

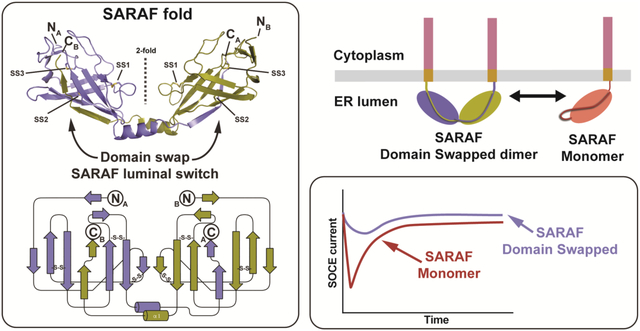
Abstract
Store Operated Calcium Entry (SOCE) plays key roles in cell proliferation, muscle contraction, immune responses, and memory formation. The coordinated interactions of a number of proteins from the plasma and endoplasmic reticulum (ER) membranes control SOCE to replenish internal Ca2+ stores and generate intracellular Ca2+ signals. SARAF, an ER resident component of the SOCE pathway having no homology to any characterized protein, serves as an important brake on SOCE. Here, we describe the X-ray crystal structure of the SARAF luminal domain, SARAFL. This domain forms a novel ten-stranded β-sandwich fold that includes a set of three conserved disulfide bonds, denoted the ‘SARAF-fold’. The structure reveals a domain-swapped dimer in which the last two β-strands (β9 and β10) are exchanged forming a region denoted the ‘SARAF luminal switch’ that is essential for dimerization. Sequence comparisons reveal that the SARAF-fold is highly conserved in vertebrates and in a variety of pathologic fungi. Förster Resonance Energy Transfer (FRET) experiments using full-length SARAF validate the formation of the domain-swapped dimer in cells and demonstrate that dimerization is reversible. A designed variant lacking the SARAF luminal switch shows that the domain swapping is essential to function and indicates that the SARAF dimer accelerates SOCE inactivation.
Keywords: Store operated calcium entry (SOCE); SARAF; X-ray crystallography; Domain swapping, β -sandwich fold, electrophysiology
Graphical Abstract
Calcium is a potent second messenger required for diverse cellular signaling processes that occur over a wide range of timescales such as vesicle release (μ s), fertilization (minutes), and proliferation and apoptosis (hours) [1, 2]. Consequently, cells use a multitude of systems to control cytoplasmic Ca2+ level changes. Signalling in both non-excitable and excitable cells is frequently initiated by stimulation of a G-protein coupled receptors and receptor tyrosine kinases [3, 4] that trigger inositol triphosphate (IP3)-mediated release of Ca2+ from intracellular stores. The resulting intracellular Ca2+ store depletion activates a process called Store Operated Calcium Entry (SOCE) that works to replenish the internal Ca2+ stores and that affects a range of responses, such as proliferation, transcription, and cell motility [5–9].
The prototypical mediator of SOCE is the calcium release-activated calcium (CRAC) channel formed by the plasma membrane (PM) pore-forming subunit, Orai, and an endoplasmic reticulum (ER) Ca2+ sensor and channel activator, STIM [10–13]. Both SOCE components have multiple isoforms of which the best studied are STIM1 and Orai1. SOCE activation involves an elegant mechanism that results in the clustering of both STIM and Orai at the ER-PM junctions [14–16]. Depletion of ER Ca2+ induces STIM1 oligomerization and translocation to the ER-PM junctions, where it binds to Orai1 and initiates Ca2+ influx [17–22]. Once SOCE is initiated, Ca2+ from CRAC channels initiates autoregulatory deactivation and inactivation processes that shape the duration and magnitude of the Ca2+ signal. Two types of Ca2+ dependent inactivation have been described: a fast process that occurs on the millisecond time scale, and a slow process that develops over multiple minutes [23]. The ER resident, single pass transmembrane SARAF [24] (for SOCE-Associated RegulAtory Factor) is a central facilitator of the slow Ca2+ dependent inactivation (SCDI) of CRAC channels.
SARAF lacks homology to any known protein. Previous studies established that the SARAF elements on either side of the membrane encode two distinct functions. The SARAF cytosolic domain is required for driving SOCE inactivation through interaction with STIM [24–26], whereas the luminal domain regulates SOCE inactivation by responding to changes in ER Ca2+ levels [24]. Here, we present the X-ray crystal structure of the SARAF ER luminal domain, SARAFL. This domain forms a domain-swapped dimer arrangement built from a novel β -sandwich fold that we term the ‘SARAF fold’. Cross-linking, analytical ultracentrifugation, and FRET experiments demonstrate that self-association of SARAFL and full-length SARAF in cells depends upon the domain-swapped element denoted as the ‘SARAF luminal switch’. Finally, CRAC current recording in cells that express wild-type SARAF or dimerization-incompetent SARAF show that self-association via the SARAF luminal switch is critical for control of CRAC currents.
Results
SARAF luminal domain dimerizes using a novel, conserved domain-swapped β -sandwich fold
The SARAF endoplasmic reticulum (ER) luminal domain contains six cysteines and bears no sequence homology to any protein of known structure (Fig. 1a and b). Therefore, we set out to define its structure to gain insight into how it might affect SOCE. Extensive screening identified a human SARAF luminal domain construct that could be expressed in Escherichia coli Shuffle Express cells, purified to homogeneity, and crystallized. This construct, denoted SARAFL, excludes the N-terminal signal peptide, encompasses luminal domain residues 30–164, and ends eight residues before the putative transmembrane helix [24, 27] (Fig. 1a). SARAFL crystals diffracted X-rays to a resolution of 1.9 Å and the structure was determined by single isomorphous replacement with anomalous scattering (SIRAS) using a single platinum derivative (K2Pt(NO)4) that diffracted X-rays to 2.15 Å (Fig. S1a, Table S1). SARAFL crystallized with two molecules in the asymmetric unit. One copy had continuous electron density from N- to C-terminus while the other showed short regions of disorder between residues 88–93 and 149–156 (Fig. S1b). Hence, the structural description focuses on the complete copy.
Fig. 1. Structure of SARAFL.

(a) SARAF schematic. Signal peptide (SP, grey), luminal (purple), transmembrane (TM, brown), and cytoplasmic (magenta) domains and amino acid boundaries of each are indicated. Extent of crystallized SARAFL construct is shown. (b) SARAFL sequence comparison from human (H. sapiens), pig (S. scrofa), mouse (M. musculus), frog (X. laevis), salmon (S. salar), and mushroom (S. commune). Invariant (blue), conserved (green) and cysteines (yellow) are highlighted. Secondary structure elements and disulfide bonds (SS1, SS2, and SS3) are shown. (c) Structure of the SARAFL domain swapped dimer. N and C termini (NA, NB, CA, and CB) and secondary structure elements of each chain are labeled. Disulfide bonds are shown as sticks and are labeled. Chains A and B are slate and deep olive, respectively. (d) SARAFL domain swapped dimer topology diagram. ‘S-S’ denotes disulfide bonds. (e) Detailed view of interactions of domain swapped β 9 and β 10 with the SARAFL core. Dashed lines indicate hydrogen bonds.
SARAFL forms a domain swapped dimer comprising a ten β -strand barrel having three conserved disulfides (Fig. 1b–d). There are two well-ordered extended loops that connect β 2-β 4 and β 6- β 8. Both are stabilized by the presence of a short intervening β -strand midway through the loop that forms β -sheet interactions with other β -strands (Fig. 1c and d). The β 3 strand in the middle of the β 2-β 4 loop makes a parallel β -sheet with β 8, whereas the β 7 strand in the β 6- β 8 loop forms an anti-parallel interaction with β 1. The β 2-β 4 and β 6- β 8 loops are further constrained by the Cys66-Cys73 and Cys114-Cys130 disulfides, respectively. The remaining disulfide, Cys83-Cys97, is buried in the core of the protein and links β 4 and β 5. There is a single β -helix, β 1, that follows β 8. This helix extends from the body of the structure and mediates a domain swap through which β -strands β 9 and β 10 complete the β -sheet fold of the other member of the dimer (Fig. 1e).
The domain swapped β 9- β 10 element comprising Gln152-Lys164 makes extensive interactions with the SARAFL core that are mediated by the formation of anti-parallel β -sheet main chain hydrogen bonds between β 9- β 5 and a short parallel β -sheet made between β 10 and β 6 and that bury 2232 Å2 per tail-body interface. (Fig.1c–e). The backbone β -sheet interactions are accompanied by a number of sidechain mediated interactions. Cross-strand hydrogen bonds are formed by His153-Tyr105 (Fig. S1c), a network comprising Ser157, Ser159, and Thr110 (Fig. S1d), and a Try163 hydroxyl and Tyr91 carbonyl interaction (Fig. 1e). Phe155 and Phe158 sidechains rest in shallow grooves on the surface the SARAFL core while the Tyr161 sidechain is buried in a largely hydrophobic pocket (Fig. 1e). Finally, the Lys164 C-terminal carboxylate forms a hydrogen bond to the amide of Gly116 and salt-bridge with the Arg57 sidechain (Fig. 1e). Despite being involved in a Ca2+-dependent process, SARAF lacks any obvious Ca2+-binding motifs [24]. Examination of the surface electrostatic potential (Fig. 2) revealed a few disperse regions of negative potential, but none that would indicate a site for Ca2+-binding, as well as a large positive patch in a pocket near SS3. In accordance with this lack of clear Ca2+-binding motifs, even though the structure was determined in the presence of 1 mM CaCl2, we found no crystallographic evidence for Ca2+ binding to SARAFL.
Fig. 2. SARAFL electrostatic surface potential.

(a) Electrostatic surface potential for the SARAFL dimer calculated in 150 mM ionic strength. (a) Electrostatic surface potential for one monomer of the SARAFL dimer. In both panels, select elements are labeled.
Searches for structural homology between SARAFL and proteins of known structure using the DALI database [28] revealed no strong matches, indicating that SARAFL has a unique fold. Relaxation of the similarity criteria to allow for a generous Z-score cutoff (>2) identified a set of β-sheet structures that include the γ -COPI appendage domain (1R4X), Xenavidin (2UYW), Avidin-related protein 2 (1WBI), and a conserved domain from Bacillus anthracis (3FBQ) (Z-scores of 2.3, 2.2, 2.1 and 2.0 respectively)(Fig. 3). However, strand connectivity analysis reveals that SARAFL is substantially different from each of these folds. Namely, SARAFL lacks the Greek-key motif of Xenavidin and Avidin related protein 2, and is not composed of strictly anti-parallel β-sheets as are the γ -COPI appendage domain and the Bacillus anthracis conserved domain. Hence, SARAFL has a novel β -sandwich architecture that we now call the SARAF-fold.
Fig. 3. Analysis of the SARAFL fold.

SARAFL structural homologs identified using DALI [28]. DALI search Z-score, RMSDCα, diagram of strand topology, and structure are shown. Cartoon representations highlighting secondary structure elements are colored by rainbow from N-terminus (blue) to C-terminus (red).
Analysis of >90 SARAFL-related sequences uncovers a set of related proteins spanning all five vertebrate classes (mammals, birds, amphibians, fish and reptiles), including some very ancient animals such as the sea lamprey (Petromyzon marinus) and coelacanth (Latimeria chalumnae). This family of SARAFL homologs all have strong conservation of the six cysteines that form the three SARAFL disulfides as well as high conservation of many residues that form the core of the SARAFL fold (Figs. 1b, 4a, and S2). We did not find SARAF-like sequences among other metazoans, but, surprisingly, identified a group of transmembrane proteins similar to SARAFL in fungi, including organisms that are pathogens of mammals, insects, or plants (Fig. S3). This group of SARAFL homologs diverges more from human SARAFL than the vertebrate sequences. For example, the exposed disulfide, SS1, lacks one of the cysteines in a small subset of the fungal sequences and the swapped β 9/β 10 strand is not well conserved. Nevertheless, the data clearly identify the presence of key elements of the core SARAFL structure (Fig. 4b). The unique nature of the SARAFL structure, together with the presence of clear homologs in vertebrates and fungi indicates that SARAFL structure represents a previously unknown, widely-occurring protein fold.
Fig. 4.

Conservation mapping of vertebrate sequences on the SARAFL structure for (a) vertebrate and (b) fungal sequences. One member of the dimer is shown in surface rendering. Select structural elements are labeled.
SARAFL dimerization depends on the swapped domain
The presence of the domain-swapped dimer in the crystals prompted us to investigate the nature of this interaction further. Characterization by size exclusion chromatography and multi-angle light scattering (SEC-MALS) [29] indicated the dominant presence of SARAFL monomers in solution at 64 μM (observed 15.84 ± 0.08 kDa, calculated 15.49 kDa) (Fig. 5a). Further probing with glutaraldehyde crosslinking revealed a dimeric species that appeared with increasing protein concentrations into the 100 μM range (Fig. 5b), suggesting that the propensity to dimerize is weak. As the domain swapped dimer observed requires an exchange of β 9 and β 10 (Fig. 1c and d), we created a SARAFL deletion construct truncated at residue 150 to remove the domain-swapped β -strands β 9 and β 10 (Δβ9/Δβ10). Expression and purification of SARAFL(Δβ9/Δβ10) yielded a protein having similar properties to SARAFL running as a monomer on gel filtration (Fig. S4a and b) and having a similar circular dichroism spectrum to SARAFL, indicative of a folded protein (Fig. S4c). Notably, the deletion of the domain-swapped strands, β 9 and β 10, dramatically diminished the ability of the protein to be crosslinked by glutaraldehyde (Fig. 5b), supporting the idea that the dimer seen in SARAFL relies on the domain swap interaction of β 9 and β 10To probe the strength of SARAFL dimer formation, we used equilibrium analytical ultracentrifugation. Fits of the SARAFL data using a single species monomer model yielded upwardly curving residuals, particularly at the highest concentration (200 μ M), that were indicative of a poor fit and the formation of a higher-order species (Fig. S4d). Accordingly, the SARAFL data could be well fit with a monomer-dimer association model (Fig. 5c and d), as indicated by the uniformly stochastic residuals. This analysis yields an estimate of the SARAFL dissociation constant in the low millimolar range (Kd ~2 mM). Such a value is entirely in line with the observation that SARAFL is monomeric under the low micromolar concentrations conditions used for SEC-MALS (Fig. 4a). In contrast to the behavior of SARAFL, equilibrium analytical sedimentation studies of SARAFL(Δβ9/Δβ10), which lacks the ability to form domain-swapped dimers, showed that this construct behaved as a monomeric protein that was well fit by a single species model (Fig. 5e). Together with the crosslinking studies, these data demonstrate that the domain swap of the SARAFL β 9/β10 element is essential for dimerization.
Fig. 5. SARAF self-association requires the SARAF luminal switch domain.

(a) SEC-MALS of 64 μM (1 mg ml−1) SARAFL (experimental Mw 15.84 kDa ± 0.08, Mw/Mn = 1.000 ± 0.007, predicted monomer Mw 15.49 kDa). (b) SDS-PAGE of glutaraldehyde crosslinked SARAFL and SARAFL (Δβ 9/Δβ 10) as a function of protein concentration indicates SARAFL dimerization. (c) Equilibrium ultracentrifugation of SARAFL at the indicated concentrations. Rotor speeds of 10K, 18K, 22K and 31K rpm are denoted by increasingly darker shades for each concentration. Residuals show fits to a monomer-dimer self-association model. (d) Calculated fraction of monomer and dimer SARAFL species as a function of concentration. (e) Equilibrium ultracentrifugation of SARAFL (Δβ 9/Δβ 10) at the indicated concentrations. Rotor speeds of 10K, 18K, 22K and 31K rpm are denoted by increasingly darker shades for each concentration. Residuals show fits to a single species model. (f) Example of images taken of cells expressing GFP-SARAF and mCherry-SARAF using DualView. The red channel is the FRET signal. (g) Bar graph describing the FRET/GFP signal ratio of acquired images as described in (f). Fluorescent tags are fused N-terminally to SARAF or SP-SARAFL-R (SARAF luminal domain (Δ165–339) with a signal peptide, SP, and an ER retention signal, R). Signals from cells expressing monomeric SP-GFP-R and SP-mCherry-R are used to establish the background signal. (h) SP-SARAFL-R (orange) inhibits the FRET signals between XFP-SARAF and (i) N-terminally tagged, SP-XFP-SARAFL-R, or C-terminally tagged, SP-SARAFL-XFP-R, luminal domains. For (g-i) *** denotes p<0.001. n for each combination is denoted above the bars.
SARAF self-associates in the ER
Given that purified SARAFL forms dimers solution, we sought to probe the extent to which such an interaction might occur in the context of a cell. We transfected HEK293 cells with equal amounts of full-length SARAF constructs bearing the fluorescent proteins, green fluorescent protein (GFP) [30] or mCherry [31], fused to the SARAF N-terminus (GFP-SARAF and mCherry-SARAF) and measured the Förster Resonance Energy Transfer (FRET) between the two constructs. We determined the amount of FRET by measuring the fluorescence emitted from mCherry under exclusive excitation of GFP, using Dual-View imaging, and quantified as the ratio of the red to green fluorescent intensities (FRET signal) (Fig 4f). We then used a similar approach with N-terminally-tagged soluble luminal domains SARAF(Δ165–339) that lacked the transmembrane anchor and that were targeted to the ER lumen by bearing both the SARAF signal peptide (SP) and a C-terminal retention signal (KDEL). FRET signals from the GFP-SARAF(Δ165–339) and mCherry-SARAF(Δ165–339) pair were substantially smaller than the GFP-SARAF and mCherry-SARAF pair but were still well above background (Fig. 5g). These results suggest that, in line with the biochemical studies, the SARAF luminal domains self-associate in the ER. This association happens whether the luminal domain is soluble form confined to the ER or is membrane anchored. The stronger FRET signals from the full-length constructs indicate that membrane anchoring enhances the luminal domain effective concentration [32, 33] and facilitates self-association.
Because of the domain swapped SARAFL dimer architecture, there is a much shorter distance between the N-termini and C-termini of the dimer partners (19.6 Å, Cα-Cα) than between the N- and C- termini of an individual subunit (66.7Å, Cα-Cα) or the N- and C-termini of the dimer partners (75.4 Å and 61.8 Å, respectively) (Fig. S4e). These constraints predict that FRET signals will be larger between constructs in which the fluorophores are placed on opposite termini of the tested pairs (i.e. N-donor and C-acceptor). In line with this prediction, FRET between the GFP-SARAFL:SARAFL-mCherry pair was ~3-fold larger than the FRET signals observed from co-expressed pairs having each fluorophore fused to the SARAFL N-terminus, GFP-SARAF and mCherry-SARAF (Fig. 5h). Furthermore, in both cases, co-expression of untagged SARAF(Δ165–339) reduced the FRET signals to background levels, indicating that the FRET signals derive from co- association of the test proteins (Fig. 5i). Together, these data strongly support the notion that the domain swapped form of the SARAFL occurs in a cellular context, forms in the full-length protein in cell membranes, that similar to in solution, self-association is reversible.
Design and characterization of SARAFL ‘Cys-lock’ mutants identifies the SARAF luminal switch domain
The crosslinking and sedimentation equilibrium studies suggest that domain-swapped dimer exchanges freely with the monomeric state. In order to characterize properties of the monomer and dimer forms separately, we set out to create a SARAFL mutant that would be incapable of domain swap. We reasoned that incorporation of a fourth disulfide bond between the swapped strand and the core of SARAFL structure could serve as a ‘Cys-lock’ that would covalently tether the swapped β 9/β 10 strand to the body of the protein. We identified a residue pair, Lys98-Ala156 on strands β 6 and β 9, as having favorable geometry to form such a disulfide when each member was mutated to cysteine. Purification of SARAFL K98C/A156C by ion exchange chromatography revealed the presence of two species corresponding to monomer and dimer forms present in a ratio of ~10:1 (Fig. S4f) and having different mobilities on size exclusion chromatography (Fig. 6a). SDS-PAGE under non-reducing conditions revealed two, separate species that ran at molecular weights consistent with the monomeric form (15.5 kDa) and the dimeric form (31.0 kDa). By contrast, both species ran identically upon addition of reducing agent (Fig. 6b), indicating that the two forms are the disulfide linked dimer (16.5 ml form) and disulfide linked monomer (18.3 ml form), respectively (Fig. 6a). Both SARAFL K98C/A156C forms produced crystals that diffracted X-rays to high resolution, 1.58Å and 2.1Å for the monomer and dimer, respectively (Table S1). Molecular replacement using the SARAFL core lacking the surface loops and the β 9/β10 strand, revealed clear electron density in both structures for the engineered K98C/A156C ‘Cys-lock’ disulfide (Fig. 6c and d). Model building and refinement revealed that the Cys-lock monomer and Cys-lock dimer protomer maintain the same overall fold as the wild-type SARAFL dimer protomer (Fig. 6e) (RMSDCα = 0.739 and 0.590 for residues 33–145 of the Cys-lock monomer and Cys-lock dimer versus SARAFL, respectively). Notably, the SARAFL K98C/A156C Cys-lock monomer has the marked difference that the tail, which extends from the core in the dimeric form, is wrapped under the bottom of SARAFL so that β 9/β 10 inserts in cis to complete the anti-parallel and mixed β -sheets of the SARAFL fold (Fig. 6d). The α1helix is retained despite this topology change of the SARAFL tail, although it shorter by one helical turn relative to the dimer form. In both the Cys-locked dimer and the Cys-locked monomer the electrostatic surface potentials were very similar to wild-type SARAFL (Fig. S5). These structural studies demonstrate that the switch from monomer to dimer requires changes only in the β 9/β 10 tail and not the SARAFL core. Accordingly, we term the β 9/β 10 tail as the ‘SARAF luminal switch domain’.
Fig. 6. Characterization and structures of Cys-locked SARAFL mutants.

(a) Size exclusion chromatography profiles monomer and dimer species of SARAFL K98C/A156C. The Cys-locked dimer species elution profile is shifted earlier, consistent with a larger hydrodynamic radius. (b) SDS -PAGE gel of purified SARAFL K98C/A156C monomer (M) and dimer (D) species. The dimer band collapses to the same size as the monomer band upon addition of reducing agent (β ME). (c) Crystal structure of the Cys-locked SARAFL K98C/A156C dimer showing the same topology and disulfide bonding as wild type SARAFL. Inset shows electron density (blue mesh, 1.5 β) for the engineered extra 4th disulfide bond (SS4). (d) Crystal structure of the Cys-locked SARAFL K98C/A156C monomer again showing the same strand arrangement and topology as wild type SARAFL with the exception of β 9 and β 10 inserting in cis into the monomer β -sheet. Inset shows electron density (blue mesh, 1.5 β) for the engineered 4th disulfide bond (SS4). (e) Superposition of wild type SARAFL (smudge green), Cys- locked SARAFL K98C/A156C dimer protomers (orange and deep teal) and Cys-locked SARAFL K98C/A156C monomer (firebrick red) showing the fold conservation across all three structures.
SARAF dimerization affects SOCE inactivation
To test the functional importance of the SARAF luminal switch, we used whole-cell patch clamp to measure SOCE currents from HEK293 cells that were co-transfected with Orai1-CFP and STIM1-mCherry along with GFP tagged version of SARAF, SARAF-GFP, or a SARAF mutant lacking the luminal switch, SARAF (Δβ9/Δβ10)-GFP. Recording conditions included 1,4-Dihydroxy-2,5-di-tert-butylbenzene, (BHQ), a reversible SERCA inhibitor, to allow for both depletion and refilling of Ca2+ stores. Because we observed a direct relationship between STIM1 expression level and Orai1 current density (Fig. S6), we limited our analysis to cells expressing STIM1-mCherry at or below a threshold of 5000 fluorescence counts/cell to ensure that any functional effects were not due to exceptionally high levels of STIM1. Cells expressing SARAF-GFP or SARAF(β 9/β10)-GFP had comparable levels of STIM1 expression (Fig. 7a and b) and similar passive membrane properties. However, we observed that the SOCE current densities 18s after addition of external Ca2+ to activate the current were roughly twice as large in cells expressing SARAF(β 9/β 10)-GFP versus SARAF-GFP (42.6±7.5 pA/pF (n=9) and 21.2±4.3 pA/pF (n=11) p<0.05, respectively) (Fig.6c). Further, the time to maximum response for SOCE current activation was faster by ~2-fold in cells expressing the SARAF luminal switch mutant (17.1±1.7s and 32.5±4.6s (n=11) and (n=9), p<0.01, for SARAF (β 9/β10)-GFP and SARAF-GFP, respectively) (Fig. 7d). These results indicate that the self-associated form of SARAF accelerates SOCE inactivation and that domain swap of the luminal switch is important for stabilizing this state.
Fig. 7. Self-associated SARAF accelerates SOCE inactivation.

(a) STIM1-mCherry and (b) SARAF-GFP expression measured by fluorescence using epifluorescence illumination of cells expressing Ora1-CFP, STIM1-mCherry and SARAF-GFP or SARAF ((Δβ 9/(Δβ 10)-GFP. (c) Average La3+-sensitive current densities of HEK293 cells expressing Orai1-CFP, STIM1-mCherry, and SARAF-GFP or SARAF ((Δβ 9/(Δβ 10)-GFP measured at the end of 60 ms hyperpolarizing pulse to −100 mV in the presence of 10 mM extracellular Ca2+. Inset shows the first 50s of recording. Stimulation protocol is shown and was applied at 0.5 Hz. (d), Time to peak measured following the application of 10 mM Ca2+ for SARAF and SARAF ((Δβ 9/(Δβ 10). n= 11 and 9, respectively. ‘*’ indicates p<0.05. ‘**’ indicates p<0.01 n.s., not statistically different. Error bars indicate s.e.m.
Discussion
SARAF is a transmembrane, ER-resident, negative regulator of SOCE [24]. Understanding how it influences SOCE has been limited due to its lack of similarity to proteins of known structure. Crystallographic determination of the structure of the SARAF luminal domain, SARAFL, shows that this domain comprises a novel, ten-strand β -sheet fold constrained by three conserved disulfides that we name the ‘SARAF-fold’. Although, the SARAF fold belongs to a class of β -sheet sandwich proteins represented by the γ -COPI appendage domain (1R4X), Xenavidin (2UYW), Avidin-related protein 2 (1WBI), and a Bacillus anthracis conserved domain (3FBQ) (Fig. 3), the SARAF fold and topology are unique. Homologs having all six cysteines that form the three SARAFL disulfides as well as many conserved core residues in the SARAFL core occur in all five vertebrate classes (mammals, birds, amphibians, fish and reptiles), including quite ancient members of this phylum, such as the sea lamprey (Petromyzon marinus) and coelacanth (Latimeria chalumnae) (Figs. 1b, 4a and S2). Although SARAFL is widespread among vertebrates, it appears to be absent from other metazoans. Intriguingly, we found a set of fungal transmembrane proteins that are also SARAFL homologs (Figs. 4b and S3). This unusual distribution of homologs in both vertebrates and fungi establishes that the previously unknown SARAF-fold is widespread. Because fungi are not known to use the SOCE pathway, it seems likely that the SARAF-fold has other functions beyond its role in SOCE regulation.
Apart from its unique fold, our structural studies revealed a second key feature of the SARAFL fold, the ability to domain-swap. The formation of intertwined protein assemblies by exchange of identical structural elements is observed in many classes of soluble and transmembrane proteins [34–37]. Although this phenomenon provides a straightforward mechanism for homo-oligomer formation, its functional relevance is often not clear [34, 37, 38]. In SARAFL, the swapped domain is a simple element comprising the β 9 and β 10 β -strands that insert into the essentially rigid, stable core of the rest of the fold.
Two structure-based protein design strategies firmly establish that the β 9/β 10 strands form the dimerization element. First, design of SARAFL mutant lacking β 9 and β 10 yielded a well-folded, stable protein that only differed from SARAFL in its inability to dimerize. Second, structure-based design to incorporate a disulfide between β 9 and the SARAFL core yielded two covalently trapped species, a covalent dimer and a self-ligated monomer. Structural studies show that apart from the topological change, these two forms are identical. The intrinsic affinity of SARAF luminal switch mediated dimerization is modest having a dissociation constant in the low millimolar range (Kd~ 2 mM). Nevertheless, the transmembrane nature of full-length SARAF clearly imposes diffusional restrictions that serve to increase the effective concentration [32, 33] and favor self-association(Fig. 5g). Importantly, overexpression of non-membrane anchored SARAFL is able to suppress the amount of dimer, indicating that full-length SARAF self-association is reversible (Fig. 5h). Hence, we term the β 9/β 10 element as the ‘SARAF luminal switch’ domain, as biochemically this is the sole element required to convert between monomeric and dimeric forms and this domain is necessary for self-association of SARAF in membranes.
A hallmark of the SOCE pathway is that it is governed by reversible self-association of the ER sensor, STIM, in response to changes in ER calcium levels [15, 17–22]. SARAF functional studies indicate that domain-swap mediated self-association is key for the ability of SARAF to accelerate inactivation of the SOCE current (Fig. 7). Whether, as with STIM1, SARAF also forms higher order oligomers remains to be elucidated. In this regard, it is worth noting that crystal packing of SARAFL shows a simple face-to-face arrangement in which the C-termini that link SARAFL to the transmembrane portion all face the same direction (Fig. S1b). Such an arrangement would be compatible with formation of higher order assemblies of SARAF dimers in the context of the membrane. Nevertheless, even though the self-associated form of SARAFL is a dimer in both the crystal structure and in solution, non-symmetric domain swapping via the SARAF luminal switch could also form higher-order domain swapped assemblies as has been observed in other domain-swapped proteins [38]. Hence, even though SARAF clearly self-associates in the membrane (Fig. 5g–i), and this association is important for function, the stoichiometry of self-organization and how such assemblies might be affected by interactions with STIM or Orai requires further investigation.
Initial characterization of SARAF demonstrated that it acts in a calcium-dependent manner, inhibiting SOCE only when ER stores were refilled with calcium [24]. SARAFL has no identifiable Ca2+-binding motifs and despite being crystallized in 1 mM CaCl2, there is no evidence for non-canonical binding sites for Ca2+ on SARAFL. Hence, how SARAF senses and responds to ER Ca2+ changes remains to be discovered. Given the absence of Ca2+ binding to SARAFL, it may be that there is some type of Ca2+-mediated interaction of SARAFL with the membrane inner leaflet or Ca2+-dependent control of SARAF by a yet to be defined ER Ca2+-sensor protein.
SOCE is a complex, multicomponent process that involves the reversible, coordinated association of proteins in both the ER and plasma membranes [15]. The ER resident transmembrane protein SARAF serves an important role in this phenomenon by fine-tuning SOCE activity in response to ER refilling with Ca2+. The discovery that the SARAF luminal domain has a unique, disulfide-bonded β -sheet protein fold, capable of domain-swap mediated self-assembly that impacts function sets a key structural framework for understanding the basic roles of SARAF in controlling SOCE and the possible roles for SARAF in cancer, neurodegenerative diseases, and cardiomyopathy [39–43].
Materials and Methods
Protein expression and purification
Human SARAFL (residues 30–164) or SARAFL((Δβ9/(Δβ10) (residues 30–150) were cloned into a modified version of the pET28 vector containing an N-terminal combination His6 and Maltose Binding protein tag followed by TEV protease site (HMT). Point mutants were introduced using site directed mutagenesis. The SARAFL or SARAFL((Δβ9/(Δβ10) constructs were transformed into SHuffle Express cells (NEB) and grown in 1L cultures of 2YT media at 37°C. The SHuffle Express cells have deletions of the genes for glutaredoxin reductase and thioredoxin reductase (Δgor ΔtrxB), which allow disulfide bonds to form in the cytoplasm. Additionally, they constitutively express a chromosomal copy of the disulfide bond isomerase DsbC allowing for rearrangement of improperly oxidized disulfides. Cultures were induced with 0.5 mM IPTG at an OD600nm of ~0.6 and moved to 24°C. Protein was expressed overnight and cells were harvested by centrifugation and then immediately flash frozen in liquid nitrogen and stored at −80°C. Frozen cell pellets were thawed on ice and resuspended in lysis buffer (25 mM HEPES pH 7.4, 300 mM KCl, 1 mM CaCl2, 10 mM Imidazole, 1 mM PMSF, 1μg ml−1 DNaseI) at a ratio 6 ml lysis buffer to 1g cell pellet. Resuspended cells were disrupted by sonication and insoluble material pelleted by centrifugation. Clarified lysate was mixed with 2 ml bed volume of Talon beads (Clonetech) and incubated while rocking for 1 hour at 4°C. Following incubation, beads and lysate were transferred to a gravity flow column at washed with 2 × 30ml lysis buffer before eluting with 15 ml lysis buffer containing 400 mM imidazole. After elution, protein was digested overnight with TEV at 4°C and buffer exchanged into 10 mM Tris, pH 8.8, 10 mM KCl over a HiPrep desalting column. Desalted SARAF was then passed over a POROS MC20 column followed by an Amylose column to remove TEV and cleaved MBP. SARAF was further purified by MonoQ ion exchange before final gel filtration in 10 mM HEPES, pH7.4, 200 mM KCl, 1 mM CaCl2 and concentrated to 10 mg ml−1 for crystallization.
The double mutant SARAFL K98C/A156C was expressed and purified following the same protocol as wild type protein with the separation of monomer and dimer populations occurring at the ion exchange step before proceeding on to gel filtration. SARAF 31–164 K98C/A156C monomer and dimer were concentrated to 10 mg ml−1 and 4mg ml−1 respectively prior to crystallization.
Crystallization and Data Collection
Crystals used for structure determination of the wild type construct were grown in 0.1 M BisTris, pH 6.5, 18–20% PEG 5000 MME in hanging drop format. Crystallization drops were set over a thin layer of vacuum grease to prevent crystals from sticking to the cover slips and facilitate harvesting. Harvested crystals were cryoprotected by sequential soaks in mother liquor plus 5%, 10%, 15% and 20% glycerol before flash cooling in liquid nitrogen. For experimental phasing, crystals were soaked with 1 mM K2Pt(NO2)4 overnight before back-soaking into mother liquor and cryoprotecting in glycerol as above. Crystals of SARAF 30–164 K98C/A156C monomer were grown in 0.1 M sodium acetate, pH 4.2–4.6 and 1.2–1.6 M sodium formate in standard hanging drop format without the use of vacuum grease. Harvested crystals were cryoprotected by sequential soaks in mother liquor plus 5%, 10%, 15% and 20% glycerol before flash cooling in liquid nitrogen. Crystals of SARAF 30–164 K98C/A156C dimer were grown in 3–5% glycerol or ethylene glycol after first treating protein to mild heating at 37°C for 5 minutes or ultracentrifigation (40000 rpm, 30min, 4°C) to remove microcrystals that spontaneously form in concentrated protein solution. Harvested crystals were cryoprotected by directly soaking in 25% ethylene glycol before flash cooling in liquid nitrogen
Data were collected at Advanced Light Source beamline 8.3.1. Native datasets of wild type SARAF were collected at a wavelength of 1.127Å and diffracted to 1.90Å. Peak, inflection and high remote datasets were collected from K2Pt(NO2)4-soaked crystals and diffracted to a resolution of 2.15Å. Native datasets for both SARAF K98C/A156C monomer and dimer were collected at a wavelength of 1.116Å and diffracted to resolutions of 1.58Å and 2.10Å respectively.
Data Processing and Structure Determination
Data from both native and K2Pt(NO2)4-soaked wild type crystals were indexed, integrated, and scaled in spacegroup C2 using autoPROC [44]. autoSHARP [45] was used to determine initial experimental phases using peak, inflection, and remote datasets from K2Pt(NO2)4-soaked crystals finding a total of 10 platinum sites. An initial model was obtained using ARP/wARP [46] and improved with iterative rounds of manual rebuilding with COOT [47] and refinement with Phenix [48]. The SARAF K98C/A156C monomer and dimer structures were phased by molecular replacement with PHASER [49] using the wild type structure with the tail and surface loops removed as a search model. As with the wild type structure, the models were improved with iterative rounds of manual rebuilding in COOT [47] and refinement in Phenix [48].
Analytical Ultracentrifugation
Sedimentation equilibrium experiments were performed at 4°C in an Optima XL-I analytical ultracentrifuge (Beckman Coulter). Prior to loading the rotor cells, 500 μl of SARAFL or SARAFL((Δ 9/(Δβ10) was dialyzed against 1 L of buffer (200 mM KCl, 10 mM HEPES, pH 7.4) overnight at 4°C. 125 μl of SARAFL or SARAFL((Δβ 9/(Δβ 10) were loaded into six chamber center pieces at three concentrations of 20 μM, 60 μM and 200 μM determined by absorbance at 280 nm [50]. 115 μl of dialysate buffer was loaded into adjacent reference chambers. Data were acquired using interference optics at rotor speeds of 10K, 18K, 22K and 31K rpm. Data acquired at multiple loading concentrations and rotor speeds were modeled globally in IgorPro using a standard monomer-dimer self-association model. Vbar and solvent density were calculated using Sednterp and the interference extinction coefficient was calculated using the formula eint = 2.733 * MW. For global fitting, Vbar, MW, solvent density and eint were held constant while Kd (for N = 2) was allowed to float.
Glutaraldehyde crosslinking
Purified SARAFL or SARAFL((Δβ9/(Δβ10) in a buffer of 10 mM HEPES, pH 7.4, 200 mM KCl at concentrations ranging from 25 μM to 100 μM, determined by absorbance at 280 nm [50] were combined with 0.01% glutaraldehyde (Aldrich) in a final volume of 10 μl and incubated at room temperature for 10 minutes. Reactions were quenched by adding 1 μl of 1 M Tris pH 8.0 to achieve a final concentration of 100 mM, Tris pH 8 and were then boiled for 10 min in reducing SDS-sample buffer prior. Samples were subsequently analyzed by SDS-PAGE.
SEC-MALS
Multi-angle light scattering (MALS) experiments were carried out at 4°C using an HPLC (Shimadzu) with UV detector connected to a miniDAWN TREOS MALS detector and an Optilab T-rEX refractometer (Wyatt Technology). 100 μl of 1mg ml−1 of purified SARAFL was injected onto a Superdex S200 10/300 GL column (GE Healthcare) equilibrated in 200 mM KCl, 10 mM HEPES, pH7.4, and eluted peak was detected online. Molecular weight was calculated at each time point during elution using a combination of UV absorbance, light scattering and differential refractive index measurements with the Astra software package (Astra 6.0, Wyatt Technology). The experimentally determined molecular weight of SARAFL of 15.84 kDa (+/− 0.509%) compares well with the 15.49kDa calculated from the protein sequence. SARAFL was monodisperse with a polydispersity ratio (Mw/Mn) of 1.000 (+/− 0.719%).
Circular Dichroism
Prior to measurement, 600 μl each of 10 μM SARAFL and 10 μM SARAFL ((Δβ 9/(Δβ 10), determined by absorbance at 280 nm [50], were dialyzed overnight at 4°C against 1 L of 30 mM sodium phosphate buffer. Circular dichroism spectra were measured with a 1 mm path length quartz cuvette using an Aviv model 215 spectropolarimeter (Aviv Biomedical) equipped with a Peltier temperature controller. Wavelength scans from 320 to 185 nm were taken at 1 nm intervals at 4°C. Each scan was performed in triplicate from the same sample and subtracted by the average of a triplicate scan of the dialysate for a matched buffer blank. Molar ellipticity was calculated as follows: θ = 100(Δm)/(Cnl), where Δm is the CD signal in millidegrees after buffer subtraction, C is the millimolar peptide concentration, n is the number of residues in the peptide, and l is the cuvette path length in centimeters.
Cell Culture
HEK293 cells were grown in DMEM supplied with fresh L-glutamine, sodium pyruvate, FBS and penicillin/streptomycin. Cells were split to 35mm plates one day prior to transfection. Transfection with 600 ng of C-terminally GFP-tagged SARAF or SARAF((Δβ9/(Δβ 10), 200 ng of C-terminally mCherry- tagged STIM1 and 200 ng of C-terminally CFP-tagged Orai1 with PEImax reagent (Polysciences) was performed 22–36 hours prior to imaging and electrophysiological experiments. Cells were transferred to poly-L-lysine covered 24 mm cover slips one night prior to the experiments.
Forster Resonance Energy Transfer
Cells were excited with 470/40 nm light for FRET measurements, fluorescent signals were collected through the objective and split using Dual-View device (565LP dichroic) to GFP (525/50) and mCherry (650/75) channels using EMCCD 512×512 (Princeton Instruments). Images were processed using SlideBook (Intelligent Imaging Innovations) software and exported as Microsoft Excel files. FRET signals were assessed by dividing the FRET (mCherry) channel by the GFP channel. GFP or mCherry were fused to the N-termini of SARAFL (XFP-SARAFL) or SARAF full length (XFP-SARAF) using the following sequence, SARAF(M1-G30)-TG-XFP-RT-SARAF(W31-K164)-KDEL or SARAF(M1-G30)-TG-XFP-RT-SARAF(W31-R339), respectively. Fusions of monomeric forms of GFP [30] or mCherry [31] were made at the end of the luminal domain (K164) of the SARAFL (SARAFL- XFP) or full length SARAF were at position K164 using the following sequence SARAF(1–164)-TG RPACKIPNDLKQKVMNH-XFP- KDEL or SARAF(M1-K164)- TGLGGGGSGGGGSGGGGSAAARPACKIPNDLKQKVMNH-XFP- LGGGGSGGGGSGGGGSAAASGLRS-SARAF(M173-R339), respectively. ER retention signal (KDEL) was added at the C-termini of all luminal SARAF constructs.
Electrophysiological recordings
Membrane currents were recorded under voltage-clamp conditions using the whole-cell patch-clamp configuration using an Axopatch 200B (Axon Instruments) amplifier. Patch pipettes were fabricated from borosilicate glass capillaries (2–5 MΩ). Signals were analog filtered using a 2 kHz low-pass Bessel filter. Data acquisition and analysis were done using pCLAMP 9 software (Molecular Devices). Current densities were calculated by normalizing currents to cell capacitance. The recording protocol consisted of a 60 ms hyperpolarizing step to −100 mV from 0 mV, followed by a 20 ms step to 0 mV, and a 120 ms ramp from −100 mV to +100 mV repeated at 0.5 Hz for 300s. All data were leak- corrected using the current in lanthanum-containing solution. The EGTA (ethylene glycol-bis(β- aminoethyl ether)-N,N,N′,N′-tetraacetic acid) internal solution contained 150 mM Cs aspartate, 8 mM MgCl2, 1.2 mM EGTA, 10 mM HEPES and 2 mM Mg-ATP. The pH was titrated to pH 7.2 with CsOH.
The external solution contained 145 mM NaCl, 2 mM MgCl2, 2.8 mM KCl, 10 mM CsCl, 10 mM HEPES and 10 mM Glucose. The pH of the external solution was titrated to pH7.4 with NaOH. 10 mM CaCl2, 100 μM EDTA (ethylenediaminetetraacetic acid) or 10 mM MgCl2, 1 mM EDTA were added to the external solution for high-Ca2+ or Ca-free solution, respectively. Fluorescence was measured from images acquired using EMCCD 1024×1024 iXonUltra camera under epifluorescence, and analyzed with VisiView software (Visitron Systems GmbH). Cells were excited using Xcite Exacte (Excelitas technologies) using the following filters: excitation filters ET470/40 for GFP and FF01–580/14 for mCherry, emitted light was filtered with BP536/40 for GFP and HQ650/75 for mCherry. Data are presented as mean +/− S.E.M.
Supplementary Material
Highlights.
SARAF luminal domain, SARAFL is a novel, (β -sandwich fold
SARAFL associates as a domain-swapped dimer using the SARAF luminal switch segment
Self-association of SARAF via the luminal switch accelerates SOCE inactivation
Acknowledgements
This work was supported by grants NIH-NHLBI R01-HL080050 and NIH-NIDCD R01-DC007664 to D.L.M., Israeli Science Foundation grant 1248/15 to E.R. US-Israel Binational Science Foundation 2015298 to D.L.M. and E.R, AHA 14POST18740062 and NIH-HLBI T32HL007731 to C.K., and an Israeli Ministry of Aliyah and Integration fellowship to A.M. E.R. is the incumbent of the Charles H. Hollenberg Professorial Chair.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers: Coordinates and structure factors for SARAFL, SARAFL.SS Monomer, and SARAFL.SS Dimer are deposited with the RCSB under accession codes 6O2U, 6O2V, and 6O2W, respectively, and will be released immediately upon publication.
Declaration of Interests
The authors declare no competing interests.
References
- [1].Clapham DE. Calcium signaling. Cell. 2007;131:1047–58. [DOI] [PubMed] [Google Scholar]
- [2].Hille B Ion Channels of Excitable Membranes. 3rd ed. Sunderland, MA: Sinauer Associates, Inc.; 2001. [Google Scholar]
- [3].Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nature Rev Mol Cell Biol. 2003;4:517–29. [DOI] [PubMed] [Google Scholar]
- [4].Carafoli E, Santella L, Branca D, Brini M. Generation, control, and processing of cellular calcium signals. Critical reviews in biochemistry and molecular biology. 2001;36:107–260. [DOI] [PubMed] [Google Scholar]
- [5].Hodeify R, Yu F, Courjaret R, Nader N, Dib M, Sun L, et al. Regulation and Role of Store-Operated Ca(2+) Entry in Cellular Proliferation In: Kozak JA, Putney JW Jr., editors. Calcium Entry Channels in Non-Excitable Cells. Boca Raton (FL)2018. p. 215–40. [Google Scholar]
- [6].Cahalan MD. STIMulating store-operated Ca(2+) entry. Nat Cell Biol. 2009;11:669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–7. [DOI] [PubMed] [Google Scholar]
- [8].Putney JW Jr. A model for receptor-regulated calcium entry. Cell calcium. 1986;7:1–12. [DOI] [PubMed] [Google Scholar]
- [9].Shim AH, Tirado-Lee L, Prakriya M. Structural and Functional Mechanisms of CRAC Channel Regulation. Journal of molecular biology. 2015;427:77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Feske S A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–85. [DOI] [PubMed] [Google Scholar]
- [11].Peinelt C Amplification of CRAC current by STIM1 and CRACM1 (Orai1). Nature Cell Biol. 2006;8:771–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Roos J STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Soboloff J STIM2 is an inhibitor of STIM1-mediated store-operated Ca2+ entry. Curr Biol. 2006;16:1465–70. [DOI] [PubMed] [Google Scholar]
- [14].Poteser M, Leitinger G, Pritz E, Platzer D, Frischauf I, Romanin C, et al. Live-cell imaging of ER- PM contact architecture by a novel TIRFM approach reveals extension of junctions in response to store-operated Ca(2+)-entry. Sci Rep. 2016;6:35656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Prakriya M, Lewis RS. Store-Operated Calcium Channels. Physiol Rev. 2015;95:1383–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Qiu R, Lewis RS. Structural features of STIM and Orai underlying store-operated calcium entry. Curr Opin Cell Biol. 2019;57:90–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bird GS, Hwang SY, Smyth JT, Fukushima M, Boyles RR, Putney JW Jr. STIM1 is a calcium sensor specialized for digital signaling. Curr Biol. 2009;19:1724–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Kawasaki T, Lange I, Feske S. A minimal regulatory domain in the C terminus of STIM1 binds to and activates ORAI1 CRAC channels. Biochem Biophys Res Commun. 2009;385:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lee KP. Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc Natl Acad Sci USA. 2009;106:14687–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wu MM, Buchanan J, Luik RM, Lewis RS. Ca2+ store depletion causes STIM1 to accumulate in ER regions closely associated with the plasma membrane. J Cell Biol. 2006;174:803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhou Y The short N-terminal domains of STIM1 and STIM2 control the activation kinetics of Orai1 channels. J Biol Chem. 2009;284:19164–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Parekh AB. Regulation of CRAC channels by Ca(2+)-dependent inactivation. Cell calcium. 2017;63:20–3. [DOI] [PubMed] [Google Scholar]
- [24].Palty R, Raveh A, Kamisky I, Meller R, Reuveny E. SARAF inactivates the store operated calcium entry machinery to prevent excess calcium refilling. Cell. 2012;149:425–38. [DOI] [PubMed] [Google Scholar]
- [25].Albarran L, Lopez JJ, Gomez LJ, Salido GM, Rosado JA. SARAF modulates TRPC1, but not TRPC6, channel function in a STIM1-independent manner. Biochem J. 2016;473:3581–95. [DOI] [PubMed] [Google Scholar]
- [26].Jha A, Ahuja M, Maleth J, Moreno CM, Yuan JP, Kim MS, et al. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. The Journal of cell biology. 2013;202:71–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zhang Z, Henzel WJ. Signal peptide prediction based on analysis of experimentally verified cleavage sites. Protein science : a publication of the Protein Society. 2004;13:2819–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Holm L, Rosenstrom P. Dali server: conservation mapping in 3D. Nucleic acids research. 2010;38:W545–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Folta-Stogniew E Oligomeric states of proteins determined by size-exclusion chromatography coupled with light scattering, absorbance, and refractive index detectors. Methods Mol Biol. 2006;328:97–112. [DOI] [PubMed] [Google Scholar]
- [30].Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 2002;296:913–6. [DOI] [PubMed] [Google Scholar]
- [31].Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nature biotechnology. 2004;22:1567–72. [DOI] [PubMed] [Google Scholar]
- [32].Jencks WP. Catalysis in Chemistry and Enzymology. Dover ed. New York, NY: Dover Publications; 1987. [Google Scholar]
- [33].Creighton TE. Proteins: Structures and Molecular Properties. 2nd ed. New York: W.H. Freeman and Company; 1993. [Google Scholar]
- [34].Wodak SJ, Malevanets A, MacKinnon SS. The Landscape of Intertwined Associations in Homooligomeric Proteins. Biophysical journal. 2015;109:1087–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Bennett MJ, Eisenberg D. The evolving role of 3D domain swapping in proteins. Structure. 2004;12:1339–41. [DOI] [PubMed] [Google Scholar]
- [36].Bennett MJ, Choe S, Eisenberg D. Domain swapping: entangling alliances between proteins. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rousseau F, Schymkowitz J, Itzhaki LS. Implications of 3D domain swapping for protein folding, misfolding and function. Advances in experimental medicine and biology. 2012;747:137–52. [DOI] [PubMed] [Google Scholar]
- [38].Mackinnon SS, Malevanets A, Wodak SJ. Intertwined associations in structures of homooligomeric proteins. Structure. 2013;21:638–49. [DOI] [PubMed] [Google Scholar]
- [39].Chen X, Long F, Cai B, Chen X, Chen G. A novel relationship for schizophrenia, bipolar and major depressive disorder Part 7: A hint from chromosome 7 high density association screen. Behavioural brain research. 2015;293:241–51. [DOI] [PubMed] [Google Scholar]
- [40].Romanuik TL, Ueda T, Le N, Haile S, Yong TM, Thomson T, et al. Novel biomarkers for prostate cancer including noncoding transcripts. Am J Pathol. 2009;175:2264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Romanuik TL, Wang G, Holt RA, Jones SJ, Marra MA, Sadar MD. Identification of novel androgen-responsive genes by sequencing of LongSAGE libraries. BMC Genomics. 2009;10:476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Twine NA, Janitz K, Wilkins MR, Janitz M. Whole transcriptome sequencing reveals gene expression and splicing differences in brain regions affected by Alzheimer’s disease. PloS one. 2011;6:e16266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Camargo A, Azuaje F. Identification of dilated cardiomyopathy signature genes through gene expression and network data integration. Genomics. 2008;92:404–13. [DOI] [PubMed] [Google Scholar]
- [44].Vonrhein C, Flensburg C, Keller P, Sharff A, Smart O, Paciorek W, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods in molecular biology. 2007;364:215–30. [DOI] [PubMed] [Google Scholar]
- [46].Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for X- ray crystallography using ARP/wARP version 7. Nat Protoc. 2008;3:1171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Edelhoch H Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 1967;6:1948–54. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.