

Abstract
Defects in the architecture or integrity of the nuclear envelope (NE) are associated with a variety of human diseases1. Micronuclei, one common nuclear aberration, are an origin for chromothripsis2, a catastrophic mutational process commonly observed in cancer3–5. Chromothripsis occurs after micronuclei spontaneously lose NE integrity, which generates chromosome fragmentation6. NE disruption exposes DNA to the cytoplasm and initiates innate immune proinflammatory signaling7. Despite its importance, the basis for the NE fragility of micronuclei has not been determined. Here, we demonstrate that micronuclei undergo defective NE assembly: Only “core” NE proteins8,9 assemble efficiently on lagging chromosomes whereas “non-core” NE proteins8,9, including nuclear pore complexes (NPCs), do not. Consequently, micronuclei fail to properly import key proteins necessary for NE and genome integrity. We show that spindle microtubules block NPC/non-core NE assembly on lagging chromosomes, causing an irreversible NE assembly defect. Accordingly, experimental manipulations that position missegregated chromosomes away from the spindle correct defective NE assembly, prevent spontaneous NE disruption, and suppress DNA damage in micronuclei. Thus, during mitotic exit in metazoan cells, chromosome segregation and NE assembly are only loosely coordinated by the timing of mitotic spindle disassembly. The absence of precise checkpoint controls may explain why errors during mitotic exit are frequent and often trigger catastrophic genome rearrangements4,5.
During telophase, the assembling NE transiently forms two domains around decondensing chromosomes1,8,9. “Core” NE proteins (e.g., the membrane protein emerin) assemble all around chromosomes and then concentrate in regions adjacent to the spindle. Concomitantly, nuclear pore complex proteins (NPCs) and other “non-core” proteins, (e.g., Lamin B receptor, LBR), assemble on the chromosome periphery away from the spindle, transiently excluded from the core domain (Extended Data Fig. 1a–c)8,9. After mitotic exit, these domains are intermingled and additional NPCs slowly assemble via an interphase assembly pathway10.
To understand the NE defect of micronuclei, we asked whether NE assembly on lagging chromosomes occurs in this same spatiotemporal pattern. In three cell lines, lagging chromosomes were generated in synchronized cells by two independent methods (see Methods). Whereas core proteins were recruited to lagging chromosomes at equivalent or higher levels than to the main chromosome mass, NPC and other non-core proteins near-completely failed to assemble (Fig. 1a, Extended Data Fig. 1b–f). Chromatin bridges extending between daughter cells displayed a similarly defective NE protein composition (Extended Data Fig. 1g, h).Thus, consistent with a recent study11, only a subset of NE proteins assemble normally on chromatin that remains within the telophase central spindle.
Figure 1. Defective NE assembly on lagging chromosomes.
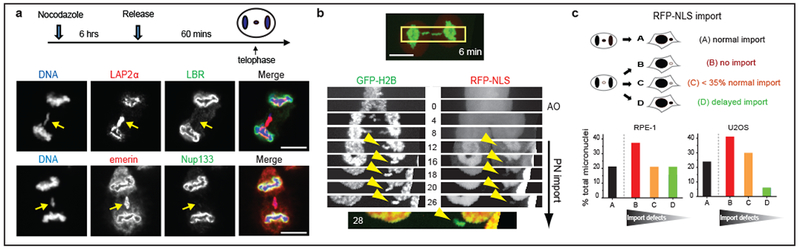
a. Non-core NE assembly defect on lagging chromosomes. Top: Experimental scheme. Bottom: Images of RPE-1 cells with lagging chromosomes (arrows, 3 experiments). Red: core NE proteins; Green: non-core proteins. b, c, Impaired nuclear import in micronuclei. b, Kymograph of RPE-1 cell (boxed region of top image) shows impaired import of RFP fused to a nuclear localization signal (RFP-NLS) in the newly formed micronuclei (arrowheads). Synchronization as in a (t=0 is anaphase onset, AO). Bottom: Merged image. Representative images of 9 cells, category “B” next panel. c, Top: Cartoon summarizing patterns of import to micronuclei (from b, see Extended Data Fig. 3a for representative quantification of import defects). Bottom: Percentage of micronuclei corresponding to the categories above (n=24, from 11 experiments for RPE-1, n=17, from 9 experiments for U2OS). Scale bars, 10 μm.
Micronuclei from lagging chromosomes also had reduced NPC/non-core protein assembly (Extended Data Fig. 2a–g). Accordingly, with some variability, micronuclei had significant nuclear import defects (Fig. 1b, c, Extended Data Fig. 3a, b, Supplementary Videos 1 and 2) and were defective in accumulating nuclear proteins such as replication protein A (RPA), a key dosage-sensitive regulator of DNA replication and repair12, and B-type lamins, known to be required to maintain NE integrity (Extended Data Figs. 2a–g, 3c–e)6,13. For individual cells, the extent of RPA and an importin beta-dependent reporter were correlated (Extended Data Fig. 3e), suggesting a general import defect at least for importin beta clients. Thus, reconciling differing prior reports6,11,14–16, micronuclei undergo aberrant NE and NPC assembly, leading to defective import and nuclear protein accumulation.
We considered the possibility that the NPC/non-core assembly defect on lagging chromosomes was due to a previously proposed “chromosome separation checkpoint”15. Under this model, the spindle midzone-centered Aurora B phosphorylation gradient17 provides a spatial cue that blocks NE assembly until membrane-free lagging chromosomes can be incorporated within the main chromosome mass to form a single nucleus. However, several observations were inconsistent with this model. First, core membrane proteins, which were not previously examined15, assemble on lagging chromosomes and chromosome bridges independent of their position relative to the spindle midzone (Extended Data Fig. 4a), indicating that an NE does form on lagging chromosomes, albeit one depleted for NPCs. Second, chromosome bridges are uniformly depleted for non-core proteins, displaying no obvious gradient (Extended Data Figs. 1g, h). Third, in late telophase, lagging chromosomes lose Aurora B-mediated histone 3 serine 10 phosphorylation (pH3S10) but do not assemble NPCs or other non-core proteins (Extended Data Fig. 4b, c). Finally, in cells where the midzone Aurora B gradient was lost after siRNA-mediated knockdown of its transport motor MKLP217, non-core NE assembly was still inhibited on lagging chromosomes (Extended Data Fig. 4d).
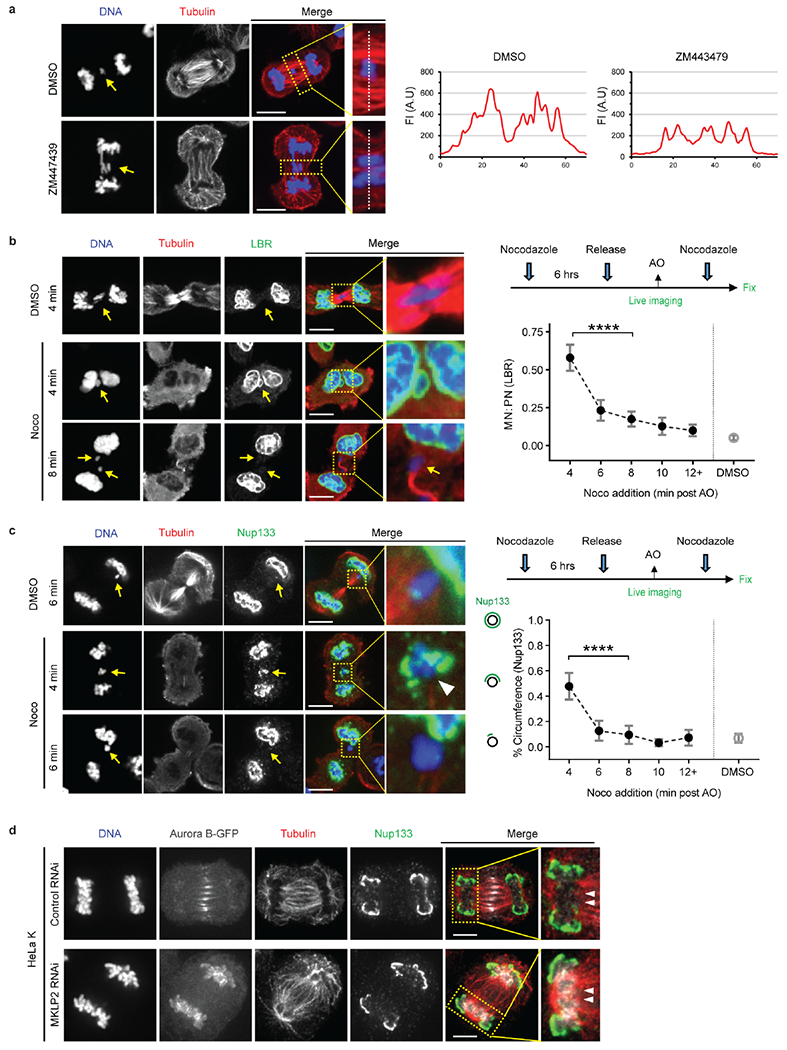
The checkpoint model requires that lagging chromosome position be monitored continuously throughout mitotic exit by Aurora B. To test this, cells were released from a mitotic arrest, imaged with high temporal resolution, treated with an Aurora B inhibitor (ZM447439 for 12 min) around anaphase/telophase, and then fixed and labeled to assess NE assembly (Extended Data Fig. 4e). Because cells release from the mitotic block with only partial synchrony, we could identify cells where Aurora B was inhibited at each stage between metaphase and telophase. Surprisingly, Aurora B inhibition restored non-core protein assembly to lagging chromosomes only if it occurred early, at or shortly after anaphase onset, but not later, ~6–8 min after anaphase onset (Fig. 2a). The irreversibility of late Aurora B inhibition cannot be explained by a partial effect of drug inhibition: Near-complete loss of Aurora B activation was observed within 3 min post inhibitor treatment and indistinguishable results were obtained using a 10-fold higher dose of the inhibitor (Extended Data Fig. 4f, g). Thus, the global activation of Aurora B can inhibit non-core NE assembly up until early anaphase, but it has no effect later, when a chromosome separation checkpoint would need to operate. Instead, lagging chromosomes become irreversibly defective for non-core NE recruitment around mid-anaphase.
Figure 2. Irreversibility of the NE assembly defect on lagging chromosomes.
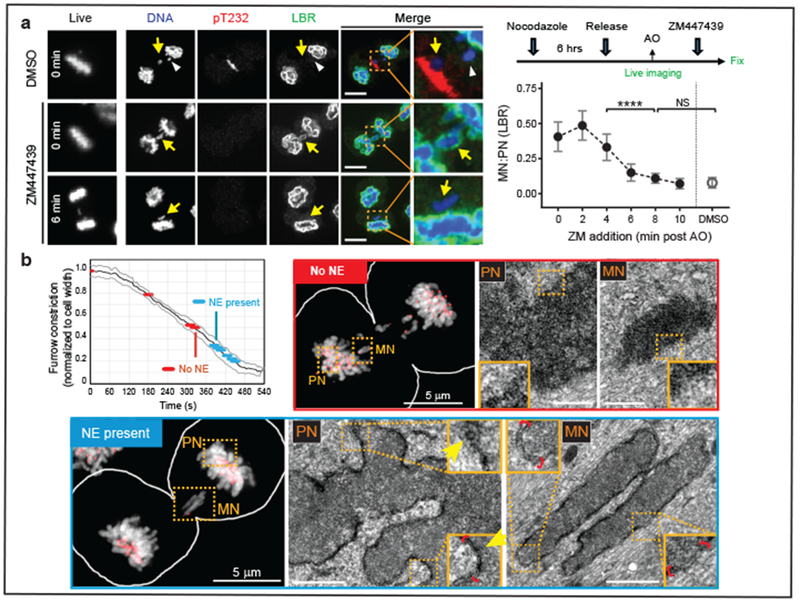
a, Aurora B inhibition after late-anaphase fails to restore non-core (LBR) assembly to lagging chromosomes (arrows). Left column: Cells at the time of ZM447439 or DMSO addition (min post AO). Right columns: Cells labeled for DNA (blue), phospho-T232 Aurora B (red) and LBR (green). In controls, lagging chromosome fail to recruit LBR even if they are located (white arrowhead) far from active Aurora B (pT232). Scale bars, 10 μm. Right, top: Experimental scheme. Right, bottom: MN/PN (primary nucleus) FI ratio for LBR in cells exposed to ZM at the indicated times (mean with 95% CI, n=40, 29, 31, 31, 43, 43, 34 each timepoint, from 5 experiments. For brevity, lagging chromosomes are designated as “MN”). **** P < 0.0001, NS: P=0.0816, two-tailed Mann-Whitney test. b, CLEM showing synchronous NE formation on all chromosomes. Top left: Time course of furrow ingression in RPE-1 cells with lagging chromosomes (black line: mean, grey lines: ±1 SD). Red and blue discs indicate stages of the 10 cells analyzed by CLEM: No NE is detected by CLEM during earlier stages of cytokinetic furrowing (red); at later stages (blue), double-membranous NE is present at both PN and MN. Perinuclear space (red brackets) is wider around MN. NPCs (arrows) are present only on PN. LM images: DNA in greyscale, kinetochores (CenpA-GFP) and centrosomes (centrin1-GFP) in red. Scale bars, 0.5 μm.
Previous studies suggested that the loss of chromosome condensation, marked by the loss of pH3S10 or condensins, might be required for NPC/NE assembly15,18,19. Accordingly, persistent mitotic condensin proteins were suggested to block NPC/NE assembly on lagging chromosomes15. To further examine this, we used high temporal resolution imaging to relate the timing of NPC/non-core assembly to condensin levels. Unexpectedly, NPC/non-core protein assembly was readily observed with high levels of the condensin subunit SMC2 (~70% maximum levels, Extended Data Fig. 5a, b). Similar results were obtained for pH3S10, although there were antibody-specific differences for detecting the loss of pH3S10 (Extended Data Fig. 4c). Additionally, ~5-fold RNAi-mediated knockdown of SMC2 failed to restore NPC/non-core NE assembly on lagging chromosomes (Extended Data Fig. 5a, c). Moreover, correlated light and electron microscopy (CLEM) experiments indicated that all chromosomes, including lagging chromosomes, appear comparably condensed when the NE assembles (Fig. 2b, bottom panels). Therefore, consistent with data from early cell fusion studies20, extensive chromosome decondensation is not required for NPC assembly, and persistent condensin-binding cannot explain the irreversible NE assembly defect on lagging chromosomes.
Considering prior work10,21,22, we hypothesized that the irreversible defect in NE assembly on lagging chromosomes might be caused by their enclosure in an NE lacking NPCs. For example, Xenopus egg extracts lacking the Nup107–160 complex can only assemble NPCs if the purified complex is added back early in the reaction, because the formation of a continuous NPC-free NE irreversibly blocks NPC assembly21. Consistent with this idea, ESCRT-III components, which promote the closure of the newly formed NE in telophase1,23, associated and dissociated from lagging chromosome with normal kinetics (Extended Data Fig. 5d, e, Supplementary Video 3). Structured illumination microscopy (SIM) revealed that lagging chromosomes were indeed enclosed by the membrane protein emerin, but not NPCs (Extended Data Fig. 5f). CLEM experiments demonstrated that double membranes form synchronously around all chromosomes, including lagging chromosomes, although membranes around lagging chromosomes lack NPCs (Fig. 2b). CLEM verified that this paucity of NPCs persisted on interphase micronuclei (Extended Data Fig. 5g), consistent with an irreversible NE assembly defect.
Although our experiments argue against spatially-patterned checkpoint regulation of NE assembly (above and Extended Data Figs. 4, 5), the effects of global Aurora B inhibition on NE assembly in early anaphase still needed to be explained (Fig. 2a, Extended Data Fig. 4g). We considered the possibility that partial restoration of NE assembly might occur as an indirect consequence of Aurora B-dependent spindle microtubule stabilization during early anaphase (Extended Data Fig. 6a)24. Indeed, in Xenopus egg extracts, microtubule stabilization near chromosomes irreversibly inhibits the formation of an NE containing NPCs22. Accordingly, we found that nocodazole and Aurora B inhibition had a remarkably similar effect on non-core NE assembly: Early anaphase microtubule disassembly by nocodazole reversed the non-core assembly defect on lagging chromosomes whereas there was minimal effect if nocodazole was added later (Extended Data Fig. 6b, c). Additionally, during normal NE assembly, non-core NE is typically excluded from the core domain adjacent to dense microtubule bundles from the central spindle (Extended Data Fig. 1b, c)8,25. However, this exclusion occurs irrespective of whether Aurora B localizes to the spindle midzone or is forced to remain on the main chromosome mass (MKLP2 knockdown, Extended Data Fig. 6d).
Because of the interdependent functions of Aurora B and microtubules24, we used paclitaxel to prevent microtubule disassembly after Aurora B inhibition (Fig. 3a) and determined whether microtubules can still exclude the non-core NE from central spindle region (Extended Data Figs. 1b, 1c, 6d). In control late-telophase cells, core and non-core proteins become intermingled on the main chromosome mass after mitotic exit as expected. By contrast, in paclitaxel-treated cells, the gap between non-core NE domains persisted and/or the core domain became exaggerated (Fig. 3a, arrowheads). Importantly, co-addition of an Aurora B inhibitor with paclitaxel had the identical effect as paclitaxel treatment alone (Fig. 3a). Thus, microtubule inhibition of non-core NE assembly is independent of Aurora B.
Figure 3. Spindle microtubules block non-core NE assembly independent of Aurora B.
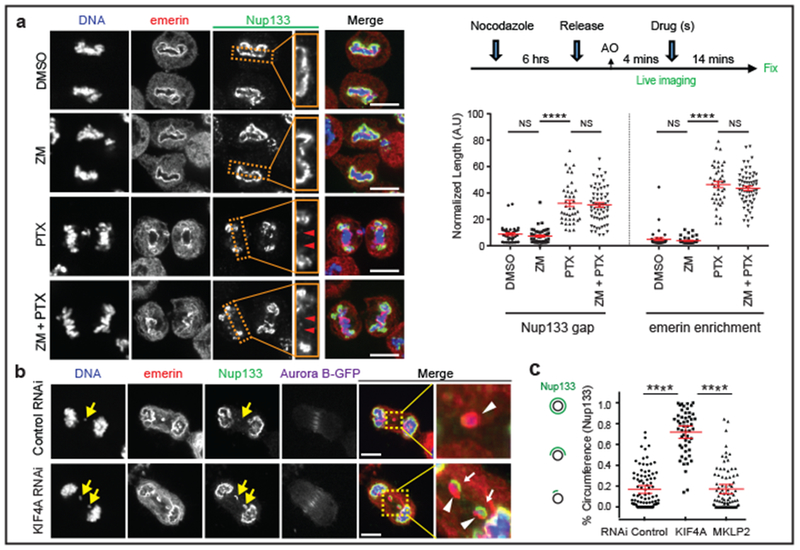
a, Microtubule stabilization by paclitaxel (PTX) inhibits non-core invasion of core domain. Left: Images of RPE-1 cells after drug treatment(s): Enlarged images from the boxed regions show the non-core (Nup133) gap on the main chromosome mass (red arrowheads). Right, top: Experimental scheme. Right, bottom: Quantification of the results (mean with 95% CI, n=40, 47, 41, 61, 40, 47, 41, 61, left to right, from 2 experiments). **** P < 0.0001, NS: P=0.2974, 0.9837, 0.2473, 0.3374 (left to right), two-tailed Mann-Whitney test. b, c, Small-scale core/non-core domain separation on lagging chromosomes after KIF4A depletion. b, Images of Aurora B-GFP-expressing HeLa K cells (2 technical replicates). Merged and enlarged image: Emerin (red, white arrowheads), Nup133 (green, white arrows). Synchronization as in Extended Data Fig. 1e. c, The fraction of lagging chromosome circumference covered with Nup133 in HeLa K cells (cartoon shown in left; mean with 95% CI, n=90, 52, 81, from 2 experiments). **** P < 0.0001, two-tailed Mann-Whitney test. Scale bars, 10 μm.
The above data suggest a model where non-core NE is inhibited in the areas with high density of microtubules. To test this hypothesis, we “loosened” spindle microtubule bundling by siRNA-mediated knockdown of the kinesin KIF4A26. As expected, KIF4A knockdown preserved general spindle organization and detectable central spindle localization of Aurora B (Extended Data Fig. 7a)26. However, KIF4A depletion partially reversed the NPC/non-core NE assembly defect on lagging chromosomes (Fig. 3b, c, Extended Data Fig. 7b–d). Interestingly, the NE at these sites often displayed a small-scale separation of core (emerin, white arrowheads) and non-core proteins (Nup133, white arrows in Fig. 3b), as occurs on the main chromosome masses in telophase. These “mini” core/non-core subdomains formed at any location within the central spindle, including the spindle midzone where Aurora B activity is high. Moreover, these partially-corrected lagging chromosomes exhibited pH3S10 at levels comparable to those of metaphase chromosomes (Extended Data Fig. 7e), confirming that NPC assembly is compatible with condensed chromatin. In further agreement, SIM after KIF4A knockdown suggested that the “core-only” region of the lagging chromosome was enriched for NE-microtubule contacts marked by ESCRT-III component CHM4B23 (Extended Data Fig. 8a–c). Interestingly, Aurora B inhibition in early anaphase has a very similar effect as KIF4A knockdown (Extended Data Fig. 8d), with mini core/non-core subdomains often forming around lagging chromosomes. Together, these data suggest that the local organization of microtubules is the key determinant of whether NPC/non-core NE can assemble.
Our microtubule-inhibition model predicts that positioning missegregated chromosomes away from the central spindle should normalize NE assembly and restore function to micronuclei. To generate peripheral missegregated chromosomes, cells were treated with an MPS1 kinase inhibitor, causing missegregation before chromosome congression was complete. In HeLa cells, MPS1 inhibition was combined with the knockdown of tubulin tyrosine ligase (TTL), which further enhances peripheral chromosome localization (Fig. 4a, Extended Data Fig. 9a)27. Strikingly, peripheral chromosomes recruited both core and non-core NE proteins, including NPCs, unlike central lagging chromosomes (Fig. 4a–c, Extended Data Fig. 9a, Supplementary Video 4). Microtubule and ESCRT-labeling23 indicated that peripheral chromosomes also had less contact with microtubules (Extended Data Fig. 9b). Micronuclei derived from peripheral chromosomes had normal levels of nuclear import and restored levels of lamin B1 and other nuclear proteins required for DNA replication and repair (Fig. 4d, Extended Data Fig. 9c, d). Moreover, micronuclei derived from peripheral chromosomes exhibited a normal extent of DNA replication (Extended Data Fig. 9e). The restoration of a functional NE on peripheral micronuclei, which occurred independent of their chromosome number, likely contributes to their larger size (Extended Data Fig. 9c–h). Finally, in HeLa cells, micronuclei from peripheral chromosomes had a ~5-fold lower rate of NE disruption and significantly less DNA damage (Fig. 4e, Extended Data Fig. 10a, b). In RPE-1 cells, peripheral chromosome localization also significantly reduced NE disruption and DNA damage (Extended Data Fig. 10c, Supplementary Video 5). However, in RPE-1 cells, the effect of peripheral chromosomes on NE disruption was partly masked because the large micronuclei from peripheral chromosomes in these cells are subject to actin-dependent NE breakage, as can occur transiently on primary nuclei13 (see discussion in Extended Data Fig. 10c, d). Thus, positioning chromosomes away from the spindle significantly restores NE assembly and function of the resulting micronuclei.
Figure 4. Peripheral localization of missegregated chromosomes corrects defects of micronuclei.
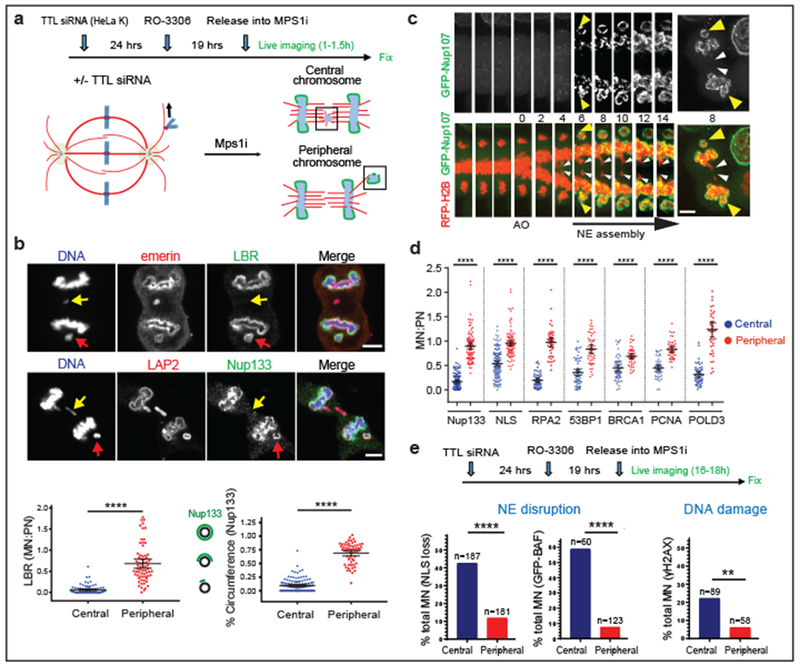
a, Experimental scheme to generate missegregated chromosomes positioned within or away from the spindle (microtubules: red; non-core NE: green). b, c, Non-core NE assembles on peripheral chromosomes but not central chromosomes. b, Top: Images of RPE-1 cells with central (yellow arrows) and peripheral (red arrows) chromosomes labeled with core (red) or non-core (green) proteins. Bottom: Quantification as in Fig. 3c [mean with 95% CI, n=83, 67 (left), n=122, 58 (right), from 2 experiments each]. **** P < 0.0001, two-tailed Mann-Whitney test. c, Similar result as in b from live-cell imaging of a HeLa K cell expressing RFP-H2B (red) and GFP-Nup107 (green) (Supplementary Video 4, representative of 15 cells). d, e, Functional restoration of MN from peripheral chromosomes. d, FI ratios for the indicated proteins, comparing MN from peripheral with central chromosomes (RPE-1 cells, scheme as in a). **** P < 0.0001, two-tailed Mann-Whitney test (mean with 95% CI, n=130, 97, 145, 112, 52, 47, 64, 48, 64, 47, 49, 45, 49, 45, left to right, from 2 experiments for the indicated proteins). e, Top: Experimental scheme. Bottom: Graphs of the results (HeLa K cells). Left: NE integrity monitored by loss of NLS-RFP (5 experiments). Middle: NE integrity monitored by hyper-accumulation of GFP-BAF (3 experiments). Right: DNA damage (3 experiments). ** P=0.0084, **** P < 0.0001, two-tailed Fisher’s exact Test. Note: Only a fraction of ruptured MN acquire DNA damage. Scale bars, 5 μm.
Here, we provide an explanation for the NE defects of micronuclei. The findings define an important mechanism underlying chromothripsis and suggest a new model for the coordination of chromosome segregation and NE assembly during normal cell division (Supplementary Video 6). We propose that during telophase, bundled spindle microtubules inhibit the recruitment of NE with NPCs/non-core proteins to lagging chromosomes. Lagging chromosomes are therefore enclosed by an NE primarily containing core proteins. Based on prior in vitro studies21,22, this NPC-deficient NE irreversibly blocks additional NPCs/non-core protein assembly. This irreversible defect is likely to be accentuated by sequestration of non-core proteins to the newly-forming daughter nuclei. Because micronuclei start out with an NE containing few NPCs, they have reduced access to interphase NPC assembly, which requires nuclear transport10. Defective nucleocytoplasmic transport then leads to defects in the accumulation of numerous proteins, including those necessary for normal DNA replication, DNA repair, and the maintenance of NE integrity (e.g., B-type lamins6,13, Extended Data Figs. 9d, 10d). The resulting spontaneous NE disruption leads to chromosome fragmentation and ultimately chromothripsis2,6,14. How microtubules inhibit non-core NE assembly remains to be determined, but could occur through a simple physical barrier effect. During telophase, NPC precursors were recently reported to assemble on fenestrated ER sheets28 (and personal communication, T. Kirchhausen) which we speculate might less readily penetrate bundles of spindle microtubules.
Our findings demonstrate that altered NE assembly on lagging chromosomes is not the consequence of a beneficial checkpoint delay, but rather a pathological outcome. During normal cell division, instead of precise and continuous monitoring of chromosome position, it appears that there is only loose coordination, with normal non-core NE assembly being dependent on timely spindle microtubule disassembly. Loose coordination between chromosome segregation and NE assembly, coupled with the irreversibility of NE assembly errors during mitotic exit, provides one explanation of why chromothripsis is common, with frequencies recently reported to be as high as 65% in some cancers4,5.
METHODS
Cell culture.
Cell lines were maintained at 37°C with 5% CO2 atmosphere in DMEM [HeLa Kyoto (HeLa K), U2OS] or DMEM:F12 without phenol red (hTERT RPE-1). All media were supplemented with 10% FBS, 100 IU ml−1 penicillin, and 100 μg ml−1 streptomycin. Stable cells lines (H2B-eGFP and TDRFP-NLS RPE-1, mRFP-H2B and GFP-BAF RPE-1, mRFP-H2B RPE-1, GFP-BAF and mRFP-LAP2β RPE-1, mRFP-H2B and mNeonGreen-PCNA RPE-1, H2B-eGFP and TDRFP-NLS HeLa K, eGFP-BAF HeLa K, TDRFP-NLS U2OS) were generated by transduction using lentivirus or retrovirus vectors carrying the genes of interest. Cells were infected for 18–24h with virus in the presence of 10 μg ml−1 polybrene, washed, and allowed to recover before antibiotic selection or fluorescence-activated cell sorting (FACS). Virus was generated by transfection of HEK293FT cells with the appropriate packaging plasmids (Lentivirus, pMD2.G and psPAX2; Retrovirus, pUMVC and pVSV-G) using Lipofectamine 3000 (Life Technologies), according to the manufacturer’s instructions. RPE-1 cells stably expressing emerin-eGFP were generated by transient transfection (Lipofectamine 3000 transfection reagent, Life Technologies) of emerin-eGFP plasmid followed by selection using G418 (500 μg/ml). HeLa K cells stably expressing mRFP-H2B and CHMP4B-eGFP (BAC), H2B-mCherry and Aurora B-eGFP (BAC) were derived from CHMP4B-eGFP HeLa K cells and Aurora B-eGFP HeLa K cells, respectively (gifts from A. Hyman)29. HeLa K cells stably expressing mRFP-H2B and mEGFP-Nup107 were derived from mEGFP-Nup107 expressing HeLa K cells (constructed by zinc finger nuclease-mediated genome-editing, from J. Ellenberg)10. U2OS cells stably expressing H2B-mCherry and 3XGFP-IBB were a gift from E. Hatch and M. Hetzer6. All cells and the derivative cell lines for this study were tested for Mycoplasma.
DNA constructs.
The following constructs were obtained from Addgene: H2B-eGFP (plasmid 11680, from G. Wahl), emerin-eGFP (plasmid 61985, from E. Schirmer), pmRFP-LAP2β-IRES-puro2b (plasmid 21047, from D. Gerlich). The construct encoding eGFP-BAF was made by recombining full-length human BAF into pLenti-CMV-neo-eGFP vector (T. Kuroda, vector plasmid Addgene 17447, from E. Campeau and P. Kaufman). The construct encoding mRFP-H2B was generated by recombining mRFP-H2B into pBABE-Puro vector (T. Kuroda, vector plasmid Addgene 1764, from H. Land, J. Morgenstern and B. Weinberg). The construct encoding TDRFP-NLS (referred to as RFP-NLS throughout the manuscript) was a gift from A. Salic. The construct encoding mNeonGreen-PCNA was made by recombining mNeonGreen-PCNA into pLENTI CMV Neo DEST vector (N. Umbreit, vector plasmid Addgene 17392, from E. Campeau and P. Kaufman) using Gateway LR Clonase II Enzyme (Invitrogen). Before Gateway cloning (Invitrogen), mNeonGreen-PCNA was first amplified by PCR using Ex Taq polymerase (Takara, primers: Forward 5’ CACCATGGTGAGCAAGGG 3’; Reverse 5’ CCTCTACAAATGTGGTATGGCTG 3’) and then the resulting PCR product was inserted into the pCR8/GW/TOPO vector (Invitrogen), according to the manufacturer’s instructions.
Antibodies for immunofluorescence.
For immunofluorescence, the following primary antibodies were used: LAP2α (1:300, Abcam, ab66588), Lap2β (1:200, Bethyl Laboratories, A304–840A), LAP2 (1:750, BD Transduction, 611000), mouse emerin (1:300, Abcam, ab54996), rabbit emerin (1:150 , Proteintech, 10351–1-AP), rabbit lamin A/C (1:1000, Abcam, ab26300), mouse lamin A (1:1000, Abcam, ab8980), rabbit LBR (1:500, Abcam, ab32535), mouse LBR (1:300, Sigma-Aldrich, SAB1400151), Nup133 (1:300, Abcam, ab155990), Nup107 (1:100, Life Technologies, 39C7), Nup62 (1:500, BD Transduction, 610497), Nup358 (1:1000, Abcam, ab64276), Tpr (1:200, Abcam, ab84516), Nup153 (1:500, Abcam, ab24700), mAb414 (1:1000 −1:2000, Abcam, ab24609), ELYS/MEL28 (1:200, Bethyl Laboratories, A300–166A), lamin B1 (1:1000, Abcam, ab16048), lamin B2 (1:1000, Abcam, ab151735), RPA1 (1:100, Cell Signaling, 2267), RPA2 (1:100, Abcam, ab2175), LSD1 (1:200, Cell Signaling, 2184), Rb (1:400, Cell Signaling, 9309), Aurora B (1:100, Abcam, ab3609), mouse phospho-H3S10 (1:10000, Abcam, ab14955), rabbit phospho-H3S10 (1:5000, Abcam, ab5176), SMC2 (1:1000, Abcam, ab10412), Aurora B Phospho pT232 (1:500, Rockland Antibodies, 600–401-677), CHMP4B (1:100, Proteintech, 13683–1-AP), CHMP2A (1:100, Proteintech, 10477–1-AP), IST1 (1:100, Proteintech, 51002–1-AP), rabbit α-Tubulin (1:1000, Abcam, ab18251), mouse α-Tubulin (1:300, Sigma-Aldrich, DM1A), CENP-A (1:300, Abcam, ab13939), rabbit γH2AX (1:500, Cell Signaling, 2577), mouse γH2AX (1:500, EMD Milipore, JBW301), FluoTag (Atto488)-X4 anti-GFP (1:250, NanoTag Biotechnologies), 53BP1 (1:100, Cell Signaling, 4937), BRCA1 (1:200, Santa Cruz, D-9) and POLD3 (1:100, Abnova, Clone 3-E2). Secondary antibodies used were Alexa Fluro 405, 488, 568 and 647 (1:1000, 1:500 for Alexa Fluro 405; Life Technologies).
SDS-PAGE and western blotting.
Cells were lysed in 2X sample buffer (100 mM TrisHCl pH 6.8, 4% SDS, 10% β-Mercaptoethanol). The whole-cell lysate was subjected to SDS–polyacrylamide gel electrophoresis on a NuPAGE 4–12% Bis-Tris gradient gels (Novex Life Technologies) and proteins were transferred onto a nitrocellulose membrane (Millipore). For Immunoblotting, the following primary antibodies were used: rabbit anti-MKLP2 (1:1000, Bethyl Laboratories, A300–879A), mouse anti-SMC2 (1:1000, Abcam, ab10412), rabbit anti-KIF4A (1:500, Bethyl Laboratories, A301–074A), rabbit anti-TTL (1:2000, Proteintech, 13618–1-AP), and mouse anti-α-Tubulin (1:15000, Sigma-Aldrich, DM1A).
RNA interference.
Cells at 50% confluency were transfected with 5 nmol (Extended Data Fig. 1h) or 40 nmol siRNA for 6h using the Lipofectamine 3000 transfection reagent (Life Technologies) according to the manufacturer’s instructions. Transfections were performed 48h before the induction of lagging chromosomes. The following siRNA sequences were used: MKLP2, 5’-AACCACCUAUGUAAUCUCAUG-3’30; KIF4A, 5’-AAGCAGAUUGAAAGCCUAGAG-3’26; TTL, 5’-CAGCCACCAAUCAGUAACU-3’27; SMC2, SMARTpool: ON-TARGETplus SMC2 siRNA L-006836–01-0005 (Dharmacon); TP53, SMARTpool: ON-TARGETplus TP53 siRNA L-003329–00-0050, (J-003329–14) GAAAUUUGCGUGUGGAGUA, (J-003329–15) GUGCAGCUGUGGGUUGAUU, (J-003329–16) GCAGUCAGAUCCUAGCGUC, (J-003329–17) GGAGAAUAUUUCACCCUUC (Dharmacon). Non-targeting Silencer Select Negative Control No. 1 siRNA (Thermo Fisher Scientific, 4390844) or Luciferase, 5’-CGUACGCGGAAUACUUCGATT-3’ was used as controls.
EdU incorporation.
Synchronized cells were continuously incubated with 10 μM EdU starting 1h after mitotic exit. Detection of EdU incorporation was performed using Click-iT Plus EdU Alexa Fluor imaging kits 647 or 594 (Life Technologies) according to the manufacturer’s instructions.
SiR-DNA labelling for live-cell imaging.
Synchronized cells were treated with 1 μM of the cell-permeable DNA dye SiR-DNA (Cytoskeleton Inc.), and 1 μM of Verapamil, a broad-spectrum efflux pump inhibitor, approximately 30 min-1h prior to imaging.
Cell cycle synchronization and generation of lagging chromosomes/micronuclei.
For the nocodazole block and release protocol to induce lagging chromosomes and micronuclei, cells were treated with 100 ng ml−1 nocodazole (Sigma-Aldrich) for 6h. Arrested cells were then collected by a shake-off procedure and washed three times with PBS prior to replating. For experiments where cells were imaged within the first 3 hours after the nocodazole block and release, mitotic cells were plated onto coverslips or imaging dishes that were pre-coated with 0.1 mg ml−1 Poly-D-lysine hydrobromide (Sigma-Aldrich) to facilitate attachment of mitotic cells. To obtain anaphase/telophase cells, cells were fixed ~40–50 min or 90 min post nocodazole release for RPE-1 and HeLa K cells, respectively. For experiments where micronucleated cells were imaged in mid-late interphase, cells after shake-off were plated onto uncoated coverslips.
For the induction of lagging chromosomes and micronuclei by MPS1 inhibition, cells were plated on coverslips or imaging dishes for 16–24h and then treated with 9 μM RO-3306 (EMD Millipore) for another 19h to arrest cells in the G2 phase. Arrested cells were then washed 7 times with pre-warmed medium containing 10% FBS and treated with the MPS1 inhibitor, 1 μM NMS-P715 (EMD Milipore). To induce a high frequency of peripheral missegregated chromosomes and their resulting micronuclei, RPE-1 cells were treated with 3 μM NMS-P715. For HeLa K cells, TTL knockdown was done with 40 nmol TTL siRNA, added 24h before the RO-3306 treatment. These G2-arrested cells (19h) were washed and released into 3 μM NMS-P715. We note that peripheral missegregated chromosomes can also be generated at a high frequency using the combination of 0.05 μM CENP-E inhibitor (GSK923295, Selleckchem) and 1 μM NMS-P715 (EMD Milipore; not shown31).
Indirect immunofluorescence microscopy.
Cells were washed in PBS and fixed with 4% paraformaldehyde for 15 min (unless otherwise stated); cells were then extracted in PBS-0.5% Triton X-100 for 5 min, washed 3 times with PBS, blocked for 30 min in PBS containing 3% BSA (PBS-BSA) and incubated with primary antibodies diluted in PBS-3% BSA for 60 min. These samples were then washed 3 times for 5 min with PBS-0.05% Triton X-100. Primary antibodies were detected using species-specific fluorescent secondary antibodies (Life Technologies). Finally, the samples were washed 3 times for 5 min with PBS-0.05% Triton X-100 prior to addition of 2.5 μg/ml Hoechst 33342 to detect DNA. Prolong Diamond Antifade or SlowFade Diamond Antifade (Life Technologies) was used for mounting of all immunofluorescence samples.
For labelling of SMC2 (Extended Data Fig. 5a–c), cells were pre-extracted with 0.5% Triton X-100 in CSK (1 M PIPES, 10 mM NaCl, 300 mM EGTA, 1 mM MgCl2) for 5 min on ice, fixed with 4% paraformaldehyde for 15 min and then processed as described above.
For labeling of CHMP4B, CHMP2A, and IST1 (Extended Data Fig. 5e) cells were fixed with methanol for 5–10 min at −20 °C and then processed as described above.
For co-staining of α-Tubulin and CHMP4B (Extended Data Fig. 9b), microtubule integrity was preserved by fixation of cells with 0.5% glutaraldehyde in MTSB (80 mM K-PIPES, 5 mM EGTA, 1 mM MgCl2 (PH6.8) for 10 min followed by addition of 0.1% NaBH4 for 10 min to quench unreacted glutaraldehyde. Cells were then washed 3 times with PBS, and processed as above.
Images were acquired on a Nikon Ti-E inverted microscope (Nikon Instruments, Melville, NY) with a Yokogawa CSU-22 spinning disk confocal head with the Borealis modification. Laser excitation was with 405 nm, 488 nm, and 561 nm and 642 nm lasers. Images were acquired using a X60 or X100 Plan Apo NA 1.4 oil objectives with a CoolSnapHQ2 CCD camera (Photometrics). Acquisition parameters, shutters, filter positions and focus were controlled by Metamorph software (Molecular Devices).
All immunofluorescence images shown in the manuscript are from a single focal plane, with the exception of Extended Data Figs. 4b, 5f, 7e, 9b, 9g which are maximum intensity projections from a z-focal plane series.
Correlative live-cell/ fixed cell imaging experiments.
For the nocodazole block and release protocol to induce lagging chromosomes and micronuclei, mitotic cells were collected and processed as above, but were plated on glass-bottomed gridded 35 mm imaging μ-dishes (ibidi) precoated with 0.1 mg ml−1 Poly-D-lysine hydrobromide (Sigma-Aldrich). For the induction of lagging chromosomes and micronuclei by MPS1 inhibition, cells were plated on ibidiTreat bottomed gridded 35 mm μ-dishes (ibidi) 6–24 h prior to RO-3306 treatment. Imaging dishes were mounted on a TE2000-E2 inverted microscope equipped with a Nikon Perfect Focus system. An Okolab cage incubator was used to maintain samples at 37°C and 5% humidified CO2. Fluorescence and DIC images of cells expressing mRFP-H2B, H2B-eGFP and RFP-NLS, H2B-eGFP and eGFP-BAF were acquired with a X20 0.5 NA Plan Fluor objective. After live-cell imaging, cells were washed with PBS, fixed and processed for immunofluorescence on the same imaging dishes. Cells of interest were identified from the recorded movies using the mRFP-H2B channel (unless otherwise stated), and their positions were determined based on the grid pattern on the dishes using the DIC images (see Extended Data Fig. 4e). Immunostained samples were then imaged by spinning disk confocal microscopy as described above.
To characterize the recruitment pattern of emerin and Nup133 onto lagging chromosomes (Extended Data Fig. 1b), or the loss of H3S10 phospohorylation or SMC2 on lagging chromosomes or interphase micronuclei (Extended Data Figs. 4b, 4c, 5b), synchronized mRFP-H2B-expressing RPE-1 cells were imaged at 2 min intervals and then subjected to fixation and labeling as described above.
For ZM447439 or nocodazole treatment experiments to test for the reversal of the non-core NE assembly defect on lagging chromosomes in telophase (Fig. 2a, Extended Data Figs. 4e, 4g, 6b, 6c, 8d), synchronized mRFP-H2B-expressing RPE-1 cells were imaged at 2 min intervals during mitosis and the drugs or vehicle controls were added during live-cell imaging. For paclitaxel/ ZM447439 experiments to analyze the non-core NE exclusion from the main chromosome mass (Fig. 3a), synchronized mRFP-H2B-expressing RPE-1 cells were imaged and treated with drugs or vehicle controls as described above. Following 12–14 min of live-cell imaging, cells were processed for indirect immunofluorescence as described above. Drug concentrations were: ZM447439 (5 μM or 50 μM, Tocris), nocodzole (10 μM, Sigma-Aldrich), paclitaxel (10 μM, Life Technologies).
For long-term correlated live-cell/fixed cell imaging experiments to assay for interphase micronuclear defects (Fig. 4, Extended Data Figs. 3d, 3e, 9, 10), RPE-1 or HeLa K cells expressing the tagged fluorescent proteins of interest were first imaged at higher temporal resolution to monitor the location of missegregated chromosomes during mitotic exit (3–5 min intervals for 1–1.5h post anaphase onset). All analyses were done only from chromosomes that remain consistently peripheral or central relative to the mitotic spindle until mitotic exit. After mitotic exit, MPS1 inhibitor (NMS-P715) was reduced to 0.5–1 μM, and cells were then imaged at 20–30 min intervals for 16–18h. When SiR-DNA was used to label DNA for long-term imaging, SiR-DNA concentration was reduced to 0.25 μM after initial 1.5h of imaging. For latrunculin A treatment (Extended Data Fig. 10c, d), latrunculin A (150 nM, Life Technologies) was added to RPE-1 cells approximately 1h post mitotic exit and maintained for the 16–18h period of live-cell imaging. After live-cell imaging, samples were processed for immunofluorescence as described above to assay for NE rupture, DNA damage or DNA replication. Alternatively, to assess the accumulation of NE proteins, nuclear proteins or the import reporter into early interphase micronuclei (Fig. 4d, Extended Data Fig. 9), cells were imaged at 20–30 min intervals for 1–3h after mitotic exit and then prepared for indirect immunofluorescence.
Image analysis for indirect immunofluorescence.
Image analysis was done with ImageJ/FIJI software. Regions of interest (ROI) were generated for lagging chromosomes/ micronuclei and the corresponding primary nuclei using customized ImageJ/FIJI macros. First, nuclear segmentation was performed using Li or Otsu thresholding from the DNA (Hoechst) image or the mRFP-H2B image. Second, the ImageJ/FIJI function “Watershed” and “Erode” were used to the refine the nuclear segmentation. Third, the segmented regions containing nuclei of interest (micronuclei and corresponding primary nuclei) were manually selected as ROI using the ImageJ/FI function “Wand Tool”. Based on these ROIs, the mean fluorescence intensities (FI) of labeled proteins were quantified from other channels. For quantification of nuclear proteins (RFP-NLS, RPA1, RPA2, LSD1, Rb, 53BP1, BRCA1, POLD3, mNeonGreen-PCNA), chromatin-associated proteins (LAP2α, GFP-BAF, γH2AX), or labelled DNA (as well as EdU incorporation), ROIs were generated based on the segmented nuclear area from the best focal plane for the micronucleus and primary nucleus. For quantification of NE or NPCs proteins, band-shaped ROIs around micronuclei and primary nuclei were generated using ImageJ/FJI function “Make band” after nuclear segmentation followed by the procedures described above.
For characterizing the assembly of NE/NPC proteins on lagging chromosomes (Fig. 1a and Extended Data Fig. 1), lagging chromosomes were scored as being positive if the mean FI (after background subtraction) of the labeled proteins on lagging chromosomes was >3 standard deviations above the mean cytoplasmic background. Only telophase cells that had a near-complete assembly of non-core NE on the main chromosome mass were scored.
For the fluorescence intensity (FI) ratios of proteins in micronuclei relative to the primary nuclei (MN/PN), the mean FI were quantified from the defined ROIs. Background-subtracted FI ratios of MN/PN were then calculated and are shown in corresponding graphs.
To characterize the levels of phospho-H3S10 and SMC2 on the main and lagging chromosomes (Extended Data Figs. 4b, 4c, 5b, 7e), images were acquired from a z-stack (~12 μm) with a 0.2 μm (or 0.5 μm) step size to cover the region of the cell that contained the chromosomes. Because both anti-phospho-H3S10 antibodies showed stronger recognition for the antigens on the surface of the chromosome mass than those within the chromosome mass, the maximum intensity projections of z-focal plane series were used to estimate the average FI of phospho-H3S10. In parallel, the average FI of phospho-H3S10 was directly quantified from the lines manually drawn along the periphery of the main chromosome mass and lagging chromosomes, yielding similar results (not shown). Note that measurements for FI of Nup133 in Extended Data Figs. 4c and 5b were taken from the same samples.
To calculate the fraction of the lagging chromosome circumference labeled for Nup133 (Figs. 3c, 4b, Extended Data Figs. 6c, 7d, 8d, 9a, 9h), we measured the length of a manually drawn line along the lagging chromosome periphery that was positive for Nup133 (FI > 3 standard deviations above the mean cytoplasmic background). The ratio of this value was then made with the circumference of the lagging chromosome determined from the DNA label. This ratio was used in circumstances (e.g., KIF4A knockdown) where some late mitotic NPC (non-core) assembly is restored on lagging chromosomes, but is restricted to a local “mini-domain” (e.g., Fig. 3c, Extended Data Fig. 7d, 8d). Only lagging chromosomes with a near-continuous emerin signal were analyzed. In parallel, the FI ratios of Nup133 for MN/PN (e.g., Extended Data Fig. 4g where measurements were taken from the same smaples as in Extended Data Fig. 8d) were quantified to verify the late mitotic NPC assembly from corresponding experiments, yielding similar results.
For analysis of the effect of Aurora B inhibition (ZM447439) on spindle microtubule density (Extended Data Fig. 6a), the FI of α-tubulin was measured along the line perpendicular to the long axis of the mitotic spindle.
For paclitaxel/ ZM447439 experiments to analyze the non-core NE exclusion on the main chromosome mass (Fig. 3a), the Nup133 gap was measured as the cumulative length of individual small discontinuities in the Nup133 signal (FI< 3 standard deviations above the mean cytoplasmic background) along the main chromosome mass. A threshold was set at ≥ 5 pixels as a definition for the lower bound for Nup133 discontinuities. The length of emerin enrichment was measured as the cumulative length of lines manually drawn along the main chromosome mass where emerin was enriched (> 2-fold emerin FI on the chromosome periphery). Only cells which had a near-continuous emerin signal on the main chromosome mass were quantified. The length of the Nup133 gaps and the emerin enrichment were normalized to DAPI signal circumference of the main chromosome mass, set to 100.
For analysis of the level of phospho-H3S10 on lagging chromsomes after KIF4A knockdown (Extended Data Fig. 7e), linescan profiles of the FI of phospho-H3S10 and Nup107 was measured along the white dashed line.
For the experiments to measure the restoration of DNA replication in peripheral chromosome-derived micronuclei (Extended Data Fig. 9e), the FI ratios of EdU in the micronucleus and primary nucleus were measured as described above, and then normalized to the DNA label FI ratio. To measure the frequency of micronuclear NE disruption (Fig. 4e, Extended Data Fig. 10a–c), micronuclei were scored as being disrupted if the FI ratio of RFP-NLS in micronuclei relative to the cytoplasm dropped to ~1 or if the FI ratio of GFP-BAF in micronuclei relative to the primary nuclei rose to > 3. For DNA damage, MN were scored as being positive if the mean FI of γH2AX in MN > 3 standard deviations above the mean FI of γH2AX in the primary nuclei. This threshold was also used to exclude rare cells where the primary nucleus had significant DNA damage (e.g., as can occur in a fraction of cells after high dose inhibition of MPS1).
Confocal live-cell microscopy.
In all confocal live-cell experiments, cells were synchronized to induce lagging chromosomes/micronuclei using the nocodazole block and release protocol, as described above. After mitotic shake-off, cells were replated onto glass-bottomed 35 mm imaging μ-dishes (ibidi) precoated with 0.1 mg ml−1 Poly-D-lysine hydrobromide (Sigma-Aldrich). All images were collected on a Nikon Ti inverted microscope equipped with a Yokogawa CSU-X1 spinning disk confocal head, Spectral Applied Precision LMM-5 with AOTF, a Hamamatsu ORCA ER cooled CCD camera and the Nikon Perfect Focus System. GFP was imaged using a 488nm laser for excitation and a 525/50nm emission filter; RFP was imaged using a 561nm laser for excitation and a 620/60 emission filter. An Okolab cage incubator was used to maintain samples at 37°C and 5% humidified CO2. Image acquisition was controlled with MetaMorph software (Molecular Devices).
For live-cell imaging to measure the kinetics of nuclear import into micronuclei (Fig. 1b, c, Extended Data Fig. 3a, b), RPE-1 cells expressing H2B-eGFP and RFP-NLS or U2OS cells expressing GFP-IBB and mCherry-H2B, or U2OS cells expressing RFP-NLS labeled with SiR-DNA were prepared for imaging as described above. To consistently track lagging chromosomes and micronuclei that move in and out of focal plane over time, images were acquired from 7 z sections with a 2 μm step size for RPE-1 or 12 z sections with 1 μm for U2OS using a X60 Plan Apo NA 1.4 objective lens at 1 min intervals during the 30 min-3h window after nocodazole release.
For live imaging of ESCRT-III dynamics (Extended Data Fig. 5d) or Nup107 dynamics (Fig. 4c, Extended Data Fig. 7c), HeLa K cells expressing CHMP4B-eGFP and mRFP-H2B or HeLa K cells expressing GFP-Nup107 and mRFP-H2B were prepared as described above. Images were collected from 10 z sections with a 2 μm step size at 1 min intervals using a X60 Plan Apo NA 1.4 objective lens during the 1–3h window after nocodazole release.
Nuclear import assay.
To quantify nuclear import (Extended Data Fig. 3a), customized ImageJ/FIJI macros were used. ROIs for lagging chromosomes/MN and PN were generated as described above in the Image analysis for indirect immunofluorescence section. Based on these ROIs, the mean FI of RFP-NLS for PN and MN pairs along with non-cell background was quantified through z stacks from time-lapse image series. The background-subtracted mean FI of RFP-NLS from the best focal plane for lagging chromosomes/MN and PN pairs was determined (best focal plane was by maximum FI, which could be different for the MN and the PN). This procedure was employed to enable the accurate measurement of the FI of MN that move above and below PN during the course of imaging. Detection accuracy was verified manually. The measured values were then corrected for photobleaching, using the bleaching coefficient described below.
We used G2 cells to calculate the rate of photobleaching in both intact micronuclei and primary nuclei under what we presume were steady state conditions. The measured bleaching coefficients were then used to correct for bleaching during the nuclear import assay. Live-cell imaging of RPE-1 cells expressing GFP-H2B and RFP-NLS was performed in cells 16h after release from the nocodazale block, using the identical imaging settings and conditions that were used for nuclear import assay. We measured the RFP-NLS FI in the cytoplasm, primary nucleus and micronucleus, subtracting background outside of the cell. To control for subtle cell-to-cell variability in RFP-NLS expression (post FACS), the FI in the primary nucleus was set to 100 in each cell. The normalized mean FI of RFP-NLS from 12 MN/PN pairs was then plotted over time. For each PN/MN pair, the plotted data was fit to an exponential curve with an offset: y=a * exp (-bx) +c, where b is the bleaching coefficient, a is the y value at t0 before bleaching, and c is the y value at plateau after bleaching (camera noise). From this analysis, there was no statistically significant difference between the average bleaching coefficients for the PN and the MN [b=0.05 (both PN and MN), P= 0.62 (NS), Wilcoxon matched-pairs test]. This bleaching correction method was validated by comparing the experimentally measured (RFP-NLS FI) values and the predicted bleaching corrected values over time using the above equation. The ratios of experimentally measured values relative to the predicted bleaching-corrected values remained constant (nearly 1).
An orthogonal wide-field imaging approach, enabling a larger sample size, confirmed defective nuclear accumulation of RFP-NLS and RPA2 in MN (Extended Data Fig. 3d, e). For these experiments, imaging was performed at 5 min intervals using a X20 objective, followed by fixation and quantitative immunofluorescence as described above.
Correlative Light and Electron Microscopy (CLEM).
For CLEM analyses of telophase cells (Fig. 2b), RPE-1 cells expressing CenpA-GFP and centrin1-GFP were treated with 3 μM nocodazole for 4 hrs, released into fresh media, and mounted in Rose chambers. Live-cell imaging (17 cells) was performed at 5 sec intervals from mid-late anaphase through completion of cytokinesis. Dynamics of furrow ingression were determined by plotting the extent of constriction (normalized to the cell width) over time (Fig. 2b, top left). This plot was used to identify the stage of cells selected for EM. Cells were fixed with 2.5% glutaraldehyde in PBS, pH7.4 for 30 min. Chromosomes were stained with Hoechst 33342 at 1 μg/ml. Multimode (DIC and fluorescence) LM (light microscope) datasets were obtained on a Nikon TE2000 microscope equipped with X100 1.45 NA Plan Apo objective lens and Zyla 4.2 sCMOS camera (Andor). All fluorescence images were deconvolved in SoftWoRx 5.0 software (Applied Precision) with lens-specific PSFs. Post-fixation, embedding, and sectioning were done as previously described32. Serial thin sections (70 nm) were imaged on JEOL 1400 microscope operated at 80 kV using side-mounted 4.0 Megapixel XR401 sCMOS camera (Advanced Microscopy Techniques Corp) controlled by AMT Capture Engine ver.7.0.
For CLEM of interphase micronuclei (Extended Data Fig. 5g, 4 cells with 3 intact MN and 4 newly disrupted MN) RPE-1 cells expressing H2B-eGFP and RFP-NLS were synchronized by the nocodazole block and release protocol and replated as described above. Prior to live-cell imaging, multiple xyz positions for subsequent analysis were marked with a diamond scribe. Images were acquired at 10 min intervals and micronuclear disruption was detected by abrupt loss of RFP-NLS. Cells selected for EM were then fixed and processed as described above. Note: similar results were obtained from the analysis of 4 cells (5 intact and 2 disrupted MN) that were collected from fixed samples (without live-cell imaging) 16h after release from the nocodazole block (not shown).
Structural illumination Microscopy (SIM).
For 3D-SIM of RPE-1 cells expressing emerin-eGFP (Extended Data Fig. 5f) or HeLa K cells expressing CHMPB-eGFP (Extended Data Fig. 8a–c), lagging chromosomes were induced by a nocodozole block and release protocol. After nocodazole release, cells were replated onto coverslips (MatTek, high tolerance coverslips) that were pre-coated with 0.1 mg ml−1 Poly-D-lysine hydrobromide (Sigma-Aldrich). To obtain anaphase/telophase cells, cells were fixed during mitotic exit (~50 min or 90 min post nocodazole release for RPE-1 and HeLa K cells, respectively) in 4% EM grade formaldehyde (Polysciences) diluted in PEM buffer [80 mM K-Pipes, 5 mM EGTA, 1 mM MgCl2 (pH 6.8)] for 5 min at 37oC. Cells were then permeabilized with PEM/0.15% Triton X-100 for 2 min and processed for immunofluorescence. Primary and secondary antibodies were diluted in PBS/0.05% Saponin and incubated for 2–3h. To minimize photobleaching of samples, CHMP4B-eGFP or emerin-eGFP signal was enhanced by labeling with FluoTag (Atto488)-X4 anti-GFP (1:250, NanoTag Biotechnologies).
SIM was performed on an OMX V4 Blaze (GE Healthcare) microscope equipped with three watercooled PCO.edge sCMOS cameras, 405 nm, 488 nm, 568 nm and 642 nm laser lines. Images were acquired with a 0.125 um step size without binning using a X60 1.42 NA Plan Apochromat objective (Olympus). For each z-section, 15 raw images (three rotations with five phases each) were acquired. To minimize the photobleaching of GFP in thick mitotic specimens, z-stack collection was limited by manual selection to the middle (3.5–5 μm) region of the cell that contained the entire lagging chromosome. Images were acquired from ~28–40 z-sections with a 0.125 μm step size. Spherical aberration was minimized by immersion oil matching33. Super-resolution images were computationally reconstructed from the raw data sets with a channel-specific, measured optical transfer function and a Wiener filter constant of 0.001 using CUDA-accelerated 3D-SIM reconstruction code based on Gustafsson et al. (2008)34. TetraSpeck beads (Thermo Fisher) or a nano-grid control slide (GE) were used to measure axial and lateral chromatic misregistration, and experimental data sets were registered using the imwarp function in MATLAB (MathWorks). 3D rendered images were generated using Imaris image analysis software (Bitplane).
Statistical analysis.
Statistical analysis was performed using Prism (GraphPad software Inc.). Student’s t-test or non-parametric tests (Mann-Whitney test) were performed based on the sample distributions. For bar graphs or scatter dot plots, bars indicate the mean value of population with 95% CI, unless otherwise specified. To determine the correlation between the accumulation level of RFP-NLS and RPA2 in MN (Extended Data Fig. 3d, e), the Spearman’s correlation analysis was used. No statistical methods were used to predetermine sample size. The experiments were not randomized and the investigators were not blinded to allocation during experiments and outcome assessment.
Reporting summary.
Further information on experimental design is available in the Nature Research Reporting Summary linked to this paper.
Data availability.
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Source data for all graphs (Figs. 1c, 2a, 2b, 3a, 3c, 4b, 4d, 4e; Extended Data Figs. 1d, 1f, 2b, 2c, 2d, 2f, 2g, 3a, 3b, 3c, 3e, 4c, 4d, 4f, 4g, 5b, 5c, 5e, 6a, 6b, 6c, 7b, 7d, 7e, 8d, 9a, 9d, 9e, 9f, 9h, 10c, 10d) are provided with the online version of the paper. All other datasets generated and/or analysed in the current study are available from the corresponding author upon reasonable request. Supplementary Figure 1 contains scanned complete images of western blots.
Extended Data
Extended Data Figure 1. Defective nuclear envelope assembly on lagging chromosomes (or chromosome bridges) generated by multiple methods.
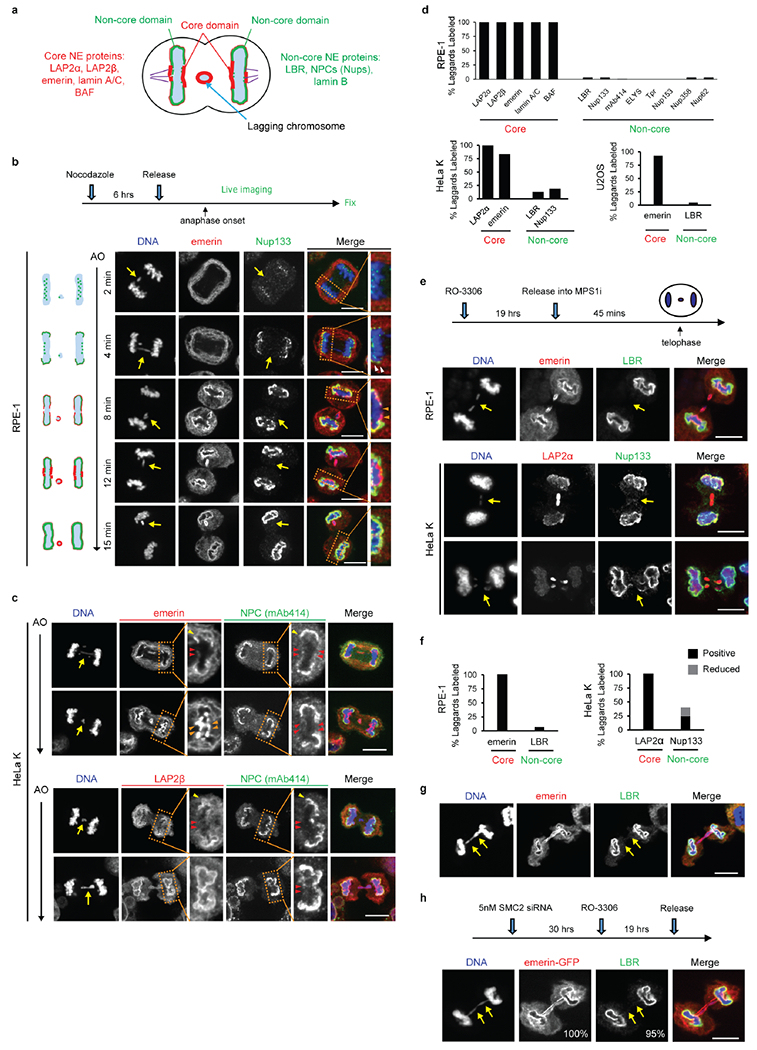
a, Cartoon showing the NE subdomains that transiently form around the main chromosome mass in a telophase cell8,9 and summarizing results reported here for lagging chromosomes. DNA is shown in light blue. Lamina-associated polypeptide 2α/β (LAP2α/β). Barrier-to-autointegration factor (BAF). Lamin B receptor (LBR). The NPC is a complex containing nucleoporins (Nups), including Nup133, Nup107, ELYS, Tpr, Nup153, Nup358 and Nup621 (see Extended Data Figs 1d, 2f).
b, Live-cell/ fixed cell imaging showing the recruitment pattern of core (emerin) and non-core (Nup133) proteins onto lagging chromosomes (yellow arrows) and the main chromosome mass at the indicated timepoints after anaphase onset (AO, t=0). Top: Experimental scheme. RFP-H2B-expressing RPE-1 cells were released from the nocodazole mitotic block, imaged on gridded coverslips at 2 min intervals, and then fixed and labeled for immunofluorescence (as in Extended Data Fig. 4e). Bottom: cartoons (left) and images of RPE-1 cells showing DNA (blue), emerin (red) and Nup133 (green). Each image is representative of 20 cells (from 2 technical replicates) from the indicated timepoints. Scale bars, 10 μm. Note: there is some variability between cells (~1–2 min) for the timing at which different proteins are recruited. During early-mid anaphase (2–4 min post AO), Nup133 is on kinetochores as previously described35 (green dots in the cartoon). ~4–6 min post AO, Nup133 begins to assemble on the chromosome periphery (enlarged image in the right most column, white arrowheads). ~6–8 min post AO, the membrane protein emerin assembles on lagging chromosome and the main chromosome mass, including the region adjacent to the central spindle (enlarged image in the right most column, orange arrowheads, microtubules are not shown). By 8–12 min post AO, emerin becomes concentrated in a recognizable “core” domain8, which is also detected as a gap in Nup133 signal (enlarged image). The peripheral localization of Nup133 (e.g., 8 min) marks the “non-core” domain, cartooned in (a). About half of the NPCs of the interphase nucleus assemble in this ~ 8–10 minute period of telophase28 (hereafter referred to as late mitotic NPC assembly). Note that nuclear pore proteins display the most obvious “non-core” gap, whereas other non-core proteins, such as LBR, more commonly display reduced signal intensity within the core domain (Extended Data Fig. 1e, top row, RPE-1), rather than absence of signal in this region. Defective Nup133 assembly on lagging chromosomes persists throughout the mitotic exit (>15 min post AO). By ~15 min post AO, the core and non-core domains become intermingled on the main nucleus, with fragments of the core domain persisting as “pore-free” islands that slowly populated by NPCs during interphase10,36.
c, Similar to RPE-1 cells (see b above), images of HeLa Kyoto (HeLa K) cells (representative of 30 cells, from 2 experiments) showing the “core” membrane proteins emerin (top two rows) and LAP2β (bottom two rows) first associate with the chromosome periphery (yellow arrowheads) contemporaneously with the non-core (NPC, mAb414 detects FG-containing nucleoporins.) proteins. ~2–4 min later, the core proteins extend into and then concentrate (emerin, orange arrowheads; LAP2β does not concentrate) in the core domain (red arrowheads). Cells were synchronized as in Extended Data Fig. 1e. Note in HeLa K cells, lagging chromosomes often exhibit a slight delay (~1–2 min) in the recruitment of core membrane proteins (emerin and LAP2β) as compared to the periphery of the main chromosome mass. Scale bars, 10 μm.
Discussion: It is thought that the NE likely assembles from a continuous network of mitotic endoplasmic reticulum (ER)1. It is therefore simplest to propose that the core and non-core subdomains are also in a continuous network, but the core domain is just a region of the continuous ER network that is missing the non-core subgroup of proteins. Supporting this idea, prior work8 and our data (b, c above) show that the core proteins initially assemble together with the non-core proteins around the chromosome periphery and only later become enriched in the regions near the microtubules. This data suggests a model that the domain partitioning could come solely from NPC precursors and LBR (which requires ELYS for recruitment37,38) being preferentially retained in fenestrated ER sheets28 that might less readily penetrate bundled spindle microtubules (see Fig. 3a). Although we favor this model, at this point, we cannot exclude the alternative that core and non-core proteins somehow partition into separate membrane compartments.
d, Quantitation of defective non-core NE protein recruitment to lagging chromosomes in different cell lines. Synchronization as in Fig. 1a (n=64, 118, 151, 124, 151, 145, 150, 69, 90, 65, 70, 60, 64, left to right, from 3 experiments for RPE-1, n=149, 110, 124, 132, from 2 experiments for HeLa K and n=44, 76, from 2 experiments for U2OS cells).
e, f, Orthogonal method (1 μM NMS-P715, MPS1i) to generate lagging chromosomes shows a similar non-core NE assembly defect as observed with the nocodazole block-and-release protocol (Fig. 1a). e, Top: Experimental scheme. Bottom: Representative images of RPE-1 and HeLa K cells. Note that in RPE-1 cells and U2OS cells there is a near absence of non-core protein on lagging chromosomes, irrespective of the method to generate them. However, in HeLa K cells, the effect is slightly less penetrant. ~60% of lagging chromosomes lack detectable non-core protein recruitment, ~15% display strongly reduced levels (scored as labeled but shown in gray bar as “reduced”, see f) , and ~25% display a clear signal (Nup133), but often only covering part of the circumference of the lagging chromosome. These subtle differences are likely explained by differences in spindle organization between cell lines (see Fig. 3 and Extended Data Fig. 6, 7, 8 below). f, Quantitation of the results in different cell lines (n=56, 78 for RPE-1, from 2 experiments; n=75, 174 for HeLa K, from 3 experiments). Scale bars, 10 μm.
g, h, Chromatin bridges (arrows) formed after nocodazole release or after partial depletion of the condensin SMC239, display core-only NE protein assembly. g, Images of an RPE-1 cell after release from a nocodazole block (representative of 30 DNA bridges, from 5 experiments). h, Top: Experimental scheme for generating chromosome bridges by partial SMC2 depletion. Bottom: Images of an emerin-GFP expressing RPE-1 cell. Percentage of cells with the indicated staining pattern are on the lower right (n=30, from 2 experiments). Scale bars, 10 μm. Note that the DNA bridges are uniformly depleted for non-core (LBR) proteins, with no evidence for a gradient (i.e. increased LBR away from the central spindle) as might be expected for the chromosome separation checkpoint hypothesis15,40. We also note that interphase chromatin bridges were previously reported to have an altered NE protein composition including reduced levels of lamin B1 and NPCs41, which is consistent with our findings here.
Extended Data Figure 2. NPC and other non-core protein assembly defect persists in interphase micronuclei in multiple cell lines.
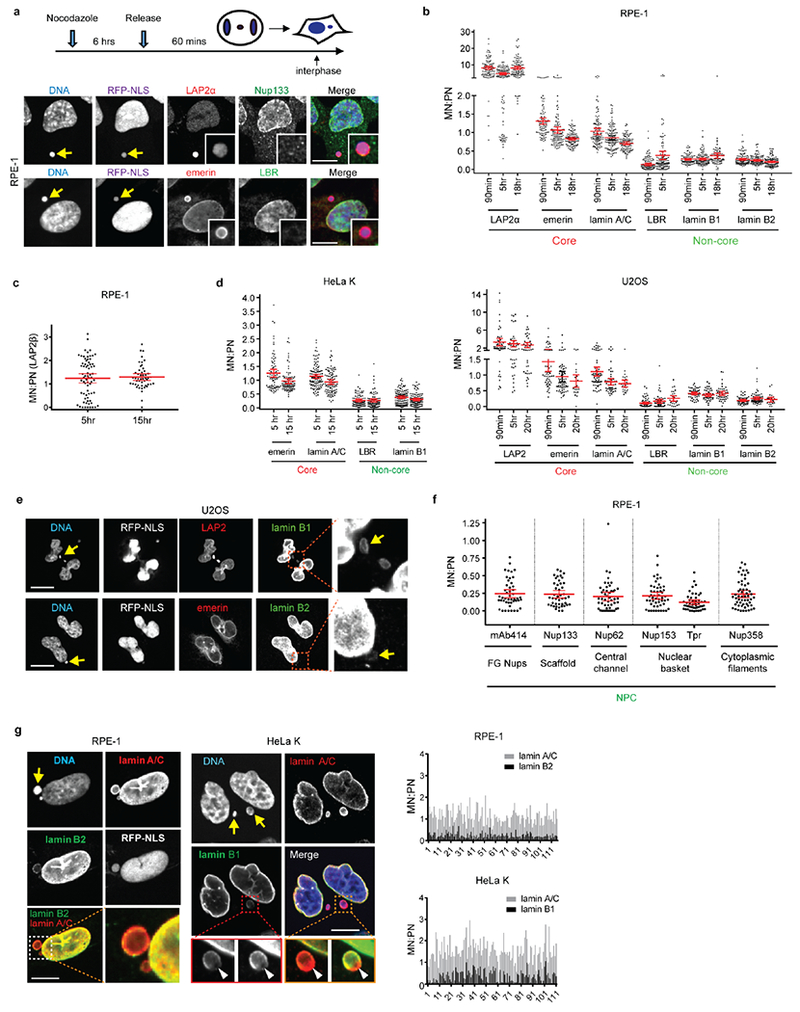
a-c, Defective non-core NE protein recruitment to micronuclei (MN) in RPE-1 cells. a, Top: Experimental scheme. Bottom: Representative images (see b for quantification) with MN (arrows, insets for enlarged images). Red letters: core NE proteins; Green: non-core proteins. b, Quantification of the results from a. The fluorescence intensity (FI) ratio of the indicated NE proteins in intact MN (RFP-NLS positive) relative to primary nucleus (PN) at the indicated timepoints after release from nocodazole block (mean with 95% CI, n=97, 120, 106, 92, 114, 104, 113, 116, 104, 114, 121, 91, 114, 89, 113, 116, 125, left to right, from 2 experiments). Scale bars, 10 μm. c, MN/PN FI ratio of the core membrane protein LAP2β in RPE-1 cells at the indicated timepoints after release from a nocodazole block (mean with 95% CI, n=70, 49, from 2 experiments).
d, Deficiency of non-core proteins persists in micronuclei in HeLa K (left) and U2OS (right). MN/PN FI ratio of the indicated NE proteins after release from a nocodazole block as in a (mean with 95% CI, n=84, 111, 90, 79, 84, 111, 90, 111, left to right for HeLa K, from 2 experiments; n= 53, 47, 45, 53, 66, 23, 64, 53, 27, 66, 54, 28, 53, 47, 46, 53, 69, 23, left to right for U2OS, from 2 experiments).
e, Representative images of U2OS cells from d showing reduced assembly of B-type lamins in MN (arrows) at 90 min post nocodazole release. Scale bars, 10 μm.
f, Reduced NPC assembly on MN. MN/PN FI ratio of the indicated NPC proteins in RPE-1 cells at indicated timepoints after release from nocodazole block (mean with 95% CI, n=47, 44, 49, 51, 49, 56, left to right, from 2 experiments).
g, Reduced accumulation of B-type lamins but not A-type lamins on intact micronuclei. Left: Representative images of RPE-1 (left) and HeLa K (right) cells. In the enlarged merged images from the orange boxed region (HeLa K), the intensity of the lamin B1 has been scaled differently to illustrate two points. First, there is a reduction of lamin B in MN but not lamin A/C, which becomes evident when the lamin B and lamin A/C on the main nucleus are scaled to the same intensity. Second, some MN display a lamin B1 gap, as has been previously reported6, (arrowheads, the lamin B1 gap formed where the lamin A/C rim is still continuous), which becomes evident when the lamin B1 intensity is scaled to a higher level. Graphs on the right: General reduction of B-type lamins in micronuclei. MN/PN FI ratio for the indicated lamins. Shown is the background-subtracted raw data for each individual micronucleus analyzed in the indicated cell lines (n=116 for RPE-1, n=111 for HeLa K, from b, d above), 5h post nocodazole release. Scale bars, 10 μm. Note that a fraction of micronuclei exhibit a reduced level of lamin A/C, which may be caused by impaired import of lamin A/C during interphase.
Extended Data Figure 3. Micronuclei have defective nuclear import and impaired accumulation of nuclear proteins.
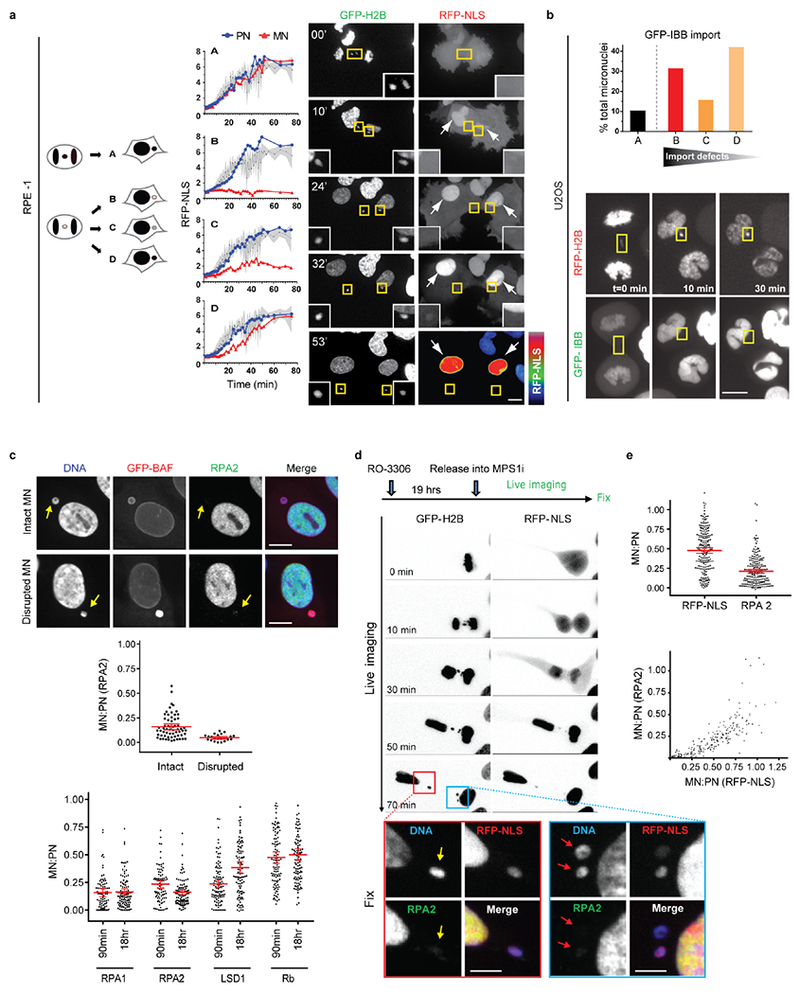
a, b, Defective nuclear import kinetics in newly formed MN visualized by high temporal resolution, confocal live-cell imaging. MN were generated in RPE-1 (a) or U2OS (b) cells as in Fig. 1a. a, Defective nuclear RFP-NLS import in RPE-1 cells. Left: Cartoon depicting categories of cells with differing levels of accumulation of the import reporter in MN as compared to the PN (see also in Fig. 1c for quantitation). Middle: Graphs showing nuclear import kinetics obtained from representative RPE-1 cells expressing GFP-H2B and RFP-NLS for PN (blue) and MN (red). Black lines indicate the mean values from 11 PN; shaded regions indicate standard deviation (SD). To control for photobleaching, the bleaching coefficient of the RFP-NLS signal was experimentally determined by imaging of cells during late interphase when nuclear import was presumed to be at steady state. A correction based on this bleaching coefficient obtained for 12 PN and MN pairs was then applied to the data presented here (see Methods). Right: Representative images from a time-lapse series from RPE-1 cells expressing GFP-H2B (left) and RFP-NLS (right). Time is in min. Arrows indicate a pair of PN with RFP-NLS import evident from 10 min. Yellow boxes show lagging chromosomes and the resulting MN with nuclear RFP-NLS accumulation defects (insets: enlarged images). A heat map of RFP-NLS intensity is shown in the bottom right panel (t=53 min) illustrating the strong import defect in the MN (see Supplementary Videos 1 and 2). b, Defective nuclear GFP-IBB import in lagging chromosomes from U2OS cells. Top: Percentage of micronuclei corresponding to the categories (a, cartoon) in U2OS cells expressing RFP-H2B and GFP-IBB (n=18 cells, from 17 experiments). Bottom: Images from a time-lapse series (boxes, lagging chromosomes and micronuclei). Scale bars, 10 μm.
c, Impaired nuclear accumulation of multiple proteins in MN relative to PN. Top: Representative images of RPE-1 cells with an intact MN or a disrupted MN (arrows, MN disruption is visualized by hyper-accumulation of GFP-BAF42. Both the intact and disrupted MN lack detectable accumulation of RPA2. Middle: MN/PN FI ratio of RPA2 (mean with 95% CI, n=62, 23, from 2 technical replicates). MN were generated in RPE-1 cells as in Extended Data Fig. 1e. Bottom: FI ratio of the indicated proteins in intact (RFP-NLS positive) MN from RPE-1 cells at the indicated timepoints after nocodazole release as in Extended Data Fig. 2a (mean with 95% CI, n=89, 99, 89, 98, 107, 100, 107, 100, left to right, from 3 experiments). Lysine-specific histone demethylase 1 (LSD1). Retinoblastoma protein (Rb). Scale bars, 10 μm.
d, e. Live-cell/fixed cell imaging showing impaired nuclear accumulation of RPA2 and RFP-NLS in MN after mitotic exit. d, Top: Experimental scheme. Bottom: Images from wide-field time-lapse series followed by fixation and confocal imaging at the end of the experiment. The confocal images (from red and blue boxed regions) illustrate reduced accumulation of RFP-NLS and RPA2 in newly formed MN (yellow and red arrows). Time post AO (t=0 min) is shown. Scale bars, 10 μm. see e for quantification, numbers, and experimental repetitions. e, Top: MN/PN FI ratio of RPA2 and RFP-NLS in RPE-1 cells ~1h post AO (mean with 95% CI, n=211, from 2 experiments). Bottom: Defective accumulation of RPA2 and RFP-NLS are correlated (r= 0.9039, P<0.0001, two-tailed Spearman’s correlation analysis).
Extended Data Figure 4. Proximity to spindle midzone Aurora B does not affect the NE assembly defect of lagging chromosomes; this non-core NE assembly defect persists through late telophase regardless of whether Aurora B is active.
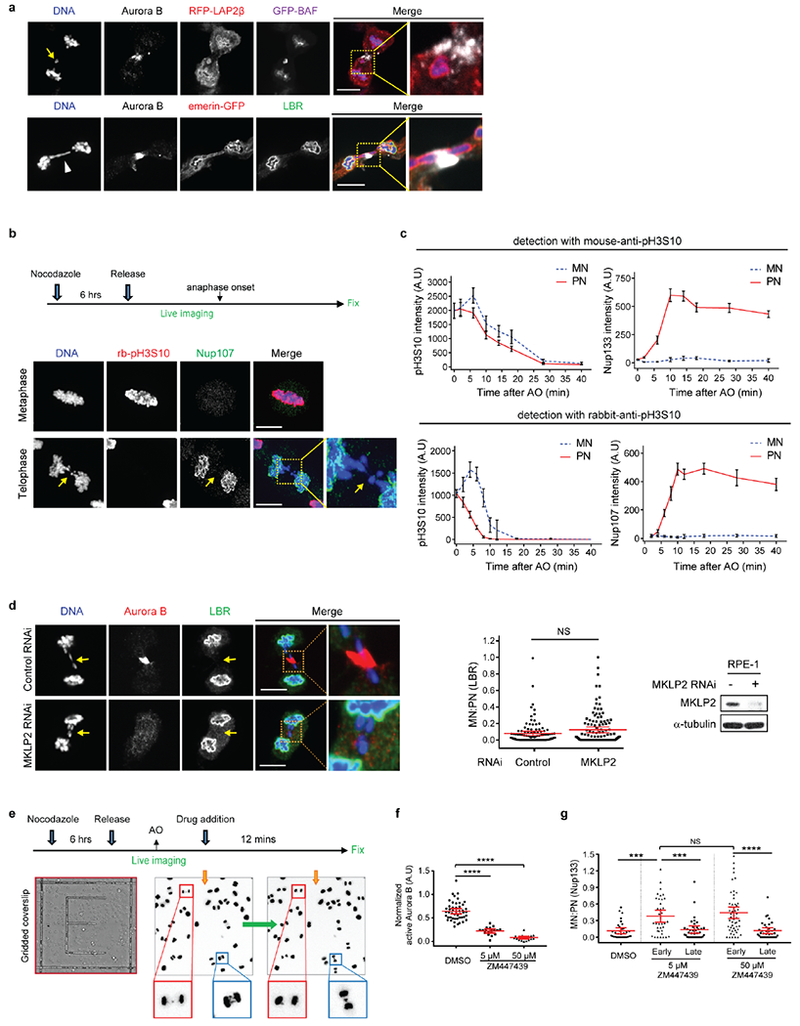
a, Core membrane proteins (LAP2β and emerin), assemble on lagging chromosomes and chromosome bridges, independent of their position relative to the spindle midzone. Top: Images of a GFP-BAF and RFP-LAP2β-expressing RPE-1 cell show recruitment of LAP2β to lagging chromosome (arrow). Bottom: Images of an emerin-GFP-expressing RPE-1 cell show recruitment of emerin to a chromosome bridge (arrowhead). Both LAP2β and emerin can assemble near the concentration of Aurora B (white, in merged images) at the spindle midzone (representative of 30 cells, from 2 experiments). Cells synchronized as in Fig. 1a. Scale bars, 10 μm.
b, c, Live-cell/ fixed cell imaging showing that loss of H3S10 phosphorylation in late telophase does not enable non-core NE assembly onto lagging chromosomes. b, Top: Experimental scheme. Bottom: Representative images of RPE-1 cells. Scale bars, 10 μm. c, FI measurements from the main chromosome mass (PN) and lagging chromosome (MN) showing loss of H3S10 phosphorylation relative to the recruitment of NPC proteins in the same cell at the indicated timepoints. Top: Phospho-H3S10 with a mouse monoclonal anti-pH3S10 antibody (mean with 95% CI, n=31, 46, 34, 27, 26, 28, 34, 26 each timepoint for MN and PN mouse-pH3S10, from 2 experiments; n=31, 46, 34, 27, 26, 28, 34, 26 each timepoint for MN and PN Nup133, from 2 experiments). Bottom: rabbit polyclonal anti-pH3S10 antibody (mean with 95% CI, n=27, 20, 33, 29, 22, 30, 22, 32, 31, 28 each timepoint for MN and PN rabbit-pH3S10, from 2 experiments; n=30, 49, 28, 32, 30, 19, 32, 32, 27 each timepoint for MN and PN Nup107, from 2 experiments). Zero timepoint measurements for both MN and PN were from metaphase chromosomes. The specificity of both anti-pH3S10 antibodies was confirmed by the near-complete loss of labeling after treatment of nocodazole-arrested mitotic cells with 50 μM ZM447439 (not shown). Note that Nup133 starts to assemble on the main chromosomes (~4–6 min post AO28, also see Extended Data Fig. 5b) with readily detected H3S10 phosphorylation.
d, MKLP2 knockdown, disrupting the transport of Aurora B to the spindle midzone, fails to restore non-core (LBR) NE assembly to lagging chromosomes (arrows). Synchronization as in Fig. 1a. Left: Representative images of control or MKLP2-depleted RPE-1 cells. Middle: MN/PN FI ratio of LBR. For MN in MKLP2 depleted cells, only cells lacking detectable midzone Aurora B were quantified (mean with 95% CI, n=118, 105, from 3 experiments). NS: Not significant (P=0.8147), two-tailed Mann-Whitney test. Right: Western blot showing MKLP2 knockdown in RPE-1 cells (2 experiments, for gel source data, see Supplementary Fig. 1). Scale bars, 10 μm.
e, Scheme of the live-cell/ fixed cell imaging protocol. RFP-H2B-expressing RPE-1 cells were plated on gridded dishes to identify cells of interest. Cells were imaged at 2 min intervals during the experiments. Representative images of two live cells (red and blue boxes) upon (left) or after (right, prior to fixation) ZM447439 addition.
f, Consistent with prior studies17,43,44, Aurora B inhibition (ZM447439, 5 μM or 50 μM) rapidly (3 min) inactivates the kinase (mean with 95% CI, n=48, 17, 19, from 2 technical replicates). **** P < 0.0001, two-tailed Mann-Whitney test. Active Aurora B was assessed by the FI ratio of phospho-Aurora B (pT232) to total Aurora B in the midbody of telophase cells 3 min after ZM or DMSO addition. RPE-1 cells were synchronized as in Fig. 1a.
g, Ten-fold higher (50 μM) Aurora B inhibitor concentration gives similar results as obtained with 5 μM inhibitor treatment (Fig. 2a), confirming that early Aurora B inhibition (2 min before-2 min after AO) but not late (6–12 min post AO) partially restores non-core protein (Nup133) recruitment to lagging chromosomes. Note that non-core NE is only partially restored on a fraction of lagging chromosomes even with the high dose of inhibitor (early 50 μM ZM447439) even though this treatment led to a uniform loss of phospho-H3S10 on all chromosomes after anaphase entry (not shown). Therefore, the persistent NE assembly defect on lagging chromosomes after Aurora B inhibition cannot be explained by residual phospho-H3S10. Instead, the partial effect is explained by the effects of Aurora B inhibition on the local organization of microtubules (see Extended Data Fig. 8d). Shown is the MN/PN FI ratio of Nup133 at the indicated time intervals and drug doses (mean with 95% CI, n=33, 37, 37, 52, 37, left to right, from 2 experiments). Experiments performed as in Fig. 2a in RPE-1 cells. For simplicity, timepoints corresponding to ± 2 min post AO were grouped as “early” treatment, whereas timepoints corresponding to 6–12 min post AO were grouped as “late” treatment. *** P=0.0001 (DMSO and early 5 μM ZM), ***P=0.0003 (early 5 μM ZM and late 5 μM ZM), **** P < 0.0001, NS: not significant (P=0.494), two-tailed Mann-Whitney test.
Extended Data Figure 5. Rather than persistent condensins, the irreversible NE assembly defect of lagging chromosomes may be caused by enclosure in an NPC-deficient NE.
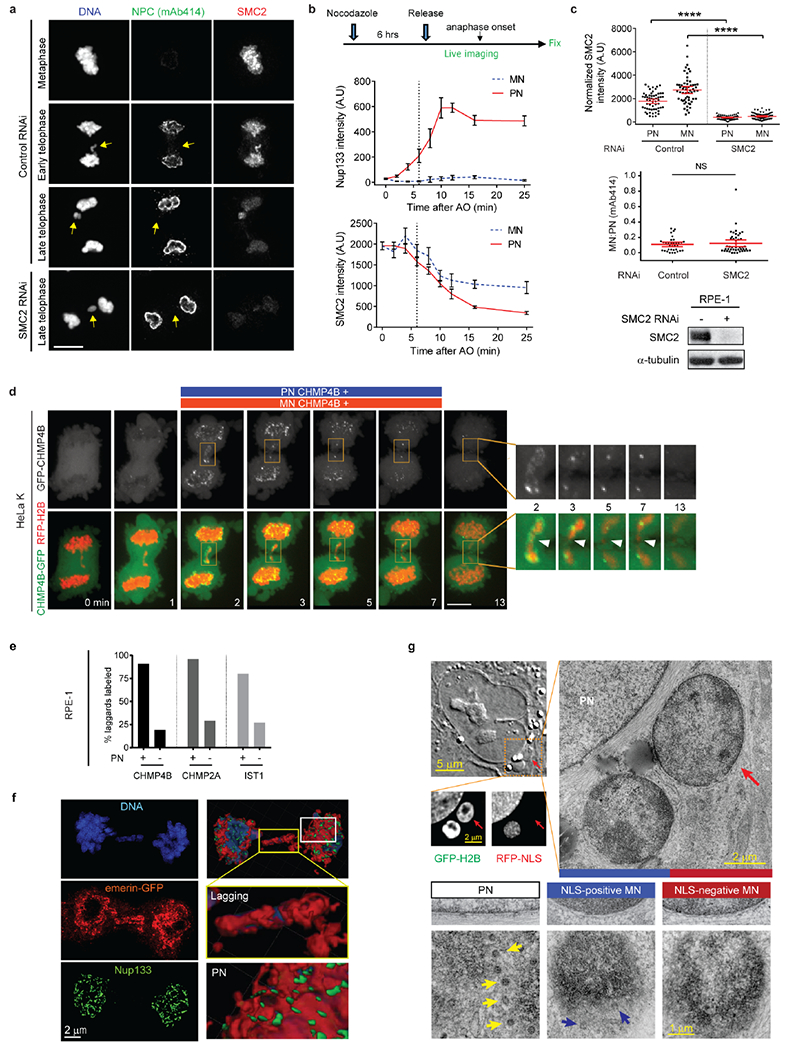
a-c, Loss of condensin (SMC2) is not required for non-core/NPC assembly. a, Representative images of control and SMC2-depleted RPE-1 cells showing that loss of SMC2 does not restore non-core assembly on lagging chromosomes (see b, c for quantification). Scale bar, 10 μm. b, Top: Experimental scheme of live-cell/ fixed cell imaging. Middle and bottom: FI of Nup133 and SMC2 on the main chromosome mass (PN) and lagging chromosomes (MN) at timepoints after AO in RPE-1 cells (mean with 95% CI, n=52, 37, 35, 44, 35, 36, 32, 46, 41 each timpoint for MN SMC2, from 3 experiments; n=52, 37, 47, 55, 40, 48, 38, 46, 41 each timepoint for PN SMC2, from 3 experiments; n=31, 28, 37, 22, 23, 25, 26, 28, 34 each timepoint for MN and PN Nup133, from 2 experiments). Zero timepoint measurements for both MN and PN were from metaphase chromosomes. Dashed lines indicate timepoints, demonstrating that Nup133 can assemble on the main chromosome mass when condensins are present at ~70% of their maximum levels in metaphase. Note that for MN (~10 min post AO), the decline in SMC2 levels is slower than on the PN, presumably because of a requirement for nuclear transport for complete chromosome decondensation. c, Top: SMC2 levels on PN and MN during telophase in control and SMC2-depleted cells (mean with 95% CI, n=58, 58, 75, 75, left to right, from 2 experiments). **** P < 0.0001, two-tailed Mann-Whitney test. Middle: MN/PN FI ratio of mAb414 to detect nucleoporins. For MN in SMC2 depleted cells, only cells with no detectable SMC2 were quantified for mAb414 (mean with 95% CI, n=35, 45, from 2 experiments). NS: Not significant (P=0.9251), two-tailed Mann-Whitney test. Bottom: Western blot showing SMC2 knockdown in RPE-1 cells (2 experiments, for gel source data, see Supplementary Fig. 1).
d, e, ESCRT-III recruitment to lagging chromosomes. d, Normal kinetics for the association and dissociation of ESCRT-III (CHMP4B-GFP) on lagging chromosomes during NE reformation. Lagging chromosomes were induced in HeLa K cells expressing CHMP4B-GFP (green) and RFP-H2B (red) by the nocodazole block-and-release protocol as in Fig. 1a and imaged at 1 min intervals on a confocal microscope (Supplementary Video 3). Left: A representative cell showing the CHMP4B residence time on lagging chromosome (referred as MN) as compared to the residence time on the PN. Right: Enlarged images of boxed regions showing lagging chromosome (white arrowheads). The duration of CHMP4B association with the PN or MN is shown by the colored bars above the time-lapse series. Similar results were obtained for chromosome bridges (not shown). Note that prior work on normal NE assembly suggests that the timely dissociation of CHMP4B is a marker for successful NE sealing and that delayed CHMP4B dissociation is a marker for defective NE sealing23. The normal ESCRT-III kinetics on lagging chromosomes or chromosome bridges therefore suggests significant NE membrane sealing on these structures (representative of 10 cells, from 6 experiments). ESCRT-III was not detected on newly formed micronuclei after mitotic exit (not shown). We note that ESCRT-III was previously reported to associate with interphase MN45, which we also observe, but only on disrupted MN (not shown). We also note that the previously described23 enrichment of ESCRT-III in the core domain of the main chromosome mass is less apparent after nocodazole release (shown here) than is observed when lagging chromosomes are generated by MPS1i treatment (see Extended Data Fig. 9b). Scale bars, 10 μm. e, ESCRT-III (CHMP4B, CHMP2A and IST1) is recruited to lagging chromosomes in telophase RPE-1 cells. Cells were labeled to detect the indicated proteins synchronized from nocodazole release as in Fig.1a. Graph showing percentage of lagging chromosomes that were positive for the indicated proteins in early telophase cell (when the main chromosome mass is positive for ESCRT III, PN+) and late telophase cell (when ESCRT III is no longer detected on the main chromosome mass, PN-) (n=44, 78, 45, 31, 67, 37, each category, from 2 experiments).
f, Near-continuous assembly of core membrane protein around the lagging chromosome. Images of 3D-SIM (left) and Imaris surface renderings (right) of an emerin-GFP-expressing RPE-1 cell. Enlarged images show lagging chromosomes (yellow box) and PN (white box): DNA (blue), emerin (red), Nup133 (green) (representative of 22 cells, from 2 technical replicates).
g, Correlative light and electron microscopy (CLEM) showing double-membranous NE (enlarged images in middle panels, cross sections) but reduced NPCs on intact (RFP-NLS positive) and newly disrupted MN (RPF-NLS negative, red arrows). An RPE-1 cell was fixed <20 min after spontaneous disruption of one of MN. Left, top: DIC and fluorescence images immediately after loss of RFP-NLS signal in one of the two MN present in the cell. Right, top: 70-nm thin section selected from the full series of the cell. Double-membranous NE is present on the PN as well as on the disrupted (red arrow) and intact MN. Middle-row panels depict NE at higher magnification. NPCs are prominent in the tangential view (bottom-row panels) of primary nucleus (yellow arrows) but not on NLS-positive MN (blue arrows). No NPC is present on the disrupted RPF-NLS negative MN. Representative of 4 cells with 3 intact and 4 newly disrupted MN, from 3 experiments (see details in the Methods).
Extended Data Figure 6. Microtubule disruption restores non-core NE assembly to lagging chromosomes only if it occurs early in anaphse.
a, Aurora B inhibition (5 μM ZM447439) reduces spindle microtubule bundling and microtubule mass. Left: Representative images of RPE-1 cells (n=20, from 2 technical replicates). Right: Linescan profiles of the FI for α-tubulin (red) along the white dashed lines (see the enlarged images from the boxed region). Scale bars, 10 μm.
b, c, Parallel effects of nocodazole and Aurora B inhibitor treatment (see Fig. 2a) on non-core NE assembly. Live-cell/ fixed cell imaging as in Fig. 2a showing that nocodazole-mediated microtubule depolymerization only allows non-core NE assembly if it occurs early in anaphase. Labeling to detect α-tubulin and LBR is in b; labeling to detect α-tubulin and Nup133 is in c. Left (b, c): Representative images of RPE-1 cells exposed to DMSO or nocodazole at the indicated times post AO (fixation and labeling of cells was performed 12 min after DMSO or nocodazole addition). Yellow arrows (b, c) indicate lagging chromosomes; White arrowhead (enlarged images in c) shows that even on a lagging chromosome where Nup133 (green) has largely been restored after nocodazole treatment, the region of this chromosome that remains in contact with residual microtubules (red) is depleted for Nup133. Right, (b, c): Experimental scheme is shown on the top (see Extended Data Fig. 4e) and quantification is shown on the bottom. “12+” includes cells that exposed to nocodazole 12–16 min post AO. (b) MN/PN FI ratio for LBR (mean with 95% CI, n=64, 62, 54, 44, 63, 69 each timepoint, from 3 experiments); (c) the fraction of the lagging chromosome circumference with Nup133 (mean with 95% CI, n=33, 29, 22, 21, 24, 64 each timepoint, from 2 experiments). **** P < 0.0001, two-tailed Mann-Whitney test. Scale bars, 10 μm.
d, Independence for the formation of the non-core gap from the normal spatial distribution of Aurora B. Images showing that a non-core protein (Nup133) is excluded from the central spindle region (white arrowheads) on the chromosome mass in HeLa K cells expressing Aurora B-GFP (white, in merged images, representative of 30 cells, from 2 technical replicates). Synchronization as in Extended Data Fig. 1e. Nup133 assembly occurred normally on the periphery of the main chromosome mass, away from the spindle, whether Aurora B localized to the central spindle or was forced to remain near the chromosome mass (MKLP2 RNAi)30. Scale bars, 10 μm.
Extended Data Figure 7. KIF4A knockdown partially restores non-core NE assembly to lagging chromosomes even when they have maximal (metaphase) levels of H3S10 phosphorylation.
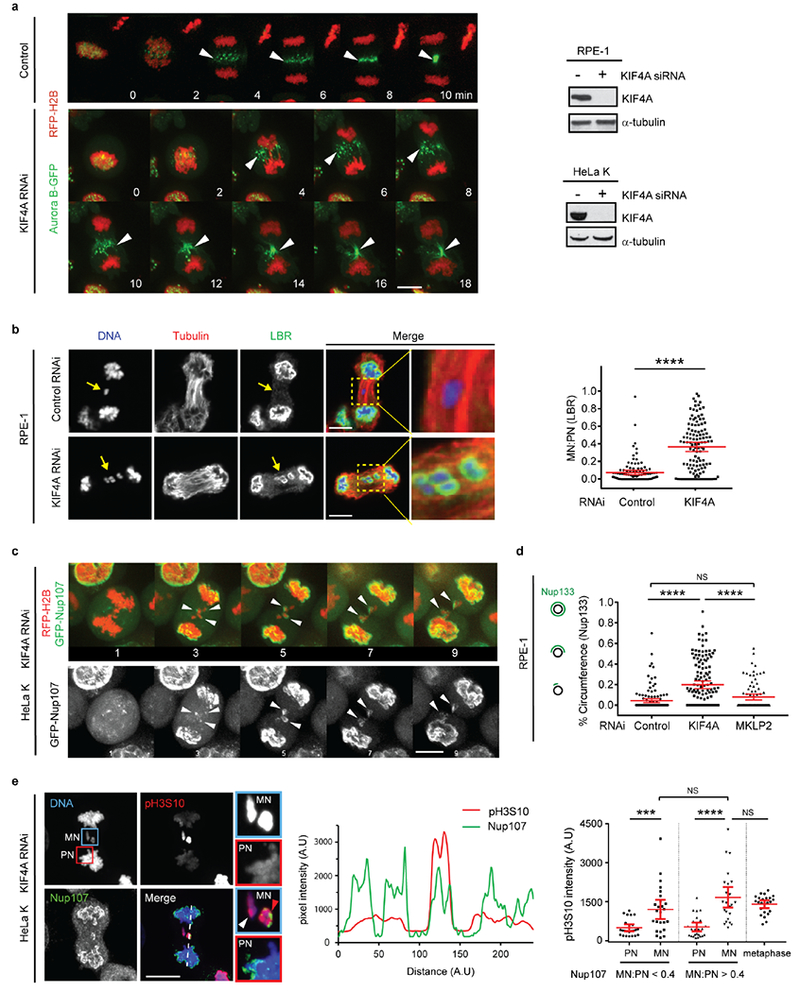
a, Consistent with previous studies26, KIF4A knockdown largely preserves the redistribution of Aurora B from centromeres to the central spindle (arrowheads) early in anaphase. Synchronization as in Fig. 1a. Left: Images from time-lapse series are shown from control (representative of 29 cells, from 2 experiments) or KIF4A-depleted (representative of 27 cells, from 3 experiments) HeLa K cells expressing Aurora B-GFP (green) and RFP-H2B (red). Time is shown in min (t=0, AO). Right: Western blots showing depletion of KIF4A protein by siRNA in RPE-1 (top, 3 experiments) and HeLa K (bottom, 3 experiments) cells (also related to Fig. 3b, c, for gel source data, see Supplementary Fig. 1). Scale bar, 10 μm.
b, Restoration of LBR to some lagging chromosomes after KIF4A depletion (RPE-1 cells). Synchronization as in Fig. 1a. Left: Representative images: DNA (blue), α-tubulin (red) and LBR (green). Right: MN/PN LBR FI ratio (mean with 95% CI, n=105, 119, from 3 experiments). **** P < 0.0001, two-tailed Mann-Whitney test. Scale bars, 10 μm. Note that the restoration of LBR is often continuous around MN with partially-restored MN/PN FI ratio (see also Fig. 2a after Aurora B inhibition), whereas the NPC assembly is commonly restored discontinuously after KIF4A depletion (see Fig. 3b, c) or Aurora B inhibition (see Extended Data Fig. 8d). This is consistent with different localization patterns of LBR and NPC in the core domain on the main chromosome mass (Extended Data Fig. 1b, c, e), which may arise from their different mobilities within the NE.
c, Live-cell imaging confirming the partial restoration of an NPC protein (Nup107) to lagging chromosomes in HeLa K cells expressing GFP-Nup107 (green) and RFP-H2B (red) after KIF4A knockdown. Synchronization as in Fig. 1a. Images from a confocal time-lapse series are shown with arrows indicating GFP-Nup107 protein recruited to lagging chromosomes (representative of 6 cells, from 3 experiments). Time is shown in min. Scale bar, 10 μm.
d, Unlike KIF4A knockdown, disruption of Aurora B spindle midzone localization by MKPL2 knockdown fails to restore the recruitment of Nup133 to lagging chromosomes. Synchronization as in Fig. 1a. Graph showing quantification of the results (mean with 95% CI, n=131, 136, 85, from 2 experiments). **** P < 0.0001, NS: Not significant (P=0.0616), two-tailed Mann-Whitney test.
e, H3S10 phosphorylation does not block NPC/non-core NE assembly. Left: Representative images of KIF4A-depleted HeLa K cells showing restoration of non-core (Nup107) assembly onto lagging chromosomes with high level of H3S10 phosphorylation (red arrowhead, enlarged images of the blue boxed region). Note that the level of phospho-H3S10 is similar between the lagging chromosomes with (red arrowhead) or without (white arrowhead) Nup107 recruitment. To illustrate the relative difference in pH3S10 levels between PN and MN, pH3S10 has been scaled differently in the pH3S10 channel and the merged channel. Cells were synchronized as in Extended Data Fig. 1e. Scale bar, 10 μm. Middle: Linescan profile of the indicated proteins along the dashed line in the merged image on the left. Right: FI of phospho-H3S10 on MN and the corresponding PN (mean with 95% CI, n=24, 25, 25, 25, 24, from 2 technical replicates). In MN that have recruited Nup107 (MN/PN FI ratio >0.4), the H3S10 phosphorylation level is comparable to metaphase chromosomes. *** P =0.0009, **** P < 0.0001, NS: Not significant (P=0.0709, P=0.6699, left to right), two-tailed Mann-Whitney test.
Extended Data Figure 8. Partitioning of the NE into core and non-core subdomains depends on the local organization of spindle microtubules.
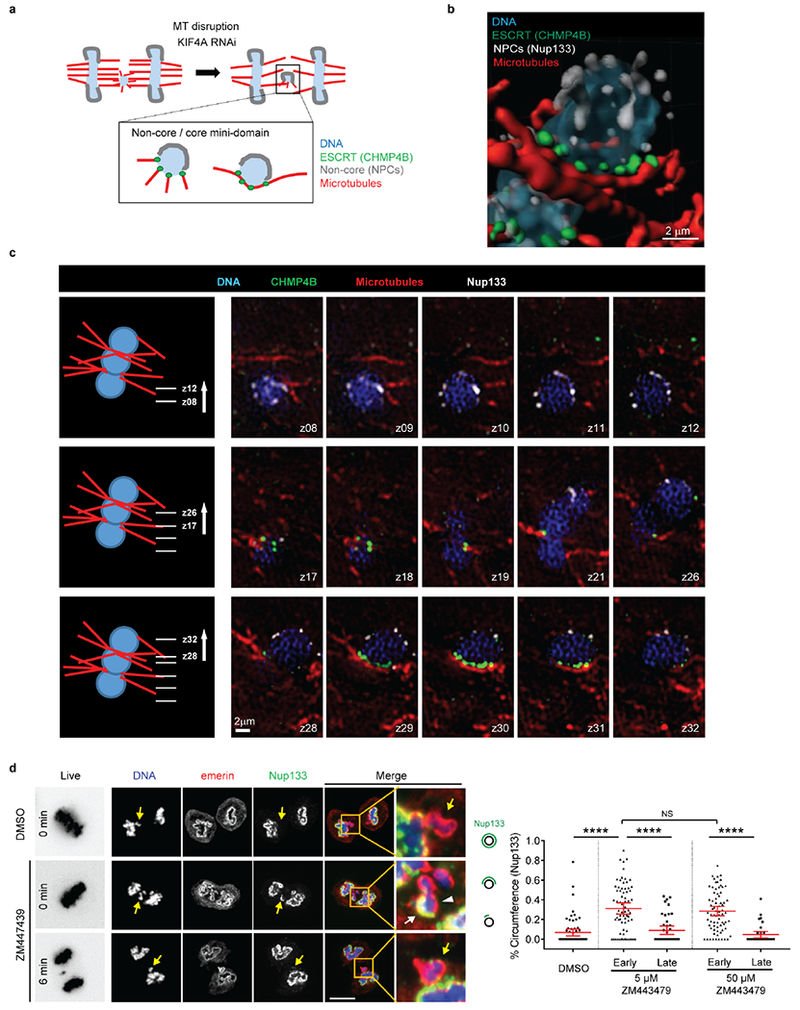
a-c. 3D-SIM showing exclusion of non-core NE assembly from the region of the lagging chromosome adjacent to CHIMP4B marked microtubules (MTs). a. Scheme summarizing the SIM results after KIF4A knockdown. The ESCRT-III complex is recruited to small membrane holes at the sites where spindle microtubules intersect the reforming NE where it is thought to be required for normal NE sealing23. Therefore, we used ESCRT-III (CHMP4B) labeling to identify the NE region on the lagging chromosome that is intersected by microtubules for the SIM experiments.
b, Imaris surface three-dimensional renderings from SIM images (from c, below) showing recruitment of Nup133 (white) to the region of a lagging chromosome depleted of CHMP4B (green) and microtubules (red) from a KIF4A-depleted HeLa K cell (representative of 4 lagging chromosomes, from 2 experiments). c, SIM images showing that similar to what occurs on the main chromosome masses during normal NE assembly23, NPCs (Nup133) are recruited to regions of the lagging chromosome lacking CHMP4-decorated microtubules. Left: A cartoon depicting the entire lagging chromosome visualized from a KIF4A-depleted anaphase/telophase cell (microtubules in red, DNA in blue, z focal planes as indicated). Right: Serial sections from a SIM z-stack of an anaphase/telophase HeLa K cell expressing CHMP4B-GFP (green) labeled for microtubules (red), Nup133 (white) and DNA (blue) after KIF4A depletion. These results are consistent with the idea that microtubules inhibit non-core NE assembly.
d, KIF4A knockdown (Fig. 3b, c, Extended Data Fig. 7b–d) and early anaphase Aurora B inhibition have a similar effect on NPCs/non-core NE assembly to lagging chromosomes. Experiments performed as in Fig. 2a in RPE-1 cells. Left panel images: left column: Representative timelapse images of cells upon ZM447439 or DMSO addition (live, 0 or 6 min post AO); right five columns: Representative images of the same cells after fixation and labeling (12 min post treatment). For the cell exposed to ZM447439 at anaphase onset (middle row), one of the two lagging chromosomes exhibits a small-scale separation of core (white arrowhead, emerin) and non-core (white arrow, Nup133) NE domains. Scale bar, 10 μm. Right: Quantification of the results (mean with 95% CI, n=64, 72, 43, 72, 34, from 2 experiments). **** P < 0.0001, NS: Not significant (P=0.6932), two-tailed Mann-Whitney test.
Extended Data Figure 9. Characterization of peripherally localized missegregated chromosomes and the resulting MN: microtubule proximity, nuclear import, and chromosome number.
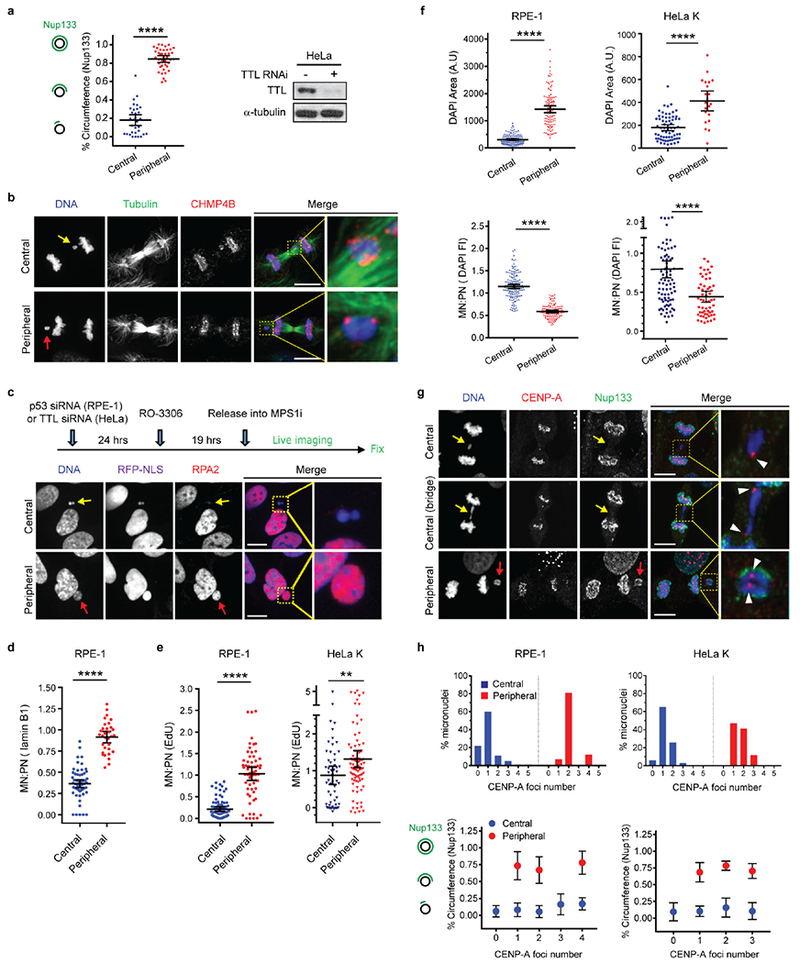
a, Restored assembly of Nup133 on micronuclei derived from peripheral chromosomes as compared to central spindle lagging chromosomes in HeLa K cells. Left: Quantification of the fraction of the lagging chromosome circumference with Nup133 (mean with 95% CI, n=32, 40, from 2 experiments). **** P < 0.0001, two-tailed Mann-Whitney test. Right: Western blots showing depletion of TTL protein by siRNA in HeLa K cells (3 experiments, for gel source data, see Supplementary Fig. 1).
b, Fewer microtubules associated with peripheral missegregated chromosomes (red arrow) as compared to MN from central missegregated chromosomes (yellow arrow). Synchronization as in Fig. 4a. Representative images of RPE-1 cells after glutaraldehyde fixation (representative of 30 for central or peripheral missegregated chromosome, from 2 technical replicates). Scale bars, 10 μm. Note the sparse microtubule density near the peripheral chromosome (bottom images) that, in 30 cells, is never associated with more than 3 CHMP4B foci.
c-e, Normal nuclear import and DNA replication in MN from peripherally missegregated chromosomes (red arrows) as compared to MN from central missegregated chromosomes (yellow arrows). c, Top: Scheme of the experiment. RPE-1 cells were treated with p53 siRNA to facilitate cell-cycle progression. After mitotic exit, micronucleated cells were imaged at 20 min intervals. Bottom: Images of fixed and labeled RPE-1 cells, 4h post the RO release at the end of the live-cell experiment. Scale bars, 10 μm. See Fig. 4d for quantification of this experiment. d, MN/PN FI ratio for lamin B1 in RPE-1 cells, 4h post the RO release (mean with 95% CI, n=60, 32, from 2 experiments). **** P < 0.0001, two-tailed Mann-Whitney test. e, Restoration of DNA replication (EdU incorporation, shown as the MN/PN FI ratio) in MN from peripheral chromosomes in RPE-1 (left) and HeLa K (right) cells, 16~18h post the RO release (mean with 95% CI, n=81, 60, from 5 experiments for RPE-1; n=55, 86, from 2 experiments for HeLa K). Note that in RPE-1 cells, consistent with prior results46, high dose of MPS1 inhibition causes a fraction of cells to delay in cell cycle progression. Therefore, we restricted the analysis to cells that progressed into the S-phase of the cell cycle as evidenced by EdU labeling of the primary nucleus. ** P < 0.0067, **** P < 0.0001, two-tailed Mann-Whitney test.
f, MN derived from peripheral chromosomes were larger (DAPI area, top) and more decondensed (the density of DAPI FI, bottom) than MN from central chromosomes. Shown are data from RPE-1 (left) and HeLa K (right) cells (mean with 95% CI, n=145, 112, from 3 experiments for DAPI area for RPE-1, n=145, 112, from 3 experiments for DAPI FI for RPE-1; n=66, 21, from 2 experiments for DAPI area for HeLa K, n=88, 58, from 3 experiments for DAPI FI for HeLa K). **** P < 0.0001, two-tailed Welch’s t test for DAPI FI, two-tailed Mann-Whitney test for DAPI area. Note: The extensive decondensation of MN from peripheral missegregated chromosomes may be a consequence of their initiation of nuclear import slightly earlier than the assembling main daughter nuclei (Fig. 4c) as well as their difference in the ratio of surface area to volume.
g, h, Micronuclear chromosome number does not influence non-core NE assembly. Comparison of chromosome number in MN from central chromosomes and peripheral chromosomes. g, Confocal images of RPE-1 cells labeled for CENP-A (a marker for centromere, arrowheads), Nup133 and DNA. Yellow arrows; central chromosomes; red arrows: peripheral chromosomes. Synchronization as in Fig. 4a. Scale bars, 10 μm. See h for quantification. h, Top: Distribution of chromosome number (CENP-A foci) in central (blue) and peripheral (red) MN. Data from RPE-1 (left) and HeLa K (right) cells are shown. Bottom: The fraction of the lagging chromosome circumference with Nup133 is shown for MN with the indicated chromosome number in RPE-1 (left) and HeLa K (right) cells (mean ± SD, n=40, 111, 20, 9, 3 each category for central chromosomes, n=0, 6, 71, 0, 11 for peripheral chromosomes, from 2 experiments for RPE-1; mean ± SD, n=2, 35, 14, 2, 0 each category for central chromosomes, n=0, 8, 6, 2, 0 for peripheral chromosomes, from 2 experiments for HeLa K). Although MN from peripheral chromosomes generally contain a higher number of chromosomes than MN from central chromosomes, non-core assembly is inhibited on MN from central chromosomes irrespective of their chromosome number. Furthermore, non-core NE assembly is significantly restored on MN from peripheral chromosomes, again irrespective of chromosome number (consistent with Fig. 4b, c).
Extended Data Figure 10. Peripheral localization of lagging chromosomes reduces MN NE disruption and DNA damage; actin-dependent peripheral MN disruption in RPE-1 cells.
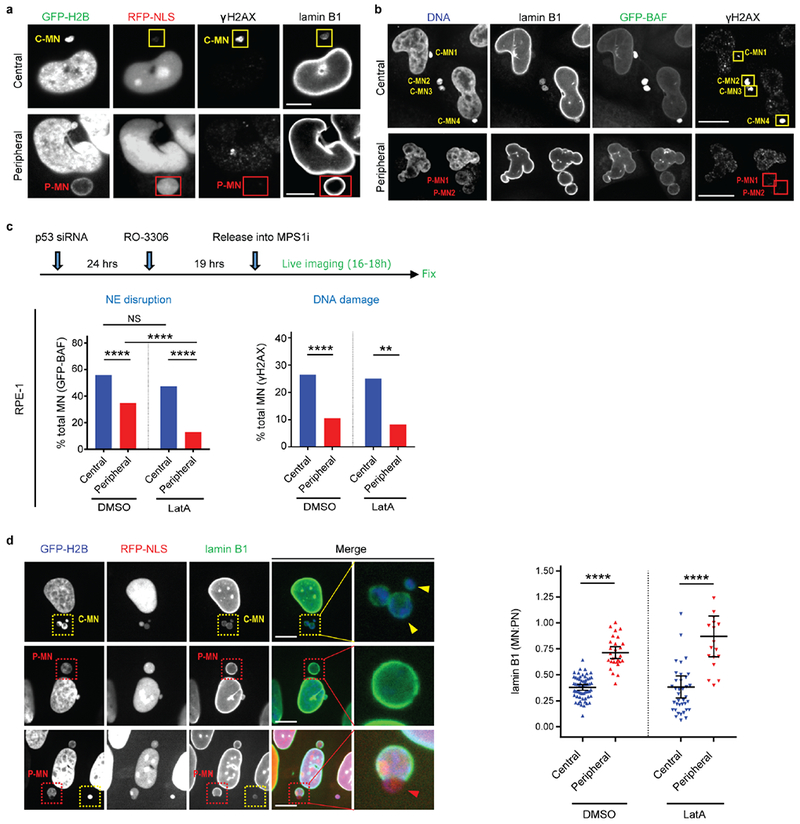
a, Peripheral localization of lagging chromosomes reduces MN disruption and DNA damage. Representative images of HeLa K cells (C-MN from central chromosomes; P-MN from peripheral chromosomes). See Fig. 4e for quantification of the results. Scale bars, 5 μm.
b, Detection of MN disruption by GFP-BAF accumulation yields similar results as in panel a for NE disruption detection by the loss of RFP-NLS. Representative images from a live-cell/ fixed cell imaging experiment (as in Fig. 4e) from HeLa K cells expressing GFP-BAF. Central MN (C-MN) show NE disruption detected by hyper-accumulation of GFP-BAF (top) whereas peripheral MN (P-MN) show infrequent disruption (bottom). NE disruption of central MN is accompanied by the acquisition of DNA damage (γH2AX, see Fig. 4e). At the end of 16–18h live imaging, cells were fixed and stained for γH2AX and lamin B1. See Fig. 4e for quantification of the results. Scale bars, 5 μm.
c, In RPE-1 cells, peripheral MN undergo actin-dependent NE disruption. Top: Experimental scheme. Chromosome missegregation was induced in RPE-1 cells expressing RFP-H2B and GFP-BAF (or GFP-H2B and RFP-NLS) as in Extended Data Fig. 9c. ~1h post mitotic exit, cells were treated with DMSO or a low dose (150 nM) of the actin assembly inhibitor latrunculin A (LatA) and imaged for 16–18h. See discussion below for the rationale. Bottom: Shown is the percentage of peripheral or central MN that underwent NE disruption (hyper-accumulation of GFP-BAF, from 4 experiments for both DMSO and LatA) or that displayed DNA damage (FI of γH2AX in MN > 3 SD of the background γH2AX in PN, from 6 experiments for DMSO and 3 experiments for LatA). NS: Not significant (P=0.0948), ** P=0.0044, **** P < 0.0001, two-tailed Fisher’s exact test [For NE disruption: n=258 for central MN (DMSO), n=176 for peripheral MN (DMSO), n=173 for central MN (LatA), n=125 for peripheral MN (LatA); for DNA damage: n=306 for central MN (DMSO), n=182 for peripheral MN (DMSO), n=128 for central MN (LatA), n=73 for peripheral MN (LatA)].
d, Peripheral MN develop discontinuities in the lamin B1 nuclear rim. Left: Representative images of RPE-1 cells ~18h post mitotic exit, as in Extended Data Fig. 9c. Yellow boxes indicate C-MN (yellow arrowheads) that have reduced levels of lamin B1. Right: Quantification of the results (mean with 95% CI, n=61, 28, 38, 18, left to right, from 2 experiments). **** P < 0.0001, two-tailed Welch’s t test for DMSO, two-tailed Mann-Whitney test for LatA. Red boxes indicate peripheral MN, one of which displays a prominent lamin B1 rim discontinuity. Red arrowhead in the enlarged image shows an NE herniation on a peripheral MN. Scale bars, 10 μm.
Discussion: Consistent with prior work, MN from lagging chromosomes undergo spontaneous disruption in a manner that is independent of actin13. By contrast, it was previously shown that the transient disruption of primary nuclei, as occurs during confined cell migration47, is mediated by actomyosin contractile forces13. In our experiments, we confirm that MN from central lagging chromosomes undergo disruption in a manner that is independent from actin (above). However, we noted a difference between RPE-1 and HeLa K cells in the behavior of MN from peripheral chromosomes. Although NE assembly appeared to be significantly restored in both HeLa K and RPE-1 cells, peripheral MN in RPE-1 cells underwent significant residual disruption (note that there is nevertheless a statistically significant reduction of the NE disruption frequency when comparing P-MN and C-MN in RPE-1 cells). We hypothesized that in the highly motile RPE-1 cells, large peripheral MN might more likely undergo actin-dependent NE breakage, essentially becoming more similar to the transient NE disruption of primary nuclei13,47. The above data (c) after Lat-A treatment in RPE-1 cells confirm that peripheral MN do undergo actin-dependent breakage. Furthermore, we show that peripheral MN in RPE-1 cells have lamin B1 gaps (d, red arrowhead)6. One possible mechanism that could generate these gaps could be residual contact between peripheral missegregated chromosomes with astral microtubules (see Extended Data Fig. 9b). Additionally, peripheral MN are more decondensed, and have larger NE surface area which may dilute lamins. The increased breakage of peripheral MN in RPE-1 cells relative to HeLa K cells may be due to higher contractile forces in RPE-1 cells.
Supplementary Material
Acknowledgements
We thank I. Cheeseman, T. Rapoport, N. Umbreit, T. Walther, and K. Xie for advice or comments on the manuscript; E. Jackson and A. Spektor for preliminary experiments; J. Ellenberg, D. Gerlich, E. Hatch, M. Hetzer, A. Hyman, T. Kuroda, and L. Shao for reagents; J. Waters and T. Lambert of the Nikon Imaging Center at Harvard Medical School, H. Elliott and C. Yapp from the Image and Data Analysis Core at Harvard Medical School for advice. We acknowledge the use of the Wadsworth Center’s Electron Microscopy Core Facility. A. K. is supported by NIH GM059363. D.P. is a HHMI investigator and is supported by R37 GM61345.
Footnotes
The authors declare no competing interests.
References
- 1.Ungricht R & Kutay U Mechanisms and functions of nuclear envelope remodelling. Nature Reviews Molecular Cell Biology 18, 229–245, doi: 10.1038/nrm.2016.153 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Zhang C-Z et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184, doi: 10.1038/nature14493 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephens PJ et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 144, 27–40, doi: 10.1016/j.cell.2010.11.055 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Notta F et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 538, 378–382, doi: 10.1038/nature19823 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ly P & Cleveland DW Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol 27, 917–930, doi: 10.1016/j.tcb.2017.08.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hatch Emily M., Fischer Andrew H., Deerinck Thomas J. & Hetzer Martin W. Catastrophic Nuclear Envelope Collapse in Cancer Cell Micronuclei. Cell 154, 47–60, doi: 10.1016/j.cell.2013.06.007 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umbreit NT & Pellman D Cancer biology: Genome jail-break triggers lockdown. Nature, doi: 10.1038/nature24146 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Haraguchi T et al. Live cell imaging and electron microscopy reveal dynamic processes of BAF-directed nuclear envelope assembly. Journal of Cell Science 121, 2540–2554, doi: 10.1242/jcs.033597 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Dechat T et al. LAP2alpha and BAF transiently localize to telomeres and specific regions on chromatin during nuclear assembly. J Cell Sci 117, 6117–6128, doi: 10.1242/jcs.01529 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Otsuka S et al. Nuclear pore assembly proceeds by an inside-out extrusion of the nuclear envelope. Elife 5, doi: 10.7554/eLife.19071 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Castro IJ, Gil RS, Ligammari L, Di Giacinto ML & Vagnarelli P CDK1 and PLK1 coordinate the disassembly and reassembly of the nuclear envelope in vertebrate mitosis. Oncotarget 9, 7763–7773, doi: 10.18632/oncotarget.23666 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toledo LI et al. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103, doi: 10.1016/j.cell.2013.10.043 (2013). [DOI] [PubMed] [Google Scholar]
- 13.Hatch EM & Hetzer MW Nuclear envelope rupture is induced by actin-based nucleus confinement. J Cell Biol 215, 27–36, doi: 10.1083/jcb.201603053 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crasta K et al. DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–58, doi: 10.1038/nature10802 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Afonso O et al. Feedback control of chromosome separation by a midzone Aurora B gradient. Science 345, 332–336, doi: 10.1126/science.1251121 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karg T, Warecki B & Sullivan W Aurora B-mediated localized delays in nuclear envelope formation facilitate inclusion of late-segregating chromosome fragments. Mol Biol Cell 26, 2227–2241, doi: 10.1091/mbc.E15-01-0026 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuller BG et al. Midzone activation of aurora B in anaphase produces an intracellular phosphorylation gradient. Nature 453, 1132–1136, doi: 10.1038/nature06923 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramadan K et al. Cdc48/p97 promotes reformation of the nucleus by extracting the kinase Aurora B from chromatin. Nature 450, 1258–1262, doi: 10.1038/nature06388 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Vagnarelli P et al. Repo-Man coordinates chromosomal reorganization with nuclear envelope reassembly during mitotic exit. Dev Cell 21, 328–342, doi: 10.1016/j.devcel.2011.06.020 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Obara Y, Chai LS, Weinfeld H & Sandberg AA Synchronization of events in fused interphase-metaphase binucleate cells: progression of the telophase-like nucleus. J Natl Cancer Inst 53, 247–259 (1974). [DOI] [PubMed] [Google Scholar]
- 21.Walther TC et al. The conserved Nup107–160 complex is critical for nuclear pore complex assembly. Cell 113, 195–206 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Xue John Z., Woo Eileen M., Postow L, Chait Brian T. & Funabiki H Chromatin-Bound Xenopus Dppa2 Shapes the Nucleus by Locally Inhibiting Microtubule Assembly. Developmental Cell 27, 47–59, doi: 10.1016/j.devcel.2013.08.002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vietri M et al. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 522, 231–235, doi: 10.1038/nature14408 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Hochegger H, Hegarat N & Pereira-Leal JB Aurora at the pole and equator: overlapping functions of Aurora kinases in the mitotic spindle. Open Biol 3, 120185, doi: 10.1098/rsob.120185 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu L, Ladinsky MS & Kirchhausen T Formation of the postmitotic nuclear envelope from extended ER cisternae precedes nuclear pore assembly. The Journal of Cell Biology 194, 425–440, doi: 10.1083/jcb.201012063 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurasawa Y, Earnshaw WC, Mochizuki Y, Dohmae N & Todokoro K Essential roles of KIF4 and its binding partner PRC1 in organized central spindle midzone formation. EMBO J 23, 3237–3248, doi: 10.1038/sj.emboj.7600347 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barisic M et al. Mitosis. Microtubule detyrosination guides chromosomes during mitosis. Science 348, 799–803, doi: 10.1126/science.aaa5175 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otsuka S et al. Postmitotic nuclear pore assembly proceeds by radial dilation of small membrane openings. Nature Structural & Molecular Biology 25, 21–28, doi: 10.1038/s41594-017-0001-9 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Poser I et al. BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nat Methods 5, 409–415, doi: 10.1038/nmeth.1199 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gruneberg U, Neef R, Honda R, Nigg EA & Barr FA Relocation of Aurora B from centromeres to the central spindle at the metaphase to anaphase transition requires MKlp2. The Journal of Cell Biology 166, 167–172, doi: 10.1083/jcb.200403084 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soto M et al. p53 Prohibits Propagation of Chromosome Segregation Errors that Produce Structural Aneuploidies. Cell Rep 19, 2423–2431, doi: 10.1016/j.celrep.2017.05.055 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Rieder CL & Cassels G Correlative light and electron microscopy of mitotic cells in monolayer cultures. Methods Cell Biol 61, 297–315 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Hiraoka Y, Sedat JW & Agard DA Determination of three-dimensional imaging properties of a light microscope system. Partial confocal behavior in epifluorescence microscopy. Biophys J 57, 325–333, doi: 10.1016/S0006-3495(90)82534-0 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gustafsson MG et al. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J 94, 4957–4970, doi: 10.1529/biophysj.107.120345 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references for Extended Data Legends
- 35.Belgareh N et al. An evolutionarily conserved NPC subcomplex, which redistributes in part to kinetochores in mammalian cells. J Cell Biol 154, 1147–1160, doi: 10.1083/jcb.200101081 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maeshima K et al. Cell-cycle-dependent dynamics of nuclear pores: pore-free islands and lamins. J Cell Sci 119, 4442–4451, doi: 10.1242/jcs.03207 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Clever M, Funakoshi T, Mimura Y, Takagi M & Imamoto N The nucleoporin ELYS/Mel28 regulates nuclear envelope subdomain formation in HeLa cells. Nucleus 3, 187–199, doi: 10.4161/nucl.19595 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mimura Y, Takagi M, Clever M & Imamoto N ELYS regulates the localization of LBR by modulating its phosphorylation state. J Cell Sci 129, 4200–4212, doi: 10.1242/jcs.190678 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hudson DF, Vagnarelli P, Gassmann R & Earnshaw WC Condensin is required for nonhistone protein assembly and structural integrity of vertebrate mitotic chromosomes. Dev Cell 5, 323–336 (2003). [DOI] [PubMed] [Google Scholar]
- 40.Maiato H, Afonso O & Matos I A chromosome separation checkpoint: A midzone Aurora B gradient mediates a chromosome separation checkpoint that regulates the anaphase-telophase transition. Bioessays 37, 257–266, doi: 10.1002/bies.201400140 (2015). [DOI] [PubMed] [Google Scholar]
- 41.Maciejowski J, Li Y, Bosco N, Campbell PJ & de Lange T Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 163, 1641–1654, doi: 10.1016/j.cell.2015.11.054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Denais CM et al. Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358, doi: 10.1126/science.aad7297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Steigemann P et al. Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 136, 473–484, doi: 10.1016/j.cell.2008.12.020 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Uehara R et al. Aurora B and Kif2A control microtubule length for assembly of a functional central spindle during anaphase. The Journal of Cell Biology 202, 623–636, doi: 10.1083/jcb.201302123 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sagona AP, Nezis IP & Stenmark H Association of CHMP4B and autophagy with micronuclei: implications for cataract formation. Biomed Res Int 2014, 974393, doi: 10.1155/2014/974393 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Santaguida S et al. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev Cell 41, 638–651 e635, doi: 10.1016/j.devcel.2017.05.022 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shah P, Wolf K & Lammerding J Bursting the Bubble - Nuclear Envelope Rupture as a Path to Genomic Instability? Trends Cell Biol 27, 546–555, doi: 10.1016/j.tcb.2017.02.008 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the paper and its Supplementary Information files. Source data for all graphs (Figs. 1c, 2a, 2b, 3a, 3c, 4b, 4d, 4e; Extended Data Figs. 1d, 1f, 2b, 2c, 2d, 2f, 2g, 3a, 3b, 3c, 3e, 4c, 4d, 4f, 4g, 5b, 5c, 5e, 6a, 6b, 6c, 7b, 7d, 7e, 8d, 9a, 9d, 9e, 9f, 9h, 10c, 10d) are provided with the online version of the paper. All other datasets generated and/or analysed in the current study are available from the corresponding author upon reasonable request. Supplementary Figure 1 contains scanned complete images of western blots.