

Abstract
Rapid advances in biology demand new tools for more active research dissemination and engaged teaching. This paper presents Synteny Explorer, an interactive visualization application designed to let college students explore genome evolution of mam- malian species. The tool visualizes synteny blocks: segments of homologous DNA shared between various extant species that can be traced back or reconstructed in extinct, ancestral species. We take a karyogram-based approach to create an interactive synteny visualization, leading to a more appealing and engaging design for undergraduate-level genome evolution education. For validation, we conduct three user studies: two focused studies on color and animation design choices and a larger study that performs overall system usability testing while comparing our karyogram-based designs with two more common genome mapping representations in an educational context. While existing views communicate the same information, study participants found the interactive, karyogram- based views much easier and likable to use. We additionally discuss feedback from biology and genomics faculty, who judge Synteny Explorer’s fitness for use in classrooms.
1 -. INTRODUCTION
It is often said that humans and chimpanzees share 99% of their DNA. The actual answer is more subtle than this: it is 99% if sequences are directly compared, and 96% when insertions and deletions are taken into account [51].
This sharing happens because past evolutionary processes have resulted in patterns of sequence conservation between today’s species. As recently as four million years ago, an animal existed that was a common ancestor to both humans and chimpanzees [42]. From this CHLCA (Chimpanzee-Human Last Common Ancestor), the genera Homo and Pan emerged. Their genomes evolved, but large sections of chromosomes maintained the same order of genes. These similar blocks of DNA exist in extant human and chimpanzee genomes and are formally called homologous synteny blocks, or HSBs. Generally, the more closely related two species are, the more HSBs they share [38]. However, these DNA “jigsaw pieces” are put together in different ways across the genomes of all animal species.
The first free-living organism to have its genome fully sequenced was the bacterium Haemophilus influenzae in 1995 [21]. Now, with the power of genomics, hundreds of genomes have been sequenced and analyzed. At first, comparing exact syntenic regions between today’s animals and long extinct ancestors was not possible, as we did not know the chromosome constitution of these extinct species. Thanks to advances in genome reconstruction [29, 31], scientists can successfully now determine and place the synteny blocks in extinct species based on the sequenced genomes of extant species.
Visualization is an effective tool for understanding complex structures, relationships, and interactions often found in scientific data. While a number of tools for searching, cataloging, analyzing, and disseminating genomic research findings have been developed, they are mainly designed to support the needs of domain experts. Little has been done to facilitate intuitive, large-scale, whole-genome comparisons for novice users or students, limiting their ability to learn about these complex evolutionary processes. Engaging students requires design that succinctly and lively conveys the necessary information. Our work presents an initial step in that direction.
This paper presents a new tool called Synteny Explorer for the purpose of teaching genome evolution between species. Within the application, the user toggles between two main views. (1) The Phylo View shows a mammalian tree of life and is used for navigation and selection. (2) The Genome View displays and evolves selected species’ chromosomes. In this latter view, synteny blocks in an ancestor species’ genome translocate, invert, undergo fission, and recombine in one or more descendant species. To show this, we provide a total of four genomic mappings: two conventional views from existing tools, and two that are designed around an interactive, karyogram-based approach using animation and spline mappings.
A large focus of this work is to study techniques and concepts for effectively communicating genome evolution and DNA conservation in an introductory, educational manner. To this end, we start by describing the iterative design process of Synteny Explorer, performed in close collaboration with domain scientists. Our application design is grounded in HCI theory, existing genomic techniques and practices, and color and animation theory. For validation, we performed three user studies. Two within-user studies focused on design choices for animation and color styles, by measuring both effectiveness and preferability. A larger, between-subjects study evaluated the four genomic mappings and the system’s overall qualitative usability. From these, we present a number of design recommendations and guide-lines. Finally, we include commentary from local biology and genomics faculty on our tool’s fitness for classroom usage. While the immediate context of our results concerns college-level students, they can also be applied in the larger visualization and scientific communities for those who wish to use genomic-based visualization with both the general public and other scientists.
2 -. BACKGROUND
In Synteny Explorer, the Phylo View uses a phylogenetic tree for navigation and selection, while the Genome View displays selected species’ genomes and is where the user maps (evolves) synteny blocks between species. We frame our background discussion using these views, with the context that the tool’s overall purpose is education.
2. 1 -. Displaying Phylogenetic Trees
The first paleontological Tree of Life was published in 1840 by Edward Hitchcock [5]. Though an incomplete illustrative metaphor for all life (for example, bacteria can change genomes through horizontal gene transfer [19]), the branching lines of phylogenetic trees are still used today to illustrate relationships between bacteria, archaea, and enkaryotic life domains. Phylogenetic trees are commonly seen in introductory biology courses, but unfortunately numerous studies show “tree thinking” is neglected in American K-12 science curricula [11] and that even undergraduate biology students can have misconceptions about reading and understanding phylogenetic trees [33, 9].
Many scientists and educators feel that tree thinking - correct interpretation of phylogenetic trees - is vital to understanding evolution [6]. One potential reason for current deficiencies is that textbooks and static tree images do not support “active learning,” which interactive systems are better suited for [47].
While explicitly research- and academic-focused tools exist for analyzing trees (such as Mesquite [32]), other interfaces use active learning and active prolonged engagement [24] for the purpose of education, collaboration, and discussion. DeepTree [7] allows museum patrons to interactively explore and “fly through” the tree of life. FloTree [13] and Build-A-Tree [23] let users interactively construct and edit trees using gamification. Phylo-Genie [45] is designed to foster a collaborative learning environment for phylogenetics. The positive user evaluations of these applications partly motivated us to include a phylogenetic tree as part of our system, which we discuss in Section 3.
2.2 -. Genomic Reconstruction and Visualization Tools
In 1998, the nematode worm Caenorhabditis elegans was the first multicellular eukaryotic animal to have its entire genome sequenced [16]. In 2004, the Human Genome Project published a representation of a whole sequenced human genome [15]. Today, there are hundreds of sequenced genomes, while thousands of species are in the process of being sequenced [27].
As chromosomes and genomes were being sequenced, the discovery of homologous synteny blocks (HSBs) between species, along with emerging patterns of synteny break, led to investigation of the role of evolutionary breakpoint regions (EBRs) in genome evolution. EBRs are locations between synteny blocks that demonstrate a point where a chromosome rearrangement has taken place in evolution. When this occurs, chromosome segments and even whole chromosomes can be recombined in different ways: for example, they can flip (called an inversion), a chromosome segment can break and then link with another chromosome (translocation), they can fuse (fusion), or split into independent chromosomes (fission). Repair of the chromo- some breakages can also be a source for generating new genes and new genetic material. Together with point mutations, chromosome rearrangements are the driving forces behind genome evolution and evolutionary processes. Both genome sequencing and reconstruction of HSBs and EBRs in ancestral species are frontiers in genomics re- search. For more information on this topic, see [29, 38].
To accommodate scientific research, a number of chromosome- specific mapping tools called synteny browsers have been developed [14, 41, 25, 17, 48, 36, 34, 20]. Evolution Highway [3] was an early tool for analyzing HSBs and EBRs across different species that lays out each chromosome section as a vertical, aligned block. One draw-back to this tool is its granularity: the user can only look at one chromosome, in depth, at a time.
In contrast, full-scale genome browsers allow whole genome visualization and comparison between species. In their paper describing the Mizbee tool [35], the authors propose a taxonomy of six genome browser layouts based on chromosome arrangement. The layouts fall into three categories depending on the positioning approach used: linear, circular, and discrete.
In linear layouts, chromosomes are arranged in a straight (some- times connected) line, like a row of bricks. If the compared genomes are separate, the lines are arranged parallel to each other and synteny mappings shown using color or line segments (see [18, 40, 4]). Linear- combined layouts place both genomes in a single row.
Circular-based layouts can also be separate, where each genome is arranged in a circular fashion and multiple circles are used. Mizbee itself uses this layout. If multiple genomes are mapped along arcs on a single circle, the layout is combined. Circos [28] is this latter circular-combined layout, and uses a chord diagram to map synteny between chromosomes. Circos is currently the most widely-used tool for genomics visualization in the genetics community; over 500 publications and links are listed on its website [2].
Finally, discrete views arrange chromosomes on a grid. Cinteny [50] was an early browser that did this in a segregated manner, where genomes are separated into distinct chromosome grids (with mappings shown via color). Alternatively, two genomes can be interleaved together, such as what is done in the Apollo browser [30].
A major issue with the above tools is they are designed for domain scientists and thus lack evaluation in the context of education. This led us to consider each of the various layouts as candidates for inclusion in Synteny Explorer. During development, we mocked up many of the described layouts from the Mizbee taxonomy and implemented the Apollo and Circos views as mappings in our tool. We also include Cinteny’s rainbow palette as one of the color schemes.
3 -. DESIGN PROCESS AND SYSTEM OVERVIEW
The design process for Synteny Explorer was iterative, spanning approximately thirteen months. We met with a number of genomics researchers with the goal of understanding their current workflow and how they envisioned genome evolution in an educational context. We defined our initial target audience as students in introductory courses in evolution, which meant that an effective application would be able to demonstrate fundamental principles of chromosome and genome evolution. For our genomicist collaborators (co-authors on this paper), the synteny browser Evolution Highway [3] was their main tool, which lays out selected chromosomes as parallel blocks and uses color to denote preserved regions. We quickly determined that this approach was too limited for our purposes. To maximize prior familiarity and conceptual understanding, we decided to show each species’ whole karyotype. involved users dragging DNA “chunks” from this view. (Middle Right) Cytogeneticists usually represent chromosomes as a single cylinder with “bands” and centromere noted. (Right) Our view removes the centromere, leaving a chromosome as a rectangular block with different colors denoting synteny blocks.
Before presenting the user with a view of genome evolution, we decided to first require them to select a set of species from a phylogenetic tree. While some early mock-ups omitted a hierarchical species-based view of the data, we decided that having a tree-based overview of animals would let users ground their prior evolutionary knowledge. A user who did not understand the tree of life, the de facto standard for showing evolutionary connections between species, would have difficulty comprehending a more advanced topic such as the synteny mappings that primarily make up genome evolution. Therefore, a basic understanding of evolution at a tree-based level became a prerequisite for use.
Our decision to include a tree-based view of animals is also rooted in the Visual Information Seeking Mantra of “overview first, zoom and filter, details on demand” [49]. That is, upon starting the application, the user sees the tree of life and the “big picture” (overview) of evolutionary change. In our system, this is the Phylo View (Figure 1). Here, the user navigates the tree and selects desired animals before switching (zooming and filtering) to the Genome View (Figure 2) where s/he can visualize species genomes and synteny block mappings between species (details on demand). This type of tree-based exploration is used in other systems (such as DeepTree [7]) to positive effect, so we felt this decision was well-justified.
Fig. 1.
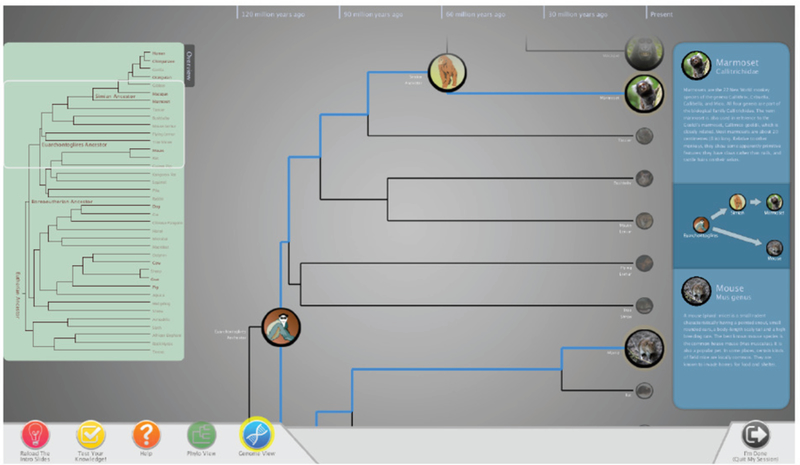
The Phylo View lets users explore a subset of the mammalian tree of life. Species selection is done by dragging icons into the blue sidebar. In this example, marmoset and mouse are selected.
Fig. 2.
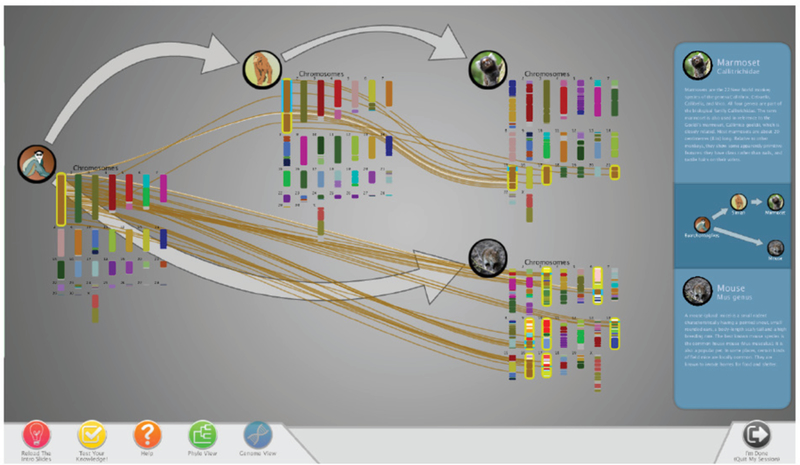
The Genome View shows the chromosomes and synteny map- pings of selected species. In this example, the Static mapping is used with the Faded color palette.
For showing chromosomes, we at first implemented artistically rendered X and V shapes (see Figure 3). After receiving feedback from the genomicists, this was changed to a bar design that omits the centromere. This was done for three reasons: (1) Chromosomes only appear paired (two arms) after replication (joined at their centromeres). (2) A bar is a standard representation for chromosomes used in many existing systems [50, 3] and is a simple and effective visual mark. (3) Current ancestral reconstruction cannot determine centromere position. Within each chromosome, color is used to denote synteny blocks and is preserved across species. For visualization of genome evolution, we focused exclusively on chromosome rearrangements. There are very few tools that visualize this dynamically, which is critical to understanding the relationship between chromosomes and speciation. We did not focus on visualization of point mutations or of small structural variants (such as insertions and deletions) because they do not facilitate understanding of the macroscale chromosome rearrangements that occur during evolution. Visualization of these types of mutations is best handled by tools such as the UCSC and ENSEMBL Genome Browsers [1, 14].
Fig. 3.
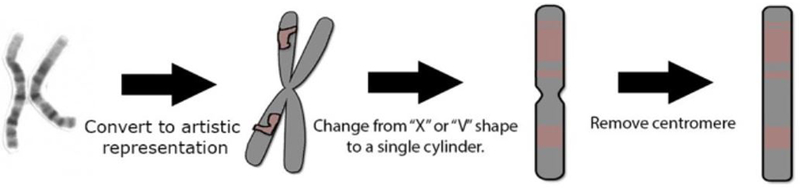
Representing chromosomes: (Left) A picture of human chromosome 1. (Middle Left) The “X”-shaped figure is familiar to many people but is inaccurate except during cell division. An early system prototype
Finally, to focus on education, we emphasized keeping the application workflow simple and straightforward, with a focus on interactivity and teaching concepts. Interactive visualization [47] and multi-media [39] have been shown to play important roles in active learning and developing higher-order thinking [43]; as such, we incorporated techniques like animated transitions (such as between the Phylo and Genome Views) and interactive highlights (strokes around currently selected chromosomes, etc.).
3.1 -. Phylo View: Navigation and Selection
Upon starting the application, the user scrolls through a set of introductory slides; these give a brief overview of the tool and usage instructions. This help menu can be reopened as needed.
The initially shown Phylo View (Figure 1) displays a subset of the tree of life, consisting of many extant mammalian species (hu- man, chimpanzee, dog, mouse, etc.) for which we have derived their genomes, as well as some important common ancestors with recon- structed genomes. For example, the Simian ancestor is the root of all ape and human species. Animal species are shown as circular icons containing a representative picture or artistic illustration. The tree is oriented from left to right and nodes are placed along the y-axis to help give a sense of the geological “deep time” scale over which macroevolution takes place [10].
We note that we include many “disabled” animal nodes in the tree. This serves two purposes: (1) to give users better context of where enabled (sequenced) animals fit in the tree and who they are related to, and (2) because new animals (both extant and extinct) can be enabled once their genomes become available.
Decoding an entire genome for ancestral reconstruction is not a trivial task, but this is being done to more and more extant species. Synteny Explorer is scalable to include the entire set of mammal genomes and can easily be extended to other vertebrates and invertebrate animals.
To select a species, users drag its icon into a right-hand sidebar. This loads the animal in the sidebar with a short set of descriptive text. When two species are loaded, a mini-tree is built showing the evolutionary path between them. In Figure 1, a user has selected the marmoset and mouse species. In addition to the two selected species, the mini-tree also shows the intermediate species: Euarchontoglires and Simian ancestors. Once two species are selected and the mini-tree is built, the option to switch into the Genome View becomes enabled in the bottom bar. This is shown as the highlighted blue button with a DNA sequence symbol on the bottom bar of the view.
3.2 -. Genome View: Details on Demand
When going to the Genome View, the species currently in the mini-tree are displayed in the main viewing area with their full genomes; the rest of the phylogenetic tree is hidden. Here, synteny mappings between the selected species can be viewed and interacted with, and a user can track how chromosomes translocate, split, and invert.
Figure 2 shows an example of the Genome View built around the marmoset and mouse. Chromosome 1 in the Euarchontoglires ancestor has been selected; it is highlighted with a yellow stroke. The brown splines coming out of this chromosome and going to the chromosomes in other species (also highlighted with yellow strokes) represent mappings between the loaded genomes. They show how the synteny blocks from chromosome 1 underwent several changes when going to the descendant Simian ancestor, marmoset, and mouse species.
In considering potential genome mappings for the Phylo View, we referenced two main sources: the layouts from the Mizbee taxonomy [35] and images from textbooks. We sketched, prototyped, and discussed a number of mockups with our genomicist collaborators, which led to many layout options being discarded. For example, with prototypes for both the linear-separated and linear-combined layouts, display space became an issue. Due to our requirement that the full genome is shown, individual chromosomes were scaled very small to fit on a normal-sized monitor. The circular-separated layout was also considered and discussed with the genomicists, but skipped in favor of using the simpler circular-combined view.
Ultimately, we implemented four representations for genome evolution, which we term mappings. The first two views, called Static and Animated, use a karyogram-based layout for displaying chromosomes (a specific type of discrete-separated layout). The other two mappings come from the research community: the Circos [28] and the Apollo [30] views.
3.2.1. Static and Animated Karyogram-based Mappings
In genetics, a karyotype is the term to describe the set and appearance of chromosomes for a eukaryotic cell. This is usually displayed using microscopic pictures of the chromosomes of a cell in metaphase, the stage of maximum DNA compression (such as in Figure 3). The depiction of these chromosomes is called a karyogram, and is the common representation for displaying genomes in biology textbooks. When deciding on a layout to use, we reasoned that a familiar chromosome representation would intuitively connect users to our system and help them remember their prior exposure to genomes (if they had any). Normally, karyograms show chromosomes as dyads ordered by size and centromere position. We show chromosomes with the bar view from Figure 3 and use the karyogram grid-based, size-ordered layout for the first two mappings, termed Static and Animated. These are shown in Figure 4a and 4b. (Variations on this view are also used by some prior systems, such as the Cinteny browser [50].)
Fig. 4.

The Genome View has four mappings for displaying synteny between species. (a, b) Static and Animated are based on karyogram layouts of chromosomes, while (c, d) Circos and Apollo are used in the scientific community. In these examples, we are showing how Chromosome 7 in the extinct Simian ancestor maps to the macaque. (a) Static uses colored splines to map syntenic relationships, while (b) Animation uses a flying effect. (c) Circos is a chord diagram with ribbons, and (d) Apollo arranges the macaque genome in an interleaved, grid-based layout, where each of its chromosomes is surrounded by contributing Simian chromosomes.
The difference between the two mappings happens when user interaction occurs. In Static (Figure 4a), clicking on a chromosome shows synteny mappings via colored splines. That is, each synteny block is connected by a single string to its destination genome and chromosome position. Splines are colored based on the block color, and inversions are noted with blue stroke highlights on the destination chromosome(s).
In contrast, when a user clicks a chromosome in the Animated mapping (Figure 4b), it splits into the appropriate synteny blocks and flies to its mapped places on the descendant species’ chromosomes. If a block inverts in going to a descendant species, it does a half spin as it flies. We choose to include this animation-based approach (as opposed to only having a Static mapping) because animation as a technique has been shown to be an effective way to enhance HCI and foster learning [12], especially in highly interactive scenarios [52].
3.2.2. Circos and Apollo Mappings from the Community
The other two mappings are implementations of existing tools: the Circos chord diagram [28] and Apollo interleaved viewer [30].
Circos (Figure 4c) was chosen because of its widespread popularity in genomics research. We felt it would serve well as a “standard” or baseline technique for evaluating the other genome mappings. Though a Circos diagram can technically compare an arbitrary number of species, we restrict each circle to only show two genomes - the connection between a single ancestor and descendant. This simplifies the visual complexity of each mapping though it means that multiple circles are used when an ancestor is shown with multiple descendants (the ancestor’s genome is duplicated on each circle). Ancestor chromosomes are oriented in a 180° arc along the left side of the circle while descendant species chromosomes are reflected on the right. Synteny connections are shown using colored ribbons. Like the Static mapping, inversions are shown by blue highlights on the destination chromosome.
Apollo (Figure 4d) arranges the chromosomes of the descendant species in a grid. Around each, all origin chromosomes that have map- pings to that particular destination are arranged. Line segments show synteny connections. We use black for normal mappings, and color the line if the block inverts. Apollo doesn’t require user interaction to see chromosome mappings; they are already shown in this initial layout. Despite the fact that this view has potential problems with spatial locality (as origin chromosomes are not arranged together, and potentially shrunk to fit around their respective destination chromosomes), Apollo was chosen as a view because it has less visual clutter compared to the Static and Circos mappings (less spline and ribbon crossings when all mappings are shown).
3.2.3. Genome Color Palettes
Within the Genome View, chromosome coloring is used to denote conserved DNA between species. In Figure 2, the brown color of Euarchontoglires’ chromosome 1 is preserved in descendant species. Since synteny blocks split, merge, and move to different chromosomes as species evolve, color intuitively indicates how much genetic fragmentation and rearrangement has taken place between two genomes without actually having to interact with them. It is, therefore, important to choose an effective color palette for the Genome View.
We experimented with five color palettes for chromosomes (Figure 5). Initially, we were inspired by spectral karyotyping (SKY), a bio- logical technique where each chromosome in a genome is “painted” with a different fluorescent color [46]. From this, we created a qualitative colormap denoted as Bright (Figure 5a). We used this palette to derive two additional schemes: Faded and Grayscale (Figure 5b and 5c). The Faded scheme uses darkened versions of Bright’s colors, and the Grayscale scheme uses monochrome ones. When one of the de- rived palettes is selected by a user (i.e., it is clicked and needs to show its synteny mapping s), we temporarily change its color to the corresponding Bright scheme to give it a “pop.”
Fig. 5.
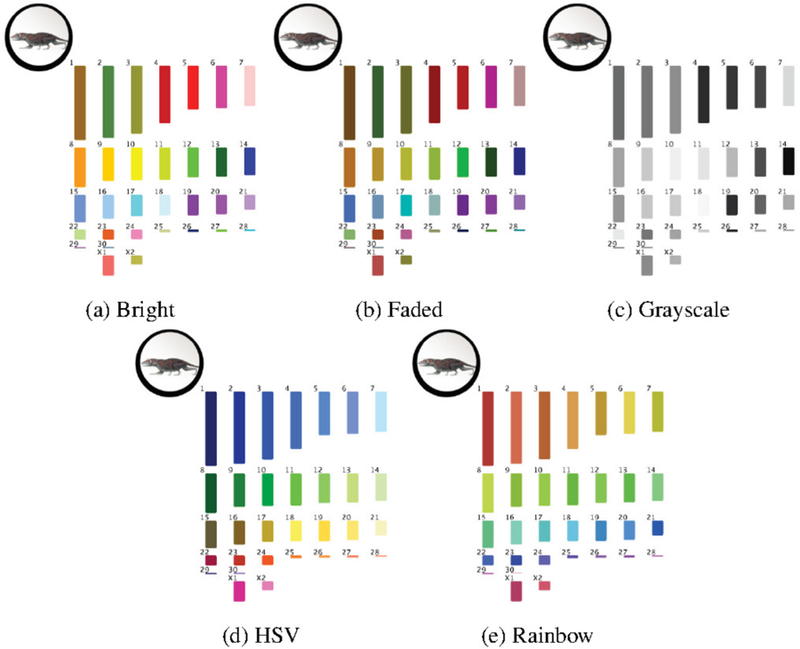
There are five color palette options for chromosomes.
For a fourth, HSY-based palette (Hue, Saturation, Value, see Figure 5d), we coordinated with a professional artist who studies color and human perception for use in scientific visualization [44]. In this scheme, each row of chromosomes in the karyogram layout comes from one dominant color (blue, green, yellow, red, and violet). Each individual chromosome color is then defined by tweaking the row’s color in the HSV space, where the human brain naturally resides. By modifying all three variables, we create a higher perceivable contrast than in a sequential, linear, or CIELAB-based color space [53].
As a fifth palette, we included the rainbow color scheme from the Cinteny genome browser [50], see Figure 5e. Though rainbow palettes are considered suboptimal [8] in the visualization community (and Cinteny’s scheme specifically has been criticized even in textbooks [371), this palette was chosen to serve as a baseline for which to mea- sure the other color schemes (similar to the Circos mapping), since it is currently used in an existing research tool.
3.2.4. Evolving the Whole Genome at Once
In the Genome View, a user clicks on an individual chromosome to map its synteny blocks to one or more descendant genomes. Our genomics collaborators recommended that we add the ability to see all chromosomal links between two species simultaneously, to provide a sense of the overall magnitude of rearrangement and fragmentation that takes place. Therefore, we implemented a “chromothripsis”-like event, invoked by clicking the gray arrow that connects two species.
Three of the four genomic mappings require this action. For the Animated view, all synteny blocks fly to their destination positions. We considered four different styles for this, based on whether the animation happens as a single action, or is broken into two discrete steps. This is shown in the three images in Figure 6: (1) Morph (re- arrange) the chromosomes to their destination genome’s position in place, then slide the rearranged genome from the ancestor to the descendant species in a separate, second step (Figure 6a). (2) Alternatively, slide the chromosomes to the destination species, then morph them into their destination mappings (Figure 6b). (3) Do the morph and slide simultaneously (Figure 6c). (4) Do the morph and slide simultaneously, but cascade the start time for each chromosome based on the technique advocated by Heer and Robertson in [22] (Figure 6c, but with staggered start times).
Fig. 6.
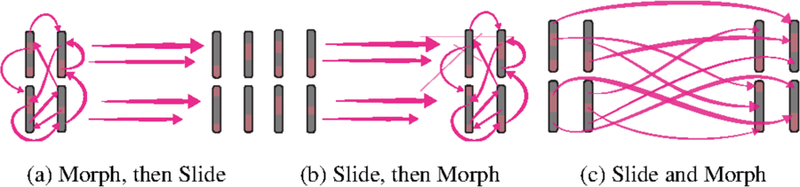
There are four ways to animate a genome chromothripsis event, as shown with these three figures. (a) and (b) break it into two distinct parts: a morph step and a slide step. In (c), the morphing and sliding happens together. For the fourth technique, the morph and slide from (c) is used, but the starting time of each chromosome is staggered.
In Static and Circos, whole genome rearrangement is shown statically. For Static all splines are shown together and for Circos all ribbons are shown inside the chord diagram. Since Apollo by default shows all synteny connections, it does not need a chromothripsis-like interaction.
3.3. Implementation Details
Synteny Explorer is built using Java and the Processing graphical library. For generating species data, we use whole genome assembly of over twenty species from eleven mammalian orders. Ancestral chromosome reconstructions were done using an algorithm by Kim et al. [26]. This reconstruction data is stored in a directory structure comprised of sets of text files, and a Wikipedia web service is used to retrieve sidebar species descriptions.
4. USER STUDIES 1 AND 2: COLOR AND ANIMATION
Since multiple color palettes and chromothripsis-like animation styles were implemented, we wanted to know which were best for our system. To answer this, we conducted two focused, within-subjects studies. Each was performed with the same set of ten participants (mean age=25.7, SD=3.09, five males, all engineering students) and the same setup: a Windows 7 computer with a 30-inch display monitor (2560 × 1600px resolution).
4.1. Color Palette User Study
For the first study, we wanted to know which color palette was best at helping users determine synteny mappings between species sans inter- action. Though interaction is an integral part of Synteny Explorer, a good color scheme should intuitively assist users in seeing conserved regions. For this study, we compared the Faded, HSV, Grayscale, and Rainbow color schemes; the Bright scheme was omitted since it uses the same base colors as the Faded scheme.
To perform the study, we generated a set of twenty species pairs, chose a single chromosome from each of them, and asked the following questions: “Where does chromosome [number] in [species A] go to in [species B]?” For each question, we loaded the required species in the Genome View (using a karyogram layout) and applied one of the color palettes. To minimize potential bias and carryover effects, question order and applied color palette were randomized. The participant viewed the monitor and stated their answer to an observer, who recorded correctness and response time. After finishing, each participant rated the color palettes on a l-to-5 rating scale for the following question: “How much do you like this color scheme?”
4.1.1. Color Study Results
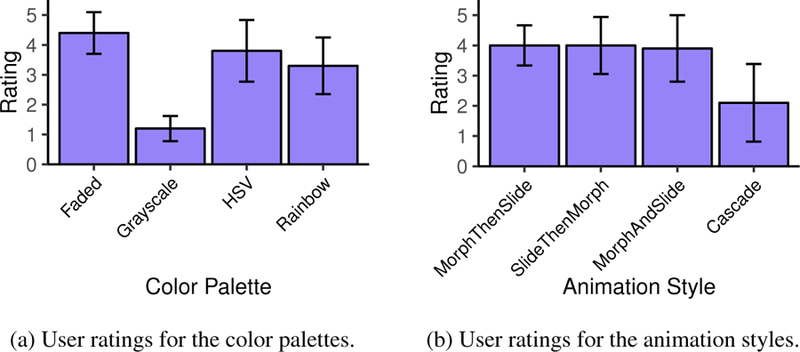
Table 1 shows each color palette’ s correctness and task completion time. The Krnskal-Wallis significance test indicates an effect of color palette on correctness. Post hoc pairwise comparisons show that Faded, HSV, and Rainbow all are significantly better than Grayscale (p < .01) for correctly observing mappings. Faded has the highest overall correctness. Within-subjects repeated-measures ANOVA shows an effect of color palette on average task completion time (p <.05). Bonferroni-Holm corrected pairwise comparisons show that Grayscale is significantly slower than the other color schemes (p < .001). Faded has the fastest task completion time.
Table 1:
Correctness and response time for the color user study.
| % Correct | Response Time (s) | |||
|---|---|---|---|---|
| Color Palette | Avg. | S.D. | Avg. | S.D. |
| Faded | 94.0 | 24.0 | 8.6 | 4.8 |
| Grayscale | 38.8 | 49.2 | 21.7 | 12.0 |
| HSV | 88.2 | 32.5 | 11.7 | 7.4 |
| Rainbow | 76.0 | 43.1 | 13.9 | 10.3 |
Ratings scale results are noted in Figure 7a. Faded has the highest score with a 4.4 average, and Grayscale the lowest at 1.2. The HSV and Rainbow palettes are rated similar to Faded, but they also received multiple negative feedback comments from users who felt they were sometimes hard to discern. As one participant stated, “on Rainbow and [HSV], for some questions they are decent, but for some they are hard. The shades between those are really difficult to tell apart.”
Fig. 7.
Qualitative scores for user studies 1 and 2, which show how well participants liked each of the color palette and animation styles.
Based on the combination of correctness, timing, and user rating results, we use the Faded color palette as the system default.
4.2. Animation Styles User Study
The animation study had a similar motivation and setup to the color study. The purpose was to determine which chromothripsis-like animation style was best for our tool. The four styles were termed MorphThenSlide, SlideThenMorph, MorphAndSlide, and Cascade. Six- teen sets of animals were retrieved, and for each the following question was asked: “Is there more genetic fragmentation going from [species A] to [species B] or from [species C] to [species D]?” where “genetic fragmentation” is defined as “the overall rearrangement of chromo somes via number of inversions, translocations, and splits.”
Like the color study, question order and chosen animation style were randomized, and response correctness and time were recorded by an observer. Unlike the color study, the user could interact with the Genome View by invoking the chromothripsis events. They then visually tracked the synteny blocks as they animated to their destination.
This led to more variability in response time, as some users invoked the animation events multiple times for some questions. After finish- ing, users rated their animation style preferences like the color study.
4.2.1. Animation Study Results
Table 2 shows each animation style’s correctness and task completion time. A chi-square significance test shows no effect of animation style on correctness of answers (χ2 = 0.8, d f = 3). However, within- subjects repeated-measures ANOVA show an effect of animation on average task completion time (p < .05). Bonferroni-Holm corrected pairwise comparisons show that Cascade and SlideThenMorph animations are slower than MorphAndSlide and MorphThenSlide (p ≤ .05). Average user ratings are shown in Figure 7b. The Cascade style rates significantly lower than the other three, which have almost identical averages. On the low preference for Cascade, multiple users noted that, due to the varying start times, it was hard to discern the magnitude of the genetic fragmentation while optically following synteny blocks across the monitor. One noted, “At first, I thought Cascade might be easiest to track, but because I have to track them over a long place, I think I lose track of them.” Another mentioned that it “stretches my view field. I’d rather see it all together instead of tracking them all over the place.” Users noted they rated the other views higher because they “liked that everything happened at the same time.” More specifically, for the MorphThenSlide and SlideThenMorph styles, some users said they “see all the changes in one place, so it’s easier to track.”
Table 2:
Correctness and response time for the animation user study.
| % Correct | Response Time (s) | |||
|---|---|---|---|---|
| Animation Style | Avg. | S.D. | Avg. | S.D. |
| MorphThenSlide | 82.5 | 38.4 | 32.3 | 14.1 |
| SlideThenMorph | 85.0 | 36.2 | 53.3 | 75.8 |
| MorphAndSlide | 82.5 | 38.4 | 34.4 | 13.5 |
| Cascade | 77.5 | 42.3 | 56.1 | 22.0 |
Based on the combination of high user rating, fast response time, and user comments, we use MorphThenSlide as the default chromothripsis animation style.
5. USER STUDY 3: EVALUATING GENOME MAPPINGS AND OVERALL SYSTEM USABILITY
After finalizing our design and verifying basic functionality, we wished to perform a formal system validation and compare the genomic mappings against each other. To do this, we conducted a large, between-subjects study with the following hypotheses:
H1 Overall, Synteny Explorer is an enjoyable and successful tool for teaching people about genome evolution. H2 The two karyogram-based mappings (Static and Animated) are easier to use and make more sense for non-experts than the mappings based on current domain tools (Circos and Apollo).
5.1. Setup
The groups in our study were based on the four genome mappings: Animated, Apollo, Circos, and Static. We used the MorphThenSlide animation style and the Faded color palette based on results from Section 4. Study participants followed the steps in Table 3 to conduct a user session. Each user, with a randomly assigned genome mapping, did a short training phase by playing with the system and learning how to navigate between views. When ready, they toggled a quiz interface that led them to navigate, load, and analyze different genomes and synteny relationships to answer questions. After this, they filled out a short survey where they could type freeform comments, rate various system aspects, and make suggestions. Finally, they could play with the other three genome mappings and give feedback.
Table 3:
Steps for a user session in Study 3.
| Step | Action |
|---|---|
| Training | The user reads the help menu and plays with the application to understand the interface. |
| Quiz | The user answers a set of questions using a randomly preselected genome mapping. |
| Survey | Upon finishing the quiz, the user fills out a survey to give feedback on the chosen mapping. |
| Playing with other Mappings | The user is shown how to toggle between the other three mappings. They can play with these as long as desired, and give feedback to the observer. |
During sessions, no audio was recorded, though an observer was present to assist the user if they felt stuck or confused (though no help was given for quiz questions) and to scribe any freeform comments that the participants had during their sessions.
For the quiz, we asked four types of multiple-choice questions. Each participant was asked to answer twelve total questions (three from each category) that asked about the following:
Q1 Pick the chromosome that has a synteny mapping to another species’ specific chromosome.
Q2 Given a set of species pairs, find the one with the most genetic fragmentation.
Q3 Count the number of chromosomes in an animal.
Q4 Count the number of inversions in a specific chromosome between two species.
For the study, there were sixty participants (mean age=26.35, SD=4.22, 20 males) from the student body at the University of California, Davis. Users completed the study in an average time of 25 minutes (SD=7.5 minutes). Users were recruited by open-invitation department emails and flyer postings. Due to the described nature of the study, a significant percent were from biological fields (20/60) and consequently had been at least exposed to the concepts in the app. The rest of the majors were representative of the university’s population. The undergraduate-to-graduate ratio was also consistent with the university’s population. Two computer stations were used. The first was the same machine from the color and animation studies, and the sec- ond was a 27-inch iMac with 2560 × 1440px display running OS X. Since Synteny Explorer is written in Java, rendering, speed, and frame rate are equivalent on both computers. For both computers, all system interactions were performed using a mouse only.
5.2. Results
Upon completing the study, we analyzed results along two main axes: quiz results data generated from application logging, and user feed- back based on comments during sessions and filled-out surveys.
5.2.1. Analyzing Quiz Results
Table 4 shows the percentage of correct answers for each mapping as well as how long each task takes on average, where a task is defined as answering one question. Chi-square tests on 720 question-answer samples (60 participants × 12 questions each) show that mapping has no significant effect on answering the quiz questions (p = 0.97); a user’s chosen mapping doesn’t affect their final quiz score. Overall, users performed well on the quiz, with a mean of 10 out of 12 questions correctly answered (SD=4). Interestingly, users in biologically-related fields did not answer more correctly (p = 0.11) than other users.
Table 4:
Correctness and task time for each mapping in Study 3.
| % Correct | Task Time (s) | |||
|---|---|---|---|---|
| Genome Mapping | Avg. | S.D. | Avg. | S.D. |
| Animated | 86.3 | 12.1 | 54.6 | 14.0 |
| Apollo | 85.1 | 14.0 | 47.6 | 19.1 |
| Circos | 84.5 | 14.9 | 44.9 | 12.2 |
| Static | 85.1 | 10.4 | 52.5 | 17.4 |
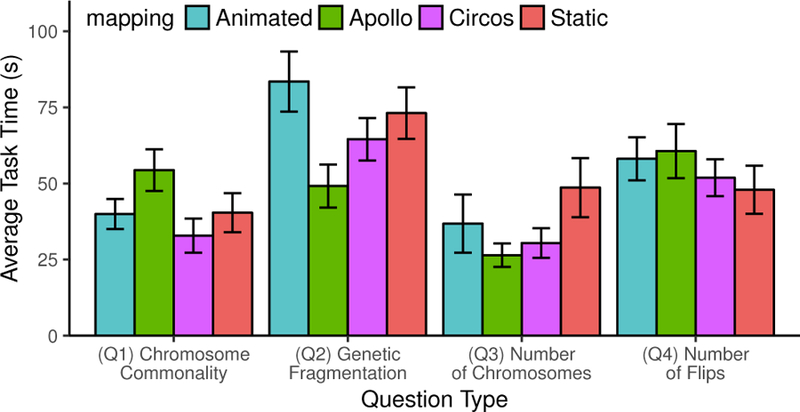
One potential reason for these high quiz scores is that our population was predominantly volunteers, and were thus particularly motivated and interested to perform well. It is possible that the types of quiz questions that we ask are too easy, though they are intentionally designed to be introductory and not to trick users. Instead, questions are a means to cause users to investigate and analyze synteny map- pings between different species. Some users commented that asking them questions helped them explore Synteny Explorer and learn more. Figure 8 shows both the type of question and the mapping influence the average task time. Q3 takes the lowest amount of time while Q2 takes the longest. For Q4, the two karyogram views (Animated and Static) take the longest on average, and Apollo is the fastest. For other questions, this is not always the case.
Fig. 8.
For each mapping in Study 3, how do question type and map- ping influence average task time?
The mean task completion time for all tasks is 50.0 seconds. One result that we expected is users with the Animated mapping would complete tasks faster, due to its intuitiveness and familiarity. How- ever, Table 4 shows Circos is the fastest at 44.9 seconds while Ani- mated is the slowest at 54.6 seconds (though all measures are within a standard deviation of each other). While two-way ANOVA shows mapping has no main significant effect on average task completion time (F3,208 = 1.432), there is a significant question type × mapping interaction effect (F9,208 = 2.082, p < 0.05, η2 = 0.083) as well as a main significant effect of question type on average task completion time (F3,208 = 14.738, p < 0.01, η2 = 0.175). Thus, Figure 8 shows this depends on the task. One possible reason Animated is slow for some tasks is that this mapping requires the user to wait and observe while the animation plays out. However, Bonferroni-Holm corrected post hoc tests show both karyogram-based mappings are slower than the non-karyogram ones, though not significantly (p = .06).
We examine the difficulty of each question type in Table 5. Our results strongly suggest a main effect of question type on correctness (F3,208 = 17.622, p < .01, η2 = 0.203). For example, no matter the genome mapping, users accurately answer Q3 with a 90% success rate. By contrast, Q2 has only a 66% success rate. Bonferroni-Holm corrected post hoc tests show a significant difference between Q2 scores and the other question types (all p < 0.01). We note that comparing the correctness and response times for Study 3’s Q3 to the animation styles study (Section 4.2) cannot be perfectly done, since in the animation- specific study, users did not have to navigate and load species in the Phylo View. There is also the possibility that the questions asked in that study are easier than the Q3 ones in Study 3, or that users became adept at answering the same type of question repeatedly.
Table 5:
Correctness and task time based on question type in Study 3.
| % Correct | Task Time (s) | |||
|---|---|---|---|---|
| Question Type | Avg. | S.D. | Avg. | S.D. |
| (Q1) Chrom Commonality | 97.0 | 9.6 | 41.9 | 22.5 |
| (Q2) Genetic Fragmentation | 66.7 | 36.0 | 67.6 | 36.9 |
| (Q3) Number of Chroms | 90.5 | 20.8 | 35.6 | 26.0 |
| (Q4) Number of Inversions | 87.0 | 18.7 | 54.6 | 25.9 |
5.2.2. Analyzing User Comments and Survey Results
We logged a total of 597 user comments, either spoken aloud by participants or entered during the survey portion of their session. To classify comments while ensuring lack of bias, five unaffiliated volunteers each independently classified all comments by type (positive, critique, etc.) and feature (Circos mapping, General comment, etc.). Each com ment’s final type/feature classification is determined by the majority rating of the group. These classification counts are shown in Table 6.
Table 6:
Survey comments summed by type of response they are and application view/feature that they refer to. Bolded numbers indicate that comment type was the maximum for its particular genome view.
| Genome View Mappings | Application Areas | |||||||
|---|---|---|---|---|---|---|---|---|
| Type of Comment | Animated | Apollo | Circos | Static | General | Phylo View | Sidebar | Slides |
| Positive | 55 | 14 | 29 | 36 | 68 | 5 | 2 | 1 |
| Suggestion | 13 | 6 | 4 | 9 | 67 | 10 | 4 | 21 |
| Confusing | 10 | 21 | 18 | 12 | 33 | 3 | 2 | 9 |
| Critique | 11 | 5 | 14 | 12 | 11 | 0 | 0 | 1 |
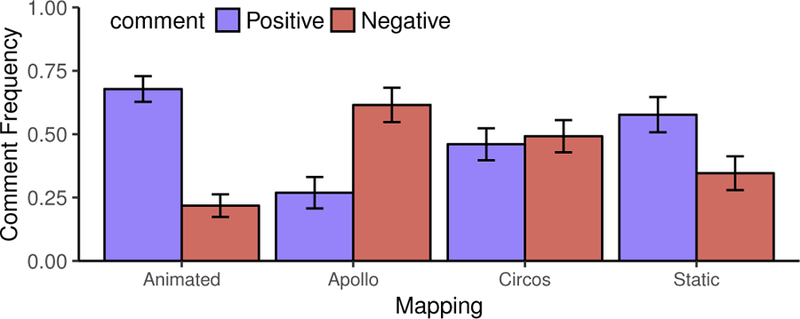
User comments help us derive a number of conclusions about our system. The Animated and Static mappings have a total of 89 positive comments and only 37 negative ones (where negative comments are either confusing or critical comments). In contrast, Apollo and Circos have 43 total positive comments to 57 negative ones. Chi-square tests confirm a significant difference in both positive (χ2 = 16.84, d f =1) and negative (χ2 = 19.75, d f = 1) comments (p < 0.01). This is shown plainly in Figure 9, where Animated and Static have much higher frequencies of positive comments than Circos and Apollo. This indicates users prefer the karyogram-based views to the conventionally-used techniques.
Fig. 9.
Frequency of positive and negative comments (the sum of con- fusing and critique comments) for each mapping in Study 3.
In considering overall user opinions of our system, many comments are grouped into a “General” application area. This includes positive comments (“I really like this application!”) and suggestions (“You should have a sidebar Clippy-type of character that tells you the actions you are doing.”). For each of the four application areas, Suggestion is the most common comment type. The reason for this is we solicited suggestions from the users as one of our survey questions; most users listed multiple ideas if they had them.
The introductory slides have only one positive comment (“The [slide] explanations were simple without being condescending, and the graphics really helped explain the concepts.”) but many suggestions, confusions, and critiques. This is one feature of the tool that we believe was not well thought-out. Some users suggested alternative introductions, such as walking the user through a hands-on tutorial, or using a movie to explain how to use the application.
While the Phylo View only has six positive comments (and nine suggestions), they support our intentions for including a phylogenetic tree in our tool. Positive comments were about how it based users’ prior knowledge (“I loved the phylogenetic tree... and the quick ability to see how closely different species were related through a common ancestor.”) and that it “showed family relationships well.” In contrast, suggestions were for more stylistic tweaks, for example asking us to include names on species icons.
In the survey, we asked users to rate their quiz session on a 1-to- 5 rating scale (similar to the color and animation studies). Figure 10 shows the ratings for three of the questions: (1) “How well do you like your mapping?” (2) “How much effort was required to take the quiz?” and (3)“How stressful was the quiz?” For the first question, all mappings are similar, with only Apollo being a little less liked than the other three. For the second and third questions, the karyogram mappings are rated slightly better (lower) than the conventional views. Another survey question asked participants if and what they learned using Synteny Explorer. 53/60 users affirmatively stated they learned something by using the tool; the other seven were biology students with extensive prior knowledge in this domain. Thirty-nine people referenced specific things that they learned, which are categorized in Figure 11a. Eighteen people simply learned that animals are genomically related, that they share DNA. This shows that our tool effectively communicates the concept of synteny conservation between species. Seven people referenced chromosomal rearrangement. One stated, “I learned how chromosome flips and recombinations lead to genomes of new species.” Another put it more colloquially: “Never heard of synteny blocks before! Did not realize that such large blocks/chunks of chromosomes could rearrange in that way.” Seven participants mentioned inversion events, and interestingly, four people stated that they learned about the existence of sex chromosomes. Comments that only mentioned common ancestors with no reference to DNA, genomes, or synteny are classified under “Phylogeny,” which was three people. At the end of their session, users were asked to pick their favorite genomic mapping (Figure 11b). The two karyogram-layout mappings are the most popular by far (Animation=26 and Static=20), compared to Circos (12) and Apollo (2). Stated reasons for preference varied among users. One user noted, “I like the circle view the best for my- self, but I think that the animated view would probably be better for teaching.” Many users commented that Animated was their favorite because it “was the easiest to understand,” “more visually intuitive,” and was “fun,” or “cool.” Similarly for Static, “the arrangement is intuitively satisfying: the way you expect to see,” and it is “probably the simplest for communicating that one chromosome breaks up and goes to different chromosomes.” Some users also recommended a hybrid view: animating synteny blocks with static trails behind them.
Fig. 10.
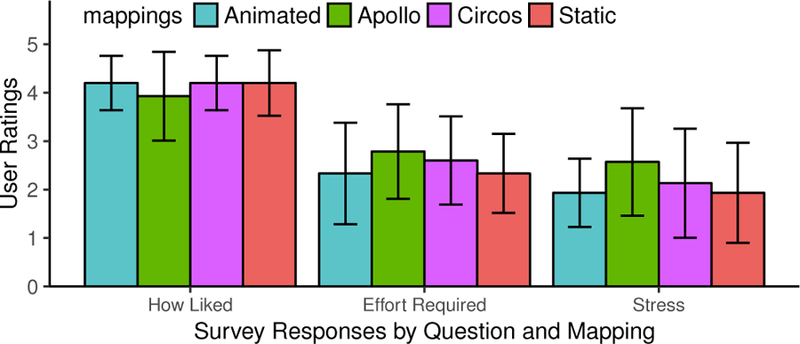
User ratings for three questions regarding their experience using the genome mappings in Study 3.
Fig. 11.
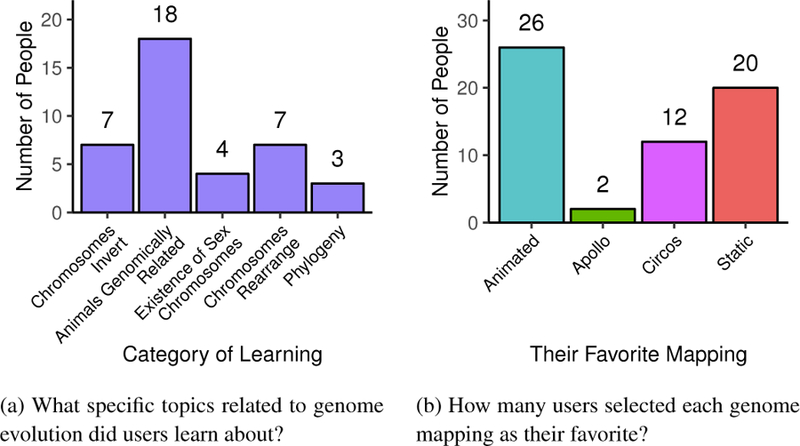
Answers for two survey questions for Study 3, assessing user learning outcomes and their preferred view
5.3. Evaluating our Hypotheses
Study results indicate that both H1 and H2 were confirmed.
For H1, on average users scored 10/12 quiz questions correctly, and 53/60 users stated they learned using our tool (the others had extensive biological backgrounds). Figure 11a shows different aspects of genome evolution that participants learned about. There is a total of 210 positive feedback comments to 162 negative ones, and Figure 10 shows all four genome mappings are rated highly.
For H2, results show the karyogram-based layouts perform equivalently on quiz questions, and are widely preferred over the conventional views. As a whole, Static and Animated have a much higher number of positive comments, a better ratio of positive-to-negative comments, are chosen by users as favorite views more often, and are seen as requiring less effort and causing less stress. Interestingly though, our results show users can use the Circos and Apollo mappings effectively for quiz tasks, suggesting these techniques are still viable for teaching genome evolution, at least to a college-aged population.
6. DISCUSSION
Ancestral genome reconstruction is at the frontier of genomics re- search. Interactively visualizing chromosome evolution from a com- mon ancestor to multiple descendant species allows for optical recognition and tracking of synteny blocks over evolutionary time spans. This opens new avenues for the teaching fundamental principles of comparative genomics and evolution. Our tool provides an initial step in creating educational visualization software and establishing design principles for teaching about synteny-level evolution. From the results of our design process and studies (and additional feedback received from faculty discussions, see below), we feel we are progressing in the right direction. To synthesize some of the lessons learned in this process, we give the following design recommendations:
DG1 Use karyogram mappings for user likability. Compared to the more spatially complex Apollo and Circos views, users highly prefer Animated and Static. They rate them as less stressful and easier to use. Quantitatively, these views do not negatively affect response time, ability to learn, or basic analytic ability.
DG2 Circos and Apollo still effective for teaching. Despite the increased complexity, negative user comments, and effort re quired, Circos and Apollo can be equally effective for teaching genome evolution to college-aged students, measured in terms of completing analytic tasks and gauging whether or not users learned. Circos, the domain standard, was actually the fastest in average task time in Study 3, albeit barely.
DG3 Use color to assist perception of synteny mappings. Of our tested palettes, the qualitative, Faded color scheme works best. Palettes that vary along a scale (Rainbow and HSV) have issues for certain chromosome color combinations. Grayscale should not be considered when interaction is not allowed.
DG4 Morph independent of sliding for animating full-genome evolution. As opposed to doing both simultaneously or in a cascading manner, independent morphing and sliding provides the best comprehension.
6.1. Feedback from Academic Faculty
This paper focuses mainly on the initial development and evaluation of Synteny Explorer. In-classroom deployment is the natural next step. To help assess our tool’s potential utility for this task, we solicited feedback of three local faculty members from our campus biology and genetics departments. We let each play with the application for as long as they liked, and then asked if they thought it would have a place in their classroom curriculum (or another course, if applicable) and how they might integrate it into their current teaching procedure(s).
Overall feedback was positive, though one instructor was not currently teaching genomics-related material. However, he liked the over- all application design and flow, namely selecting animals from a tree and then diving into a more detailed view: “Let me show a text description of each animal’s physical features instead of its genome and it’d be great for my class!” The other two professors had different strategies for how to integrate Synteny Explorer into their curriculum. The first especially liked how the Animated mapping illustrated concepts. “I would use this as a discussion point for evolutionary mechanisms in [my evolutionary ecology class], for example, why did this inversion or change happen between chimp and human?” He said he would use the tool to demo genome evolutions to the class during discussion. He further suggested this tool be could be used in the department’s Introduction to Phylogenies course.
The second faculty member, who teaches karyotype evolution in his genetics courses, saw a more hands-on deployment: “Students can use it independently as a learning tool through which they can investigate evolutionary events. Teachers can use it as a demonstration tool during lecture to explain karyotypic changes. It illustrates very well how breakages, fusions, inversions, and transposition diversify the karyotypes of two organisms with a common ancestor. I would take advantage of its interactive design, to write quizzes and tests based on use of the tool.” In contrast to the prior professor, he felt that switching be- tween the four genome mappings was useful in showing students how the karyogram-based view of textbooks is transformed to the views more commonly found in research applications: “The different visualization modes, such as color schemes, dynamic versus static changes, linear versus circular, are also important pedagogical features. For example, the use of circular graphing versus linear graphing in genomics can be exemplified and taught.”
Based on feedback from these sessions, we are working towards deploying Synteny Explorer in a classroom setting for a future study.
6.2. Current Target Audience Considerations
Synteny Explorer’s initial target audience was defined as undergraduate biology students. After going through the design process and analyzing study results, we believe an additional two groups may currently benefit from this tool and its associated designs.
First, we feel graduate students that attend courses in genome evolution can benefit from this interactive approach. Visualization allows for deeper dives into the evolutionary history of individual chromosomes, a dynamic view of the rates of chromosome rearrangements in different mammalian lineages, and an intuitive exploration of the different organizations of extant mammalian genomes and reconstructed genomes of ancestral species. This can help them gain a deeper sense of genome reconstruction and change between species.
Second, based on discussion with our genomics collaborators, we feel our visual interactions and karyogram designs can be successfully used by faculty and genomic researchers to help identify important evolutionary changes in chromosomes of different species. As sequencing and assembly of additional genomes become available, new data can be added because the tool is scalable.
6.3. Future Work and Expanding To New Domains
There are many elements regarding this work that can be further investigated and addressed, either in future versions of Synteny Explorer or in work done by others. For example, we have only implemented a subset of layouts from the Mizbee taxonomy [35] that we felt would be most appropriate for education, but other layout options exist. There are also HCI-specific optimizations that can be made, such as a better tool introduction method than our set of explanatory slides.
Our study results, analysis, and discussion should be noted with the following considerations: The foci of our three studies were students in college, where chromosome evolution is usually taught, but it has potential to bias our work to well-educated, analytically-minded users. The studies themselves were performed under controlled settings (one user and a proctor, a large monitor, etc.) and our analysis reflects that. For Study 3, the average session time was over 25 minutes.
For deployment in a different domain, such as a public museum exhibit or via a website, there are many new factors to consider. The age of users will potentially be much younger and their education levels certainly more varied. In a busy setting, many people may try to interact with the display simultaneously. In this context, a teaching tool must ensure a succinct, collaborative, and engaging user experience that is still interesting, interactive, and educational [24]. The quiz interface might not be a very effective feature in such a setting. Thus, components and interactions will probably have to be stream-lined and/or redesigned depending on the intended space and engagement patterns of users.
7. CONCLUSIONS
While the community has come up with ways to visualize genome evolution for research, in terms of student education it has thus far not received adequate attention. In a collaborative effort with genomics researchers, we have developed Synteny Explorer for this purpose.
The development of this interactive visualization tool provides an opportunity to transform and enhance scientific research and education for genome evolution. Visually following one or many synteny blocks containing tens of genes over hundreds of millions of years between different species illustrates several very important principles of evolution. Based on our results, what we have created in this project can also serve as a guide for future, design-based work. This supports inquiry and analysis in both visualization-based applications and exhibits in informal learning environments such as museums.
ACKNOWLEDGMENTS
The authors wish to thank Francesca Samsel of the Center for Ag ile Technology, University of Texas at Austin, for assistance with color palette choices. This research was sponsored in part by the UC Davis RISE program, US National Science Foundation via grants DRL-1323214, IIS-1528203, and IIS-1320229, and U.S. Department of Energy via grant DE-FC02–12ER26072.
REFERENCES
- [1].Ucsc genome browser http://genome.ucsc.edu/ Accessed September 4, 2015.
- [2].Circos circular genome data visualization http://circos.ca/, 2015. Accessed February 15, 2016.
- [3].Evolution highway http://eh-demo.ncsa.uiuc.edu/, 2015.
- [4].Albers D, Dewey C, and Gleicher M Sequence surveyor: Leveraging overview for scalable genomic alignment visualization. Visualization and Computer Graphics, IEEE Transactions on, 17(12):2392–2401, 2011. [DOI] [PubMed] [Google Scholar]
- [5].Archibald J Edward hitchcocks pre-darwinian (1840)tree of life. Jour- nal of the History of Biology, 42(3):561–592, 2009. [DOI] [PubMed] [Google Scholar]
- [6].Baum DA and Offner S Phylogenics & tree-thinking. The American Biology Teacher, 70(4):222–229, 2008. [Google Scholar]
- [7].Block F, Horn M, Phillips B, et al. The deeptree exhibit: Visualizing the tree of life to facilitate informal learning. Visualization and Computer Graphics, IEEE Transactions on, 18(12):2789–2798, 2012. [DOI] [PubMed] [Google Scholar]
- [8].Borland D and Taylor R II Rainbow color map (still) considered harm- ful. IEEE Computer Graphics and Applications, (2):14–17, 2007. [DOI] [PubMed] [Google Scholar]
- [9].Catley K and Novick L Seeing the wood for the trees: an analysis of evolutionary diagrams in biology textbooks. BioScience, 58(10):976–987, 2008. [Google Scholar]
- [10].Catley K and Novick L Digging deep: Exploring college students’ knowledge of macroevolutionary time. Journal of Research in Science Teaching, 46(3):311–332, 2009. [Google Scholar]
- [11].Catley K, Phillips B, and Novick L Snakes and eels and dogs! oh, my! evaluating high school students tree-thinking skills: an entry point to understanding evolution. Research in Science Education, 43(6):2327–2348, 2013. [Google Scholar]
- [12].Chan M and Black J When can animation improve learning? some im- plications on human computer interaction and learning. In World Conference on Educational Multimedia, Hypermedia and Telecommunications, volume 2005, pages 933–938, 2005. [Google Scholar]
- [13].Chua K, Qin Y, Block F, et al. Flotree: A multi-touch interactive simula- tion of evolutionary processes. In Proceedings of the 2012 ACM International Conference on Interactive Tabletops and Surfaces, pages 299–302. ACM, 2012. [Google Scholar]
- [14].Clamp M, Andrews D, Barker D, et al. Ensembl 2002: accommodating comparative genomics. Nucleic Acids Research, 31(1):38–42, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].I. H. G. S. Consortium. Finishing the euchromatic sequence of the human genome. Nature, 431(7011):931–945, 2004. [DOI] [PubMed] [Google Scholar]
- [16].Consortium S et al. Genome sequence of the nematode C. elegans: A platform for investigating biology. Science, 282:2012–2018, 1998. [DOI] [PubMed] [Google Scholar]
- [17].Crabtree J, Angiuoli S, Wortman J, and White O Sybil: methods and software for multiple genome comparison and visualization. In Gene Function Analysis, pages 93–108. Springer, 2007. [DOI] [PubMed] [Google Scholar]
- [18].Darling A, Mau B, Blattner F, and Perna N Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Research, 14(7):1394–1403, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Doolittle W Uprooting the tree of life. Scientific American, 282(2):90, 2000. [DOI] [PubMed] [Google Scholar]
- [20].Ferstay JA, Nielsen CB, and Munzner T Variant view: Visualiz- ing sequence variants in their gene context. Visualization and Computer Graphics, IEEE Transactions on, 19(12):2546–2555, 2013. [DOI] [PubMed] [Google Scholar]
- [21].Fleischmann R, Adams M, White O, et al. Whole-genome ran- dom sequencing and assembly of haemophilus influenzae rd. Science, 269(5223):496–512, 1995. [DOI] [PubMed] [Google Scholar]
- [22].Heer J and Robertson G Animated transitions in statistical data graphics. Visualization and Computer Graphics, IEEE Transactions on, 13(6):1240–1247, 2007. [DOI] [PubMed] [Google Scholar]
- [23].Horn M, Atrash Leong Z, Block F, Diamond J, Evans EM, Phillips B, and Shen C Of bats and apes: an interactive tabletop game for natural history museums. In Proceedings of the SIGCHI Conference on Human Factors in Computing Systems, pages 2059–2068. ACM, 2012. [Google Scholar]
- [24].Humphrey T, Gutwill J, et al. Fostering active prolonged engagement San Francisco, CA: The Exploratorium, 2005. [Google Scholar]
- [25].Hunt E, Hanlon N, Leader D, et al. The visual language of synteny. Omics: A Journal of Integrative Biology, 8(4):289–305, 2004. [DOI] [PubMed] [Google Scholar]
- [26].Kim J, Larkin D, Cai Q, et al. Reference-assisted chromosome as- sembly. Proceedings of the National Academy of Sciences, 110(5):1785–1790, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Koepfli K-P, Paten B, and OBrien S The genome 10k project: A way forward. Annu. Rev. Anim. Biosci, 3(1):57–111, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Research, 19(9):1639–1645, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Larkin D, Pape G, Donthu R, et al. Breakpoint regions and homolo- gous synteny blocks in chromosomes have different evolutionary histo- ries. Genome Research, 19(5):770–777, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lewis S, Searle S, Harris N, et al. Apollo: a sequence annotation editor. Genome Biol, 3(12):1–14, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ma J, Zhang L, Suh B, et al. Reconstructing contiguous regions of an ancestral genome. Genome Research, 16(12):1557–1565, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Maddison W and Maddison D Mesquite: a modular system for evolu- tionary analysis 2001.
- [33].Meir E, Perry J, Herron J, and Kingsolver J College students’ mis- conceptions about evolutionary trees. The American Biology Teacher, 69(7):e71–e76, 2007. [Google Scholar]
- [34].Meyer M, Munzner T, DePace A, and Pfister H Multeesum: a tool for comparative spatial and temporal gene expression data. Visualization and Computer Graphics, IEEE Transactions on, 16(6):908–917, 2010. [DOI] [PubMed] [Google Scholar]
- [35].Meyer M, Munzner T, and Pfister H Mizbee: a multiscale synteny browser. Visualization and Computer Graphics, IEEE Transactions on, 15(6):897–904, 2009. [DOI] [PubMed] [Google Scholar]
- [36].Muffato M, Louis A, Poisnel C-E, and Crollius H Genomicus: a database and a browser to study gene synteny in modern and ancestral genomes. Bioinformatics, 26(8):1119–1121, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Munzner T Visualization Analysis and Design CRC Press, 2014. [Google Scholar]
- [38].Murphy W, Larkin D, Everts-van der Wind A, et al. Dynamics of mammalian chromosome evolution inferred from multispecies comparative maps. Science, 309(5734):613–617, 2005. [DOI] [PubMed] [Google Scholar]
- [39].Nusir S, Alsmadi I, Al-Kabi M, and Shardqah F Designing an inter- active multimedia learning system for the children of primary schools in jordan. In Global Engineering Education Conference (EDUCON), 2011 IEEE, pages 45–51. IEEE, 2011. [Google Scholar]
- [40].O’Brien TM, Ritz AM, Raphael BJ, and Laidlaw DH Gremlin: an interactive visualization model for analyzing genomic rearrangements. Visualization and Computer Graphics, IEEE Transactions on, 16(6):918–926, 2010. [DOI] [PubMed] [Google Scholar]
- [41].Pan X, Stein L, and Brendel V Synbrowse: a synteny browser for com- parative sequence analysis. Bioinformatics, 21(17):3461–3468, 2005. [DOI] [PubMed] [Google Scholar]
- [42].Patterson N, Richter D, Gnerre S, et al. Genetic evidence for complex speciation of humans and chimpanzees. Nature, 441(7097):1103–1108, 2006. [DOI] [PubMed] [Google Scholar]
- [43].Prince M Does active learning work? a review of the research. Journal of Engineering Education, 93:223–232, 2004. [Google Scholar]
- [44].Samsel F, Petersen M, Geld T, et al. Colormaps that improve perception of high-resolution ocean data. In Proceedings of the 33rd Annual ACM Conference Extended Abstracts on Human Factors in Computing Systems, pages 703–710. ACM, 2015. [Google Scholar]
- [45].Schneider B, Strait M, Muller L, et al. Phylo-genie: engaging students in collaborative’tree-thinking’through tabletop techniques. In Proceed- ings of the SIGCHI Conference on Human Factors in Computing Systems, pages 3071–3080. ACM, 2012. [Google Scholar]
- [46].Schröck E, Du Manoir S, Veldman T, et al. Multicolor spectral kary- otyping of human chromosomes. Science, 273(5274):494–497, 1996. [DOI] [PubMed] [Google Scholar]
- [47].Schweitzer D and Brown W Interactive visualization for the active learning classroom. In ACM SIGCSE Bulletin, volume 39, pages 208–212. ACM, 2007. [Google Scholar]
- [48].Shaer O, Kol G, Strait M, et al. G-nome surfer: a tabletop interface for collaborative exploration of genomic data. In Proceedings of the SIGCHI Conference on Human Factors in Computing Systems, pages 1427–1436. ACM, 2010. [Google Scholar]
- [49].Shneiderman B The eyes have it: a task by data type taxonomy for in- formation visualizations. In Visual Languages, 1996. Proceedings., IEEE Symposium on, pages 336–343, September 1996. [Google Scholar]
- [50].Sinha AU and Meller J Cinteny: flexible analysis and visualization of synteny and genome rearrangements in multiple organisms. BMC bioin- formatics, 8(1):82, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].The Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature, 437(7055):69–87, 2005. [DOI] [PubMed] [Google Scholar]
- [52].Tversky B, Morrison J, and Betrancourt M Animation: can it facili- tate? International Journal of Human-Computer Studies, 57(4):247–262, 2002. [Google Scholar]
- [53].Ware C Information visualization: perception for design Elsevier, 2012. [Google Scholar]