are also present on HCN channels. Consequently, the HCN1-alkylphenol association is likely similar in nature, but likely unique in detail, to the alkylphenol interaction with GABAA-Rs, and that the alkylphenol:HCN1 association can be defined and optimized.
In the present study, we demonstrate that H-bonding, ring planarity and/or π electron interactions and coupling to ring adducts at the 2 and 6 positions all contribute to HCN1 inhibition at a site with a volume ≥800 Å3. Such characteristics are consistent with alkylphenols acting at a defined site within the membrane-embedded core of HCN1 channels. Examination of HCN sequences in conjunction with the cryo-EM structure of HCN1, shows multiple sites of divergence within this region. Together, the data suggest that alkylphenol-mediated inhibition of HCN1 channel opening occurs via a defined and “druggable” site which, if correct, may open the way to generation of a novel class of high-potency, non-opioid, therapeutics for neuropathic pain.
2. Materials and Methods
2.1. Molecular Biology
Methods for heterologous expression were as previously described [22]. Briefly, cDNA encoding murine HCN1 channels were sub-cloned into a pGH19 vector and amplified in STBL2 cells (Invitrogen Corporation, Grand Island, NY, USA). cRNA was transcribed from NheI linearized DNA using T7 RNA polymerase (Message Machine; Ambion, Houston, TX, USA).
2.2. Electrophysiology
Recordings were made from Xenopus oocytes (Ecocyte, Austin, TX) 1–3 days after cRNA injection (1–50 ng/oocyte). Cells were maintained in L-15 media without Ficoll (Specialty Media, Phillipsburg, NJ) at 16 °C until use.
Two-electrode voltage clamp (TEVC) data were acquired using a Warner Instruments (Hamden, CT) OC-725C amplifier, filtered at 1 kHz (Frequency Devices Bessel model 902, Ottawa, IL) digitized at 2 kHz by an ITC-18 interface (Instrutech Corporation, Port Washington, NY) and recorded with Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany).
Microelectrodes were fabricated from 1B120-F4 borosilicate glass (World Precision Instruments, Sarasota, FL) and had resistances of 0.1 – 0.5 MΩ (I passing) and 1 – 4 MΩ (V sensing) when filled with 3 M KCl. An active virtual ground was used to clamp the bath. Ag-AgCl ground wire(s) were connected to the bath solution by 3 M KCl-2% agar salt bridges placed downstream of, but close to, the oocyte. Oocytes were bathed in a recording solution of (in mM): 107 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES-free acid pH 7.4 (NaOH). Recordings were obtained at room temperature. In all cases, voltages are reported as the command potentials. The observed potential was within 1% of the command.
2.3. Paradigms and analysis
The holding potential was −30 mV. Tail currents were recorded at 0 mV. Steady-state activation properties were determined from tail current activation curves (see Riegelhaupt et al. for a discussion on the criteria adopted to eliminate collection and analysis bias - [34]). Fits of these relations with the Boltzmann function (equation 1 where A1 is the current offset and A2 the maximal amplitude and V the step voltage) yielded the V1/2 (the activation mid-point) and the slope factor (equal to RT/ZF where R, T and F have their usual meaning and Z is an approximation of the gating charge associated with activation).
| 1. |
Gating of HCN channels is slowed in the presence of in the presence of inverse agonists. To ensure the determined Boltzmann parameters reflect thermodynamic behavior free of kinetic contamination, we routinely determined the gating parameters using step durations of 3 s and 5 s. In no case was there a systematic difference in fit parameters. This observation is consistent with activation being equilibrated at all voltages under all conditions tested. The two determinations were averaged and treated as a single measurement. Data reported as shifts in the V1/2 (ΔV1/2) were determined with respect to vehicle control recordings performed before and after the experimental determinations. Standard errors around vehicle populations were less than 1 mV.
Concentration response data were fit with the Hill equation (equation 2), where ΔV1/2 is as defined above, ΔV1/2 max the maximal change as determined from the fit, IC50 the concentration of ligand required for half maximal inhibition of gating and h the Hill coefficient. In all fits, ΔV1/2 in the absence of ligand was constrained to zero. For several ligands, estimates of IC50 were determined assuming ΔV1/2 max and h were equal to those obtained for DTB-P (see thin green lines on Figure 3 and discussion thereof).
Figure 3: Inhibition by 2,6-di-alkyl phenyl derivatives reveals hydrogen bond potential, alkyl side chain identity and the presence or absence of π electrons (and/or ring planarity) all contribute to ligand association with HCN1 channels.

Panels show the shift in the V1/2 as a function of concentration of each of the indicated ligands. Thick blue lines are the fit of the Hill equation to the DTB-P data, reproduced to facilitate visual comparison. Solid thick green lines are fits of the Hill function to ligands (other than DTB-P) that clearly demonstrated full (DIP-P) or partial (DE-P, DIP-CH, DTB-CH – see text and Figure 5) inverse agonist behavior. The dashed green lines in the DIP-P and DTB-P panels are scaled representations of the Hill fits to DIP-CH and DTB-CH, respectively with the cyclohexanol fit lines scaled according to the ratio ΔV1/2 max Phenol / ΔV1/2 max Cyclohexanol with all other terms of the cyclohexanol fit unaltered. The thin green lines are fits of the Hill equation to ligands whose efficacy is poorly determined (DM-PT, DE-PIC, DE-CH, DIP-PT, DIP-PIC, DIP-PF, DSB-P) wherein the ΔV1/2 max and h were held equal to the DTB-P values, with only the IC50 allowed to optimize. The number of independent determinations in each panel were: (phenols) 14, 44, 64, 165, 122; (thiols) 67, 45; (isocyanates) 15, 45, 47; (fluoro) 61; (cyclohexanols) 14, 25, 100, 53, 19.
| 2. |
2.4. Modeling of alkylphenol and alkylcyclohexanol coupling to channel gating
To consider the mechanistic basis by which chemical modification(s) of the alkylphenol structure perturbs coupling of the ligands to HCN1, we examined models of the form shown schematically in Figure 1.
Figure 1: Alkylphenol and alkylcyclohexanol interaction with HCN1 channel gating modeled as, respectively, inverse agonist and partial inverse agonist coupling to channel opening.

CR, CA and O represent the closed-resting, closed-activated and open states; KV and L the equilibrium constants describing activation and opening; I and H are an inverse agonist and a partial inverse agonist; p and q represent the number of binding events of I and H; KC and KO represent the association constants for I and H to closed and open channels with reference to I and H denoted by the appropriate superscript. For simplicity, only activation and opening transitions between un-liganded states are shown but all CR-CA and CA-O transitions are permissible. When H and I associate with common site(s) the model contains 3 states per plane, 9 states in total (S=1) and 15 states per plane, 45 states in total (S=4). When H and I associate with distinct site(s) the model contains 4 states per plane, 12 states in total (S=1) and 25 states per plane, 75 states in total (S=4). Importantly, as the equilibrium constants are the only free parameters and the number of equilibrium constants is independent of the expansion of the number of states, each of these models should be equally well determined if they are equally able to describe the data.
Here, CR, CA and O represent the closed-resting, closed-activated and open states, of the channel. L is the voltage independent equilibrium constant that describes opening of activated channels. Activation of voltage sensors is described by the voltage-dependent equilibrium constant KV (equation 3) where KV0 is KV in the absence of an applied field.
| 3. |
I and H are full and partial inverse agonists, respectively. Consistent with previous work wherein we showed DIP-P was indifferent to the activation status of the voltage sensors [25], here we assume neither I nor H alter KV. Which is to say both I and H are considered to bind equally well (or poorly) to CR and CA. I binds tightly to CR and CA (as defined by the association constant KCI) but poorly to O (as defined by the association constant KOI) and thus acts as a full inverse agonist of opening. For the partial inverse agonist, H, the loss of efficacy must arise from proportionally weaker binding to C than to O when compared to binding of I. While it is critical to understand how such partial behavior arises, particularly when considering rational drug design, it is not possible to a priori intuit whether it arises due to a loss of function (a proportionally greater decrement in KC versus KO) or a gain of function (a proportionally greater improvement in KO versus KC).
Within this overall scheme, we examined two limiting conditions. Specifically, we assumed I and H bind to common site(s) or to distinct site(s). Here, the definition of common site(s) includes pocket(s) with over-lapping structures, occupancy of which are mutually-exclusive. In contrast, distinct sites are, de facto, non-overlapping. In the common site model, the sum of p and q cannot exceed S (where p and q represent the number of binding events of I and H, respectively, and S is the number of sites per channel) and the equation determining the change in the V1/2 (ΔV1/2) due to the presence of I and/or H is given by equation 4. In the distinct site model, p and q can both equal S and ΔV1/2 is given by equation 5.
| 4. |
| 5. |
In both equations, A = [H]*KOH, B = [I]*KOI, C = [H]*KCH and D = [I]*KCI. Note that in the presence of only H or I (and the absence of both) equations 4 and 5 become equivalent. It is the behavior of gating in the combined presence of H and I that distinguishes between these two mechanisms.
To allow us to simultaneously fit the behavior of iso-propyl and tert-butyl analogues, we defined two sets of equilibrium constants wherein KOH, KOI, KCH and KCI describe tert-butyl association and QOH, QOI, QCH and QCI describe iso-propyl association. Versions of equations 4 and 5 describing individual ligand effects and additivity were generated by substitution of the appropriate equilibrium constants (and corresponding ligand concentration terms). In simultaneous fits to the iso-propyl and tert-butyl data, all eight binding constants and L were free parameters. Importantly, equations 4 and 5 show that the degrees of freedom in the common and distinct site models are identical and that this is independent of S. As such, measures of the goodness of fit are directly comparable. As is evident from inspection of equations 4 and 5, in models where inhibitors do not discriminate between CR and CA, KV does not contribute to their influence on ΔV1/2.
We applied only three constraints. First, S (the number of sites per channel), was held to be either 1 or 4. While arbitrary, this is consistent with the tetrameric nature of HCN channels. An S of 1 is consistent with the site being at the four-fold axis of symmetry, that is, at a point of juxtaposition of all four subunits. An S of 4 is consistent with the sites lying within or between channel subunits and distant (with respect to ligand size) from the central axis. Second, the gating valence Z was held equal to 2.82. This value was determined from fits of the Boltzmann function to control GV relations. While not arbitrary, it is well understood that, in a complex system such as exists for HCN gating, the slope of such a fit is a secondary, and imprecise, measure that will likely underestimate the total charge moved [35, 36]. However, given that our three-state gating scheme is a clear simplification in and of itself ([37] and references therein) and in the model Z serves only as a proportionality term (equations 4 and 5), an error in the estimation of Z will have no impact on the qualitative integrity of our analysis (assuming Z is constant). Is an assumption of constancy of Z reasonable? At high concentrations, DIP-P and DTB-P both decrease the steepness of the gating relation (i.e., the Boltzmann slope term gets larger, meaning the estimated value of Z would get smaller – not shown). Would use of experimentally determined values of Z under each condition be better? We think not; while a change in the GV slope could imply Z has changed, it is highly improbable that the fundamental motions of the sensors (and of the net charge moved) is radically altered by the presence of an alkylbenzene. A more reasonable interpretation of changes in the activation slope is that this reflects altered co-operativity between subunits, an effect the Boltzmann equation does not account for. For these reasons, we consider use of a stable measure of charge moved to be the conservative assumption. Third, and finally, values of all equilibrium constants were constrained to be >0, a physically reasonable expectation.
To examine the secondary butyl analogue of DTB-P (2,6-di-sec-butyl phenol; DSB-P) we defined a third set of equilibrium constants, JO and JC. As responses to the DSB-P were only observed at high concentrations and were, therefore, relatively poorly determined, these data were not included in the simultaneous fit of iso-propyl and tert-butyl responses as per Figure 5. For the DSB-P fit, all parameters other than JO and JC were held equal to the values used and/or determined for and from the fits shown in Figure 5 and reported in Table 4.
Figure 5: Alkylphenols and alkylcyclohexanols associate with HCN1 channels via four common, radially-distributed, sites.

A,B. ΔV1/2 as a function of concentration of iso-propyl (A) and tert-butyl (B) phenols and cyclohexanols. Data are reproduced from Figure 3. Solid blue lines are simultaneous fits of the common site model (S=4) to the data in these two panels and to the data in panels C-E.
C-E. ΔV1/2 as a function of concentration of DIP-CH (C), DTB-CH (D) or DIP-P (E), in the presence of 3 μM DTB-P. The solid blue lines are as defined in panels A and B. The dashed blue lines are the predicted behavior of the distinct site model at S=4 using the association constants determined from the fit of the common site model. The green lines report the mean value of ΔV1/2 (solid) and SEM (dashed) elicited by 3 μM DTB-P in paired controls for each additivity condition. The number of independent determinations in C, D and E were 27, 80 and 33, respectively.
E. Association constants determined from the fits of the common site model with S=4 (as per A-E and reported in Table 4) transformed to free energy terms according to equation 6. The reported value of the opening equilibrium constant (L) is from this same fit.
Table 4:
Model determined association constants for phenol and cyclohexanol binding to HCN1 channels.
| Constant | DTB-P | DTB-CH | Fold Change |
|---|---|---|---|
| KC | 24.6 ± 4.5 | 4.6 ± 2.0 | 5.4 |
| KO | 5.5 ± 2.6 | 3.1 ± 1.5 | 1.8 |
| QC | 24.8 ± 4.4 | 6.5 ± 2.2 | 3.8 |
| Qo | 7.4 ± 2.1 | 4.2 ± 1.5 | 1.8 |
| JC | 0.4 ± 0.3 | no effect | -- |
| JO | 0.1 ± 0.2 | no effect | -- |
Constants and their errors have been divided by 104 for clarity.
Association constants determined from the fits of the models were transformed to free energy terms according to equation 6.
| 6. |
It is important to note that within both classes of models the maximal ΔV1/2 is limited to RT/ZF * Ln 1/1+L (equations 4 and 5) and, ipso facto, the maximal observed response of the most effective inverse agonist will strongly influence the estimate of L. This arises because the inverse agonists are stabilizing closing and, therefore, can only register an inhibitory voltage shift to the extent permitted by opposing the effect of a favorable opening transition (large L) that normally drags the activation transition forward. What does this mean with respect to the behavior expected within competition experiments? Intuitively, for a channel behaving according to the common site model, the main phenotype will be for the partial inverse agonist to have no effect in the face of an intermediate concentration of the full inverse agonist (or suppress that if high enough concentrations of the partial inverse agonist can be achieved and its maximal response is lower than that of the included concentration of full inverse agonist). In contrast, while the actions of partial and full inverse agonists acting via distinct sites will be (as expected) energetically additive, this will only be revealed as an additive effect on the ΔV1/2 at sub-maximal concentrations of the full inverse agonist. Together, these considerations reveal examination of ligands of unknown properties need to be considered in the face of a low concentration of DTB-P. We selected 3 μM.
Theoretically, an independent measure of L can be determined from the maximal open probability (PMAX). While conceptually straightforward, determining PMAX of HCN1 channels is difficult. First, the single channel conductance of HCN channels is very low, making direct analysis of single channel records problematic. Second, wild-type HCN1 channels cluster and gating is slow making noise analysis similarly challenging. We have previously shown that deletion of the N and C termini of HCN1 generates HCN1 channels that are amenable to non-stationary fluctuation analysis (NSFA; [25]). When determined in excised inside-out patches in high K+ recording conditions (to maximize the current amplitude), the PMAX of HCN1-ΔNvΔC is 0.88, which corresponds to a value of L of 7.3 (equation 7).
| 7. |
However, it should be noted that analysis of HCN1-ΔNvΔC in the excised patch configuration as used for NSFA will underestimate the likely value of L of wtHCN1 as the effect of at least three factors that stabilize gating (internal cAMP, internal 4,5-PIP2, and a physiological level of external Na+ and K+) are lost. This disruption is reflected in a 40 mV hyperpolarization of gating of HCN1 upon patch excision. If these positive regulators all acted via enhancing L (stabilizing opening), equation 7 reveals L would be 93-fold higher in intact cells than reported by NSFA in excised patches. Further, it is clear that while the effect of 4,5-PIP2 seems to involve, at least in part, a surface charge contribution, the positive effects of cAMP (5–7 mV right-shift in the V1/2 for HCN1) and physiological external Na+ and K+ concentrations (10–12 mV) do indeed arise from a stabilization of opening [38–41] (and our unpublished observations). Thus, the model determined value of L of 125 is completely consistent with independent experimental evidence wherein endogenous positive modulation of L accounts for 25 mV of the observed 40 mV difference between recording modes.
Equation 8 determines the value of L in an intact cell under TEVC (LT) as a function of the value of L determined from NSFA in excised inside out patches (LI) and V(T-I), the difference in V1/2 between these modes. Equation 8 assumes KV0 is unaltered by patch excision.
| 8. |
2.5. Physicochemical parameter estimates
We found determinations of ligand physicochemical parameters used in this study to be sensitive to the analysis engine used (not shown but compare values in Tables 1 and 2 to those of Krasowski et al. and Woll et al. [42, 43]). To provide a unified set of values, we redetermined chemical properties using a single engine, the Maestro 2018–2 package from Schrödinger. LigPrep was used to generate all possible states at a target pH of 7.4 ± 0.2. QikProp was used to calculate the reported properties of all generated states. Properties are reported in Tables 1 (phenyl derivatives) and 2 (cyclohexanol derivatives). LogP is log of the calculated octanol/water partition coefficient. Values for all ligands in the matrix are provided to facilitate cross-study comparisons.
Table 1:
Physicochemical properties of 2,6-di-alkyl substituted phenyls.
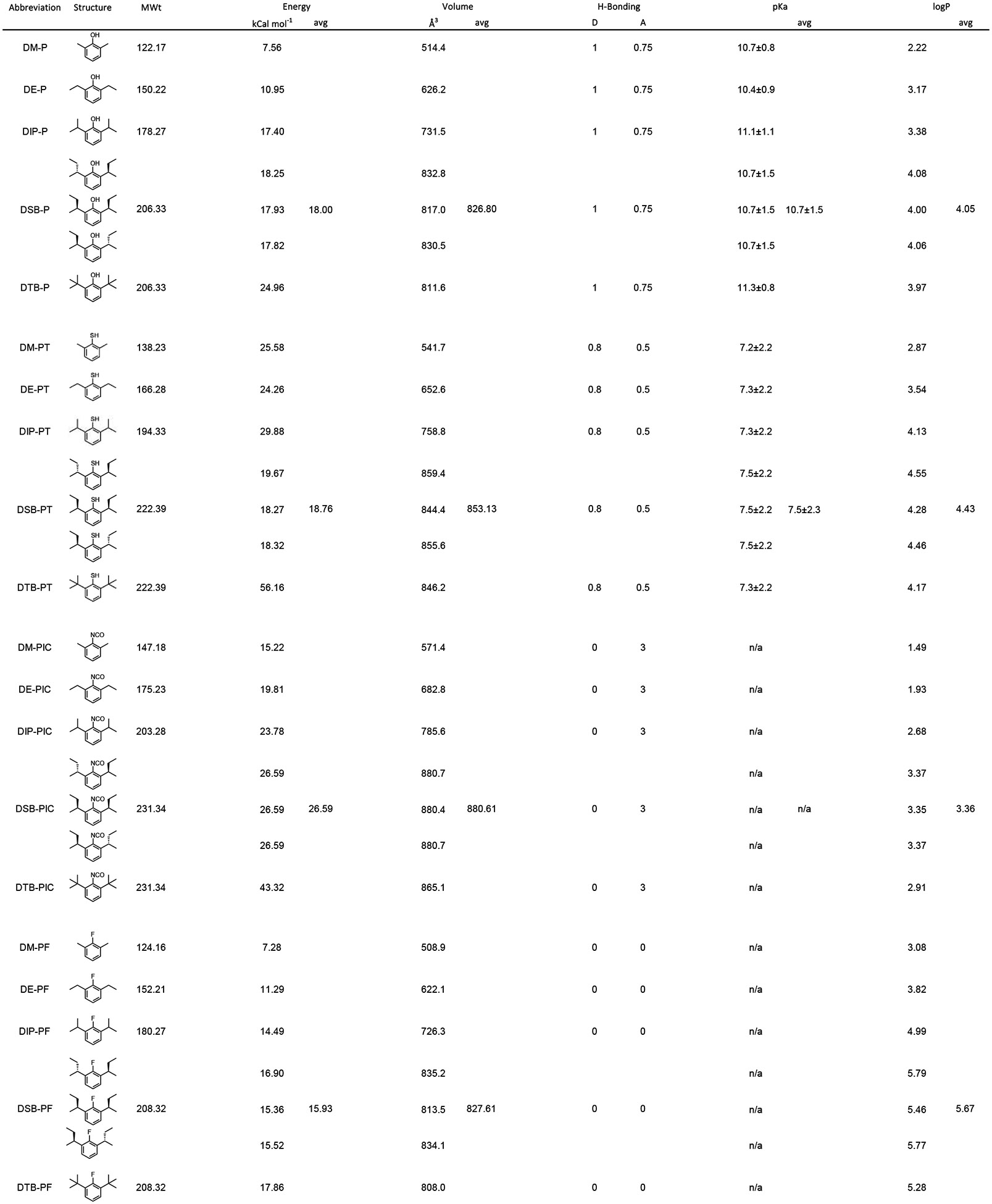 |
H-bonding: D donor, A Acceptor; logP: log of the calculated octanol/water partition coefficient
Table 2:
Physicochemical properties of 2,6-di-alkyl substituted cyclohexanols.
 |
H-bonding: D donor, A Acceptor; logP: log of the calculated octanol/water partition coefficient
2.6. Data and statistical analysis
Data analysis was performed in PulseFit (HEKA Elektronik) or using custom analysis routines written in IgorPro (Wavemetrics Corporation, Lake Oswego, OR). Data were typically collected and analyzed independently by two or more experimenters with some recordings randomly selected for independent reanalysis. Data are presented as mean ± SEM with the numbers of observations per data set reported in figure legends. Errors around Hill fit parameters and the equilibrium constants determined from fits of the gating model are non-linear regression approximations of the standard deviation.
Fits of the Hill equation were to the individual data. To minimize the effect of different population sizes on the global minimization, fits of the models were to mean behavior. As all models have the same degrees of freedom and were fit to a common data set, the goodness of fits are directly comparable and allow for robust model discrimination. A two-tailed t-test was used to compare two populations. Comparisons between multiple populations and a control were by one-way ANOVA using Dunnett’s test for multiple comparisons. Statistical tests were performed in SigmaStat 4.0 (Systat Software).
2.7. Chemicals and reagents
Phenols:
2,6-di-methyl phenol (DM-P; Sigma-Aldrich Cat. # D175005), 2,6-di-ethyl phenol (DE-P; Sigma-Aldrich Cat. # CBR02039), 2,6-di-iso-propyl phenol (DIP-P; Sigma-Aldrich Cat. # D126608), 2,6-di-tert-butyl phenol (DTB-P; Sigma-Aldrich Cat. # D48400), 2,6-di-sec-butyl phenol (DSB-P; Acros Organics, Fairlawn, NJ, USA Cat. #AC15283).
Isocyanates:
2,6-di-methyl phenyl-isocyanate (DM-PIC; Sigma-Aldrich, Cat. # 252336), 2,6-diethyl phenyl-isocyanate (DE-PIC; Alfa Aesar, Ward Hill, MA, USA Cat. # L11339), 2,6-di-isopropyl phenyl-isocyanate (DIP-PIC; Sigma-Aldrich Cat. # 376930).
Thiols:
2,6-di-methyl phenyl-thiol (DM-PT; Sigma-Aldrich Cat. # 306940); 2,6-di-iso-propyl phenyl-thiol (DIP-PT) was synthesized within the Milstein Chemistry Core Facility at Weill Cornell Medicine. LCMS, 1H-NMR, and 13C-NMR analysis conformed to literature precedent [44] and confirmed purity at >99%.
Fluoro:
2-Fluoro-1,3-di-iso-propylbenzene was synthesized as previously described [43]. For clarity, here we use a nomenclature that accords with the other molecules studied, that is, 2,6-di-iso-propyl phenyl-fluorine (DIP-PF).
Cyclohexanols:
2,6-di-methyl cyclohexanol (DM-CH) was obtained from Acros Organics (Thermo Fisher Scientific, New Jersey, USA; Cat. # 149810050); 2,6-di-ethyl cyclohexanol (DE-CH), 2,6-di-iso-propyl cyclohexanol (DIP-CH) and 2,6-di-sec-butyl cyclohexanol (DSBCH) were as previously described [45]. 2,6-di-tert-butyl cycloxhexanol (DTB-CH) was synthesized within the Milstein Chemistry Core Facility at Weill Cornell Medicine. LCMS, 1H-NMR, and 13C-NMR analysis conformed to literature precedent [45] and confirmed purity at >99%.
All ligands were prepared as stock solutions in DMSO then dispersed in the bath solution with vigorous stirring for at least 60 minutes. In the absence of any other excipient, the final DMSO concentration was 0.2% (V/V). 2-Hydroxypropyl-β-cyclodextrin (HPβCD; Sigma-Aldrich, St. Louis, MO;H107) can increase the solubility of ligands such as DTB-P and DIP-P by ≥200-fold and do so with no effect on pharmacokinetics ([22]; and references therein). In an attempt to extend the range of the concentration responses we could examine, in some experiments we included HPβCD. While the presence of HPβCD did not qualitatively alter the behavior of any ligand, its presence did cause a modest right-shift in the inverse agonist concentration response curves when it was included. However, as the right-shift was not demonstrably outside the normal scatter observed in the data between recordings from different donor frogs on different days and was far smaller than the variance in the model fit parameters, the different vehicle conditions were combined.
3. Results
3.1. H-bond interactions with position 1 confer potency
Figure 2 shows representative families of voltage-clamp recordings (Figure 2A) in either vehicle or vehicle plus 10 μM of one of four 2,6-di-iso-propyl phenyl derivatives (Figure 2B, also Figure 3). Inspection of the records suggests that systematic perturbation of the H-bonding capability of the adduct at the 1-position leads to a disruption of inhibition of HCN1 gating. This inference is confirmed by inspection of the activation curves (Figure 2C).
Figure 2: Hydrogen bond functionality at position 1 is important for alkylbenzene inhibition of HCN1 gating.

A. Representative voltage-clamp recordings of HCN1 currents (left) following incubation for 20 min in the absence or presence of 10 μM of the indicated reagent. Tail currents (right) are shown on expanded scales. In each case the black trace is the current recorded at an activation potential of −65 mV. To permit comparison, all recordings were obtained on the same day from distinct oocytes from a single donor frog. Scale bars are 2 μA and 1s (left) and 200 nA and 50 ms (right).
B. Schematic representation of 2,6-di-iso-propyl phenyls. Substitutions at the 1-position (as per the legend) describe molecules whose effects are reported in A and C.
C. Normalized steady-state activation curves constructed from the records shown in A. The smooth lines are fits of the Boltzmann function. Symbols represent molecules as described in B.
Diminished inhibition could be due to reduced efficacy, a weakening of potency, or both. To address this, we constructed complete concentration-response relations for the four ligands (Figure 3 central column, first four rows). Inspection confirms there is a loss of potency upon 1-position substitution. The progressive loss of potency (DIP-P > DIP-PT > DIP-PIC >>> DIP-PF) evident in Figure 3 and 4A (symbols within the dashed ellipse – see also Table 3) does not track the ability of the ligands to act as either a H-bond donor (DIP-P > DIP-PT >>> DIP-PIC ~ DIPPF) or acceptor (DIP-PIC > DIP-P > DIP-PT >>> DIP-PF) (Table 1).
Figure 4: Inverse agonist potency of 2,6-di-alkyl phenyl analogues as a function of molecular volume.

The observed aqueous IC50 (A) and the
partition-corrected IC50 (B) plotted as a function of
the calculated molecular volume. Except where no inflection was observable in
the concentration response curve, IC50’s were determined from
the fits shown in Figure 3 (see also Table 3). For ineffective molecules, the
IC50 was set equal to 100 mM. The partition-corrected
IC50 is the aqueous IC50 multiplied by the calculated
partition coefficient, P (as per
Tables 1 and 2). In both A and B, the
dashed ellipse encircles the data for the iso-propyl family of
reagents. As their IC50s are at best ill-defined, values for DM-P
( ), DM-PIC
(
), DM-PIC
( ), DM-CH
(
), DM-CH
( ), DE-CH
(
), DE-CH
( ) and DSBCH
(
) and DSBCH
( ) are omitted from
B.
) are omitted from
B.
Table 3:
Hill equation parameters.
| Ligand | Hill Fit Paramters (mean ± SD) | ||
|---|---|---|---|
| IC50, μM | ΔV1/2 max, mV | h | |
| DM-PT | 408 ± 55 | -- | -- |
| DE-P | 25.1 ±2.7 | −22.1 ± 1.0 | 1.95 ± 0.44 |
| DE-PIC | 240 ± 20 | -- | -- |
| DE-CH | 2995 ± 723 | -- | -- |
| DIP-P | 5.8 ± 1.2 | −39.8 ± 2.3 | 0.85 ± 0.11 |
| DIP-PT | 22.2 ± 1.8 | -- | -- |
| DIP-PIC | 43.7 ± 3.8 | -- | -- |
| DIP-PF | 267 ± 24 | -- | -- |
| DIP-CH | 35.0 ± 21.0 | −18.9 ± 3.4 | 0.76 ± 0.19 |
| DTB-P | 4.8 ± 0.8 | −42.4 ± 2.8 | 0.97 ± 0.13 |
| DTB-CH | 11.4 ± 4.2 | −9.9 ± 1.5 | 1.28 ± 0.47 |
| DSB-P | 355 ± 38 | -- | -- |
Where not reported, parameters were constrained to those determined for DTB-P as described in Section 2.3 and Figure 3. For all ligands, fitted parameters were significantly different from those of DTB-P and DIP-P (P < 0.05: one-way ANOVA).
It should be noted, however, that changing the 1-position adduct will alter not just the H-bonding profile of the ligand but other aspects of its physical chemistry. Thus, the four ligands here will have modestly different volumes, pKa’s and markedly divergent hydrophobicities (Table 1), and each of these properties is examined below.
3.2. Compensating for partitioning reveals the 1-position adduct influences potency through action as an H-bond acceptor
Due to partitioning, at an equivalent aqueous concentration, more hydrophobic molecules will have a higher intra-plasma membrane concentration. As alkylphenols act by coupling to the membrane embedded core of HCN1 channels [25], we next asked whether partitioning per se alters either the rank order and/or energetic strength of 1-position adduct substitutions.
To address this, we assumed the within-membrane concentration of each ligand at its aqueous IC50 can be represented by the aqueous IC50 multiplied by the calculated partition coefficient (P). The dashed ellipse-encircled symbols in Figure 4B plot the partition-corrected IC50 values for the four ligands considered in Figure 2 (and DIP-CH). The progressive loss of potency (DIP-PIC ~ DIP-P >> DIP-PT >>> DIP-PF) qualitatively tracks closer to the ability of the ligand to act as an H-bond acceptor (DIP-PIC > DIP-P > DIP-PT >>> DIP-PF) than a donor (DIP-P > DIP-PT >>> DIP-PIC ~ DIP-PF) with the most glaring potency-acceptor inconsistency being the behavior of isocyanate compared to hydroxyl. While isocyanate can theoretically accept 3 H-bonds but hydroxyl only one, DIP-PIC and DIP-P exhibit essentially identical potency. We posit that the similarity of DIP-PIC and DIP-P is consistent with the 1-adduct acting as an H-bond acceptor in an environment where there is only a single H-bond donor available. Such a conclusion is inconsistent with ligand coupling to water and suggests the bond is a ligand-protein interaction.
3.3. Are the effects of alterations in position 1 chemistry energetically consistent with disruption of an H-bond?
Replacement of the 1-OH by F eliminates the capacity of this adduct to engage in H-bonding - either as a donor or an acceptor. Importantly, it does so with little effect on most other physicochemical properties of the molecule [43, 46]. Consequently, we can discretely ascribe alterations in the IC50 to disruption of H-bonding. Based on the data in Figures 3 and 4, we estimate that the shift in IC50 in response to replacing OH with F is between 46 to 1,870-fold as determined from the ratios of the observed IC50s for DIP-P and DIP-PF (Figure 4A) and the partition-corrected IC50s for those ligands (Figure 4B), respectively. These changes correspond to a 2.3 to 4.5 kCal mol−1 destabilization of association; energetically, the observed loss of potency is entirely consistent with the disruption of an H-bond.
3.4. Do differences in pKa influence the H-bonding profile of the phenyl and cyclohexanol derivatives?
As pKa’s of the phenol and cyclohexanol derivatives are orders of magnitude larger than the experimental pH, the concentrations of ionized species will be very low. Differential presentation of the hydroxyls is unlikely to contribute to the observed behavior. While the pKa of sulphydryls is in the experimental range, as the site of action of the ligands appears to be in the largely anhydrous membranous space, differential ionization is unlikely to be a factor beyond influencing the partitioning coefficient – an effect already accounted for in the data in Tables 1 and 2 and Figure 4B.
3.5. Does the H-bond contribute to coupling energy?
At the highest concentrations, DIP-P (and DTB-P) elicit an ~ −40 mV shift in the V1/2 of HCN1 gating. While we were unable to reach concentrations sufficient to obviously saturate the concentration response curves for DIP-PT, DIP-PIC or DIP-PF, the first two appear to trend towards an efficacy similar to that of DIP-P (and DTB-P). In contrast, the concentration response relation of DIP-PF appears to be flatter than the hydroxyl, thiol or isocyanate iso-propyl analogues, and it appears it may tend to a plateau less than −40 mV; this suggests the interaction energy at the 1-position does not absolutely partition towards initial binding but, rather, the 1-position contributes modestly to coupling energy.
3.6. The alkylbenzene site is large
To examine the role of ligand volume, we systematically and symmetrically varied the nature of the alkyl adducts on positions 2 and 6 and did so in the background of not only a hydroxyl on position 1 (top row) but also in the background of both a thiol (second row) and an isocyanate (third row).
Inspection of the hydroxyl-containing substituents appears to show a U-shaped dependence of potency on the alkyl identity (Figure 3). Iso-propyl and tert-butyl derivatives are comparable with respect to potency but inclusion of either smaller (ethyl) or more extended (sec-butyl) side-chains results in a marked right-shift in the concentration-response curves. A plot of IC50 as a function of molecular volume supports this impression (Figure 4A). Although we do not have access to the complete alkyl adduct series for 1-position thiol or isocyanate families, these partial series exhibit a comparable trend to higher potency as the alkyl group tends towards the iso-propyl : tert-butyl apparent optimum (Figures 3, 4A).
These data suggest that there is a narrowly-defined pocket that alkylbenzene derivatives dock within. If the alkyl groups are too small the ligand does not orient correctly while if the groups get above a threshold, the ability of the ligands to enter the pocket is diminished. Such a conclusion would be consistent with the well described “cut-off” effect of anesthetics at other molecular targets [47–50] with the caveat that the changes considered here (increasing the number of carbons in the 2 and 6 alkyl adducts) increases not only ligand volume, they increase ligand hydrophobicity and, as such, increases the ligand’s partition coefficient.
Figure 4B plots the partition-corrected IC50 values of the matrix of ligands as a function of their molecular volume. When we consider only those ligands where a clear inflection and rising phase is discernable in the concentration response relation (disregarding DM-P, DM-PIC, DM-CH, DE-CH, and DSB-CH) the dependence on volume below the apparent iso-propyl : tert-butyl optimum largely disappears.
There is one anomaly within this analysis. DM-PT exhibits a clear inverse agonist activity whereas DM-P and DM-PIC do not. Given that both hydroxyl and isocyanate are better H-bond acceptors than is a thiol, and hydroxyl a better donor (Table 1), this preference for thiol in the di-methyl family is unexpected. As DM-PT is more hydrophobic (logP 2.87) than either DM-P (logP 2.22) or DM-PIC (logP 1.49; Table 1), it is possible that the apparent discontinuity reflects an edge effect wherein DM-PT reaches an effective within-membrane concentration whereas what would otherwise be effective molecules (DM-P and DM-PIC) do not. However, this is unlikely to be the sole determinant. Thus, the observations that DE-PIC is effective despite a logP of 1.93 would argue against inadequate ligand partitioning into the membrane being the sole reason DM-P and DM-PIC are ineffective in comparison with DM-PT. The basis for the additional advantage afforded to DM-PT (or disadvantage of the hydroxyl and isocyanate dimethyl derivatives) is unclear.
What about the apparent suppression of effectiveness at alkyl sizes above that of tert-butyl? The response curve for DSB-P suggests it could be either a low potency and high efficacy full inverse agonist (the data in Figure 3 represent the foot of a curve that has a right-shifted IC50 of ~355 μM as shown by the thin green line) or a partial inverse agonist that has a higher potency and lower efficacy (the data in Figure 3 represent the maximal ΔV1/2 with an IC50 of ~30 μM). Given the low aqueous solubility of butyl phenols (~200 μM; [51]) we were unable to reliably address this question by simply increasing the range of the concentration response curve.
Importantly, as an IC50 of either 30 μM or 355 μM is markedly larger than that of DTB-P (IC50 = 4.8 μM) the upturn seen at larger volumes in Figure 4 is likely to be real. This conclusion receives further support from the additivity/competition approach adopted below, that suggests DSB-P is indeed a low potency full inverse agonist. Together, these data are consistent with the notion that, while large (≥ 800 Å3), the inverse agonist alkylphenol site has a defined volume and geometry.
3.7. Alkylbenzenes act via four, radially-arrayed, structurally-defined, sites
The observation that various congeners of DIP-P and DTB-P may exhibit partial inverse agonist behavior has important implications with respect to understanding the molecular interactions between this class of molecules and HCN1 and, hence, the development of a SAR profile for the presumptive inverse agonist site(s). Accordingly, we next examined whether the behavior of corresponding cyclohexanols and alkyl adduct-modified ligands could further inform us about the nature of the alkylbenzene:HCN1 channel interaction. Specifically, we sought to determine the answers to four fundamental questions: 1) Are the cyclohexanols and the short and long alkyl group phenols truly partial inverse agonists? 2) Does the behavior of cyclohexanols and phenols further validate the notion of a structurally-defined alkylbenzene site on HCN1 as opposed to a generalized perturbation effect? 3) Can the data inform us as to the stoichiometry and location of the presumptive alkylbenzene site(s)? 4) Can we elucidate the mechanistic basis for the loss of efficacy in the cyclohexanols?
The simplest ligand to consider is DE-P. The observed ΔV1/2 appears to a saturate at 100 μM. Indeed, in paired experiments we find the ΔV1/2 in the face of 100 and 300 μM to be indistinguishable (−21.6 mV ± 4.2, n = 8 and −21.2 mV ± 4.6, n = 9; respectively; P = 0.95, two-tailed t-test). Importantly, the aqueous solubility limit of DE-P (~10 mM; https://comptox.epa.gov/dashboard/dsstoxdb/results?search=DTXSID5061401#comments) is two-orders of magnitude higher than the functional saturation observed here. That is, DE-P is indeed a partial inverse agonist with a maximal efficacy ~2-fold lower than the maximal effect seen with DIP-P and DTB-P (see also Table 3). These data show the 2 and/or 6 adducts can “sense” changes in channel structure upon channel opening and, in this case, the effect of the smaller alkyl adduct is to reduce coupling energy.
With respect to the cyclohexanols, the maximal ΔV1/2 determined from fits of the Hill equation suggest there to be a >2-fold decrease in efficacy in the di-iso-propyl background and a >4-fold decrease in the tert-butyl background (Figure 3 and Table 3). This decrement is associated with little (tert-butyl) or only modest (iso-propyl) decrement in potency (see the green dashed lines in Figure 3). However, the low aqueous solubility of these cyclohexanols raises the concern that the apparent saturation of their concentration response curves is a function of ligand availability and not channel responsiveness. To address this, we presented HCN1 channels with a submaximal concentration of DTB-P in the absence and presence of increasing concentrations of DIP-CH and DTB-CH (as well as DIP-P, DSB-P and DE-CH) and considered the results within the context of the gating models outlined in Figure 1.
The symbols in Figure 5A–B reproduce the observed concentration response data (from Figure 3) for DIP-CH and DIP-P (Figure 5A) and DTB-CH and DTB-P (Figure 5B). Figure 5C–E present the shift in the V1/2 elicited by increasing concentrations of DIP-CH (Figure 5C), DTB-CH (Figure 5D) and DIP-P (Figure 5E) in the presence of 3 μM DTB-P. The solid blue lines in Figure 5A–E represent the simultaneous fit of the common site model at S=4.
Qualitatively, the behaviors of DIP-CH and DTB-CH are exactly those expected for partial inverse agonists while DIP-P behaves exactly as expected for a second full inverse when all act at the same site(s) as DTB-P (Methods). Quantitatively, the optimized values of the ligand association constants are physically reasonable and the errors around each term are relatively modest (Table 4; Figure 5F). In contrast, the fit of the common site model at S=1 is inferior (χ2 is 147 vs. 91 for S=4) and L adopts a physically unreasonable value of 7 * 108 ± 5 * 108.
We examined the reasonableness of the distinct-site models in two ways. First, we used the parameters reported in Table 4 and Figure 5F to predict the behavior of the distinct-site model at S=4. As anticipated (Methods) models of this class are, by definition, additive in the domain of inverse agonist concentrations where the ΔV1/2 is submaximal; the dashed blue lines in Figure 5C–E show poor or no correspondence to the observed data. Second, we fit the distinct-site model to the data. The fits were inferior to those of the single site model (χ2 at S=4 was 263 and 809 at S=1). Furthermore, at both S=1 and S=4, one or more ligand association constants dropped to zero while, at S=1, L was unreasonably large and very poorly defined (4.1 * 105 ± 9.3 * 108). It is important to note that as the distinct-site model is conceptually identical to a generalized membrane perturbation model, failure of the distinct model provides additional evidence for the existence of specific, structurally-defined, alkylbenzene site(s) on HCN1.
Consistent with the above findings, DE-CH exhibited neither additivity nor competition with 3 μM DTB-P (data not shown). While DE-CH appears to be only weakly effective as an inverse agonist (displaying an inflexion in the concentration response curve only at the highest concentration achieved - Figure 3) the lack of effect of DE-CH in the presence of DTB-P provides an additional control for potential non-specific effects of loading the cell membrane with these small molecules.
The simplest interpretation of these findings is that DTB-P (and DIP-P) act as full inverse agonists by interacting with four radially-arrayed binding sites lying within or between each HCN subunit, and that cyclohexanols (and ligands with altered alkyl adducts) associate with these same sites but generate less coupling energy than do the iso-propyl and tert-butyl phenols.
3.8. What is the basis for degradation of coupling energy upon conversion of a phenol to a cyclohexanol?
Figure 5F shows this arises from a preferential degradation in the association of the ligand with the closed state with a more modest disruption of open state affinity. In the background of iso-propyl and tert-butyl ligand families, the effect of ring saturation on closed and open state association is largely independent of the alkyl chain background (as shown by the comparable shifts in ΔG - Figure 6F). This is consistent with these alkyl groups contributing comparably to channel-ligand association energies.
Figure 6: DSB-P is a low potency, high efficacy, inverse agonist.

ΔV1/2 as a function of concentration of DSB-P in the absence (A) and presence (B) of 3 μM DTB-P. Data in A are from Figure 3. The data in B are from 32 independent recordings. In both panels, the solid blue lines are simultaneous fits of the common site model (S=4) to the data in these two panels with all constants other than JOI and JcI held equal to the values determined in Figure 5 (see Table 4). JOI and JcI as determined from these fits were 881 ± 1,801 and 3,696 ± 2,948, respectively. The dashed blue line in B is the predicted behavior for the distinct site model at S=4 with parameters as described above. Discrete fits of the four models yielded χ2 values of 165 and 167 (distinct site) and 600 and 128 (common site) at S equal to 1 and 4, respectively. The symbols and red line in A are the observed ΔV1/2 for DSB-P and IC50-adjusted DTB-P fit line (IC50 set to 355 μM) reproduced from the DSB-P panel of Figure 3. In B, the green lines report the mean value of ΔV1/2 (solid) and SEM (dashed) elicited by 3 μM DTB-P in paired controls.
3.9. Extended alkyl chains disrupt ligand association with HCN1
Figure 6 shows the results of an additivity experiment directly comparable to that in Figure 5. While the achievable concentration range of DSB-P is a limiting factor and the fits are, consequently, poorly determined, the data are consistent with DSB-P being a low affinity full inverse agonist. As with the data for DIP-CH, DTB-CH and DIP-P, the behavior of DSB-P was best described by the common-site model S=4.
4. Discussion
Four key chemical characteristics define congeners of DIP-P - H-bond propensity, ring saturation/planarity, hydrophobicity, and molecular volume. We find that: 1) There is an alkylbenzene site on HCN1 channels that is relatively large, having a volume ≥ 800 Å3; 2) The site contains an H-bond donor that interacts with the 1-position adduct on the ligand in a manner that contributes to initial binding energy; 3) The 1-position adduct is, at best, poorly involved with generation of coupling energy indicating the site’s hydrogen bond donor is relatively immobile (with respect to ligand) during channel opening; 4) The presence of a π electron cloud is coupled to the generation of coupling energy suggesting that these electrons (or the imposed ring planarity) is important in establishing a preferential association between the ligands and the closed state(s) of the channel; 5) Substituents at the 2 and 6 positions of the ligand (and possibly others excluding the 1-position) interact with aspects of the channel that are structurally dynamic during opening; 6) The data are most readily reconciled with the existence of four radially-arrayed alkylbenzene sites suggesting the sites lie within each HCN1 subunit or at the inter-subunit interface and these sites are distant (in terms of ligand geometry) from the central axis of the channel; 7) Addition of adducts capable of forming more specific interactions than permitted by alkyl chemistry may better exploit the state dependent channel surface and generate a higher potency class of inverse agonists.
4.1. Reasonableness of the common site, S=4, gating parameters
The best test of a model is to have it predict data not included in the fit. While we cannot readily gain independent measures of the association constants, we can independently estimate L. Based on the maximal open probability (PMAX) we estimate L to be between 7.3 and 679 (Methods). That our model-determined measure of L (125) falls within this range suggests the model determined parameters are consistent with the energetics of HCN1 gating and drug effects thereon.
Does our use of a simple three-state gating scheme undermine the validity of this analysis? While the model shown in Figure 1 does not reproduce the nuanced kinetic behavior of HCN channels ([37] and references therein), the general form of the relationships between L, PMAX and ΔV1/2 in more complex, kinetically-faithful, models will be comparable to equations 7 and 8 (albeit such solutions will contain proportionality terms based on the presence of additional coupled equilibria). Any quantitative impact on the estimates of L from such an increase in the complexity of the basal gating model will equivalently impact its determination from the additivity data in Figure 5 and the NSFA studies. Accordingly, we posit that the analysis of inverse agonists and partial inverse agonists predicts a physiologically-reasonable determination of the fully-activated open-closed equilibrium which will remain true with any such model expansion.
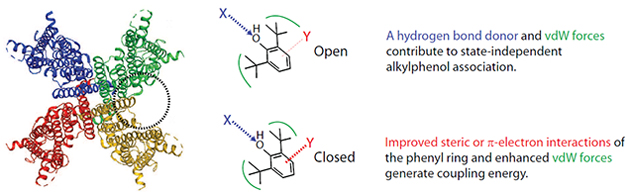
4.2. How does ring planarity and/or the π electron cloud contribute to coupling energy?
What is the likely basis for the preferential loss of closed state binding upon ring saturation? One possibility is that the π electrons form an electrostatic bond with the channel that is stronger in the closed state than in the open state. The ~1 kCal mol−1 degradation in Kc on shifting from the phenolic structure to the cyclohexanol is energetically consistent with such a scenario [52]. An alternative scenario is that the rigid structure of the phenolic ring acts as a better “foot-in-the-door” than does the more flexible cyclohexanol. Distinguishing between these mechanisms could be aided by use of fluorine for ring-hydrogen substitution analogues [53, 54] in the background of either DIP-P or DTB-P, but those studies are beyond the scope of the current manuscript.
4.3. Do the chemical characteristics identified here correspond to known alkylbenzene binding sites?
The existence of spatially-defined cavities (i.e., presumptive functional binding sites) that can accommodate small anesthetic molecules have been identified in a variety of proteins, including firefly luciferase, human serum albumin, and both ligand- and voltage-gated ion channels [55–57]. Modeling based on molecular mechanics–quantum mechanics calculations suggest that anesthetic-protein complexes share a set of common characteristics, or “binding motifs”; these motifs are “polar and nonpolar interactions within an amphiphilic binding cavity, including the presence of weak hydrogen bond interactions with amino acids and water molecules” [58]. The results presented herein suggest the existence of a spatially-defined cavity in HCN1 channels that accommodates small lipophilic molecules and that the underlying interactions conform to the previously proposed “binding motifs.”
4.4. Mapping the isoform divergence onto the HCN1 cryo-EM structure
HCN1 is the isoform most sensitive to alkylphenols with HCN2, 3 and 4 being progressively less sensitive albeit with some dependence on the alkyl side chain identity (iso-propyl versus tert-butyl) [22]. With respect to trying to identify key residues that determine the potency and efficacy of alkylphenol inhibition, this profile suggests residues that are unique to HCN1 may not be the complete picture but that locations that follow a divergence in conservation may also be important.
Figure 7 shows within the transmembrane structure of HCN1-ΔNvΔC, the minimal HCN1 channel that retains alkylphenol sensitivity, there are residues unique to HCN1 and others divergent between the four isoforms. While our hypothesis is that residues contacting, or within, the S5-S6 motif are most likely to contribute to the site, it is clear that there are too many divergent locations to justify a site-directed analysis (with all its inherent caveats of interpretation). Accordingly, identification of the alkylbenzene sites will likely be best determined using photolabeling and chimeric approaches combined with molecular docking strategies once the open and closed architectures of the HCN1 channel are known [55]. The results presented here will allow for rationale interpretation of the findings of any, and all, such studies.
Figure 7: Cryo-EM structure of apo-state of alkylbenzene sensitive HCN1 channel core.

A. Amino acid sequence alignment for HCN1–4. Sequences shown are for the membrane embedded core region only, and correspond to mHCN1-ΔNvΔC as previously described [25]. mHCN1-ΔNvΔC contains 34 unique residues (with respect to mHCN2, hHCN3, and mHCN4) and 50 residues that are divergent from at least one other isoform. Magenta – residue identity unique to mHCN1; teal and green – locations where mHCN1-ΔNvΔC diverges from at least one other isoform (teal shows commonality with mHCN1 while green highlights the divergent residue(s) in HCN2–4; yellow - four-fold conservation. Sequence alignment obtained using Clustal Omega(1.2.4) (https://www.ebi.ac.uk/Tools/msa/clustalo/) for mouse (m)HCN1 (O88704.1), mHCN2 (O88703.1), human (h)HCN3 (Q9P1Z3.2), and mHCN4 (O70507.2).
B. Top-down (left panel) and side view (right panel) of the hHCN1 tetramer. As neither the divergent amino terminus nor the post-S6 C-terminus are involved in alkylbenzene inhibition of HCN1, these sequences have been eliminated from the structure for clarity. Note, the mouse and human HCN1 sequences are identical in the region defined by mHCN1-ΔNvΔC. HCN1 unique residues are shown as magenta spheres. Structure based on atomic coordinates of HCN1 in the unbound state from Lee & MacKinnon 2017 and deposited in the protein data bank (uniprot.org) under accession code 5U6O. In the right panel two of the opposing hHCN1 subunits removed for clarity.
C. Top-down (left panel) and side-view (right panel) of hHCN1 as in B now showing the residues in mHCN1 that are common to at least one (but not all) other mHCN1 isoform (teal spheres).
4.5. Alkylphenols as novel antihyperalgesics and addiction potential
Might propofol, and by extension other alkylphenols, interact with other signaling pathways, especially those centered on the opiate receptor system? In vitro, propofol has no direct effect on G-protein coupled inwardly rectifying K+ channels (GIRKs), nor does a clinically relevant concentration of propofol (2 μM) potentiate D-Ala2,N-Me-Phe4,Gly5-ol]enkephalin (DAMGO)-evoked currents generated by GIRK2 channels [59]; importantly, and in contrast, supratherapeutic propofol concentrations (up to 100 μM propofol, a 50×-clinically relevant concentration) inhibit DAMGO-evoked currents generated by chimeric mu opiate (μOR-Gqi5) receptors [60]. Thus, there is no evidence that propofol potentiates mu opiate receptor (mOR) function at the molecular level [61]. As to other alkylphenols, DTB-P, like propofol, inhibits HCN1 gating [22], but unlike propofol, it has minimal activity at GABAA receptors [42] although it increases open channel probability (Po) of α3β glycine receptors [23]; there are no published data on the effects of DTB-P on G-protein coupled receptors in general, and mORs in particular. Despite increasing Po of α3β glycine receptors, α3β glycine receptors do not contribute to either DTB-P-mediated mechanical or thermal anti-hyperalgesia following chronic constriction nerve injury [23], an effect comparable to our observations using a partial sciatic nerve ligation model of neuropathic pain [22].
With regards to analgesia and modulation of pain pathways in vivo, propofol suppresses activity of spinal cord dorsal horn neurons in response to noxious stimulation in goats [62] and rats [63], and this effect appears to be partly GABAergic in origin [63]. In human subjects, however, a meta-analysis of thirty-nine clinical trials with a combined subject population of 4,520 patients indicated that propofol-based anesthetics do not result in less postoperative pain than those utilizing volatile anesthetics [64], suggesting that propofol per se does not produce meaningful analgesia that extends into the immediate postoperative period. Such an observation does not preclude the development of alkylphenols as anti-hyperalgesics as anti-hyperalgesia is not the same as analgesia, reflecting the fact that pain - “an unpleasant sensory and emotional experience associated with actual or potential tissue damage” is not equivalent to hyperalgesia – “increased pain from a stimulus that normally provokes pain” (https://www.iasp-pain.org/terminology?navItemNumber=576). The in vivo data comport with the in vitro data and suggest that propofol, and presumably related alkylphenols, should have a good therapeutic index with respect to the risk of addiction as the relevant circuitry underlying opioid-dependent addiction [65, 66] and propofol mechanisms of action [67, 68] do not overlap.
Our data suggest that a spatially defined cavity that accommodates alkylphenols exists in the transmembrane or interfacial region of HCN1. Whether the binding site is similar to comparable binding sites in other channels, including GABAA [26, 69], TRPA1 [70, 71], and glycine [23], receptors remains to be determined. Given the size of the pocket and the lack of specific chemical interactions with alkyl adducts, the findings suggest it may be possible to develop a higher potency analogue of DTB-P with drug-like properties, particularly as the HCN1 cryo-EM structure is now available [33]. Such experiments have the potential to reveal a new, opioid-independent approach to treating neuropathic pain.
Acknowledgements
This work was supported by the Dept. of Anesthesiology, Weill Cornell Medical School, the Blakeslee Foundation (ACH), and the NIH (KAW and RGE; Grant number GM55876) and Akelos, Inc., New York, NY. We thank Dr. C. D. Hall, Department of Chemistry, University of Florida, Gainesville, FL for preparation of several of the cyclohexanols; David J. Huggins and Mayako Michino of the Tri-Institutional Therapeutics Discovery Institute for their assistance in ligand-analysis using the Maestro molecular modeling software; Jan Rheinberger for guidance in the use of Pymol for visualization of the HCN1 structure; and Alex Proekt, M.D., Ph.D., for his ever-insightful thoughts on statistical analysis.
Abbreviations:
- GABAA-Rs
GABAA receptors
- HCN
hyperpolarization cyclic nucleotide gated channel
- NSFA
non-stationary fluctuation analysis
- PMAX
maximal open probability
- SAR
structure-activity relationship
- TEVC
two-electrode voltage clamp
- DM-P
2,6-di-methyl phenol
- DM-PT
2,6 di-methyl phenyl-thiol
- DM-PIC
2,6-di-methyl phenyl-isocyanate
- DM-PF
2,6-di-methyl phenyl-fluorine
- DM-CH
2,6-di-methyl cyclohexanol
- DE-P
2,6-di-ethyl phenol
- DE-PT
2,6 di-ethyl phenyl-thiol
- DE-PIC
2,6 di-ethyl phenyl-isocyanate
- DE-PF
2,6-di-ethyl phenyl-flurorine
- DE-CH
2,6 di-ethyl cyclohexanol
- DIP-P
2,6 di-iso-propyl phenol (propofol)
- DIP-PT
2,6-di-iso-propyl phenyl-thiol
- DIP-PIC
2,6 di-iso-propyl phenyl-isocyanate
- DIP-PF
2,6-di-iso-propyl phenyl-fluorine
- DIP-CH
2,6 di-iso-propyl cyclohexanol
- DSB-P
2,6-di-sec-butyl phenol
- DSB-PT
2,6-di-sec-butyl phenyl-thiol
- DSB-PIC
2,6 di-sec-butyl phenyl-isocyanate
- DSB-PF
2,6-di-sec-butyl phenyl-fluorine
- DSB-CH
2,6 di-sec-butyl cyclohexanol
- DTB-P
2,6-di-tert-butyl phenol
- DTB-PT
2,6-di-tert-butyl phenyl-thiol
- DTB-PIC
2,6 di-tert-butyl phenyl-isocyanate
- DTB-PF
2,6 di-tert-butyl phenyl-fluorine
- DTB-CH
2,6 di-tert-butyl cycloxhexanol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
PAG and GRT are co-inventors on patents related to the development of alkylphenols for the treatment of neuropathic pain and serve on the Scientific Advisory Board for Akelos, Inc., a research-based biotechnology company that has secured a licensing agreement for the use of those patents.
References
- [1].Costigan M, Scholz J, Woolf CJ, Neuropathic pain: a maladaptive response of the nervous system to damage, Annu Rev Neurosci 32 (2009) 1–32. 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson AH, Yarnitsky D, Freeman R, Truini A, Attal N, Finnerup NB, Eccleston C, Kalso E, Bennett DL, Dworkin RH, Raja SN, Neuropathic pain, Nat Rev Dis Primers 3 (2017) 17002 10.1038/nrdp.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Freynhagen R, Bennett MI, Diagnosis and management of neuropathic pain, BMJ 339 (2009) b3002 10.1136/bmj.b3002. [DOI] [PubMed] [Google Scholar]
- [4].Seretny M, Currie GL, Sena ES, Ramnarine S, Grant R, MacLeod MR, Colvin LA, Fallon M, Incidence, prevalence, and predictors of chemotherapy-induced peripheral neuropathy: A systematic review and meta-analysis, Pain 155(12) (2014) 2461–70. 10.1016/j.pain.2014.09.020. [DOI] [PubMed] [Google Scholar]
- [5].Bouhassira D, Lanteri-Minet M, Attal N, Laurent B, Touboul C, Prevalence of chronic pain with neuropathic characteristics in the general population, Pain 136(3) (2008) 380–7. 10.1016/j.pain.2007.08.013. [DOI] [PubMed] [Google Scholar]
- [6].van Hecke O, Austin SK, Khan RA, Smith BH, Torrance N, Neuropathic pain in the general population: a systematic review of epidemiological studies, Pain 155(4) (2014) 654–62. 10.1016/j.pain.2013.11.013. [DOI] [PubMed] [Google Scholar]
- [7].Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, Gilron I, Haanpaa M, Hansson P, Jensen TS, Kamerman PR, Lund K, Moore A, Raja SN, Rice AS, Rowbotham M, Sena E, Siddall P, Smith BH, Wallace M, Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis, Lancet Neurol 14(2) (2015) 162–73. 10.1016/S1474-4422(14)70251-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G, Finnerup NB, Haanpää M, Hansson P, Hüllemann P, Jensen TS, Freynhagen R, Kennedy JD, Magerl W, Mainka T, Reimer M, Rice AS, Segerdahl M, Serra J, Sindrup S, Sommer C, Tölle T, Vollert J, Treede RD, Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles, Pain 158(2) (2017) 261–272. 10.1097/j.pain.0000000000000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].West SJ, Bannister K, Dickenson AH, Bennett DL, Circuitry and plasticity of the dorsal horn--toward a better understanding of neuropathic pain, Neuroscience 300 (2015) 254–75. 10.1016/j.neuroscience.2015.05.020. [DOI] [PubMed] [Google Scholar]
- [10].Tibbs GR, Posson DJ, Goldstein PA, Voltage-Gated Ion Channels in the PNS: Novel Therapies for Neuropathic Pain?, Trends Pharmacol Sci 37(7) (2016) 522–42. 10.1016/j.tips.2016.05.002. [DOI] [PubMed] [Google Scholar]
- [11].Chaplan SR, Guo HQ, Lee DH, Luo L, Liu C, Kuei C, Velumian AA, Butler MP, Brown SM, Dubin AE, Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain, J Neurosci 23(4) (2003) 1169–78. 10.1523/JNEUROSCI.23-04-01169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luo L, Chang L, Brown SM, Ao H, Lee DH, Higuera ES, Dubin AE, Chaplan SR, Role of peripheral hyperpolarization-activated cyclic nucleotide-modulated channel pacemaker channels in acute and chronic pain models in the rat, Neuroscience 144(4) (2007) 1477–85. 10.1016/j.neuroscience.2006.10.048. [DOI] [PubMed] [Google Scholar]
- [13].Jiang YQ, Xing GG, Wang SL, Tu HY, Chi YN, Li J, Liu FY, Han JS, Wan Y, Axonal accumulation of hyperpolarization-activated cyclic nucleotide-gated cation channels contributes to mechanical allodynia after peripheral nerve injury in rat, Pain 137(3) (2008) 495–506. 10.1016/j.pain.2007.10.011. [DOI] [PubMed] [Google Scholar]
- [14].Takasu K, Ono H, Tanabe M, Spinal hyperpolarization-activated cyclic nucleotide-gated cation channels at primary afferent terminals contribute to chronic pain, Pain 151(1) (2010) 87–96. 10.1016/j.pain.2010.06.020. [DOI] [PubMed] [Google Scholar]
- [15].Noh S, Kumar N, Bukhanova N, Chen Y, Stemkowsi PL, Smith PA, The heart-rate-reducing agent, ivabradine, reduces mechanical allodynia in a rodent model of neuropathic pain, Eur J Pain 18(8) (2014) 1139–47. 10.1002/j.1532-2149.2014.00460.x. [DOI] [PubMed] [Google Scholar]
- [16].Young GT, Emery EC, Mooney ER, Tsantoulas C, McNaughton PA, Inflammatory and neuropathic pain are rapidly suppressed by peripheral block of hyperpolarisation-activated cyclic nucleotide-gated ion channels, Pain 155(9) (2014) 1708–19. 10.1016/j.pain.2014.05.021. [DOI] [PubMed] [Google Scholar]
- [17].Descoeur J, Pereira V, Pizzoccaro A, Francois A, Ling B, Maffre V, Couette B, Busserolles J, Courteix C, Noel J, Lazdunski M, Eschalier A, Authier N, Bourinet E, Oxaliplatin-induced cold hypersensitivity is due to remodelling of ion channel expression in nociceptors, EMBO molecular medicine 3(5) (2011) 266–78. 10.1002/emmm.201100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang H, Dougherty PM, Enhanced excitability of primary sensory neurons and altered gene expression of neuronal ion channels in dorsal root ganglion in paclitaxel-induced peripheral neuropathy, Anesthesiology 120(6) (2014) 1463–75. 10.1097/ALN.0000000000000176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Aromolaran KA, Goldstein PA, [EXPRESS] Ion channels and neuronal hyperexcitability in chemotherapy-induced peripheral neuropathy; cause and effect?, Mol Pain 13 (2017) 1744806917714693 10.1177/1744806917714693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tu H, Zhang L, Tran TP, Muelleman RL, Li YL, Diabetes alters protein expression of hyperpolarization-activated cyclic nucleotide-gated channel subunits in rat nodose ganglion cells, Neuroscience 165(1) (2010) 39–52. 10.1016/j.neuroscience.2009.10.002. [DOI] [PubMed] [Google Scholar]
- [21].McClure KJ, Maher M, Wu N, Chaplan SR, Eckert WA III, Lee DH, Wickenden AD, Hermann M, Allison B, Hawryluk N, Breitenbucher JG, Grice CA, Discovery of a novel series of selective HCN1 blockers, Bioorg Med Chem Lett 21(18) (2011) 5197–201. 10.1016/j.bmcl.2011.07.051. [DOI] [PubMed] [Google Scholar]
- [22].Tibbs GR, Rowley TR, Sanford RL, Herold KF, Proekt A, Hemmings HC, Andersen OS, Goldstein PA, Flood P, HCN1 channels as targets for anesthetic and non-anesthetic propofol analogs in the amelioration of mechanical and thermal hyperalgesia in a mouse model of neuropathic pain, J Pharmacol Exp Ther 345 (2013) 363–373. 10.1124/jpet.113.203620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Acuña MA, Yévenes GE, Ralvenius WT, Benke D, Di Lio A, Lara CO, Muñoz B, Burgos CF, Moraga-Cid G, Corringer PJ, Zeilhofer HU, Phosphorylation state-dependent modulation of spinal glycine receptors alleviates inflammatory pain, J Clin Invest 126(7) (2016) 2547–60. 10.1172/JCI83817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Resta F, Micheli L, Laurino A, Spinelli V, Mello T, Sartiani L, Di Cesare Mannelli L, Cerbai E, Ghelardini C, Romanelli MN, Mannaioni G, Masi A, Selective HCN1 block as a strategy to control oxaliplatin-induced neuropathy, Neuropharmacology 131 (2018) 403–413. 10.1016/j.neuropharm.2018.01.014. [DOI] [PubMed] [Google Scholar]
- [25].Lyashchenko AK, Redd KJ, Yang J, Tibbs GR, Propofol inhibits HCN1 pacemaker channels by selective association with the closed states of the membrane embedded channel core, J Physiol 583(Pt 1) (2007) 37–56. 10.1113/jphysiol.2007.136465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jayakar SS, Zhou X, Chiara DC, Dostalova Z, Savechenkov PY, Bruzik KS, Dailey WP, Miller KW, Eckenhoff RG, Cohen JB, Multiple propofol-binding sites in a γ-aminobutyric acid type A receptor (GABAAR) identified using a photoreactive propofol analog, J Biol Chem 289(40) (2014) 27456–68. 10.1074/jbc.M114.581728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Forman SA, Miller KW, Mapping General Anesthetic Sites in Heteromeric γ-Aminobutyric Acid Type A Receptors Reveals a Potential For Targeting Receptor Subtypes, Anesth Analg 123(5) (2016) 1263–1273. 10.1213/ANE.0000000000001368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Nury H, Van Renterghem C, Weng Y, Tran A, Baaden M, Dufresne V, Changeux JP, Sonner JM, Delarue M, Corringer PJ, X-ray structures of general anaesthetics bound to a pentameric ligand-gated ion channel, Nature 469(7330) (2011) 428–31. 10.1038/nature09647. [DOI] [PubMed] [Google Scholar]
- [29].Franks NP, General anaesthesia: from molecular targets to neuronal pathways of sleep and arousal, Nat Rev Neurosci 9(5) (2008) 370–86. 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- [30].Hemmings HC Jr., Akabas MH, Goldstein PA, Trudell JR, Orser BA, Harrison NL, Emerging molecular mechanisms of general anesthetic action, Trends Pharmacol Sci 26(10) (2005) 503–10. 10.1016/j.tips.2005.08.006. [DOI] [PubMed] [Google Scholar]
- [31].Brosnan RJ, Pham TL, GABAA Receptor Modulation by Phenyl Ring Compounds Is Associated with a Water Solubility Cut-Off Value, Pharmacology 98(1–2) (2016) 13–9. 10.1159/000444935. [DOI] [PubMed] [Google Scholar]
- [32].Long SB, Tao X, Campbell EB, MacKinnon R, Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment, Nature 450(7168) (2007) 376–82. 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- [33].Lee CH, MacKinnon R, Structures of the Human HCN1 Hyperpolarization-Activated Channel, Cell 168(1–2) (2017) 111–120 e11. 10.1016/j.cell.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Riegelhaupt PM, Tibbs GR, Goldstein PA, HCN and K2P Channels in Anesthetic Mechanisms Research, Methods Enzymol 602 (2018) 391–416. 10.1016/bs.mie.2018.01.015. [DOI] [PubMed] [Google Scholar]
- [35].Bezanilla F, Stefani E, Voltage-dependent gating of ionic channels, Annu Rev Biophys Biomol Struct 23 (1994) 819–46. 10.1146/annurev.bb.23.060194.004131. [DOI] [PubMed] [Google Scholar]
- [36].Bezanilla F, The voltage sensor in voltage-dependent ion channels, Physiol Rev 80(2) (2000) 555–92. 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- [37].Lyashchenko AK, Redd KJ, Goldstein PA, Tibbs GR, cAMP Control of HCN2 Channel Mg2+ Block Reveals Loose Coupling between the Cyclic Nucleotide-Gating Ring and the Pore, PLoS One 9(7) (2014) e101236 10.1371/journal.pone.0101236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Azene EM, Xue T, Li RA, Molecular basis of the effect of potassium on heterologously expressed pacemaker (HCN) channels, J Physiol 547(Pt 2) (2003) 349–56. 10.1113/jphysiol.2003.039768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shin KS, Maertens C, Proenza C, Rothberg BS, Yellen G, Inactivation in HCN channels results from reclosure of the activation gate: desensitization to voltage, Neuron 41(5) (2004) 737–44. 10.1016/S0896-6273(04)00083-2. [DOI] [PubMed] [Google Scholar]
- [40].Craven KB, Zagotta WN, Salt bridges and gating in the COOH-terminal region of HCN2 and CNGA1 channels, J Gen Physiol 124(6) (2004) 663–77. 10.1085/jgp.200409178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen S, Wang J, Zhou L, George MS, Siegelbaum SA, Voltage sensor movement and cAMP binding allosterically regulate an inherently voltage-independent closed-open transition in HCN channels, J Gen Physiol 129(2) (2007) 175–88. 10.1085/jgp.200609585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Krasowski MD, Jenkins A, Flood P, Kung AY, Hopfinger AJ, Harrison NL, General anesthetic potencies of a series of propofol analogs correlate with potency for potentiation of γ-aminobutyric acid (GABA) current at the GABAA receptor but not with lipid solubility, J Pharmacol Exp Ther 297(1) (2001) 338–51. [PubMed] [Google Scholar]
- [43].Woll KA, Weiser BP, Liang Q, Meng T, McKinstry-Wu A, Pinch B, Dailey WP, Gao WD, Covarrubias M, Eckenhoff RG, Role for the Propofol Hydroxyl in Anesthetic Protein Target Molecular Recognition, ACS Chem Neurosci 6(6) (2015) 927–35. 10.1021/acschemneuro.5b00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Denmark SE, Hartmann E, Kornfilt DJ, Wang H, Mechanistic, crystallographic, and computational studies on the catalytic, enantioselective sulfenofunctionalization of alkenes, Nat Chem 6(12) (2014) 1056–64. 10.1038/nchem.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hall AC, Griffith TN, Tsikolia M, Kotey FO, Gill N, Humbert DJ, Watt EE, Yermolina YA, Goel S, El-Ghendy B, Hall CD, Cyclohexanol analogues are positive modulators of GABAA receptor currents and act as general anaesthetics in vivo, Eur J Pharmacol 667(1–3) (2011) 175–81. 10.1016/j.ejphar.2011.05.058. [DOI] [PubMed] [Google Scholar]
- [46].Qiu L, Lin J, Liu Q, Wang S, Lv G, Li K, Shi H, Huang Z, Bertaccini EJ, The Role of the Hydroxyl Group in Propofol-Protein Target Recognition: Insights from ONIOM Studies, J Phys Chem B 121(24) (2017) 5883–5896. 10.1021/acs.jpcb.7b02079. [DOI] [PubMed] [Google Scholar]
- [47].Jenkins A, Greenblatt EP, Faulkner HJ, Bertaccini E, Light A, Lin A, Andreasen A, Viner A, Trudell JR, Harrison NL, Evidence for a common binding cavity for three general anesthetics within the GABAA receptor, J Neurosci 21(6) (2001) RC136 10.1523/JNEUROSCI.21-06-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Krasowski MD, Nishikawa K, Nikolaeva N, Lin A, Harrison NL, Methionine 286 in transmembrane domain 3 of the GABAA receptor β subunit controls a binding cavity for propofol and other alkylphenol general anesthetics, Neuropharmacology 41(8) (2001) 952–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Krasowski MD, Hong X, Hopfinger AJ, Harrison NL, 4D-QSAR analysis of a set of propofol analogues: mapping binding sites for an anesthetic phenol on the GABAA receptor, J Med Chem 45(15) (2002) 3210–21. 10.1021/jm010461a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kash TL, Jenkins A, Harrison NL, Molecular volume determines the activity of the halogenated alkane bromoform at wild-type and mutant GABA(A) receptors, Brain Res 960(1–2) (2003) 36–41. 10.1016/S0006-8993(02)03748-4. [DOI] [PubMed] [Google Scholar]
- [51].Brewster ME, Braunstein AJ, Bartruff MSM, Kibbey C, Huang M-J, Pop E, Bodor N, Solubilization and electrochemical stabilization of substituted phenols through the use of 2-hydroxypropyl-β-cyclodextrin, Supramolecular Chemistry 4(1) (1994) 69–76. DOI: 10.1080/10610279408029864. [DOI] [Google Scholar]
- [52].Neel AJ, Hilton MJ, Sigman MS, Toste FD, Exploiting non-covalent π interactions for catalyst design, Nature 543(7647) (2017) 637–646. 10.1038/nature21701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Ahern CA, Eastwood AL, Lester HA, Dougherty DA, Horn R, A cation-π interaction between extracellular TEA and an aromatic residue in potassium channels, J Gen Physiol 128(6) (2006) 649–57. 10.1085/jgp.200609654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Van Arnam EB, Dougherty DA, Functional probes of drug-receptor interactions implicated by structural studies: Cys-loop receptors provide a fertile testing ground, J Med Chem 57(15) (2014) 6289–300. 10.1021/jm500023m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Franks NP, Structural comparisons of ligand-gated ion channels in open, closed, and desensitized states identify a novel propofol-binding site on mammalian gamma-aminobutyric acid type A receptors, Anesthesiology 122(4) (2015) 787–94. 10.1097/ALN.0000000000000588. [DOI] [PubMed] [Google Scholar]
- [56].Chen Q, Xu Y, Tang P, X-Ray Crystallographic Studies for Revealing Binding Sites of General Anesthetics in Pentameric Ligand-Gated Ion Channels, Methods Enzymol 603 (2018) 21–47. 10.1016/bs.mie.2018.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Bu W, Liang Q, Zhi L, Maciunas L, Loll PJ, Eckenhoff RG, Covarrubias M, Sites and Functional Consequence of Alkylphenol Anesthetic Binding to Kv1.2 Channels, Mol Neurobiol 55(2) (2018) 1692–1702. 10.1007/s12035-017-0437-2. [DOI] [PubMed] [Google Scholar]
- [58].Bertaccini EJ, Trudell JR, Franks NP, The common chemical motifs within anesthetic binding sites, Anesth Analg 104(2) (2007) 318–24. 10.1213/01.ane.0000253029.67331.8d. [DOI] [PubMed] [Google Scholar]
- [59].Yamakura T, Lewohl JM, Harris RA, Differential effects of general anesthetics on G protein-coupled inwardly rectifying and other potassium channels, Anesthesiology 95(1) (2001) 144–53. [DOI] [PubMed] [Google Scholar]
- [60].Minami K, Sudo Y, Shiraishi S, Seo M, Uezono Y, Analysis of the effects of anesthetics and ethanol on μ-opioid receptor, Journal of pharmacological sciences 112(4) (2010) 424–31. 10.1254/jphs.10003FP. [DOI] [PubMed] [Google Scholar]
- [61].Minami K, Uezono Y, The recent progress in research on effects of anesthetics and analgesics on G protein-coupled receptors, J Anesth 27(2) (2013) 284–92. 10.1007/s00540-012-1507-2. [DOI] [PubMed] [Google Scholar]
- [62].Antognini JF, Wang XW, Piercy M, Carstens E, Propofol directly depresses lumbar dorsal horn neuronal responses to noxious stimulation in goats, Can J Anaesth 47(3) (2000) 273–9. 10.1007/BF03018926. [DOI] [PubMed] [Google Scholar]
- [63].Takazawa T, Furue H, Nishikawa K, Uta D, Takeshima K, Goto F, Yoshimura M, Actions of propofol on substantia gelatinosa neurones in rat spinal cord revealed by in vitro and in vivo patch-clamp recordings, Eur J Neurosci 29(3) (2009) 518–28. 10.1111/j.1460-9568.2008.06607.x. [DOI] [PubMed] [Google Scholar]
- [64].Peng K, Liu HY, Wu SR, Liu H, Zhang ZC, Ji FH, Does Propofol Anesthesia Lead to Less Postoperative Pain Compared With Inhalational Anesthesia?: A Systematic Review and Meta-analysis, Anesth Analg 123(4) (2016) 846–58. 10.1213/ANE.0000000000001504. [DOI] [PubMed] [Google Scholar]
- [65].Rosen LG, Sun N, Rushlow W, Laviolette SR, Molecular and neuronal plasticity mechanisms in the amygdala-prefrontal cortical circuit: implications for opiate addiction memory formation, Frontiers in neuroscience 9 (2015) 399 10.3389/fnins.2015.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Langlois LD, Nugent FS, Opiates and Plasticity in the Ventral Tegmental Area, ACS Chem Neurosci 8(9) (2017) 1830–1838. 10.1021/acschemneuro.7b00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zhang H, Wang W, Zhao Z, Ge Y, Zhang J, Yu D, Chai W, Wu S, Xu L, The action sites of propofol in the normal human brain revealed by functional magnetic resonance imaging, Anat Rec (Hoboken) 293(12) (2010) 1985–90. 10.1002/ar.21069. [DOI] [PubMed] [Google Scholar]
- [68].Song XX, Yu BW, Anesthetic effects of propofol in the healthy human brain: functional imaging evidence, J Anesth 29(2) (2015) 279–88. 10.1007/s00540-014-1889-4. [DOI] [PubMed] [Google Scholar]
- [69].Woll KA, Murlidaran S, Pinch BJ, Henin J, Wang X, Salari R, Covarrubias M, Dailey WP, Brannigan G, Garcia BA, Eckenhoff RG, A Novel Bifunctional Alkylphenol Anesthetic Allows Characterization of GABAA Receptor Subunit Binding Selectivity in Synaptosomes, J Biol Chem 291(31) (2016) 20473–20486. 10.1074/jbc.M116.736975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ton HT, Phan TX, Abramyan AM, Shi L, Ahern GP, Identification of a putative binding site critical for general anesthetic activation of TRPA1, Proc Natl Acad Sci U S A 114(14) (2017) 3762–3767. 10.1073/pnas.1618144114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Woll KA, Skinner KA, Gianti E, Bhanu NV, Garcia BA, Carnevale V, Eckenhoff RG, Gaudet R, Sites Contributing to TRPA1 Activation by the Anesthetic Propofol Identified by Photoaffinity Labeling, Biophys J 113 (2017) 1–5. 10.1016/j.bpj.2017.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]