

Abstract
Dysferlin has been implicated in acute membrane repair processes, whereas myoferlin’s activity is maximal during the myoblast fusion stage of early skeletal muscle cell development. Both proteins are similar in size and domain structure; however, despite the overall similarity, myoferlin’s known physiological functions do not overlap with those of dysferlin. Here we present for the first time the X-ray crystal structure of human myoferlin C2A to 1.9 Å resolution bound to two divalent cations, and compare its 3D structure and membrane binding activities to that of dysferlin C2A. We find that while dysferlin C2A binds membranes in a Ca2+-dependent manner, Ca2+-binding was the rate-limiting kinetic step for this interaction. Myoferlin C2A, on the other hand, binds two calcium ions with an affinity 3-fold lower than that of dysferlin C2A; and, surprisingly, myoferlin C2A binds only marginally to phospholipid mixtures with a high fraction of phosphatidylserine.
Keywords: Ferlin proteins, C2 domains, X-ray Crystallography
Graphical Abstract
1. Introduction
The ability to modify and sculpt phospholipid membranes in a regulated manner is a critical process for almost all cell types. These processes involve the fusion of neurotransmitter-containing vesicles for cell-to-cell communication in the case of neurons, gene transfer in the case of viruses, and maintenance of membrane integrity in the case of membrane repair. Dysferlin (DYSF) is a 237 kDa type II integral membrane protein that facilitates the Ca2+-dependent aspects of membrane repair. Dysferlin is expressed in all mammalian tissues, but a mutant phenotype is more prevalent in human skeletal muscle. Prolonged membrane damage stimulates a cascade of immune factors that ultimately results in extensive muscle inflammation and damage [1, 2, 3]. Failure to adequately repair this damage can result in a gradual and progressive loss of muscle tissue. Indeed, mutations in the DYSF gene have been linked to Limb-Girdle Muscular Dystrophy Type 2B (LGMD-2B) or Miyoshi Myopathy (MM) in humans [4]. Dysferlin rapidly responds to injury by sensing Ca2+ influx through the site of damage, and then facilitates Ca2+-dependent patch-repair [5, 6]. Dysferlin-deficient muscle fibers demonstrate a primary defect in Ca2+-dependent, vesicle-mediated membrane repair [7]. Given the association of dysferlin with LGMD-2B and MM, and its importance in maintaining the integrity of the cell membrane, this protein has been hypothesized to play a critical role as a facilitator of membrane repair in skeletal muscles.
Myoferlin is a 230 kDa type II integral membrane protein that is encoded by the MYOF gene and is a paralog of dysferlin [8]. The protein is expressed in myoblasts, endothelial cells, cardiac and skeletal muscles. Myoferlin expression can also be detected in pulmonary tissue and at very low levels in kidney, placenta, and brain [9]. Myoferlin-null mice myoblasts are not able to form syncytial myotubes, and as a result, they cannot generate the large myofibers needed to form skeletal muscles [10]. Myoferlin mRNA is upregulated in patients diagnosed with Duchenne muscular dystrophy [11, 12, 13]. Indeed, increased levels of myoferlin measured in Duchenne muscular dystrophy patients suggest a role for myoferlin in muscle regeneration and muscle repair [10]. Aberrant myoferlin expression has also been linked to cancer progression, as the protein is over-expressed in numerous cancers (e.g. pancreas, lung, liver, breast, kidney) [14, 15, 16, 17, 18]. Its depletion results in impairment of caveole formation, perturbed EGFR and VEGFR2 activity, altered metabolism and mitochondrial function, reduced exosome formation and overall decrease of proliferative, migratory, and invasive properties of tumors [19, 20, 21, 22, 23].
The overall domain structure of both dysferlin and myoferlin is arranged as seven tandem C2 domains labeled C2A-C2G (Fig. 1a), a Ca2+-dependent, membrane-binding four-helix bundle domain known as FerA [24], and a Trp-Arg-rich DysF domain [25]. Both dysferlin and myoferlin possess a single C-terminal transmembrane helix that is embedded within a patching vesicle. The multiple C2 motifs are the most characteristic features of the ferlin proteins. C2 domains are typically Ca2+-dependent phospholipid binding domains with an approximate length of 130 amino acids [26]. Among all C2 domains of dysferlin, only the C2A domain strongly binds to lipids in a Ca2+-dependent manner [27]. The other C2 domains show weaker or Ca2+-independent binding to phospholipids [27, 28]. In addition to C2A, C2E and C2F domains have been shown to play a core roll in the function of ferlin protein activity [29, 30].
Figure 1:

A. Domain schematic of the dysferlin and myoferlin proteins. C2 domains are shown as boxes, FerA is depicted as a helical cartoon. The domains highlighted in red are the two C2A domains under consideration B. Structure-based alignment of dysferlin C2A and myoferlin C2A [31].β-strands are labeled above and highlighted as blue-shaded boxes. Residues observed to coordinate Ca2+ are boxed. Hydrophobic residues at the Ca2+ binding site that interact with the membrane are shown in red.
Although roles for dysferlin and myoferlin in normal maturation and function of muscle cells have been posited, a lack of knowledge about the characteristics and specific functions of the various domains of these proteins is a major obstacle in understanding their respective mechanism of action. The physiological differences between dysferlin and myoferlin are supported by the fact that, despite their overall similarity, they do not appear to be functionally interchangeable [32]. Our analysis of the C2A domains from both proteins shows that dysferlin C2A binds phospholipid in the presence of Ca2+ slower than other C2 domains. In addition myoferlin binds Ca2+ with a much lower affinity than dysferlin C2A; and, its Ca2+-dependent phospholipid-binding activity is much reduced relative to dysferlin C2A. These findings define crucial differences between myoferlin and dysferlin that may explain why these two paralogs cannot fully complement the function of the other.
2. Results
2.1. X-ray Crystal Structure of Human Myoferlin C2A
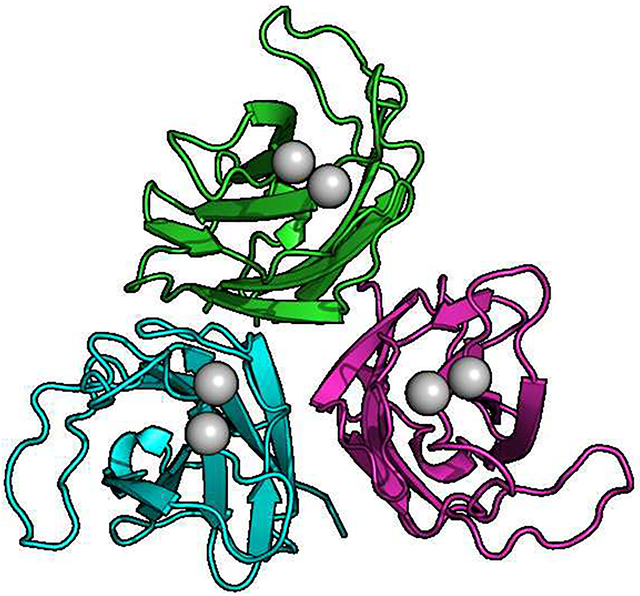
Human myoferlin C2A (residues 1–125) was crystallized in 0.1 M BisTris, pH 6.5, 20% PEG 3350, 0.3 M SrCl2 or CaCl2. Strontium ion has been used as a surrogate for Ca2+ in many Ca2+-binding proteins, including the C2B domain of synaptotagmin 1 [33]. We completed the Sr2+-bound form for phasing purposes, which we did not utilize. Further, the quality of the Sr2+-bound crystals were better than the Ca2+-bound form of myoferlin C2A crystals. The cell constants of the two data sets were almost identical. Crystals were grown at 23 °C (Fig. 2A) in space group P212121 with three molecules in the asymmetric unit (Table 1). X-ray data were collected at SSRL (Stanford Synchrotron Radiation Lightsource). The crystals diffracted to 1.93 Å and were solved by molecular replacement (Fig. 2) using the NMR-derived structure of Ca2+-free myoferlin C2A as the search model (2DMH [34]). Previously, we solved the crystal structures of dysferlin C2A and dysferlin C2Av1 (C2A variant 1) [35]. Both dysferlin C2A domains crystallized with three molecules in the asymmetric unit; and interestingly, the inner DysF domain of dysferlin also crystallized with three molecules in the asymmetric unit [25]. While it is tempting to speculate about the oligomerization state of ferlins based on crystallographic observations, currently there are no other experimental indications that implicates trimerization with ferlin function.
Figure 2:

A. The asymmetric unit of human myoferlin C2A crystallized with Sr2+. B. Single myoferlin C2A domain highlighting Phe-17 and Ile-75 putative membrane-anchoring residues. C. Close-up of Ca2+-binding residues of myoferlin C2A. Calcium ion ‘I’ is analogous to the calcium ion found in dysferlin C2A bound to the high-affinity Ca2+-binding pocket [35]. D. Dysferlin C2A structure (chain E of pdb code: 4IHB) showing the single calcium ion observed in dysferlin C2A. The residues that make up the loops 1, 2, and 3 are shown as sticks. E. Distance-difference matrix comparing liganded vs. un-liganded myoferlin C2A. Loop 3 residues are boxed. F. Superposition of myoferlin 2DMH (yellow, without divalent cation) and myoferlin from this X-ray crystal structure (green, with Sr2+). Calcium ion binding loops are labeled.
Table 1:
Crystallographic summary of myoferlin C2A
| Resolution Range (Å) | 41.14 – 1.93 (1.99 – 1.93) |
| Space Group | P 21 21 21 |
| Unit Cell(Å) | 47.35, 83.11, 94.28 |
| Total Reflections | 333686 (32892) |
| Unique Reflections | 28689 (2825) |
| Multiplicity | 11.6 (11.6) |
| Completeness (%) | 99.52 (99.82) |
| Mean I/σI | 9.5 (1.7) |
| Wilson B-factor | 20.0 |
| Rmerge | 0.112 (0.645) |
| Rmeas | 0.135 (0.777) |
| Rpim | 0.060 (0.348) |
| CC1/2 | 0.907 (0.779) |
| Reflections used in Refinement | 28580 (2825) |
| Reflections used for Rfree | 2793 (260) |
| Rwork | 0.181 (0.232) |
| Rfree | 0.192 (0.244) |
| Number of non-hydrogen atoms | 3496 |
| ligands | 6 |
| solvent | 470 |
| Protein residues | 387 |
| RMS (bonds, Å) | 0.017 |
| RMS (angles, °) | 1.87 |
| Ramachandran favored (%) | 97.9 |
| Ramachandran allowed (%) | 2.1 |
| Ramachandran outliers (%) | 0 |
| Rotamer outliers (%) | 0 |
| Clashscore | 0.97 |
| Average B-factor (Å2) | 29.00 |
| macromolecules (Å2) | 26.94 |
| ligands (Å2) | 30.82 |
| solvent (Å2) | 31.89 |
Myoferlin C2A consists of eight β-strands with a type-2 (P-type) C2 domain topology. The β-strands form three loops (Loops 1, 2, and 3) at the apex of the C2 domain that shape the divalent binding pocket. In myoferlin C2A, loop 3 possesses three of the residues that bind Ca2+ (Asp-71, Phe-72, Glu-73). Loop 2 possesses one of the Ca2+-binding residues (Asn-40) (sites I and II, Fig 2C). The phospholipid-binding residues of C2 domains are typically linked to the same loops that bind Ca2+. Phe-17 on loop 1 and Ile-75 on loop 3 (Fig. 2B and C) are located at the apex of their respective loops and should act as phospholipid-anchoring residues for this C2A domain (Fig. 2B) [36].
An NMR solution structure of the un-liganded human myoferlin C2A was initially deposited with the PDB by the RIKEN Structural Initiative (RSGI) as 2DMH in 2006 [34]. As such, no accompanying biochemical data was published. We can, therefore, compare the 20 NMR structures of the un-liganded myoferlin C2A domain with our ligand-bound X-ray structure of myoferlin C2A within the experimental limits of the two structures (root-mean-square deviation of atomic positions [RMSD] = 0.96 ± 0.06 Å, over all Cα atoms). No Ca2+ or divalent cation was reported in the NMR structure of myoferlin C2A [34]. It is possible that the myoferlin C2A used for the NMR structure could have access to calcium ions from the buffer solution. However the rotamers of the acidic residues coordinating the divalent cations in the crystal structure versus the solution-state structure are clearly The most obvioust. Therefore, it is unlikely that the NMR structure contained bound divalent cations.
The most obvious difference between the two structures is the deviation of loop 3 (Figs.2E,F). The difference-distance matrix of myoferlin C2A without Ca2+ (2DMH) was subtracted from the difference-distance matrix of the crystal structure of myoferlin C2A with bound cations. A difference- distance matrix is generated by calculating the distances between all pairs of equivalent Cα atoms in each molecule and then subtracting those distances. This allows a means of quantitatively assessing any structural similarity or differences between two structures. (Fig 2E). The residues that form loop 3 (residues 71–79) deviate significantly, indicating a significant difference between the two loops. Interestingly, there is little difference in the loop 1 (residues 12–22) conformation between un-liganded and liganded structures of myoferlin C2A, thus suggesting an unusually rigid loop 1 structure (Fig. 2E,F).
2.2. differences in Ca2+ Binding Between Myoferlin C2A and Dysferlin C2A
Despite the overall similarity between the 3D structures of myoferlin and dysferlin C2A (RMSD = 1.91 Å, over all Cα atoms), the manner in which Ca2+ is coordinated between the two domains is different. Using the dysferlin C2A structure as a reference, the first Ca2+-binding site in myoferlin C2A (Site I, Fig. 2C) utilizes a reduced ligation shell with only three oxygen atoms contributed by the domain. The high affinity site in dysferlin C2A, on the other hand, binds Ca2+ with one additional oxygen atom contributed by Asp-18 on loop 1 [35]. Therefore, relative to the same binding site in dysferlin C2A, the myoferlin C2A site I is incomplete (Fig. 2C). The more typical loop 1 aspartate (Asp-18 in dysferlin C2A) has been substituted with Gly-18 in myoferlin C2A. The majority of protein sequences identified as myoferlin in the Genbank database utilize a glycine at this locus (Fig. S3). The ligation shell for the second binding site in myoferlin C2A (Site II, Fig. 2C) is similar to that of dysferlin C2A [35].
Comparing NMR solution structures to a crystal structure can be problematic due to differences in buffer conditions and data collection temperatures; however, some key differences can be concluded. Superposition of the 20 NMR-derived ligand-free structures shows heterogeneity in conformation of loop 3, but less variation in loop 1 (Fig. 3a). In other C2 domains, loop 1 is flexible, and therefore not modeled in X-ray structures, or it is completely absent from domain as in the case of otoferlin C2A [37]. Loop 1 in myoferlin C2A appears more rigid than other loop 1 sequences in C2 domains. Its rigidity can be traced to a difference in the primary sequence of a conserved stretch of residues that make up loop 1. In (Type-1) synaptotagmin C2 domains, the …SDPYVK… motif, located at the C-terminal end of the loop 1 sequence is essential for Ca2+ binding, and is a hallmark of the C2 motif. Mutations of synaptotagmin C2 domains within this conserved motif eliminate Ca2+-binding in vitro [38, 39], as well as Ca2+-dependent exocytosis in Drosophila in vivo [40]. In myoferlin C2A, the corresponding sequence is …PDPIVS…, where the first serine in the conserved sequence has been been replaced by a proline (Pro-20); Pro is conserved at this locus in the majority of myoferlin sequences (Fig. S2, S3). In general proline augments the local structural rigidity of loops, and with two proline residues in close proximity within the same loop, we would therefore expect them to significantly decrease the conformational entropy of that loop. Indeed, in the NMR-derived, un-liganded domain, the average RMSD for backbone atoms for the residues in loop 1 (residues 10–22) is 0.52 Å, while the average RMSD between the 20 computed NMR structures is 0.78 Å A for loop 3 (residues 71–79). The average RMSD value is 0.42 Å for all structures calculated using backbone atoms for the whole domain. Similarly the relative rigidity of myoferlin C2A loop 1 is also reflected in the crystallographic B-factor of the liganded form of the domain. For loop 1 amino acids (residues 10–22), the average refined B-factor is 30.86 Å2, while loop 3 (residues 71–79) is 44.26 Å2. The refined B-factor for all atoms in myoferlin C2A domain, averaged from all three molecules in the asymmetric unit, is 26.94 Å2 (Fig. 3b, Table 1).
Figure 3:

Superimposed structures of myoferlin C2A with and without divalent cation. a. myoferlin C2A from the NMR deposited structure (2DMH) b. Superimposed molecules from the myoferlin C2A X-ray crystal structure (6EEL).
2.3. Lipid-Binding Model for Dysferlin and Myoferlin C2A
As peripheral membrane binding domains, C2 domains often possess bulky hydrophobic residues at the tips of loops 1 and 3 to use as membrane-anchors [36, 41]. Myoferlin C2A possesses one bulky hydrophobic residue on loop 1 (Phe-17) and one hydrophobic residue on loop 3 (Ile-75), which is analogous to other C2 domains with membrane anchors. Dysferlin C2A has a single residue on loop 3 that could serve as a membrane anchor (Met-75); however, the membrane-binding potential of loop 1 is less clear. The sequence of Loop 1 of dysferlin C2A is His-Thr-Pro-Asp-Thr-Asp-Ile-Ser-Asp; therefore, it does possess a hydrophobic residue on loop 1 (Ile-19, Fig. 2 D). If Ile-19 does indeed interact with membrane, it is positioned differently from other loop 1 binding residues. To ensure that the Ca2+-binding pockets remained intact after mutagenesis, we measured the Ca2+-binding properties of mutant dysferlin C2A and mutant myoferlin C2A. When we mutated Ile-19 on dysferlin C2A to an alanine residue (I19A), in addition to M75A, Ca2+-binding of the domain was abrogated (Fig. S5). It is likely that this residue may affect the overall shape of the calcium ion binding pocket; therefore, Ile-19 was excluded from further consideration. These results cannot, however, completely exclude Ile-19 as a membrane interacting residue without further experimentation. Our Ca2+-binding measurements confirmed that the Ca2+-binding pockets of the mutant C2 domains that we tested (dysferlin M75A and myoferlin F17A/I75A) remained functional and displayed similar binding values to that of the wild type C2 domains (Table 2). To test the membrane-anchoring potential of the loop residues of myoferlin and dysferlin C2A, we used a co-sedimentation assay to compare wild type and alanine mutations of the possible membrane-anchoring residues.
Table 2:
Summary of the ITC analysis of dysferlin C2A, dysferlin C2A M75A, myoferlin wildtype and myoferlin C2A F17A/I75A. Dysferlin C2A was fitted with a multiple site model, and myoferlin C2A was fitted with an independent site model. For myoferlin C2A wildtype number of binding sites was set to n=2 due to availability of myoferlin C2A wildtype Ca2+-bound structural information.
| Domain | n1 | n2 | ΔH1 (kcal/mol) |
ΔH2 (kcal/mol) |
ΔG1 (kcal/mol) |
ΔG2 (kcal/mol) |
TΔS1 (kcal/mol) |
TΔS2 (kcal/mol) |
Kd1 (μM) |
Kd2 (μM) |
|---|---|---|---|---|---|---|---|---|---|---|
| Dys C2A WT | 0.16 ± 0.06 | 2.68 ± 0.33 | 18.28 ± 2–28 | −1.94 ± 0.29 | −16.7 ± 1442 | −5.70 ± 1.22 | 34.98 ± 12.24 | 3.76 ± 1.18 | 0.70 ± 0.59 | 45.09 ± 4.30 |
| Dys C2A M75A | 0.30 ± 0.07 | 3.52 ± 0.17 | 47.78 ± 0.04 | −1.80 ± 0.18 | −9.65 ± CK42 | −5.53 ± 0.28 | 57.43 ± 0.42 | 3.73 ± 0.22 | 0.05 ± 0.03 | 64.14 ± 16−38 |
| Myo C2A WT | 2 | - | −0.89 ± 0.05 | - | −5.16 ± 1.02 | - | 4.27 ± 1.02 | - | 128.44 ± 49.44 | - |
| Myo C2A F17A/I75A | 1.79 ± 0.45 | - | −0.47 ± 0.18 | - | −4.71 ± −0.47 | - | 4.24 ± 0.43 | 312.23 ± 129.63 | - |
Since there was no prior evidence for number of Ca2+ binding sites in myoferlin C2A mutant, the curve was fitted to a model allowing for a variable ligand occupancy for an unbiased calculation of the number of ligand binding sites.
Dysferlin C2A did not significantly bind uncharged 100% PC (phosphotidylcholine) vesicles either with or without Ca2+ (Fig. 6). Upon exposure to a phospholipid surface with a large fraction of phosphoserine (60:40 PC:PS, phosphatidylcholine:phosphatidylserine), the Ca2+-dependent component of binding appears. Thus, the Ca2+-dependent enhancement, in the presence of Ca2+, is ~ 5X greater relative to the Ca2+-independent binding activity of the domain in 60:40 PC:PS (Fig. 6). The binding of the dysferlin C2A M75A mutation was reduced in 60:40 lipids with or without Ca2+, suggesting that Met-75 is essential for membrane anchoring (Fig. 6). Myoferlin C2A, on the other hand, showed negligible binding throughout the range of phospholipids with or without Ca2+ (Fig. 6). Consistent with previously published data on the membrane binding specificity of myoferlin C2A [11], we did notice a minor component of Ca2+-dependent phospholipid binding; however, in only very negatively-charged phospholipid mixtures (50%:50% PC:PS). We did not observe significant binding of myoferlin C2A to membranes containing up to 5% PIP2 (phosphatidylinositol 4,5-bisphosphate) (data not shown). The F17A/I75A mutation of myoferlin C2A displayed a minor Ca2+-dependent effect relative to its Ca2+-independent phospholipid binding component in the 50%:50% PC:PS lipid mixture; however, the effect was weak in this assay (Fig. 6).
Figure 6:

Co-sedimentation assay results for dysferlin C2A and myoferlin C2A versus phospholipid. Error bars are standard deviation from at least 4 experiments. The ‘*’ above the myoferlin C2A wild type data denotes a significant statistical difference (p<0.05) between the two datasets.
2.4. Kinetics of Ca2+-Dependent Phospholipid Binding by Dysferlin C2A
To study the kinetics of Ca2+-dependent phospholipid binding in dysferlin C2A, we used stopped-flow fluorescence spectrometry to determine the rate of binding of the C2 domains to a lipid surface in the presence of Ca2+ [42]. Similar to our co-sedimentation results, myoferlin C2A did not show appreciable binding to phospholipid by this method. Mixing of either Ca2+/dysferlin C2A with liposomes, or lipids/dysferlin C2A with Ca2+, resulted in changes in the fluorescence signals between the FRET acceptor incorporated into the vesicle and the donor Trp of dysferlin C2A (Fig. 7). Raw data traces from the stopped-flow spectrometer were fitted with single exponentials to obtain kinetic values for either the lipid-dependence or Ca2+-dependence of membrane binding by dysferlin C2A (Fig.7A,C). Concerning the differences between experimental conditions, several features emerge from our kinetic measurements compared to similar experiments performed using synaptotagmin C2 domains. Ca2+-dependent membrane binding by dysferlin C2A was markedly slower than that of the C2A domains of synaptotagmins 1 and 7, but was roughly comparable to that of the C2 domain from cytosolic phospholipase A2 [41, 43]. Surprisingly, the observed rates (kobs) for dysferlin C2A were almost invariant with respect to liposome concentration; however, kobs exhibited a robust, linear dependence on [Ca2+], suggesting a first-order dependence of lipid binding on [Ca2+] in the concentration range measured. In this range of [Ca2+] concentrations, the kon for [Ca2+] was 1.32 × 105 M−1s−1. This observation represents a striking contrast with other C2 domains, such as the C2A domain of synaptotagmin 1, which binds Ca2+ with very rapid, diffusion-limited kinetics, i.e., ~108 M−1s−1 [44]. Lipid binding, not Ca2+ binding, is the rate-limiting step in the case of synaptotagmin 1 C2A. Therefore for dysferlin C2A, Ca2+-binding appears to be kinetically limiting, a feature that distinguishes it from C2 domains of other proteins.
Figure 7:

A. Representative dysferlin C2A stopped-flow traces; binding was monitored as a function of [liposome]. B. Plot of kobs from panel A. C. Same as in panel A, except that binding was monitored as a function of [Ca2+]. D. Plot of kobs from panel C. Error bars are the standard error from at least three independent experiments.
3. Discussion
Dysferlin (237 kDa) and myoferlin (230 kDa) participate in membrane fusion at different stages of muscle development and physiology. Dysferlin has been implicated in maintaining the integrity of cell membranes [45], while myoferlin is instrumental in the early stages of muscle development[46]. Overall dysferlin and myoferlin are 68% similar (56% identical) at the primary sequence level. Indeed, the first C2A domains of both proteins are 57% similar (42% identical). Given the sequence identity shared between these two large proteins, one would naturally expect that they should function similarly or at least complement each other’s function. However based on our results, the C2A domains of myoferlin and dysferlin share relatively few biophysical properties. Here we show that the C2A domain of dysferlin has slow Ca2+-dependent phospholipid-binding kinetics compared to other C2 domains. Further, both myoferlin and dysferlin not only bind Ca2+ differently, but they also have distinct lipid-binding avidities that likely reflect their different roles in muscle physiology.
The overall tertiary structure of myoferlin C2A is similar to other Type-2 (P-type) ferlin C2 domains. Both myoferlin and dysferlin C2A domains bind two calcium ions; however, each domain utilizes calcium ions for different purposes. Dysferlin C2A possesses one high affinity site and one low affinity site (Table 2). The high affinity Ca2+ site in dysferlin C2A is primarily structural, and its occupancy at this locus increases the likelihood of proper domain folding in C2A [35]. The lower affinity site in dysferlin C2A responds to environmental Ca2+, as it alters the surface electrostatics of the domain, and prepares the domain for peripheral membrane attachment [35]. C2 domains coordinate calcium ions through a hexadentate arrangement of oxygen atoms contributed by both main-chain carbonyl oxygen atoms and side-chain oxygen atoms [47]. Most Ca2+-binding C2 protein motifs have evolved to provide four equatorial and one axial oxygen atom. The sixth coordinating atom is typically contributed by phospholipids [48]; hence, providing a link between Ca2+ binding and phospholipid binding. Dysferlin C2A has this property; however, myoferlin is different. The affinity for Ca2+ of the myoferlin C2A domain is surprisingly low (KD=128 μM), and its low Ca2+-binding affinity can be explained by two unique structural features. First, myoferlin C2A has substituted a glycine residue for the negatively-charged residue that is typically located on loop 1. In fact, glycine at this locus on loop 1 is conserved in the vast majority of sequences labeled as myoferlin (Fig. S3). In the myoferlin C2A crystal structure, the hexadentate arrangement of oxygen atoms for the ”I” calcium ion (Fig. 2 C,D) is contributed by the side chains of Asp-21 and Asn-40, and the carbonyl oxygen from Lys-19. The remaining coordinating atoms in the crystal structure are provided by one water molecule, and/or two residues (Asp-94 and Gln-95) from symmetry-related molecules. The coordination of calcium ion ”II” is similar to the lower affinity calcium ion in dysferlin C2A.
The second adaptation of myoferlin can be explained by the rigidity of loop 1. The substitution of the first serine with a proline residue in the conserved …SDPYVK… motif likely contributes to the reduced calcium ion affinity. Since loop 1 in myoferlin C2A does not contribute side chain oxygen atoms to Ca2+ binding, the change in the conformation of loop 1 cannot be linked to Ca2+ binding as with other C2 domains. To bind calcium ion at all, the protein must, therefore, conserve the overall shape of the Ca2+ binding pocket via an alternative mechanism. Indeed, the entire loop 1 sequence is flanked by proline residues. The N-terminus of loop 1 begins with Pro-13 while the C-terminus ends with Pro-20, Asp-21, Pro-22. Therefore, the conformation of loop 1 in myoferlin C2A is dominated by the restricted ϕ torsional angles accessible by its flanking proline residues. A similar conclusion has been made in the ‘RGD’ loop of the HIVZ2 Tat protein [49]. The restricted conformational space of loop 1 is also reflected in the thermodynamic values obtained from cation binding (Table 2, Fig. 5). In the dysferlin C2A case, the relatively large enthalpic penalty for ions to bind at the high affinity site is related to the folding dynamics of the domain as a function of Ca2+ occupancy (Table 2). The enthalpic barrier to binding is compensated by a net favorable ΔS term. The net favorable ΔS term includes the entropy changes due to the ordering of binding loops, in addition to the entropy changes due to the displacement of water molecules from the Ca2+ binding site (Table 2, Fig. 5). Indeed, the ΔG of unfolding for dysferlin C2A is near 0 kcal/mol without Ca(2+) [35], so; the domain samples a larger ensemble of unfolded states without Ca2+. In myoferlin C2A, we measured a small enthalpic term complemented by a small, but favorable entropic term upon Ca2+ binding. Therefore, the thermodynamic values we measured for myoferlin C2A Ca2+ binding are consistent with the relatively small changes to the shape of the binding pocket, consistent with a more rigid binding pocket.
Figure 5:

Graphical summary from Table 2 of the thermodynamic parameters obtained from the ITC analysis of Ca2+ binding in myoferlin C2A and dysferlin C2A and the lipid-binding mutants.
We predicted that myoferlin C2A should associate with phospholipid membranes in a similar manner to dysferlin and synaptotagmin C2A domains, as it has hydrophobic side chains located in homologous positions on loops 1 and 3. We also predicted that the C2 domains of myoferlin and dysferlin would prefer binding to negatively-charged phospholipids similar to other C2 domains [50]. We measured avid dysferlin C2A association with a mixture of 60:40 PC:PS in a Ca2+-dependent manner; myoferlin, on the other hand, did not show analogous degrees of binding to any of the phospholipid mixtures we tested with or without Ca2+ (Fig. 6). We did measure weak Ca2+-dependent binding of myoferlin C2A with 50:50 PC:PS membranes[11](Fig. 6), so a negatively-charged phospholipid membrane is a component of myoferlin C2A phospholipid interaction. Therefore even weak binding to anionic membranes likely reflects at least some electrostatic contribution to membrane docking for myoferlin C2A [41]. Indeed, a surface electrostatic representation of Ca2+-bound myoferlin versus Ca2+-bound dysferlin C2A shows a clear difference in the strength of the electrostatic surface potential relative to dysferlin C2A (Fig. 8a, 8b). The relatively weak surface electrostatics of myoferlin C2A is also reflected in the myoferlin C2A purification protocol as we could not use ion-exchange chromatography as with dysferlin C2A (Fig. 8b). In addition, we could not detect Ca2+-dependent phospholipid binding of the myoferlin C2A double-mutant (Fig. 6); hence, these residues may contribute to a more tenuous protein:phospholipid association under our experimental conditions. Marty, et al. report a moderate Ca2+-dependent, phospholipid-ordering effect of myoferlin C2A using a fluorescence polarization technique [51]. It is possible that the exposed basic residues on the surface of myoferlin C2A loosely interact with negatively charged phospholipids to alter the order of lipids. Indeed, weak, tenuous protein-phospholipid interactions have been hypothesized in similar domains [52]. Another possibility to explain the anomalously weak interaction of myoferlin C2A with PS-containing membranes is that myoferlin C2A may be more selective for a specific membrane curvature or a different phospholipid packing arrangement relative to dysferlin C2A. For example, the C2 domains of synaptotagmin tend to bind to phospholipid mixtures that accentuate both electrostatics and highly-curved surfaces [53].
Figure 8:

Electrostatic representation of (a) dysferlin C2A with Ca2+ and (b) myoferlin C2A with Ca2+. Blue regions reflect positively-charged zones on the surface of the domain. Red regions reflect negatively-charged zones on the surface. Both molecules were contoured to ± 3 kT/e.
Dysferlin and myoferlin are the most closely related members of ferlin protein family [54]. Despite their similarities, however, they are expressed in different stages of muscle cell growth and development and are associated with different diseases. The mechanism by which myoferlin interacts with Ca2+ and phospholipids has not been completely elucidated. In this study, we investigated the structural and functional characteristics of myoferlin C2A and compared it to the more extensively studied dysferlin C2A. We found significant differences between these seemingly similar domains that may partially explain the disparate physiological activity between dysferlin and myoferlin. We showed that myoferlin C2A binds calcium ions, but with 3-fold lower affinity than that of dysferlin C2A. We also showed that the phospholipid-binding activity of dysferlin C2A in the presence of Ca2+ is much higher than myoferlin C2A. We identified and established the significance of phospholipid-interacting residues of the dysferlin (Met-75). Our M75A mutant of dysferlin C2A reduced Ca2+-dependent phospholipid-binding activity of dysferlin C2A, while the Ca2+-binding activity was preserved. The association of myoferlin C2A to PC:PS (50:50) liposomes in the presence of Ca2+ was also reduced in the F17A and I75A mutants, albeit weakly. Further, we studied the kinetics of dysferlin C2A interaction with phospholipids. Our stopped-flow spectrometry results indicate that the kinetics of the dysferlin C2A Ca2+-dependent phospholipid interaction are mainly a function of Ca2+ concentration. In dysferlin C2A while increasing lipid concentration provoked only small increases in kobs, we observed a steep linear dependence of kobs on [Ca2+]. To the best of our understanding, this indicates that formation of the C2A-Ca2+ complex, i.e. Ca2+-binding, is rate-limiting, rather than binding of the complex to lipids. Dysferlin C2A represents the first example of a C2 domain for which membrane association is dependent on the concentration of the C2A-Ca2+ complex, though the lipid versus Ca2+ dependence of membrane association kinetics has not been formally established for most C2 domains. Presumably, elevated cytoplasmic calcium ion concentration is among the stimulatory signals for dysferlin’s action, so the protein should be capable of differentiating between the elevation of transient myocyte cytoplasmic calcium ion concentration and those transients associated with sarcolemmal damage. A “slow” Ca2+ sensor that is only activated by prolonged Ca2+ transients could serve this function. The Ca2+-sensitive kinetics that we observed for C2A may allow dysferlin to act as a Ca2+ sensor for sarcolemmal damage in an environment where large Ca2+ transients are commonplace. Finally, our crystal structure of divalent-bound myoferlin C2A together with our thermodynamics analysis suggests that unlike dysferlin C2A, loop 1 in myoferlin C2A is less flexible and there is likely a conformational change in its loop 3 upon Ca2+ binding. Future studies will include the analysis of the other domains of ferlin family members to understand their role in the total activity of these proteins, and why mutations lead to disease.
4. Methods and Materials
4.1. Cloning, Expression and Purification of C2A Domains
The cloning of dysferlin C2A was described previously [35]. The myoferlin C2A gene (1–125) was assembled by Genewiz based on the human myoferlin sequence from GENBANK:AAF27176.1. The C-terminus of myoferlin C2A was determined based on the alignment of myoferlin with the dysferlin C2A crystal structure (4IHB). The primary sequence for myoferlin C2A was reverse-translated and optimized for E. coli expression. The resulting nucleotide sequence was cloned into the pET-based p202 expression vector for subsequent expression. Dysferlin and myoferlin C2A wildtype and mutant domain expression plasmids were transformed into BL21(DE3) competent cells and spread onto LB agar plates containing 50 μg/mL kanamycin and incubated at 37 °C overnight. Isolated colonies were picked and used to start a 50 mL Luria Broth (LB) containing 50 μg/mL kanamycin and the culture was incubated overnight at 37°C while shaking at 250 rpm. 10 mL of this culture was used to inoculate a 1 L Terrific Broth (TB) culture also containing 50 μg/mL kanamycin at 37 °C and 250 rpm. At OD600 = 2.0, the culture was cooled to 18 °C and the protein expression was induced with the addition of 400 μL of 1 M isopropyl β-D-1-thiogalactopyranoside (IPTG). After 12 hours of incubation, cells were harvested using a Beckman JLA-8.1000 rotor at 5000 rpm (6227 x g) and flash frozen in liquid nitrogen until ready for use. Next, cells were thawed in lysis buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 5 mM CaCl2), ruptured using a Microfluidizer, and spun using a Beckman JA-20 rotor at 19,500 rpm (45.900 x g) for 45 min. The supernatant was passed through a Ni-NTA affinity column which was equilibrated with lysis buffer. The column then was washed with 150 mL lysis buffer, followed by a wash in lysis buffer plus 30 mM imidazole. Finally, His6-MBP-C2A was eluted with 80 mL lysis buffer including 300 mM imidazole. The resulting fusion protein was cleaved with Tobacco Etch Virus Protease (TEV) overnight at 4°C. At this step, all proteins were buffer-exchanged into 20 mM HEPES pH 7.4, 50 mM NaCl, and 5 mM CaCl2 for further purification using ion-exchange chromatography. In the case of dysferlin, SP-Sepharose column was used to separate C2A from MBP, TEV protease, and uncleaved fusion proteins. A 0 to 1 M NaCl gradient was applied to the column and dysferlin C2A wildtype and mutant domains were eluted in fractions starting at a conductivity of 38 mS/cm. In the case of myoferlin, protein solutions were loaded onto QAE-Sepharose column and myoferlin C2A wildtype or mutant myoferlin C2A domains came off in the flow-through. We tried SP-Sepharose column for myoferlin and still myoferlin C2A domain mostly came off in the flow-through. In either case, the solution containing the protein of interest was concentrated and loaded onto a Superdex 75 column to remove the remaining contaminations. Purity was assessed using SDS PAGE Stain-Free gels from BioRad (Fig. S1) and protein concentrations were quantitated by OD280 using each proteins calculated extinction coefficient.
4.2. Crystallization and Data Collection
Purified myoferlin C2A domain (10 mg/mL) was crystallized in 0.1 M Bis-Tris, pH 6.5, 20% PEG 3350, 0.3 M SrCl2. Crystals were grown at 23 °C. The crystals were captured into nylon loops and frozen in liquid N2. Initial data sets were collected on a Rigaku ScreenMachine. Subsequent data sets were collected at SSRL beamline 7–1. The wavelength of the final data sets was 0.9796 Å, and the data were collected at 90 K. X-ray data were processed with imosflm [55], and the data were scaled using AIMLESS as a part of the CCP4 package [56]. The X-ray crystal structure was solved using molecular replacement techniques (Phaser) [57] and subsequently refined using Phenix[58].
4.3. Isothermal Titration Calorimetry
The Isothermal Titration Calorimetry (ITC) buffer, containing 150 mM KCl, and 20 mM HEPES pH 7.4, was passed through a Bio-Rad Chelex 100 resin to remove cation impurities and filtered using 0.22 μm membrane filter. The Ca2+ stock solution was prepared by diluting sterile and filtered 2.0 M CaCl2 stock solution purchased from RPI (Research Products International) with the ITC buffer. Additionally, the proteins were buffer exchanged into the ITC buffer using PD-10 columns from GE Healthcare. Each experiment was repeated at least 3 times with 200 rpm mixing at 15 °C. The best results obtained using a protein concentration ranging from 150–400 μM and a titrant (Ca2+) concentration of either 5 or 10 mM. All experimental repeats corresponding to each protein sample were performed using a constant CaCl2 concentration. The analysis was performed using NanoAnalyze software. We fitted the raw data using a multiple binding sites model for dysferlin C2A and an independent model for myoferlin C2A (with n=2).
4.4. Liposome Preparation
1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamineN-(5-dimethylamino-1-naphthalenesulfonyl) (dansyl-PE) were purchased fromAvanti Polar Lipids. Phospholipids in chloroform were mixed to obtain the desired molar ratio and residual chloroform was then evaporated using nitrogen gas. Dried phospholipids were put under vacuum overnight to remove the chloroform residues. The phospholipids were then resuspended in a buffer(200 mM NaCl and 20 mM HEPES, pH 7.4) that was passed through Bio-Rad Chelex 100 resin to remove divalent cations. The phospholipids were allowed to rehydrate over 15 minutes while vortexed and then were sonicated. 100nm liposomes were prepared by extrusion through 100 nm polycarbonate filters using an Avanti Mini Extruder. The size of vesicles was confirmed to be 100 ± 12 nm, as determined using a Malvern Zetasizer Nano ZS (data not shown). Liposome concentration was calculated assuming a population of unilamellar liposomes of 5 nm thickness and 100 nm outer diameter at .71 nm2 per lipid headgroup. This resulted in an estimate of 8.0 × 104 lipid molecules per liposome, e.g., 10 nm [liposomes] = 0.8 mM [lipid].
4.5. Stopped-flow Rapid Mixing Experiments
Kinetics experiments were carried out using an Applied Photophysics SX.18MV stopped-flow spectrometer as described [44]. Large (~100 nm) unilamellar liposomes containing 65% POPC, 30% POPS, and 5% dansyl-PE were prepared, and FRET experiments were carried out by excitation of tryptophan at 285 nm and measuring the emission of dansyl-PE using a 523 nm band-pass filter. All experimental data represent three independent experiments.
4.6. Sedimentation Assay
Co-sedimentation experiments were performed as described previously[24, 59]. In a 100 μL total reaction volume, liposomes with various POPC/POPS ratio were mixed with protein and either EGTA or Ca2+. The proteins were also buffer exchanged into the Chelex-treated buffer used in preparation of liposomes to removes any Ca2+ contamination. The final concentration of each component was as follows: 1 mM liposomes, 4 μM protein, and either 0.2 mM EGTA or 1 mM CaCl2. The samples were allowed to incubate for 15 minutes in 37 °C and then centrifuged at 65,000 rpm (183,000 x g) using a Beckman TLA-100 rotor for 45 minutes in a Beckman Optima MAX-E table-top ultracentrifuge. In most cases, analysis of the supernatant is preferable to analysis of the pellet because it requires reduced sample handling, produces more robust results, and is amenable to large scale comparisons with other C2 domains [53]. Therefore, equal amounts of the supernatants were loaded to SDS-PAGE and were imaged and quantified using a Bio-Rad Criterion Stain Free Imaging System Gel Imager and Image Lab software (Fig S4). This technology does not require staining and destaining steps, providing a highly sensitive and reliable method in protein quantification that is equal or better than Coomassie staining. Each experiment was repeated at least 3 times, and the errors are presented as standard deviation.Students t-test statistical analysis was performed to test the significance of the difference observed between the measurements.
4.7. Equilibrium Co-sedimentation Control Experiment
The co-sedimentation assay has been a common biochemical technique to study protein-phospholipid interactions, especially in ferlins and synap-totagmins. In co-sedimentation experiments, additional controls need to be considered to assure that the observed interaction is a direct result of protein vesicle interaction. We considered three control experiments for each protein: the input (protein only), protein+EGTA control, and protein+Ca2+ control (Fig S4). These controls lack lipid vesicles and reveal aggregation due to possible interaction of proteins with either EGTA or Ca2+. We observed no detectable depletion in the amount of protein in the supernatant in our control experiments, which strongly suggests that C2A proteins do not aggregate or pellet when combined with Ca2+ or EGTA.
4.8. RMSD calculations
All RMSD (root-mean-square deviation) values were computed using the method as implemented in PyMOL [60].
4.9. Accession Numbers
Coordinates and structure factors have been deposited in the Protein Data Bank with accession number 6EEL.
Supplementary Material
Figure 4:

Representative ITC thermograms and their corresponding fitted curves of Ca2+ binding to wild type a. dysferlin C2A (red) and b. myoferlin C2A (green).
Highlights.
The initial C2 domains of dysferlin and myoferlin are 57% similar (42% identical). Despite this similarity, myoferlin overexpression does not completely rescue the null dysferlin phenotype
Unlike dysferlin C2A, myoferlin binds two Ca2+ with equivalent affinity.
Unlike dysferlin C2A, the membrane binding loop 1 of myoferlin C2A is relatively rigid
Unlike dysferlin C2, myoferlin C2A binds lipids with different kinetics than other known C2 domains.
The first X-ray structure of myoferlin C2A bound to divalent cation to 1.9A resolution.
4.10. Acknowledgments
Research reported in this publication was supported in part by the Jain Foundation and The National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number R01AR063634. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. We also would like to thank Dr. Michael Latham, Dr. Mariana Fiori, Matthew Dominguez, Jacob Gendelman, and Jon McCord for their help in reviewing this publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. References
- [1].Han R, Muscle membrane repair and inflammatory attack in dysferlinopathy, Skeletal Muscle 1 (1) (2011) 10. doi: 10.1186/2044-5040-1-10. URL 10.1186/2044-5040-1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Han R, Frett EM, Levy JR, Rader EP, Lueck JD, Bansal D,Moore SA, Ng R, de Bernabé DB-V, Faulkner JA, Campbell KP, Genetic ablation of complement C3 attenuates muscle pathology in dysferlin-deficient mice, Journal of Clinical Investigation 120 (12) (2010) 4366–4374. doi: 10.1172/jci42390.URL 10.1172/jci42390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rawat R, Cohen TV, Ampong B, Francia D, Henriques-Pons A,Ho man EP, Nagaraju K, Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle, The American Journal of Pathology 176 (6) (2010) 2891–2900. doi: 10.2353/ajpath.2010.090058.URL 10.2353/ajpath.2010.090058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Weiler T, Bashir R, Anderson LV, Davison K, Moss JA, Britton S, Nylen E, Keers S, Vafiadaki E, Greenberg CR, Bushby CR,Wrogemann K, Identical mutation in patients with Limb Girdle Muscular Dystrophy Type 2B or Miyoshi Myopathy suggests a role for modifier gene(s), Hum. Mol. Genet 8 (5) (1999) 871–877. [DOI] [PubMed] [Google Scholar]
- [5].Davenport NR, Sonnemann KJ, Eliceiri KW, Bement WM, Membrane dynamics during cellular wound repair, Molecular Biology of the Cell 27 (14) (2016) 2272–2285. doi: 10.1091/mbc.e16-04-0223.URL 10.1091/mbc.e16-04-0223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Davenport NR, Bement WM, Cell repair: Revisiting the patch hypothesis, Communicative & Integrative Biology 9 (6) (2016) e1253643. doi: 10.1080/19420889.2016.1253643.URL 10.1080/19420889.2016.1253643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R,McNeil PL, Campbell KP, McNeil PL, Defective membrane repair in dysferlin-deficient muscular dystrophy, Nature 423 (6936) (2003) 168–172. [DOI] [PubMed] [Google Scholar]
- [8].Lek A, Evesson FJ, Sutton RB, North KN, Cooper ST, Ferlins: Regulators of vesicle fusion for auditory neurotransmission, receptor trafficking and membrane repair, Traffic 13 (2) (2011) 185–194. doi: 10.1111/j.1600-0854.2011.01267.x. URL [DOI] [PubMed] [Google Scholar]
- [9].Davis DB, Delmonte AJ, Ly CT, McNally EM, Myoferlin, a candidate gene and potential modifier of muscular dystrophy, Hum. Mol. Genet 9 (2) (2000) 217–226. [DOI] [PubMed] [Google Scholar]
- [10].Demonbreun AR, Posey AD, Heretis K, Swaggart KA, Earley JU, Pytel P, McNally EM, Myoferlin is required for insulin-like growth factor response and muscle growth, The FASEB Journal 24 (4) (2010) 1284–1295. doi: 10.1096/fj.09-136309.URL 10.1096/fj.09-136309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Davis DB, Doherty KR, Delmonte AJ, McNally EM, Calciumsensitive phospholipid binding properties of normal and mutant ferlin C2 domains, Journal of Biological Chemistry 277 (25) (2002) 22883–22888. doi: 10.1074/jbc.m201858200.URL 10.1074/jbc.m201858200 [DOI] [PubMed] [Google Scholar]
- [12].Doherty KR, Demonbreun AR, Wallace GQ, Cave A, Posey AD, Heretis K, Pytel P, McNally EM, The endocytic recycling protein EHD2 interacts with myoferlin to regulate myoblast fusion, Journal of Biological Chemistry 283 (29) (2008) 20252–20260. doi: 10.1074/jbc.m802306200.URL 10.1074/jbc.m802306200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bernatchez PN, Acevedo L, Fernandez-Hernando C, Murata T,Chalouni C, Kim J, Erdjument-Bromage H, Shah V, Gratton J-P, McNally EM, Tempst P, Sessa WC, Myoferlin regulates vascular endothelial growth factor receptor-2 stability and function, Journal of Biological Chemistry 282 (42) (2007) 30745–30753. doi: 10.1074/jbc.m704798200.URL 10.1074/jbc.m704798200 [DOI] [PubMed] [Google Scholar]
- [14].Leung C, Yu C, Lin MI, Tognon C, Bernatchez P, Expression of myoferlin in human and murine carcinoma tumors, The American Journal of Pathology 182 (5) (2013) 1900–1909. doi: 10.1016/j.ajpath.2013.01.041.URL 10.1016/j.ajpath.2013.01.041 [DOI] [PubMed] [Google Scholar]
- [15].Turtoi A, Blomme A, Bellahcene A, Gilles C, Hennequiere V,Peixoto P, Bianchi E, Noel A, Pauw ED, Lifrange E, Delvenne P,Castronovo V, Myoferlin is a key regulator of EGFR activity in breast cancer, Cancer Research 73 (17) (2013) 5438–5448. doi: 10.1158/0008-5472.can-13-1142.URL 10.1158/0008-5472.can-13-1142 [DOI] [PubMed] [Google Scholar]
- [16].Song D, Ko G, Lee J, Lee J, Lee G-W, Kim H, Yang J, Heo R,Roh G, Han S-Y, Kim D, Myoferlin expression in non-small cell lung cancer: Prognostic role and correlation with VEGFR-2 expression, Oncology Letters 11 (2) (2015) 998–1006. doi: 10.3892/ol.2015.3988.URL 10.3892/ol.2015.3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Song D, Ko G, Lee J, Lee J, Yang J, Kim M, An H,Kang M, Jeon K, Kim D, Prognostic role of myoferlin expression in patients with clear cell renal cell carcinoma, Oncotarget 8 (51). doi: 10.18632/oncotarget.21645.URL 10.18632/oncotarget.21645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hermanns C, Hampl V, Holzer K, Aigner A, Penkava J, Frank N,Martin DE, Maier KC, Waldburger N, Roessler S, GoppeltStruebe M, Akrap I, Thavamani A, Singer S, Nordheim A, Gudermann T, Muehlich S, The novel MKL target gene myoferlin modulates expansion and senescence of hepatocellular carcinoma, Oncogene 36 (24) (2017) 3464–3476. doi: 10.1038/onc.2016.496.URL 10.1038/onc.2016.496 [DOI] [PubMed] [Google Scholar]
- [19].Li R, Ackerman WE, Mihai C, Volakis LI, Ghadiali S, Kniss DA, Myoferlin depletion in breast cancer cells promotes mesenchymal to epithelial shape change and stalls invasion, PLoS ONE 7 (6) (2012) e39766. doi: 10.1371/journal.pone.0039766.URL 10.1371/journal.pone.0039766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Volakis LI, Li R, Ackerman WE, Mihai C, Bechel M, Summerfield TL, Ahn CS, Powell HM, Zielinski R, Rosol TJ, Ghadiali SN, Kniss DA, Loss of myoferlin redirects breast cancer cell motility towards collective migration, PLoS ONE 9 (2) (2014) e86110. doi: 10.1371/journal.pone.0086110.URL 10.1371/journal.pone.0086110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fahmy K, Gonzalez A, Arafa M, Peixoto P, Bellahcène A, Turtoi A,Delvenne P, Thiry M, Castronovo V, Peulen O, Myoferlin plays a key role in VEGFA secretion and impacts tumor-associated angiogenesis in human pancreas cancer, International Journal of Cancer 138 (3) (2015) 652–663. doi: 10.1002/ijc.29820.URL 10.1002/ijc.29820 [DOI] [PubMed] [Google Scholar]
- [22].Blomme A, Costanza B, de Tullio P, Thiry M, Simaeys GV,Boutry S, Doumont G, Valentin ED, Hirano T, Yokobori T, Gofflot S, Peulen O, Bellahcène A, Sherer F, Go CL, Cavalier E,Mouithys-Mickalad A, Jouret F, Cusumano PG, Lifrange E, Muller RN, Goldman S, Delvenne P, Pauw ED, Nishiyama M, Castronovo V, Turtoi A, Myoferlin regulates cellular lipid metabolism and promotes metastases in triple-negative breast cancer, Oncogene 36 (15) (2016) 2116–2130. doi: 10.1038/onc.2016.369.URL 10.1038/onc.2016.369 [DOI] [PubMed] [Google Scholar]
- [23].Blomme A, Fahmy K, Peulen O, Costanza B, Fontaine M, Struman I, Baiwir D, de Pauw E, Thiry M, Bellahcène A, Castronovo V, Turtoi A, Myoferlin is a novel exosomal protein and functional regulator of cancer-derived exosomes, Oncotarget 7 (50). doi: 10.18632/oncotarget.13276.URL 10.18632/oncotarget.13276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Harsini FM, Chebrolu S, Fuson KL, White MA, Rice AM,Sutton RB, FerA is a membrane-associating four-helix bundle domain in the ferlin family of membrane-fusion proteins, Scientific Reports 8 (1). doi: 10.1038/s41598-018-29184-1.URL 10.1038/s41598-018-29184-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sula A, Cole AR, Yeats C, Orengo C, Keep NH, Crystal structures of the human dysferlin inner DysF domain, BMC Structural Biology 14 (1) (2014) 3. doi: 10.1186/1472-6807-14-3.URL 10.1186/1472-6807-14-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shao X, Li C, Fernandez I, Zhang X, Sudhof TC, Rizo J, Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch, Neuron 18 (1) (1997) 133–142. [DOI] [PubMed] [Google Scholar]
- [27].Therrien C, Fulvio SD, Pickles S, Sinnreich M, Characterization of lipid binding specificities of dysferlin C2 domains reveals novel interactions with phosphoinositides, Biochemistry 48 (11) (2009) 2377–2384. doi: 10.1021/bi802242r.URL 10.1021/bi802242r [DOI] [PubMed] [Google Scholar]
- [28].Abdullah N, Padmanarayana M, Marty NJ, Johnson CP, Quantitation of the Ca2+ and membrane binding properties of the C2 domains of dysferlin, Biophysical Journal 106 (2) (2014) 382–389. doi: 10.1016/j.bpj.2013.11.4492.URL 10.1016/j.bpj.2013.11.4492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lek A, Evesson FJ, Lemckert FA, Redpath GM, Lueders AK, Turnbull L, Whitchurch CB, North KN, Cooper ST, Calpains, cleaved mini-dysferlinC72, and L-type channels underpin calcium-dependent muscle membrane repair, J. Neurosci 33 (12) (2013) 5085–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Redpath GM, Woolger N, Piper AK, Lemckert FA, Lek A, Greer PA, North KN, Cooper ST, Calpain cleavage within dysferlin exon 40a releases a synaptotagmin-like module for membrane repair, Mol. Biol. Cell 25 (19) (2014) 3037–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Pei J, Kim B-H, Grishin NV, PROMALS3d: a tool for multiple protein sequence and structure alignments, Nucleic Acids Research 36 (7) (2008) 2295–2300. doi: 10.1093/nar/gkn072.URL 10.1093/nar/gkn072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lostal W, Bartoli M, Roudaut C, Bourg N, Krahn M, Pryadkina M,Borel P, Suel L, Roche JA, Stockholm D, Bloch RJ, Levy N,Bashir R, Richard I, Lack of correlation between outcomes of membrane repair assay and correction of dystrophic changes in experimental therapeutic strategy in dysferlinopathy, PLoS ONE 7 (5) (2012) e38036. doi: 10.1371/journal.pone.0038036.URL 10.1371/journal.pone.0038036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cheng Y, Sequeira SM, Malinina L, Tereshko V, Sollner TH, Patel DJ, Crystallographic identification of Ca2+ and Sr2+ coordination sites in synaptotagmin I C2B domain, Protein Sci. 13 (10) (2004) 2665–2672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Nagashima T, Hayashi F, Yokoyama S, Solution structure of the first C2 domain of human myoferlin (October 2006). doi: 10.2210/pdb2dmh/pdb. URL 10.2210/pdb2dmh/pdb [DOI]
- [35].Fuson K, Rice A, Mahling R, Snow A, Nayak K, Shanbhogue P,Meyer AG, Redpath GM, Hinderliter A, Cooper ST, Sutton RB, Alternate splicing of dysferlin C2A confers Ca2+-dependent and Ca2+-independent binding for membrane repair, Structure 22 (1) (2014) 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chapman ER, Davis AF, Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers, Journal of Biological Chemistry 273 (22) (1998) 13995–14001. doi: 10.1074/jbc.273.22.13995.URL 10.1074/jbc.273.22.13995 [DOI] [PubMed] [Google Scholar]
- [37].Helfmann S, Neumann P, Tittmann K, Moser T, Ficner R,Reisinger E, The crystal structure of the C2A domain of otoferlin reveals an unconventional top loop region, Journal of Molecular Biology 406 (3) (2011) 479–490. doi: 10.1016/j.jmb.2010.12.031.URL 10.1016/j.jmb.2010.12.031 [DOI] [PubMed] [Google Scholar]
- [38].Shields MC, Bowers MR, Fulcer MM, Bollig MK, Rock PJ, Sutton RB, Vrailas-Mortimer AD, Lochmüller H, Whittaker RG, Horvath R, Reist NE, Drosophila studies support a role for a presynaptic synaptotagmin mutation in a human congenital myasthenic syndrome, PLOS ONE 12 (9) (2017) e0184817. doi: 10.1371/journal.pone.0184817.URL 10.1371/journal.pone.0184817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ubach J, Zhang X, Shao X, Südhof TC, Rizo J, Ca2+ binding to synaptotagmin: how many calcium ions bind to the tip of a C2-domain?, The EMBO Journal 17 (14) (1998) 3921–3930. doi: 10.1093/emboj/17.14.3921.URL 10.1093/emboj/17.14.3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].DiAntonio A, Schwarz TL, The effect on synaptic physiology of synaptotagmin mutations in Drosophila, Neuron 12 (4) (1994) 909–920. [DOI] [PubMed] [Google Scholar]
- [41].Brandt DS, Coffman MD, Falke JJ, Knight JD, Hydrophobic contributions to the membrane docking of synaptotagmin 7 C2A domain: Mechanistic contrast between isoforms 1 and 7, Biochemistry 51 (39) (2012) 7654–7664. doi: 10.1021/bi3007115.URL 10.1021/bi3007115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hui E, Bai J, Chapman ER, Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I, Biophys. J 91 (5) (2006) 1767–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nalefski EA, Wisner MA, Chen JZ, Sprang SR, Fukuda M,Mikoshiba K, Falke JJ, C2 domains from different Ca2+ signaling pathways display functional and mechanistic diversity, Biochemistry 40 (10) (2001) 3089–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Davis AF, Bai J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER, Kinetics of synaptotagmin responses to Ca2+ and assembly with the core SNARE complex onto membranes, Neuron 24 (2) (1999) 363–376. doi: 10.1016/s0896-6273(00)80850-8.URL 10.1016/s0896-6273(00)80850-8 [DOI] [PubMed] [Google Scholar]
- [45].Bansal D, Campbell KP, Dysferlin and the plasma membrane repair in muscular dystrophy, Trends in cell biology 14 (4) (2004) 206–213. [DOI] [PubMed] [Google Scholar]
- [46].Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, Hadhazy M, McNally EM, Normal myoblast fusion requires myoferlin, Development 132 (24) (2005) 5565–5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Falke JJ, Drake SK, Hazard AL, Peersen OB, Molecular tuning of ion binding to calcium signaling proteins, Q. Rev. Biophys 27 (3) (1994) 219–290. [DOI] [PubMed] [Google Scholar]
- [48].Verdaguer N, Corbalan-Garcia S, Ochoa WF, Fita I, Gómez-Fernández JC, Ca2+ bridges the C2 membrane-binding domain of protein kinase Cα directly to phosphatidylserine, The EMBO Journal 18 (22) (1999) 6329–6338. doi: 10.1093/emboj/18.22.6329.URL 10.1093/emboj/18.22.6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu J, Tan H, Rost B, Loopy proteins appear conserved in evolution, Journal of Molecular Biology 322 (1) (2002) 53–64. doi: 10.1016/s0022-2836(02)00736-2.URL 10.1016/s0022-2836(02)00736-2 [DOI] [PubMed] [Google Scholar]
- [50].Zhang X, Rizo J, Südhof TC, Mechanism of phospholipid binding by the C2A-domain of synaptotagmin 1, Biochemistry 37 (36) (1998) 12395–12403. doi: 10.1021/bi9807512.URL 10.1021/bi9807512 [DOI] [PubMed] [Google Scholar]
- [51].Marty NJ, Holman CL, Abdullah N, Johnson CP, The C2 domains of otoferlin, dysferlin, and myoferlin alter the packing of lipid bilayers,Biochemistry 52 (33) (2013) 5585–5592. doi: 10.1021/bi400432f. URL 10.1021/bi400432f [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].H. M> Khan, T. He, E. Fuglebakk, C. Grauffel, B. Yang, M. F. Roberts, A. Gershenson, N. Reuter, A role for weak electrostatic interactions in peripheral membrane protein binding, Biophysical Journal 110 (6) (2016) 1367–1378. doi: 10.1016/j.bpj.2016.02.020.URL 10.1016/j.bpj.2016.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hui E, Johnson CP, Yao J, Dunning FM, Chapman ER, Synaptotagmin-mediated bending of the target membrane is a critical step in Ca2+-regulated fusion, Cell 138 (4) (2009) 709–721. doi: 10.1016/j.cell.2009.05.049.URL 10.1016/j.cell.2009.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lek A, Lek M, North KN, Cooper ST, Phylogenetic analysis of ferlin genes reveals ancient eukaryotic origins, BMC Evolutionary Biology 10 (1) (2010) 231. doi: 10.1186/1471-2148-10-231.URL 10.1186/1471-2148-10-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Battye TGG, Kontogiannis L, Johnson O, Powell HR, Leslie AGW, iMOSFLM: a new graphical interface for diffraction-image processing with mosflm, Acta Crystallographica Section D Biological Crystallography 67 (4) (2011) 271–281. doi: 10.1107/s0907444910048675. URL 10.1107/s0907444910048675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P,Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A,McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS, Overview of the CCP4 suite and current developments, Acta Crystallographica Section D Biological Crystallography 67 (4) (2011) 235–242. doi: 10.1107/s0907444910045749. URL 10.1107/s0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software J Appl Crystallogr 40(Pt 4) (2007) 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW,Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH, PHENIX: a comprehensive python-based system for macromolecular structure solution, Acta Crystallographica Section D Biological Crystallography 66 (2) (2010) 213–221. doi: 10.1107/s0907444909052925.URL 10.1107/s0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang P, Wang CT, Bai J, Jackson MB, Chapman ER, Mutations in the effector binding loops in the C2A and C2B domains of synaptotagmin I disrupt exocytosis in a nonadditive manner, J. Biol. Chem 278 (47) (2003) 47030–47037. [DOI] [PubMed] [Google Scholar]
- [60].Schrödinger LLC, The PyMOL molecular graphics system, version 1.8 (November 2015).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.