

Abstract
Background
Actinic keratoses are a skin disease caused by long‐term sun exposure, and their lesions have the potential to develop into squamous cell carcinoma. Treatments for actinic keratoses are sought for cosmetic reasons, for the relief of associated symptoms, or for the prevention of skin cancer development. Detectable lesions are often associated with alteration of the surrounding skin (field) where subclinical lesions might be present. The interventions available for the treatment of actinic keratoses include individual lesion‐based (e.g. cryotherapy) or field‐directed (e.g. topical) treatments. These might vary in terms of efficacy, safety, and cosmetic outcomes.
Objectives
To assess the effects of topical, oral, mechanical, and chemical interventions for actinic keratosis.
Search methods
We searched the following databases up to March 2011: the Cochrane Skin Group Specialised Register, CENTRAL in The Cochrane Library, MEDLINE (from 2005), EMBASE (from 2010), and LILACS (from 1982). We also searched trials registers, conference proceedings, and grey literature sources.
Selection criteria
Randomised controlled trials (RCTs) comparing the treatment of actinic keratoses with either placebo, vehicle, or another active therapy.
Data collection and analysis
At least two authors independently abstracted data, which included adverse events, and assessed the quality of evidence. We performed meta‐analysis to calculate a weighted treatment effect across trials, and we expressed the results as risk ratios (RR) and 95% confidence intervals (CI) for dichotomous outcomes (e.g. participant complete clearance rates), and mean difference (MD) and 95% CI for continuous outcomes (e.g. mean reduction in lesion counts).
Main results
We included 83 RCTs in this review, with a total of 10,036 participants. The RCTs covered 18 topical treatments, 1 oral treatment, 2 mechanical interventions, and 3 chemical interventions, including photodynamic therapy (PDT). Most of the studies lacked descriptions of some methodological details, such as the generation of the randomisation sequence or allocation concealment, and half of the studies had a high risk of reporting bias. Study comparison was difficult because of the multiple parameters used to report efficacy and safety outcomes, as well as statistical limitations. We found no data on the possible reduction of squamous cell carcinoma.
The primary outcome 'participant complete clearance' significantly favoured four field‐directed treatments compared to vehicle or placebo: 3% diclofenac in 2.5% hyaluronic acid (RR 2.46, 95% CI 1.66 to 3.66; 3 studies with 420 participants), 0.5% 5‐fluorouracil (RR 8.86, 95% CI: 3.67 to 21.44; 3 studies with 522 participants), 5% imiquimod (RR 7.70, 95% CI 4.63 to 12.79; 9 studies with1871 participants), and 0.025% to 0.05% ingenol mebutate (RR 4.50, 95% CI 2.61 to 7.74; 2 studies with 456 participants).
It also significantly favoured the treatment of individual lesions with photodynamic therapy (PDT) compared to placebo‐PDT with the following photosensitisers: aminolevulinic acid (ALA) (blue light: RR 6.22, 95% CI 2.88 to 13.43; 1 study with 243 participants, aminolevulinic acid (ALA) (red light: RR 5.94, 95% CI 3.35 to 10.54; 3 studies with 422 participants), and methyl aminolevulinate (MAL) (red light: RR 4.46, 95% CI 3.17 to 6.28; 5 studies with 482 participants). ALA‐PDT was also significantly favoured compared to cryotherapy (RR 1.31, 95% CI 1.05 to 1.64).
The corresponding comparative risks in terms of number of participants completely cleared per 1000 were as follows: 313 with 3% diclofenac compared to 127 with 2.5% hyaluronic acid; 136 with 0.5% 5‐fluorouracil compared to 15 with placebo; 371 with 5% imiquimod compared to 48 with placebo; 331 with ingenol mebutate compared to 73 with vehicle; 527 to 656 with ALA/MAL‐PDT treatment compared to 89 to 147 for placebo‐PDT; and 580 with ALA‐PDT compared to 443 with cryotherapy.
5% 5‐fluorouracil efficacy was not compared to placebo, but it was comparable to 5% imiquimod (RR 1.85, 95% Cl 0.41 to 8.33).
A significant number of participants withdrew because of adverse events with 144 participants affected out of 1000 taking 3% diclofenac in 2.5% hyaluronic acid, compared to 40 participants affected out of 1000 taking 2.5% hyaluronic acid alone, and 56 participants affected out of 1000 taking 5% imiquimod compared to 21 participants affected out of 1000 taking placebo.
Based on investigator and participant evaluation, imiquimod treatment and photodynamic therapy resulted in better cosmetic outcomes than cryotherapy and 5‐fluorouracil.
Authors' conclusions
For individual lesions, photodynamic therapy appears more effective and has a better cosmetic outcome than cryotherapy. For field‐directed treatments, diclofenac, 5‐fluorouracil, imiquimod, and ingenol mebutate had similar efficacy, but their associated adverse events and cosmetic outcomes are different. More direct comparisons between these treatments are needed to determine the best therapeutic approach.
Plain language summary
Interventions for actinic keratoses
Actinic keratoses are a skin disease caused by long‐term sun exposure. Damaged skin shows small, red, rough, scaly, flat spots called actinic keratoses or lesions, which feel like patches of dry skin. Symptoms such as bleeding and pain can be associated with actinic keratoses. Moreover, actinic keratoses have the potential to develop into skin cancer if left untreated. The reasons for treatment may include cosmetic appearance, relief of symptoms, or prevention of skin cancer. Treatment can be directed either at individual lesions or to larger areas of the skin where several visible and less visible lesions occur (field‐directed treatment).
This systematic review included results from 83 randomised controlled clinical trials evaluating 24 treatments, with a total of 10,036 participants diagnosed with actinic keratosis. We included 18 topical creams or gels applied to a skin area by the participants: adapalene gel, aretinoid methyl sulfone (Ro 14‐9706), betulin‐based oleogel, calcipotriol (vitamin D), colchicine, diclofenac, 2‐(difluoromethyl)‐dl‐ornithine (DFMO), 5‐fluorouracil, ß‐1,3‐D‐glucan, imiquimod, ingenol mebutate (PEP005), isotretinoin, masoprocol, nicotinamide, resiquimod, sunscreen, DL‐α‐tocopherol (vitamin E), and tretinoin. One treatment, etretinate, was taken orally. Clinical staff administered two mechanical treatments (carbon dioxide and Er:YAG laser resurfacing) on a skin area, and they administered three chemical treatments: cryotherapy on individual lesions, photodynamic therapy on individual lesions or a skin area, and trichloroacetic acid peel on a skin area.
The clinical effects resulting from the treatment of actinic keratoses were reported differently from one study to another. In spite of this inconsistency, it can be concluded that several good treatment options exist for the treatment of actinic keratoses. Actinic keratoses were successfully treated with cryotherapy, diclofenac, 5‐fluorouracil, imiquimod, ingenol mebutate, photodynamic therapy, resurfacing, and trichloroacetic acid peel. These different treatments were generally comparably effective. Skin irritation was associated with some of these treatments, such as diclofenac and 5‐fluorouracil, but other side‐effects were uncommon. The final cosmetic appearance varies from one treatment to another. Imiquimod treatment and photodynamic therapy resulted in better cosmetic appearance than treatment with cryotherapy and 5‐fluorouracil.
Treatment with photodynamic therapy gives better therapeutic and cosmetic results than cryotherapy for individual lesions. For field‐directed treatments, diclofenac, 5‐fluorouracil, imiquimod, and ingenol mebutate are good options associated with different side‐effects and cosmetic results. Thus, the choice of treatment option for actinic keratosis depends on the number of lesions, the individual's desired results, and tolerance to the treatments.
Background
Description of the condition
Disease definition
Actinic keratoses are scaly lesions on the skin resulting from abnormal growth of atypical epidermal keratinocytes. They are localised at the surface of the skin on the sun‐exposed parts of the face or hands, particularly among older fair‐skinned individuals. Actinic keratoses are markers for increased rate of non‐melanoma skin cancer (Ramsay 2003) and shows the morphological and histological features of squamous cell carcinoma (Cockerell 2000; Feldman 2011). An actinic keratosis could be considered a precancerous lesion or carcinoma in situ based on the fact that the majority of invasive squamous cell carcinomas arise from actinic keratoses. Actinic keratoses are confined to the epidermis, whereas squamous cell carcinoma extends more deeply into the dermis. Thus, to limit the morbidity and mortality associated with squamous cell carcinoma, treatment of actinic keratoses is strongly recommended.
Actinic keratosis is also known as solar keratosis, senile keratosis, senile hyperkeratosis, keratoma senile, keratosis senilis, and actinic cheilitis (actinic keratosis on the lip) (Marks 1993; Rigel 2008; Schwartz 1997).
Clinical Features
The conventional clinical actinic keratosis lesion is a pink, red, or brown scaly patch on the skin, less than one centimetre in diameter (Roewert‐Huber 2007). Often, the scaliness of a lesion can be felt before it can be seen; this may progress into thickened or hypertrophic (increased bulk, due to an increase in lesion size) lesions. Actinic keratoses can be clinically graded with grade 1, slightly palpable; grade 2, moderately thick and visible; and grade 3, very thick and hyperkeratotic (Cockerell 2000; Olsen 1991). Accurate clinical diagnosis requires careful observation under adequate lighting conditions and palpation of the lesion texture (Marks 1993). Actinic keratoses are diagnosed histologically with a skin biopsy (Cockerell 2000; Marks 1993). Detectable actinic keratosis lesions are often associated with field change where the surrounding skin is also altered, and subclinical lesions may be present (Vatve 2007).
There are different classifications based on the clinical appearance of actinic keratoses: atrophic, hyperkeratotic, bowenoid, acantholytic, lichenoid, and pigmented (Rigel 2008; Roewert‐Huber 2007). Atrophic actinic keratoses are dry, scaly‐appearing lesions on a reddened base (due to dilated blood capillaries) without distinct margins. Hyperkeratotic actinic keratoses are papules and plaques with scale or scale‐crust that also possibly have cutaneous horns or conical masses. Bowenoid actinic keratoses are scaling red plaques with sharply‐established borders that simulate Bowen's Disease (a solitary red plaque with distinct borders) in that the abnormal cells are found throughout the depth of the epidermis. Acantholytic actinic keratoses have focal acantholysis (separation from other cells) occasionally accompanied by clefts. Lichenoid actinic keratoses show dense band‐like infiltration of lymphocytes in the papillary dermis and vacuolar alteration at the dermoepidermal junction. Pigmented actinic keratoses have a hyperpigmented or reticulated appearance. Differential diagnosis of actinic keratosis includes Bowen's disease, squamous cell carcinoma, keratoacanthoma, basal cell carcinoma, seborrhoeic keratosis, and lentigo maligna (Holmes 2007).
Symptoms of actinic keratosis include tenderness, itchiness, burning, and a sandpaper‐like texture. Over time, lesions may remain unchanged, proliferate, regress, reappear, or develop into squamous cell carcinoma. Microscopically, actinic keratosis lesions show abnormal tissue development (dysplasia) in the skin cells (keratinocytes). During early development of a lesion, the lower layers of the epidermis show the most dysplastic keratinocytes. As a lesion develops, the dysplastic cells permeate the epidermis and form conical‐shaped scales when the surface of the epidermis is reached. Acceleration of growth of the epidermal layer and abnormal cellular maturation leads to excessive production of immature adherent scales with a sandpaper or gritty feel (Marks 1993). The lower skin layer (dermis) undergoes patchy inflammation as seen by an increased number of white blood cells (lymphocytes) noted in the dermis (Marks 1993).
Pathogenesis and epidemiology
The anatomical distribution of actinic keratosis lesions correlates with areas of the body that receive the most long‐term, chronic, and intense exposure to ultraviolet radiation in sunlight (Marks 1993; Schwartz 1997). More than 80% of the lesions occur on the head, neck, back of the hands, and forearms (Salasche 2000). Chronic exposure to ultraviolet (UV) radiation, mainly UVB (290 to 320 nm), is the major agent leading to mutagenesis (disordered regulation of growth) in keratinocytes (Callen 1997). In fact, mutations in the p53 tumour suppressor gene have been found in 53% of those with actinic keratoses and 69% of squamous cell carcinoma biopsies (Nelson 1994). Ultraviolet radiation can also contribute to suppression of the immune system, resulting in a decreased ability to eliminate over‐proliferating cells (Holmes 2007). Moreover, UV light could directly activate human papillomavirus replication. The virus, in turn, degrades a proapoptotic protein BAk, also preventing elimination of tumour cells (Holmes 2007). Thus, sunlight initiates and promotes the formation of non‐melanoma skin cancer.
The cause of actinic keratosis involves an interaction between skin colour (melanin protects by absorbing UVB radiation); advancing age (cumulative sun exposure and decrease in the effectiveness of the immune system); gender (actinic keratosis is more prevalent in men); history of severe sunburn in childhood; and sun exposure, which is influenced by latitude and the integrity of the ozone layer (Holmes 2007; Lebwohl 2003; Salasche 2000). Other factors may include occupation (working outdoors), socioeconomic status, and diet (Lebwohl 2003; Marks 1993; Salasche 2000; Schwartz 1997). Immunosuppressive therapy, e.g. in organ transplant recipients, and exhibition of genetic diseases of skin hypopigmentation (low pigmentation), such as xeroderma pigmentosum or albinism (Holmes 2007; Moy 2000), are also risk factors.
The first National Health and Nutrition Examination Survey (NHANES I) found that in healthy white people in the US, the age‐adjusted prevalence rate for actinic keratoses was 6.5%. This increases significantly with advancing age: In 65‐ to 74 year‐old men with high sun exposure, the prevalence rate was 55.4% and 18.5% for low sun exposure (Engel 1988). In Australia, where prevalence of actinic keratosis is the highest, as many as 40% of white adults may have an actinic keratosis. For younger adults, aged 30 to 39 years, the rate was 22% for men and 8% for women. In older adults aged 60 to 69 years, 83% of men and 64% of women have an actinic keratosis. For this population of adults, 42% developed at least 1 new lesion within the year (Frost 2000). Although known to be precancerous, the probability of a lesion undergoing malignant transformation to a squamous cell carcinoma is not clear, but ranges from 0.025% to 16% per year (Glogau 2000; Jeffes 2000).
Description of the intervention
An actinic keratosis may potentially become cancerous; therefore, monitoring is advised. Because of the prevalence of actinic keratoses among an ageing population, treatment has been sought by an increasing number of people (Warino 2006). Reasons for treatment include prevention of cancer development; relief of symptoms, such as bleeding; and improvement of cosmetic appearance. Interventions for actinic keratoses could be divided into individual treatment of lesion and field‐directed treatment, i.e. applied to an area of sun‐damaged skin where there may be multiple lesions. Individual lesion treatment (spot) might relieve symptoms or cosmetic concerns, whereas field‐directed treatment might be more appropriate for prevention of transformation into squamous cell carcinoma. Most of the field‐directed treatments are topical treatments where efficacy depends on patient compliance.
Behaviour modifications, including limiting sun exposure between 10am and 4pm, the use of sunscreens with a SPF (sun protection factor) rating of at least 15, and the use of protective clothing, are the best methods for the prevention of actinic keratosis and will help reduce the need for treatment (Schwartz 1997; Wilkerson 1984).
Various strategies for the treatment of actinic keratoses have been developed; these include physician‐administered cryotherapy for a few lesions, and topical 5‐fluorouracil, topical imiquimod, topical masoprocol, topical diclofenac in 2.5% hyaluronic acid gel, and photodynamic therapy for large numbers of lesions. Salicylic acid may also be used for early lesions, while dermabrasion and laser resurfacing are beneficial when there is coexistent photodamage or multiple recalcitrant lesions. Excision (removal of the lesion, often using a scalpel blade) and chemical peels (use of a caustic agent that causes the lesion to slough off) are both appropriate for hyperkeratotic or recalcitrant lesions. Interferon and oral retinoids are uncommon treatments, and they are still under development. These treatments have varying efficacies and adverse effect profiles (Dinehart 2000; Ibrahim 2009; Marks 1993; Wilkerson 1984).
Thus, the factors to consider when making decisions about treatment include efficacy, tolerability, number of lesions to treat, spot or field‐directed treatment, compliance, history of skin cancer, immunosuppression, previous treatment history, and cosmetic appearance.
How the intervention might work
Topical Interventions
Diclofenac gel
One topical treatment for actinic keratoses is the non‐steroidal anti‐inflammatory drug (NSAID) diclofenac in 2.5% hyaluronic acid gel. The hyaluronic acid vehicle contributes to the success of this treatment by delivering and then retaining diclofenac at the epidermis, protecting against UV radiation and its cosmetic properties (Brown 2005). Although the precise mechanisms of action are not clear, diclofenac is thought to target several aspects of actinic keratosis pathophysiology. One mechanism that has been proposed is the inhibition of cyclooxygenase 2 (COX‐2) (Hemmi 2002), which leads to a reduction in prostaglandin synthesis (Rivers 2004). This COX‐2 inhibition or other mechanisms may be responsible for diclofenac’s inhibition of cell differentiation in vitro, induction of apoptosis in vitro and in vivo, alteration of cell proliferation, and inhibition of angiogenesis (Adamson 2002; Alam 1995; Lu 1995; Seed 1997). Diclofenac has also been shown to activate the nuclear hormone receptors, peroxisome proliferator‐activated receptors (PPARs), in vitro; these receptors are involved in many cellular functions including cell differentiation and apoptosis (Adamson 2002).
5‐Fluorouracil (5‐FU)
This topical agent causes a decrease in cell proliferation and an induction of cell death, particularly in cells with high mitotic (cell division) rates. This occurs through the inhibition of thymidylate synthetase, which blocks the methylation reaction of deoxyuridylic acid to thymidylic acid, thereby, interfering with DNA and RNA synthesis (Berman 2006; Chakrabarty 2004; Eaglstein 1970; Robins 2002b).
Imiquimod
This topical treatment for actinic keratoses is a synthetic compound belonging to the imidazoquinolone family of drugs (Hemmi 2002). It acts as an immune modulator by activating toll‐like receptors, ultimately resulting in the modulation of the mRNA expression of many immunomodulatory genes, which induces the production of cytokines by monocytes, macrophages, and epidermal keratinocytes (Correale 2002; Stanley 1999). This has the effect of enhancing innate and acquired immune responses, which leads to strong antiviral and antitumoural activity (Vidal 2006). Imiquimod also induces pro‐apoptotic pathways through a variety of mechanisms (Amini 2010).
Chemical Interventions
Cryotherapy
Cryotherapy is often the treatment of choice for individual actinic keratosis lesions (Goldberg 2010). It uses liquid nitrogen to freeze and destroy the epidermis containing actinic keratoses (Goldberg 2010), with efficacy increasing as a function of freezing duration (Thai 2004).
Photodynamic therapy (PDT)
Photodynamic therapy involves the selective accumulation of a photosensitising agent in premalignant or malignant cells (Gold 2008; Juarranz 2008). This is achieved by the application of 5‐aminolevulinic acid (5‐ALA) or MAL (ALA methyl ester), which are precursors to protoporphyrin IX (PpIX), a potent photosensitiser (Fink‐Puches 1997). This causes an excess of PpIX, which selectively accumulates in neoplastic cells. Subsequently, the photosensitiser is activated by visible light, causing the generation of reactive oxygen species in the presence of oxygen. These reactive oxygen species [mainly singlet oxygen (ˈO₂)] start a cascade of biochemical events that induce damage and the death of neoplastic cells through an apoptotic mechanism (Juzeniene 2007; Moan 1991).
Why it is important to do this review
The existing evidence for use of the various treatment agents for actinic keratoses is varied, and there are concerns regarding adverse events and cosmetic outcomes. It is vital to critically assess data in terms of the benefits as well as the risks associated with treatment.
Objectives
To assess the effects of interventions for actinic keratoses.
Methods
Criteria for considering studies for this review
Types of studies
This review included randomised controlled trials comparing the treatment of actinic keratoses to either placebo, vehicle, other current therapies, or variation in treatment conditions (e.g. different concentrations of the active ingredient or types of light sources for phototherapy). We included cross‐over trials and parallel and intraindividual (e.g. left‐ or right‐side comparison) studies.
Types of participants
We included participants with clinical signs of actinic keratoses as assessed by a medical practitioner or histological diagnosis. Diagnostic criteria, such as the Marks definition (Marks 1993) or the Salasche or Schwartz characterisation (Salasche 2000; Schwartz 1997), were acceptable, as was the diagnosis of actinic keratoses by a dermatologist using the terms 'actinic keratosis', 'solar keratosis', 'senile keratosis', 'senile hyperkeratosis', 'keratoma senile', or 'keratosis senilis'. We included studies with immunocompetent and immunosuppressed participants.
Types of interventions
We considered the following interventions:
prescription‐based topical treatments, e.g. diclofenac in hyaluronic gel, 5‐fluorouracil, or imiquimod;
prescription‐based oral drugs, e.g. oral retinoids;
mechanical interventions, e.g. curettage, dermabrasion, or resurfacing;
chemical interventions, e.g. chemical peels, cryotherapy, or photodynamic therapy; and
combinations of topical and oral treatments with mechanical or chemical interventions.
The comparators were vehicle, placebo, another active compound or intervention, or a variation of the treatment (duration, concentration, etc).
Types of outcome measures
For actinic keratoses, the outcomes can be expressed per lesion or per participant. Because the participants or body parts of the participants (intraindividual design), not the lesions, were generally randomised, only per‐participant outcomes could be included in meta‐analyses. Thus, the included outcomes in this review were outcomes reported per participant.
Efficacy outcomes for studies on actinic keratoses are generally based on the clearance of individual lesions. Lesions present at baseline are generally identified, graded (grade I: slightly palpable, better felt than seen; grade II: moderately thick, easily seen and felt; and grade 3: very thick, hyperkeratotic, or both), and mapped. Use of transparencies and photography might help with this process. Sometimes distinction is made between lesions present at the baseline and new lesions appearing during the study. At the end of the study, the assessors evaluate the clearance, or not, of the lesions.
Ideally, complete clearance of actinic keratosis lesions at follow‐up would be measured (i.e. number of participants with 100% clearance of target (present at baseline) or all actinic keratosis lesions).
A second outcome measurement, such as partial clearance, is also often used. The definition of partial clearance is subjective but frequently indicates the number of participants with 75% or more of actinic keratosis lesions being completely cleared, i.e. a reduction in the number of lesions by at least 75%.
Alternatively, the mean reduction of total number of lesions at baseline per participant is also used, i.e. the difference between the mean number of lesions at baseline and the mean number of lesions at assessment. The results are then presented as absolute mean or mean percentage of reduction in lesion counts compared to baseline.
We only included outcomes expressed as number of participants experiencing adverse events in this review.
Cosmetic outcomes are really varied from global assessment to individual characteristics, such as changes in pigmentation. We only included outcomes expressed as number of participants or mean per participant in this review.
Primary outcomes
Efficacy outcomes
Subjective assessment: global degree of improvement in symptoms or signs as rated by a medical practitioner or participant, or global improvement indices (GII) for completely improved or cleared.
Objective assessment: participant complete (100%) or partial (> 75%) clearance.
Objective assessment: mean reduction in lesion counts (absolute number or percentage).
Secondary outcomes
Safety and cosmetic outcomes
Withdrawal due to adverse events.
Skin irritation.
Minor adverse events excluding skin irritation.
Cosmetic outcomes: cosmetic changes, including pigmentation and scarring.
Search methods for identification of studies
We aimed to identify all relevant randomised controlled trials (RCTs) regardless of language or publication status (published, unpublished, in press, and in progress).
Electronic searches
We searched the following databases up to 23 March 2011:
the Cochrane Skin Group Specialised Register using the terms: ((actinic or solar or senile) and keratos*) or hyperkeratos*;
the Cochrane Central Register of Controlled Trials (CENTRAL) in The Cochrane Library using the search strategy in Appendix 1:
PUBMED/MEDLINE via OVID (from 2005) using the strategy in Appendix 2;
EMBASE via OVID (from 2010) using the strategy in Appendix 3; and
LILACS (Latin American and Caribbean Health Science Information database, from 1982) using the strategy in Appendix 4.
The UK and US Cochrane Centres have an ongoing project to systematically search MEDLINE and EMBASE for reports of trials, which are then included in the Cochrane Central Register of Controlled Trials. Searching has currently been completed in MEDLINE from inception to 2004 and in EMBASE from inception to 2009. Further searches of these two databases were undertaken for this review by the Cochrane Skin Group to cover the years not searched by the UK and US Cochrane Centres for CENTRAL.
A final prepublication search for this review was undertaken on 4 April 2012. Although it has not been possible to incorporate RCTs identified through this search within this review, relevant references are listed under 'Studies awaiting classification'. They will be incorporated into the next update of the review.
Trials Registers
We searched the following trials registers on 10 March 2011 using the search terms ((actinic, senile, or solar) and keratos) or hyperkeratos.
The metaRegister of Controlled Trials (www.controlled‐trials.com).
The US National Institutes of Health Ongoing Trials Register (www.clinicaltrials.gov).
The Australian New Zealand Clinical Trials Registry (www.anzctr.org.au).
The World Health Organisation International Clinical Trials Registry platform (www.who.int/trialsearch).
The Ongoing Skin Trials Register (www.nottingham.ac.uk/ongoingskintrials).
Searching other resources
Unpublished literature
We conducted online searches (via pharmaceutical company websites, the U.S. Food and Drug Administration (FDA) website, or both) for the following products and drug companies:
3M/Graceway Pharmaceuticals (imiquimod, Aldara, or Zyclara);
Actavis Mid‐Atlantic LLC (imiquimod);
Allergan (5‐fluorouracil, Fluoroplex);
Apotex (imiquimod);
Dermik/Sanofi Aventis (5‐fluorouracil, Carac);
DUSA Pharmaceuticals (aminolevulinic acid, Levulan Kerastick);
Galderma (adapelene, Differin);
ICN (5‐fluorouracil, Efudex);
Leo Pharmaceuticals (calcipotriol, Dovonex, or Daivonex);
Mochida Pharmaceuticals (imiquimod, Beselna);
PharmaDerm/NycoMed US (diclofenac, Solaraze);
Pharmacia & Upjohn (5‐fluorouracil);
Photocure ASA/Galderma (methyl aminolevulinate, Metvix, or Metvixia);
Roche (etretinate, Tegison);
Stiefel/GlaxoSmithKline (isotretinoin, Isotrex, or Isotrexin); and
URL Pharma (colchicine, Colcrys).
Conference proceedings
We scanned the conference proceedings of the British Association of Dermatologists and the European Academy of Dermatology from 2007 to 2011 for further references to relevant trials. We examined the conference proceedings for 2009 and 2010 of the Annual Meeting of the American Academy of Dermatology, the Annual Meeting of the European Society for Dermatological Research, the Congress of the European Association of Dermatol‐Oncology, the Annual Meeting of the British Association of Dermatologists, and the Annual Meeting of the Australasian College of Dermatologists. We scanned the conference proceedings for the 2012 Annual meeting of the American Academy of Dermatology.
Language restrictions
We imposed no language restrictions when we searched for publications. We electronically translated articles published in languages other than English.
Adverse effects
We did not perform a separate search for adverse effects of interventions for actinic keratoses. We looked at reports of adverse events or side‐effects in the RCTs identified as a result of our searches, as part of our secondary outcomes.
Data collection and analysis
Selection of studies
At least two authors (WB and MP) independently checked titles and abstracts identified from the searches. We obtained the full text of all studies of possible relevance for independent assessment by two authors (MP and WB). The authors decided which trials fit the inclusion criteria and recorded their methodological quality (MP and WB). They resolved any disagreement by discussion between the authors and a third party arbitrator (AG). Previous contributors also participated in this process in earlier versions of the review.
Data extraction and management
At least two authors extracted and summarised, using data collection forms, the details of eligible trials. One author (MP) double‐checked and entered data. The authors were not blinded to the names of the trial authors, journals, or institutions.
Assessment of risk of bias in included studies
Assessment of risk of bias included the Review Manager 5.1 'Risk of bias' assessment tool shown in the 'Risk of bias' tables. In addition, GradePro "quality of evidence" was also used for selected outcomes, and the results are shown in the overview tables for five selected interventions.
Measures of treatment effect
We performed a meta‐analysis for each treatment comparison to calculate a weighted treatment effect across trials. We expressed the results as a risk ratio (RR) with 95% confidence intervals (CI) for dichotomous outcomes, and a mean difference (MD) with 95% CI for continuous outcomes. We calculated the number needed to treat (NNT) for significantly different dichotomous outcomes using the following formula: NNT = Ι 1/ ACR * (1‐RR)Ι where the risk ratios (RR) from the meta‐analysis and the moderate assumed control risk (ACR) calculated in GRADEpro was used. For ACR, a mean baseline risk from the study was used for analysis with only one study; and low, median, or high control‐group risk were used based on the variation in the included studies in meta‐analysis. This previous method would not be applicable to outcomes with an ACR of 0%, i.e. no event in the control group, because of the numerical problems that would ensue.
Unit of analysis issues
The unit of analysis was the participant. We analysed cross‐over trials using data from the first phase only and pooled, where possible, with parallel‐design studies. We divided results from withinparticipant trials (intraindividual, e.g. split face) into 2 categories: 1) outcomes expressed as number of participants (e.g. participant complete clearance), which could not be included in meta‐analyses, were only reported in the text; and 2) outcomes expressed as mean with standard deviation (e.g. mean reduction in lesion counts = mean of reductions observed in each participant), which could be included in meta‐analyses using the inverse‐variance method. We combined together data from studies with multiple treatments when appropriate (e.g. "all treatment groups" versus "placebo"), or we split the data from the shared group. If studies were using more than one outcome included in this review, we included all outcomes in the analyses.
Assessment of heterogeneity
We assessed heterogeneity using an I² statistic value expressed as a percentage. We excluded results from meta‐analyses with an I² statistic value of 80% or higher. We explored reasons for heterogeneity in studies, and if necessary, sensitivity analyses examined the effects of excluding a study, e.g. those studies with lower methodological quality.
Many studies do not distinguish between the physical location of actinic keratosis lesions on the body. This can introduce heterogeneity, as actinic keratoses of the face and scalp are often more effectively treated by certain topical formulations than lesions located elsewhere. In some studies, pretreatment of lesions to remove hyperkeratosis essentially negated the differences encountered by lesion location, as lower response has been associated with greater hyperkeratosis. Most of the studies included limited their investigation to grade one or two lesions, i.e. minimally to moderate thick lesions. However, when comparing efficacy results from two separate studies using the same treatment, studies incorporating pretreatment of any kind may have accounted for different efficacy rates.
Data synthesis
A random‐effects model was prespecified for all meta‐analyses. The Mantel‐Haenszel method was used for dichotomous outcomes (e.g. cure rates), and an inverse variance model was used for continuous outcomes (e.g. mean reduction in lesion counts).
Subgroup analysis and investigation of heterogeneity
Where appropriate, we undertook subgroup analysis (subgroups of participants) in an attempt to decrease heterogeneity between studies (for example, when different dosing regimens were used or to keep information separated, i.e. when blue or red light was used for photodynamic therapy). In addition, if data were presented for several assessment time points or anatomical locations, we performed subgroup analyses.
Results
Description of studies
See the 'Characteristics of included studies', 'Characteristics of excluded studies', and 'Characteristics of ongoing studies'.
Results of the search
We identified 1001 references from searching bibliographic and trials databases, as well as 28 references through other sources. After removing duplicate references and ongoing studies without results, we had 469 records to screen. We excluded 318 records based on titles and abstracts as they did not meet our eligibility criteria (non‐randomised studies, reviews, not interventions to cure). We assessed the full texts of the remaining 151 records. We then excluded a further 55 records, leaving 96 studies. We included 83 of these in our qualitative analysis; 12 are listed under studies awaiting classification, and 1 is an ongoing study. We included 75 studies in our meta‐analysis.
The PRISMA study flow chart in Figure 1 summarises the results of the search for studies.
1.

Study flow diagram.
(Please note that in all tables in the results section, the "X" means that the associated outcome was reported, and when there was no participant withdrawal it is specified between parentheses.)
Included studies
We included 83 randomised studies in the review, encompassing 10,036 participants in total.
Design
We only included the randomised (participants or right/left side in intraindividual studies) clinical trials if the interventions were covered by this review and if they reported numerical results for at least one of the review outcomes. This criterion excluded the outcome 'withdrawal due to adverse events', which is generally reported in all studies.
Some studies had more than one design. The design of the studies is summarised in the following table.
| Placebo/vehicle‐controlled |
Active‐ controlled1 |
|
| Parallel groups | 46 studies (including part I of 1 cross‐over study) |
17 studies |
| Intraindividual2 | 12 studies | 10 studies |
1. Active‐controlled = compared to another treatment, which could be a different treatment or the same treatment at a different concentration, duration, or types of light used for photodynamic therapy.
2. Intraindividual = within‐patients, i.e. different body parts of the same participant received different treatments in parallel (not sequentially).
Sample sizes
Studies ranged in sample size from 4 to 492 participants (124 + 127, mean + SD).
Interventions
The interventions assessed in the studies included the following.
Topical treatments
Adapalene gel
Aretinoid methyl sulfone (Ro 14‐9706)
Betulin‐based oleogel
Calcipotriol (vitamin D)
Colchicine
Diclofenac
2‐(Difluoromethyl)‐dl‐ornithine (DFMO)
5‐fluorouracil (5‐FU)
ß‐1,3‐D‐glucan
Imiquimod
Ingenol mebutate (PEP005)
Isotretinoin
Masoprocol
Nicotinamide
Resiquimod
Sunscreen
DL‐α‐tocopherol (vitamin E)
Tretinoin
A total of 60 studies investigated topical treatments.
Oral treatments
Etretinate
One study investigated oral treatment.
Mechanical interventions
Resurfacing (carbon dioxide and Er:YAG lasers)
Two studies investigated mechanical interventions.
Chemical interventions
Cryotherapy
Photodynamic therapy (using a variety of different parameters)
Chemical peel (trichloroacetic acid)
A total of 37 studies investigated chemical interventions.
Interventions in the included studies could also be segregated based on clinical (e.g. PDT or cryotherapy) or participant (e.g. topical cream) administration, as well as treatments for individual lesions (e.g. cryotherapy) or field‐directed treatments (e.g. topical cream).
Participants
Participants in the studies were generally in good health, but a few studies specifically recruited participants with a history of non‐melanoma skin cancer. We included studies with organ transplant recipients (immunosuppressed), but these were analysed separately. Responsiveness to immunomodulators may decrease with increasing age, so the age of participants might influence the efficacy of treatments using them. In the included studies, most of the participants were men with mean ages of 60 to 70 years. Lesions were generally grade I (slightly palpable, better felt than seen) or II (moderately thick, easily seen and felt). The location of actinic keratosis lesions, i.e. lesions difficult to access for cream application, could also influence participant compliance and ultimately the efficacy of participant‐administered treatments. Lesions were located on the head only (i.e. face, forehead, temples, cheeks, scalp, ear, lips, and neck) in 59 studies, on only non‐head locations (upper and lower extremities, legs, arms, elbow, forearms, hands, dorsa of hands, shoulder, décolleté, chest, trunk, and back) in 9 studies, and on both head and non‐head locations (including the term "other") in 22 studies. One study did not specify the location of the lesions. In general, lesions were more often located on the face and scalp, which are easy to reach.
Outcomes
Efficacy outcomes
The included studies reported several efficacy outcomes. A lot of the studies did not specify if only target (baseline) lesions or all lesions [i.e. target and subclinical lesions (new lesions appearing during the study)] were included in their analysis. Most of the studies reported more than one outcome. Some of these outcomes corresponded to our primary outcomes or could be transformed into our primary outcomes, whereas others did not meet our criteria for this review. We have summarised the primary and other outcomes in the following table.
| Number of studies | Outcomes | Equivalence or transformed into outcome |
| Primary outcomes | ||
| 12 | Global improvement indices expressed per participant (Investigator, participant, or both) |
Physician global assessment improvement, global therapeutic response or treated area, investigator assessment scale, investigator global assessment, overall response |
| 53 | Participant complete clearance (number of participants, rate, proportion, percentages) | Complete responders, total clearance, response to treatment, proportion of participants achieving total clearance, field complete clearance, complete remission, complete response of lesional area, participant's complete resolution, complete clearing, number of participants with 100% clearance, complete participant response, target lesion number score = 0, complete healing, cumulative lesion number score = 0, 100% lesions cleared, percentage of participants who experienced 100% clearance of all target lesions, number of participants with all cleared lesions |
| 20 | Participant partial (> 75%) clearance (number of participants, rate, proportion, percentages) | At least 75% reduction in the number of lesions, at least 75% of lesions cleared, percentage of participants who experienced 75% or greater clearance of all target lesions, therapy responders with at least 75% of clearing of the lesions, participant partial (> 80%) clearance rates |
| 50 | Mean reduction in lesion counts (absolute values or percentages) | Mean reduction in the number of actinic keratoses, mean changes of lesion counts, mean numbers of lesions at baseline and assessment time point, mean percentage reduction in the number of actinic keratoses, average changes in lesion counts, mean per cent changes from baseline for all actinic keratoses, mean per cent lesions cleared |
| Other outcomes | ||
| 3 | Global improvement indices expressed as scores | Physician global assessment, global improvement score |
| 29 | Lesion complete response (per lesions) | Reduction rate in number of actinic keratoses, clearance of individual lesions, rate of totally healed lesions, number of lesions with 0% of remaining area, complete clinical clearance rate on lesion basis, complete clearance rate of lesions, individual lesion clearance, lesion counts at baseline and assessment, percentage lesion reduction, proportion of baseline lesions cleared at the end of treatment, lesions remitted, total lesion counts |
| 9 | Median per cent reduction of baseline lesions | Median per cent changes from baseline for all actinic keratoses |
| 6 | Participant histological clearance | Histological clearance, histological confirmation |
| 5 | Recurrence | ‐ |
| 3 | Participant partial (> 50%) clearance |
Participant with 50% or greater reduction, clearance = resolution of > 50% of the lesions |
| 5 | Reduction in lesion size | Overall reduction in lesion area, partial remission (50% size reduction of 75% of lesions), mean diameter of target lesion at baseline and assessment |
| 2 | Median number of lesions at baseline and assessment time point | ‐ |
| 1 | Participant partial (> 66%) clearance |
‐ |
| 1 | Participant partial (% not specified) clearance |
‐ |
| 1 | Total lesion number score (0 = 0 lesions, 1 = 1 to 3 lesions, 2 = 2 to 4 lesions, 3 = > 6 lesions) | ‐ |
| 1 | Negative predictive value, i.e. ratio between histological and clinical clearance | ‐ |
| 1 | Participant's perception of efficacy | ‐ |
| 1 | Efficacy on a visual analogue scale for field‐directed treatment | ‐ |
| 1 | Relapse | ‐ |
Safety outcomes
There was a lot of variability in the safety outcomes reported by the included studies. Some studies provided briefly qualitative observations on adverse events, whereas others gave detailed quantitative description of adverse events. Intraparticipant studies have limitations in assessing adverse events other than application site and local skin reactions. Adverse events might influence a participant's compliance as well as the maintenance of the blinding. In turn, poor compliance and unblinding could compromise the evaluation of the treatment efficacy. Moreover, adverse events are an important factor in a physician's decision about appropriate treatment for their patients, and a more standardised report of adverse events would be beneficial. The safety outcomes that were our prespecified secondary outcomes found in the included studies, as well as other outcomes, are summarised in the following table.
| Number of studies | Outcomes | Equivalence or transformed into outcome |
| Secondary outcomes | ||
| 77 | Withdrawal due to adverse events | None lost = all participants completed the trial/study, or lost participants were all justified by other reasons |
| 15 | Skin irritation (per participant) | Application site irritation, local irritation, facial irritation, graphical representation of irritation, number of participants reporting relative irritation between treatments |
| 31 | Minor adverse events excluding skin irritation (number or percentages of participants) | Most frequent adverse events, number of participants reporting individual adverse events, participants with eye irritation, percentages of participants reporting adverse events for only 1 treatment arm or pooled data, specific treatment‐related adverse events |
| Other outcomes | ||
| 16 | Application site reactions in general (number or percentages of participants experiencing reactions in general) |
Adverse events at treatment sites |
| 15 | Application site reactions for specific reactions (number or percentages of participants experiencing specific reactions) |
Adverse events at treatment sites |
| All = 6 Severe = 3 |
Local skin/adverse reactions ‐ in general (number of percentages of participants) |
Local adverse events |
| All = 33 Severe = 12 |
Local skin/adverse reactions for specific reactions (number of percentages of participants) |
Local skin reactions reported for only 1 treatment arm or pooled data, graphical representation of local skin reactions |
| 20 | Participants experiencing at least 1 adverse event (number or percentages of participants) |
Number or percentage of participants reporting adverse events, graphical representation of percentages of participants experiencing adverse events |
| 11 | Treatment‐related adverse events in general (number or percentages of participants) | ‐ |
| 31 | Serious adverse events (treatment‐related or not) | ‐ |
| 6 | Serious adverse events‐detection of basal cell carcinoma (presence or not per participant) | ‐ |
| 7 | Serious adverse events ‐ detection of squamous cell carcinoma (presence or not per participant) | ‐ |
| 24 | Clinical laboratory tests | ‐ |
| 2 | Incidences of application site reactions (number of events) |
‐ |
| 1 | Application site reactions reported per lesions | ‐ |
| 18 | Local tolerability (severe, moderate, mild, absent) | Severity of local skin reactions, global severity rating of local reactions, side‐effects (skin reactions) on a scale, irritation severity, severity of facial irritation, severity of local adverse events, grading of individual local reactions, physician's grading of erythema |
| 2 | Number of reports of skin irritations | Incidence of local skin reactions |
| 2 | Number of participants with strong, moderate, weak, or no inflammatory reaction | ‐ |
| 1 | Local phototoxic reactions | ‐ |
| 2 | Number of treatment‐related adverse events (incidence) | ‐ |
| 1 | Qualitative report on treatment‐related adverse events | ‐ |
| 22 | Qualitative report on skin irritation (types and severity) | Comparison of severity of adverse events between treatments |
| 5 | Number of reports of adverse events (incidences) | ‐ |
| 1 | Number of reports of serious adverse events | ‐ |
| 12 | New actinic keratosis lesions | Subclinical lesions, increase in number of lesions during the study |
| 7 | Pain score | Mean visual analogue scale for pain |
| 2 | Skin discomfort on a visual analogue scale | ‐ |
| 1 | Duration of discomfort | ‐ |
| 1 | Erythema measured by skin reflectance meter | ‐ |
| 1 | Graft rejection (organ transplant participants) |
‐ |
| 1 | Detection of Bowen's disease | ‐ |
| 1 | Incidence of new non‐melanoma skin cancer | ‐ |
The evaluation of the 'skin irritation' outcome was restricted, as only 15 studies had outcomes containing explicitly the term 'irritation'. Several studies reported application site, local skin reactions, or both, which generally included signs and symptoms of skin irritation, such as burning/stinging, erythema, oedema, pruritus, and scaling. We could have included these skin irritation signs and symptoms as more specific 'skin irritation' outcomes if a universal definition of skin irritation existed. Because of the exclusion of skin irritation in the 'minor adverse events' outcome, these reactions as well as the number of participants reporting at least one adverse event, related or not to the treatment (which could include skin irritation), could not be included in any of our secondary outcomes.
Cosmetic outcomes
Only a few studies reported cosmetic outcomes and were varied. In general, cosmetic evaluation was performed on cleared lesions. The cosmetic outcomes that were our prespecified secondary outcomes found in the included studies, as well as other outcomes, are summarised in the following table.
| Number of studies | Outcomes | Equivalence or transformed into outcome |
| Cosmetic outcomes reported per participant | ||
| 4 | Changes in pigmentation | Hypopigmentation, hyperpigmentation |
| 3 | Global cosmetic outcome of "good", "very good", or "excellent" | Final cosmetic results, overall cosmetic outcome |
| 2 | Cosmetic appearance score | Total score for cosmetic appearance (erythema, desquamation, induration), cosmetic appearance scores by participant and investigator on a 7‐point scale (‐3 = much worse to +3 = much better) |
| 4 | Skin quality | Decrease in roughness/dryness/scaliness of the skin, normal skin surface, decrease of scarring |
| 4 | Improvement in photodamage or photoageing score | Investigator global integrated photodamage, photodamage score (fine lines, mottled pigmentation, tactile roughness, sallowness), photoageing score (global appearance, fine wrinkles, mottled hyperpigmentation, coarse wrinkles, rosy glow) |
| 2 | Significantly ‐ or much‐ improved cosmetic outcome | ‐ |
| 1 | Decreased infiltration and disappearance of crust | ‐ |
| 1 | Proportion of participants with improvement of surface with actinic damage | Note: the number of participants was not given and could not be included in the analysis |
| Other outcomes | ||
| 5 | Cosmetic outcomes per cleared lesions | ‐ |
| 2 | Total thickness score | ‐ |
| 1 | Changes in pigmentation per lesions | ‐ |
Other outcomes
The studies sometimes reported additional outcomes, and they are summarised in the following table. They rarely reported important outcomes, such as compliance, (7 studies) compared to the number of studies investigating participant‐administered treatments (63 studies, including 3 daylight photodynamic therapy studies).
| Number of studies | Outcomes |
| 10 | Participant's satisfaction |
| 8 | Rest periods or temporary interruption during treatment |
| 7 | Compliance |
| 6 | Participant's preference |
| 2 | Biological and immunological outcomes |
| 1 | Skin concentrations of drug and products due to its mechanism of action |
| 1 | Investigator's preference |
| 1 | Lesion severity index |
| 1 | Quality of life on a visual analogue scale |
| 1 | Number of spray cooling for photodynamic therapy |
In 2011, we contacted the following authors to get clarification on the studies included.
| Author | Topic | Clarification |
| Kurt Gebauer | Type of analysis used in the study Gebauer 2003 | Intention‐to‐treat |
| Joseph Jorizzo | Type of analysis used in the studies Jorizzo 2002 and Jorizzo 2006 | The type of analysis could not be confirmed |
| Iraji Fariba | Outcome presented was 'lesions complete response' or 'participant complete clearance' in the study Fariba 2006 | No response received |
| Emil Tanghetti | Type of analysis used in the study Tanghetti 2007 | Intention‐to‐treat |
Excluded studies
Generally, we excluded studies if they were not randomised clinical trials on interventions to cure actinic keratosis lesions (actinic keratoses). In addition, we excluded some randomised studies for the reasons cited in the tables of excluded studies in the 'Characteristics of excluded studies' section. The following table summarises the main reasons for the exclusion of these studies.
| Data not separated for actinic keratoses | Did not meet review criteria for outcomes | Unacceptable or unclear randomisation | No numerical values, graphical data, or not enough information | Prevention of actinic keratosis lesions | Follow‐up reports on included studies |
|
Alberts 2004 Green 1998 Humphreys 1996 |
Apalla 2010b Babilas 2007 Babilas 2008 Bartels 2009 Biecha‐Thalharnmer 2003 Braathen 2009 Dirschka 2010 Edwards 1986 Epstein 2006 Ericson 2004 Jury 2005 Kurwa 1999 Morales 2010 Puizina‐Ivic 2008a Radakovic‐Fijan 2005 Shuttleworth 1989 Smith 2006 Sotiriou 2011 Wulf 2006 |
Alexiades‐Armenakas 2003 Babilas 2006 Berlin 2008 Gold 2006 Goldman 2003 Griffin 1991 Grimaître 2000 Marrero 1998 Tsoukas 2010 Valeant 2004 |
Apalla 2010a Breza 1976 de Sévaux 2003 Dermik 2003 Gupta 2004 Robins 2002a Rosen 2010 Simmonds 1973 Spencer 2010 Touma 2004 Weinstock 2010 Yamauchi 2002 |
Apalla 2010c Elmets 2010 Naylor 1995 Wennberg 2008 |
Fowler 2002 Hanke 2011 Stockfleth 2004 Szeimies 2010a |
In 2011, we tried to contact the following author to get clarification on the studies excluded.
| Author | Topic | Clarification |
| Barbara A. Gilchrest | 1) Number of treatment arms and number of participants allocated 2) Mean numbers of lesions and their standard error of the mean (SEM) for the 3 groups, i.e. 1, 2, and 3 hours |
1) 3 groups of 6 participants each incubated for 1, 2, or 3 hours 2) not received |
Risk of bias in included studies
Please refer to the 'Risk of bias' tables for each included study, which are part of the 'Characteristics of included studies' tables, and the summary figure, Figure 2 ('Risk of bias' graph: review authors' judgements about each 'Risk of bias' item presented as percentages across all included studies).
2.
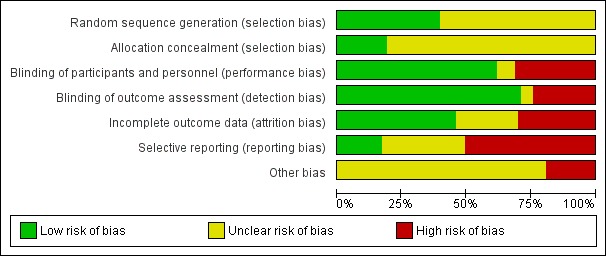
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
Ffity studies were judged to be at low risk of bias with regard to the method used to generate the randomisation sequence, which were stratification (Alberts 2000; Foote 2009; Freeman 2003; Hauschild 2009a; Hauschild 2009b; Pariser 2003; Pariser 2008; Szeimies 2002; Thompson 1993), computer‐generated randomisation schedule (Gebauer 2009; Huyke 2009; Jorizzo 2004; Jorizzo 2006; Jorizzo 2010; Korman 2005; Lebwohl 2004; Loven 2002; Ooi 2006; Ostertag 2006; Szeimies 2004; Szeimies 2009; Szeimies 2010b; Wiegell 2011a), permuted block randomisation (Anderson 2009; Chen 2003; Kang 2003; McEwan 1997; Moloney 2010; Szeimies 2008; Wiegell 2011a), shuffling of envelopes or drawing of lots (Wiegell 2008; Wiegell 2009), and random digits table or number generator (Seckin 2009; Shaffelburg 2009).
Only 16 studies stated the methods used for allocation concealment before the treatments were assigned, and we judged these studies at low risk of bias. Eight studies used opaque sealed envelopes (Chen 2003; Freeman 2003; Moloney 2010; Szeimies 2004; Tarstedt 2005; Wiegell 2008; Wiegell 2009; Wiegell 2011a). Two studies assigned the next sequential number (Korman 2005; Shaffelburg 2009). An external person (pharmacist, sponsor, or CRO) handled the randomisation process in six studies (Krawtchenko 2007; Pariser 2008; Siller 2009; Stockfleth 2002; Swanson 2010a; Van der Geer 2009).
Blinding
Double‐blind or assessor‐blind were used in 58 and 10 studies, respectively. Nine studies were open. In some of these studies, blinding was difficult because of the nature of the treatments being compared (e.g. surgical treatment versus topical treatment). Some authors also reported that adverse events, such as the local skin reactions associated with some treatments, might have compromised the blinding. In these cases, different investigators could have been involved in the treatment/safety assessment and the efficacy assessment in order to keep the part of the assessment blinded. Additionally, the use of photography in the evaluation process could help to keep the assessor blinded. The evaluation of the risk of bias for participants, personnel and assessors took into consideration the type of blinding, and when possible, the possibility of unblinding. Of our 83 included studies, we judged 48 as at low risk of bias for both these domains, 19 studies, at high risk of bias, and 3, as unclear for both domains.
Incomplete outcome data
Three studies used intention‐to‐treat (ITT) analyses, and 25 studies used per‐protocol (PP) analyses. Nine studies used both types of analysis. The type of analysis was undetermined in 12 studies. Most studies adequately recorded characteristics of participants not completing the study. We considered studies where < 20% of enrolled participants dropped out as acceptable, and only three studies (Alirezai 1994; Persaud 2002; Zeichner 2009) exceeded 20%. For the meta‐analyses, we favoured data from ITT analyses over PP analyses, and we converted PP data to ITT data when possible. The evaluation of the risk of bias took into consideration the type of analysis, the number of dropouts, if the reasons for the dropouts were given, and possible discrepancy in the data presented.
Selective reporting
We judged 14 studies as at low risk of bias based on the following criteria: 1) The study protocols were available, and all the prespecified outcomes were presented; 2) the same data were presented in different formats (abstract, protocol with data, product insert, and published report); or 3) non‐significant outcomes were reported.
We judged 42 studies as at high risk of bias based on the following criteria: 1) Not all prespecified outcomes in the protocol or methods section were presented (e.g. the percentage in mean reduction in lesion counts was stated, but only absolute counts were presented); or 2) when the outcomes were incompletely reported and could not be entered in meta‐analysis (e.g. the standard deviations associated with mean reduction in lesion counts were not reported and the statistical significance between treatments was impossible to determine). We encountered this last example frequently. A few studies only gave data for only one treatment arm or pooled together for different treatment arms. For example, they did not always report adverse events separately for the different treatments. Of course, separate reports were impossible for studies using intraindividual study design.
Twenty‐seven studies reported unclear risk of bias. We refer the readers to the 'Risk of bias' tables for each included study for additional information on possible publication bias.
Effects of interventions
We presented the data and analyses of the included studies in two sections.
A) Overviews of the results with five selected outcomes (three primary and two secondary outcomes) expressed as comparative risks and risk ratios (RR) for five selected interventions in immunocompetent participants.
B) Results expressed as risk ratios (RR), number needed to treat (NNT), and mean difference (MD) presented for all interventions and all reported primary and secondary outcomes.
A) Overviews of selected interventions
Because of the variety of data presented for the different outcomes, we made a selection based on the data most frequently presented. For example, 'participant complete clearance' has been reported for target, subclinical, and all lesions, but most of the included studies reported data for all lesions. Thus, to be able to compare the different treatments and keep the summary table simple, we only included 'participant complete clearance' for all lesions. When data were presented for different cycles of treatments, only data for one cycle were included. Selections specific for one treatment are described in the comments section of the overview tables.
Diclofenac in 2.5% hyaluronic acid
Table 67 is an overview for 3% diclofenac in 2.5% hyaluronic acid.
1.
Overview for 3% diclofenac in 2.5% hyaluronic acid
| Diclofenac in 2.5% hyaluronic acid compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk With comparator |
Corresponding risk With intervention |
|||||
| Participant complete clearance | ||||||
| 3% diclofenac in 2.5% hyaluronic acid/2.5% hyaluronic acid | Study population | RR 2.46 (1.66 to 3.66) | 420 (3 studies) | ⊕⊕⊕⊝ moderate | For all lesions, data from 30, 60, and 90 day treatments pooled together, assessment at 30 days after the end of treatment (Analysis 6.5) | |
| 127 per 1000 | 313 per 1000 (211 to 466) | |||||
| Moderate | ||||||
| 132 per 1000 | 325 per 1000 (219 to 483) | |||||
| 3% diclofenac in 2.5% hyaluronic acid/5% imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT/2.5% hyaluronic acid + ALA‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean reduction in lesion counts | ||||||
| 3% diclofenac in 2.5% hyaluronic acid/2.5% hyaluronic acid | The mean reduction in lesion counts in the control groups was 2.5 lesions | The mean reduction of lesion counts in the intervention groups was 2.55 higher (1.56 to 3.53 higher) | ‐ | 345 (2 studies) | ⊕⊕⊕⊕ high | Data from 30, 60, and 90 day treatments pooled together, assessment 30 days after the end of treatment (Analysis 6.12) |
| 3% diclofenac in 2.5% hyaluronic acid/ 5% imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT/2.5% hyaluronic acid + ALA‐red light PDT | See comment | See comment | Not estimable | 10 (1 study) | ⊕⊕⊝⊝ low | Intraindividual study: at 6 weeks; diclofenac/hyaluronic acid (HA) + ALA‐PDT = 10.13, HA + ALA‐PDT= 9.9, at 6 months; diclofenac/HA + ALA‐PDT = 11.56, HA + ALA‐PDT = 10.56, at 12 months; diclofenac/HA + ALA‐PDT = 12.5, HA + ALA‐PDT = 8.8 |
| Mean percentage of reduction in lesion counts | ||||||
| All comparisons | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Withdrawal due to adverse events | ||||||
| 3% diclofenac in 2.5% hyaluronic acid/2.5% hyaluronic acid | Study population | RR 3.59 (1.92 to 6.7) | 592 (4 studies) | ⊕⊕⊕⊕ high | (Analysis 6.13) Additional data from intraindividual study: no participant withdrew because of adverse events (N = 20). GRADE = low. |
|
| 40 per 1000 | 144 per 1000 (77 to 269) | |||||
| Moderate | ||||||
| 43 per 1000 | 154 per 1000 (83 to 288) | |||||
| 3% diclofenac in 2.5% hyaluronic acid/5% imiquimod | 0 per 1000 | 0 per 1000 | Not estimable | 49 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| 3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT/2.5% hyaluronic acid + ALA‐red light PDT | 0 per 1000 | 0 per 1000 | Not estimable | 10 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| Skin irritation | ||||||
| 3% diclofenac in 2.5% hyaluronic acid/2.5% hyaluronic acid | See comment | See comment | Not estimable | 20 (1 study) | ⊕⊕⊝⊝ low | Intraindividual study reported irritation only on the diclofenac treated side of 8 out of 20 participants |
| 3% diclofenac in 2.5% hyaluronic acid/5% imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT/2.5% hyaluronic acid + ALA‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
In summary, diclofenac was significantly more efficacious than its vehicle, 2.5% hyaluronic acid. It was also associated with more adverse events, based on the number of participants who withdrew because of adverse events and the number of participants who experienced skin irritation. Diclofenac treatment in 2.5% hyaluronic acid combined with ALA‐PDT might increase the long‐term efficacy compared to ALA‐PDT with 2.5% hyaluronic acid.
5‐fluorouracil (5‐FU)
Table 68 is an overview for 5‐fluorouracil.
2.
Overview for 5‐fluorouracil
| 5‐fluorouracil (5‐FU) compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk With comparator |
Corresponding risk With intervention |
|||||
| Participant complete clearance | ||||||
| 0.5% 5‐FU/Vehicle | Study population | RR 8.86 (3.67 to 21.40) | 522 (3 studies) | ⊕⊕⊕⊕ high | Data from 1, 2, and 4 week treatments were pooled together (Analysis 9.1) | |
| 15 per 1000 | 136 per 1000 (56 to 328) | |||||
| Moderate | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | 71 per 1000 | 291 per 1000 (116 to 731) | RR 4.08 (1.63 to 10.23) | 142 (1 study) | ⊕⊕⊝⊝ low | 1 cycle (Analysis 62.1) |
| 0.5% 5‐FU/ALA‐PDT | 292 per 1000 | 499 per 1000 (239 to 1000) | RR 1.71 (0.74 to 3.98) | 48 (1 study) | ⊕⊝⊝⊝ very low | Data from blue light and pulsed dye laser were pooled (Analysis 11.1) |
| 0.5% 5‐FU/5.0% 5‐FU | See comment | See comment | Not estimable | 21 (1 study) | ⊕⊝⊝⊝ very low | Intraindividual study: 0.5% and 5.0% 5‐FU = 9/21 |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU /10% masoprocol | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/5% Imiquimod | Study population | RR 1.85 (0.41 to 8.33) | 89 (2 studies) | ⊕⊝⊝⊝ very low | (Analysis 13.1) | |
| 600 per 1000 | 1000 per 1000 (246 to 1000) | |||||
| Moderate | ||||||
| 555 per 1000 | 1000 per 1000 (230 to 1000) | |||||
| 5% 5‐FU/Carbon dioxide laser resurfacing | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Er:YAG laser resurfacing | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Cryotherapy | 680 per 1000 | 959 per 1000 (721 to 1000) | RR 1.41 (1.06 to 1.87) | 49 (1 study) | ⊕⊕⊝⊝ low | Only data after the treatment (Analysis 14.1) |
| 5% 5‐FU/Trichloroacetic acid peel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean reduction in lesion counts | ||||||
| 0.5% 5‐FU/Vehicle | The mean reduction in lesion counts in the control groups was 4 lesions | The mean reduction in lesion counts in the intervention groups was 5.40 higher (2.94 to 7.86 higher) | ‐ | 142 (1 study) | ⊕⊕⊕⊝ moderate | Data from 1, 2, and 4 week treatment were pooled. (Analysis 9.2) Results from another study (N = 177) with no SD: placebo: 2.7 lesions, 5‐FU = 8.8 lesions, GRADE = moderate |
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | The mean reduction in lesion counts in the control groups was 6.6 lesions | The mean reduction in lesion counts in the intervention groups was 2 higher (0.49 lower to 4.49 higher) | 142 (1 study) | ⊕⊕⊕⊝ moderate | 1 cycle (Analysis 62.2) | |
| 0.5% 5‐FU/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5% 5‐FU/5.0% 5‐FU | See comment | See comment | Not estimable | 21 (1 study) | ⊕⊕⊝⊝ low | Intraindividual study: results with no SD: 0.5% 5‐FU = 8.8 lesions, 5.0% 5‐FU = 6.1 lesions |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | The mean reduction in lesion counts in the control groups was 11.1 lesions | The mean reduction in lesion counts in the intervention groups was 1.2 higher (3.24 lower to 5.64 higher) | ‐ | 19 (1 study) | ⊕⊕⊝⊝ low | (Analysis 12.1) |
| 5% 5‐FU /10% masoprocol | The mean reduction in lesion counts in the control groups was 11.3 lesions | The mean reduction in lesion counts in the intervention groups was 1.5 higher (2.36 lower to 5.36 higher) | ‐ | 49 (1 study) | ⊕⊕⊝⊝ low | (Analysis 15.2) |
| 5% 5‐FU/5% Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Carbon dioxide laser resurfacing | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Er:YAG laser resurfacing | See comment | See comment | Not estimable | 55 (1 study) | ⊕⊕⊝⊝ low | Results with no SD: number of lesions at 3 months:5‐FU = 13.2, resurfacing = 13.8, at 6 months:5‐FU = 12.5, resurfacing = 13.9, at 12 months: 5‐FU = 12.4, resurfacing = 14.2 |
| 5% 5‐FU/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Trichloroacetic acid peel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean percentage of reduction in lesion counts | ||||||
| 0.5% 5‐FU/Vehicle | The mean percentage of reduction in lesion counts ranged across control groups from 28.8 per cent | The mean percentage of reduction in lesion counts in the intervention groups was 33.60 higher (22.88 to 44.32 higher) | ‐ | 142 (1 study) | ⊕⊕⊕⊝ moderate | Data from 1 week treatment.(Analysis 9.3) Results from two other studies with no SD 1) (N = 207) placebo = 21.6%, 5‐FU = 69.5%, GRADE = low, 2)(N = 177) placebo = 34.4%, 5‐FU = 78.5%, GRADE = moderate |
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | The mean percentage of reduction in lesion counts in the control groups was 45.6 per cent | The mean percentage of reduction in lesion counts in the intervention groups was 21.4 higher (5.1 to 37.7 higher) | ‐ | 142 (1 study) | ⊕⊕⊕⊝ moderate | (Analysis 62.3) |
| 0.5% 5‐FU/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5% 5‐FU/5.0% 5‐FU | See comment | See comment | Not estimable | 21 (1 study) | ⊕⊕⊝⊝ low | Intraindividual study: results with no SD: 0.5% 5‐FU = 67% and 5.0% 5‐FU = 47% |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU /10% masoprocol | The mean percentage of reduction in lesion counts in the control groups was 77.6 percent | The mean percentage of reduction in lesion counts in the intervention groups was 20 higher (11.82 to 28.18 higher) | ‐ | 49 (1 study) | ⊕⊕⊕⊝ moderate | (Analysis 15.3) |
| 5% 5‐FU/5% Imiquimod | See comment | See comment | Not estimable | 39 (1 study) | ⊕⊕⊝⊝ low | Results with no SD: 5% 5‐FU = 94%, 5% imiquimod = 66% |
| 5% 5‐FU/Carbon dioxide laser resurfacing | The mean percentage of reduction in lesion counts in the control groups was 92 percent | The mean percentage of reduction in lesion counts in the intervention groups was 8.80 lower (20.76 lower to 3.16 higher) | ‐ | 14 (1 study) | ⊕⊝⊝⊝ very low | (Analysis 16.1 ) |
| 5% 5‐FU/Er:YAG laser resurfacing | See comment | See comment | Not estimable | 55 (1 study) | ⊕⊕⊝⊝ low | Results with no SD: at 6 months: 5‐FU = 79.2%, resurfacing 94.5%, at 12 months: 5‐FU = 76.6%, resurfacing = 91.1% |
| 5% 5‐FU/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Trichloroacetic acid peel | The mean percentage of reduction in lesion counts in the control groups was 89 per cent | The mean percentage of reduction in lesion counts in the intervention groups was 5.8 lower (15.38 lower to 3.78 higher) | ‐ | 18 (1 study) | ⊕⊝⊝⊝ very low | (Analysis 18.1) |
| Withdrawal due to adverse events | ||||||
| 0.5% 5‐FU/Vehicle | 0 per 1000 | N/A (5/119 = 42/1000) | RR 5.41 (0.3 to 96.18) | 177 (1 study) | ⊕⊝⊝⊝ very low | Data from 1, 2, and 4 week treatments were pooled.(Analysis 9.4) Another study reported 24/207 participants withdrew because of adverse events and 12 of them were in 4 week 5‐FU group. GRADE = low |
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | See comment | See comment | Not estimable | 142 (1 study) | ⊕⊕⊝⊝ low | There were no participant withdrawals in the first part of this three part study (incomplete data were given for the whole study). |
| 0.5% 5‐FU/ALA‐PDT | 0 per 1000 | N/A (1/12 = 83/1000) | RR 5.77 (0.25 to 131.92) | 36 (1 study) | ⊕⊕⊝⊝ low | Data from blue light and pulsed dye laser were pooled (Analysis 11.2) |
| 0.5% 5‐FU/5.0% 5‐FU | See comment | See comment | Not estimable | 21 (1 study) | ⊕⊕⊝⊝ low | Intraindividual study: 16/21 discontinued treatment but did not withdraw: 4 because of 0.5%, 8 because of 5.0% , 4 because of both creams. |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | See comment | See comment | Not estimable | 19 (1 study) | ⊕⊕⊝⊝ low | Intraindividual study: 1 participant withdrew because of irritation but associated treatment was not specified. |
| 5% 5‐FU /10% masoprocol | 0 per 1000 | N/A (1/30 = 33/1000) | RR 2.71 (0.12 to 63.84) | 57 (1 study) | ⊕⊕⊝⊝ low | (Analysis 15.4) |
| 5% 5‐FU/5% Imiquimod | 0 per 1000 | 0 per 1000 | Not estimable | 89 (2 studies) | ⊕⊕⊝⊝ low | There were no participant withdrawals due to adverse events. |
| 5% 5‐FU/Carbon dioxide laser resurfacing | 250 per 1000 | 45 per 1000 (2 to 817) | RR 0.18 (0.01 to 3.27) | 17 (1 study) | ⊕⊕⊝⊝ low | (Analysis 16.2) |
| 5% 5‐FU/Er:YAG laser resurfacing | 0 per 1000 | N/A (1/27 = 37/1000) | RR 3.11 (0.13 to 73.11) | 55 (1 study) | ⊕⊕⊝⊝ low | (Analysis 17.1) |
| 5% 5‐FU/Cryotherapy | 0 per 1000 | 0 per 1000 | Not estimable | 49 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| 5% 5‐FU/Trichloroacetic acid peel | 0 per 1000 | 0 per 1000 | Not estimable | 18 (1 study) | ⊕⊕⊝⊝ low | There were no participant withdrawals due to adverse events. |
| Skin irritation | ||||||
| 0.5% 5‐FU/Vehicle | 654 per 1000 | 948 per 1000 (830 to 1000) | RR 1.45 (1.27 to 1.65) | 384 (2 studies) | ⊕⊕⊕⊝ moderate | Data from 1, 2, and 4 week treatments were pooled (Analysis 9.5) |
| 0.5% 5‐FU with cryotherapy/Vehicle with cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5% 5‐FU/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5% 5‐FU/5.0% 5‐FU | 1000 per 1000 | 1000 per 1000 | ‐ | 21 (1 study) | ⊕⊕⊕⊝ moderate | Intraindividual study: All participants reported facial irritation in association with both creams |
| 5% 5‐FU with 0.05% tretinoin /5% 5‐FU with placebo | See comment | See comment | Not estimable | 19 (1 study) | ⊕⊕⊕⊝ moderate | Intraindividual study: 12 had more irritation with tretinoin, 4 had more with placebo, and 3 had equal irritation. |
| 5% 5‐FU /10% masoprocol | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/5% Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Carbon dioxide laser resurfacing | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Er:YAG laser resurfacing | 429 per 1000 | 703 per 1000 (429 to 1000) | RR 1.64 (1 to 2.69) | 55 (1 study) | ⊕⊕⊝⊝ low | At the end of treatment (Analysis 17.2) |
| 5% 5‐FU/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% 5‐FU/Trichloroacetic acid peel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
In summary, 0.5% and 5% 5‐fluorouracil treatments resulted in similar efficacy and safety based on 1 study comparing them directly. 5‐Fluorouracil was significantly more efficacious than vehicle and cryotherapy, but similar to ALA‐PDT (see PDT overview table: Table 69) and carbon dioxide laser resurfacing. More studies are needed to confirm its superiority to masoprocol and imiquimod and its long‐term inferiority to Er:YAG laser resurfacing. In 1 study, additional treatment with 5‐fluorouracil increased the efficacy of cryotherapy with vehicle, but the efficacy (illustrative comparative risks) of cryotherapy alone in this study seemed much lower than other studies investigating cryotherapy (see cryotherapy overview table: Table 70). On the other hand, additional treatment with tretinoin did not improve the efficacy of 5‐fluorouracil. In general, 5‐fluorouracil treatment did not lead to withdrawal because of adverse events; however, substantial skin irritation was associated with this intervention.
3.
Overview for photodynamic therapy
| Photodynamic therapy compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk With comparator |
Corresponding risk With intervention |
|||||
| Participant complete clearance | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) |
83 per 1000 | 500 per 1000 (71 to 1000) | RR 6 (0.85 to 42.59) | 24 (1 study) | ⊕⊝⊝⊝ very low | (Analysis 48.1) |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) |
500 per 1000 | 500 per 1000 (225 to 1000) | RR 1 (0.45 to 2.23) | 24 (1 study) | ⊕⊝⊝⊝ very low | (Analysis 50.1) |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) |
97 per 1000 | 602 per 1000 (279 to 1000) | RR 6.22 (2.88 to 13.43) | 243 (1 study) | ⊕⊝⊝⊝ very low | 1 treatment. (Analysis 47.1) Additional intraindividual study: ALA‐PDT: 16/35, placebo‐PDT = 2/35. GRADE = moderate |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) |
500 per 1000 | 85 per 1000 (10 to 590) | RR 0.17 (0.02 to 1.18) | 24 (1 study) | ⊕⊝⊝⊝ very low | (Analysis 50.1) |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | 474 per 1000 | 237 per 1000 (118 to 469) | RR 0.5 (0.25 to 0.99) | 72 (1 study) | ⊕⊕⊝⊝ low | Data from assessment at 8 weeks after the end of treatment (Analysis 49.2) |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) |
471 per 1000 | 235 per 1000 (118 to 475) | RR 0.5 (0.25 to 1.01) | 68 (1 study) | ⊕⊝⊝⊝ very low | Data from assessment at 8 weeks after the end of treatment (Analysis 49.2) |
| 0.5h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | 735 per 1000 | 235 per 1000 (125 to 449) | RR 0.32 (0.17 to 0.61) | 68 (1 study) | ⊕⊕⊝⊝ low | Data from assessment at 8 weeks after the end of treatment (Analysis 49.2) |
| 1h ALA‐red light PDT /2h ALA‐red light PDT (individual lesions) | 471 per 1000 | 475 per 1000 (292 to 772) | RR 1.01 (0.62 to 1.64) | 72 (1 study) | ⊕⊝⊝⊝ very low | Data from assessment at 8 weeks after the end of treatment (Analysis 49.2) |
| 1h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | 735 per 1000 | 471 per 1000 (324 to 699) | RR 0.64 (0.44 to 0.95) | 72 (1 study) | ⊕⊕⊝⊝ low | Data from assessment at 8 weeks after the end of treatment (Analysis 49.2) |
| 2h ALA‐red light PDT/4h ALA‐red light PDT (individual lesions) | 735 per 1000 | 471 per 1000 (309 to 706) | RR 0.64 (0.42 to 0.96) | 68 (1 study) | ⊕⊕⊝⊝ low | Data from assessment at 8 weeks after the end of treatment (Analysis 49.2) |
| 3‐4h ALA‐red light PDT/3 to 4h placebo‐red light PDT (individual lesions) |
89 per 1000 | 527 per 1000 (297 to 935) | RR 5.94 (3.35 to 10.54) | 422 (3 studies) | ⊕⊕⊕⊕ high | 1 treatment (Analysis 47.1) |
| 3% diclofenac in 2.5% hyaluronan gel + 4h ALA‐red light PDT /2.5% hyaluronan gel + 4h ALA‐red light PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 4h ALA‐red light PDT/Cryotherapy (individual lesions) |
443 per 1000 | 580 per 1000 (465 to 726) | RR 1.31 (1.05 to 1.64) | 297 (1 study) | ⊕⊕⊝⊝ low | (Analysis 51.1) |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) |
See comment | See comment | Not estimable | 25 (1 study) | ⊕⊕⊝⊝ low | Intraindividual study: ALA‐PDT + 5% imiquimod = 2/25; ALA‐PDT + placebo = 2/25 |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) |
See comment | See comment | Not estimable | 16 (1 study) | ⊕⊕⊕⊝ moderate | Intraindividual study: ALA‐PDT = 6/16, MAL‐PDT = 7/16 |
| MAL‐PDT | ||||||
| All day 16% MAL‐daylight PDT /All day 8% MAL‐daylight PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2h MAL‐daylight PDT /3h MAL‐daylight PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2.5‐4h MAL‐red light PDT /2.5‐4h placebo‐red light PDT (individual lesions) |
147 per 1000 | 656 per 1000 (466 to 924) | RR 4.46 (3.17 to 6.28) | 482 (5 studies) | ⊕⊕⊕⊝ moderate | (Analysis 52.1) |
| 3h MAL‐red light LED PDT /3h MAL‐broad visible + water‐filtered infrared A PDT (individual lesions) |
500 per 1000 | 575 per 1000 (380 to 865) | RR 1.15 (0.76 to 1.73) | 80 (1 study) | ⊕⊕⊝⊝ low | Data from assessment at 12 weeks after the end of treatment.(Analysis 53.1) |
| 3h MAL‐red light LED PDT /3h MAL‐daylight PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Single 3h MAL‐red light PDT /Multiple 3h MAL‐red light PDT [2 treatments 1 week apart] (individual lesions) |
755 per 1000 | 883 per 1000 (777 to 1000) | RR 1.17 (1.03 to 1.33) | 211 (1 study) | ⊕⊕⊝⊝ low | (Analysis 57.1) |
| 3h MAL‐red light PDT /Cryotherapy (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean reduction in lesion counts | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT /2h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3‐4h ALA‐red light PDT /3‐4h placebo‐red light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid gel + 4h ALA‐red light PDT /2.5% hyaluronic acid gel + 4h ALA‐red light PDT (field‐directedtreatments) |
See comment | See comment | Not estimable | 10 (1 study) | ⊕⊕⊝⊝ low | Intraindividual study: at 6 weeks; diclofenac/hyaluronic acid (HA) + ALA‐PDT = 10.13, HA + ALA‐PDT= 9.9, at 6 months:; diclofenac/HA + ALA‐PDT = 11.56, HA + ALA‐PDT = 10.56, at 12 months; diclofenac/HA + ALA‐PDT = 12.5, HA + ALA‐PDT = 8.8 |
| 4h ALA‐red light PDT /Cryotherapy (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) |
See comment | See comment | Not estimable | 25 (1 study) | ⊕⊕⊝⊝ low | Results from intraindividual study without SD: ALA‐PDT + 5% imiquimod = 19.9 lesions; ALA‐PDT + placebo = 16.0 lesions |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) |
The mean reduction in lesion counts in the control groups was 5.6 lesions | The mean reduction in lesion counts in the intervention groups was 0.6 higher (1.28 lower to 2.48 higher) | ‐ | 15 (1 study) | ⊕⊕⊝⊝ low | (Analysis 59.1) |
| MAL‐PDT | ||||||
| All day 16% MAL‐daylight PDT /All day 8% MAL‐daylight PDT (field‐directedtreatments) |
The mean reduction in lesion counts in the control groups was 14.5 lesions | The mean reduction in lesion counts in the intervention groups was 0.3 higher (3.77 lower to 4.37 higher) | ‐ | 29 (1 study) | ⊕⊕⊝⊝ low | (Analysis 56.1) |
| 2h MAL‐daylight PDT /3h MAL‐daylight PDT (field‐directedtreatments) |
The mean reduction in lesion counts in the control groups was 9.7 lesions | The mean reduction in lesion counts in the intervention groups was 0.1 higher (3.17 lower to 3.37 higher) | 120 (1 study) | ⊕⊕⊝⊝ low | (Analysis 55.1) | |
| 2.5‐4h MAL‐red light PDT /2.5‐4h placebo‐red light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light LED PDT /3h MAL‐broad visible + water‐filtered infrared A PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light LED PDT /3h MAL‐daylight PDT (field‐directedtreatments) |
The mean reduction in lesion counts in the control groups was 8.4 lesions | The mean reduction in lesion counts in the intervention groups was 0.4 lower (3.23 lower to 2.43 higher) | ‐ | 29 (1 study) | ⊕⊕⊝⊝ low | (Analysis 54.1) |
| Single 3h MAL‐red light PDT /Multiple 3h MAL‐red light PDT [2 treatments 1 week apart] (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light PDT /Cryotherapy (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean percentage of reduction in lesion count | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT /2h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3‐4h ALA‐red light PDT /3‐4h placebo‐red light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3% diclofenac in 2.5% hyaluronic acid gel + 4h ALA‐red light PDT /2.5% hyaluronic acid gel + 4h ALA‐red light PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 4h ALA‐red light PDT /Cryotherapy (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) |
See comment | See comment | Not estimable | 25 (1 study) | ⊕⊕⊝⊝ low | Results from intraindividual study without SD: ALA‐PDT + 5% imiquimod = 86.7%; ALA‐PDT + placebo = 73.1% |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) |
‐ | See comment | ‐ | ‐ | ‐ | Not reported |
| MAL‐PDT | ||||||
| All day 16% MAL‐daylight PDT /All day 8% MAL‐daylight PDT (field‐directedtreatments) |
See comment | See comment | Not estimable | 29 (1 study) | ⊕⊕⊝⊝ low | Data with no SD: 16% MAL‐daylight PDT = 76.9%, 8% MAL‐daylight PDT = 79.5%. |
| 2h MAL‐daylight PDT /3h MAL‐daylight PDT (field‐directedtreatments) |
The mean percentage of reduction in lesion counts in the control groups was 74.6 percent | The mean percentage of reduction in lesion counts in the intervention groups was 2.6 higher (6.46 lower to 11.66 higher) | ‐ | 120 (1 study) | ⊕⊕⊝⊝ low | (Analysis 55.2) |
| 2.5‐4h MAL‐red light PDT /2.5‐4h placebo‐red light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light LED PDT /3h MAL‐broad visible + water‐filtered infrared A PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light LED PDT /3h MAL‐daylight PDT (field‐directedtreatments) |
See comment | See comment | Not estimable | 29 (1 study) | ⊕⊕⊝⊝ low | Data with no SD: MAL‐red light LED PDT = 71%, MAL‐daylight PDT = 79%. |
| Single 3h MAL‐red light PDT /Multiple 3h MAL‐red light PDT [2 treatments 1 week apart] (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3h MAL‐red light PDT /Cryotherapy (individual lesions) |
See comment | See comment | Not estimable | 240 (2 studies) | ⊕⊝⊝⊝ very low | Intraindividual studies with no SD: at 12 weeks: MAL‐PDT = 84.4%, cryotherapy = 74.5%, at 24 weeks: MAL‐PDT = 75‐86.7%, cryotherapy = 83.9‐87% |
| Withdrawal due to adverse events | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) |
0 per 1000 | 0 per 1000 | Not estimable | 24 (1 study) | ⊕⊕⊝⊝ low | There were no participant withdrawals due to adverse events. |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) |
83 per 1000 | 28 per 1000 (1 to 621) | RR 0.33 (0.01 to 7.45) | 24 (1 study) | ⊕⊕⊝⊝ low | (Analysis 50.3) |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) |
0 per 1000 | 0 per 1000 | Not estimable | 271 (2 studies) | ⊕⊕⊝⊝ low | There were no participant withdrawals due to adverse events. |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) |
83 per 1000 | 28 per 1000 (1 to 621) | RR 0.33 (0.01 to 7.45) | 24 (1 study) | ⊕⊕⊝⊝ low | (Analysis 50.3) |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 72 (1 study) | ⊕⊝⊝⊝ very low | No details were given for the reasons for withdrawal. |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) |
See comment | See comment | Not estimable | 68 (1 study) | ⊕⊝⊝⊝ very low | No details were given for the reasons for withdrawal. |
| 0.5h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 68 (1 study) | ⊕⊝⊝⊝ very low | No details were given for the reasons for withdrawal. |
| 1h ALA‐red light PDT /2h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 72 (1 study) | ⊕⊝⊝⊝ very low | No details were given for the reasons for withdrawal. |
| 1h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 72 (1 study) | ⊕⊝⊝⊝ very low | No details were given for the reasons for withdrawal. |
| 2h ALA‐red light PDT /4h ALA‐red light PDT (individual lesions) | See comment | See comment | Not estimable | 68 (1 study) | ⊕⊝⊝⊝ very low | No details were given for the reasons for withdrawal. |
| 3‐4h ALA‐red light PDT /3‐4h placebo‐red light PDT (individual lesions) |
0 per 1000 | 0 per 1000 | Not estimable | 391 (3 studies) | ⊕⊕⊕⊕ high | There were no participant withdrawals due to adverse events. |
| 3% diclofenac in 2.5% hyaluronic acid gel + 4h ALA‐red light PDT /2.5% hyaluronic acid gel + 4h ALA‐red light PDT (field‐directedtreatments) |
0 per 1000 | 0 per 1000 | Not estimable | 10 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| 4h ALA‐red light PDT /Cryotherapy (individual lesions) |
0 per 1000 | 0 per 1000 | Not estimable | 255 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | 0 per 1000 | 0 per 1000 | Not estimable | 30 (1 study) | ⊕⊕⊝⊝ low | There were no participant withdrawals due to adverse events. |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) |
0 per 1000 | 0 per 1000 | Not estimable | 25 (1 study) | ⊕⊕⊝⊝ low | There were no participant withdrawals due to adverse events. |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) |
0 per 1000 | 0 per 1000 | Not estimable | 15 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| MAL‐PDT | ||||||
| All day 16% MAL‐daylight PDT /All day 8% MAL‐daylight PDT (field‐directedtreatments) |
See comment | See comment | Not estimable | 29 (1 study) | ⊕⊕⊕⊝ moderate | One of 30 participants withdrew because of adverse events unrelated to treatments. |
| 2h MAL‐daylight PDT /3h MAL‐daylight PDT (field‐directedtreatments) |
0 per 1000 | 0 per 1000 | Not estimable | 120 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| 2.5‐4h MAL‐red light PDT /2.5‐4h placebo‐red light PDT (individual lesions) |
0 per 1000 | N/A (3/130 = 23/1000) | RR 2 (0.23 to 17.74) | 191 (2 studies) | ⊕⊝⊝⊝ very low | (Analysis 52.3) Two additional studies with no participant withdrawals because of adverse events (N = 211). GRADE = low |
| 3h MAL‐red light LED PDT /3h MAL‐broad visible + water‐filtered infrared A PDT (individual lesions) |
0 per 1000 | 0 per 1000 | Not estimable | 78 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| 3h MAL‐red light LED PDT /3h MAL‐daylight PDT (field‐directedtreatments) |
0 per 1000 | 0 per 1000 | Not estimable | 29 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| Single 3h MAL‐red light PDT /Multiple 3h MAL‐red light PDT [2 treatments 1 week apart] (individual lesions) |
9 per 1000 | 3 per 1000 (0 to 77) | RR 0.34 (0.01 to 8.17) | 211 (1 study) | ⊕⊕⊝⊝ low | (Analysis 57.2) |
| 3h MAL‐red light PDT /Cryotherapy (individual lesions) |
11 per 1000 | 10 per 1000 (1 to 67) | RR 0.94 (0.14 to 6.36) | 379 (2 studies) | ⊕⊕⊕⊝ moderate | (Analysis 58.1) Two additional intraindividual studies: 4 of 119 and 2 of 121 participants withdrew because of adverse events and one of them was related to MAL‐PDT. GRADE = low |
| Skin irritation | ||||||
| ALA‐PDT | ||||||
| 1h ALA‐blue light PDT /1h ALA‐pulsed dye laser PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐blue light PDT /0.5% 5‐FU (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 14‐18h ALA‐blue light PDT /14‐18h placebo‐blue light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐pulsed dye laser PDT /0.5% 5‐FU (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/1h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 0.5h ALA‐red light PDT/4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT/2h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 1h ALA‐red light PDT/4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 2h ALA‐red light PDT/4h ALA‐red light PDT (individual lesions) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3 to 4h ALA‐red light PDT /3 to 4h placebo‐red light PDT (individual lesions) |
0 per 1000 | N/A (77/217 = 355/1000) | RR 59.72 (3.75 to 952.48) | 300 (2 studies) | ⊕⊕⊕⊝ moderate | Data for ALA‐PDT was given separately for two studies but not for placebo. Data from assessment after treatment (Analysis 47.7) |
| 3% diclofenac in 2.5% hyaluronic acid gel + 4h ALA‐red light PDT /2.5% hyaluronic acid gel + 4h ALA‐red light PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 4h ALA‐red light PDT /Cryotherapy (individual lesions) |
101 per 1000 | 371 per 1000 (220 to 627) | RR 3.69 (2.19 to 6.23) | 297 (1 study) | ⊕⊕⊝⊝ low | Assessment one day after the treatment (Analysis 51.2) |
| ALA‐red light PDT (individual lesions)/5% imiquimod (field‐directedtreatment) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐blue light PDT + 5% imiquimod / ALA‐blue light PDT + placebo (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐PDT versus MAL‐PDT | ||||||
| 5h ALA‐red light PDT /3h MAL‐red light PDT (field‐directedtreatments) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| MAL‐PDT | ||||||
| All comparisons | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
4.
Overview for cryotherapy
| Cryotherapy compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/ Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ‐ | With comparator | With intervention | ‐ | ‐ | ‐ | ‐ |
| Participant complete clearance | ||||||
| Cryotherapy /Betulin‐based oleogel | 643 per 1000 | 784 per 1000 (489 to 1000) | RR 1.22 (0.76 to 1.97) | 28 (1 study) | ⊕⊝⊝⊝ very low | (Analysis 42.1) |
| Cryotherapy/cryotherapy with betulin‐based oleogel | 714 per 1000 | 786 per 1000 (514 to 1000) | RR 1.1 (0.72 to 1.69) | 28 (1 study) | ⊕⊝⊝⊝ very low | (Analysis 61.1) |
| Cryotherapy/5% 5‐FU | 958 per 1000 | 680 per 1000 (518 to 901) | RR 0.71 (0.54 to 0.94) | 49 (1 study) | ⊕⊕⊝⊝ low | Assessment after treatment (Analysis 43.1) |
| Vehicle with cryotherapy/0.5% 5‐FU with cryotherapy | 292 per 1000 | 70 per 1000 (29 to 178) | RR 0.24 (0.1 to 0.61) | 142 (1) | ⊕⊕⊝⊝ low | 1 cycle (Analysis 63.1) |
| Cryotherapy /Imiquimod | 846 per 1000 | 677 per 1000 (499 to 931) | RR 0.8 (0.59 to 1.10) | 51 (1 study) | ⊕⊝⊝⊝ very low | 5% imiquimod (Analysis 44.1) |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | Study population | RR 0.2 (0.05 to 0.73) | 311 (2 studies) | ⊕⊕⊕⊕ high | Pooled data (5% and 3.75% imiquimod)(Analysis 64.1) Results from an additional intraindividual study: cryotherapy + vehicle = 5/27, cryotherapy+imiquimod = 8/27 GRADE = moderate |
|
| 287 per 1000 | 57 per 1000 (14 to 209) | |||||
| Moderate | ||||||
| 264 per 1000 | 53 per 1000 (13 to 193) | |||||
| Cryotherapy /ALA‐red light PDT | 581 per 1000 | 442 per 1000 (354 to 558) | RR 0.76 (0.61 to 0.96) | 297 (1 study) | ⊕⊕⊝⊝ low | (Analysis 46.1) |
| Cryotherapy/MAL‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean reduction in lesion counts | ||||||
| Cryotherapy /Betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/cryotherapy with betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Vehicle + cryotherapy/0.5% 5‐FU + cryotherapy | The mean reduction in lesion counts in the control groups was 8.6 lesions | The mean reduction in lesion counts in the intervention groups was 2 lower (4.49 lower to 0.49 higher) | ‐ | 142 (1) | ⊕⊕⊕⊝ moderate | 1 cycle (Analysis 63.2) |
| Cryotherapy /Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy /ALA‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/MAL‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Mean percentage of reduction in lesion counts | ||||||
| Cryotherapy /Betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/cryotherapy with betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Vehicle with cryotherapy/0.5% 5‐FU with cryotherapy | The mean percentage of reduction in lesion counts in the control groups was 67 percent | The mean percentage of reduction in lesion counts in the intervention groups was 21.4 lower (37.7 to 5.1 lower) | ‐ | 142 (1) | ⊕⊕⊕⊝ moderate | (Analysis 63.3) |
| Cryotherapy /Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | See comment | See comment | ‐ | 301 (2 studies) | ⊕⊝⊝⊝ very low | High heterogeneity (I2=86%) between 3.75% (parallel group, MD ‐34.10, 95% CI ‐41.38 to ‐26.82)) and 5.0% (intraindividual, MD ‐11.20, 95% CI ‐26.53 to 4.13) imiquimod studies. (Analysis 64.4) |
| Cryotherapy /ALA‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/MAL‐red light PDT | See comment | See comment | Not estimable | 240 (2 studies) | ⊕⊝⊝⊝ very low | Intraindividual studies with no SD: at 12 weeks: cryotherapy = 74.5%, MAL‐PDT= 84.4%, at 24 weeks: cryotherapy = 83.9‐87%, MAL‐PDT = 75‐86.7% |
| Withdrawal due to adverse events | ||||||
| Cryotherapy /Betulin‐based oleogel | 0 per 1000 | 0 per 1000 | Not estimable | 28 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| Cryotherapy/cryotherapy with betulin‐based oleogel | 0 per 1000 | 0 per 1000 | Not estimable | 28 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| Cryotherapy/5% 5‐FU | 0 per 1000 | 0 per 1000 | Not estimable | 49 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| Vehicle with cryotherapy/0.5% 5‐FU with cryotherapy | See comment | See comment | Not estimable | 142 (1 study) | ⊕⊕⊝⊝ low | There were no participant withdrawals due to adverse events in the first part of this three part study (incomplete data were given for the whole study). |
| Cryotherapy /Imiquimod | 0 per 1000 | 0 per 1000 | Not estimable | 51 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | Study population | RR 0.93 (0.28 to 3.07) | 312 (2 studies) | ⊕⊕⊕⊝ moderate | Pooled data (5% and 3.75% imiquimod) (Analysis 64.6) | |
| 33 per 1000 | 30 per 1000 (9 to 100) | |||||
| Moderate | ||||||
| 21 per 1000 | 20 per 1000 (6 to 64) | |||||
| Cryotherapy /ALA‐ red light PDT | 0 per 1000 | 0 per 1000 | Not estimable | 297 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| Cryotherapy/MAL‐ red light PDT | 11 per 1000 | 11 per 1000 (2 to 75) | RR 1.06 (0.16 to 7.16) | 379 (2 studies) | ⊕⊕⊕⊝ moderate | (Analysis 45.2) Two additional intraindividual studies: 4 of 119 and 2 of 121 participants withdrew because of adverse events and one of them was related to MAL‐PDT. GRADE = low |
| Skin irritation | ||||||
| Cryotherapy /Betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/cryotherapy with betulin‐based oleogel | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy/5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Vehicle with cryotherapy/0.5% 5‐FU with cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy /Imiquimod | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy with vehicle /Cryotherapy with imiquimod | Study population | RR 0.39 (0.1 to 1.54) | 311 (2 studies) | ⊕⊕⊕⊝ moderate | Pooled data (5% and 3.75% imiquimod) (Analysis 64.7) |
|
| 83 per 1000 | 32 per 1000 (8 to 128) | |||||
| Moderate | ||||||
| 125 per 1000 | 49 per 1000 (13 to 192) | |||||
| Cryotherapy /ALA‐red light PDT | 372 per 1000 | 100 per 1000 (59 to 171) | RR 0.27 (0.16 to 0.46) | 297 (1 study) | ⊕⊕⊝⊝ low | Assessment one day after the treatment (Analysis 46.2) |
| Cryotherapy/MAL‐red light PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
Imiquimod
Table 71 is an overview for imiquimod.
5.
Overview for imiquimod
| Imiquimod compared to interventions for actinic keratoses in immunocompetent participants | ||||||
| Intervention/Comparison intervention | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| ‐ | With comparator | With intervention | ‐ | ‐ | ‐ | ‐ |
| Participant complete clearance | ||||||
| 2.5% imiquimod/placebo | 62 per 1000 | 277 per 1000 (148 to 518) | RR 4.49 (2.4 to 8.39) | 486 (2 studies) | ⊕⊕⊕⊕ high | (Analysis 20.1) |
| 3.75% imiquimod/placebo | Study population | RR 6.45 (3.87 to 10.73) | 730 (3 studies) | ⊕⊕⊕⊕ high | (Analysis 20.1) | |
| 53 per 1000 | 343 per 1000 (206 to 571) | |||||
| Moderate | ||||||
| 50 per 1000 | 322 per 1000 (193 to 536) | |||||
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | 33 per 1000 | 301 per 1000 (111 to 820) | RR 9.12 (3.36 to 24.79) | 247 (1 study) | ⊕⊕⊕⊝ moderate | For all lesions (Analysis 65.1) |
| 5% imiquimod/placebo | Study population | RR 7.70 (4.63 to 12.79) | 1871 (9 studies) | ⊕⊕⊕⊕ high | (Analysis 20.1) | |
| 48 per 1000 | 371 per 1000 (223 to 617) | |||||
| Moderate | ||||||
| 32 per 1000 | 246 per 1000 (148 to 409) | |||||
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod /5% 5‐FU | See comment | See comment | ‐ | 89 (2 studies) | ⊕⊝⊝⊝ very low | The two studies was associated with high heterogeneity (I²= 93%) and the results could not be pooled together. One study favoured 5‐FU (RR 0.31, 95% CI 0.14 to 0.67] whereas the other did not (RR 0.88, 95% CI 0.73 to 1.06] (Analysis 22.1) |
| 5% imiquimod/Cryotherapy | 680 per 1000 | 843 per 1000 (619 to 1000) | RR 1.24 (0.91 to 1.7) | 51 (1 study) | ⊕⊝⊝⊝ very low | (Analysis 23.1) |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | 91 per 1000 | 225 per 1000 (64 to 796) | RR 2.48 (0.70 to 8.76) | 64 (1 study) | ⊕⊕⊝⊝ low | For all lesions.(Analysis 65.1) Results from an additional intraindividual study: cryotherapy + imiquimod side (8/27 = 30%), cryotherapy alone side (5/27 = 19%), GRADE = low |
| 5% imiquimod/ALA‐PDT | ‐ | Not reported | ||||
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | See comment | See comment | Not estimable | 25 (1 study) | ⊕⊕⊝⊝ low | Intraindividual study: ALA‐PDT + 5% imiquimod = 2/25; ALA‐PDT + placebo = 2/25 |
| Mean reduction in lesion counts | ||||||
| 2.5% imiquimod/placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3.75% imiquimod/placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/placebo | The mean reduction in lesion counts in the control groups was 0.6 lesions | The mean reduction in lesion counts in the intervention groups was 2.20 higher (1.05 lower to 5.45 higher) | ‐ | 12 (1 study) | ⊕⊕⊝⊝ low | (Analysis 19.5) Results from an additional intraindividual study with no SD (N = 21): 5% imiquimod: 3.9 lesions, placebo = 0.5 lesions, GRADE = very low |
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod /5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | See comment | See comment | Not estimable | 25 (1 study) | ⊕⊕⊝⊝ low | Results from intraindividual study without SD: ALA‐PDT + 5% imiquimod= 19.9 lesions; ALA‐PDT + placebo= 16.0 lesions |
| Mean percentage of reduction in lesion counts | ||||||
| 2.5% imiquimod/placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 3.75% imiquimod/placebo | The mean percentage of reduction in lesion counts in the control groups was 21.1 per cent | The mean percentage of reduction in lesion counts in the intervention groups was 46.90 higher (36.68 to 57.12 higher) | ‐ | 247 (1 study) | ⊕⊕⊕⊝ moderate | (Analysis 20.3) |
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | The mean percentage of reduction in lesion counts in the control groups was 43.3 per cent | The mean percentage of reduction in lesion counts in the intervention groups was 34.1 higher (26.82 to 41.38 higher) | ‐ | 247 (1 study) | ⊕⊕⊕⊝ moderate | For all lesions (Analysis 65.2) |
| 5% imiquimod/placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod /5% 5‐FU | See comment | See comment | Not estimable | 39 (1 study) | ⊕⊕⊝⊝ low | Results with no SD: 5% imiquimod = 66%, 5% 5‐FU = 94% |
| 5% imiquimod/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | The mean percentage of reduction in lesion counts in the control groups was 62 per cent | The mean percentage of reduction in lesion counts in the intervention groups was 11.2 higher (4.13 lower to 26.53 higher) | ‐ | 27 (1 study) | ⊕⊕⊝⊝ low | For all lesions.(Analysis 65.2) Results from an additional intraindividual study: cryotherapy‐5% imiquimod = 73.2+27.1%, cryotherapy + vehicle = 62.0+30.3%. GRADE = moderate |
| 5% imiquimod/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | See comment | See comment | Not estimable | 25 (1 study) | ⊕⊕⊝⊝ low | Results from intraindividual study without SD: ALA‐PDT + 5% imiquimod = 86.7% ; ALA‐PDT + placebo = 73.1 % |
| Withdrawal due to adverse events | ||||||
| 2.5% imiquimod/placebo | 19 per 1000 | 9 per 1000 (2 to 50) | RR 0.5 (0.09 to 2.7) | 486 (2 studies) | ⊕⊕⊕⊝ moderate | (Analysis 20.5) |
| 3.75% imiquimod/placebo | 19 per 1000 | 17 per 1000 (4 to 73) | RR 0.92 (0.22 to 3.93) | 483 (2 studies) | ⊕⊕⊕⊝ moderate | (Analysis 20.5) |
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | 32 per 1000 | 41 per 1000 (11 to 150) | RR 1.3 (0.36 to 4.73) | 247 (1 study) | ⊕⊕⊝⊝ low | (Analysis 65.3) |
| 5% imiquimod/placebo | Study population | RR 2.59 (1.59 to 4.23) | 2290 (8 studies) | ⊕⊕⊕⊝ moderate | (Analysis 20.5) Four small sample size studies with no participant withdrawal are not included in meta‐analysis: pooled data, imiquimod 0/79 and placebo 0/31. Additional two intraindividual studies: no participant withdrew because of adverse events (0/42) GRADE = very low (both studies had more than 20% participant lost). | |
| 21 per 1000 | 56 per 1000 (34 to 91) | |||||
| Moderate | ||||||
| 5 per 1000 | 13 per 1000 (8 to 22) | |||||
| High | ||||||
| 0 per 1000 | 0 per 1000 (0 to 0) | |||||
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | 0 per 1000 | 0 per 1000 | Not estimable | 49 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| 5% imiquimod /5% 5‐FU | 0 per 1000 | 0 per 1000 | Not estimable | 50 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| 5% imiquimod/Cryotherapy | 0 per 1000 | 0 per 1000 | Not estimable | 51 (1 study) | ⊕⊕⊕⊝ moderate | There were no participant withdrawals due to adverse events. |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | 30 per 1000 | 10 per 1000 (0 to 246) | RR 0.34 (0.01 to 8.13) | 65 (1 study) | ⊕⊕⊝⊝ low | (Analysis 65.3) |
| 5% imiquimod/ALA‐PDT | 0 per 1000 | 0 per 1000 | Not estimable | 30 (1 study) | ⊕⊕⊝⊝ low | There were no participant withdrawals due to adverse events. |
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | 0 per 1000 | 0 per 1000 | Not estimable | 25 (1 study) | ⊕⊕⊝⊝ low | There were no participant withdrawals due to adverse events. |
| Skin irritation | ||||||
| 2.5% imiquimod/placebo | 6 per 1000 | 21 per 1000 (4 to 117) | RR 3.45 (0.63 to 18.97) | 486 (2 studies) | ⊕⊕⊕⊝ moderate | (Analysis 20.6) |
| 3.75% imiquimod/placebo | 6 per 1000 | 30 per 1000 (6 to 159) | RR 4.86 (0.92 to 25.83) | 484 (2 studies) | ⊕⊕⊕⊝ moderate | (Analysis 20.6) |
| Cryotherapy + 3.75% imiquimod/Cryotherapy + vehicle | 8 per 1000 | 56 per 1000 (7 to 445) | RR 6.72 (0.84 to 53.83) | 247 (1 study) | ⊕⊕⊝⊝ low | (Analysis 65.4) |
| 5% imiquimod/placebo | 5 per 1000 | 18 per 1000 (4 to 79) | RR 3.68 (0.86 to 15.74) | 708 (3 studies) | ⊕⊕⊕⊝ moderate | (Analysis 20.6) Additional intraindividual study: similar mild irritation between the two treatment sides (N = 20) GRADE = very low |
| 5% imiquimod/3% diclofenac in 2.5% hyaluronic acid | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/5% 5‐FU | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| 5% imiquimod/Cryotherapy | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Cryotherapy + 5% imiquimod/Cryotherapy + vehicle | 121 per 1000 | 194 per 1000 (61 to 622) | RR 1.6 (0.5 to 5.13) | 64 (1 study) | ⊕⊕⊝⊝ low | (Analysis 65.4) |
| 5% imiquimod/ALA‐PDT | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| ALA‐PDT + 5% imiquimod/ALA‐PDT + placebo | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
In summary, imiquimod was significantly more efficacious than vehicle, but similar to cryotherapy and 3% diclofenac in 2.5% hyaluronic acid (based on another efficacy outcome presented below). More studies are needed to confirm its inferiority to 5% 5‐fluorouracil. Additional treatment with 3.75%, but not 5%, imiquimod increased the efficacy of cryotherapy, but the efficacy (illustrative comparative risks) of cryotherapy with vehicle in this study seemed much lower than other studies investigating cryotherapy alone (see cryotherapy overview table: Table 70). More data are needed to be able to compare 5% imiquimod to photodynamic therapy, and additional treatment with imiquimod did not improve the efficacy of photodynamic therapy. Treatment with 5% imiquimod resulted in a larger number of participant withdrawals due to adverse events than treatment with 2.5% and 3.75% imiquimod.
Cryotherapy
Table 70 is an overview for cryotherapy.
In summary, cryotherapy had similar efficacy to betulin‐based oleogel and imiquimod. Cryotherapy was significantly inferior to 5‐fluorouracil and ALA‐PDT. No conclusion could be made on its efficacy compared to MAL‐PDT based on our primary outcomes. Additional treatment with 5‐fluorouracil or imiquimod might increase the efficacy of cryotherapy, but these studies had generally lower efficacy associated with cryotherapy with vehicle treatment than the other studies with cryotherapy alone. Cryotherapy was generally not associated with withdrawal due to adverse events and had less skin irritation than ALA‐PDT3.
Photodynamic therapy (PDT)
Table 69 is an overview for photodynamic therapy.
In summary, similar efficacy was obtained for the two photosensitising agents, ALA and MAL, under similar photodynamic therapy conditions. The use of ALA/MAL with blue or red light PDT resulted in similar results, which were significantly different than vehicle with blue or red light PDT. Longer incubation (4 hours [h]) with ALA resulted in better results compared to shorter incubation time (0.5, 1, and 2 hours). Consequently, 4‐hour incubation with ALA followed by PDT was significantly more efficacious than cryotherapy, but 1‐hour incubation with ALA followed by PDT (blue light or pulsed dye laser) was not significantly different than 0.5% 5‐fluorouracil. Additional treatment with 5% imiquimod did not improve the efficacy of ALA‐PDT. With MAL‐PDT, similar efficacy was observed for red light, broad visible light with water‐filtered infrared A, and daylight. With daylight PDT, no difference was found between 16% and 8% MAL or between 2‐hour and 3‐hour incubation with MAL before daylight exposure. Based on our primary outcomes, no conclusion could be made on MAL‐red light PDT efficacy compared to cryotherapy or the benefit of multiple versus single treatment. Photodynamic therapy generally did not lead to withdrawal because of adverse events. Based on the only two studies reporting skin irritation, incubation with ALA might cause skin irritation.
B) All interventions
This section is addressed as planned in 'Criteria for considering studies for this review' and 'Types of interventions':
(1) Prescription‐based topical treatments (2) Prescription‐based oral drugs (3) Mechanical interventions (4) Chemical interventions (5) Combinations of topical and oral treatments with mechanical or chemical interventions
(1) Prescription‐based topical treatments
Prescription‐based topical treatments, which are generally field‐directed treatments, were addressed in alphabetical order: adapalene gel, aretinoid methyl sulfone (Ro 14‐9706), betulin‐based oleogel, calcipotriol, colchicine, diclofenac, 2‐(Difluoromethyl)‐dl‐ornithine (DFMO), 5‐fluorouracil (5‐FU), β‐1,3‐D‐glucan, imiquimod, ingenol mebutate (PEP005), isotretinoin, masoprocol, nicotinamide, resiquimod, sunscreen, DL‐α‐tocopherol (vitamin E), and tretinoin.
Adapalene gel
This intervention was addressed by only 1 study (Kang 2003), which compared the efficacy of 0.1% adapalene gel, 0.3% adapalene gel, and vehicle gel for the treatment of actinic keratoses on the face, ears, scalp, arms, and back of the hands. Participants were treated with adapalene gel or placebo daily for four weeks, followed by twice‐daily applications for up to nine months. The assessment was performed at the end of treatment. There was no major source of possible bias.
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 90 participants) |
Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 90 participants) |
| Kang 2003 | Investigator | ‐ | ‐ | Absolute values |
With regard to the outcome 'global Improvement Indices (investigator): completely improved or cleared', we detected no significant difference in efficacy between adapalene and placebo treatments (Analysis 1.1), or between 0.1% and 0.3% adapalene (Analysis 2.1). The proportion of participants who had positive outcomes (clear, marked, moderate, or slight improvements) was higher in participants treated with adapalene (52/60) than those treated with placebo (21/30), and the proportion of participants graded unchanged or worse was higher in those treated with placebo (9/30) than those that were adapalene‐treated (8/60) (Kang 2003).
1.1.
Comparison 1 Adapalene gel versus placebo, Outcome 1 Global Improvement Indices (investigator)‐cleared.
2.1.
Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 1 Global Improvement Indices (investigator)‐cleared.
Mean changes [reduction (‐) for adapalene and increase (+) for placebo] in the number of actinic keratoses from baseline were the means of measuring efficacy. Compared to placebo, both 0.1% and 0.3% adapalene gel resulted in a significant reduction in mean lesion counts [0.1% = MD ‐2.00, 95% CI ‐2.73 to ‐1.27, and 0.3% = MD ‐4.00, 95% CI ‐4.73 to ‐3.27; Analysis 1.2]. The 0.3% adapalene gel was significantly more efficient than 0.1% adapalene gel in reducing the number of lesions [MD ‐2.00, 95% CI ‐2.46 to ‐1.54; Analysis 2.2].
1.2.
Comparison 1 Adapalene gel versus placebo, Outcome 2 Mean changes in lesion counts.
2.2.
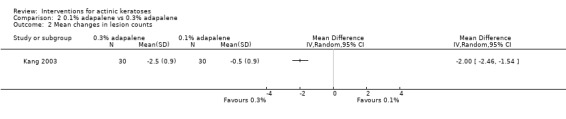
Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 2 Mean changes in lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 90 participants) |
Skin irritation |
Minor adverse events excluding skin irritation (N = 90 participants) |
Cosmetic outcome (N = 42 participants) |
| Kang 2003 | x | ‐ | x | Graph |
Three participants in the 0.3% adapalene group had to withdraw because of these adverse events: skin irritation, dermatitis, and eye dryness. This number of participants was not significantly different from the placebo group (Analysis 1.3) or the 0.1% adapalene treated‐group (Analysis 2.3).
1.3.
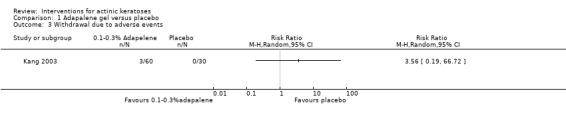
Comparison 1 Adapalene gel versus placebo, Outcome 3 Withdrawal due to adverse events.
2.3.
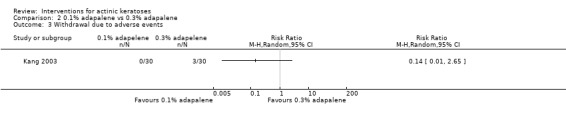
Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 3 Withdrawal due to adverse events.
Dermatitis was the only minor adverse event reported quantitatively. Dermatitis was significantly more frequent in the participants treated with adapalene (20/60) than with placebo (3/30) [RR 3.33, 95% CI 1.08 to 10.34; Analysis 1.4], corresponding to a NNT for an additional harmful outcome of 4.3. In contrast, the number of participants experiencing dermatitis was similar in the 0.1% and 0.3% adapalene‐treated groups (Analysis 2.4).
1.4.
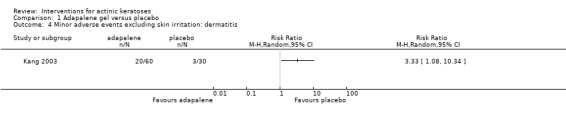
Comparison 1 Adapalene gel versus placebo, Outcome 4 Minor adverse events excluding skin irritation: dermatitis.
2.4.
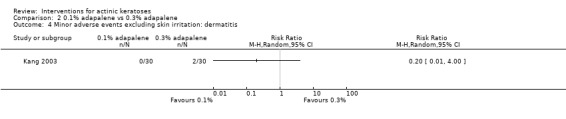
Comparison 2 0.1% adapalene vs 0.3% adapalene, Outcome 4 Minor adverse events excluding skin irritation: dermatitis.
Kang 2003 graphically reported improvements in the following clinical features of photoageing of the skin: mottled hyperpigmentation, fine wrinkles, coarse wrinkles, and rosy glow. The authors stated that a significant difference between adapalene and placebo was detected for global appearance, mottled hyperpigmentation, fine wrinkles, and rosy glow, but not for coarse wrinkles. The exact percentages of participants with improvement in mottled hyperpigmentation were given (55% in the 0.1% group, 65% in the 0.3%, and 25% in the placebo group), but only a subpopulation of participants were evaluated, and the number of participants for each treatment group was not given. Thus, no statistical analysis was performed on this data.
To summarise, adapalene gel was more efficient than placebo in treating actinic keratoses. In addition, 0.3% adapalene gel gave better results than 0.1% adapalene gel, based on the mean reduction in lesion counts without an increase in adverse events.
Aretinoid methyl sulfone (Ro 14‐9706)
Ro 14‐9706 versus 0.05% tretinoin
This intervention was addressed by only 1 intraindividual study (Misiewicz 1991), which compared the efficacy of 0.05% Ro 14‐9706 and 0.05% tretinoin applied twice‐daily for 16 weeks for the treatment of facial actinic keratoses. Assessment was performed at the end of the 16‐week treatment. There was no major source of possible bias.
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 25 participants) |
Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 25 participants) |
| Misiewicz 1991 | x | ‐ | ‐ | Percentages |
Because of the study design, i.e. intraindividual study, the data for 'global improvement indices' for participants receiving the two different interventions could not be included in a meta‐analysis, but is presented in the following table.
| Number of participants with the following improvements | Ro14‐9706 | Tretinoin |
| Complete response | 0/25 | 2/25 |
| Partial response | 12/25 | 10/25 |
| No response | 13/25 | 11/25 |
| Worsening | 0/25 | 2/25 |
Areas treated with tretinoin cream showed an initial increase in the number of lesions (weeks 3 to 9), which eventually decreased after week 10. Ro 14‐9706 showed no initial increase in number of actinic keratoses lesions, but a gradual decline over time. The resulting mean percentage of reduction in lesion counts was significantly higher in the group treated with Ro 14‐9706 than the group treated with tretinoin (MD 7.50, 95% CI 6.57 to 8.43; Analysis 3.1).
3.1.
Comparison 3 Arotinoid Methyl Sulfone (Ro 14‐9706) versus Tretinoin, Outcome 1 Mean percentage of reduction in lesion counts.
Secondary outcomes
Misiewicz 1991 reported none of our secondary outcomes.
To summarise, Ro 14‐9706 treatment showed better overall reduction in lesion counts, whereas tretinoin treatment, which showed an initial increase in lesions, resulted in more participants with complete response.
Betulin‐based oleogel
Studies using betulin‐based oleogel used this treatment as a comparator for cryotherapy. The results will be discussed in the cryotherapy section below.
Calcipotriol (vitamin D)
This intervention was addressed by only one study (Seckin 2009), which compared the efficacy of calcipotriol (vitamin D) to placebo treatment for actinic keratoses on the face and scalp. One treatment was applied on 1 randomised side of the face, and the other treatment on the other side twice daily for 12 weeks. Assessment was performed at the end of the 12‐week treatment. There was possible attrition and reporting bias associated with this study.
Primary outcomes
| Study | Global improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 8 participants) |
| Seckin 2009 | ‐ | ‐ | ‐ | Absolute values, percentages |
Mean changes [reduction (‐) for calcipotriol and increase (+) for placebo] in the number of actinic keratoses from baseline were the means of measuring efficacy. In contrast to placebo, calcipotriol reduced the number of lesions; however, the overall effect was not statistically different (Analysis 4.1). In addition, no statistical difference in the mean percentage of reduction in lesion counts was detected by the authors (Seckin 2009).
4.1.
Comparison 4 Calcipotriol (vitamin D) versus placebo, Outcome 1 Mean changes in lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 8 participants) |
Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (N = 8 participants) |
| Seckin 2009 | x (none lost) | ‐ | ‐ | x |
There were no participant withdrawals due to adverse events.
The reduction of the total score for cosmetic appearance, which rated erythema, desquamation, and induration of a target lesion, was not different between the calcipotriol and placebo groups (Analysis 4.2).
4.2.
Comparison 4 Calcipotriol (vitamin D) versus placebo, Outcome 2 Cosmetic outcomes: Reduction in total cosmetic appearance score.
To summarise, the data available for treatment of actinic keratoses with calcipotriol could not demonstrate its superiority for efficacy or cosmetic outcomes compared to placebo.
Colchicine
This intervention was addressed by only 1 study (Akar 2001), which compared the efficacy of 1% colchicine to 0.5% colchicine for actinic keratosis lesions. Both 0.5% and 1% colchicine were applied twice daily for 10 days on the face, scalp, and upper extremities. Assessment was performed at four weeks. There was no major source of possible bias.
Primary outcomes
| Study | Global improvement indices for completely improved or cleared |
Participant complete clearance (N = 16 participants) |
Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 16 participants) |
| Akar 2001 | ‐ | x | ‐ | Absolute values |
In general, 0.5% and 1% colchicine treatments resulted in similar efficacy, with 7/8 and 6/8 participants completely cleared (Analysis 5.1) and similar mean reduction in lesion counts for all lesions (Analysis 5.2) or by anatomical locations (Analysis 5.3).
5.1.

Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 1 Participant complete clearance.
5.2.
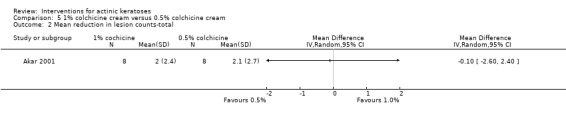
Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 2 Mean reduction in lesion counts‐total.
5.3.
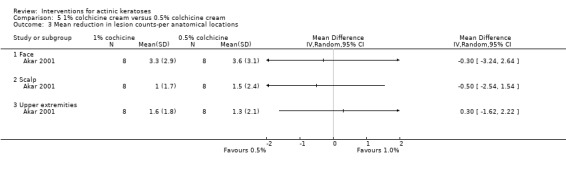
Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 3 Mean reduction in lesion counts‐per anatomical locations.
Secondary outcomes
| Study | Withdrawal due to adverse events | Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (N = 16 participants) |
| Akar 2001 | ‐ | ‐ | ‐ | x |
The final cosmetic results in successful cases were good for both colchicine concentrations, as supported by the quantitative analysis of the number of participants with decreased infiltration and disappearance of crust at one month (Analysis 5.4), which showed no difference between the two treatments.
5.4.
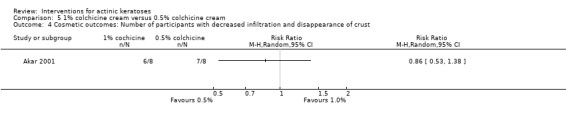
Comparison 5 1% colchicine cream versus 0.5% colchicine cream, Outcome 4 Cosmetic outcomes: Number of participants with decreased infiltration and disappearance of crust.
To summarise, 0.5% and 1% colchicine had similar efficacy and cosmetic outcomes and showed high efficacy (81% of participants completely cleared); however, these conclusions were based on a small sample size.
Diclofenac
Diclofenac versus vehicle
This intervention was addressed by 7 studies (Fariba 2006; Gebauer 2003; McEwan 1997; Rivers 2002; Solaraze study 2; Ulrich 2010; Wolf 2001) comparing 3% diclofenac in 2.5% hyaluronic acid gel to 2.5% hyaluronic acid gel, for the treatment of actinic keratoses. The characteristics of these studies are presented in the following table. There was possible attrition bias (McEwan 1997; Ulrich 2010), reporting bias (Solaraze study 2; Ulrich 2010), and other bias (Fariba 2006; McEwan 1997).
| Characteristic | McEwan 1997 | Wolf 2001 | Rivers 2002 | Solaraze study 2 | Gebauer 2003 | Fariba 2006 | Ulrich 2010 |
| Study design | Parallel‐group | Parallel‐group | Parallel‐group | Parallel‐group | Parallel‐group | Intraindividual | Parallel‐group |
| Anatomical locations | Face, scalp, ear, neck, lower arm/elbow, hand, lower leg/knee | Forehead, central face, scalp, arms, hands | Forehead, central face, scalp, dorsum of hands | Face, scalp, forehead, arm, forearm, back of hands | Head/neck, hands, or arms | Face or scalp | Face, scalp, hands |
| Diclofenac concentration (%) | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| Frequency of treatment | Twice daily | Twice daily | Twice daily | Twice daily | Twice daily | Twice daily | Twice daily |
| Duration given (days) | 56 to 168 | 90 | 30, 60 | 90 | 90 | 90 | 112 |
| Assessment | At the end of 24‐week treatment | 4 weeks after the end of treatment | 4 weeks after the end of treatment | 4 weeks after the end of treatment | At the end of 12‐week treatment and 4 weeks after the end of treatment | At the end of 12‐week treatment | At the end of 16‐week treatment |
In Ulrich 2010, participants were immunosuppressed (organ transplant patients). Three of the included studies (Rivers 2002; Solaraze study 2; Wolf 2001) were part of the Solaraze product insert, and the number of participants experiencing at least one adverse event was pooled and reported in the Solaraze study 2.
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 312 participants) |
Participant complete clearance (N = 490 participants) |
Participant partial (> 75%) clearance (N = 28 participants) |
Mean reduction (changes) in lesion counts (N = 345 participants) |
| McEwan 1997 | ‐ | x | ‐ | ‐ |
| Wolf 2001 | Investigator and participant | x | ‐ | ‐ |
| Rivers 2002 | Investigator and participant | x | ‐ | Absolute values |
| Solaraze study 2 | ‐ | x | ‐ | ‐ |
| Gebauer 2003 | ‐ | x | ‐ | Absolute values |
| Fariba 2006 | ‐ | ‐ | ‐ | ‐ |
| Ulrich 2010 | ‐ | x | x | Percentages |
Efficacy measurements using investigator and participant global improvement indices for the outcome 'completely improved' showed the superiority of the 3% diclofenac in 2.5% hyaluronic acid gel over 2.5% hyaluronic acid gel alone for 60‐day treatment [investigator: RR 3.06, 95% CI 1.21 and 7.77, NNT= 4.8; participant: RR 2.86, 95% CI 1.12 to 7.32, NNT = 5.3] and 90‐day treatment [investigator: RR 2.50, 95% CI 1.37 to 4.55, NNT = 3.6; participant: RR 2.44, 95% CI 1.28 to 4.64], but not for 30‐day treatment [investigator and participant: RR 4.00, 95% CI 0.89 to 17.89] (Analysis 6.1; Analysis 6.2).
6.1.
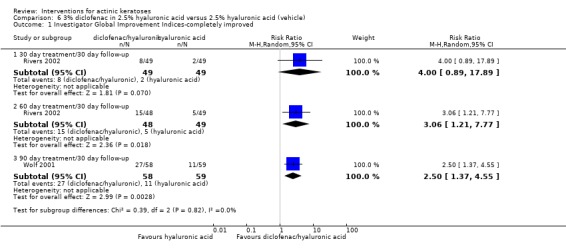
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 1 Investigator Global Improvement Indices‐completely improved.
6.2.
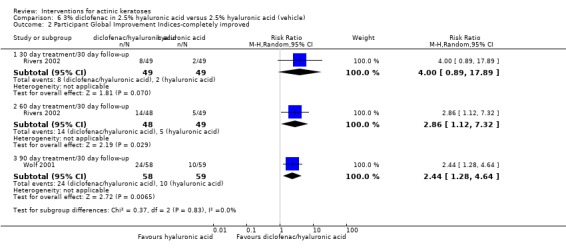
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 2 Participant Global Improvement Indices‐completely improved.
We performed seven meta‐analyses for the outcome 'participant complete clearance'.
Analysis for efficacy assessment at the end of treatment: The 2 studies that reported the efficacy assessment at the end of treatment used a treatment period longer than 30 days and showed the superiority of the diclofenac treatment over 2.5% hyaluronic acid gel alone [RR 1.95, 95% CI 1.21 to 3.13, NNT = 7.1] (Analysis 6.3).
Analysis for efficacy assessment after a 30‐day follow‐up for target lesions, i.e. present at baseline: This was similar to the global improvement indices for completely improved 60‐ and 90‐day treatments, but not 30‐day treatment. Diclofenac/hyaluronic acid showed superiority over the vehicle for target lesions [(60 days: RR 3.27, 95% CI 1.30 to 8.21, NNT = 4.3) (90 days: RR 2.87, 95% CI 1.84 to 4.48, NNT = 3.4) (Analysis 6.4)].
Analysis for efficacy assessment after a 30‐day follow‐up for all lesions, i.e. target and new lesions: Again, participant complete clearance was significantly different for 60‐day therapy (RR 3.83, 95% CI 1.37 to 10.71, NNT = 4.3) and 90‐day therapy (RR 2.20, 95% CI 1.40 to 3.44, NNT = 4.5), but not after 30 days of therapy (RR 3.50, 95% CI 0.76 to 16.01). The pooled RR was 2.46 (95% CI 1.66 to 3.66, NNT = 5.4) (Analysis 6.5). The small sample sizes resulted in no significant difference in the RRs for participant complete clearance between the different anatomical locations.
Analysis for 30‐day treatment with subgroups by anatomical locations (Analysis 6.6).
Analysis for 60‐day treatment with subgroups by anatomical locations (Analysis 6.7).
Analysis for 90‐day treatment with subgroups by anatomical locations: In contrast to locations on the face or forehead, the RRs for scalp, arm/forearms, and back of the hands did not favour diclofenac for 90‐day treatment over vehicle, because of the variability between studies (Analysis 6.8).
Analysis of immunosuppressed participants with efficacy assessment after a 30‐day follow‐up (Analysis 6.9): In immunosuppressed participants, statistically significant superiority of 16‐week treatment with diclofenac over placebo could not be demonstrated despite a large effect for participant complete clearance (RR 5.78, 95% CI 0.38 to 87.35; Analysis 6.9) or partial (> 75%) clearance (RR 3.55, 95% CI 0.57 to 21.94; Analysis 6.10). This was probably due to the small number of participants involved in this single study. Further studies are needed to be able to conclude on the efficacy of diclofenac in immunosuppressed participants.
6.3.
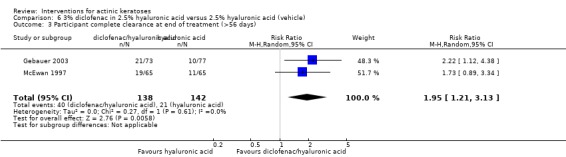
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 3 Participant complete clearance at end of treatment (>56 days).
6.4.
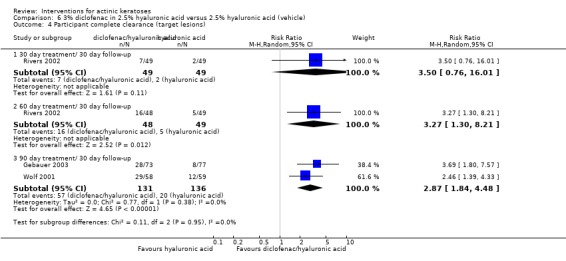
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 4 Participant complete clearance (target lesions).
6.5.
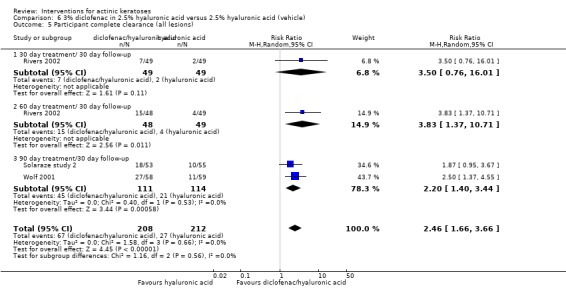
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 5 Participant complete clearance (all lesions).
6.6.
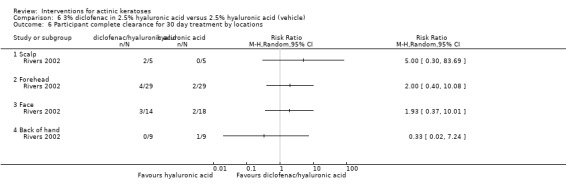
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 6 Participant complete clearance for 30 day treatment by locations.
6.7.
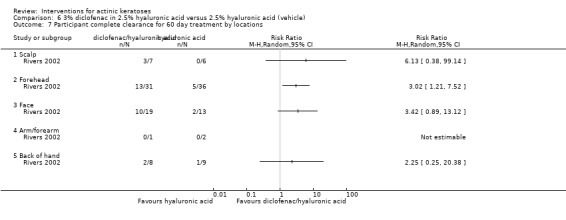
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 7 Participant complete clearance for 60 day treatment by locations.
6.8.
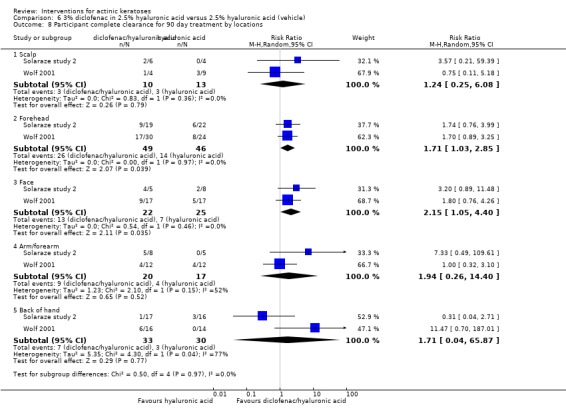
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 8 Participant complete clearance for 90 day treatment by locations.
6.9.
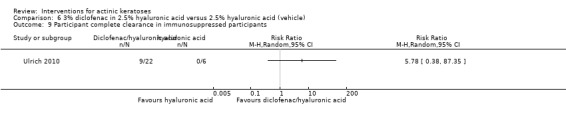
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 9 Participant complete clearance in immunosuppressed participants.
6.10.
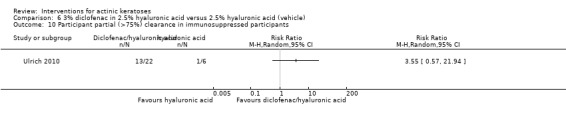
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 10 Participant partial (>75%) clearance in immunosuppressed participants.
The healing properties of diclofenac seem to continue after treatment. There was no significant difference in the mean reduction of lesion counts at the end of 60‐ to 90‐day treatment between diclofenac and vehicle (2.5% hyaluronic acid gel) (Analysis 6.11). In contrast, a significantly better reduction in lesion counts was achieved by the diclofenac treatment compared to 2.5% hyaluronic acid gel alone after a 30‐day follow‐up (MD of at least 2.00; Analysis 6.12). For the immunosuppressed participants, a mean reduction of 53% in the lesion counts was observed for diclofenac, whereas a mean increase of 17% was observed for 2.5% hyaluronic acid gel alone. A statistical analysis could not be performed because standard deviations were not provided.
6.11.
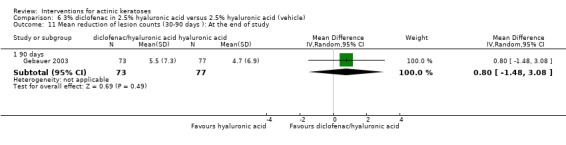
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 11 Mean reduction of lesion counts (30‐90 days ): At the end of study.
6.12.
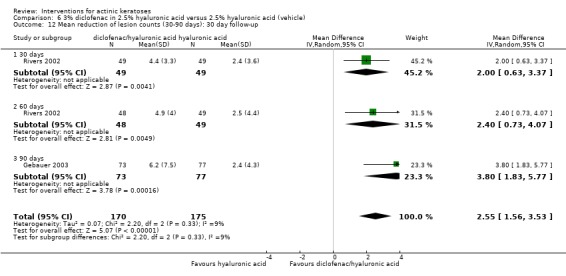
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 12 Mean reduction of lesion counts (30‐90 days): 30 day follow‐up.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 644 participants) |
Skin irritation (N = 20 participants) |
Minor adverse events excluding skin irritation (N = 462 participants) |
Cosmetic outcome (N = 32 participants) |
| McEwan 1997 | x | ‐ | ‐ | ‐ |
| Wolf 2001 | x | ‐ | In general (excluding dermatology because it could include skin irritation) and specific adverse events based on body system | ‐ |
| Rivers 2002 | x | ‐ | x | ‐ |
| Solaraze study 2 | ‐ | ‐ | ‐ | ‐ |
| Gebauer 2003 | x | ‐ | x | ‐ |
| Fariba 2006 | x (intraindividual) | x | ‐ | ‐ |
| Ulrich 2010 | x | ‐ | ‐ | x |
None of the participants (N = 20) in the intraindividual study by Fariba 2006 withdrew because of adverse events. In contrast, significantly more participants withdrew because of adverse events in the 3% diclofenac in 2.5% hyaluronic acid group compared to the 2.5% hyaluronic acid group for the other studies (RR 3.59, 95% CI 1.92 to 6.70; Analysis 6.13), corresponding to a NNT of 9.4 for an additional harmful outcome. In the immunosuppressed participants, 2 out of 24 participants in the diclofenac with 2.5% hyaluronic acid group withdrew because of adverse events, whereas none of the 8 participants receiving 2.5% hyaluronic acid alone withdrew.
6.13.
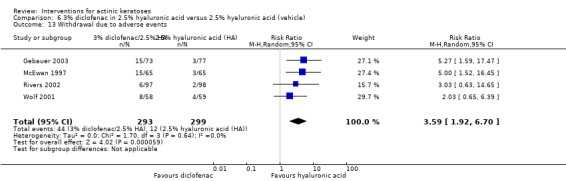
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 13 Withdrawal due to adverse events.
Fariba 2006 reported irritation only on the side treated with diclofenac in 8 of 20 participants.
Minor adverse events were reported for several body systems, and only the number of participants experiencing minor adverse events related to metabolic and nutritional disorders was significantly higher for diclofenac/hyaluronic acid (RR 5.09, 95% CI 1.16 to 22.22; Analysis 6.28), corresponding to a NNT of 7.2 for an additional harmful outcome. Unfortunately, the authors of the study (Wolf 2001) did not give the details of the adverse events related to metabolic and nutritional disorders. A large number of specific minor adverse events have been reported by only one study, and none of them were significantly different between the two groups. One of the minor adverse events reported by the three studies was dry skin. Dry skin was significantly more frequent in the diclofenac/hyaluronic acid group (RR 2.40, 95% CI 1.20 to 4.78; Analysis 6.20), corresponding to a NNT of 4.4 for an additional harmful outcome. Two studies reported two adverse events related to the nervous system, hyperaesthesia and paraesthesia, which were localised to treatment sites by Rivers 2002. The number of participants experiencing both neurological adverse events were not different between the two treatment groups.
6.28.
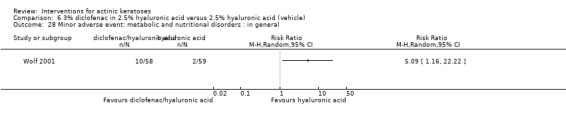
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 28 Minor adverse event: metabolic and nutritional disorders : in general.
6.20.
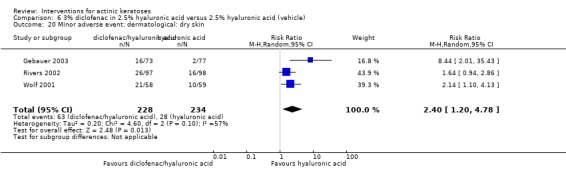
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 20 Minor adverse event: dermatological: dry skin.
Ulrich 2010 mentioned that all immunosuppressed participants on the diclofenac treatment group had "cosmetically appealing results" four weeks after the end of the study, but did not mention anything about the hyaluronic acid (vehicle) group.
To summarise, diclofenac was in general significantly more effective than hyaluronic acid alone, but it was associated with significantly more withdrawals due to adverse events. Unfortunately, the data reported by the included studies did not allow comparison of efficacy and safety between immunosuppressed and immunocompetent participants.
Diclofenac versus imiquimod
This intervention was addressed by 1 open‐label study (Kose 2008), which compared the efficacy of 3% diclofenac in 2.5% hyaluronic acid (once daily for 12 weeks) and 5% imiquimod (3 times per week for 12 weeks) for the treatment of actinic keratoses on the face and scalp. Assessment was performed at the end of the 12‐week treatment. There was possible performance, detection, and reporting bias in this study.
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 49 participants) |
Participant complete clearance | Participant partial (> 75%) clearance | Mean reduction (changes) in lesion counts |
| Kose 2008 | Investigators and participants | ‐ | ‐ | ‐ |
No significant difference was found between diclofenac or imiquimod either by the investigator global improvement indices (Analysis 7.1) or by the participant global improvement indices (Analysis 7.2).
7.1.
Comparison 7 3% diclofenac in 2.5% hyaluronic acid versus 5% imiquimod, Outcome 1 Investigator Global Improvement Indices‐Complete improvement.
7.2.
Comparison 7 3% diclofenac in 2.5% hyaluronic acid versus 5% imiquimod, Outcome 2 Participant Global Improvement Indices‐Complete improvement.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 49 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Kose 2008 | x | ‐ | ‐ | ‐ |
There were no participant withdrawals due to adverse events.
To summarise, diclofenac and imiquimod treatments were equivalent.
Other comparisons
The efficacy of diclofenac in combination with photodynamic therapy will be discussed in the phototherapy section.
2‐(Difluoromethyl)‐dl‐ornithine (DFMO)
This intervention was addressed by 1 intraindividual study (Alberts 2000), which compared the efficacy of 10% 2‐(Difluoromethyl)‐dl‐ornithine (DFMO) with placebo for the treatment of actinic keratosis. The creams were applied to the randomised forearms twice daily for six months. Assessment was performed at the end of the 24‐week treatment. There was possible reporting bias in this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 42 participants) |
| Alberts 2000 | ‐ | ‐ | ‐ | Absolute values and percentages |
The mean numbers of lesions at baseline were high [DFMO‐treated arms: 28.1 + 17.1 (SD); placebo‐treated arms: 29.2 + 18.7], and the reduction rates of lesion counts were relatively low: 23.5% for DFMO and 2.4% for placebo. Moreover, because of the large variability associated with this efficacy outcome, the mean difference (MD) of the absolute mean reduction in lesion counts did not reach statistical significance (MD 5.90, 95% CI ‐3.84 to 15.64; Analysis 8.1).
8.1.
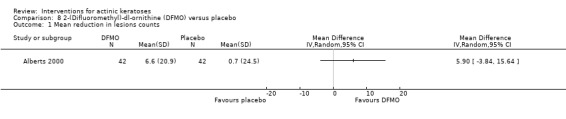
Comparison 8 2‐(Difluoromethyl)‐dl‐ornithine (DFMO) versus placebo, Outcome 1 Mean reduction in lesions counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 42 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Alberts 2000 | x | ‐ | ‐ | ‐ |
Two participants for this intraindividual study withdrew because of adverse events.
To summarise, with severe actinic keratosis, DFMO had limited efficacy and is associated with severe inflammatory reactions.
5‐Fluorouracil (5‐FU)
5‐Fluorouracil versus placebo
This intervention was addressed by 3 studies (Jorizzo 2002;Jorizzo 2004; Weiss 2002). Jorizzo 2002 and Weiss 2002 compared 0.5% 5‐fluorouracil to vehicle cream applied daily for 1, 2, or 4 weeks on lesions located on the face or frontal scalp, and the data were part of the Carac product insert. Jorizzo 2004 reported results from an assessment 4 weeks after 1 week of treatment with either 0.5% 5‐fluorouracil or vehicle cream prior to cryotherapy treatment on lesions on the face, scalp, ears, neck, and lips. Assessment was performed at four weeks after the end of treatment. There was possible performance, detection, attrition, and reporting bias associated with Jorizzo 2002 and Weiss 2002 studies. The latter also has other possible sources of bias.
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 351 participants) |
Participant complete clearance (N = 528 participants) |
Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 528 participants) |
| Jorizzo 2002 | Scores (not included) | x | ‐ | Absolute values |
| Jorizzo 2004 | Scores (not included) | x | ‐ | Absolute values |
| Weiss 2002 | ‐ | x | ‐ | Absolute values |
The data for participant complete clearance from these studies was separated into subgroups based on duration of treatment. Subgroup analyses (Analysis 9.1) showed that treatment with 0.5% 5‐fluorouracil treatment resulted in a significantly higher number of completely cleared participants than the placebo cream when applied for 1 week (NNT = 15.4), 2 weeks (NNT = 7.1), and 4 weeks (NNT = 3.2), resulting in an overall significantly better efficacy for 5‐fluorouracil (RR 8.86, 95% CI 3.67 to 21.40, NNT = 8.5; Analysis 9.1). When participant complete clearance for the different treatment durations were compared, daily application of 0.5% 5‐fluorouracil for 4 weeks was found to have significantly higher efficacy than treatment for 1 week and 2 weeks (RR 0.39, 95% CI 0.19 to 0.81 and RR 0.56, 95% CI 0.36 to 0.87, respectively; Analysis 10.1). No difference was found between treatment for one week and treatment for two weeks. We must be cautious in our interpretation of these results because the design of the studies did not blind the participants and assessor for the treatment duration.
9.1.
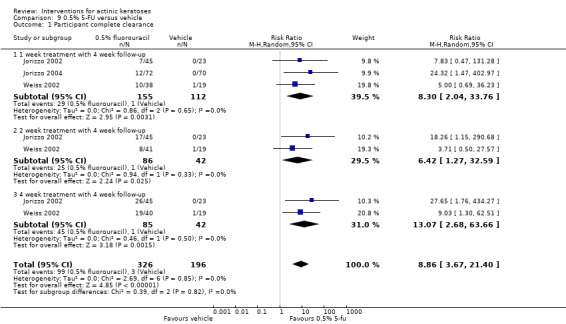
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 1 Participant complete clearance.
10.1.
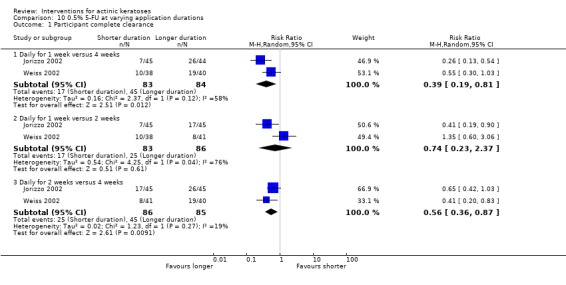
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 1 Participant complete clearance.
Mean reduction in lesion counts was presented as absolute values, percentages, or both. In Jorizzo 2002, only the percentages without the associated standard deviations were presented, as shown in the following table. Jorizzo 2004 presented both absolute values and percentages (table) with their associated standard deviation. Analysis of the absolute values showed a significant reduction in lesion counts with 1 week of 5‐fluorouracil compared to placebo (MD 5.40, 95% CI 2.94 to 7.86; Analysis 9.2). Finally, Weiss 2002 presented the absolute values (placebo: 2.7, 1 week: 8.8, 2 weeks: 11.7, and 4 weeks: 11.1) and percentages (table) without their associated standard deviations. In all studies, the mean percentages of reduction in lesion counts were higher in the 5‐fluorouracil‐treated groups than placebo.
9.2.
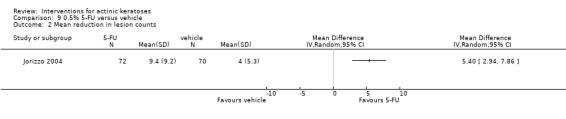
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 2 Mean reduction in lesion counts.
| Mean percentage of reduction in lesion counts | Placebo |
5‐fluorouracil (1 week) |
5‐fluorouracil (2 weeks) |
5‐fluorouracl (4 weeks) |
5‐fluorouracl (pooled) |
| Jorizzo 2002 (N = 207 participants) | 21.6% | 69.5% | 86.1% | 91.7% | 82.4% |
| Jorizzo 2004 (N = 144 participants) | 28.8% + 32.6% (SD) |
62.4% + 32.6% (SD) |
N/A | N/A | 62.4% |
| Weiss 2002 (N = 177 participants) | 34.4% | 78.5% | 83.6% | 88.7% | 83.6% |
N/A = not available
Based on the data from Jorizzo 2004, the mean percentage of reduction in lesion counts for 1 week of treatment with 0.5% 5‐fluorouracil compared to vehicle was statistically significant in favour of 5‐fluorouracil (MD 33.60, 95% CI 22.88 to 44.32; Analysis 9.3).
9.3.
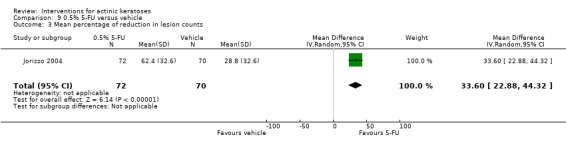
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 3 Mean percentage of reduction in lesion counts.
Secondary outcomes
Because the safety analysis in Jorizzo 2004 included cryotherapy treatment, this study was excluded from this section. All the safety outcomes, except 'skin irritation' in general, were pooled together in the Carac product insert and were reported as in the Jorizzo 2002 study.
| Study |
Withdrawal due to adverse events (N = 384 participants) |
Skin irritation (N = 384 participants) |
Minor adverse events excluding skin irritation (N = 384 participants) |
Cosmetic outcome |
| Jorizzo 2002 | x | x | In general (excluding dermatology because it could include skin irritation) and specific adverse events based on body system | ‐ |
| Weiss 2002 | x | x | In general (excluding dermatology because it could include skin irritation) and specific adverse events based on body system | ‐ |
Analysis of data reported by Weiss 2002 showed that the number of participants who withdrew because of adverse events had a tendency to be higher in the 5‐fluorouracil‐treated group compared to the placebo‐treated group (Analysis 9.4). The number of withdrawals due to adverse events had a tendency to increase with longer treatment with 5‐fluorouracil (Analysis 10.2), but this was not statistically significant. Similar to Weiss 2002, Jorizzo 2002 had 4 treatments arms (1, 2, and 4 weeks of 5‐fluorouracil and placebo). In this study, a total of 24 participants out of 207 withdrew because of adverse events, but the authors only mentioned that 12 of them (50%) were in the 4‐week group (N = 45). Together, these data suggest that severe adverse events are indeed associated with 4‐week treatment with 5‐fluorouracil.
9.4.
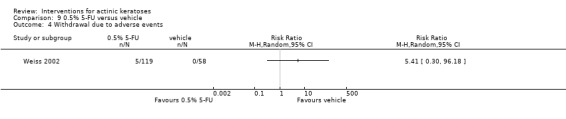
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 4 Withdrawal due to adverse events.
10.2.
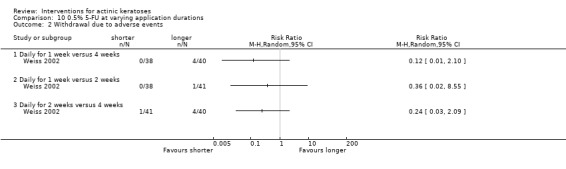
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 2 Withdrawal due to adverse events.
Similarly, the number of participants experiencing facial irritation was significantly higher in the 5‐fluorouracil‐treated group than in the group treated with placebo (RR 1.45, 95% CI 1.27 to 1.65, NNT = 3; Analysis 9.5) without any difference between treatment durations (Analysis 10.3). The number of participants experiencing skin irritation was slightly lower in the 1‐week group than the 2 other 5‐fluorouracil groups, i.e. 2 and 4 weeks of treatments. Irritation related to treatment was mostly of mild to moderate severity.
9.5.
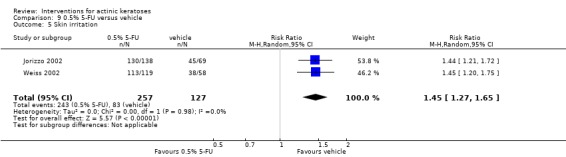
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 5 Skin irritation.
10.3.
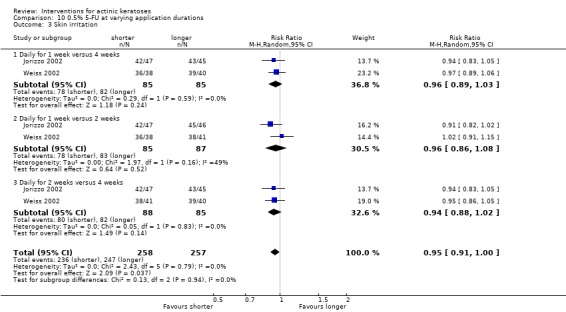
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 3 Skin irritation.
None of the analyses for our outcome 'minor adverse events' resulted in significant differences between 5‐fluorouracil and vehicle‐treated participants (Analysis 9.6; Analysis 9.7; Analysis 9.8; Analysis 9.9; Analysis 9.10; Analysis 9.11; Analysis 9.12; Analysis 9.13; Analysis 9.14; Analysis 9.15; Analysis 9.16). Moreover, no difference was detected between the different 5‐fluorouracil treatment durations (Analysis 10.4; Analysis 10.5; Analysis 10.6; Analysis 10.7; Analysis 10.8; Analysis 10.9; Analysis 10.10; Analysis 10.11; Analysis 10.12).
9.6.
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 6 Minor adverse event excluding skin irritation: body as a whole : in general.
9.7.
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 7 Minor adverse event excluding skin irritation: body as a whole : allergy.
9.8.
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 8 Minor adverse event excluding skin irritation: body as a whole : "flu" or common cold.
9.9.
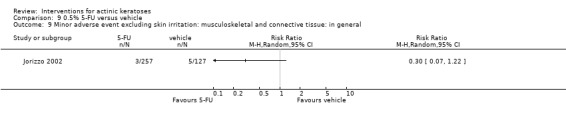
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 9 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: in general.
9.10.
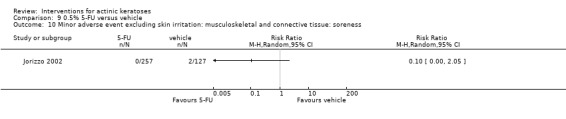
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 10 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: soreness.
9.11.
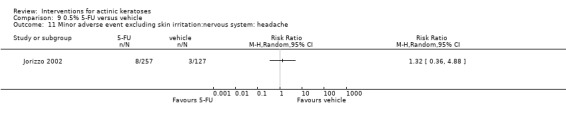
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 11 Minor adverse event excluding skin irritation:nervous system: headache.
9.12.
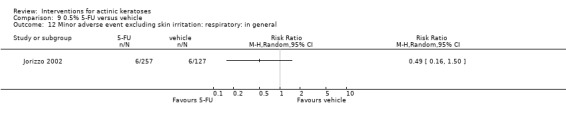
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 12 Minor adverse event excluding skin irritation: respiratory: in general.
9.13.
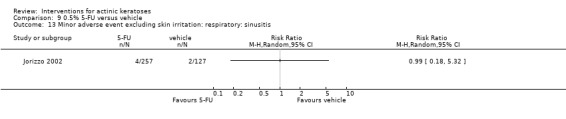
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 13 Minor adverse event excluding skin irritation: respiratory: sinusitis.
9.14.
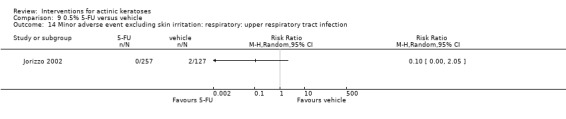
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 14 Minor adverse event excluding skin irritation: respiratory: upper respiratory tract infection.
9.15.
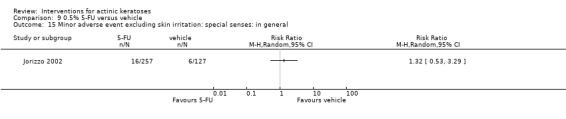
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 15 Minor adverse event excluding skin irritation: special senses: in general.
9.16.
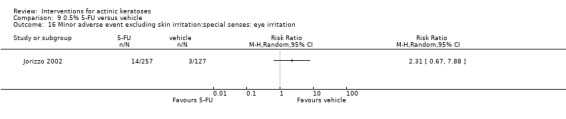
Comparison 9 0.5% 5‐FU versus vehicle, Outcome 16 Minor adverse event excluding skin irritation:special senses: eye irritation.
10.4.
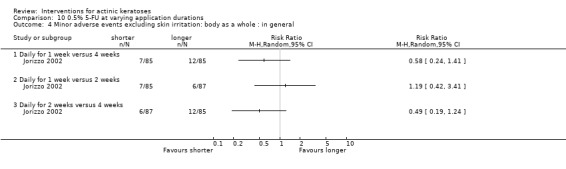
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 4 Minor adverse events excluding skin irritation: body as a whole : in general.
10.5.
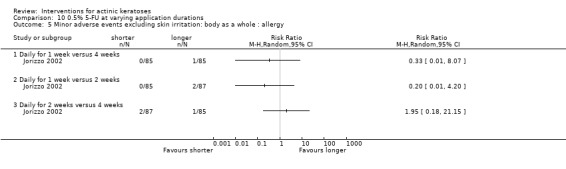
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 5 Minor adverse events excluding skin irritation: body as a whole : allergy.
10.6.
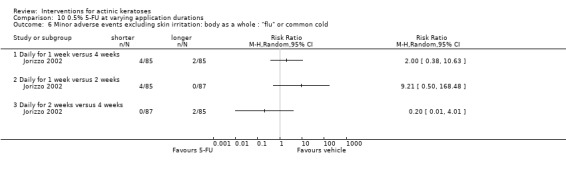
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 6 Minor adverse events excluding skin irritation: body as a whole : "flu" or common cold.
10.7.
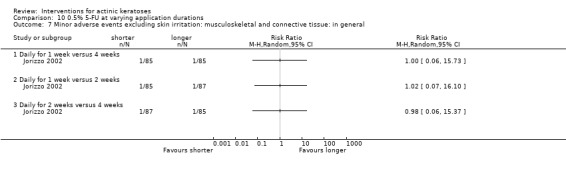
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 7 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.
10.8.
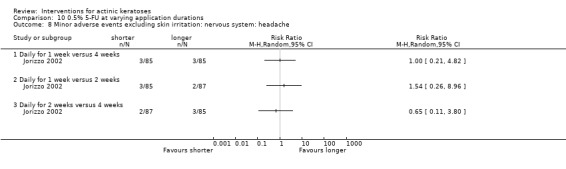
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 8 Minor adverse events excluding skin irritation: nervous system: headache.
10.9.
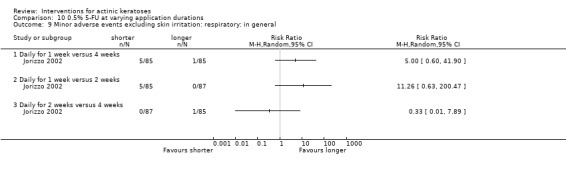
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 9 Minor adverse events excluding skin irritation: respiratory: in general.
10.10.
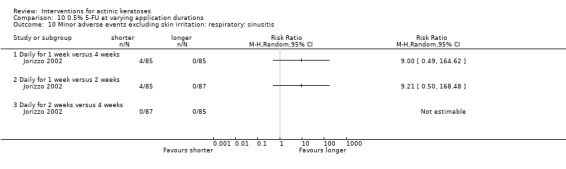
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 10 Minor adverse events excluding skin irritation: respiratory: sinusitis.
10.11.
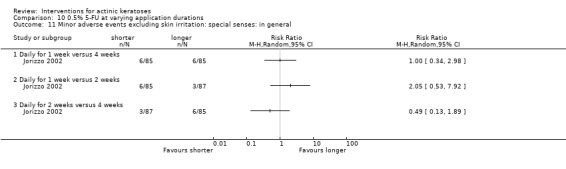
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 11 Minor adverse events excluding skin irritation: special senses: in general.
10.12.
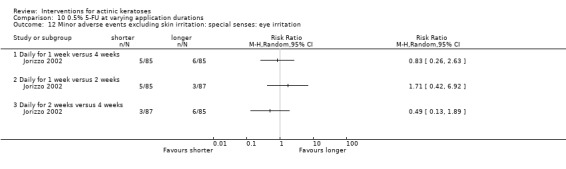
Comparison 10 0.5% 5‐FU at varying application durations, Outcome 12 Minor adverse events excluding skin irritation: special senses: eye irritation.
To summarise, 5‐fluorouracil was more efficient than vehicle to treat actinic keratoses. Four‐week treatment gave better results than one‐ and two‐week treatments, which were comparable. Treatment with 0.5% 5‐fluorouracil for 4 weeks could lead to more adverse events as shown by the number of withdrawals due to adverse events. Significant facial irritation was associated with 0.5% 5‐fluorouracil treatment.
Different concentrations of 5‐fluorouracil
This intervention was addressed by one study (Loven 2002), which used a right/left withinparticipant design (intraindividual) to compare the efficacy of 0.5% to 5% 5‐fluorouracil cream or the treatment of actinic keratoses on the face, anterior bald scalp, or forehead. The creams were applied once daily on 1 side of the face with 0.5% cream and twice daily on the other side with 5% cream for 4 weeks. Assessment was performed at 4 weeks after the end of treatment. There was possible performance and reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 21 participants) |
Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 21 participants) |
| Loven 2002 | ‐ | x | ‐ | Absolute values and percentages |
Due to the intraindividual design of the study, no analysis could be performed for the participant‐based outcome 'participant complete clearance'; however, a similar total clearance rate was obtained for the 2 treatments, i.e. approximately 43% (9/21) (Loven 2002).
After treatment with 0.5% 5‐fluorouracil, participants had a mean reduction of 8.8 lesions, corresponding to 67%, and after 5% 5‐fluorouracil, the mean reduction was 6.1 (47%). The authors reported a significant difference (P = 0.044) between the 2 treatments for the absolute mean reduction in lesion counts and graphically represented the standard deviations associated with lesion counts at baseline and week 8. An analysis could not be performed because the numerical values of the standard deviations were not provided.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 21 participants) |
Skin irritation (N = 21 participants) |
Minor adverse events excluding skin irritation (N = 21 participants) |
Cosmetic outcome |
| Loven 2002 | x | x | x | ‐ |
Sixteen of 21 participants discontinued treatment but did not withdraw from the study, because of irritation: 4 participants discontinued because of treatment with 0.5% 5‐fluorouracil cream, 8 because of 5% cream, and 4 because of both creams.
All participants reported facial irritation in association with both creams.
Eye irritation was reported in 5 out of 21 participants and nasal congestion in 3 out of 21 participants. It was not mentioned if these events were associated with a particular treatment side.
To summarise, 0.5% 5‐fluorouracil cream might be more efficient than the 5% cream and is associated with similar skin irritation.
5‐fluorouracil with tretinoin
This intervention was addressed by 1 intraindividual study (Bercovitch 1987) comparing 5% fluorouracil treatment combined with 0.05% tretinoin and 5% fluorouracil treatment combined with placebo for treatment of actinic keratoses. 5% fluorouracil was applied twice daily on both forearms and hands, and 0.05% tretinoin cream was applied nightly on a randomised forearm/hand and placebo on the other forearm/hand up to 12 weeks. Assessment was performed at the end of the 12‐week treatment. There was possible reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 20 participants) |
| Bercovitch 1987 | ‐ | ‐ | ‐ | Absolute values |
The additional treatment with tretinoin did not make any difference in the mean reduction of lesion counts by 5% fluorouracil (Analysis 12.1).
12.1.
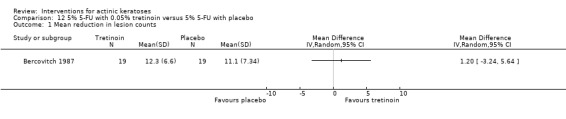
Comparison 12 5% 5‐FU with 0.05% tretinoin versus 5% 5‐FU with placebo, Outcome 1 Mean reduction in lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 20 participants) |
Skin irritation (N = 20 participants) |
Minor adverse events excluding skin irritation | Cosmetic outcome |
| Bercovitch 1987 | x | x (relative) | ‐ | ‐ |
Twelve participants experienced more irritation on the side treated with tretinoin cream, four on the side treated with placebo, and three had equal irritation. One participant withdrew from the study due to irritation, but it was not mentioned if it was due to one treatment in particular.
To summarise, additional treatment with tretinoin did not improve the efficacy of the 5‐fluorouracil treatment and was associated with more skin irritation.
5‐fluorouracil versus imiquimod
This comparison is discussed in the imiquimod section below, and the results presented in Table 68 correspond to Analysis 13.1.
13.1.

Comparison 13 5% 5‐FU versus 5% imiquimod, Outcome 1 Participant complete clearance.
5‐fluorouracil versus masoprocol
This intervention was addressed by 1 study (Kulp‐Shorten 1993), comparing 5% 5‐fluorouracil and 10% masoprocol for the treatment of actinic keratoses. Both creams were applied twice daily for four weeks on the head or neck. Assessment was performed four weeks after the end of treatment. There was possible bias (other) associated with this study.
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 57 participants) |
Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 49 participants) |
| Kulp‐Shorten 1993 | Investigator | ‐ | ‐ | Absolute values and percentages |
Analysis of 'investigator global improvement indices' for cleared participants showed a strong and significant risk ratio favouring 5‐fluorouracil over masoprocol treatments (RR 3.60, 95% CI 1.57 to 8.26; Analysis 15.1). Two (NTT = 2.1) participants need to be treated to result in 1 clearance with 5‐fluorouracil, whereas a larger number would be needed for masoprocol.
15.1.
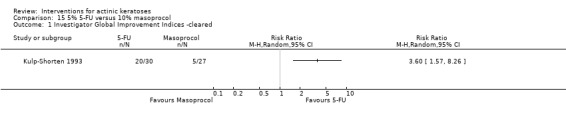
Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 1 Investigator Global Improvement Indices ‐cleared.
No significant difference was detected with the absolute values for mean reduction in lesion counts (Analysis 15.2). In contrast, the mean percentages were significantly different and supported the superiority of 5‐fluorouracil in the treatment of actinic keratoses (MD 20.00, 95% CI 11.82 to 28.18; Analysis 15.3).
15.2.
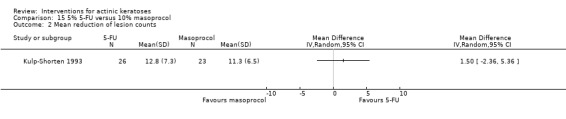
Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 2 Mean reduction of lesion counts.
15.3.
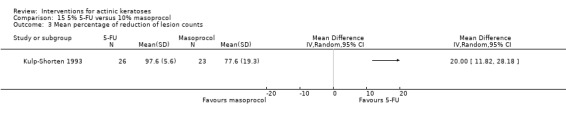
Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 3 Mean percentage of reduction of lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 57 participants) |
Skin irritation |
Minor adverse events excluding skin irritation (N = 57 participants) |
Cosmetic outcome |
| Kulp‐Shorten 1993 | x | ‐ | Graph | ‐ |
Only 1 participant in the 5‐fluorouracil group withdrew because of adverse events (Analysis 15.4).
15.4.
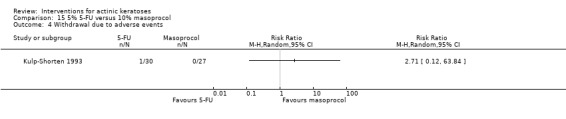
Comparison 15 5% 5‐FU versus 10% masoprocol, Outcome 4 Withdrawal due to adverse events.
Minor adverse events were presented graphically based on their severity as percentages of participants experiencing different adverse events, such as necrosis and contact dermatitis. Based on these data and the incidences of these events, the authors concluded that masoprocol treatment was better tolerated than the 5‐fluorouracil treatment, and they made a correlation with the number of participants that failed to complete 28 days of treatment. Indeed, a significantly higher percentage (65.5%) of participants treated with 5‐fluorouracil failed to complete 28 days of treatment than participants treated with masoprocol (16%).
To summarise, 2 out of 3 efficacy outcome measurements supported the superiority of 5% 5‐fluorouracil treatment over 10% masoprocol treatments for actinic keratosis. Masoprocol treatment may be associated with better tolerability.
Other comparisons
The comparisons between 5‐fluorouracil and cryotherapy, photodynamic therapy, resurfacing, and chemical peel are discussed in their respective sections. The results presented in Table 68 correspond to the following analyses: 1) cryotherapy (Analysis 14.1); 2) photodynamic therapy (Analysis 11.1; Analysis 11.2); 3) carbon dioxide laser resurfacing (Analysis 16.1; Analysis 16.2); 4) Er:YAG laser resurfacing (Analysis 17.1; Analysis 17.2); and 5) trichloroacetic acid peel (Analysis 18.1).
14.1.
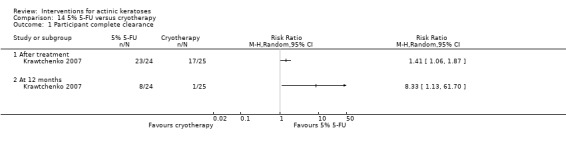
Comparison 14 5% 5‐FU versus cryotherapy, Outcome 1 Participant complete clearance.
11.1.
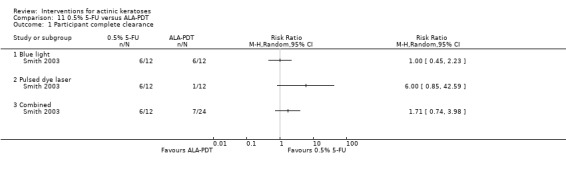
Comparison 11 0.5% 5‐FU versus ALA‐PDT, Outcome 1 Participant complete clearance.
11.2.
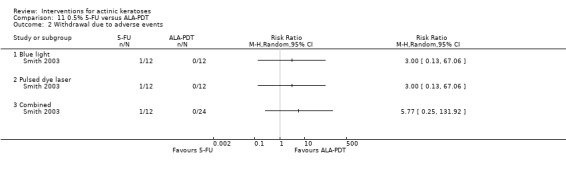
Comparison 11 0.5% 5‐FU versus ALA‐PDT, Outcome 2 Withdrawal due to adverse events.
16.1.
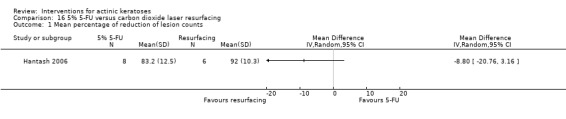
Comparison 16 5% 5‐FU versus carbon dioxide laser resurfacing, Outcome 1 Mean percentage of reduction of lesion counts.
16.2.

Comparison 16 5% 5‐FU versus carbon dioxide laser resurfacing, Outcome 2 Withdrawal due to adverse events.
17.1.
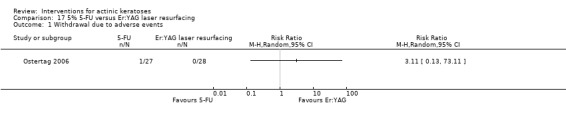
Comparison 17 5% 5‐FU versus Er:YAG laser resurfacing, Outcome 1 Withdrawal due to adverse events.
17.2.
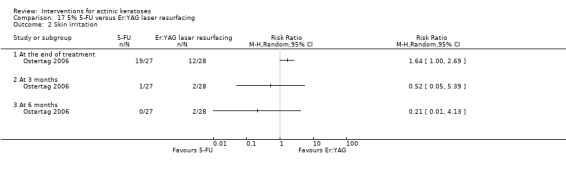
Comparison 17 5% 5‐FU versus Er:YAG laser resurfacing, Outcome 2 Skin irritation.
18.1.
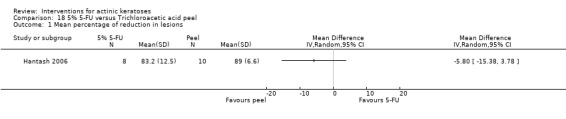
Comparison 18 5% 5‐FU versus Trichloroacetic acid peel, Outcome 1 Mean percentage of reduction in lesions.
β‐1,3‐D‐glucan
This intervention was addressed by one study (Tong 1996), which compared β‐1,3‐D‐glucan with placebo for the treatment of solar keratoses. β‐1,3‐D‐glucan was applied twice daily for 7 days to 1 arm with placebo on the other arm. Assessment was performed seven weeks after the end of treatment. There was possible reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 20 participants) |
| Tong 1996 | ‐ | ‐ | ‐ | Absolute values |
The mean number of lesions at baseline were respectively 22.5 and 23.9, and the mean reductions after 8 weeks were 5.7 and 8.3 for β‐1,3‐D‐glucan and placebo treatment. Based on the graphical representation of the data (means and standard deviations) over time provided by the authors, β‐1,3‐D‐glucan treatment was not more effective than placebo at reducing actinic keratosis lesions. A statistical analysis was not possible because the numerical values of the standard deviations were not given by the authors.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 20 participants) |
Skin irritation |
Minor adverse events excluding skin irritation (N = 20 participants) |
Cosmetic outcome |
| Tong 1996 | x (none lost) | ‐ | x | ‐ |
There were no participant withdrawals due to adverse events.
The participants did not report any minor adverse events.
To summarise, β‐1,3‐D‐glucan treatment showed no benefits in treating solar keratoses.
Imiquimod cream
Imiquimod versus placebo
This intervention was addressed by 18 studies (Alomar 2007; Chen 2003; Gebauer 2009; Hanke 2010; Jorizzo 2007; Jorizzo 2010; Korman 2005; Lebwohl 2004; NCT00828568 Taro; NCT00828568 Taro; Persaud 2002; Ooi 2006; Ortonne 2010; Stockfleth 2002; Swanson 2010a; Szeimies 2004; Ulrich 2007; Zeichner 2009) comparing 2.5% to 5% imiquimod cream and placebo in the treatment of actinic keratoses. Two studies had an intraindividual design (Persaud 2002; Zeichner 2009), whereas all the other studies had a parallel design. In only one study the participants were immunosuppressed (organ transplant patients, Ulrich 2007). Dosing regimens were varied and included 2.5%, 3.75%, and 5% imiquimod and 8 dosing regimens with and without repetition of the treatment schedule, which are summarised in the following table. There were possible performance (Gebauer 2009; Hanke 2010; Ortonne 2010), detection (Hanke 2010; Jorizzo 2007; Ortonne 2010), attrition (Chen 2003; Lebwohl 2004; Persaud 2002; Szeimies 2004; Zeichner 2009), reporting (Alomar 2007; Jorizzo 2007; Jorizzo 2010; Korman 2005; Lebwohl 2004; Persaud 2002; Szeimies 2004; Ulrich 2007), and other (Jorizzo 2007) biases.
| Study | Anatomical locations | Imiquimod percentage | Number of doses/week | Number of weeks | Number of doses | Time of assessment |
| Persaud 2002 | Face, arms, legs | 5 | 3 | 8 or less | 24 or less | 8 weeks after the end of treatment |
| Stockfleth 2002 | Face, scalp, forehead, dorsal forearm, neck, back of hands | 5 | 3 | 12 or less | 36 or less | At the end of the 12‐week treatment |
| Chen 2003 | Face, forehead and temples, cheeks | 5 | 3 | 3 or 6 | 9 or 18 | 4 weeks after the end of treatment |
| Lebwohl 2004 | Face or scalp | 5 | 2 | 16 or less | 32 or less | 8 weeks after the end of treatment |
| Szeimies 2004 | Face or bald scalp | 5 | 3 | 16 or less | 48 or less | 8 weeks after the end of treatment |
| Korman 2005 | Face or bald scalp | 5 | 3 | 16 | 48 | 8 weeks after the end of treatment |
| Ooi 2006 | Scalp, extremities, or upper trunk | 5 | 3 | 16 or less | 48 or less | At the end of treatment |
| Alomar 2007 | Face or bald scalp | 5 | 3 | 4 or 8 | 12 or 24 | 4 weeks after the end of treatment |
| Jorizzo 2007 | Head | 5 | 3 | 4 or 8 | 12 or 24 | 4 weeks after the end of treatment |
| Ulrich 2007 | Face, forehead, or bald scalp | 5 | 3 | 16 | 48 | 8 weeks after the end of treatment |
| Gebauer 2009 | Dorsal of 1 or both forearms and hands | 5 | 2,3,5,7 | 8 | 16,24,40,56 | 8 weeks after the end of treatment |
| Zeichner 2009 | Head | 5 | 1 | 24 | 24 | 4 weeks after the end of treatment |
| Hanke 2010 | Face or bald scalp | 2.5, 3.75 | 7 | 6 (3 on, 3 off, 3 on) | 42 | 8 weeks after the end of treatment |
| Jorizzo 2010 | Face | 3.75 | 7 | 4 (2 on, 2 off, 2 on) | 28 | 20 weeks after the end of treatment |
| Ortonne 2010 | Head (bald scalp or face) | 5 | 3 | 8 (4 on, 4 off, 4 on) | 24 | At week 20 (6 weeks after the end of treatment) |
| Swanson 2010a | Face or bald scalp | 2.5, 3.75 | 7 | 4 (2 on, 2 off, 2 on) | 28 | 8 weeks after the end of treatment |
| NCT00828568 Aldara | Face or bald scalp | 5 | 2 | 16 | 32 | 8 weeks after the end of treatment |
| NCT00828568 Taro | Face or bald scalp | 5 | 2 | 16 | 32 | 8 weeks after the end of treatment |
Three types of subgroup analyses were performed: 1) by number of doses (from 9 to 56 doses) for 5% imiquimod (in the Analyses 19); 2) by imiquimod concentrations (in the Analyses 20); and 3) by frequency of application, i.e. number per week (in the Analyses 21).
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 20 participants) |
Participant complete clearance (N = 3637 participants) |
Participant partial (> 75%) clearance (N = 2914 participants) |
Mean reduction in lesion counts (N = 315 participants) |
| Persaud 2002 | ‐ | ‐ | ‐ | Absolute values |
| Stockfleth 2002 | ‐ | x | ‐ | ‐ |
| Chen 2003 | ‐ | x | x | Absolute values |
| Lebwohl 2004 | ‐ | x | x | ‐ |
| Szeimies 2004 | ‐ | x | x | ‐ |
| Korman 2005 | ‐ | x | x | ‐ |
| Ooi 2006 | ‐ | x | ‐ | ‐ |
| Alomar 2007 | ‐ | x | x | ‐ |
| Jorizzo 2007 | ‐ | x | x | ‐ |
| Ulrich 2007 | ‐ | x | x | ‐ |
| Gebauer 2009 | ‐ | x | x | ‐ |
| Zeichner 2009 | Investigator | ‐ | ‐ | ‐ |
| Hanke 2010 | ‐ | x | x | ‐ |
| Jorizzo 2010 | ‐ | x | ‐ | Percentages |
| Ortonne 2010 | ‐ | ‐ | ‐ | Absolute values |
| Swanson 2010a | ‐ | x | x | ‐ |
| NCT00828568 Aldara | ‐ | x | ‐ | ‐ |
| NCT00828568 Taro | ‐ | x | ‐ | ‐ |
Only one intraindividual study, Zeichner 2009, presented the number of participants with global improvement indices for complete clearance. One participant out of 15 was completely cleared on the imiquimod‐treated side, whereas none of the participants showed complete clearance on the placebo‐treated side. Thus, this dosing regimen was not very effective.
Overall, the risk ratio for participant complete clearance favoured 5% imiquimod treatment over placebo for immunocompetent (RR 6.91, 95% CI 4.25 to 11.26; Analysis 19.1) as well as in immunosuppressed participants (RR 18.50, 95% CI 1.19 to 286.45; Analysis 19.2). Eight immunocompetent participants (NNT = 7.7) must be treated with 5% imiquimod to obtain 1 complete clearance. No immunosuppressed participant in the control group was completely cleared, so the corresponding number, i.e. NNT could not be calculated for this population.
19.1.
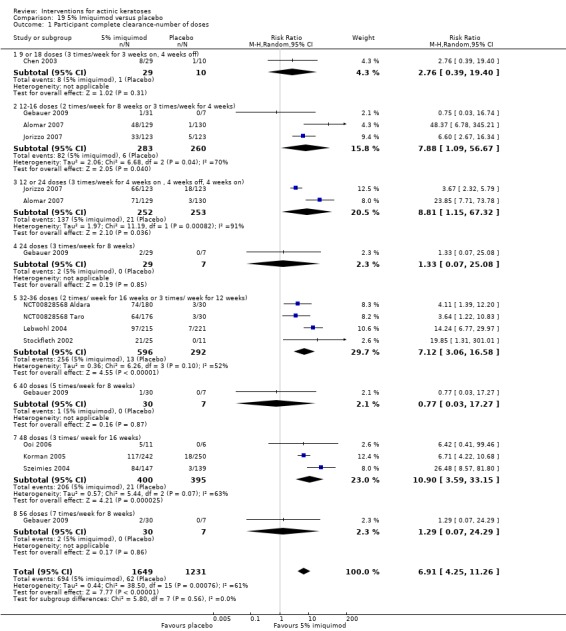
Comparison 19 5% Imiquimod versus placebo, Outcome 1 Participant complete clearance‐number of doses.
19.2.
Comparison 19 5% Imiquimod versus placebo, Outcome 2 Participant complete clearance in immunosuppressed participants.
However, 5% imiquimod was not statistically favoured in 4 of the 8 dosing regimens: 9 or 18 doses (3 times/week for 3 weeks on, 4 weeks off), 24 doses (3 times/week for 8 weeks), 40 doses (5 times/week for 8 weeks), and 56 doses (7 times/week for 8 weeks). Increasing the number of doses did not result in an increase in the values of the RRs, suggesting that the number of doses might not be a determining factor for the efficacy of imiquimod. Despite these subgroup analyses, substantial heterogeneity was associated with most of the subgroups of pooled studies. The heterogeneity was particularly high (I² statistic = 91%) for the 2 studies, with 1 or 2 courses 3 times/week for 4 weeks on, 4 weeks off, 4 weeks on (Alomar 2007; Jorizzo 2007). The two studies were similar in design. Alomar 2007 was performed in Europe and included participants with five to nine lesions, whereas Jorizzo 2007 was performed in North America and included participants with four to eight lesions. In the European study, 47/126 participants were cleared after 1 course and did not receive a second course, compared to 32/121 in the American study. This difference might explain the heterogeneity associated with these studies.
Slightly different results were obtained for participant partial (> 75%) clearance (Analysis 19.3). One additional dosing regimen did not reach significant difference (12 or 24 doses, 4 weeks on, 4 weeks off). In immunosuppressed participants, the results for partial clearance (RR 23.50, 95% CI 1.53 to 360.94; Analysis 19.4) were similar to complete clearance (Analysis 19.2).
19.3.
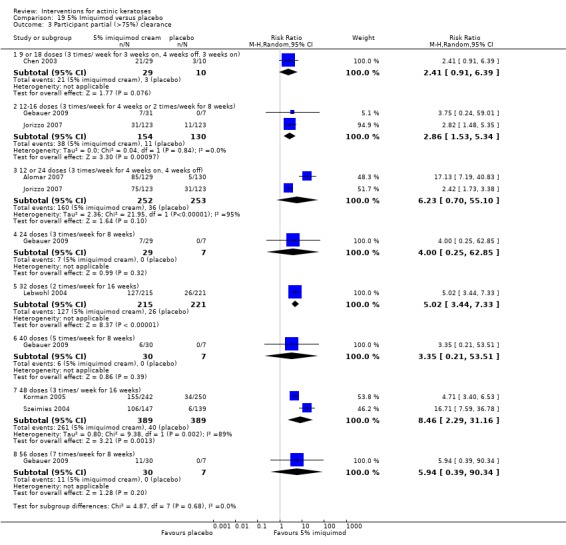
Comparison 19 5% Imiquimod versus placebo, Outcome 3 Participant partial (>75%) clearance.
19.4.
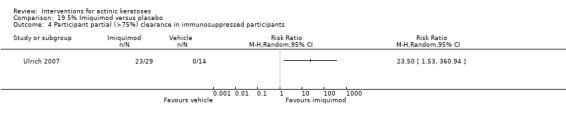
Comparison 19 5% Imiquimod versus placebo, Outcome 4 Participant partial (>75%) clearance in immunosuppressed participants.
The number of participants with complete clearance was significantly higher in the imiquimod‐treated group than the placebo‐treated group for the 3 concentrations, i.e. 2.5% (RR 4.49, 95% CI 2.40 to 8.39, NNT = 4.6), 3.75% (RR 6.45, 95% CI 3.87 to 10.73, NNT = 3.7), and 5% (RR 7.70, 95% CI 4.63 to 12.79, NTT = 4.7). Based on the result for the subgroup difference test (P = 0.42; Analysis 20.1), the efficacy of the 3 concentrations compared to placebo was not significantly different despite the fact that the magnitude of the effect increased with the concentration of imiquimod used to treat the actinic keratoses. In contrast, the analysis of the participant partial clearance, which included fewer studies, showed a significant difference between the concentrations of imiquimod (test for subgroup differences: P = 0.01; Analysis 20.2). It is also worth noting that the two studies (Gebauer 2009; Ooi 2006) not including lesions on the face did not favour imiquimod (Analysis 20.1).
20.1.
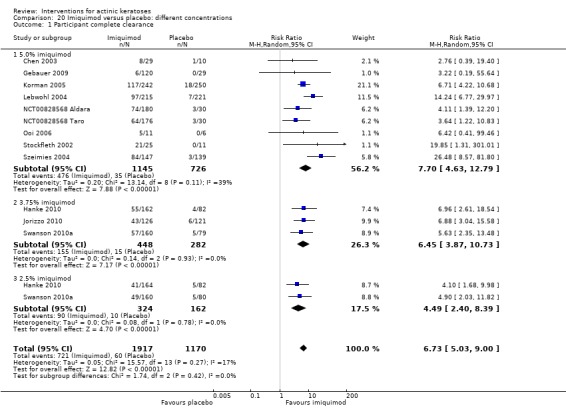
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 1 Participant complete clearance.
20.2.
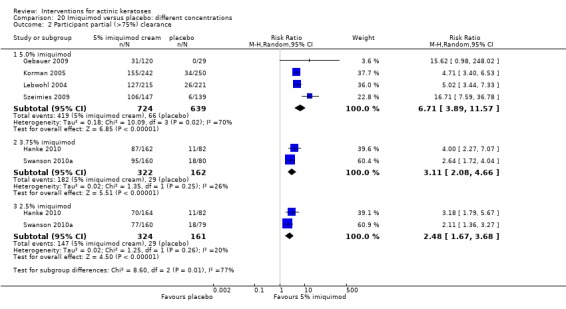
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 2 Participant partial (>75%) clearance.
The amplitude of the clearance effect increased when the frequency of application was increased from 2 to 3 times per week for both complete clearance (Analysis 21.1) from (RR 5.36, 95% CI 2.03 to 14.16) to (RR 8.38, 95% CI 3.79 to 18.52) and for partial clearance (Analysis 21.2) from (RR 4.99, 95% CI 3.43 to 7.26) to (RR 7.65, 95% CI 2.51 to 23.32), but the difference between the RRs between 2 and 3 times were not significantly different.
21.1.
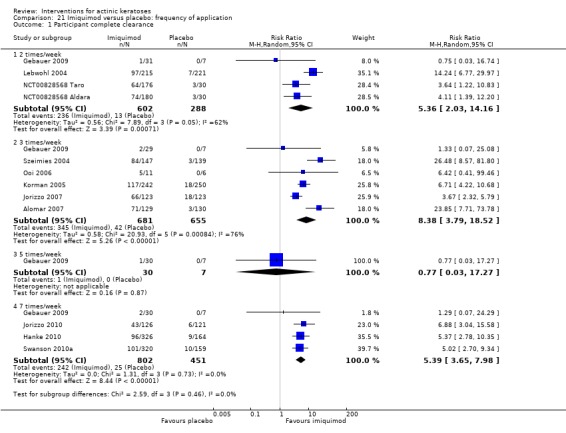
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 1 Participant complete clearance.
21.2.
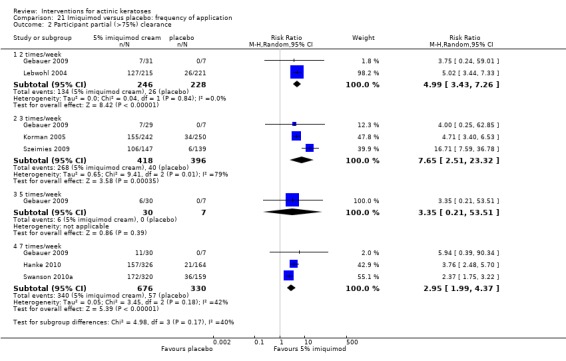
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 2 Participant partial (>75%) clearance.
Also, no correlation was found for the subgroup analysis of the number of weeks of treatment (not shown).
A funnel plot (Figure 3) for all the studies reporting 'participant complete clearance' suggests that there was no publication bias for this outcome.
3.
Funnel plot of comparison: 15 Imiquimod versus placebo: different concentrations, outcome: 15.1 Participant complete clearance.
Two studies (Ortonne 2010; Persaud 2002) with 24 doses of 5% imiquimod or placebo presented mean reduction in lesion counts as absolute values. Ortonne 2010, but not Persaud 2002, provided the associated standard deviations allowing statistical analysis. The mean reduction in lesion counts were similar for the imiquimod group (2.8 + 2.1 and 3.9) and for the placebo group (0.6 + 2.6 and 0.5), but the RR did not significantly favour imiquimod based on Ortonne 2010 alone (Analysis 19.5). In contrast, the mean percentage of reduction in lesion counts provided by Jorizzo 2010 with 3.75% imiquimod supported the superiority of imiquimod over placebo for treatment of actinic keratoses (MD 46.90, 95% CI 36.68 to 57.12; Analysis 20.3).
19.5.
Comparison 19 5% Imiquimod versus placebo, Outcome 5 Mean reduction in lesion counts.
20.3.
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 3 Mean percentage of reduction in lesion counts.
Secondary outcomes
In the Jorizzo 2010 study, some lesions also had cryotherapy treatment, and safety outcomes were only reported for the comparison between cryotherapy with and without imiquimod treatment. Thus, this study was not included in the secondary outcomes presented in this section.
| Study |
Withdrawal due to adverse events (N = 3444 participants) |
Skin irritation (N = 1677 participants) |
Minor adverse events excluding skin irritation (N = 700 participants) |
Cosmetic outcome (N = 1691 participants) |
| Persaud 2002 | x (none lost ‐ intraindividual not included) |
‐ | ‐ | ‐ |
| Stockfleth 2002 | x (none lost) | ‐ | ‐ | ‐ |
| Chen 2003 | x (none lost) | ‐ | ‐ | ‐ |
| Lebwohl 2004 | x | ‐ | ‐ | x |
| Szeimies 2004 | x | x | ‐ | x |
| Korman 2005 | x | ‐ | ‐ | x (imiquimod, not included) |
| Ooi 2006 | x (none lost) | ‐ | x | ‐ |
| Alomar 2007 | x | ‐ | ‐ | ‐ |
| Jorizzo 2007 | x | ‐ | ‐ | |
| Ulrich 2007 | x | ‐ | x (imiquimod only, not included) |
x (qualitative, not included) |
| Gebauer 2009 | x | ‐ | x | ‐ |
| Zeichner 2009 | x (none lost ‐ intraindividual not included) |
Qualitative (not included) | ‐ | ‐ |
| Hanke 2010 | x | x | x | x |
| Ortonne 2010 | x (none lost) | ‐ | ‐ | ‐ |
| Swanson 2010a | x | x | ‐ | x |
| NCT00828568 Taro | x | x | ‐ | ‐ |
| NCT00828568 Taro | x | x | ‐ | ‐ |
All of the studies reported the number of participants who withdrew because of adverse events. Six out of 17 studies reported that no participants withdrew due to adverse events (i.e. "none lost" in the previous table), which are not included in the pooled risk ratio of meta‐analysis because of the absence of events. All these studies used 5% imiquimod applied 3 times per week and had a very small sample size (< 50 participants) compared to the other studies. Thus, we have to be careful about the interpretation of the analysed data. When comparing 5% imiquimod application to placebo, there was no significant difference in withdrawals due to adverse effects except at 48 doses. At 48 doses, when 2 studies were combined, there was a significant difference in favour of placebo (RR 2.69, 95% CI 1.48 to 4.90, NNT=16.7; Analysis 19.6).
19.6.
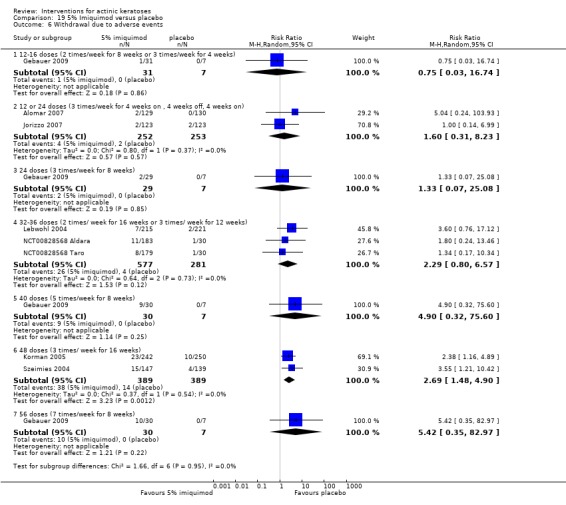
Comparison 19 5% Imiquimod versus placebo, Outcome 6 Withdrawal due to adverse events.
When 8 of the studies using 5% imiquimod were pooled together (imiquimod N = 1338, placebo N = 952), the number of participant withdrawals due to adverse events was significantly higher in the 5% imiquimod‐treated group than the placebo‐treated group (RR 2.59, 95% CI 1.59 to 4.23, NNT = 27; Analysis 20.5). The 4 studies with a parallel design (Chen 2003; Ooi 2006; Ortonne 2010; Stockfleth 2002), not included in the calculation of the pooled RR because of the lack of event, represented only 79 participants in the imiquimod group and 31 participants in the placebo group. Thus, we could conclude that 5% imiquimod treatment results in a higher number of participants withdrawn because of adverse events compared to placebo. In contrast, there was no significant difference for 3.75% and 2.5% imiquimod compared to placebo (Analysis 20.5).
20.5.
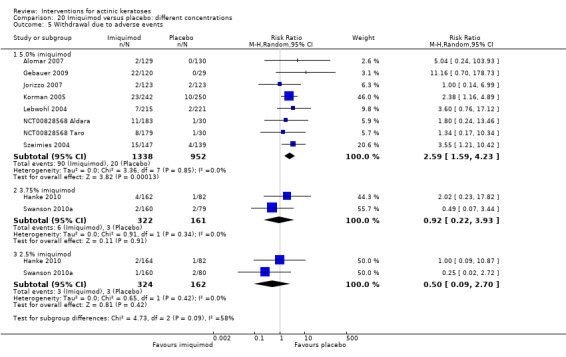
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 5 Withdrawal due to adverse events.
For all frequencies of weekly application, there was a tendency to have more participants withdraw because of adverse events in the imiquimod group; a significant difference (RR 2.47, 95% CI 1.42 to 4.30, NTT = 27.2; Analysis 21.3) was reached in the 3 times per week group. Five studies (imiquimod N = 670, placebo N = 649) were included in the calculation of the pooled risk ratio, but 4 smaller studies were not included because of absence of withdrawal in the intervention and control arms (imiquimod N = 79, placebo N = 31). Finally, there was no difference in the number of participants who withdrew because of adverse events in the immunosuppressed participants (Analysis 19.7).
21.3.
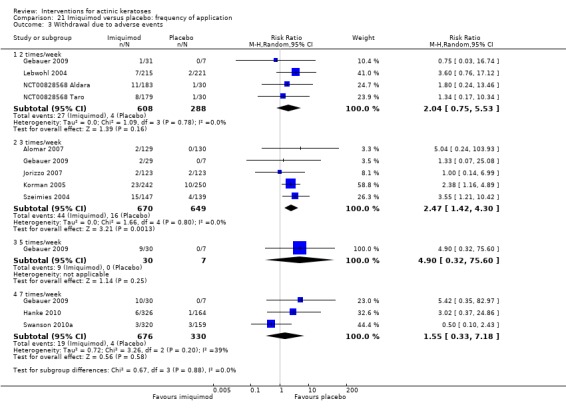
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 3 Withdrawal due to adverse events.
19.7.
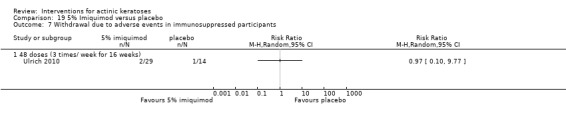
Comparison 19 5% Imiquimod versus placebo, Outcome 7 Withdrawal due to adverse events in immunosuppressed participants.
In the studies reporting skin irritation, no significant difference was observed for the separate analysis of the different concentrations. However, the pooled risk ratio did favour placebo, i.e. more participants treated with imiquimod experienced skin irritation compared to participants treated with placebo (RR 3.93, 95% CI 1.56 to 9.88, NNT = 60; Analysis 20.6). In the intraindividual study Zeichner 2009, the participants experienced similar mild irritation with 5% imiquimod and placebo.
20.6.
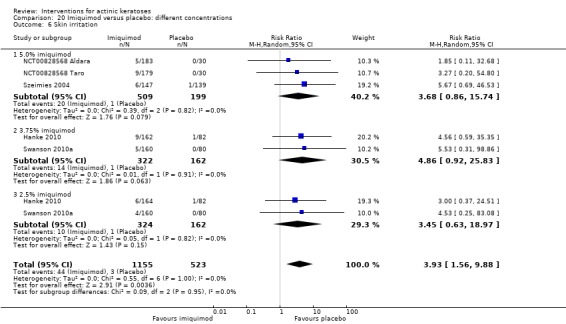
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 6 Skin irritation.
Only one study, Gebauer 2009, reported the number of participants experiencing 'minor adverse events excluding skin irritation' in general for different body systems, i.e. body as a whole, digestive system, and nervous system. None of the data were significantly different between the 5% imiquimod‐ and placebo‐treated groups. Few studies reported the number of participants experiencing specific minor adverse events. The following adverse events affecting the body as a whole (pyrexia), the haemic and lymphatic system (lymphadenopathy), the musculoskeletal system (myalgia), the nervous system (fatigue), the respiratory system (cough, sinusitis, and upper respiratory tract infection), and the urogenital system (urinary tract infection) were not different between imiquimod and placebo groups. The number of participants treated with imiquimod experiencing "flu" or "cold"‐like symptoms or "headache" was generally not different from placebo‐treated participants except for application 7 times per week reported in 1 study by Hanke 2010 (RR 19.68, 95% CI 1.20 to 323.89; Analysis 21.5).
21.5.
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 5 Minor adverse events excluding skin irritation: body as a whole:"flu" or "cold".
Only a few studies gave quantitative cosmetic outcomes (Lebwohl 2004; Szeimies 2004); a significant decrease in roughness, dryness, and scaliness of the skin was associated with 5% imiquimod treatment compared to placebo (RR 3.23, 95% CI 1.86 to 5.58, NNT = 2.6; Analysis 19.13). In addition, overall cosmetic outcomes were significantly or much improved with 2.5% (RR 2.25, 95% CI 1.62 to 3.14, NNT = 3.1) and 3.75% (RR 2.71, 95% CI 2.05 to 3.58, NNT = 2.3) imiquimod compared to placebo (Analysis 20.16).
19.13.
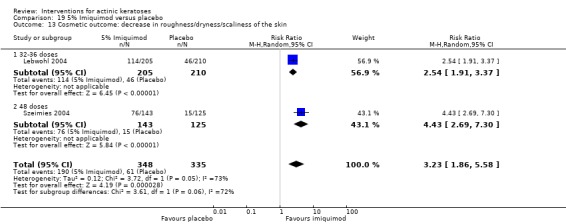
Comparison 19 5% Imiquimod versus placebo, Outcome 13 Cosmetic outcome: decrease in roughness/dryness/scaliness of the skin.
20.16.
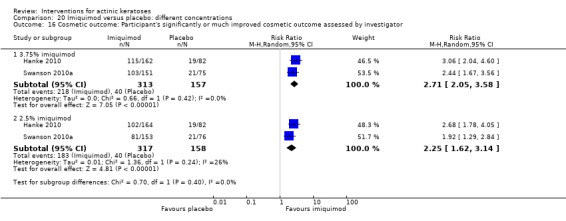
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 16 Cosmetic outcome: Participant's significantly or much improved cosmetic outcome assessed by investigator.
To summarise, the efficacy of imiquimod compared to placebo was significantly better based on the participant complete and partial clearance as well as the mean percentage of reduction in lesion counts, but not for the absolute mean reduction in lesion counts. The amplitude of the effect was independent of the number of doses of 5% imiquimod, imiquimod concentrations, or frequency of application on a weekly basis. The number of withdrawals due to adverse events in the imiquimod group compared to placebo was statistically significant in the 48‐dose group (although not in the 56‐dose group) compared with the lower doses and in the 5% compared to the 2.5% and 3.75% imiquimod concentrations. Significantly better cosmetic outcomes were obtained with imiquimod treatment.
Imiquimod versus diclofenac
This comparison was reported in the diclofenac section above.
Imiquimod versus 5‐fluorouracil
This intervention, which was addressed by 2 studies, 1 assessor‐blinded (Tanghetti 2007) and 1 open study (Krawtchenko 2007), compared the efficacy of 5% imiquimod and 5% 5‐fluorouracil for the treatment of actinic keratoses. In Krawtchenko 2007, imiquimod was applied on the head, neck, or décolleté 3 times per week for 4 weeks on and 4 weeks off, once or twice, and 5‐fluorouracil was applied twice daily for 4 weeks. Assessment was performed 4 and 8 weeks after the end of treatment for 5‐fluorouracil and imiquimod, respectively. Tanghetti 2007 applied imiquimod on the face, forehead, or scalp twice weekly for 16 weeks and 5‐fluorouracil twice daily for 2 to 4 weeks. Assessment was performed at 8 weeks after the end of treatment. There were possible performance (Krawtchenko 2007; Tanghetti 2007), detection (Krawtchenko 2007), and reporting (Tanghetti 2007) biases.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 89 participants) |
Participant partial (> 75%) clearance |
Mean reduction in lesion counts (N = 50 participants) |
| Tanghetti 2007 | ‐ | x | ‐ | ‐ |
| Krawtchenko 2007 | ‐ | x | ‐ | Percentages |
With regard to the outcome 'participant complete clearance', no pooled RR can be calculated because of the high heterogeneity (I² statistic = 93%; Analysis 22.1) associated with the 2 studies. The data from Krawtchenko 2007, which had no blinding specified, did not favour any treatment (RR 0.88, 95% CI 0.73 to 1.06). In contrast, the data from Tanghetti 2007, which was assessor‐blinded, significantly favoured 5‐fluorouracil (RR 0.31, 95% CI 0.14 to 0.67). The variability in the dosing regimen might explain the considerable heterogeneity associated with participant complete clearance. Tanghetti 2007 also supported 5‐fluorouracil superiority by reporting the mean percentage of reduction in lesion counts of 94% and 66% for 5‐fluorouracil and imiquimod, respectively. However, the authors did not provide the standard deviation associated with these values to determine statistically the significance of this difference between the treatments.
22.1.
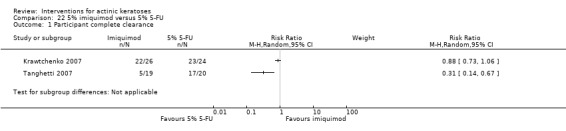
Comparison 22 5% imiquimod versus 5% 5‐FU, Outcome 1 Participant complete clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 89 participants) |
Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (N = 50 participants) |
| Tanghetti 2007 | x (none lost) | ‐ | ‐ | ‐ |
| Krawtchenko 2007 | x (none lost) | ‐ | ‐ | x |
There were no participant withdrawals due to adverse events.
The percentage of participants with a general cosmetic outcome assessed as excellent by the investigator was clearly better for imiquimod (21/26 = 81%) than 5‐fluorouracil (1/24 = 4%) (RR 19.38, 95% CI 2.82 to 133.26, NNT = 1.3; Analysis 22.2). Moreover, the skin quality was better in the imiquimod group than the 5‐fluorouracil group (RR 1.45, 95% CI 1.00 to 2.11, NNT = 3.8; Analysis 22.3).
22.2.
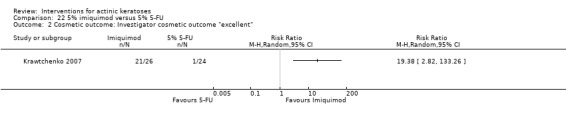
Comparison 22 5% imiquimod versus 5% 5‐FU, Outcome 2 Cosmetic outcome: Investigator cosmetic outcome "excellent".
22.3.
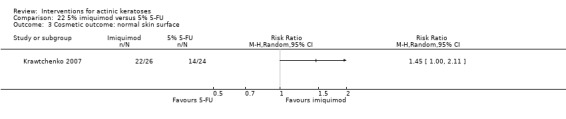
Comparison 22 5% imiquimod versus 5% 5‐FU, Outcome 3 Cosmetic outcome: normal skin surface.
To summarise, the superiority of 5‐fluorouracil over imiquimod in treating actinic keratoses needs to be supported by additional data. Imiquimod treatment seemed to result in better cosmetic outcomes than 5‐fluorouracil.
Other comparisons
The efficacy of imiquimod compared with cryotherapy will be discussed in the cryotherapy section below, and the results presented in the additional Table 71 correspond to Analysis 23.1. Similarly, comparison with photodynamic therapy will be discussed in the phototherapy section.
23.1.
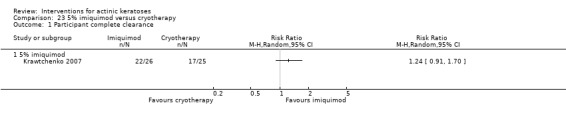
Comparison 23 5% imiquimod versus cryotherapy, Outcome 1 Participant complete clearance.
Ingenol mebutate (PEP005)
This intervention was addressed by three studies (Anderson 2009; Siller 2009; Swanson 2010b). These three studies investigated the efficacy of ingenol mebutate applied once daily for two to three consecutive days or once weekly for two weeks, i.e. two days one week apart (Siller 2009), compared to vehicle for the treatment of actinic keratoses. Treatments were applied to the arms, shoulder, chest, and scalp in both Anderson 2009 and Siller 2009. In addition, the treatments were also applied to the back in Anderson 2009 and the face in Siller 2009, whereas only non‐head locations were investigated in Swanson 2010b. Assessment was performed 8 (Anderson 2009; Swanson 2010b) and 12 (Siller 2009) weeks after the first day of treatment. There was possible reporting (Anderson 2009) and other (Siller 2009) bias.
Because different concentrations (0.025%, 0.01%, and 0.05%) of ingenol mebutate and dosing regimens were used in these studies, subgroup analyses were performed for the different ingenol mebutate concentrations (in the Analyses 25) and the number of applications for 0.05% ingenol mebutate (i.e. number of doses or days, in the Analyses 26). Analyses of pooled data were also performed (in the Analyses 24).
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 477 participants) |
Participant partial (> 75%) clearance (N = 285 participants) |
Mean reduction in lesion counts |
| Anderson 2009 | ‐ | x | x | ‐ |
| Siller 2009 | ‐ | ‐ | x | ‐ |
| Swanson 2010b | ‐ | x | x (percentage not specified and data not included in analysis) | ‐ |
Participant complete clearance was evaluated for target lesions (i.e. present at baseline) as well as for all lesions (i.e. target and new lesions). For both, the number of participants completely cleared was significantly higher in the ingenol mebutate group compared to vehicle (target: RR 3.61, 95% CI 1.86 to 7.02, NNT = 2.9; Analysis 24.1) (all lesions: RR 4.50, 95% CI 2.61 to 7.74, NNT = 3.4; Analysis 24.2), which corresponds to 383 per 1000 participants for ingenol mebutate and 73 per 1000 participants for vehicle achieving complete clearance.The amplitude of the effect had a tendency to increase with the concentration of ingenol mebutate (Analysis 25.1; Analysis 25.2), but not with the number of applications of 0.05% ingenol mebutate (Analysis 26.1; Analysis 26.2).
24.1.

Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 1 Participant complete clearance of target lesions.
24.2.
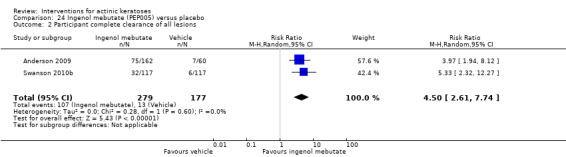
Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 2 Participant complete clearance of all lesions.
25.1.
Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 1 Participant complete clearance of target lesions.
25.2.
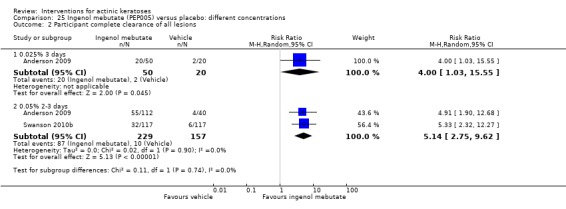
Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 2 Participant complete clearance of all lesions.
26.1.
Comparison 26 0.05% Ingenol mebutate (PEP005) versus placebo: number of doses, Outcome 1 Participant complete clearance of target lesions.
26.2.
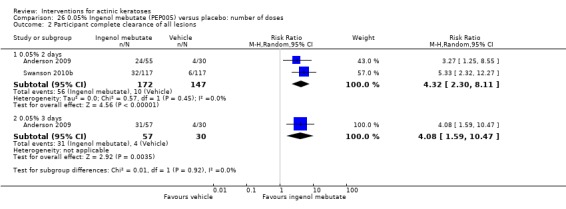
Comparison 26 0.05% Ingenol mebutate (PEP005) versus placebo: number of doses, Outcome 2 Participant complete clearance of all lesions.
Similar results were obtained for participant partial clearance with a RR of 2.88, 95% CI 1.81 to 4.58 (Analysis 24.3), corresponding to a NNT of 2.8. A possible dependence on the ingenol mebutate concentration (Analysis 25.3), but not on the number of applications for 0.05% ingenol mebutate (Analysis 26.3), was also observed.
24.3.
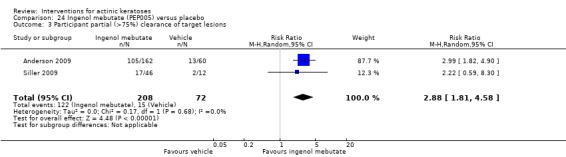
Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 3 Participant partial (>75%) clearance of target lesions.
25.3.
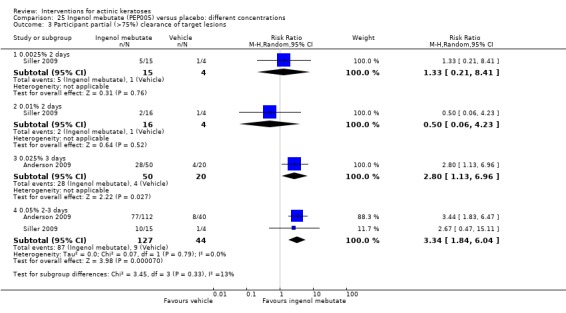
Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 3 Participant partial (>75%) clearance of target lesions.
26.3.
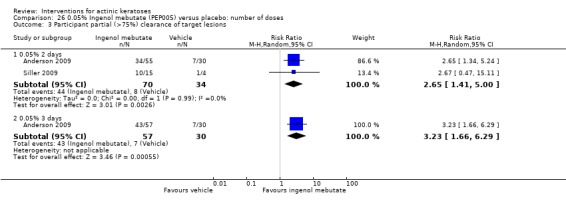
Comparison 26 0.05% Ingenol mebutate (PEP005) versus placebo: number of doses, Outcome 3 Participant partial (>75%) clearance of target lesions.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 540 participants) |
Skin irritation |
Minor adverse events excluding skin irritation (N = 222 participants) |
Cosmetic outcome (N = 540 participants) |
| Anderson 2009 | x | ‐ | x | ‐ |
| Siller 2009 | x | ‐ | ‐ | x |
| Swanson 2010b | x | ‐ | ‐ | x |
No withdrawal due to adverse events was reported in Anderson 2009 and Siller 2009. However, in Swanson 2010b, 1 out of 255 participants withdrew because of an adverse event (pain), but the associated treatment was not specified.
No statistical analyses were performed for minor adverse events because only one participant experienced the individual minor adverse events reported, as shown in the following table, and no statistical significance could be reached. However, based on the incidences, more reports of minor adverse events were associated with the 0.05% ingenol mebutate.
| Body system | Minor adverse event | Placebo (N = 60) | 0.025% ingenol mebutate for 3 days (N = 50) | 0.05% ingenol mebutate for 3 days (N = 57) | 0.05% ingenol mebutate for 2 days (N = 55) |
| Body as a whole | Chills | 0 | 0 | 1 | 0 |
| Body as a whole | Fever | 0 | 0 | 0 | 1 |
| Body as a whole | Flu or cold | 0 | 0 | 0 | 1 |
| Dermatologic | Contact dermatitis | 0 | 0 | 1 | 0 |
| Dermatologic | Impetigo | 0 | 0 | 1 | 0 |
| Hemic and lymphatic | Traumatic hematoma | 0 | 0 | 1 | 0 |
| Metabolic and nutritional disorders | Increase in creatine phosphokinase | 0 | 0 | 0 | 1 |
| Musculoskeletal and connective tissue | Muscle spasms | 1 | 0 | 0 | 1 |
| Nervous system | Headache | 0 | 0 | 1 | 0 |
| Renal and urogenital | Proteinuria | 0 | 0 | 0 | 1 |
| Respiratory | Nasal congestion | 0 | 1 | 0 | 0 |
| Incidences | |||||
| Each treatment arm | 1 | 1 | 5 | 5 | |
| 3 days versus 2 days | 6 | 5 | |||
| 0.025% versus 0.05% | 1 | 10 | |||
There was no scarring, but some pigmentation changes occurred in some participants treated with ingenol mebutate. These changes were not significantly different compared to vehicle (Analysis 24.4).
24.4.
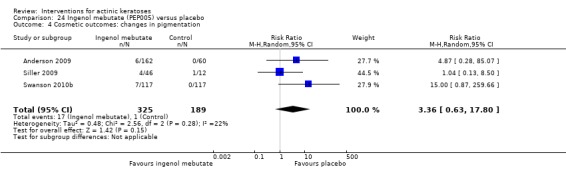
Comparison 24 Ingenol mebutate (PEP005) versus placebo, Outcome 4 Cosmetic outcomes: changes in pigmentation.
To summarise, ingenol mebutate was significantly more efficient than vehicle in treating actinic keratoses. When a higher concentration was used (i.e. 0.05%), ingenol mebutate generally resulted in better efficacy. Increasing the number of applications from two to three times did not result in an increase in the number of participants cleared. No significant difference was observed between ingenol mebutate and placebo for adverse events. Thus, ingenol mebutate treatment was relatively safe and efficient for actinic keratosis treatment.
Isotretinoin
This intervention was addressed by 1 study (Alirezai 1994) comparing the efficacy and safety of 0.1% isotretinoin and vehicle cream applied twice daily for 24 weeks for the treatment of actinic keratoses of the face, scalp, and upper extremities. Assessment was performed at the end of treatment. There was possible attrition and other bias in this study.
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 100 participants) |
Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction (changes) in lesion counts (N = 100 participants) |
| Alirezai 1994 | Investigator | ‐ | ‐ | Absolute values |
The number of participants experiencing complete clearance, partial clearance, no clearance, and worsening were determined by an investigator global evaluation at the end of treatment. The numbers of participants with complete clearance were low with both isotretinoin and placebo for the three anatomical locations, and the associated risk ratios did not favour any treatment (Analysis 27.1).
27.1.
Comparison 27 Isotretinoin versus vehicle, Outcome 1 Investigator global improvement indices‐completely cleared.
The mean reduction of lesion counts for lesions on the face (MD 2.20, 95% CI 1.97 to 2.43) and upper extremities (MD 1.90, 95% CI 1.28 to 2.52), but not on the scalp, did favour isotretinoin over placebo (Analysis 27.2).
27.2.
Comparison 27 Isotretinoin versus vehicle, Outcome 2 Mean reduction of lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 100 participants) |
Skin irritation (N = 92 participants) |
Minor adverse events excluding skin irritation | Cosmetic outcome |
| Alirezai 1994 | x | x (in general and severe) | ‐ | ‐ |
Two of 50 participants in the isotretinoin group withdrew because of adverse events, but it was not statistically different compared to the placebo group (0/50) (Analysis 27.3).
27.3.
Comparison 27 Isotretinoin versus vehicle, Outcome 3 Withdrawal due to adverse events.
Local irritation on the face, but not on the scalp or upper extremities (Alirezai 1994), was significantly more frequent for the isotretinoin‐treated group than for the placebo‐treated group for all intensities (RR 1.57, 95% CI 1.23 to 2.01, NNT = 3.0; Analysis 27.4) as well as severe irritation (RR 17.09, 95% CI 2.35 to 124.10, NNT = 3.1; Analysis 27.5).
27.4.
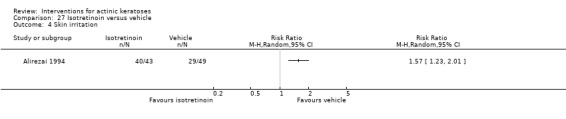
Comparison 27 Isotretinoin versus vehicle, Outcome 4 Skin irritation.
27.5.
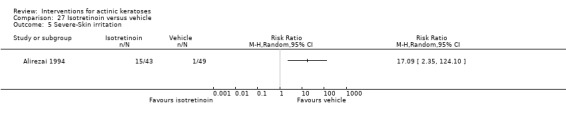
Comparison 27 Isotretinoin versus vehicle, Outcome 5 Severe‐Skin irritation.
To summarise, 0.1% isotretinoin with the dosing regimen used was able to significantly reduce actinic keratoses counts on the face or upper extremities but was not sufficient to result in significant participant complete clearance. Isotretinoin treatment was associated with significant local irritation on the face.
Masoprocol
Masoprocol versus vehicle
This intervention was addressed by 1 study (Olsen 1991) comparing 10% masoprocol cream to vehicle cream for the treatment of actinic keratoses. Masoprocol or placebo creams were applied on the head and neck once or twice daily for a maximum of 28 days and follow‐up assessment was done at 4 weeks after the last application of the study drug. There was possible attrition and other bias associated with this study.
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 154 participants) |
Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction in lesion counts (N = 154 participants) |
| Olsen 1991 | Investigator | ‐ | ‐ | Absolute values |
Masoprocol‐treated participants had a complete cure rate of 12/113 (11%), which was similar to the cure rate of the vehicle cream; 2/41 (5%), as globally assessed by the investigator. Thus, the RR associated with investigator global improvement indices for cured participant did not significantly favour masoprocol.
In contrast, mean reduction in lesion counts was significantly higher for masoprocol than for vehicle‐treated groups (MD 7.30, 95% CI 5.77 to 8.83; Analysis 28.2).
28.2.
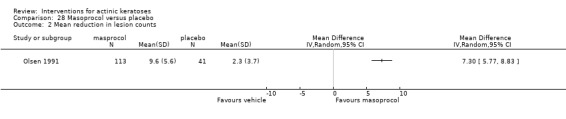
Comparison 28 Masoprocol versus placebo, Outcome 2 Mean reduction in lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 176 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Olsen 1991 | x | ‐ | ‐ | ‐ |
Two of 131 participants in the masoprocol group withdrew because of adverse events, but it was not statistically different to the placebo group (0/45) (Analysis 28.3).
28.3.
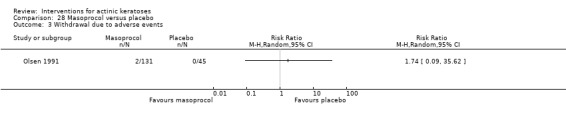
Comparison 28 Masoprocol versus placebo, Outcome 3 Withdrawal due to adverse events.
To summarise, 10% masoprocol with the dosing regimen used was able to significantly reduce actinic keratoses counts but was not sufficient to result in significant participant complete clearance as globally assessed by the investigator. Substantial local skin reactions were also associated with masoprocol treatment compared to vehicle.
Masoprocol versus 5‐fluorouracil
This comparison was presented in the 5‐fluorouracil section above.
Nicotinamide
This intervention was addressed by 1 study (Moloney 2010) investigating the efficacy of 1% nicotinamide twice daily compared to placebo for the treatment of non‐hyperkeratotic actinic keratoses on the face, scalp, and upper limbs. Assessment was performed at three and six months after the beginning of the treatment. There was possible reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean reduction in lesion counts (N = 30 participants) |
| Moloney 2010 | ‐ | ‐ | ‐ | Percentages |
Mean percentage of reduction in lesion counts was assessed at three and six months. At 3 months, the associated RR favoured nicotinamide that reduced by 21.8 + 10% the number of lesions compared to 10 + 12% for placebo (MD 11.80, 95% CI 3.92 to 19.68; Analysis 29.1). The superiority of nicotinamide was lost at six months (Analysis 29.1).
29.1.
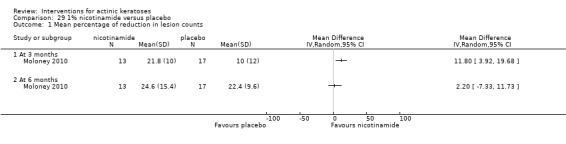
Comparison 29 1% nicotinamide versus placebo, Outcome 1 Mean percentage of reduction in lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 30 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Moloney 2010 | x | ‐ | ‐ | ‐ |
None of the 13 participants in the nicotinamide group withdrew because of adverse events; it was not statistically different to the placebo group, which had 2 withdrawals out of 17 participants (Analysis 29.2).
29.2.
Comparison 29 1% nicotinamide versus placebo, Outcome 2 Withdrawal due to adverse events.
In summary, 1% nicotinamide had very limited short‐term efficacy at the dosing regimen used.
Resiquimod
This intervention was addressed by 1 study (Szeimies 2008) investigating different concentrations (0.01%, 0.03%, 0.06%, and 0.1%) of resiquimod for the treatment of actinic keratoses on the face or bald scalp. The cream was applied once daily three times per week for four weeks on and eight weeks off, once or twice, i.e. one or two treatment cycles depending on the participant response to treatment. Assessment was performed at eight weeks after the end of treatment. There was possible other bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 132 participants) |
Participant partial (> 75%) clearance (N = 132 participants) |
Mean reduction in lesion counts |
| Szeimies 2008 | ‐ | x | x | ‐ |
Results from individual analyses of participant complete and partial clearance for pairs of resiquimod concentrations are summarised in the following table.
| Higher versus lower resiquimod concentrations | |||
|
Participant complete clearance (after 1 cycle) |
Participant complete clearance (after 1 or 2 cycles) |
Participant partial clearance (after 1 or 2 cycles) |
|
| 0.1% vs 0.01% | > | > | > |
| 0.1% vs 0.03% | > | < | = |
| 0.1% vs 0.06% | > | > | > |
| 0.06% vs 0.01% | > | = | = |
| 0.06% vs 0.03% | < | < | < |
| 0.03% vs 0.01% | > | > | > |
< : significantly inferior, < : tendency to be inferior, = : equal, >: tendency to be superior, > : significantly superior, vs = versus
For participant complete clearance, the efficacy of 0.1% resiquimod was generally superior to the other lower concentrations after 1 treatment cycle (0.1% vs 0.01%: RR 2.45, 95% CI 1.64 to 3.65, NNT = 1.7; Analysis 30.1) (0.1% vs 0.03%: RR 1.34, 95% CI 1.09 to 1.66, NNT = 4.0; Analysis 31.1) (0.1% vs 0.06%: RR 1.76, 95% CI 1.30 to 2.38, NNT = 2.3; Analysis 32.1). After the second cycle of treatment, the differences between resiquimod concentrations were lost.
30.1.
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 1 Participant complete clearance.
31.1.
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 1 Participant complete clearance.
32.1.
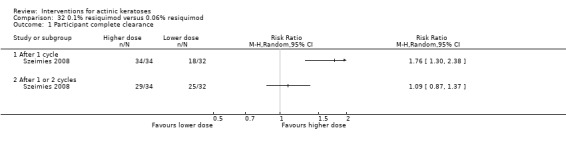
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 1 Participant complete clearance.
No significant difference was detected between the resiquimod concentrations used with the outcome 'participant partial clearance' (Analysis 30.2; Analysis 31.2; Analysis 32.2; Analysis 33.2; Analysis 34.2; Analysis 35.2).
30.2.
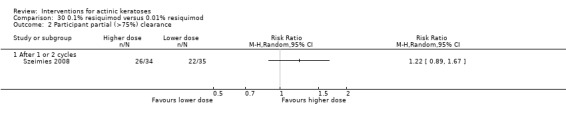
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 2 Participant partial (>75%) clearance.
31.2.
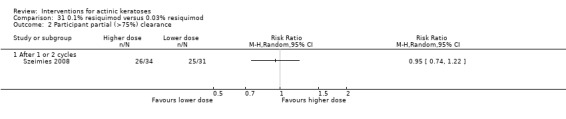
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 2 Participant partial (>75%) clearance.
32.2.
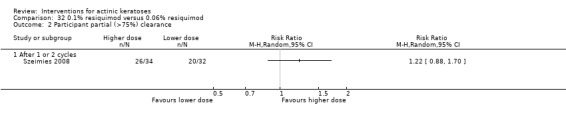
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 2 Participant partial (>75%) clearance.
33.2.
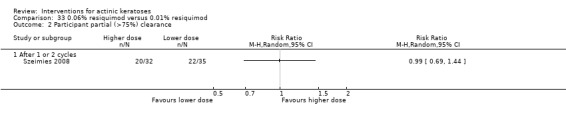
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 2 Participant partial (>75%) clearance.
34.2.
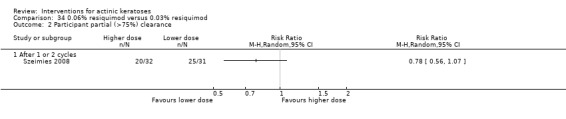
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 2 Participant partial (>75%) clearance.
35.2.
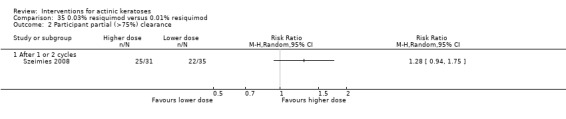
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 2 Participant partial (>75%) clearance.
In general, higher concentrations had a tendency to be more effective. The results obtained with 0.03% and 0.06% resiquimod suggest that these 2 concentrations might have been "switched" or "mislabelled" (Analysis 34.1; Analysis 34.2).
34.1.
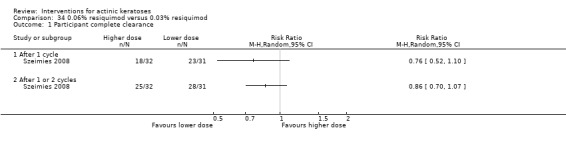
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 1 Participant complete clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 132 participants) |
Skin irritation |
Minor adverse events excluding skin irritation (N = 132 participants) |
Cosmetic outcome |
| Szeimies 2008 | x | ‐ | In general by body system and individual adverse event | ‐ |
There were significantly more participants in the 0.1% resiquimod group who withdrew because of adverse events compared to those in the 0.01% (RR 27.77, 95% CI 1.72 to 449.47, NNT = not applicable; Analysis 30.3) and 0.03% (RR 2.96, 95% CI 1.08 to 8.13, NNT = 4.0; Analysis 31.3) resiquimod groups. A significant difference was also found between 0.06% and 0.01% resiquimod (RR 22.91, 95% CI 1.40 to 375.77, NNT = not applicable; Analysis 33.3).
30.3.
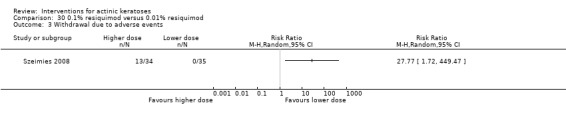
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 3 Withdrawal due to adverse events.
31.3.
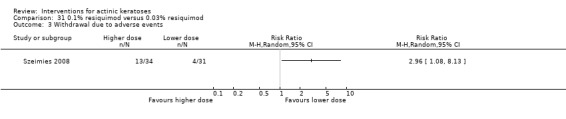
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 3 Withdrawal due to adverse events.
33.3.
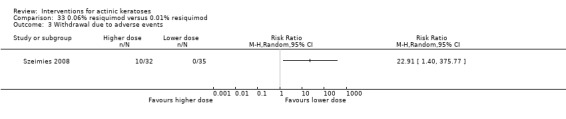
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 3 Withdrawal due to adverse events.
Results from individual analyses of minor adverse events excluding skin irritation for pairs of resiquimod concentrations are summarised in the following table.
| Minor adverse events excluding skin irritation | |||||||
| Higher versus lower resiquimod concentrations | |||||||
|
Musculoskeletal and connective tissue (in general) |
Nervous system (in general) |
Skin and subcutaneous tissue disorders (in general) |
|||||
| 0.1% vs 0.01% | > | > | > | ||||
| 0.1% vs 0.03% | > | = | > | ||||
| 0.1% vs 0.06% | = | = | < | ||||
| 0.06% vs 0.01% | > | > | > | ||||
| 0.06% vs 0.03% | > | = | > | ||||
| 0.03% vs 0.01% | > | > | < | ||||
| Body as a whole | Musculoskeletal and connective tissue | Nervous system | |||||
| Fatigue | Rigors | Arthralgia | Myalgia | Headache | Lethargy | Psychiatric disorders | |
| 0.1% vs 0.01% | > | > | > | > | > | > | > |
| 0.1% vs 0.03% | = | > | > | > | = | > | = |
| 0.1% vs 0.06% | > | < | < | > | = | = | < |
| 0.06% vs 0.01% | > | > | > | > | > | > | > |
| 0.06% vs 0.03% | < | > | > | = | = | > | > |
| 0.03% vs 0.01% | > | > | = | > | > | > | > |
< : significantly less participants, < : tendency to have less participants, = : equal number of participants, > : tendency to have more participants, > : significantly more participants
The numbers of participants experiencing adverse events related to musculoskeletal, connective tissue, and skin and subcutaneous tissue disorders, were similar between the different resiquimod concentrations. In contrast, the numbers of participants with adverse events associated with the nervous system in general were significantly lower in the 0.01% resiquimod group compared to all the other groups, which had similar number of participants (0.03%: RR 9.03, 95% CI 1.20 to 68.22, NNT = 4.3; Analysis 35.9) (0.06%: RR 10.94, 95% CI 1.48 to 80.73, NNT = 3.5; Analysis 33.9) (0.1%: RR 10.29, 95% CI 1.39 to 76.12, NNT = 3.7; Analysis 30.9). Headache, the only individual adverse event with significant difference between 2 resiquimod concentrations (0.06% vs 0.01%: RR 18.55, 95% CI 1.11 to 308.90, NNT = not applicable; Analysis 33.10), is a main contributor to the nervous system‐related adverse events in this study, with 6/8 participants in the 0.03% group, 8/10 in the 0.06% group, and 7/10 in the 0.1% group suffering from it.
35.9.
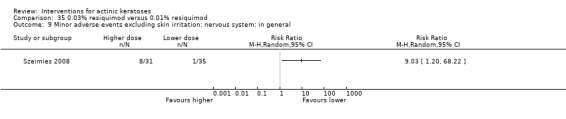
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.
33.9.
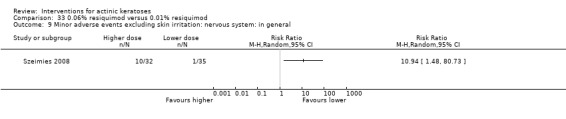
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.
30.9.
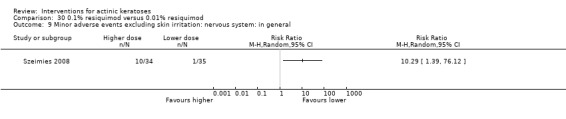
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.
33.10.
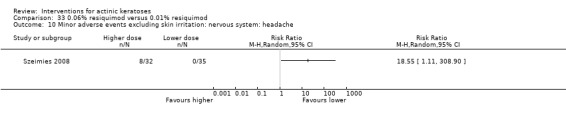
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.
To summarise, 0.1% resiquimod was more effective than the other lower concentrations only if the participants were treated with 1 cycle, i.e. once per day 3 times per week for 4 weeks on and 8 weeks off. Treatment with 0.01% resiquimod was generally associated with less adverse events compared to the other 3 concentrations used.
Sunscreen
Sunscreen is not generally a treatment for actinic keratosis, but it is a means of preventing actinic keratoses. However, one study investigated the role of sunscreen in the cure of existing lesions.
Sunscreen SPF 17 (8% 2‐ethyl‐hexyl p‐methoxycinnamate/2% 4‐tert‐butyl‐4‐methoxy‐4‐dibenzoylmethane) versus placebo
This intervention was addressed by one study (Thompson 1993) comparing sunscreen or placebo creams applied as needed daily for seven months to treat solar keratoses on the head, neck, forearms, and hands. Assessment was performed at the end of the seven‐month treatment. There was possible attrition bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 431 participants) |
| Thompson 1993 | ‐ | ‐ | ‐ | Absolute values |
Mean changes [reduction (‐) or increase (+)] in lesion counts (Analysis 36.1) were assessed at the end of treatment. The sunscreen‐treated group showed a small mean decrease in lesion counts (‐0.6 + 4.34, SD), whereas the placebo‐treated group showed a mean increase in lesion counts (1 + 4.46, SD), demonstrating that sunscreen application could not only prevent but also treat actinic keratoses. The resulting mean difference of ‐1.60 (95% CI ‐2.43 to ‐0.77) favoured the use of sunscreen.
36.1.
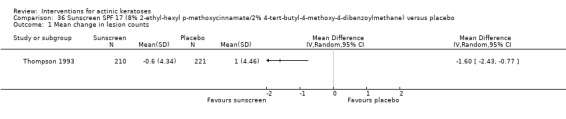
Comparison 36 Sunscreen SPF 17 (8% 2‐ethyl‐hexyl p‐methoxycinnamate/2% 4‐tert‐butyl‐4‐methoxy‐4‐dibenzoylmethane) versus placebo, Outcome 1 Mean change in lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 588 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Thompson 1993 | x | ‐ | ‐ | ‐ |
The authors of the Thompson 1993 study mentioned that 28 and 32 participants in the placebo and sunscreen groups, respectively, withdrew from the study because of skin reactions. We were unable to perform statistical analysis as the participants who withdrew were not grouped by individual reason, and some participants had multiple reasons. Because the number of participants in each treatment group was similar for withdrawal due to skin reactions, withdrawal in general, and who completed the study, we can assume that there was no significant difference in the number of participants who withdrew because of adverse events between the placebo and sunscreen groups.
To summarise, sunscreen might help to treat actinic keratoses in addition to its preventive role, but the efficacy was limited.
DL‐α‐tocopherol (vitamin E)
This intervention was addressed by 1 study (Foote 2009) comparing 12.5% DL‐α‐tocopherol (vitamin E) and placebo applied twice daily for 6 months on the right/left arms for treatment of actinic keratoses. Assessment was performed at the end of the six‐month treatment. There was possible other bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 42 participants) |
| Foote 2009 | ‐ | ‐ | ‐ | Absolute values |
No significant difference in mean reduction in lesion counts (Analysis 37.1) was observed.
37.1.
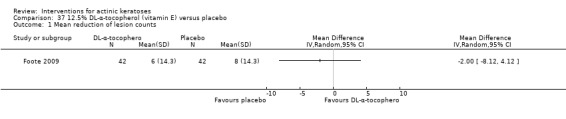
Comparison 37 12.5% DL‐α‐tocopherol (vitamin E) versus placebo, Outcome 1 Mean reduction of lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 48 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Foote 2009 | x | ‐ | ‐ | ‐ |
In this intraindividual study, 2 of the 48 participants withdrew from the study because of unrelated illness.
To summarise, vitamin E at the dosing regimen used was not more efficient than placebo to treat actinic keratoses.
Tretinoin
Tretinoin with 5‐fluorouracil
This comparison was presented in the 5‐fluorouracil section above.
Tretinoin versus arotinoid methyl sulfone (Ro 14‐9706)
This comparison was presented in the arotinoid methyl sulfone section above
(2) Prescription‐based oral drugs
Only one intervention for the treatment of actinic keratoses was given orally: etretinate.
Etretinate
This intervention was addressed by one study (Moriarty 1982) investigating the efficacy of etretinate for the treatment of actinic keratoses by comparing it to placebo treatment. Two parts were involved in this double‐blind cross‐over study, and only the first part is presented in this review. The first part involved oral etretinate, a 225 mg tablet 3 times daily for 2 months for 1 group, and the other group taking placebo with the same regimen. Assessment was performed at the end of the two‐month treatment. The anatomical locations of the lesions analysed were not specified. There was possible attrition bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 50 participants) |
Participant partial (> 75%) clearance | Mean (changes) reduction in lesion counts |
| Moriarty 1982 | ‐ | x | ‐ | ‐ |
Complete remission rates (converted to participant complete clearance) after part 1 were better in the etretinate group (5/25 = 20%) compared to placebo (0/25 = 0%), but it was not statistically significant (RR 11.00, 95% CI 0.64 to 188.95; Analysis 38.1).
38.1.
Comparison 38 Etretinate versus placebo, Outcome 1 Participant complete clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 50 participants) |
Skin irritation |
Minor adverse events excluding skin irritation (N = 50 participants) |
Cosmetic outcome |
| Moriarty 1982 | x (maybe) | ‐ | x | ‐ |
Five (etretinate = 3, placebo = 2) participants out of 50 dropped out of the study, but the reasons were not specified.
Because the adverse events were reported for both parts of the study, the quantitative data were not included in this review. Adverse effects were consistent with vitamin A‐type side‐effects (i.e. dry mouth, skin rash, desquamation, etc) and were experienced within the first three to four weeks of starting treatment by a large number of participants, but were reversed by reducing dosage. Many participants (17/44 = 39%, at the end of the 2 parts of the cross‐over study) required reduction in dosage due to toxicity of etretinate (hepatotoxicity), but response was still maintained when dosage was reduced. Hyperlipidaemia (raised serum lipid levels) associated with etretinate was not assessed in this study.
To summarise, etretinate at the dosing regimen used was not statistically more efficient than placebo to treat actinic keratoses and was associated with adverse events.
(3) Mechanical interventions
The only mechanical intervention reported in the included studies was laser resurfacing, and the different types of laser resurfacing are presented in alphabetical order: carbon dioxide and Er:YAG laser resurfacing. Both interventions are field‐directed treatments.
Carbon dioxide laser resurfacing
This intervention was addressed by 1 study (Hantash 2006) comparing the efficacy of 2 passes of carbon dioxide laser resurfacing with 5‐fluorouracil applied twice daily for 3 weeks and with trichloroacetic acid peel in the treatment of actinic keratoses on the face. Assessment was performed at 12 weeks after the end of the treatment. There was possible performance, detection, and attrition bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 27 participants) |
| Hantash 2006 | ‐ | ‐ | ‐ | Percentages |
The mean percentage of reduction in lesion counts showed a tendency to favour resurfacing compared to 5‐fluorouracil treatment (Analysis 39.1) or trichloroacetic acid peel (Analysis 40.1), but the differences were not statistically significant.
39.1.
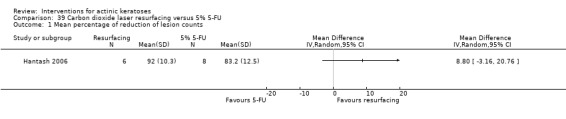
Comparison 39 Carbon dioxide laser resurfacing versus 5% 5‐FU, Outcome 1 Mean percentage of reduction of lesion counts.
40.1.
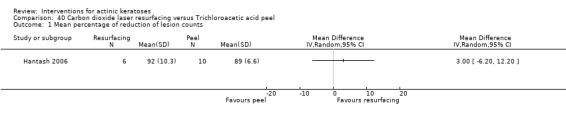
Comparison 40 Carbon dioxide laser resurfacing versus Trichloroacetic acid peel, Outcome 1 Mean percentage of reduction of lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 27 participants) |
Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (N = 27 participants) |
| Hantash 2006 | x | ‐ | ‐ | x |
Two of 8 participants in the carbon dioxide laser resurfacing group withdrew because of adverse events (incomplete treatment due to intolerance), whereas no participants withdrew in the trichloroacetic acid peel (0/10) and 5‐fluorouracil (0/9) groups. However, there was no statistically significant difference (Analysis 39.2; Analysis 40.2) between the treatments.
39.2.

Comparison 39 Carbon dioxide laser resurfacing versus 5% 5‐FU, Outcome 2 Withdrawal due to adverse events.
40.2.
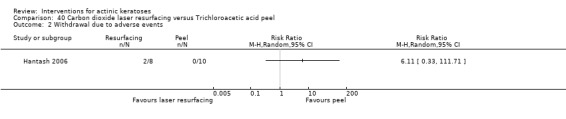
Comparison 40 Carbon dioxide laser resurfacing versus Trichloroacetic acid peel, Outcome 2 Withdrawal due to adverse events.
No postinflammatory pigmentary alteration or scarring was noted in the three treatment arms.
To summarise, the small sample size used in this study did not allow us to conclude on the superiority for efficacy or safety of carbon dioxide laser resurfacing over fluorouracil or trichloroacetic acid peel.
Er:YAG laser resurfacing
This intervention was addressed by 1 study (Ostertag 2006) comparing the efficacy of Er:YAG laser resurfacing and 5% 5‐fluorouracil applied twice daily for 4 to 7 weeks for the treatment of actinic keratoses on the face, scalp, or both. Assessments were performed at 3, 6, and 12 months after the end of treatment. There was possible reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 55 participants) |
| Ostertag 2006 | ‐ | ‐ | ‐ | Absolute values and percentages |
A statistical analysis could not be performed because the associated standard deviations were not provided with the mean reductions. The means in Analysis 41.1 suggested that the 2 treatments were equally efficient at reducing actinic keratosis lesions, whereas the mean percentages in Analysis 41.2 suggested better efficacy for laser resurfacing at 6 and 12 months. A statistical significance was stated by the authors.
41.1.
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 1 Mean reduction in lesion counts.
| Mean reduction in lesion counts | ||||
|---|---|---|---|---|
| Study | Intervention | At 3 months | At 6 months | At 12 months |
| Ostertag 2006 | 5‐fluorouracil | 13.2 | 12.5 | 12.4 |
| Ostertag 2006 | Er:YAG laser resurfacing | 13.8 | 13.9 | 14.2 |
41.2.
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 2 Mean percentage of reduction in lesion counts.
| Mean percentage of reduction in lesion counts | |||
|---|---|---|---|
| Study | Assessment | Resurfacing | 5‐FU |
| Ostertag 2006 | At 6 months | 94.4% | 79.2% |
| Ostertag 2006 | At 12 months | 91.1% | 76.6% |
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 55 participants) |
Skin irritation (N = 55 participants) |
Minor adverse events excluding skin irritation (N = 55 participants) |
Cosmetic outcome (N = 55 participants) |
| Ostertag 2006 | x | Overtime | Overtime | x |
One participant withdrew due to an adverse event (death) in the 5‐fluorouracil‐treated group, which was not significantly different to the Er:YAG laser resurfacing group (Analysis 41.3).
41.3.
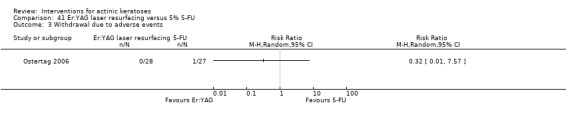
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 3 Withdrawal due to adverse events.
The adverse events (skin irritation and minor adverse events) could be categorised into 3 groups: 1) adverse events present only after treatment; 2) adverse events developing after the treatment, i.e. during the follow‐up period; and 3) adverse events present after the treatment and at follow‐up. Infection was present only at the end of the treatment.
The number of participants who developed an infection was not significantly different between the two treatments but was higher at most time points with laser resurfacing. Acne and milia developed during the follow‐up period. The number of participants with acne or milia was higher in the laser resurfacing group. The exception was acne at 12 months, which was similar between the two groups. The number of participants experiencing pain, crustea, and irritation tended to be higher in the fluorouracil treated‐group at the end of treatment, but it became higher in the laser resurfacing group during follow‐up. Only the number of participants with crustea was significantly different at the end of treatment (RR 0.46, 95% CI 0.27 to 0.79, NNT = 2.4; Analysis 41.6).
41.6.
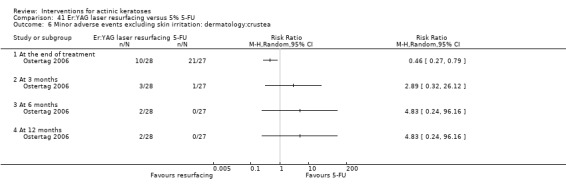
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 6 Minor adverse events excluding skin irritation: dermatology:crustea.
In terms of cosmetic outcomes, hypopigmentation got worse over time for laser resurfacing, significantly favouring 5‐fluorouracil at 12 months (RR 11.57, 95% CI 1.61 to 83.00; Analysis 41.10), corresponding to a NNT of 2.6 for an additional harmful outcome with laser re‐surfacing. Scarring was seen only in the laser resurfacing group but was not significantly different than in the fluorouracil group. In contrast, significantly more participants improved on the photoageing score with the laser resurfacing at 6 months (RR 1.57, 95% CI 1.01 to 2.43, NNT = 3.5) and 12 months (RR 1.70, 95% CI 1.01 to 2.88, NNT = 3.3) (Analysis 41.12) based on evaluation by 2 blinded investigators.
41.10.
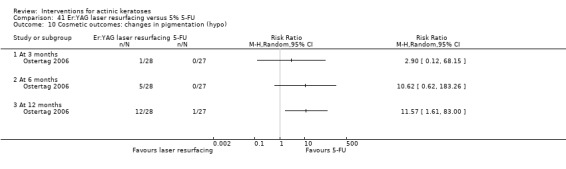
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 10 Cosmetic outcomes: changes in pigmentation (hypo).
41.12.
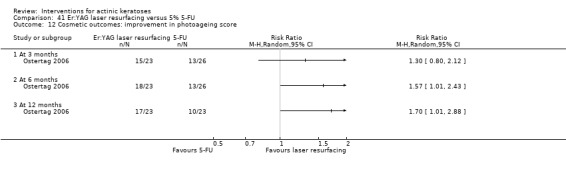
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 12 Cosmetic outcomes: improvement in photoageing score.
To summarise, the superiority of Er:YAG laser resurfacing over 5‐fluorouracil still needs to be demonstrated. More adverse events were associated with Er:YAG laser resurfacing compared to 5‐fluorouracil; however, overall ageing scores were better with Er:YAG laser resurfacing.
(4) Chemical interventions
Chemical interventions included studies on cryotherapy, photodynamic therapy, and trichloroacetic acid peel, which are presented in alphabetical order.
Cryotherapy
Cryotherapy was either compared with or combined with topical treatments or other chemical interventions, e.g. photodynamic therapy. Thus, the results are presented in two corresponding sections. Within each section, the comparisons are presented in alphabetical order of the comparison treatment. Cryotherapy is a lesion‐directed treatment for detectable lesions, whereas topical treatments are generally field‐directed treatments, which treat both detectable and subclinical lesions. Photodynamic therapy can be used for single lesion or field‐directed treatments. Cryotherapy and photodynamic therapy are provider‐administered, whereas topical treatments are administered by participants, and their efficacy is highly dependent on the compliance of the participants. These factors might influence the treatment efficacy.
Comparisons with topical treatments
Cryotherapy compared to betulin‐based oleogel
This intervention was addressed by one study (Huyke 2009) comparing cryotherapy with liquid nitrogen to betulin‐based oleogel alone on the face, scalp, or other locations. Cryotherapy of participant lesions was performed once on lesions on the face and twice on lesions on the rest of the body, whereas betulin‐based oleogel was applied twice daily for an unspecified duration. Assessment was performed at three months after the beginning of the treatment. There was possible performance and detection bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 30 participants) |
Participant partial (> 75%) clearance (N = 30 participants) |
Mean (changes) reduction in lesion counts |
| Huyke 2009 | ‐ | x | x | ‐ |
Similar participant complete or partial (> 75%) clearance rates were observed for the 2 treatments (Analysis 42.1; Analysis 42.2).
42.1.
Comparison 42 Cryotherapy versus betulin‐based oleogel, Outcome 1 Participant complete clearance.
42.2.
Comparison 42 Cryotherapy versus betulin‐based oleogel, Outcome 2 Participant partial (>75%) clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 30 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Huyke 2009 | x (none lost) | ‐ | ‐ | ‐ |
There were no participant withdrawals due to adverse events.
To summarise, the regimens used in this study for cryotherapy with liquid nitrogen and betulin‐based oleogel had similar efficacy for the treatment of actinic keratoses.
Cryotherapy compared to 5‐fluorouracil
This intervention was addressed by 1 study (Krawtchenko 2007) comparing cryotherapy with liquid nitrogen performed once or twice with 2‐week intervals to 5% 5‐fluorouracil applied twice daily for 4 weeks on the head, neck, and décolleté. Assessment was performed at 4 (5‐fluorouracil) or 6 (cryotherapy) weeks after the end of treatment and at 1‐year follow‐up. There was possible performance and detection bias.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 49 participants) |
Participant partial (> 75%) clearance | Mean (changes) reduction in lesion counts |
| Krawtchenko 2007 | ‐ | x | ‐ | ‐ |
5% 5‐fluorouracil was significantly more effective than cryotherapy to completely clear participants of lesions after the treatment (RR 0.71, 95% CI 0.54 to 0.94, NNT = 3.6) as well as at 12‐month follow‐up (RR 0.12, 95% CI 0.02 to 0.89, NNT = 3.4; Analysis 43.1).
43.1.
Comparison 43 Cryotherapy versus 5% 5‐FU, Outcome 1 Participant complete clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 49 participants) |
Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (N = 49 participants) |
| Krawtchenko 2007 | x (none lost) | ‐ | ‐ | x |
There were no participant withdrawals due to adverse events.
The same percentage (4%) of the participants in the 5% 5‐fluorouracil group and cryotherapy group showed excellent cosmetic outcome as assessed by the investigator. Significantly more participants in the 5‐fluorouracil group had better skin appearance (RR 0.27, 95% CI 0.11 to 0.72, NNT = 2.3; Analysis 43.3).
43.3.
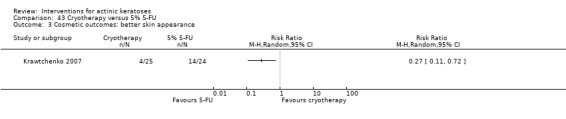
Comparison 43 Cryotherapy versus 5% 5‐FU, Outcome 3 Cosmetic outcomes: better skin appearance.
To summarise, cryotherapy was less efficacious than 5% 5‐fluorouracil at treating actinic keratoses.
Cryotherapy compared to imiquimod
This intervention was addressed by 1 study (Krawtchenko 2007) comparing cryotherapy and 5% imiquimod. Cryotherapy was performed once or twice with a 2‐week interval, whereas imiquimod was applied 3 times per week for 4 weeks followed by 4 weeks rest, and repeated if needed. There was possible performance and detection bias.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 51 participants) |
Participant partial (> 75%) clearance | Mean (changes) reduction in lesion counts |
| Krawtchenko 2007 | ‐ | x | ‐ | ‐ |
No significant difference was found in the number of participants completely cleared between 5% imiquimod applied for a total of 4 weeks and cryotherapy treatments, but there were more participants with clearance with imiquimod (22//26 compared with 17/25 on cryotherapy), which may have been due to the additional treatment of subclinical lesions (RR 0.80, 95% CI 0.59 to 1.10; Analysis 44.1).
44.1.
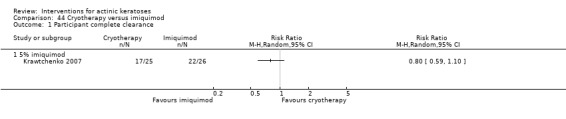
Comparison 44 Cryotherapy versus imiquimod, Outcome 1 Participant complete clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 51 participants) |
Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (N = 51 participants) |
| Krawtchenko 2007 | x (none lost) | ‐ | ‐ | x |
There were no participant withdrawals due to adverse events.
Assessment by the investigator showed that 4% and 81% of the participants had excellent cosmetic outcomes for cryotherapy and imiquimod treatments, respectively (RR 0.05, 95% CI 0.01 to 0.34, NNT = 1.3; Analysis 44.2). In particular, the skin quality was better with imiquimod treatment (RR 0.19, 95% CI 0.08 to 0.47, NNT = 1.5; Analysis 44.3).
44.2.
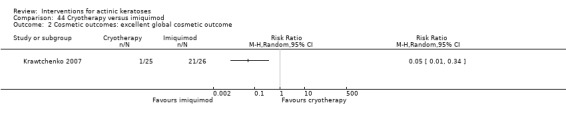
Comparison 44 Cryotherapy versus imiquimod, Outcome 2 Cosmetic outcomes: excellent global cosmetic outcome.
44.3.
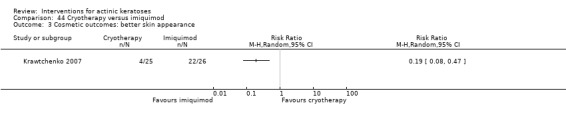
Comparison 44 Cryotherapy versus imiquimod, Outcome 3 Cosmetic outcomes: better skin appearance.
To summarise, cryotherapy and 5% imiquimod had similar efficacy, but imiquimod had significantly better cosmetic outcome.
Comparisons with photodynamic treatments
Cryotherapy versus 5‐aminolaevulinic acid (ALA)‐ photodynamic therapy (PDT)
This intervention was addressed by one open study (Hauschild 2009b) comparing cryotherapy with photodynamic therapy (PDT) using red light and auto‐adhesive ALA patches. Both interventions treated participant individual lesions on the head once, and no prior lesion preparation was performed. Assessment was performed 12 weeks after the end of treatment. There was possible performance, detection, attrition, and reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 255 participants) |
Participant partial (> 75%) clearance | Mean (changes) reduction in lesion counts |
| Hauschild 2009b | ‐ | x | ‐ | ‐ |
Analysis of participant complete clearance clearly favoured the ALA‐PDT treatment over cryotherapy (RR 0.76, 95% CI 0.61 to 0.96, NNT = 7.2; Analysis 46.1).
46.1.
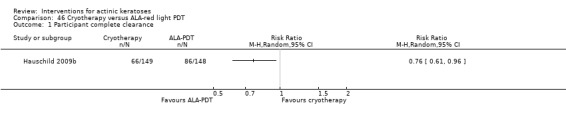
Comparison 46 Cryotherapy versus ALA‐red light PDT, Outcome 1 Participant complete clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 297 participants) |
Skin irritation (N = 297 participants) |
Minor adverse events excluding skin irritation | Cosmetic outcome |
| Hauschild 2009b | x (none lost) | x | Qualitative | ‐ |
There were no participant withdrawals due to adverse events.
Significantly more participants treated with ALA‐PDT experienced skin irritation during (RR 0.63, 95% CI 0.54 to 0.74, NNT = 3.2) and 1 day after (RR 0.27, 95% CI 0.16 to 0.46, NNT = 3.7) treatment compared to cryotherapy (Analysis 46.2).
46.2.

Comparison 46 Cryotherapy versus ALA‐red light PDT, Outcome 2 Skin irritation.
The minor adverse events reported in the cryotherapy group were eyelid oedema and swollen face, whereas pyoderma and emotional distress were documented for ALA‐PDT group. Headaches were reported in both groups.
In summary, in this single study, ALA‐PDT treatment was superior to cryotherapy for efficacy outcomes, but more skin irritation was associated with ALA‐PDT.
Cryotherapy versus methyl aminolevulinate (MAL)‐photodynamic therapy (PDT)
This intervention was addressed by 4 studies (Freeman 2003; Kaufmann 2008; Morton 2006; Szeimies 2002) comparing cryotherapy and PDT with 16% MAL for the treatment of actinic keratoses. All studies were open and used red light PDT. Characteristics of the studies are presented in the following table. There was possible performance, detection, and reporting bias for all studies, attrition bias for all studies except Morton 2006, and other bias for Freeman 2003 and Kaufmann 2008.
| Characteristic | Szeimies 2002 | Freeman 2003 | Morton 2006 | Kaufmann 2008 |
| Study design | Parallel | Parallel | Intraindividual | Intraindividual |
| Anatomical locations | Face, scalp, others (< 10%) |
Face or scalp | Face and scalp | Upper and lower extremities (98%), trunk, neck |
| Prior preparation of lesions (scale and crust removal) | Cryotherapy: yes PDT: yes |
Cryotherapy: no PDT: yes |
PDT: yes | PDT: yes (except mild lesions = 12%) |
| Number of treatment cycle | 1 (face and scalp) or 2 (other locations) | Cryotherapy : 1 PDT: 2 |
1 or 2 | 1 or 2 |
| Number of weeks between treatments | 1 | 1 | 12 | 12 |
| Number of freeze‐thaw cycles per treatment | 2 | 1 | 2 | 2 |
| Total freezing time (sec) | 24 + 18 | 12 to 26 | 16 | 20 + 14 |
| Individual lesion or field‐directed treatment (MAL) | Individual lesions | Individual lesions | Individual lesions | Individual lesions |
| Occlusion time with 16% MAL (hour) | 3 | 3 | 3 | 3 |
| PDT intensity (mW/cm²) |
70 to 200 | 50 to 250 | N/A | N/A |
| PDT dose (J/cm²) |
75 | 75 | 37 | 37 |
| Type of light source | Non‐coherent light | (CureLIght) | LED (Aktilite CL 128 lamp) | LED (Aktilite CL 128 lamp) |
| Time of assessment | 12 weeks after the end of treatment | 12 weeks after the end of treatment | 12 weeks after the end of treatment | 12 weeks after the end of treatment |
Primary outcomes
Most of the studies presented 'lesion complete response' as an efficacy outcome, which was not included in this review.
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 240 participants) |
| Szeimies 2002 | ‐ | ‐ | ‐ | ‐ |
| Freeman 2003 | ‐ | ‐ | ‐ | ‐ |
| Morton 2006 | ‐ | ‐ | ‐ | Percentages |
| Kaufmann 2008 | ‐ | ‐ | ‐ | Percentages |
Morton 2006 and Kaufmann 2008 presented the percentages without the associated standard deviations. Thus, no statistical analysis could be performed. Based on these percentages presented in Analysis 45.1 and the data presented in the overview tables for cryotherapy and photodynamic therapy, the two treatments seem to have similar efficacy.
45.1.
Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 1 Mean percentage of reduction in lesion counts.
| Mean percentage of reduction in lesion counts | |||
|---|---|---|---|
| Study | Assessment at | Cryotherapy | MAL‐PDT |
| Kaufmann 2008 | 12 weeks | N/A | N/A |
| Kaufmann 2008 | 24 weeks | 87% | 75% |
| Morton 2006 | 12 weeks | 74.5% | 84.4% |
| Morton 2006 | 24 weeks | 83.9% | 86.7% |
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 619 participants) |
Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (see table below) |
| Szeimies 2002 | x | ‐ | ‐ | x |
| Freeman 2003 | x | ‐ | Only for MAL‐PDT and not included in the analysis | x |
| Morton 2006 | x | ‐ | Intraindividual study not included in meta‐analysis | x |
| Kaufmann 2008 | x | ‐ | Intraindividual study not included in meta‐analysis | x |
In the parallel‐group studies, there was no difference in the number of participants who withdrew because of adverse events (Analysis 45.2). In the intraindividual studies, 4 of 119 (Morton 2006) and 2 of 121 (Kaufmann 2008) participants withdrew because of adverse events and 1 of them was related to MAL‐PDT treatment.
45.2.
Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 2 Withdrawal due to adverse events.
Kaufmann 2008 mentioned that the types of adverse events observed were mainly photosensitivity reaction (43% of 121 participants) and cold exposure injury (62% of 121 participants) for the MAL‐PDT and cryotherapy groups, respectively. Similar qualitative observation was mentioned by Morton 2006 (N = 119).
The types of cosmetic outcomes reported by the four studies are summarised in the following table.
| Parameter |
Szeimies 2002 (N = 122 participants) |
Freeman 2003 ( N = ? participants) |
Morton 2006 | Kaufmann 2008 |
| Evaluation by investigator | X | X | X | X |
| Evaluation by participant | X | X | N/A | N/A |
| Outcome | 1) excellent or good 2) fair or poor |
Excellent | 1) excellent 2) good 3) fair 4) poor |
1) excellent 2) good 3) fair 4) poor |
| Reported per participant | X (only for participants with > 75% reduction of total lesions) |
X (only for participants with 100% reduction of total lesions) |
N/A | N/A |
| Reported per lesion | N/A | X | X | X |
Because participants or right/left sides were randomised and not the lesions, only the data reported by participants were analysed. Freeman 2003 reported the percentages of completely cleared participants with excellent cosmetic outcome, but the number of participants completely cleared was not specified and the standard deviations associated with the percentages were not provided. Thus, no statistical analysis could be performed on these data. Similar percentages were obtained for investigator and participant assessments for MAL‐PDT (83% vs 76%) and cryotherapy (51% vs 56%). The authors reported significant differences between MAL‐PDT and cryotherapy groups. The investigator (RR 0.84, 95% CI 0.74 to 0.95, NNT = 6.5; Analysis 45.3) and participant (RR 0.93, 95% CI 0.86 to 1.01, NNT = 14.6; Analysis 45.4) evaluations in the Szeimies 2002 study also supported a better cosmetic outcome in the MAL‐PDT group.
45.3.
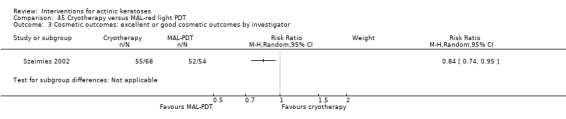
Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 3 Cosmetic outcomes: excellent or good cosmetic outcomes by investigator.
45.4.
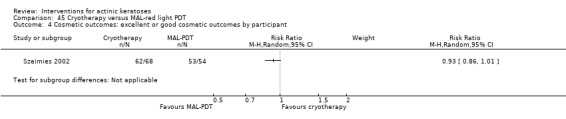
Comparison 45 Cryotherapy versus MAL‐red light PDT, Outcome 4 Cosmetic outcomes: excellent or good cosmetic outcomes by participant.
To summarise, because most of the efficacy outcomes reported could not be included in our analyses, it is difficult to determine the relative efficacy of MAL‐PDT and cryotherapy. Data from one study suggested equivalence between the two treatments. MAL‐PDT treatment seems to result in better cosmetic outcomes than cryotherapy.
Photodynamic therapy
Photodynamic therapy employs light sources and photosensitising agents that may differ between studies. As this is a relatively new treatment method, testing different combinations of variables is necessary to attempt to identify the optimal PDT treatment form and regimen. Light sources vary from polychromatic to pulsed laser. Photosensitising agents aminolevulinic acid (ALA) and newer methyl‐aminolevulinic acid (MAL) were both used, depending on the study. Thus, results are presented in two sections: photodynamic therapy with ALA and photodynamic therapy with MAL. Within these sections, the results are presented in the following order: 1) comparisons between ALA or MAL and placebo, 2) comparisons with different photodynamic therapy variables, and 3) comparisons with other treatments. Photodynamic therapy could be used to treat individual lesions or a field.
Photodynamic therapy (PDT) with 5‐aminolaevulinic acid (ALA)
ALA‐PDT versus placebo‐PDT
This intervention was addressed by five studies (Hauschild 2009a; Hauschild 2009b; Jeffes 2001; Piacquadio 2004; Szeimies 2010b) investigating the use of aminolevulinic acid (ALA) and photodynamic therapy (PDT) compared to placebo‐PDT to treat actinic keratoses. Characteristics of the studies are presented in the following table. There was possible performance (Hauschild 2009b; Jeffes 2001; Piacquadio 2004), detection (Hauschild 2009b; Piacquadio 2004), attrition (Hauschild 2009b; Piacquadio 2004), reporting (Hauschild 2009a; Hauschild 2009b; Piacquadio 2004), and other (Piacquadio 2004) bias.
| Blue light | Red light | |||
| Characteristic | Jeffes 2001 | Piacquadio 2004 |
Hauschild 2009a and Hauschild 2009b |
Szeimies 2010b |
| Study design | Assessor‐blinded intraindividual |
Assessor‐blinded parallel |
Double‐blinded parallel |
Double‐blinded parallel |
| Anatomical locations | Face and scalp | Face or scalp | Head | Face, bald scalp, or both |
| Prior preparation of lesions (e.g. scale and crust removal) | N/A | N/A | No | Yes |
| Number of treatment cycle | 1 or 2 | 1 or 2 | 1 | 1 or 2 |
| Number of weeks between treatments | 8 | 8 | N/A | 12 |
| Individual lesion or field‐directed treatment | Individual lesions | Individual lesions | Individual lesions | Individual lesions |
| ALA formulation | 20% cream | 20% cream | Patch containing 8 mg | BF‐200 gel |
| Occlusion time (hour) | 14 to 18 | 14 to 18 | 4 | 3 |
| PDT intensity (mW/cm²) |
3, 5, 10 | 10 | N/A | Aktilite: 50‐70 PhotoDyn 750: 196 |
| PDT dose (J/cm²) |
2, 5,10 | N/A | 37 | Aktilite: 37 PhotoDyn 750: 170 |
| Illumination time (seconds) | N/A | 1000 | N/A | Aktilite: N/A PhotoDyn 750: 900 |
| Type of light source | Non‐laser fluorescent (Dusa BLU‐417) |
visible (Blu‐U) |
LED (Aktilite CL 128 lamp or Omnilux) |
LED (Aktilite CL 128 lamp) or incoherent (PhotoDyn 750) |
| Time of assessment | 8 weeks after the end of each treatment | 8 weeks after the end of each treatment | 12 weeks after the end of each treatment | 12 weeks after the end of each treatment |
Subgroup analyses were performed to compare blue and red light photodynamic therapies. In addition, one study (Piacquadio 2004) provided efficacy data for individual anatomical locations, i.e. face or scalp, allowing additional subgroup analysis for blue light ALA or placebo with photodynamic treatment.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 701 participants) |
Participant partial (> 75%) clearance (N = 243 participants) |
Mean (changes) reduction in lesion counts |
| Jeffes 2001 | ‐ | intraindividual and not included in meta‐analysis | ‐ | ‐ |
| Piacquadio 2004 | ‐ | x | x | ‐ |
| Hauschild 2009a; Hauschild 2009b | ‐ | x | ‐ | ‐ |
| Szeimies 2010b | ‐ | x | ‐ | ‐ |
Most of the studies gave a second treatment to uncured lesions after the first treatment, and they provided efficacy outcomes 8 to 12 weeks after the first treatment (1 treatment) and after 4 to 12 weeks after the last treatment (1 or 2 treatments). Thus, separate comparisons were performed for the number of treatments received.
In Jeffes 2001, lesions treated with ALA were completely cleared in 45.7% (16/35) of the participants after 1 treatment using blue light PDT, whereas lesions treated with placebo were completely cleared in only 5.7% (2/35). Similarly, the number of participants with complete clearance was significantly higher in the ALA‐PDT group than placebo‐PDT group for both blue and red light after one treatment (Analysis 47.1). The amplitude of the effect was similar between blue (RR 6.22, 95% CI 2.88 to 13.43, NNT = 2.0; Analysis 47.1) and red light (RR 5.94, 95% CI 3.35 to 10.54, NNT = 2.0; Analysis 47.1), but a larger increase in the RR associated with blue light treatment following an additional treatment on uncured lesions was observed (blue light: RR 9.33, 95% CI 3.59 to 24.26, NNT = 1.8; and red light: RR 6.20, 95% CI 2.40 to 15.99, NNT = 2.0; Analysis 47.2). This difference might be explained by the fact that only one study with red light performed a second treatment: Szeimies 2010b used two light sources to reflect more medical practices. A lower efficacy was obtained with the ALA/PhotoDyn 750 lamp (26/49 = 53%) than with ALA/Aktilite CL 128 (27/31 = 87%). The PhotoDyn lamp was used in 60% of the ALA and placebo participants, resulting in lower efficacy than the other 2 studies using only the Aktilite lamp after the first treatment.
47.1.
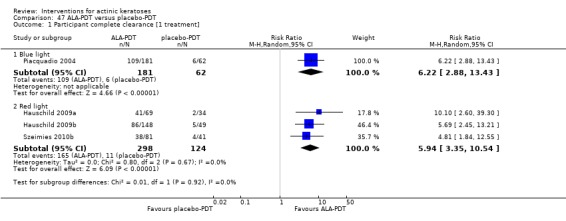
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 1 Participant complete clearance [1 treatment].
47.2.
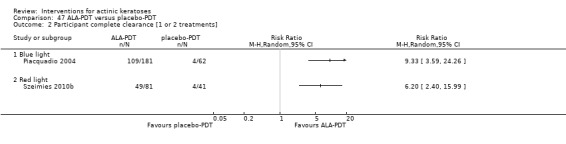
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 2 Participant complete clearance [1 or 2 treatments].
Similar results were obtained with participant partial clearance for blue light ALA‐PDT with a RR of 4.38, 95% CI 2.47 to 7.79, NNT = 1.8 for 1 treatment (Analysis 47.4) and a RR of 6.51, 95% CI 3.22 to 13.15, NNT = 1.6 for 1 or 2 treatments (Analysis 47.5). There was no difference in the RRs for participants completely (Analysis 47.3) or partially (Analysis 47.6) cleared of lesions on the face or scalp. For both outcomes and both sites, ALA‐PDT was significantly better than placebo‐PDT.
47.4.
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 4 Participant partial (> 75%) clearance [1 treatment].
47.5.
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 5 Participant partial (>75%) clearance[1 or 2 treatments].
47.3.

Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 3 Participant complete clearance [1 or 2 treatments] by anatomical location.
47.6.
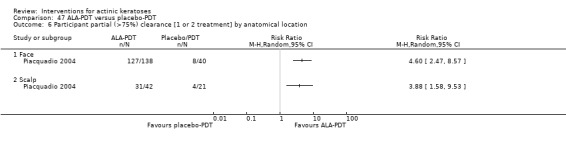
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 6 Participant partial (>75%) clearance [1 or 2 treatment] by anatomical location.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 701 participants) |
Skin irritation (N = 300 participants) |
Minor adverse events excluding skin irritation (N = 543 participants) |
Cosmetic outcome (see table below) |
| Jeffes 2001 | x (none lost) | ‐ | ‐ | x |
| Piacquadio 2004 | x (none lost) | ‐ | x | x |
|
Hauschild 2009a Hauschild 2009b |
x (none lost) | x | x | x |
| Szeimies 2010b | x (none lost) | ‐ | Intraindividual study not included in meta‐analysis | x |
There were no participant withdrawals due to adverse events.
The number of participants experiencing skin irritation was significantly higher in the ALA‐PDT group compared to placebo‐PDT during illumination (RR 8.94, 95% CI 4.62 to 17.31, NNT = 1.3; Analysis 47.7) and after the treatment (RR 59.72, 95% CI 3.75 to 952.48, NNT = not applicable; Analysis 47.7).
47.7.
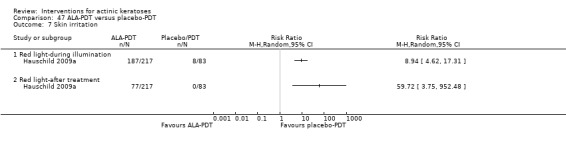
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 7 Skin irritation.
None of the adverse events reported for blue light photodynamic therapy [injury (Analysis 47.8), hypertension (Analysis 47.9), skin hypertrophy (Analysis 47.11) and headache (Analysis 47.12)] were significantly different between the two treatments. For red light photodynamic therapy, Hauschild 2009a and Hauschild 2009b reported skin discolouration in one participant in the ALA group, which was not significantly different between ALA and placebo‐treated participants (Analysis 47.10).
47.8.
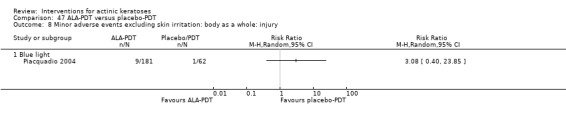
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 8 Minor adverse events excluding skin irritation: body as a whole: injury.
47.9.
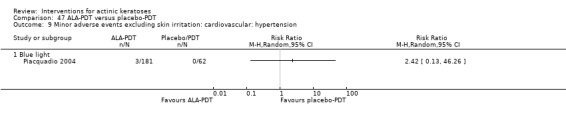
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 9 Minor adverse events excluding skin irritation: cardiovascular: hypertension.
47.11.
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 11 Minor adverse events excluding skin irritation: dermatology: skin hypertrophy.
47.12.

Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 12 Minor adverse events excluding skin irritation: nervous system: headache.
47.10.
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 10 Minor adverse events excluding skin irritation: dermatology: skin discolouration.
The types of cosmetic outcomes reported by the five studies are summarised in the following table.
| Parameter | Jeffes 2001 | Piacquadio 2004 | Hauschild 2009a; Hauschild 2009b |
Szeimies 2010b (N = 114 participants) |
| Evaluation by investigator | x (not specified) | x | x | x |
| Evaluation by participant | N/A | N/A | x | N/A |
| Outcome | Changes in pigmentation | Changes in pigmentation | 1) excellent 2) good 3) fair 4) poor |
General outcome: 1) very good or good 2) unsatisfactory/impaired outcome Skin quality (qualitative) |
| Reported per participant | N/A | N/A | N/A | x |
| Reported per lesion | x | x | x (cleared lesions only) | N/A |
Cosmetic outcomes were reported by all studies, but only Szeimies 2010b reported its outcome per participant. The cosmetic outcomes assessed by the investigator were very good or good in 49% of ALA‐PDT and 27% in placebo‐PDT groups, which was significantly different (RR 1.83, 95% CI 1.03 to 3.25, NNT = 4.5; Analysis 47.13).
47.13.
Comparison 47 ALA‐PDT versus placebo‐PDT, Outcome 13 Cosmetic outcome: very good or good general cosmetic outcome.
To summarise, ALA‐PDT was more effective than placebo‐PDT, and the efficacy is similar for blue or red light photodynamic therapy. For red light photodynamic therapy, using Aktilite CL 128 lamp gave better results than PhotoDyn 750 lamp. ALA treatment was generally associated with more skin irritation than placebo; however, ALA‐PDT resulted in better cosmetic outcomes.
ALA‐PDT: comparison between types of light source
This intervention was addressed by one study (Smith 2003) investigating ALA with one hour incubation followed by illumination with blue light or pulsed dye laser (PDL) for field‐directed treatment on the face or scalp, twice with a month interval. Assessment was performed at four weeks after the end of treatment. There was possible performance, detection, and reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 24 participants) |
Participant partial (> 75%) clearance (N = 24 participants) |
Mean (changes) reduction in lesion counts |
| Smith 2003 | ‐ | x | x | ‐ |
More participants receiving ALA‐blue light PDT compared to ALA‐PDL had complete (6/12 compared to 1/12) (Analysis 48.1) or partial (>75%) (9/12 compared to 5/12) (Analysis 48.2) clearance, respectively; however, this was not statistically significant.
48.1.
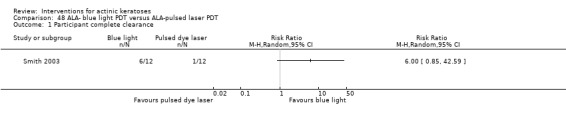
Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 1 Participant complete clearance.
48.2.
Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 2 Participant partial (>75%) clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 24 participants) |
Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (N = 24 participants) |
| Smith 2003 | x (none lost) | ‐ | ‐ | x |
There were no participant withdrawals due to adverse events.
None of the three cosmetic outcomes reported, i.e. improvements in global response, tactile roughness, and mottled hyperpigmentation, were significantly different between the two light sources (Analysis 48.3; Analysis 48.4; Analysis 48.5).
48.3.
Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 3 Cosmetic outcome: improvement in global response.
48.4.
Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 4 Cosmetic outcome: improvement in tactile roughness.
48.5.
Comparison 48 ALA‐ blue light PDT versus ALA‐pulsed laser PDT, Outcome 5 Cosmetic outcome: improvement in mottled hyperpigmentation.
To summarise, insufficient data were provided to determine the superiority of one source of light over the other for field‐directed treatment of actinic keratoses with ALA‐PDT.
ALA‐PDT: comparison for different incubation times with ALA
This intervention was addressed by 1 study (Hauschild 2009c) comparing the efficacy of self‐adhesive ALA patch treating individual lesions for different incubation times (0.5, 1, 2, and 4 hours) before PDT (red light) treatment to treat actinic keratoses on the head and face. Assessments were performed at 4 and 8 weeks after the end of treatment. There was possible attrition and reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 140 participants) |
Participant partial (> 75%) clearance | Mean (changes) reduction in lesion counts |
| Hauschild 2009c | ‐ | x | ‐ | ‐ |
Efficacy was assessed at four (Analysis 49.1) and eight weeks (Analysis 49.2), and participant complete clearance was analysed for subgroups of the different combinations between shorter and longer incubation times. At 4 weeks, analyses of participant complete clearance did not favour shorter or longer times except for comparison between the shortest (0.5 hours) and the longest (4 hours), which favoured the longest incubation time (RR 0.50, 95% CI 0.26 to 0.95, NNT = 3.8). In contrast, all comparisons favoured the longer incubation times with the exception of 1 hour versus 2 hours at week 8 (Analysis 49.2). Thus, a longer incubation with ALA gave better long‐term results.
49.1.
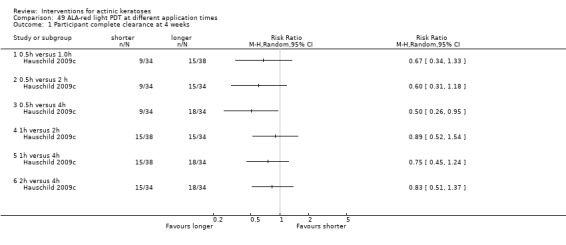
Comparison 49 ALA‐red light PDT at different application times, Outcome 1 Participant complete clearance at 4 weeks.
49.2.
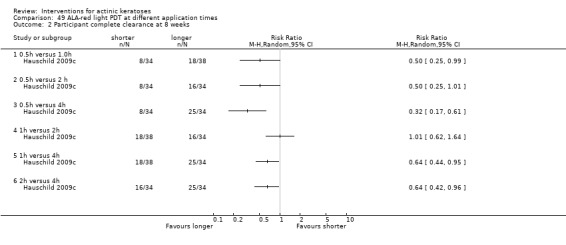
Comparison 49 ALA‐red light PDT at different application times, Outcome 2 Participant complete clearance at 8 weeks.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 149 participants) |
Skin irritation |
Minor adverse events excluding skin irritation (N = 149 participants) |
Cosmetic outcome |
| Hauschild 2009c | x (maybe) | ‐ | x | ‐ |
Of 149 participants, 9 were not included in the final efficacy analysis and 3 of them terminated the study prematurely; however, the authors did not give more details about the reasons or associated treatments.
Five of 149 participants experienced adverse events related to treatment, which were 3 headaches (1 in each of the 0.5‐, 2‐, and 4‐hour groups), 1 nose bleed (in the 4‐hour group), and a mild increase in alanine transaminase (1 in the 0.5‐hour group). None of these adverse events were significantly associated with the incubation time (mild increase in alanine transaminase: Analysis 49.3; headache: Analysis 49.4; and nose bleed: Analysis 49.5). Other adverse events were reported but not in relation to the incubation groups.
49.3.
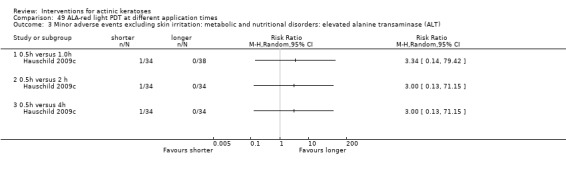
Comparison 49 ALA‐red light PDT at different application times, Outcome 3 Minor adverse events excluding skin irritation: metabolic and nutritional disorders: elevated alanine transaminase (ALT).
49.4.
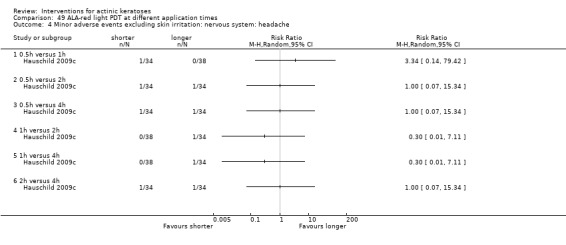
Comparison 49 ALA‐red light PDT at different application times, Outcome 4 Minor adverse events excluding skin irritation: nervous system: headache.
49.5.
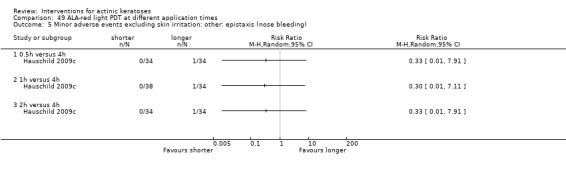
Comparison 49 ALA‐red light PDT at different application times, Outcome 5 Minor adverse events excluding skin irritation: other: epistaxis (nose bleeding).
To summarise, longer incubation with ALA resulted in an increase in long‐term efficacy.
ALA‐PDT versus 5‐fluorouracil
This intervention was addressed by 1 study (Smith 2003) comparing ALA‐PDT field‐directed treatment (twice with a 1‐month interval) using 2 different types of light sources (blue light and pulse dye laser) with 0.5% fluorouracil applied once or twice daily for 4 weeks on the face or scalp (field‐directed treatment). Assessment was performed four weeks after the end of treatment. There was possible performance, detection, and reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 36 participants) |
Participant partial (> 75%) clearance (N = 36 participants) |
Mean (changes) reduction in lesion counts |
| Smith 2003 | ‐ | x | x | ‐ |
Analyses of participant complete (Analysis 50.1) and partial (> 75%) (Analysis 50.2) clearance showed that the PDT treatments with blue light and the pulse dye laser (PDL) were comparable to 5‐fluorouracil. However, a tendency to favour 5‐fluorouracil over ALA‐PDT with pulsed dye laser could be observed for both outcomes (complete: RR 0.17, 95% CI 0.02 to 1.18; partial: RR 0.56, 95% CI 0.26 to 1.17), but this was not statistically significant.
50.1.
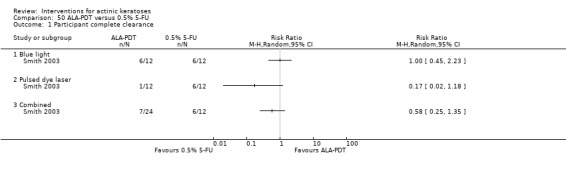
Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 1 Participant complete clearance.
50.2.
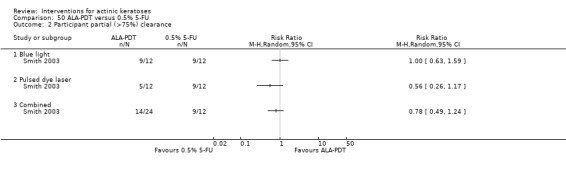
Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 2 Participant partial (>75%) clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 36 participants) |
Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (N = 36 participants) |
| Smith 2003 | x | ‐ | ‐ | x |
One of 12 participants in the 5‐fluorouracil group withdrew because of adverse events compared to none of the 24 participants in the ALA‐PDT groups, which was not significantly different (Analysis 50.3).
50.3.
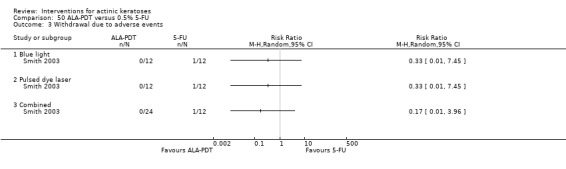
Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 3 Withdrawal due to adverse events.
None of the 3 cosmetic outcomes reported improvements in global response (RR 0.74, 95% CI 0.44 to 1.25; Analysis 50.4). Tactile roughness (RR 0.92, 95% CI 0.52 to 1.61; Analysis 50.5) and mottled hyperpigmentation (RR 0.65, 95% CI 0.34 to 1.26; Analysis 50.6) were significantly different between 5‐fluorouracil and ALA‐PDT administered with the 2 light sources, but there was a general tendency to favour 5‐fluorouracil treatment. However, this was not statistically significant.
50.4.
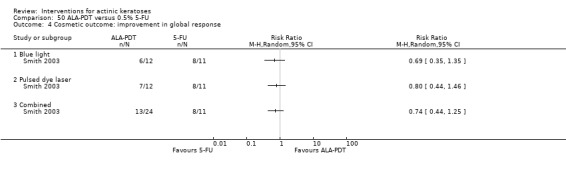
Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 4 Cosmetic outcome: improvement in global response.
50.5.
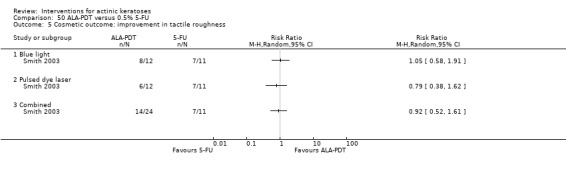
Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 5 Cosmetic outcome: improvement in tactile roughness.
50.6.

Comparison 50 ALA‐PDT versus 0.5% 5‐FU, Outcome 6 Cosmetic outcome: improvement in mottled hyperpigmentation.
To summarise, no statistical difference could be observed between 5‐fluorouracil treatments and ALA with photodynamic therapy because of the small sample of this study. However, 5‐fluorouracil treatment had a tendency to result in better outcomes.
ALA‐PDT and imiquimod
This intervention was addressed by 1 intraindividual study (Sotiriou 2009) comparing 2 treatments of ALA‐red light PDT performed at a 15‐day interval on individual lesions and a dosing cycle of 5% imiquimod once per day 3 times per week for 4 weeks on, 4 weeks off, repeated if needed on the dorsal side of the hands and forearms (field‐directed treatment). Assessments were performed 4 and 24 weeks after the end of treatment. There was possible performance and detection bias.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance | Mean (changes) reduction in lesion counts |
| Sotiriou 2009 | ‐ | ‐ | ‐ | ‐ |
The study by Sotiriou 2009 reported "lesion complete response" as an efficacy outcome, which was not one of our primary outcomes.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 30 participants) |
Skin irritation | Minor adverse events excluding skin irritation |
Cosmetic outcome (N = 30 participants) |
| Sotiriou 2009 | X (none lost) | ‐ | ‐ | x |
There were no participant withdrawals due to adverse events.
The authors of the Sotiriou 2009 study reported no significant difference in the investigator‐assessed excellent cosmetic outcome for lesions in the two treatment groups.
To summarise, the efficacy of ALA‐PDT and imiquimod could not be compared.
ALA‐PDT versus cryotherapy
This comparison was discussed in the cryotherapy section above, and the results presented in Table 69 correspond to Analysis 51.1 and Analysis 51.2 .
51.1.
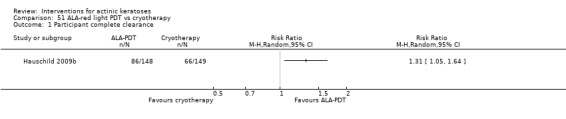
Comparison 51 ALA‐red light PDT vs cryotherapy, Outcome 1 Participant complete clearance.
51.2.
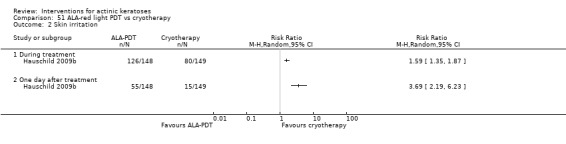
Comparison 51 ALA‐red light PDT vs cryotherapy, Outcome 2 Skin irritation.
Photodynamic therapy (PDT) with methyl‐aminolevulinic (MAL)
MAL‐PDT versus placebo‐PDT
This intervention was addressed by seven studies (Dragieva 2004a; Freeman 2003; Pariser 2003; Pariser 2008; Photocure‐Australian 2004; Photocure‐US 2004; Szeimies 2009) investigating the use of methyl‐aminolevulinic (MAL) and photodynamic therapy (PDT) compared to placebo‐PDT to treat actinic keratoses. The Dragieva 2004a study was performed with immunocompromised participants (organ transplants recipients). Characteristics of the studies are presented in the following table. There was possible performance (Dragieva 2004a; Freeman 2003), detection (Dragieva 2004a; Freeman 2003), attrition (Freeman 2003; Pariser 2003; Photocure‐Australian 2004; Photocure‐US 2004), reporting (Freeman 2003; Pariser 2003; Photocure‐Australian 2004; Photocure‐US 2004), and other (Freeman 2003) biases.
| Red light | ||||||
| Characteristic | Freeman 2003 | Pariser 2003 | Dragieva 2004a | Photocure‐Australian 2004; Photocure‐US 2004 | Pariser 2008 | Szeimies 2009 |
| Study design | Double‐blinded parallel |
Double‐blinded parallel |
Double‐blinded intraindividual |
Double‐blinded parallel |
Double‐blinded parallel |
Double‐blinded parallel |
| Anatomical locations | Face or scalp | Face and scalp | Face or scalp, neck, extremities | Face and scalp | Face and scalp | Face and scalp, hand (< 1%) |
| Prior preparation of lesions (e.g. scale and crust removal) | Yes | Yes | Yes | Yes | Yes | Yes |
| Number of treatment cycle | 2 | 2 | 2 | 2 | 2 | 2 |
| Number of weeks between treatments | 1 | 1 | 1 | 1 | 1 | 1 |
| Individual lesion or field‐directed treatment | Individual lesions | Individual lesions | Field‐directed treatment | Individual lesions | Individual lesions | Individual lesions |
| MAL formulation | 16% cream | 16% cream | N/A | 16.8% cream | 16.8% cream | 16% cream |
| Occlusion time (hour) | 3 | 3 | 3 | 2.5 to 4 | 3 | 3 |
| PDT intensity (mW/cm²) |
50 to 250 | 50 to 200 | 80 | N/A | N/A | 56 to 83 |
| PDT dose (J/cm²) |
75 | 75 | 75 | 75 | 37 | 37 |
| Illumination time (seconds) | 600 | 480 | N/A | N/A | 480 | 540 |
| Type of light source | Broadband (CureLight) |
Broadband non‐coherent light | Broadband non‐coherent (Waldmann PDT 1200) |
Broadband (CureLight) |
Light‐emitting diode (LED) (Aktilite CL 128) |
Light‐emitting diode (LED) (Aktilite CL 128) |
| Time of assessment | 12 weeks after the end of treatment | 12 weeks after the end of treatment | 16 weeks after the end of treatment | 12 weeks after the end of treatment | 12 weeks after the end of treatment | 12 weeks after the end of treatment |
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 499 participants) |
Participant partial (> 75%) clearance (N = 191 participants) |
Mean (changes) reduction in lesion counts |
| Freeman 2003 | ‐ | ‐ | ‐ | ‐ |
| Pariser 2003 | ‐ | x | ‐ | ‐ |
| Dragieva 2004a | ‐ | x | ‐ | ‐ |
| Photocure‐Australian 2004; Photocure‐US 2004; | ‐ | x | x | ‐ |
| Pariser 2008 | ‐ | x | ‐ | ‐ |
| Szeimies 2009 | ‐ | x | ‐ | ‐ |
Freeman 2003 reported only lesion complete response, which is not included in this review.
In immunocompetent participants, pooled RR for participant complete clearance favoured MAL/red light PDT (RR 4.46, 95% CI 3.17 to 6.28, NNT = 1.9; Analysis 52.1). Similarly, pooled RR (Photocure‐Australian 2004; Photocure‐US 2004) for participant partial (> 75%) clearance also favoured MAL‐PDT over placebo‐PDT (RR 3.28, 95% CI 1.73 to 6.23, NNT = 1.8; Analysis 52.2). In immunosuppressed participants, 13 out of 17 participants were completely cleared on the MAL‐PDT‐treated side and none on the placebo‐PDT‐treated side, supporting the superiority of MAL photodynamic therapy in these organ transplants patients.
52.1.
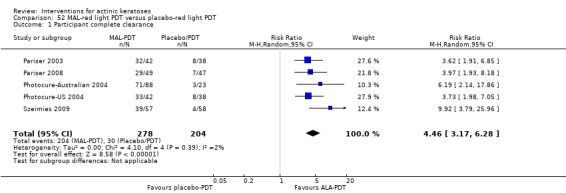
Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 1 Participant complete clearance.
52.2.
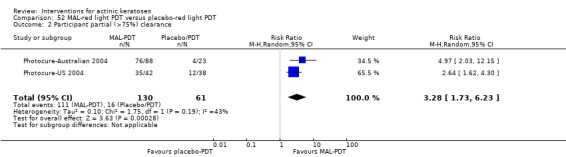
Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 2 Participant partial (>75%) clearance.
No publication bias was detected for the studies with immunocompetent participants based on the funnel plot (Figure 4).
4.
Funnel plot of comparison: 50 MAL‐PDT (red light) versus placebo‐PDT (red light), outcome: 50.1 Participant complete clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 402 participants) |
Skin irritation |
Minor adverse events excluding skin irritation (N = 115 participants) |
Cosmetic outcome (see text below) |
| Freeman 2003 | x | ‐ | ‐ | x |
| Pariser 2003 | x | ‐ | ‐ | x |
| Dragieva 2004a | ‐ | ‐ | ‐ | ‐ |
| Photocure‐Australian 2004; Photocure‐US 2004; | ‐ | ‐ | ‐ | x |
| Pariser 2008 | x (none lost) | ‐ | ‐ | ‐ |
| Szeimies 2009 | x (none lost) | ‐ | x | ‐ |
The pooled risk ratio for two of the studies showed no significant difference in the number of participants who withdrew because of adverse events between MAL‐PDT‐ and placebo‐PDT‐treated groups (Analysis 52.3). In addition, two other studies had no withdrawals due to adverse events in both treatment groups. These data together suggest that there is no difference between the two groups.
52.3.
Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 3 Withdrawal due to adverse events.
Szeimies 2009 reported one event of headache (one participant; Analysis 52.4) and three events of eyelid oedema in the MAL‐PDT group.
52.4.
Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 4 Minor adverse event: nervous system: headache.
Excellent cosmetic outcomes were observed for MAL‐PDT in 81% to 93% of participants completely cleared [Freeman 2003 (N = the number of participants evaluated was not given); Pariser 2003 (N = 32)], but in the absence of data reported for placebo‐PDT, these values could not be compared. No significant difference was observed for hyperpigmentation (N = 191; Analysis 52.5).
52.5.
Comparison 52 MAL‐red light PDT versus placebo‐red light PDT, Outcome 5 Cosmetic outcome: hyperpigmentation.
To summarise, MAL‐PDT was clearly more efficient than placebo‐PDT to treat actinic keratoses.
MAL‐PDT: comparisons between types of light source
This intervention was addressed by two studies (von Felbert 2010; Wiegell 2008). Wiegell 2008 compared field‐directed treatment using MAL‐PDT with light‐emitting diode (LED) red light and field‐directed treatment using MAL‐PDT with daylight (sun) on the face or scalp (field‐directed treatment). After removal of crust and hyperkeratoses, MAL cream was applied for three hours. After 30 minutes occlusion, the daylight‐treated side was exposed to outside daylight for 2.5 hours, and then the red light side, which stayed under occlusion for 3 hours, was treated with a LED lamp. von Felbert 2010 compared individual lesion treatment (one or two treatments) using MAL‐PDT with red light LED or a broadband visible plus water‐filtered infrared A on the face or scalp. Each treatment group was further separated into two subgroups: with and without cooling spray during illumination. Assessments were performed at 12 (von Felbert 2010; Wiegell 2008), 24 (von Felbert 2010), and 48 weeks (von Felbert 2010). There was possible performance (Wiegell 2008) and attrition (von Felbert 2010) bias.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 80 participants) |
Participant partial (> 75%) clearance (N = 80 participants) |
Mean (changes) reduction in lesion counts (N = 30 participants) |
| Wiegell 2008 | ‐ | ‐ | ‐ | Absolute values and percentages |
| von Felbert 2010 | ‐ | x | x | ‐ |
No difference in the mean reduction in lesion counts was found between red (8.0 + 5.6, mean + SD, 71%) and daylight (8.4 + 5.4, 79%) (Analysis 54.1).
54.1.
Comparison 54 MAL‐red light LED PDT versus MAL‐daylight PDT, Outcome 1 Mean reduction in lesion counts.
At 12 months, the number of participants with complete (RR 1.50, 95% CI 0.90 to 2.51; Analysis 53.1) clearance had a tendency to be higher in the MAL‐PDT using red light LED as the illumination source, although this was not statistically significant, compared to broadband visible plus water‐filtered infrared A. In contrast, no tendency could be observed for partial clearance (RR 1.03, 95% CI 0.85 to 1.25; Analysis 53.2).
53.1.
Comparison 53 MAL‐red light LED PDT versus MAL‐broad visible + water‐filtered infrared A PDT (1 or 2 treatments), Outcome 1 Participant complete clearance.
53.2.
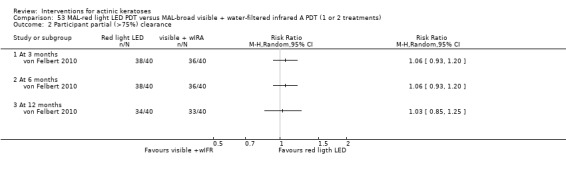
Comparison 53 MAL‐red light LED PDT versus MAL‐broad visible + water‐filtered infrared A PDT (1 or 2 treatments), Outcome 2 Participant partial (>75%) clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 110 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Wiegell 2008 | x (none lost) | ‐ | ‐ | ‐ |
| von Felbert 2010 | x (none lost) | ‐ | ‐ | ‐ |
There were no participant withdrawals due to adverse events.
It is worth noting that the authors of the Wiegell 2008 study reported a pain score significantly lower during daylight exposure than red light exposure. The adverse events were more severe in the sun‐exposed side for 42% of the participants and more severe in the red light side for 21% following treatment (Wiegell 2008).
To summarise, performing MAL‐PDT with daylight exposure resulted in similar efficacy to MAL‐PDT with red light treatment. However, a tendency for better results with red light LED compared to broad visible light with water filtered infrared A was observed.
MAL‐PDT: comparison for different incubation times with MAL
This intervention was addressed by 1 study (Wiegell 2011a) comparing the efficacy of field‐directed treatment MAL‐PDT for different illumination times with daylight in the presence of 16% MAL cream. Sunscreen was applied for 15 minutes to the treatment area on the face and scalp, and crusts and scales were gently removed before MAL application. After 30 minutes occlusion with MAL, participants were exposed to the sun for 1.5 or 2.5 hours, resulting in exposure to MAL for 2 and 3 hours. All lesions present in the area were treated, but only grade 1 lesions were included in the data analysis by the authors of the study. Assessment was performed 12 weeks after the end of treatment. There was possible performance bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 120 participants) |
| Wiegell 2011a | ‐ | ‐ | ‐ | Absolute values and percentages |
No difference was found between 2 and 3 hours MAL incubation with daylight PDT for mean reduction of lesion counts (MD 0.10, 95% CI ‐3.17 to 3.37; Analysis 55.1) or mean percentage reduction in lesion counts (MD 2.60, 95% CI ‐6.46 to 11.66; Analysis 55.2). The latter had a tendency to favour the shortest incubation time, but this was not statistically significant.
55.1.
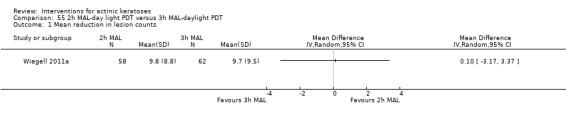
Comparison 55 2h MAL‐day light PDT versus 3h MAL‐daylight PDT, Outcome 1 Mean reduction in lesion counts.
55.2.
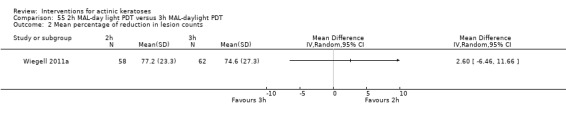
Comparison 55 2h MAL‐day light PDT versus 3h MAL‐daylight PDT, Outcome 2 Mean percentage of reduction in lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 120 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Wiegell 2011a | x (none lost) | ‐ | ‐ | ‐ |
There were no participant withdrawals due to adverse events.
To summarise, similar efficacy was obtained for 2‐ or 3‐hour incubation with 16% MAL with sun exposure for 1.5 and 2.5 hours, respectively.
MAL‐PDT: comparison for different concentrations of MAL
This intervention was addressed by 1 intraindividual study (Wiegell 2009) comparing the efficacy of field‐directed treatment with MAL‐PDT for different MAL concentrations (16% versus 8%) with daylight PDT for actinic keratoses on the face or scalp. Sunscreen was applied for 15 minutes to the treatment area, and crusts and scales were gently removed before MAL application. The participants were then instructed to spend as much time as possible outside for the rest of the day and wash off the cream at bedtime. The light dose was measured by a dosimeter. Assessment was performed 12 weeks after the end of treatment. There was possible reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 29 participants) |
| Wiegell 2009 | ‐ | ‐ | ‐ | Absolute values and percentages |
Similar efficacy was obtained for the 2 concentrations of MAL, i.e. mean reduction in lesion counts of 14.8 + 8.2 (mean + SD, 76.9%) for 16% MAL and 14.5 + 7.6 (79.5%) for 8% MAL (Analysis 56.1).
56.1.
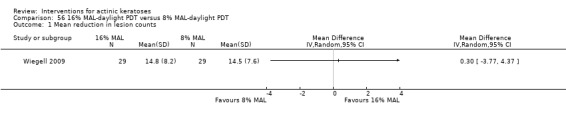
Comparison 56 16% MAL‐daylight PDT versus 8% MAL‐daylight PDT, Outcome 1 Mean reduction in lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 30 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Wiegell 2009 | x | ‐ | ‐ | ‐ |
One of 30 participants withdrew because of unrelated adverse events (terminal illness).
To summarise, 8% and 16% MAL treatments gave similar results with daylight photodynamic therapy to treat actinic keratoses.
MAL‐PDT: comparison between single and multiple MAL‐PDT treatment
This intervention was addressed by one study (Tarstedt 2005) comparing the efficacy of one MAL‐PDT treatment with red light and three‐hour incubation compared to the efficacy of multiple MAL‐PDT treatments, which involved two treatment sessions one week apart, on individual lesions on the face and scalp. Lesions not cleared after 12 weeks were retreated. Assessment was performed 12 weeks after the end of each cycle of treatment. There was possible performance, detection, and attrition bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 211 participants) |
Participant partial (> 75%) clearance | Mean (changes) reduction in lesion counts |
| Wiegell 2009 | ‐ | x | ‐ | ‐ |
The number of participants achieving complete clearance was significantly higher in the single MAL‐PDT treatment group compared to the multiple MAL‐PDT treatment (RR 1.17, 95% CI 1.03 to 1.33; Analysis 57.1).
57.1.
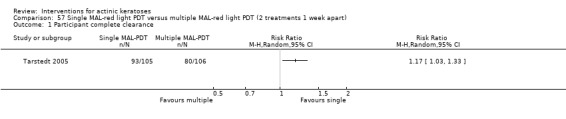
Comparison 57 Single MAL‐red light PDT versus multiple MAL‐red light PDT (2 treatments 1 week apart), Outcome 1 Participant complete clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 211 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Tarstedt 2005 | x | ‐ | ‐ | Per lesion (not included in analysis) |
The number of participants who withdrew because of adverse events was not significantly different between single MAL‐PDT and multiple MAL‐PDT (Analysis 57.2).
57.2.
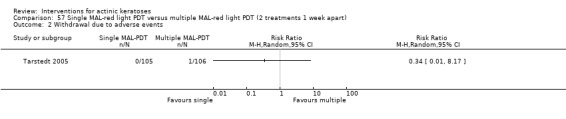
Comparison 57 Single MAL‐red light PDT versus multiple MAL‐red light PDT (2 treatments 1 week apart), Outcome 2 Withdrawal due to adverse events.
To summarise, multiple MAL‐PDT treatments were associated with more adverse events and were less efficacious than a single treatment.
MAL‐PDT versus cryotherapy
This comparison was discussed in the cryotherapy section above and the results presented in Table 69 correspond to Analysis 45.1 and Analysis 58.1.
58.1.
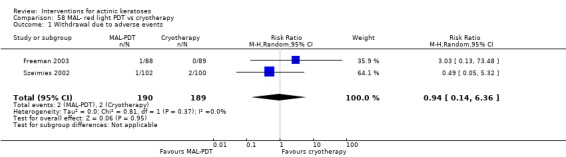
Comparison 58 MAL‐ red light PDT vs cryotherapy, Outcome 1 Withdrawal due to adverse events.
ALA‐PDT versus MAL‐PDT
This intervention was addressed by 1 intraindividual study (Moloney 2007) comparing 20% ALA incubated for 5 hours and 20% MAL incubated for 3 hours before PDT under identical conditions for field‐directed treatment of extensive actinic keratoses on the scalp. Assessment was performed four weeks after the end of treatment. There was possible reporting bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 16 participants) |
Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 15 participants) |
| Moloney 2007 | ‐ | Field complete clearance | ‐ | Absolute values |
Because of the intraindividual design of the Moloney 2007 study, participant complete clearance could not be included in meta‐analysis, but there was no significant difference between the effectiveness of the 2 treatments in curing actinic keratosis lesions based on participant complete clearance (ALA: 6/16 and MAL: 7/16) and mean reduction in lesion counts (Analysis 59.1).
59.1.
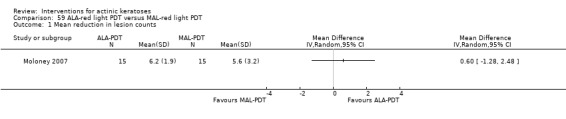
Comparison 59 ALA‐red light PDT versus MAL‐red light PDT, Outcome 1 Mean reduction in lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 16 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Moloney 2007 | x (none lost) | ‐ | ‐ | ‐ |
There were no participant withdrawals due to adverse events.
To summarise, there was no significant difference between the effectiveness of MAL and ALA treatments to treat extensive actinic keratoses.
MAL‐PDT versus 5‐fluorouracil
This intervention was addressed by 1 intraindividual study (Perrett 2007) comparing 3‐hour incubation with MAL followed by red light PDT with 5% 5‐fluorouracil twice daily for 3 weeks for treatment of individual actinic keratosis lesions and carcinoma in situ on the forearms and hands of organ transplant participants (immunosuppressed). Assessments were performed at 4, 12, and 24 weeks after the end of treatment. Data for efficacy but not safety outcomes were available separately for actinic keratoses. There was possible performance and detection bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 4 participants) |
Participant partial (> 75%) clearance | Mean (changes) reduction in lesion counts |
| Perrett 2007 | ‐ | x | ‐ | ‐ |
Because of the intraindividual design of the study, the data for the participant complete clearance could not be included in a meta‐analysis. Thus, the efficacy results at one, three, and six months after treatments are presented in the following table.
| Assessment at (months) | MAL‐PDT | 5‐fluorouracil |
| 1 | 4/4 | 0/4 |
| 3 | 4/4 | 1/4 |
| 6 | 4/4 | 1/4 |
Based on this small sample size study, MAL‐PDT seemed to be more effective at treating actinic keratoses in organ transplant participants than 5‐fluorouracil under the conditions used.
Secondary outcomes
Because of the pooled data for carcinoma in situ and actinic keratoses, none of our secondary outcomes could be taken from the study by Perrett 2007.
To summarise, despite the small sample size used in Perrett 2007, efficacy data suggested that MAL‐PDT was more efficacious than 5‐fluorouracil to treat actinic keratoses in immunosuppressed participants
Trichloroacetic acid peel
Trichloroacetic acid peel versus 5‐fluorouracil
This intervention was addressed by 1 study (Hantash 2006) comparing trichloroacetic acid peel with 5% 5‐fluorouracil applied twice daily for 3 weeks on the face. Assessment was performed 12 weeks after the end of treatment. There was possible performance and detection bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared | Participant complete clearance | Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 18 participants) |
| Hantash 2006 | ‐ | ‐ | ‐ | Percentages |
Analysis of mean percentage of reduction in lesion counts did not significantly favour any treatment, but there was a tendency to favour the chemical peel (MD 5.80, 95% CI ‐3.78 to 15.38; Analysis 60.1).
60.1.
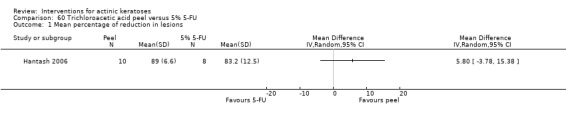
Comparison 60 Trichloroacetic acid peel versus 5% 5‐FU, Outcome 1 Mean percentage of reduction in lesions.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 19 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Hantash 2006 | x (none lost) | ‐ | ‐ | ‐ |
There were no participant withdrawals due to adverse events.
To summarise, additional data are needed to confirm the superiority of the trichloroacetic acid chemical peel over 5‐fluorouracil to treat actinic keratoses.
Trichloroacetic acid peel versus carbon dioxide laser resurfacing
This comparison was presented in the laser resurfacing section above.
(5) Combinations of topical and oral treatments with mechanical or chemical interventions
Cryotherapy combined with betulin‐based oleogel
This intervention was addressed by one study (Huyke 2009) comparing cryotherapy with liquid nitrogen to cryotherapy combined with betulin‐based oleogel on the face, scalp, or other locations. Cryotherapy of participant lesions was performed once on lesions on the face and twice on lesions on the rest of the body, whereas betulin‐based oleogel was applied twice daily for an unspecified duration. Assessment was performed at three months after the beginning of the treatment. There was possible performance and detection bias associated with this study.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 30 participants) |
Participant partial (> 75%) clearance (N = 30 participants) |
Mean (changes) reduction in lesion counts |
| Huyke 2009 | ‐ | x | x | ‐ |
Additional treatment with betulin‐based oleogel did not significantly change participant complete (Analysis 61.1) or partial (> 75%) clearance rates (Analysis 61.2) obtained with cryotherapy.
61.1.
Comparison 61 Cryotherapy versus cryotherapy with betulin‐based oleogel, Outcome 1 Participant complete clearance.
61.2.
Comparison 61 Cryotherapy versus cryotherapy with betulin‐based oleogel, Outcome 2 Participant partial (>75%) clearance.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 30 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Huyke 2009 | x (none lost) | ‐ | ‐ | ‐ |
There were no participant withdrawals due to adverse events.
To summarise, the use of betulin‐based oleogel after cryotherapy did not improve the efficacy of the cryotherapy.
Cryotherapy combined with 5‐fluorouracil
This intervention was addressed by 2 studies (Jorizzo 2004; Jorizzo 2006) comparing 0.5% 5‐fluorouracil or placebo applied daily to lesions on the face, scalp, ears, neck, and lips for 7 days combined with cryotherapy at week 4 for uncured lesions for 1 (Jorizzo 2004) to 3 (Jorizzo 2006) cycles. Assessment was performed at 4 weeks after the end of treatment. There was possible reporting (Jorizzo 2006) and other (Jorizzo 2006) bias.
Primary outcomes
| Study | Global Improvement indices for completely improved or cleared |
Participant complete clearance (N = 144 participants) |
Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 144 participants) |
| Jorizzo 2004 | ‐ | x | ‐ | Absolute values and percentages |
| Jorizzo 2006 | ‐ | x | ‐ | Absolute values and percentages |
Pretreatment with 0.5% 5‐fluorouracil before cryotherapy for 1 (RR 4.08, 95% CI 1.63 to 10.23, NNT = 4.6) or 2 (RR 3.27, 95% CI 1.82 to 5.88, NNT = 2.8), but not for 3 cycles, resulted in higher participant complete clearance (Analysis 62.1) compared to placebo combined with cryotherapy.
62.1.
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 1 Participant complete clearance at 6 months.
The absolute mean reduction in lesion counts and their associated standard deviations were calculated from the mean lesion counts at baseline and the end of the 3 different treatment cycles. The standard deviation associated with the mean percentage of reduction in lesion counts was only reported for the first treatment cycle in Jorizzo 2004. Thus, statistical analysis of this outcome could not be performed. This difference in efficacy between 0.5% 5‐fluorouracil with cryotherapy and vehicle with cryotherapy was supported by the mean percentage of reduction in lesion counts presented in the following table and the significant mean difference for the first cycle (MD 21.40, 95% CI 5.10 to 37.70; Analysis 62.3), but not by the analysis of mean reduction of lesion counts (Analysis 62.2).
62.3.
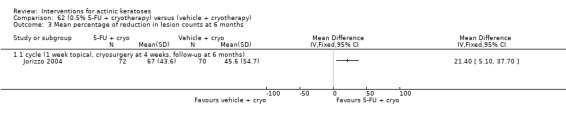
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 3 Mean percentage of reduction in lesion counts at 6 months.
62.2.
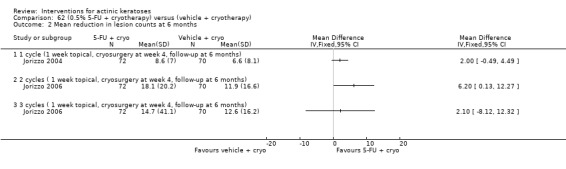
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 2 Mean reduction in lesion counts at 6 months.
| Mean percentage of reduction in lesion counts | |||
| Study | Number of cycles |
Vehicle + cryotherapy (mean + SD) |
5‐FU + cryotherapy (mean + SD) |
| Jorizzo 2004 | 1 | 45.6% + 54.7% | 67% + 43.6% |
| Jorizzo 2006 | 2 | 57.8% | 86.3% |
| Jorizzo 2006 | 3 | 65.7% | 77.8% |
The results presented in the additional Table 70 comparing vehicle with cryotherapy and 5‐FU with cryotherapy correspond to Analysis 63.1, Analysis 63.2, and Analysis 63.3.
63.1.

Comparison 63 (vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy), Outcome 1 Participant complete clearance at 6 months.
63.2.
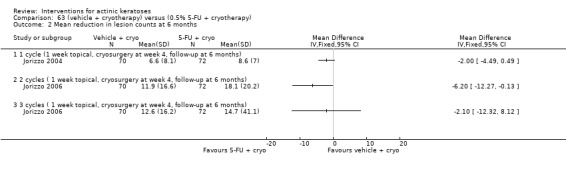
Comparison 63 (vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy), Outcome 2 Mean reduction in lesion counts at 6 months.
63.3.
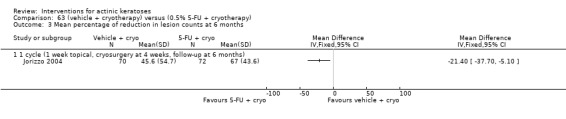
Comparison 63 (vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy), Outcome 3 Mean percentage of reduction in lesion counts at 6 months.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 144 participants) |
Skin irritation |
Minor adverse events excluding skin irritation (N = 144 participants) |
Cosmetic outcome |
| Jorizzo 2004 | x (none lost) | ‐ | x (eye irritation) | ‐ |
| Jorizzo 2006 | x | ‐ | x (including Jorizzo 2004) | ‐ |
In the first treatment cycle of the Jorizzo 2004 and Jorizzo 2006 study, there were no participant withdrawals due to adverse events. Insufficient information was provided to determine how many participants were lost due to adverse events for the whole study.
None of the adverse events reported were significantly different between cryotherapy alone and cryotherapy combined with 5‐fluorouracil. In general, eye irritation (Analysis 62.10) and conjunctivitis (Analysis 62.9) were the most commonly‐reported adverse reactions for both groups and the same numbers of participants in each group experiencing it.
62.10.
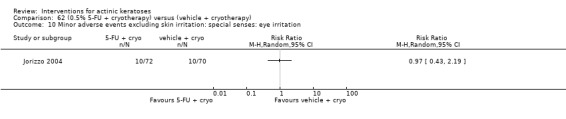
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 10 Minor adverse events excluding skin irritation: special senses: eye irritation.
62.9.
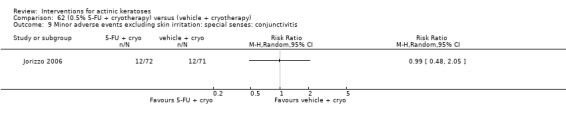
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 9 Minor adverse events excluding skin irritation: special senses: conjunctivitis.
To summarise, the efficacy of cryotherapy could be increased with pretreatment with 0.5% 5‐fluorouracil if used for 1 or 2, but not 3, cycles.
Cryotherapy combined with imiquimod
This intervention was addressed by three studies (Jorizzo 2010; NCT00774787; Tan 2007). The studies compared cryotherapy followed by vehicle and cryotherapy followed with imiquimod treatment. In Jorizzo 2010, 4 to 14 lesions were treated with cryotherapy, and 5 lesions were left untreated before randomisation. The method used to select which lesions were treated with cryotherapy was not specified. Thus, the data from this study comparing cryotherapy with imiquimod and imiquimod alone could not be used in our analyses. NCT00774787 had an intraindividual study design, whereas all the other studies had a parallel‐group design. The anatomical locations, dosing regimens, and assessment time are presented in the following table. There was possible performance (NCT00774787), attrition (Tan 2007), reporting (Jorizzo 2010; NCT00774787; Tan 2007), and other (Tan 2007) biases.
| Study | Anatomical locations | Cryotherapy (followed or not with placebo) | Cryotherapy followed by imiquimod | Imiquimod alone | Time of assessment |
| Tan 2007 | Face or scalp | Once | 5% imiquimod 2 times/week for 8 weeks |
No | 4 weeks after the end of treatment |
| Jorizzo 2010 | Face | Once | 3.75% imiquimod 3 times/week for 2 weeks on/2 weeks off/2 weeks on |
3.75 %imiquimod 3 times/week for 2 weeks on/ 2 weeks off/2 weeks on |
20 weeks after the end of treatment |
| NCT00774787 | Face or bald scalp | Once | 5% imiquimod 3 times/week for 4 weeks |
No | 4 to 8 weeks after the end of treatment |
Primary outcomes
| Study | Global improvement indices for completely improved or cleared |
Participant complete clearance (N = 339 participants) |
Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 274 participants) |
| Tan 2007 | ‐ | x | ‐ | ‐ |
| Jorizzo 2010 | ‐ | x | ‐ | Percentages |
| NCT00774787 | ‐ | Intraindividual study | ‐ | Percentages |
The primary outcomes were further divided into 3 outcomes: 1) for target lesions, i.e. cryotherapy‐treated lesions visible at baseline; 2) for subclinical lesions, i.e. lesions not visible at baseline but visible during the study; and 3) all lesions, i.e. target and subclinical lesions.
More participants had complete clearance on the cryotherapy combined with imiquimod side (8/27 = 30%) than the side that had cryotherapy alone (5/27 = 19%) in the intraindividual study. Cryotherapy combined with imiquimod had a tendency (but this was not statistically significant) to result in more participants with target lesions (cryotherapy‐treated: RR 0.62, 95% CI 0.36 to 1.04; Analysis 64.2) or subclinical lesions (RR 0.57, 95% CI 0.33 to 1.01; Analysis 64.3) completely cured compared to cryotherapy. By contrast, there was statistically significant complete clearance of all lesions in participants in Jorizzo 2010 (RR 0.20, 95% CI 0.05 to 0.73; Analysis 64.1); however, this could be due to the fact that the analysis in Jorizzo 2010 included the 5 lesions untreated with cryotherapy.
64.2.
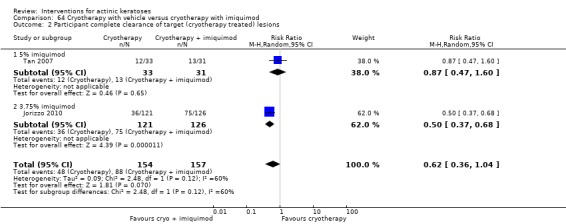
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 2 Participant complete clearance of target (cryotherapy treated) lesions.
64.3.
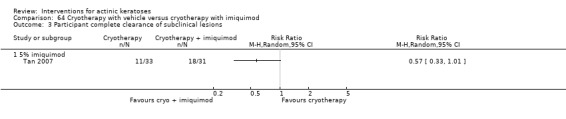
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 3 Participant complete clearance of subclinical lesions.
64.1.
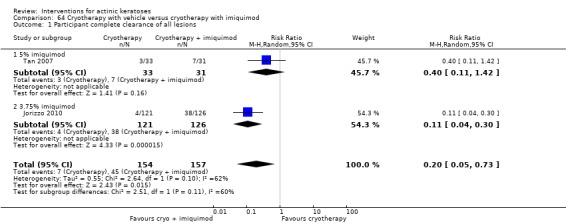
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 1 Participant complete clearance of all lesions.
The combined cryotherapy with 3.75% imiquimod therapy was also significantly favoured compared with the cryotherapy‐only treated side for mean percentages of reduction for all lesions (MD ‐34.10, 95% CI ‐41.38, to ‐26.82; Analysis 64.4) but not when 5.0% imiquimod was used (MD ‐11.20, 95% CI ‐26.53 to 4.13; Analysis 64.4). The results from the 2 studies could not be pooled due to the high heterogeneity between the 2 studies (I² statistic = 86%). It is worth noting that the study favouring the combined therapy had a parallel design, whereas the study not favouring the combined therapy was an intraindividual study. Only the study with 3.75% imiquimod reported the mean percentage of reduction for target lesions only (MD ‐10.80, 95% CI ‐17.37 to ‐4.23; Analysis 64.5), which favoured the combined therapy.
64.4.
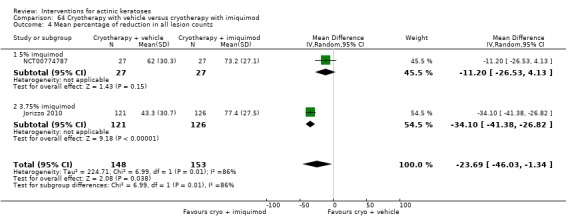
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 4 Mean percentage of reduction in all lesion counts.
64.5.
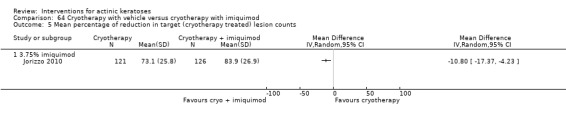
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 5 Mean percentage of reduction in target (cryotherapy treated) lesion counts.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 339 participants) |
Skin irritation (N = 312 participants) |
Minor adverse events excluding skin irritation (N = 312 participants) |
Cosmetic outcome (N = 274 participants) |
| Tan 2007 | x | x | x | ‐ |
| Jorizzo 2010 | x | x | x (only for 2 groups: cryotherapy with placebo and cryotherapy with imiquimod) | x (only for 2 groups: cryotherapy with placebo and cryotherapy with imiquimod) |
| NCT00774787 | x | ‐ | Pooled data not included | x |
In the intraindividual study, NCT00774787, there were no participant withdrawals due to adverse events. The pooled risk ratio for Tan 2007 and Jorizzo 2010 showed no difference between cryotherapy with vehicle and cryotherapy with imiquimod groups (RR 0.93, 95% CI 0.28 to 3.07; Analysis 64.6).
64.6.
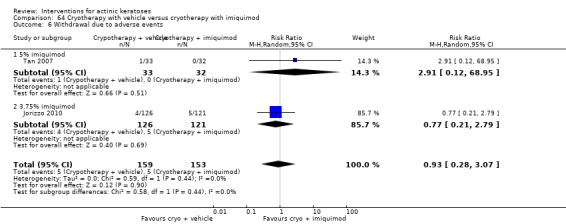
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 6 Withdrawal due to adverse events.
The number of participants experiencing skin irritation had a tendency to be higher in the group receiving additional treatment with imiquimod compared to cryotherapy alone (RR 0.39, 95% CI 0.10 to 1.54; Analysis 64.7).
64.7.
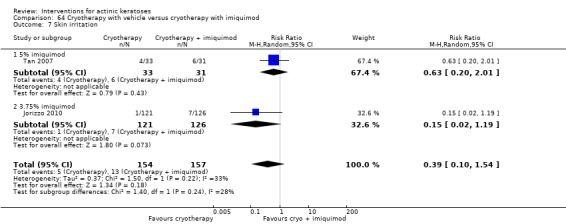
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 7 Skin irritation.
The number of participants experiencing fatigue (RR 0.09, 95% CI 0.01 to 1.69; Analysis 64.8), nausea (RR 0.09, 95% CI 0.01 to 1.69; Analysis 64.9), and myalgia (RR 0.21, 95% CI 0.02 to 1.76; Analysis 64.10) tended to be higher with additional imiquimod treatment, whereas the 3 respiratory adverse events, upper respiratory tract infection (RR 1.34, 95% CI 0.51 to 3.48; Analysis 64.11), bronchitis (RR 5.21, 95% CI 0.62 to 43.92; Analysis 64.12), and sinusitis (RR 11.45, 95% CI 0.64 to 204.88; Analysis 64.13) tended to be higher in the cryotherapy alone group. None of the minor adverse events were statistically significant. One case of skin infection due to cryotherapy had been reported by Tan 2007, but the treatment group (i.e. placebo or imiquimod) was not specified.
64.8.
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 8 Minor adverse events excluding skin irritation: body as a whole: fatigue.
64.9.
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 9 Minor adverse events excluding skin irritation: digestive: nausea.
64.10.
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 10 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
64.11.
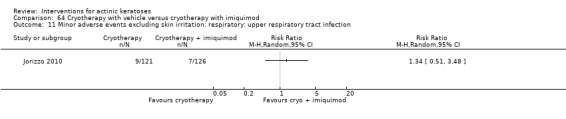
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 11 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection.
64.12.
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 12 Minor adverse events excluding skin irritation: respiratory: bronchitis.
64.13.
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 13 Minor adverse events excluding skin irritation: respiratory: sinusitis.
Additional imiquimod treatment with cryotherapy significantly improved the cosmetic outcome compared to cryotherapy alone in all individual cosmetic outcomes reported by Jorizzo 2010 (fine lines, tactile roughness, mottled pigmentation, and sallowness) as well as global photoageing score (RR 0.37, 95% CI 0.25 to 0.56, NNT = 3.1; Analysis 64.15). Cosmetic outcome assessments by participant and investigator in NCT00774787 showed similar results for the additional use of imiquimod with cryotherapy. In this study, analysis of participant assessment favoured the additional use of imiquimod, whereas analysis of investigator assessment did not favour its use. This could be explained by the fact that no placebo was used in this study reporting this cosmetic outcome, making the participants unblinded to the treatment and maybe biased towards the additional treatment with imiquimod; in contrast, the assessor was blinded.
64.15.
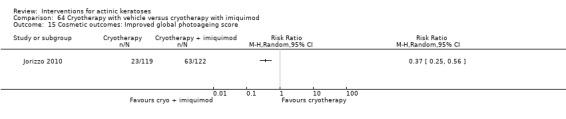
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 15 Cosmetic outcomes: Improved global photoageing score.
The results presented in the additional Table 71 for imiquimod comparisons correspond to Analysis 65.1, Analysis 65.2,Analysis 65.3, and Analysis 65.4.
65.1.
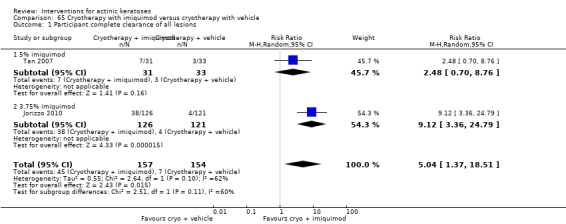
Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 1 Participant complete clearance of all lesions.
65.2.

Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 2 Mean percentage of reduction in all lesion counts.
65.3.
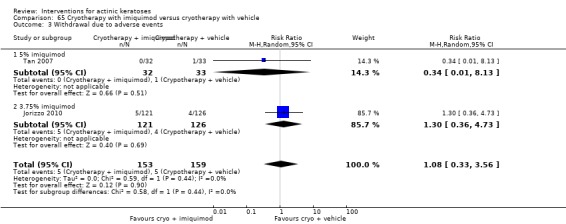
Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 3 Withdrawal due to adverse events.
65.4.
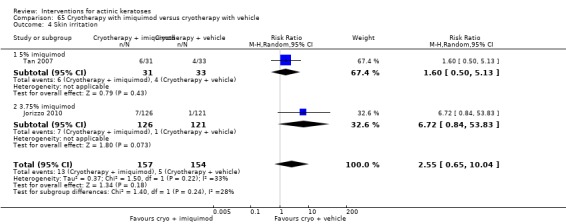
Comparison 65 Cryotherapy with imiquimod versus cryotherapy with vehicle, Outcome 4 Skin irritation.
To summarise, combination of cryotherapy and imiquimod treatments resulted in significantly better efficacy compared to the cryotherapy alone. Use of imiquimod cream after cryotherapy increased in general the number of participants experiencing adverse events, but resulted in significantly better cosmetic outcome.
ALA‐PDT combined with diclofenac in 2.5% hyaluronic acid gel pretreatment
This intervention was addressed by 1 study (Van der Geer 2009) investigating the efficacy of field‐directed treatment of ALA‐red light PDT on lesions on the dorsal side of the hands pretreated with diclofenac in 2.5% hyaluronic acid gel or 2.5% hyaluronic acid gel, twice daily for 4 weeks. Two weeks after diclofenac treatment, ALA was incubated for 4 hours then PDT with red light fractions at 80 J/cm² was performed for 16 minutes. Assessments were performed 6 weeks, and 6 and 12 months after the end of treatment. There was possible reporting bias associated with this study.
Primary outcomes
| Study |
Global Improvement indices for completely improved or cleared (N = 9 participants) |
Participant complete clearance | Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 9 participants) |
| Van der Geer 2009 | Mean scores | ‐ | ‐ | Absolute values |
The values provided for mean reduction in lesion counts at 6 weeks, 6 and 12 months (Analysis 66.2), and mean global improvement indices scores at 6 months (Analysis 66.1) were all lower in the vehicle group. The authors stated there was a significant difference in the mean number of lesions at 12 months (P = 0.017), but not in the mean reduction of lesion counts (P = 0.34) between the diclofenac and vehicle groups. However, it was impossible to test if these differences were statistically significant because measurement of variability, i.e. standard deviations or standard errors of the mean, were not provided.
66.2.
Comparison 66 (3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT) versus (2.5% hyaluronic acid + ALA‐red light PDT), Outcome 2 Mean reduction of lesion counts.
| Mean reduction of lesion counts | ||||
|---|---|---|---|---|
| Study | Intervention | At 6 weeks | At 6 months | At 12 months |
| Van der Geer 2009 | Diclofenac in 2.5% hyaluronic acid + ALA‐PDT | 10.13 | 11.56 | 12.5 |
| Van der Geer 2009 | 2.5% hyaluronic acid + ALA‐PDT | 9.9 | 10.56 | 8.8 |
66.1.
Comparison 66 (3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT) versus (2.5% hyaluronic acid + ALA‐red light PDT), Outcome 1 Global Improvement Indices (‐2 to 4) at 6 months.
| Global Improvement Indices (‐2 to 4) at 6 months | |||
|---|---|---|---|
| Study | Intervention | Patient | Investigator |
| Van der Geer 2009 | Diclofenac in 2.5% hyaluronic acid + ALA‐PDT | 3.3 | 3.4 |
| Van der Geer 2009 | 2.5% hyaluronic acid + ALA‐PDT | 2.4 | 2.7 |
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 9 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Van der Geer 2009 | x (none lost) | ‐ | ‐ | ‐ |
There were no participant withdrawals due to adverse events.
To summarise, pretreatment with diclofenac in 2.5% hyaluronic acid gel did not increase the efficacy of ALA‐PDT treatment of actinic keratoses.
ALA‐PDT combined with imiquimod
This intervention was addressed by 1 intraindividual study (Shaffelburg 2009) investigating 2 ALA‐blue light PDT treatments with an interval of 4 weeks directed to a field of lesions.This was followed after another 4 weeks by 5% imiquimod or placebo applied once per day on the face (field‐directed treatment), on 2 days per week for 16 weeks [ALA‐blue light PDT followed with imiquimod versus ALA‐blue PDT followed with placebo]. Assessments were performed at baseline and months 1, 2, 3, 4, 6, and 12 of the study. There was possible reporting bias.
Primary outcomes
| Study | Global improvement indices for completely improved or cleared |
Participant complete clearance (N = 25 participants) |
Participant partial (> 75%) clearance |
Mean (changes) reduction in lesion counts (N = 25 participants) |
| Shaffelburg 2009 | ‐ | x | ‐ | Absolute values and percentages |
The participant complete clearance was similar with (2/25) or without (2/25) additional imiquimod treatment after ALA‐PDT. The mean reduction in lesion counts was 19.9 (86.7%) for the imiquimod‐treated group and 16.0 (73.1%) for the placebo group. However, in the absence of standard deviations or standard errors of the mean values, no statistical analysis was possible to determine the significance of these data.
Secondary outcomes
| Study |
Withdrawal due to adverse events (N = 25 participants) |
Skin irritation | Minor adverse events excluding skin irritation | Cosmetic outcome |
| Shaffelburg 2009 | X (none lost) | ‐ | ‐ | ‐ |
There were no participant withdrawals due to adverse events.
To summarise, an additional treatment with imiquimod after ALA‐PDT did not improve the efficacy of the treatment for actinic keratosis.
Discussion
Summary of main results
Primary outcomes
Actinic keratoses remain unchanged, proliferate, regress, reappear, or develop into squamous cell carcinoma. Thus, comparison to a placebo control group gives a better estimate of the efficacy of an intervention for actinic keratoses. Significant estimate effects compared to vehicle or placebo were obtained for the following interventions.
1) For all reported efficacy outcomes: diclofenac (3/3 outcomes, number of studies (n) = 6, number of participants (N) = 723), 5‐fluorouracil (3/3 outcomes, n = 3, N = 528), ingenol mebutate (2/2 outcomes, n = 3, N = 540), sunscreen (1/1 outcome, n = 1, N = 588), ALA‐PDT (2/2 outcomes, n = 4, N = 814), and MAL‐PDT (2/2 outcomes, n = 5, N = 486).
2) For 50% or more of the reported efficacy outcomes: adapalene (1/2 outcomes, n = 1, N = 90), imiquimod (3/5 outcomes, n = 17, N = 3417), isotretinoin (1/2 outcomes, n = 1, N = 100), and masoprocol (1/2 outcomes, n = 1, N = 176).
3) For none of the reported efficacy outcomes: calcipotriol (vitamin D, 0/2 outcomes, n = 1, N = 9), DFMO (0/1 outcome, n = 1, N = 48), β‐1,3‐D‐glucan (0/1 outcome, n = 1, N = 20), nicotinamide (0/1 outcome, n = 1, N = 30), DL‐α‐tocopherol (vitamin E, 0/1 outcome, n = 1, N = 48), and etretinate (0/1 outcome, n = 1, N = 50).
Studies that compare the different concentrations of an intervention were included in our analysis. These studies were conducted for adapalene, colchicine, 5‐fluorouracil, imiquimod, ingenol mebutate, and MAL. Adalpene was the only intervention to demonstrate a difference in efficacy as a result of different concentrations.
The photosensitiser incubation time and light source were variables that were considered by several studies conducted on photodynamic therapy. One study comparing 0.5‐, 1‐, 2‐, and 4‐hour incubations with ALA showed that longer incubation before the photodynamic therapy resulted in better efficacy than a shorter incubation for 4 of the 6 possible comparisons between the different incubation times (see overview for photodynamic therapy in Table 69). In contrast, a similar efficacy was found for two‐ and three‐hour incubation with MAL before photodynamic therapy using daylight. No difference in efficacy was detected between the different light sources for photodynamic therapy (ALA: blue vs red light, blue light vs pulsed dye laser, and MAL: red light LED vs daylight, red light vs broadband visible plus water‐filtered infrared A).
We analysed interventions that investigated the efficacy of combined interventions, which generally combined field‐directed therapy with treatment for individual lesions. Pretreatment with 0.5% 5‐fluorouracil before cryotherapy and imiquimod after cryotherapy significantly improved the efficacy of cryotherapy (see overview for cryotherapy in Table 70). In contrast, no improvement in efficacy was detected when the following interventions were combined: additional tretinoin treatment to 5‐fluorouracil, additional betulin‐based oleogel to cryotherapy, pretreatment with diclofenac before ALA‐PDT, and imiquimod treatment after MAL‐PDT.
Several studies compared the efficacy of two different interventions. These interventions may be field‐directed treatments applied to a large area of clinical and subclinical lesions (topical creams, resurfacing, field‐directed photodynamic therapy, and chemical peel), or treatments that specifically target clinical lesions, (cryotherapy and individual lesion‐directed photodynamic therapy). Topical aretinoid methyl sulfone (Ro 14‐9706) was significantly more efficacious than topical tretinoin for only 1 of 2 outcomes reported by one study. Topical 5‐fluorouracil was more efficacious than topical masoprocol (2/3 outcomes, n = 1, N = 49) and cryotherapy (1/1 outcome, n = 1, N = 49), but had similar efficacy to topical imiquimod (2/2, n = 2, N = 89), carbon dioxide laser resurfacing (1/1 outcome, n = 1, N = 14), Er:YAG laser resurfacing (2/2 outcomes, n = 1, N = 55), ALA‐PDT for individual lesions (2/2 outcomes, n = 1, N = 36), and trichloroacetic acid peel (1/1 outcome, n = 1, N = 18) based on the data provided. However, more data are needed to be able to conclude on the difference in efficacy between 5‐fluorouracil and MAL‐PDT. Topical imiquimod efficacy was also similar to topical diclofenac (1/1 outcome, n = 1, N = 49) and cryotherapy (1/1 outcome, n = 1, N = 51) efficacies, but more data are needed to be able to compare imiquimod to ALA‐PDT for individual lesions. Betulin‐based oleogel and cryotherapy had similar efficacy (2/2 outcomes, n = 1, N = 28). Cryotherapy showed lower efficacy compared to ALA‐PDT for individual lesions (1/1 outcome, n = 1, N = 72), but more data are needed for the comparison with MAL‐PDT for individual lesions. However, field‐directed treatments with ALA‐PDT and MAL‐PDT had similar efficacy (2/2 outcomes, n = 1, N = 15). Based on these comparisons, these interventions could be ranked based on their relative efficacy as follows: (5‐fluorouracil = imiquimod = carbon dioxide laser resurfacing = Er:YAG laser resurfacing = ALA‐PDT = MAL‐PDT = trichloroacetic acid peel = diclofenac ) > masoprocol (cryotherapy = betulin‐based oleogel). The relative efficacy between masoprocol and cryotherapy was not investigated in any of the studies included. In summary, the comparisons of different interventions showed that these interventions were generally comparable.
In our review, carbon dioxide laser resurfacing has been shown to have a similar efficacy to 5‐fluorouracil and trichloroacetic acid peel to treat actinic keratosis. However, the efficiency of carbon dioxide laser resurfacing to prevent short‐term (within 12 months) recurrence of actinic keratoses has been questioned (Fulton 1999). Because recurrence, prophylaxis of actinic keratoses, or both, were not in the prespecified outcomes of our review, we will not further discuss this matter, but there might be a need for a future review on the subject.
The relative efficacy of the interventions on various anatomical locations was poorly reported. The majority of studies that investigated different regions of the skin grouped the locations together for each outcome. The only significant difference was reported between lesions on the face and upper extremities during isotretinoin treatment. There was also a decreased tendency to favour imiquimod for the lesions on the face. In summary, there was insufficient data to determine the difference in the lesions' location in this meta‐analysis.
For three interventions, the efficacy relative to vehicle/placebo was investigated in immunosuppressed participants. Data from only one study with a small sample was usually included for immunosuppressed participants in the analyses, whereas data from several studies was generally pooled for immunocompetent participants. Thus, it is difficult to compare directly the calculated risk ratios and their 95% CI between studies including immunocompetent versus immunosuppressed participants. A comparison of the unweighted 'participant complete clearance' rates suggests that a similar efficacy is achieved for the two populations. In immunocompetent participants, diclofenac resulted in a 32% (67/208) complete clearance, whereas vehicle had a 13% (27/212) clearance. In immunosuppressed participants the same rates were 41% (9/66) for diclofenac and 0% (0/6) for vehicle. Imiquimod (5%) resulted in 42% (694/1649) vs 62% (18/29) complete clearance, whereas vehicle resulted in 5% (62/1231) and 0% (0/14) for the immunocompetent and immunosuppressed groups, respectively. In PDT, 76% (13/17) of immunosuppressed participants and 74% (204/278) of immunocompetent participants were completely cleared with MAL‐PDT compared to 14% (30/204) and 0% (0/17) for placebo‐PDT. In summary, the treatments were equally efficacious in immunosuppressed and immunocompetent participants. One ongoing study (NCT01525329) is comparing treatment with MAL‐PDT alone and in combination with 5% 5‐fluorouracil in both immunocompetent and immunosuppressed participants.
Secondary outcomes
In general, the number of participants withdrawn because of adverse events was not significantly different between interventions. The only exceptions were the following:
3% diclofenac in 2.5% hyaluronic acid compared to 2.5% hyaluronic acid (see overview for diclofenac in Table 67),
5% imiquimod compared to placebo (see overview for imiquimod Table 71), and
0.06% to 0.1% resiquimod compared to 0.01% to 0.03% resiquimod.
The studies reporting skin irritation indicate that diclofenac (see overview for diclofenac in Table 67), 5‐fluorouracil (see overview for 5‐fluorouracil in Table 68), tretinoin, isotretinoin, and ALA (see overview for photodynamic therapy in Table 69) treatments are associated with significant skin irritation. Topical treatments were associated with different adverse effects than photodynamic therapy and cryotherapy. Topical treatments were associated with "flu" or "cold" symptoms, headache, and conjunctivitis or eye irritation. Photodynamic therapy and cryotherapy were associated with photosensitivity reaction and cold exposure injury, respectively. Most of the minor adverse events that were quantitatively reported were not significantly different between the two interventions that were compared. The only exceptions were the dermatitis associated with adapalene, the metabolic and nutritional disorder and dry skin associated with diclofenac, the "flu" or "cold" symptoms and headache with daily application of imiquimod, and the headache associated with concentrations of resiquimod superior to 0.01%.
Finally, the included studies reported varied cosmetic outcomes. In general, it could be concluded that imiquimod treatment and photodynamic therapy resulted in better cosmetic outcomes than cryotherapy and 5‐fluorouracil.
Overall completeness and applicability of evidence
The physician's decision about which treatment to prescribe will depend on each patient's case and their treatment aims. Different interventions might be more effective for cosmetic outcomes, symptom relief, or prevention of squamous cell carcinoma. In addition, the efficacy, cost, adverse events, length of treatment, ease of treatment, personal preference of the participant, participant compliance, severity of actinic damage, past treatment experiences, and other factors must all be taken into consideration, and the most appropriate treatment will vary from person to person. The completeness of this review will be discussed based on these factors influencing the choice of an intervention for treating actinic keratoses.
Several of these factors (cost, ease of treatment, participant preference, participant compliance, past treatment experiences) were not included in this review. Some of these factors were presented as outcomes in a few studies, and these are summarised in a table in the Included studies section under 'other outcomes'.
This review included several efficacy outcomes. However, the exclusion of an important efficacy outcome, such as 'lesion complete response', limited the evaluation of the relative efficacy of several interventions because some studies only reported this efficacy outcome (see Effects of interventions).
Adverse event reporting was complicated by the use of the generic "skin irritation" outcome. Many studies chose to use categories such as "application site reactions" and "local skin/adverse reactions" instead of "skin irritation".
The interventions could also be compared based on their length of treatment. A wide variety of treatment options are available for actinic keratosis. Treatments such as cryotherapy or photodynamic therapy are performed once or twice by clinical staff, whereas the duration of topical treatments administered by patients varies from two applications within a few days for ingenol mebutate to daily application for seven months with sunscreen. A decision based on patient compliance and preference could easily be made. The effect of changing the length of a specific treatment was reported in this review where the information was available, but we did not compare different interventions based on their length of treatment. We did not compare continuous therapy with interval/pulse/cycle therapy. The readers are referred to a recent review (Martin 2011), which discusses the efficacy of short‐course and interval/pulse/cycle therapies for 5‐fluorouracil, imiquimod, and ALA/MAL‐PDT. Outcomes reported over time showed differences between the assessments, but variations in the time of the assessment (follow‐up period) were generally not taken into consideration in our evaluation of the relative efficacy between interventions. Long‐term (> one year) outcomes were not included in our meta‐analysis, but references to these studies were included in the 'Characteristics of included studies' tables.
The severity of the lesions at baseline was not accounted for in our analysis. The studies reported the severity of lesions in different terms; some used the lesion grade, whereas others reported the number of lesions. In some studies, the number of lesions was a criterion for inclusion or exclusion of participants. This information can be found for the individual studies in the 'Characteristics of included studies' tables in the 'participants' sections.
Many patients wish to have their actinic keratoses treated in order to prevent their development into squamous cell carcinoma. The detection of squamous cell carcinoma, basal cell carcinoma, or Bowen's disease was not included in our outcomes, and little is known about their prevention by treating actinic keratoses. This outcome should be addressed in the future. One ongoing study (NCT01453179) will evaluate this issue for 5% imiquimod and 3% diclofenac in hyaluronic acid. However, it is worth noting that the studies included in our analysis did not specify if cancerous lesions were present in the treatment area. This makes the interpretation of this data difficult.
Quality of the evidence
The quality of the evidence presented in this review was evaluated in the 'Risk of bias' graph (Figure 2) and the 'Risk of bias' tables associated with each included study. The major factors decreasing the quality of evidence from the studies on interventions for actinic keratoses are as follows:
lack of reporting the methods used for allocation sequence generation and allocation concealment;
blinding of studies comparing physically‐distinct interventions;
the use of per‐protocol (PP) analysis instead of intention‐to‐treat (ITT) analysis (but PP data were converted as much as possible to ITT for meta‐analyses); and
incomplete reporting of an outcome.
We also noticed another issue with reporting the diagnosis criteria for inclusion of the participants (see 'Characteristics of included studies' tables). Many studies did not specify whether a clinical or histological diagnosis was used to include the participants in the clinical trials.
There were two major limitations in our assessment of the effects of interventions for actinic keratoses. The first was that data from numerous intraindividual studies could not be included in the meta‐analyses. The second was the frequent omission of standard deviations in the reporting of the outcome "mean reduction in lesion counts" for both absolute values and percentages, which prevented the statistical analysis of the data. Without the standard deviations, it is difficult to determine if there is a difference between the intervention and the control intervention.
The studies included in the different meta‐analyses were generally very consistent, and only a few examples of high heterogeneity were observed. Many of the analyses only included data from an individual study; this included 76% of the efficacy outcomes, 91% of the safety outcomes, and 87% of the cosmetic outcomes.
The frequency of high‐quality studies varied based on treatment. Our inclusion criteria fit more studies on imiquimod than 5‐fluorouracil or cryotherapy. In contrast, 5‐fluorouracil and cryotherapy have been compared to more interventions than imiquimod. The nature of cryotherapy does not allow for double‐blinded prospective trials, with the exception of studies investigating the combined therapy with a topical treatment, which resulted in lower quality evidence (see overview for cryotherapy in Table 70).
There were several biases that affected the quality of the evidence. Certain treatments incurred adverse skin reactions (e.g. imiquimod, 5‐fluorouracil, ALA, etc) that may indirectly introduce bias into the clinical assessment. Moreover, when comparing self‐administered and clinically‐administered interventions, such as 5‐fluorouracil and photodynamic therapy, the compliance of the participants could have an influence on the outcomes and could introduce selection bias if participants were included or excluded based on their compliance. Reporting bias could also result from the method used to assess efficacy outcomes. Several studies reported the observation of new actinic keratoses during the clinical trial, and most of the studies did not specify whether they were studying all lesions or a specific subpopulation of clinical, subclinical, or new lesions.
Potential biases in the review process
This review included a broad variety of interventions for actinic keratoses and a large number of outcomes. The search for corresponding studies resulted in a large number of studies, which produced a considerable amount of information. Data extraction sheets were really useful tools for the organisation of this information. However, important details for comparison between studies could have been missed or overlooked in this process. The amount of information presented meant that we were unable to evaluate all of the factors influencing the outcomes, including the methods used for assessment. We searched multiple databases as well as websites and grey literature for randomised clinical trials on all interventions (not prophylaxis) for actinic keratoses in any language. We also contacted pharmaceutical companies to request additional information, but this correspondence was not always successful. There is a possibility that studies may be missing. For example, a study on comparison between 5% 5‐fluorouracil and placebo for solar keratoses was registered in the metaRegister of Controlled Trials (mRCT) in 2001 to 2002, but no publication of this study was found.
Our analysis only included randomised controlled trials due to the large scope of this review; all other trial designs were excluded. The randomised clinical trials were only included if all the interventions were covered by this review and if they reported numerical results for at least one of the review outcomes. This criterion excluded the outcome 'Withdrawal due to adverse events', which is generally reported in all the studies. Because the terminology used for the different outcomes was not always consistent in the studies (see the Included studies section under 'Outcomes'), the interpretation of the outcomes' definitions could have introduced some bias in the review process.
Inconsistency in terminology could also have led to misinterpretation during the data extraction process. Some of the review outcomes have been redefined to try to avoid this problem. However, a more precise definition of some outcomes was not always possible. There does not seem to be a common definition of "skin irritation", and many studies included adverse event outcomes that included irritation, symptoms of irritation, or both. The lack of consensus on which symptoms constitute "skin irritation" and the potential for different interpretations by the authors of the studies has resulted in our decision to only include outcomes with the word "irritation" in the label. This adverse event category may also include a pooling of different outcomes, as the skin irritation was not identified by the site (application site vs local irritation).
We separated the studies into two populations of participants: immunocompetent and immunosuppressed. We considered participants to be immunocompetent by default if the studies did not specifically include immunosuppressed participants. However, in 40% of these studies, immunosuppressed participants could have been enrolled based on the inclusion and exclusion criteria reported. It is possible that the immunocompetent population included few immunosuppressed participants.
Data from intention‐to‐treat analysis was used in the analysis for the review whenever possible to reduce the attrition bias and increase consistency between studies included in the same meta‐analysis. The information provided in the studies did not always allow for the conversion of data from per‐protocol analysis to intention‐to‐treat analysis. In some studies, the authors did not observe a significant difference in the outcomes when analysis was conducted with per‐protocol analysis, as opposed to intention‐to‐treat analysis. Thus, the estimated effect size calculated in the meta‐analyses should not be affected and our conclusions should remain unchanged. Several studies presented data in a graphical format that was not included in our analysis, either because of unsuccessful correspondence with the authors, time limitations, or both. Statistical limitations due to outcome without event, such as exclusion from meta‐analysis or calculation for number needed to treat (NNT) based on assumed control risk (ACR), also restricted our analyses and conclusions.
Agreements and disagreements with other studies or reviews
In a systematic review of randomised controlled trials using 0.5% and 5% 5‐fluorouracil for treatment of actinic keratoses, 9 studies were identified (Kaur 2010). We included seven of these studies in our review and excluded two because there was no clear mention of randomisation of the treatments in the published reports. Despite this difference, the authors arrived at a similar conclusion to our review about the participant complete clearance of the 2 concentrations of 5‐fluorouracil. In another systematic review (Askew 2009), the authors estimated that about 500 of 1000 of participants receiving 5‐fluorouracil for treatment of actinic keratoses could expect complete clearance. An average of our illustrative comparative risks based on study population for 0.5% and 5% 5‐fluorouracil treatments (see overview for 5‐fluorouracil in Table 68) resulted in 577 of 1000 participants with complete clearance. A similar conclusion was reached by using different methodologies.
A meta‐analysis of 5 full journal publications of randomised double‐blind clinical trials comparing 5% imiquimod to vehicle concluded that about 50% of participants (500 of 1000) treated with 5% imiquimod achieved complete clearance (Hadley 2006). In this review, the method used to pool data was not specified (but the numbers given suggested that the numbers were added together without any weighting) and a fixed‐effect model was used to calculate the relative risk. Our review calculated a lower percentage of 25% to 38% (253 to 382 of 1000; see overview for imiquimod in Table 71) based on 9 studies using a random‐effects model to calculate the pooled risk ratio (RR). In contrast to our review, Hadley 2006 did not find any difference in the number of withdrawals due to adverse events between 5% imiquimod and vehicle, but a difference was detected for withdrawal in general, with significantly more in the imiquimod group. Hadley et al detected a greater proportion of local, treatment‐related, and overall adverse effects in the 5% imiquimod group, but did not observe any differences in serious adverse events. Thus, 5% imiquimod treatment is generally associated with more adverse events than vehicle.
In a systematic review on photodynamic therapy in the treatment of pre‐cancerous skin conditions and cancers (Fayter 2010), the authors reached the following conclusions for the treatment of actinic keratosis: 1) "The only clear evidence of effectiveness" came from the comparison of ALA/MAL‐PDT and placebo‐PDT, and 2) "Uncertainties still exist around PDT’s effectiveness compared with other topical treatments." Our analyses led to similar conclusions (see the overview for photodynamic therapy in Table 69).
In our review, the efficacy of ALA‐PDT was superior to cryotherapy based on 'participant complete clearance', but the efficacy of MAL‐PDT compared to cryotherapy could not be assessed. A meta‐analysis of 'lesion complete response' was performed in the Fayter 2010 review to compare MAL‐PDT to cryotherapy. Unfortunately, this analysis included 4 studies with high heterogeneity (I² statistic = 88%), and no definitive conclusion could be made on their relative efficacy. The authors of this review also concluded that the improved cosmetic outcomes obtained for PDT compared to cryotherapy might be due to bias, because in most of the studies the assessors were not blinded.
Authors' conclusions
Implications for practice.
The treatment of actinic keratoses is generally recommended to limit the morbidity and mortality of squamous cell carcinoma. Surprisingly, there was no evidence in the included studies that treating actinic keratoses prevented squamous cell carcinoma. Only a few studies reported the observation of squamous cell carcinoma, basal cell carcinoma, or both. In these studies, it was not specified if the cell carcinoma was observed in the treated area. Thus, it was impossible to correlate treatment of actinic keratoses with prevention of cell carcinoma. Of course, this lack of information on prevention of squamous cell carcinoma could have been a consequence of our criteria, which included interventions to treat actinic keratoses but not prophylaxis of cancers. As mentioned previously, this review did not cover long‐term follow‐up studies that could give useful information on recurrence of actinic keratoses as well as prevention of squamous cell carcinoma. We did include the recurrence rates, appearance of new actinic keratoses or incidence of cancer if they were provided in the tables of 'Characteristics of included studies'. Because of the importance of this issue, a systematic review with these long‐term outcomes must be performed, and we suggest that randomised clinical trials on interventions for actinic keratoses include observation of squamous cell carcinoma for a follow‐up period of at least one year as an efficacy outcome.
Based on the evidence presented in this review, there are many effective options available for the treatment of actinic keratoses. The most effective treatment options were diclofenac, 5‐fluorouracil, imiquimod, ingenol mebutate, laser resurfacing, trichloracetic acid peel, ALA‐PDT, and MAL‐PDT. Other treatment options should not be ruled out as they are still effective, and many have reduced side‐effects, which may be preferable or better suited to certain patients.
Ultimately, the decision about which treatment option to use should be agreed upon by both the physician and the patient, based on which intervention suits the participant's specific situation. Certain treatments are better for treating diffuse actinic damage, while others are better for individual lesions. Moreover, the appropriate treatments would depend on the patient's wishes, whether it is cosmetic, symptom relief, or prevention of squamous cell carcinoma. If the risk associated with treatment is greater than the potential benefit, observation without treatment may also be an option.
Implications for research.
Our review did not directly compare the methodology used by the studies to evaluate the efficacy outcomes of the interventions for actinic keratoses. Some studies did not give any details on their methodology, whereas others described in detail how individual lesions were mapped, photographed, and followed throughout the study. Mapping of the lesions allowed the investigators to make a distinction between baseline lesions and new or subclinical lesions. For several studies, it was not clear if the efficacy assessment included only target (baseline) lesions or all lesions, which could greatly influence the final outcome. Thus, we recommend that the authors of studies describe in details the methodology used to evaluate the efficacy of the interventions investigated and specify which lesions (baseline/target, subclinical/new, or all lesions) are included in these evaluations.
A clear definition of the lesions being treated is particularly important when comparing individual lesion‐based and field‐directed treatments, as well as to show that new lesions appeared in response to some treatments. An increase in the number of lesions during treatment was observed for imiquimod (see the 'Notes' section of the 'Characteristics of included studies' tables for Chen 2003;Korman 2005;Lebwohl 2004; and Tan 2007), 5‐fluorouracil (Jorizzo 2006; Tanghetti 2007), and tretinoin (Misiewicz 1991). This unmasking of lesions during treatment might have important implications for treatment of actinic keratoses and its associated recurrence. Long‐term randomised clinical trials comparing lesion‐based and field‐directed treatments are needed to address this issue.
Diclofenac in 2.5% hyaluronic acid has been compared directly to 5% 5‐fluorouracil (1 excluded study: Smith 2006) and 5% imiquimod for the treatment for actinic keratosis. Diclofenac and 5% imiquimod are both associated with significant adverse events based on the related withdrawals, and 5‐fluorouracil treatment is associated with significant skin irritation based on our analyses. It would be advantageous to perform randomised clinical trials comparing diclofenac with other interventions in order to clearly assess its safety outcomes. Similarly, the new treatment ingenol mebutate (PEP005) has only been compared to placebo, and comparison with other interventions for actinic keratosis is needed to evaluate its efficacy and safety compared to established therapy. As mentioned in the summary of main results, additional data are also needed to support or confirm the conclusion of some included studies.
Photodynamic therapy is a newer form of treatment that presents good results in clinical trials. Several studies tried to determine the optimal treatment regimen, output, and photosensitising agents, but most studies did not observe significant changes in efficacy in the variations studied. A few studies investigated the use of daylight for photodynamic therapy using the photosensitiser MAL, and one study showed an efficacy equivalent to MAL‐red light PDT. This source of light could be more convenient, more cost effective and easily applicable as field‐directed treatment. This is a good prospective area for further research. One ongoing study (NCT01475071) is comparing daylight PDT with conventional PDT.
What's new
| Date | Event | Description |
|---|---|---|
| 23 October 2019 | Amended | Edited the published note about the updating of the review. |
History
Protocol first published: Issue 4, 2003 Review first published: Issue 12, 2012
| Date | Event | Description |
|---|---|---|
| 8 June 2016 | Amended | This review is going to be updated. We have written a published note to say that because the scope of the review has been reduced to make it more manageable a new protocol and then a new review will be written. |
| 25 January 2011 | Amended | Change in authors |
| 11 June 2008 | Amended | Converted to new review format. |
| 9 January 2007 | New citation required and conclusions have changed | Substantive amendment |
Notes
This review is being updated by way of a new protocol and then a review, because the scope of the review has been reduced to make it more manageable. The citation for the new protocol is as follows: Foley K, Gupta AK, Martin G, Tweed JA, Villanueva E, Carviel J. Topical treatments and photodynamic therapy for actinic keratosis of the face and scalp. Cochrane Database of Systematic Reviews 2019, Issue 10. Art. No.: CD013452. DOI: 10.1002/14651858.CD013452.
Acknowledgements
The Cochrane Skin Group editorial base wishes to thank Dedee Murrell who was the Key Editor for this review; Jo Leonardi‐Bee and Philippa Middleton who were the Statistical and Methods Editors, respectively; the clinical referees, Hywel Williams and Stephen Keohane; and the consumer referee, Jack Tweed.
The following people contributed to earlier versions of this review; the authors would like to acknowledge their contribution: Kimberley Inniss, Robert Wainwright, Melody Chow, Elizabeth Cooper, Kristy Johnson, and Zbys Fedorowicz. The authors would also like to thank Chris Drummond‐Main and Fiona C. Simpson for their input.
Appendices
Appendix 1. CENTRAL (Cochrane Library) search strategy
#1 (actinic and keratos*) or (solar and keratos*) or (senile and keratos) or (hyperkeratos*) #2 MeSH descriptor Keratosis, Actinic explode all trees #3 (#1 OR #2)
Appendix 2. MEDLINE (OVID) search strategy
1. randomized controlled trial.pt. 2. controlled clinical trial.pt. 3. randomized.ab. 4. placebo.ab. 5. clinical trials as topic.sh. 6. randomly.ab. 7. trial.ti. 8. 1 or 2 or 3 or 4 or 5 or 6 or 7 9. (animals not (human and animals)).sh. 10. 8 not 9 11. actinic keratos$.mp. or exp Keratosis, Actinic/ 12. solar keratos$.mp. 13. senile keratos$.mp. 14. hyperkeratos$.mp. 15. 11 or 12 or 13 or 14 16. 10 and 15
Appendix 3. EMBASE (OVID) search strategy
1. random$.mp. 2. factorial$.mp. 3. (crossover$ or cross‐over$).mp. 4. placebo$.mp. or PLACEBO/ 5. (doubl$ adj blind$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name] 6. (singl$ adj blind$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name] 7. (assign$ or allocat$).mp. 8. volunteer$.mp. or VOLUNTEER/ 9. Crossover Procedure/ 10. Double Blind Procedure/ 11. Randomized Controlled Trial/ 12. Single Blind Procedure/ 13. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 14. actinic keratos$.mp. or exp Keratosis, Actinic/ 15. solar keratos$.mp. 16. senile keratos$.mp. 17. hyperkeratos$.mp. 18. 14 or 15 or 16 or 17 19. 13 and 18
Appendix 4. LILACS search strategy
((Pt RANDOMIZED CONTROLLED TRIAL OR Pt CONTROLLED CLINICAL TRIAL OR Mh RANDOMIZED CONTROLLED TRIALS OR Mh RANDOM ALLOCATION OR Mh DOUBLE‐BLIND METHOD OR Mh SINGLE‐BLIND METHOD OR Pt MULTICENTER STUDY) OR ((tw ensaio or tw ensayo or tw trial) and (tw azar or tw acaso or tw placebo or tw control$ or tw aleat$ or tw random$ or (tw duplo and tw cego) or (tw doble and tw ciego) or (tw double and tw blind)) and tw clinic$)) AND NOT ((CT ANIMALS OR MH ANIMALS OR CT RABBITS OR CT MICE OR MH RATS OR MH PRIMATES OR MH DOGS OR MH RABBITS OR MH SWINE) AND NOT (CT HUMAN AND CT ANIMALS)) [Words] and ((actinic$ or solar or senil$) and (keratos$ or queratosis)) or hyperkeratos$ or hiperqueratos$ [Words]
Data and analyses
1.
Adapalene gel versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global Improvement Indices (investigator)‐cleared | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean changes in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 0.1% adapalene gel | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 0.3% adapalene gel | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: dermatitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
2.
0.1% adapalene vs 0.3% adapalene
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global Improvement Indices (investigator)‐cleared | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean changes in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: dermatitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
3.
Arotinoid Methyl Sulfone (Ro 14‐9706) versus Tretinoin
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean percentage of reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
4.
Calcipotriol (vitamin D) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean changes in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Cosmetic outcomes: Reduction in total cosmetic appearance score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
5.
1% colchicine cream versus 0.5% colchicine cream
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean reduction in lesion counts‐total | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Mean reduction in lesion counts‐per anatomical locations | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 Face | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Scalp | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Upper extremities | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Cosmetic outcomes: Number of participants with decreased infiltration and disappearance of crust | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
6.
3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Investigator Global Improvement Indices‐completely improved | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 30 day treatment/30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.89, 17.89] |
| 1.2 60 day treatment/30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 3.06 [1.21, 7.77] |
| 1.3 90 day treatment/30 day follow‐up | 1 | 117 | Risk Ratio (M‐H, Random, 95% CI) | 2.50 [1.37, 4.55] |
| 2 Participant Global Improvement Indices‐completely improved | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 30 day treatment/30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.89, 17.89] |
| 2.2 60 day treatment/30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 2.86 [1.12, 7.32] |
| 2.3 90 day treatment/30 day follow‐up | 1 | 117 | Risk Ratio (M‐H, Random, 95% CI) | 2.44 [1.28, 4.64] |
| 3 Participant complete clearance at end of treatment (>56 days) | 2 | 280 | Risk Ratio (M‐H, Random, 95% CI) | 1.95 [1.21, 3.13] |
| 4 Participant complete clearance (target lesions) | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 30 day treatment/ 30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 3.5 [0.76, 16.01] |
| 4.2 60 day treatment/ 30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 3.27 [1.30, 8.21] |
| 4.3 90 day treatment/ 30 day follow‐up | 2 | 267 | Risk Ratio (M‐H, Random, 95% CI) | 2.87 [1.84, 4.48] |
| 5 Participant complete clearance (all lesions) | 3 | 420 | Risk Ratio (M‐H, Random, 95% CI) | 2.46 [1.66, 3.66] |
| 5.1 30 day treatment/ 30 day follow‐up | 1 | 98 | Risk Ratio (M‐H, Random, 95% CI) | 3.5 [0.76, 16.01] |
| 5.2 60 day treatment/ 30 day follow‐up | 1 | 97 | Risk Ratio (M‐H, Random, 95% CI) | 3.83 [1.37, 10.71] |
| 5.3 90 day treatment/30 day follow‐up | 2 | 225 | Risk Ratio (M‐H, Random, 95% CI) | 2.20 [1.40, 3.44] |
| 6 Participant complete clearance for 30 day treatment by locations | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Forehead | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.4 Back of hand | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Participant complete clearance for 60 day treatment by locations | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Forehead | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.4 Arm/forearm | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.5 Back of hand | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Participant complete clearance for 90 day treatment by locations | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 8.1 Scalp | 2 | 23 | Risk Ratio (M‐H, Random, 95% CI) | 1.24 [0.25, 6.08] |
| 8.2 Forehead | 2 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [1.03, 2.85] |
| 8.3 Face | 2 | 47 | Risk Ratio (M‐H, Random, 95% CI) | 2.15 [1.05, 4.40] |
| 8.4 Arm/forearm | 2 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.94 [0.26, 14.40] |
| 8.5 Back of hand | 2 | 63 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [0.04, 65.87] |
| 9 Participant complete clearance in immunosuppressed participants | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Participant partial (>75%) clearance in immunosuppressed participants | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Mean reduction of lesion counts (30‐90 days ): At the end of study | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 11.1 90 days | 1 | 150 | Mean Difference (IV, Random, 95% CI) | 0.80 [‐1.48, 3.08] |
| 12 Mean reduction of lesion counts (30‐90 days): 30 day follow‐up | 2 | 345 | Mean Difference (IV, Random, 95% CI) | 2.55 [1.56, 3.53] |
| 12.1 30 days | 1 | 98 | Mean Difference (IV, Random, 95% CI) | 2.00 [0.63, 3.37] |
| 12.2 60 days | 1 | 97 | Mean Difference (IV, Random, 95% CI) | 2.40 [0.73, 4.07] |
| 12.3 90 days | 1 | 150 | Mean Difference (IV, Random, 95% CI) | 3.80 [1.83, 5.77] |
| 13 Withdrawal due to adverse events | 4 | 592 | Risk Ratio (M‐H, Random, 95% CI) | 3.59 [1.92, 6.70] |
| 14 Minor adverse event: body as a whole : in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15 Minor adverse event: body as a whole : "flu" | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16 Minor adverse event:: body as a whole : infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17 Minor adverse event: cardiovascular: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18 Minor adverse event: cardiovascular: sinus bradycardia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19 Minor adverse event: dermatological: bursitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 20 Minor adverse event: dermatological: dry skin | 3 | 462 | Risk Ratio (M‐H, Random, 95% CI) | 2.40 [1.20, 4.78] |
| 21 Minor adverse event: dermatological: herpes zoster | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 22 Minor adverse event: dermatological: rash vesiculobullous | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 23 Minor adverse event::dermatological: seborrhoea | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 24 Minor adverse event: dermatological: skin exfoliation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 25 Minor adverse event: dermatological: ulcerated skin | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 26 Minor adverse event: digestive : in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 27 Minor adverse event: hemic and lymphatic: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 28 Minor adverse event: metabolic and nutritional disorders : in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 29 Minor adverse event: musculoskeletal and connective tissue: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 30 Minor adverse event: musculoskeletal and connective tissue: hypokinesia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 31 Minor adverse event: nervous system: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 32 Minor adverse event: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 33 Minor adverse event: nervous system: hyperaesthesia | 2 | 345 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.30, 2.60] |
| 34 Minor adverse event: nervous system: paraesthesia | 2 | 345 | Risk Ratio (M‐H, Random, 95% CI) | 2.53 [0.57, 11.20] |
| 35 Minor adverse event: respiratory: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 36 Minor adverse event: respiratory: bronchitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 37 Minor adverse event: respiratory: pharyngitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 38 Minor adverse event: respiratory: upper respiratory tract infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 39 Minor adverse event: special senses: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 40 Minor adverse event: urogenital: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
6.14.
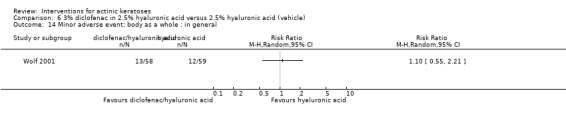
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 14 Minor adverse event: body as a whole : in general.
6.15.
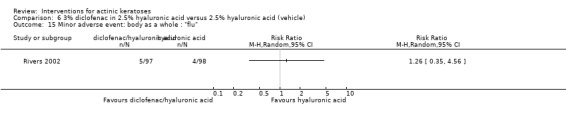
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 15 Minor adverse event: body as a whole : "flu".
6.16.
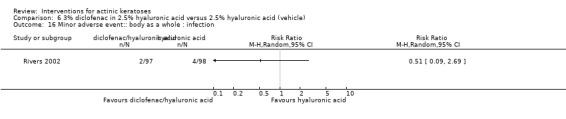
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 16 Minor adverse event:: body as a whole : infection.
6.17.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 17 Minor adverse event: cardiovascular: in general.
6.18.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 18 Minor adverse event: cardiovascular: sinus bradycardia.
6.19.
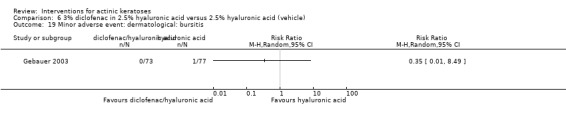
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 19 Minor adverse event: dermatological: bursitis.
6.21.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 21 Minor adverse event: dermatological: herpes zoster.
6.22.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 22 Minor adverse event: dermatological: rash vesiculobullous.
6.23.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 23 Minor adverse event::dermatological: seborrhoea.
6.24.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 24 Minor adverse event: dermatological: skin exfoliation.
6.25.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 25 Minor adverse event: dermatological: ulcerated skin.
6.26.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 26 Minor adverse event: digestive : in general.
6.27.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 27 Minor adverse event: hemic and lymphatic: in general.
6.29.
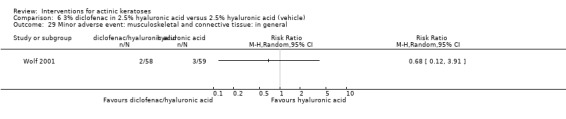
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 29 Minor adverse event: musculoskeletal and connective tissue: in general.
6.30.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 30 Minor adverse event: musculoskeletal and connective tissue: hypokinesia.
6.31.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 31 Minor adverse event: nervous system: in general.
6.32.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 32 Minor adverse event: nervous system: headache.
6.33.
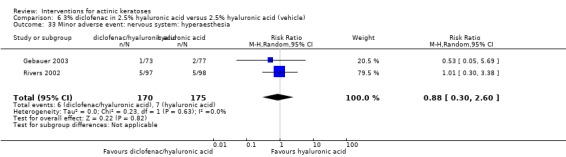
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 33 Minor adverse event: nervous system: hyperaesthesia.
6.34.
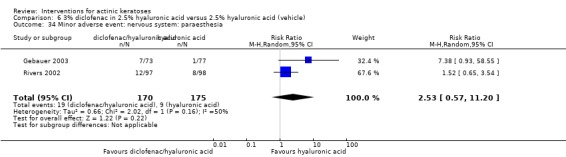
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 34 Minor adverse event: nervous system: paraesthesia.
6.35.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 35 Minor adverse event: respiratory: in general.
6.36.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 36 Minor adverse event: respiratory: bronchitis.
6.37.
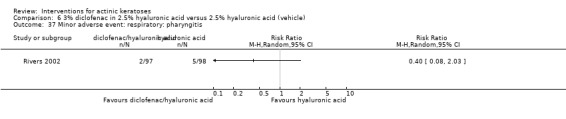
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 37 Minor adverse event: respiratory: pharyngitis.
6.38.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 38 Minor adverse event: respiratory: upper respiratory tract infection.
6.39.
Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 39 Minor adverse event: special senses: in general.
6.40.

Comparison 6 3% diclofenac in 2.5% hyaluronic acid versus 2.5% hyaluronic acid (vehicle), Outcome 40 Minor adverse event: urogenital: in general.
7.
3% diclofenac in 2.5% hyaluronic acid versus 5% imiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Investigator Global Improvement Indices‐Complete improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant Global Improvement Indices‐Complete improvement | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
8.
2‐(Difluoromethyl)‐dl‐ornithine (DFMO) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean reduction in lesions counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
9.
0.5% 5‐FU versus vehicle
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 3 | 522 | Risk Ratio (M‐H, Random, 95% CI) | 8.86 [3.67, 21.40] |
| 1.1 1 week treatment with 4 week follow‐up | 3 | 267 | Risk Ratio (M‐H, Random, 95% CI) | 8.30 [2.04, 33.76] |
| 1.2 2 week treatment with 4 week follow‐up | 2 | 128 | Risk Ratio (M‐H, Random, 95% CI) | 6.42 [1.27, 32.59] |
| 1.3 4 week treatment with 4 week follow‐up | 2 | 127 | Risk Ratio (M‐H, Random, 95% CI) | 13.07 [2.68, 63.66] |
| 2 Mean reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Mean percentage of reduction in lesion counts | 1 | 142 | Mean Difference (IV, Random, 95% CI) | 33.60 [22.88, 44.32] |
| 4 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Skin irritation | 2 | 384 | Risk Ratio (M‐H, Random, 95% CI) | 1.45 [1.27, 1.65] |
| 6 Minor adverse event excluding skin irritation: body as a whole : in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse event excluding skin irritation: body as a whole : allergy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse event excluding skin irritation: body as a whole : "flu" or common cold | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse event excluding skin irritation: musculoskeletal and connective tissue: soreness | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse event excluding skin irritation:nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse event excluding skin irritation: respiratory: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse event excluding skin irritation: respiratory: sinusitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14 Minor adverse event excluding skin irritation: respiratory: upper respiratory tract infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15 Minor adverse event excluding skin irritation: special senses: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16 Minor adverse event excluding skin irritation:special senses: eye irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
10.
0.5% 5‐FU at varying application durations
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Daily for 1 week versus 4 weeks | 2 | 167 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.19, 0.81] |
| 1.2 Daily for 1 week versus 2 weeks | 2 | 169 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.23, 2.37] |
| 1.3 Daily for 2 weeks versus 4 weeks | 2 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 0.56 [0.36, 0.87] |
| 2 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Skin irritation | 2 | 515 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.91, 1.00] |
| 3.1 Daily for 1 week versus 4 weeks | 2 | 170 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.89, 1.03] |
| 3.2 Daily for 1 week versus 2 weeks | 2 | 172 | Risk Ratio (M‐H, Random, 95% CI) | 0.96 [0.86, 1.08] |
| 3.3 Daily for 2 weeks versus 4 weeks | 2 | 173 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.88, 1.02] |
| 4 Minor adverse events excluding skin irritation: body as a whole : in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: body as a whole : allergy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Minor adverse events excluding skin irritation: body as a whole : "flu" or common cold | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: respiratory: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: respiratory: sinusitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: special senses: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: special senses: eye irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 Daily for 1 week versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 Daily for 1 week versus 2 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 Daily for 2 weeks versus 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
11.
0.5% 5‐FU versus ALA‐PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
12.
5% 5‐FU with 0.05% tretinoin versus 5% 5‐FU with placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
13.
5% 5‐FU versus 5% imiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 2 | 89 | Risk Ratio (M‐H, Random, 95% CI) | 1.85 [0.41, 8.33] |
14.
5% 5‐FU versus cryotherapy
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
15.
5% 5‐FU versus 10% masoprocol
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Investigator Global Improvement Indices ‐cleared | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean reduction of lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Mean percentage of reduction of lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
16.
5% 5‐FU versus carbon dioxide laser resurfacing
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean percentage of reduction of lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
17.
5% 5‐FU versus Er:YAG laser resurfacing
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Skin irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
18.
5% 5‐FU versus Trichloroacetic acid peel
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean percentage of reduction in lesions | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
19.
5% Imiquimod versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance‐number of doses | 11 | 2880 | Risk Ratio (M‐H, Random, 95% CI) | 6.91 [4.25, 11.26] |
| 1.1 9 or 18 doses (3 times/week for 3 weeks on, 4 weeks off) | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 2.76 [0.39, 19.40] |
| 1.2 12‐16 doses (2 times/week for 8 weeks or 3 times/week for 4 weeks) | 3 | 543 | Risk Ratio (M‐H, Random, 95% CI) | 7.88 [1.09, 56.67] |
| 1.3 12 or 24 doses (3 times/week for 4 weeks on , 4 weeks off, 4 weeks on) | 2 | 505 | Risk Ratio (M‐H, Random, 95% CI) | 8.81 [1.15, 67.32] |
| 1.4 24 doses (3 times/week for 8 weeks) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.07, 25.08] |
| 1.5 32‐36 doses (2 times/ week for 16 weeks or 3 times/ week for 12 weeks) | 4 | 888 | Risk Ratio (M‐H, Random, 95% CI) | 7.12 [3.06, 16.58] |
| 1.6 40 doses (5 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.03, 17.27] |
| 1.7 48 doses (3 times/ week for 16 weeks) | 3 | 795 | Risk Ratio (M‐H, Random, 95% CI) | 10.90 [3.59, 33.15] |
| 1.8 56 doses (7 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.29 [0.07, 24.29] |
| 2 Participant complete clearance in immunosuppressed participants | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Participant partial (>75%) clearance | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 9 or 18 doses (3 times/ week for 3 weeks on, 4 weeks off. 3 weeks on) | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 2.41 [0.91, 6.39] |
| 3.2 12‐16 doses (3 times/week for 4 weeks or 2 times/week for 8 weeks) | 2 | 284 | Risk Ratio (M‐H, Random, 95% CI) | 2.86 [1.53, 5.34] |
| 3.3 12 or 24 doses (3 times/week for 4 weeks on, 4 weeks off) | 2 | 505 | Risk Ratio (M‐H, Random, 95% CI) | 6.23 [0.70, 55.10] |
| 3.4 24 doses (3 times/week for 8 weeks) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [0.25, 62.85] |
| 3.5 32 doses (2 times/week for 16 weeks) | 1 | 436 | Risk Ratio (M‐H, Random, 95% CI) | 5.02 [3.44, 7.33] |
| 3.6 40 doses (5 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 3.35 [0.21, 53.51] |
| 3.7 48 doses (3 times/ week for 16 weeks) | 2 | 778 | Risk Ratio (M‐H, Random, 95% CI) | 8.46 [2.29, 31.16] |
| 3.8 56 doses (7 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 5.94 [0.39, 90.34] |
| 4 Participant partial (>75%) clearance in immunosuppressed participants | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Mean reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 6 Withdrawal due to adverse events | 8 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 12‐16 doses (2 times/week for 8 weeks or 3 times/week for 4 weeks) | 1 | 38 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.03, 16.74] |
| 6.2 12 or 24 doses (3 times/week for 4 weeks on , 4 weeks off, 4 weeks on) | 2 | 505 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.31, 8.23] |
| 6.3 24 doses (3 times/week for 8 weeks) | 1 | 36 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.07, 25.08] |
| 6.4 32‐36 doses (2 times/ week for 16 weeks or 3 times/ week for 12 weeks) | 3 | 858 | Risk Ratio (M‐H, Random, 95% CI) | 2.29 [0.80, 6.57] |
| 6.5 40 doses (5 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 4.90 [0.32, 75.60] |
| 6.6 48 doses (3 times/ week for 16 weeks) | 2 | 778 | Risk Ratio (M‐H, Random, 95% CI) | 2.69 [1.48, 4.90] |
| 6.7 56 doses (7 times/week for 8 weeks) | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 5.42 [0.35, 82.97] |
| 7 Withdrawal due to adverse events in immunosuppressed participants | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 48 doses (3 times/ week for 16 weeks) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: body as a whole: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: body as a whole: "flu" or "cold" | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: digestive: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: digestive: nausea | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: nervous system: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 12‐16 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 24‐28 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 40 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.4 56 doses | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Cosmetic outcome: decrease in roughness/dryness/scaliness of the skin | 2 | 683 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [1.86, 5.58] |
| 13.1 32‐36 doses | 1 | 415 | Risk Ratio (M‐H, Random, 95% CI) | 2.54 [1.91, 3.37] |
| 13.2 48 doses | 1 | 268 | Risk Ratio (M‐H, Random, 95% CI) | 4.43 [2.69, 7.30] |
19.8.
Comparison 19 5% Imiquimod versus placebo, Outcome 8 Minor adverse events excluding skin irritation: body as a whole: in general.
19.9.
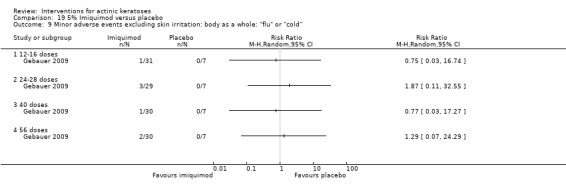
Comparison 19 5% Imiquimod versus placebo, Outcome 9 Minor adverse events excluding skin irritation: body as a whole: "flu" or "cold".
19.10.
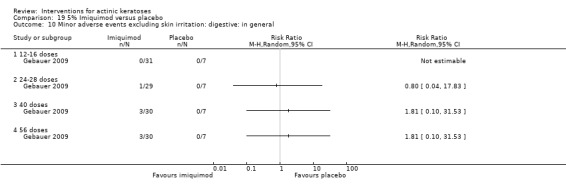
Comparison 19 5% Imiquimod versus placebo, Outcome 10 Minor adverse events excluding skin irritation: digestive: in general.
19.11.
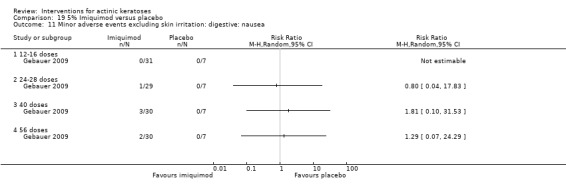
Comparison 19 5% Imiquimod versus placebo, Outcome 11 Minor adverse events excluding skin irritation: digestive: nausea.
19.12.
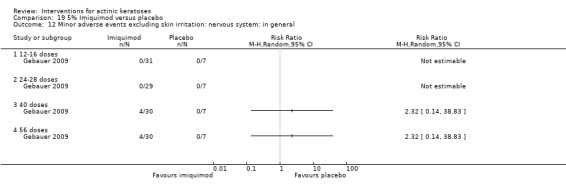
Comparison 19 5% Imiquimod versus placebo, Outcome 12 Minor adverse events excluding skin irritation: nervous system: in general.
20.
Imiquimod versus placebo: different concentrations
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 12 | 3087 | Risk Ratio (M‐H, Random, 95% CI) | 6.73 [5.03, 9.00] |
| 1.1 5.0% imiquimod | 9 | 1871 | Risk Ratio (M‐H, Random, 95% CI) | 7.70 [4.63, 12.79] |
| 1.2 3.75% imiquimod | 3 | 730 | Risk Ratio (M‐H, Random, 95% CI) | 6.45 [3.87, 10.73] |
| 1.3 2.5% imiquimod | 2 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 4.49 [2.40, 8.39] |
| 2 Participant partial (>75%) clearance | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 5.0% imiquimod | 4 | 1363 | Risk Ratio (M‐H, Random, 95% CI) | 6.71 [3.89, 11.57] |
| 2.2 3.75% imiquimod | 2 | 484 | Risk Ratio (M‐H, Random, 95% CI) | 3.11 [2.08, 4.66] |
| 2.3 2.5% imiquimod | 2 | 485 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [1.67, 3.68] |
| 3 Mean percentage of reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 3.75% imiquimod | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Minor adverse events excluding skin irritation: body as a whole: 'flu" or "cold" | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 5.0% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Withdrawal due to adverse events | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 5.0% imiquimod | 8 | 2290 | Risk Ratio (M‐H, Random, 95% CI) | 2.59 [1.59, 4.23] |
| 5.2 3.75% imiquimod | 2 | 483 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.22, 3.93] |
| 5.3 2.5% imiquimod | 2 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.09, 2.70] |
| 6 Skin irritation | 5 | 1678 | Risk Ratio (M‐H, Random, 95% CI) | 3.93 [1.56, 9.88] |
| 6.1 5.0% imiquimod | 3 | 708 | Risk Ratio (M‐H, Random, 95% CI) | 3.68 [0.86, 15.74] |
| 6.2 3.75% imiquimod | 2 | 484 | Risk Ratio (M‐H, Random, 95% CI) | 4.86 [0.92, 25.83] |
| 6.3 2.5% imiquimod | 2 | 486 | Risk Ratio (M‐H, Random, 95% CI) | 3.45 [0.63, 18.97] |
| 7 Minor adverse events excluding skin irritation: body as a whole: pyrexia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: hemic and lymphatic: lymphadenopathy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: nervous system: fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: nervous system: headache | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 5.0% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.3 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: respiratory: cough | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Minor adverse events excluding skin irritation: respiratory: sinusitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 14.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15 Minor adverse events excluding skin irritation: urogenital: urinary tract infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15.1 3.75% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 15.2 2.5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 16 Cosmetic outcome: Participant's significantly or much improved cosmetic outcome assessed by investigator | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 16.1 3.75% imiquimod | 2 | 470 | Risk Ratio (M‐H, Random, 95% CI) | 2.71 [2.05, 3.58] |
| 16.2 2.5% imiquimod | 2 | 475 | Risk Ratio (M‐H, Random, 95% CI) | 2.25 [1.62, 3.14] |
20.4.
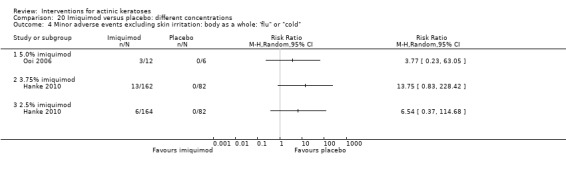
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: 'flu" or "cold".
20.7.
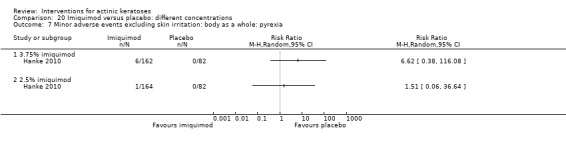
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 7 Minor adverse events excluding skin irritation: body as a whole: pyrexia.
20.8.
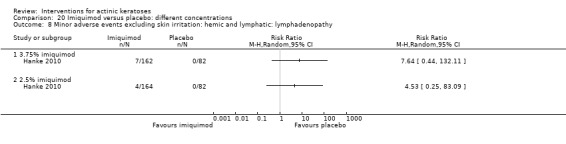
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 8 Minor adverse events excluding skin irritation: hemic and lymphatic: lymphadenopathy.
20.9.
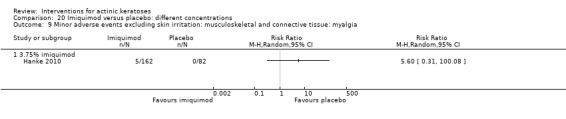
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 9 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
20.10.
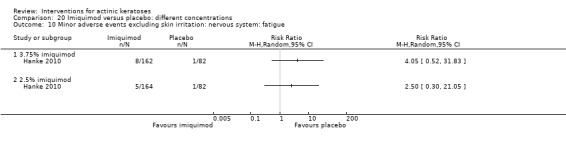
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 10 Minor adverse events excluding skin irritation: nervous system: fatigue.
20.11.
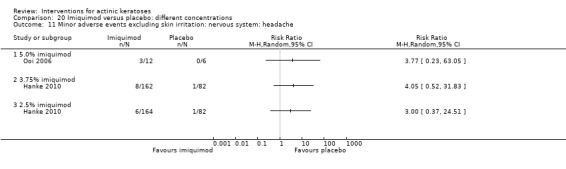
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 11 Minor adverse events excluding skin irritation: nervous system: headache.
20.12.
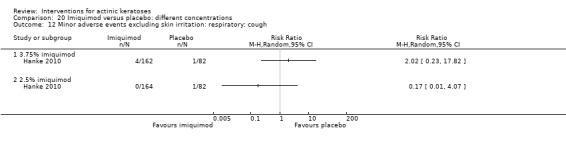
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 12 Minor adverse events excluding skin irritation: respiratory: cough.
20.13.
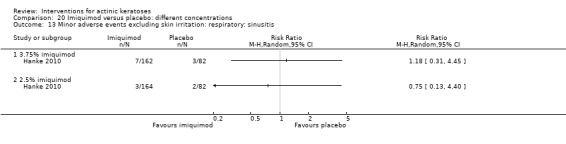
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 13 Minor adverse events excluding skin irritation: respiratory: sinusitis.
20.14.
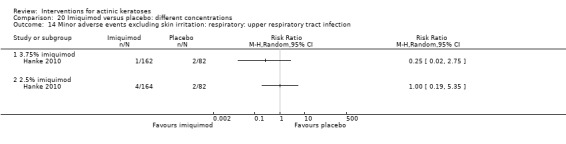
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 14 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection.
20.15.
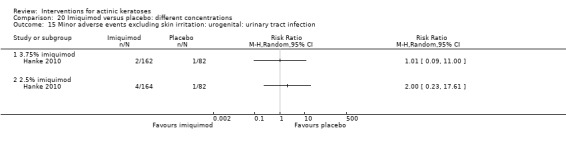
Comparison 20 Imiquimod versus placebo: different concentrations, Outcome 15 Minor adverse events excluding skin irritation: urogenital: urinary tract infection.
21.
Imiquimod versus placebo: frequency of application
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 2 times/week | 4 | 890 | Risk Ratio (M‐H, Random, 95% CI) | 5.36 [2.03, 14.16] |
| 1.2 3 times/week | 6 | 1336 | Risk Ratio (M‐H, Random, 95% CI) | 8.38 [3.79, 18.52] |
| 1.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.03, 17.27] |
| 1.4 7 times/week | 4 | 1253 | Risk Ratio (M‐H, Random, 95% CI) | 5.39 [3.65, 7.98] |
| 2 Participant partial (>75%) clearance | 6 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 2 times/week | 2 | 474 | Risk Ratio (M‐H, Random, 95% CI) | 4.99 [3.43, 7.26] |
| 2.2 3 times/week | 3 | 814 | Risk Ratio (M‐H, Random, 95% CI) | 7.65 [2.51, 23.32] |
| 2.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 3.35 [0.21, 53.51] |
| 2.4 7 times/week | 3 | 1006 | Risk Ratio (M‐H, Random, 95% CI) | 2.95 [1.99, 4.37] |
| 3 Withdrawal due to adverse events | 10 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 2 times/week | 4 | 896 | Risk Ratio (M‐H, Random, 95% CI) | 2.04 [0.75, 5.53] |
| 3.2 3 times/week | 5 | 1319 | Risk Ratio (M‐H, Random, 95% CI) | 2.47 [1.42, 4.30] |
| 3.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 4.90 [0.32, 75.60] |
| 3.4 7 times/week | 3 | 1006 | Risk Ratio (M‐H, Random, 95% CI) | 1.55 [0.33, 7.18] |
| 4 Minor adverse events excluding skin irritation:body as a whole: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 2 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 5 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: body as a whole:"flu" or "cold" | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 2 times/week | 1 | 38 | Risk Ratio (M‐H, Random, 95% CI) | 0.75 [0.03, 16.74] |
| 5.2 3 times/week | 2 | 54 | Risk Ratio (M‐H, Random, 95% CI) | 2.67 [0.36, 19.83] |
| 5.3 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.03, 17.27] |
| 5.4 7 times/week | 2 | 527 | Risk Ratio (M‐H, Random, 95% CI) | 5.20 [0.28, 95.18] |
| 6 Minor adverse events excluding skin irritation: digestive: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 5 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Minor adverse events excluding skin irritation: digestive: nausea | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 5 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.3 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: nervous system: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 3 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 7 times/week | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: nervous system: headache | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 3 times/week | 1 | 18 | Risk Ratio (M‐H, Random, 95% CI) | 3.77 [0.23, 63.05] |
| 9.2 5 times/week | 1 | 37 | Risk Ratio (M‐H, Random, 95% CI) | 1.81 [0.10, 31.53] |
| 9.3 7 times/week | 2 | 527 | Risk Ratio (M‐H, Random, 95% CI) | 4.48 [0.86, 23.31] |
21.4.
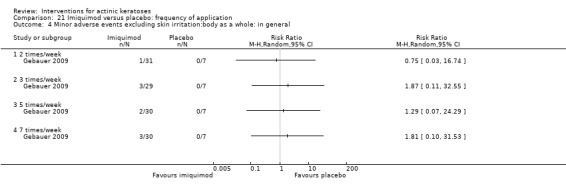
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 4 Minor adverse events excluding skin irritation:body as a whole: in general.
21.6.
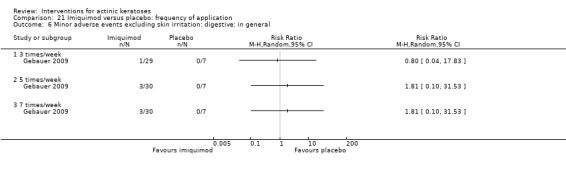
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 6 Minor adverse events excluding skin irritation: digestive: in general.
21.7.
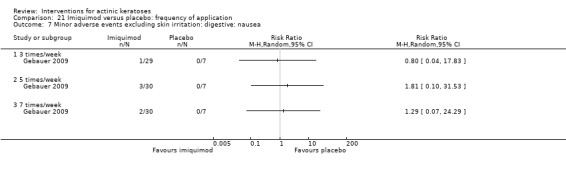
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 7 Minor adverse events excluding skin irritation: digestive: nausea.
21.8.
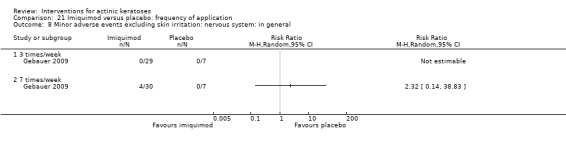
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 8 Minor adverse events excluding skin irritation: nervous system: in general.
21.9.
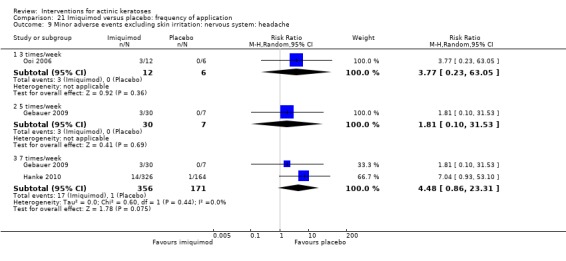
Comparison 21 Imiquimod versus placebo: frequency of application, Outcome 9 Minor adverse events excluding skin irritation: nervous system: headache.
22.
5% imiquimod versus 5% 5‐FU
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2 Cosmetic outcome: Investigator cosmetic outcome "excellent" | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Cosmetic outcome: normal skin surface | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
23.
5% imiquimod versus cryotherapy
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
24.
Ingenol mebutate (PEP005) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance of target lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant complete clearance of all lesions | 2 | 456 | Risk Ratio (M‐H, Random, 95% CI) | 4.50 [2.61, 7.74] |
| 3 Participant partial (>75%) clearance of target lesions | 2 | 280 | Risk Ratio (M‐H, Random, 95% CI) | 2.88 [1.81, 4.58] |
| 4 Cosmetic outcomes: changes in pigmentation | 3 | 514 | Risk Ratio (M‐H, Random, 95% CI) | 3.36 [0.63, 17.80] |
25.
Ingenol mebutate (PEP005) versus placebo: different concentrations
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance of target lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 0.025% 3 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 0.05% 2‐3 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant complete clearance of all lesions | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 0.025% 3 days | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 4.0 [1.03, 15.55] |
| 2.2 0.05% 2‐3 days | 2 | 386 | Risk Ratio (M‐H, Random, 95% CI) | 5.14 [2.75, 9.62] |
| 3 Participant partial (>75%) clearance of target lesions | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 0.0025% 2 days | 1 | 19 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.21, 8.41] |
| 3.2 0.01% 2 days | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.06, 4.23] |
| 3.3 0.025% 3 days | 1 | 70 | Risk Ratio (M‐H, Random, 95% CI) | 2.8 [1.13, 6.96] |
| 3.4 0.05% 2‐3 days | 2 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 3.34 [1.84, 6.04] |
| 4 Cosmetic outcomes: changes in pigmentation | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 0.01% 2 days | 1 | 20 | Risk Ratio (M‐H, Random, 95% CI) | 1.47 [0.08, 25.88] |
| 4.2 0.05% 2 days | 2 | 253 | Risk Ratio (M‐H, Random, 95% CI) | 4.86 [0.48, 49.39] |
25.4.
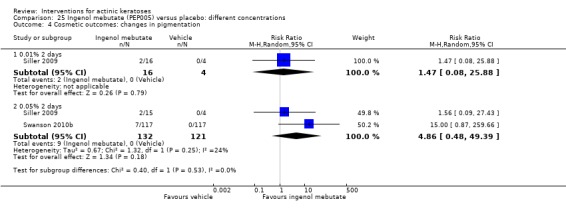
Comparison 25 Ingenol mebutate (PEP005) versus placebo: different concentrations, Outcome 4 Cosmetic outcomes: changes in pigmentation.
26.
0.05% Ingenol mebutate (PEP005) versus placebo: number of doses
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance of target lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 0.05% 2 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 0.05% 3 days | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant complete clearance of all lesions | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 0.05% 2 days | 2 | 319 | Risk Ratio (M‐H, Random, 95% CI) | 4.32 [2.30, 8.11] |
| 2.2 0.05% 3 days | 1 | 87 | Risk Ratio (M‐H, Random, 95% CI) | 4.08 [1.59, 10.47] |
| 3 Participant partial (>75%) clearance of target lesions | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 0.05% 2 days | 2 | 104 | Risk Ratio (M‐H, Random, 95% CI) | 2.65 [1.41, 5.00] |
| 3.2 0.05% 3 days | 1 | 87 | Risk Ratio (M‐H, Random, 95% CI) | 3.23 [1.66, 6.29] |
27.
Isotretinoin versus vehicle
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Investigator global improvement indices‐completely cleared | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Upper extremities | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean reduction of lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2.1 Face | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Scalp | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Upper extremities | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Skin irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Severe‐Skin irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
28.
Masoprocol versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global improvement indices‐cured | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Mean reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
28.1.
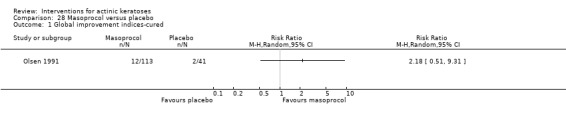
Comparison 28 Masoprocol versus placebo, Outcome 1 Global improvement indices‐cured.
29.
1% nicotinamide versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean percentage of reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 1.1 At 3 months | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 6 months | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
30.
0.1% resiquimod versus 0.01% resiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
30.4.
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
30.5.
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.
30.6.
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.
30.7.
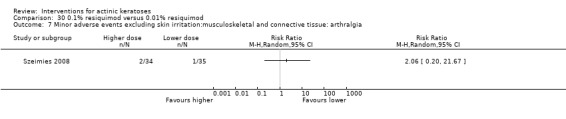
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.
30.8.

Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
30.10.
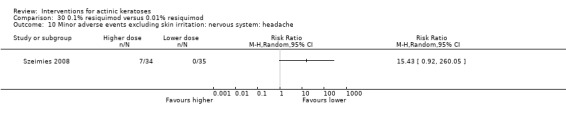
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.
30.11.
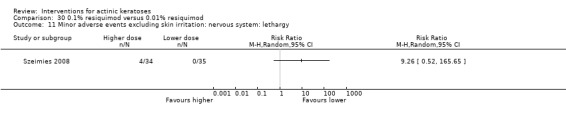
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.
30.12.
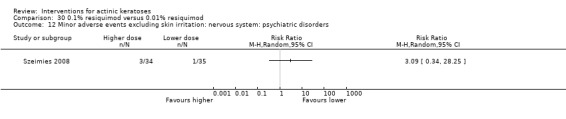
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.
30.13.
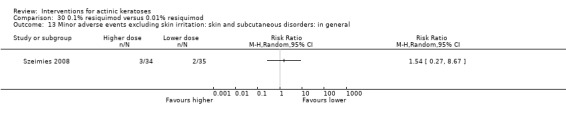
Comparison 30 0.1% resiquimod versus 0.01% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.
31.
0.1% resiquimod versus 0.03% resiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
31.4.
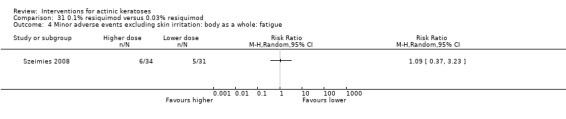
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
31.5.
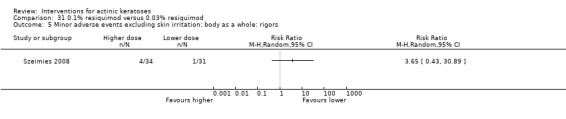
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.
31.6.
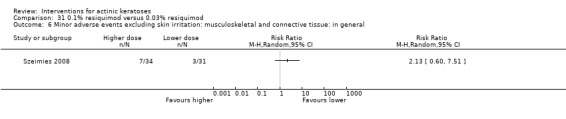
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.
31.7.
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.
31.8.
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
31.9.
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.
31.10.
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.
31.11.
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.
31.12.
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.
31.13.
Comparison 31 0.1% resiquimod versus 0.03% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.
32.
0.1% resiquimod versus 0.06% resiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
32.3.
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 3 Withdrawal due to adverse events.
32.4.
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
32.5.
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.
32.6.
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.
32.7.
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.
32.8.
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
32.9.
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.
32.10.
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.
32.11.
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.
32.12.
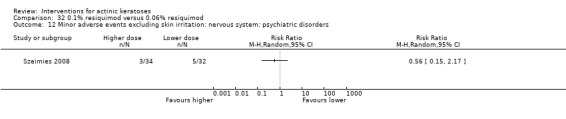
Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.
32.13.

Comparison 32 0.1% resiquimod versus 0.06% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.
33.
0.06% resiquimod versus 0.01% resiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
33.1.
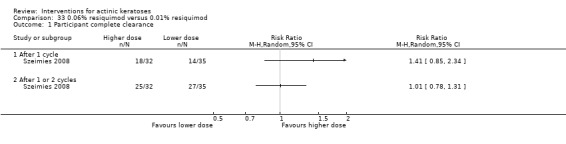
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 1 Participant complete clearance.
33.4.
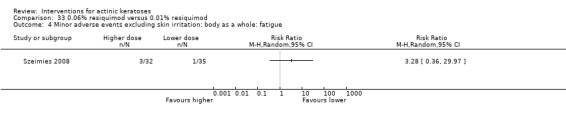
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
33.5.
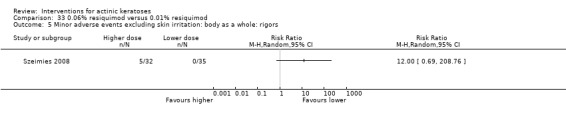
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.
33.6.
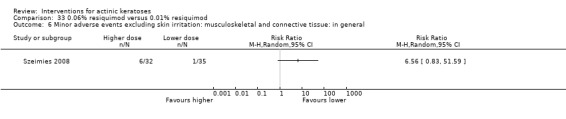
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.
33.7.
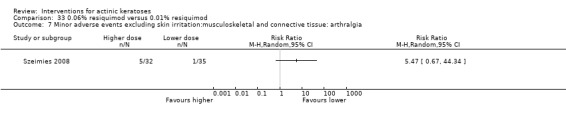
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.
33.8.
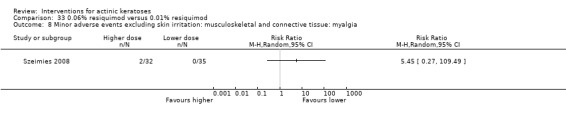
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
33.11.
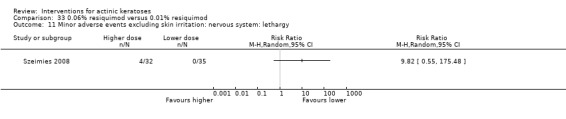
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.
33.12.
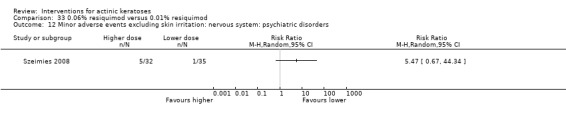
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.
33.13.
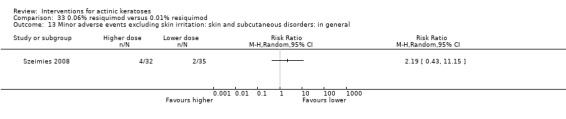
Comparison 33 0.06% resiquimod versus 0.01% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.
34.
0.06% resiquimod versus 0.03% resiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation:skin and subcutaneous disorders: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
34.3.
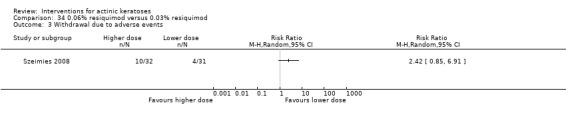
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 3 Withdrawal due to adverse events.
34.4.
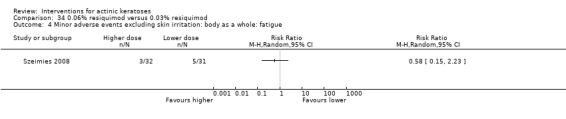
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
34.5.
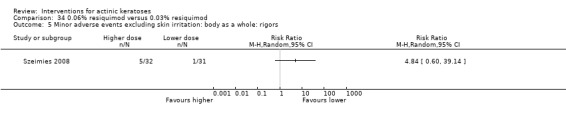
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.
34.6.
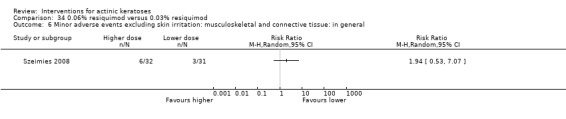
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.
34.7.
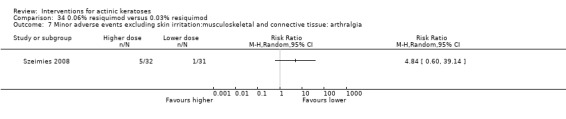
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.
34.8.
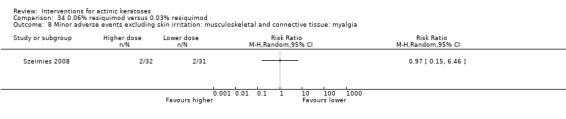
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
34.9.
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 9 Minor adverse events excluding skin irritation: nervous system: in general.
34.10.
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.
34.11.
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.
34.12.
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.
34.13.
Comparison 34 0.06% resiquimod versus 0.03% resiquimod, Outcome 13 Minor adverse events excluding skin irritation:skin and subcutaneous disorders: in general.
35.
0.03% resiquimod versus 0.01% resiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After 1 cycle | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 After 1 or 2 cycles | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Minor adverse events excluding skin irritation: body as a whole: fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: body as a whole: rigors | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: nervous system: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: nervous system: lethargy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
35.1.
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 1 Participant complete clearance.
35.3.
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 3 Withdrawal due to adverse events.
35.4.
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 4 Minor adverse events excluding skin irritation: body as a whole: fatigue.
35.5.
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 5 Minor adverse events excluding skin irritation: body as a whole: rigors.
35.6.
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 6 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: in general.
35.7.
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 7 Minor adverse events excluding skin irritation:musculoskeletal and connective tissue: arthralgia.
35.8.
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 8 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia.
35.10.
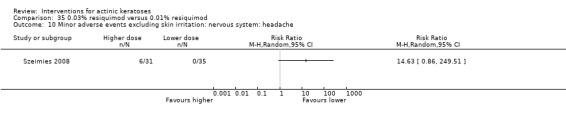
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 10 Minor adverse events excluding skin irritation: nervous system: headache.
35.11.

Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 11 Minor adverse events excluding skin irritation: nervous system: lethargy.
35.12.
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 12 Minor adverse events excluding skin irritation: nervous system: psychiatric disorders.
35.13.
Comparison 35 0.03% resiquimod versus 0.01% resiquimod, Outcome 13 Minor adverse events excluding skin irritation: skin and subcutaneous disorders: in general.
36.
Sunscreen SPF 17 (8% 2‐ethyl‐hexyl p‐methoxycinnamate/2% 4‐tert‐butyl‐4‐methoxy‐4‐dibenzoylmethane) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean change in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
37.
12.5% DL‐α‐tocopherol (vitamin E) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean reduction of lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
38.
Etretinate versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
39.
Carbon dioxide laser resurfacing versus 5% 5‐FU
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean percentage of reduction of lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
40.
Carbon dioxide laser resurfacing versus Trichloroacetic acid peel
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean percentage of reduction of lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
41.
Er:YAG laser resurfacing versus 5% 5‐FU
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean reduction in lesion counts | Other data | No numeric data | ||
| 2 Mean percentage of reduction in lesion counts | Other data | No numeric data | ||
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Skin irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: dermatology: acne | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Minor adverse events excluding skin irritation: dermatology:crustea | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.4 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Minor adverse events excluding skin irritation: dermatology: infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: dermatology: milia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: dermatology:pain | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 At the end of treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9.2 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Cosmetic outcomes: changes in pigmentation (hypo) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Cosmetic outcomes: scarring | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11.2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Cosmetic outcomes: improvement in photoageing score | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
41.4.
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 4 Skin irritation.
41.5.
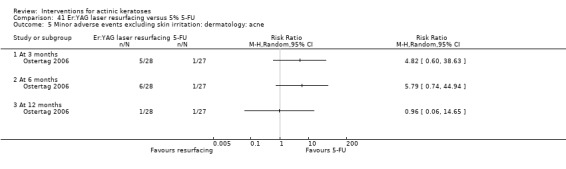
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 5 Minor adverse events excluding skin irritation: dermatology: acne.
41.7.
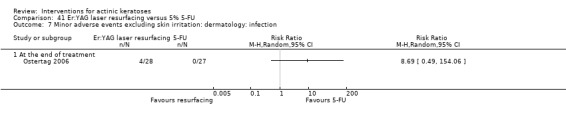
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 7 Minor adverse events excluding skin irritation: dermatology: infection.
41.8.
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 8 Minor adverse events excluding skin irritation: dermatology: milia.
41.9.
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 9 Minor adverse events excluding skin irritation: dermatology:pain.
41.11.
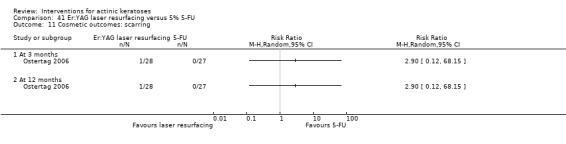
Comparison 41 Er:YAG laser resurfacing versus 5% 5‐FU, Outcome 11 Cosmetic outcomes: scarring.
42.
Cryotherapy versus betulin‐based oleogel
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
43.
Cryotherapy versus 5% 5‐FU
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 After treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Cosmetic outcomes: excellent global cosmetic outcome | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Cosmetic outcomes: better skin appearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
43.2.
Comparison 43 Cryotherapy versus 5% 5‐FU, Outcome 2 Cosmetic outcomes: excellent global cosmetic outcome.
44.
Cryotherapy versus imiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Cosmetic outcomes: excellent global cosmetic outcome | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Cosmetic outcomes: better skin appearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
45.
Cryotherapy versus MAL‐red light PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean percentage of reduction in lesion counts | Other data | No numeric data | ||
| 2 Withdrawal due to adverse events | 2 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.16, 7.16] |
| 3 Cosmetic outcomes: excellent or good cosmetic outcomes by investigator | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4 Cosmetic outcomes: excellent or good cosmetic outcomes by participant | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only |
46.
Cryotherapy versus ALA‐red light PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Skin irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 During treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 One day after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
47.
ALA‐PDT versus placebo‐PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance [1 treatment] | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Blue light | 1 | 243 | Risk Ratio (M‐H, Random, 95% CI) | 6.22 [2.88, 13.43] |
| 1.2 Red light | 3 | 422 | Risk Ratio (M‐H, Random, 95% CI) | 5.94 [3.35, 10.54] |
| 2 Participant complete clearance [1 or 2 treatments] | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Red light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Participant complete clearance [1 or 2 treatments] by anatomical location | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Participant partial (> 75%) clearance [1 treatment] | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Participant partial (>75%) clearance[1 or 2 treatments] | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Participant partial (>75%) clearance [1 or 2 treatment] by anatomical location | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Face | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Scalp | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7 Skin irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7.1 Red light‐during illumination | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 7.2 Red light‐after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 8 Minor adverse events excluding skin irritation: body as a whole: injury | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 9 Minor adverse events excluding skin irritation: cardiovascular: hypertension | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Minor adverse events excluding skin irritation: dermatology: skin discolouration | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10.1 Red light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 11 Minor adverse events excluding skin irritation: dermatology: skin hypertrophy | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 12 Minor adverse events excluding skin irritation: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 13 Cosmetic outcome: very good or good general cosmetic outcome | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
48.
ALA‐ blue light PDT versus ALA‐pulsed laser PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3 Cosmetic outcome: improvement in global response | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4 Cosmetic outcome: improvement in tactile roughness | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Cosmetic outcome: improvement in mottled hyperpigmentation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
49.
ALA‐red light PDT at different application times
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance at 4 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 0.5h versus 1.0h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 0.5h versus 2 h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.4 1h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.5 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.6 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant complete clearance at 8 weeks | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 0.5h versus 1.0h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 0.5h versus 2 h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.4 1h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.5 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.6 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Minor adverse events excluding skin irritation: metabolic and nutritional disorders: elevated alanine transaminase (ALT) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 0.5h versus 1.0h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 0.5h versus 2 h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Minor adverse events excluding skin irritation: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 0.5h versus 1h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 0.5h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 1h versus 2h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.5 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.6 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Minor adverse events excluding skin irritation: other: epistaxis (nose bleeding) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 0.5h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 1h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 2h versus 4h | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
50.
ALA‐PDT versus 0.5% 5‐FU
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Cosmetic outcome: improvement in global response | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 4.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5 Cosmetic outcome: improvement in tactile roughness | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 5.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Cosmetic outcome: improvement in mottled hyperpigmentation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6.1 Blue light | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.2 Pulsed dye laser | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6.3 Combined | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
51.
ALA‐red light PDT vs cryotherapy
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Skin irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 During treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 One day after treatment | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
52.
MAL‐red light PDT versus placebo‐red light PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 5 | 482 | Risk Ratio (M‐H, Random, 95% CI) | 4.46 [3.17, 6.28] |
| 2 Participant partial (>75%) clearance | 2 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 3.28 [1.73, 6.23] |
| 3 Withdrawal due to adverse events | 2 | 191 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.23, 17.74] |
| 4 Minor adverse event: nervous system: headache | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Cosmetic outcome: hyperpigmentation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
53.
MAL‐red light LED PDT versus MAL‐broad visible + water‐filtered infrared A PDT (1 or 2 treatments)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 At 3 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 At 6 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 At 12 months | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
54.
MAL‐red light LED PDT versus MAL‐daylight PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
55.
2h MAL‐day light PDT versus 3h MAL‐daylight PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 2 Mean percentage of reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
56.
16% MAL‐daylight PDT versus 8% MAL‐daylight PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
57.
Single MAL‐red light PDT versus multiple MAL‐red light PDT (2 treatments 1 week apart)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Withdrawal due to adverse events | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
58.
MAL‐ red light PDT vs cryotherapy
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Withdrawal due to adverse events | 2 | 379 | Risk Ratio (M‐H, Random, 95% CI) | 0.94 [0.14, 6.36] |
59.
ALA‐red light PDT versus MAL‐red light PDT
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean reduction in lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
60.
Trichloroacetic acid peel versus 5% 5‐FU
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mean percentage of reduction in lesions | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected |
61.
Cryotherapy versus cryotherapy with betulin‐based oleogel
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Participant partial (>75%) clearance | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
62.
(0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance at 6 months | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean reduction in lesion counts at 6 months | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Mean percentage of reduction in lesion counts at 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 1 cycle (1 week topical, cryosurgery at 4 weeks, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Minor adverse events excluding skin irritation: body as a whole: allergic reaction | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 5 Minor adverse events excluding skin irritation: dermatology: hyperesthesia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 6 Minor adverse events excluding skin irritation: dermatology: skin discoloration | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 7 Minor adverse events excluding skin irritation: dermatology: vesiculobullous rash | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 8 Minor adverse events excluding skin irritation: digestive: cheilitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: special senses: conjunctivitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: special senses: eye irritation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected |
62.4.
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 4 Minor adverse events excluding skin irritation: body as a whole: allergic reaction.
62.5.
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 5 Minor adverse events excluding skin irritation: dermatology: hyperesthesia.
62.6.
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 6 Minor adverse events excluding skin irritation: dermatology: skin discoloration.
62.7.
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 7 Minor adverse events excluding skin irritation: dermatology: vesiculobullous rash.
62.8.
Comparison 62 (0.5% 5‐FU + cryotherapy) versus (vehicle + cryotherapy), Outcome 8 Minor adverse events excluding skin irritation: digestive: cheilitis.
63.
(vehicle + cryotherapy) versus (0.5% 5‐FU + cryotherapy)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance at 6 months | 2 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 1.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 1.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2 Mean reduction in lesion counts at 6 months | 2 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2.1 1 cycle (1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 2 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 3 cycles ( 1 week topical, cryosurgery at week 4, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Mean percentage of reduction in lesion counts at 6 months | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3.1 1 cycle (1 week topical, cryosurgery at 4 weeks, follow‐up at 6 months) | 1 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
64.
Cryotherapy with vehicle versus cryotherapy with imiquimod
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance of all lesions | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.20 [0.05, 0.73] |
| 1.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.11, 1.42] |
| 1.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.11 [0.04, 0.30] |
| 2 Participant complete clearance of target (cryotherapy treated) lesions | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.36, 1.04] |
| 2.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.47, 1.60] |
| 2.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.50 [0.37, 0.68] |
| 3 Participant complete clearance of subclinical lesions | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 3.1 5% imiquimod | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Mean percentage of reduction in all lesion counts | 2 | 301 | Mean Difference (IV, Random, 95% CI) | ‐23.69 [‐46.03, ‐1.34] |
| 4.1 5% imquimod | 1 | 54 | Mean Difference (IV, Random, 95% CI) | ‐11.20 [‐26.53, 4.13] |
| 4.2 3.75% imiquimod | 1 | 247 | Mean Difference (IV, Random, 95% CI) | ‐34.10 [‐41.38, ‐26.82] |
| 5 Mean percentage of reduction in target (cryotherapy treated) lesion counts | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5.1 3.75% imiquimod | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Withdrawal due to adverse events | 2 | 312 | Risk Ratio (M‐H, Random, 95% CI) | 0.93 [0.28, 3.07] |
| 6.1 5% imiquimod | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 2.91 [0.12, 68.95] |
| 6.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.21, 2.79] |
| 7 Skin irritation | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 0.39 [0.10, 1.54] |
| 7.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 0.63 [0.20, 2.01] |
| 7.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 0.15 [0.02, 1.19] |
| 8 Minor adverse events excluding skin irritation: body as a whole: fatigue | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 9 Minor adverse events excluding skin irritation: digestive: nausea | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 10 Minor adverse events excluding skin irritation: musculoskeletal and connective tissue: myalgia | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 11 Minor adverse events excluding skin irritation: respiratory: upper respiratory tract infection | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 12 Minor adverse events excluding skin irritation: respiratory: bronchitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 13 Minor adverse events excluding skin irritation: respiratory: sinusitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 14 Minor adverse events excluding skin irritation: special senses: conjunctivitis | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 15 Cosmetic outcomes: Improved global photoageing score | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 16 Cosmetic outcomes: Improved fine lines | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 17 Cosmetic outcomes: Improved tactile roughness | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 18 Cosmetic outcomes: Improved mottled pigmentation | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 19 Cosmetic outcomes: Improved sallowness | 1 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 20 Cosmetic outcomes: cosmetic appearance score | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 20.1 Investigator | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 20.2 Participant | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
64.14.
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 14 Minor adverse events excluding skin irritation: special senses: conjunctivitis.
64.16.
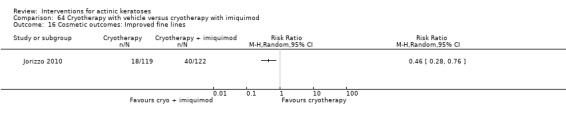
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 16 Cosmetic outcomes: Improved fine lines.
64.17.
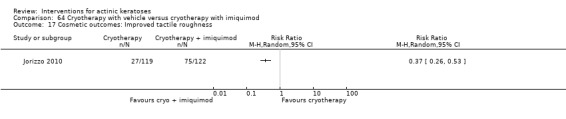
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 17 Cosmetic outcomes: Improved tactile roughness.
64.18.
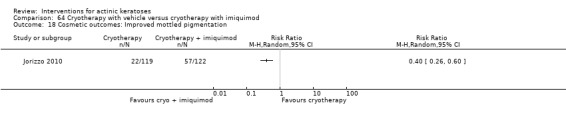
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 18 Cosmetic outcomes: Improved mottled pigmentation.
64.19.
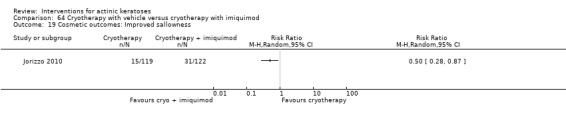
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 19 Cosmetic outcomes: Improved sallowness.
64.20.
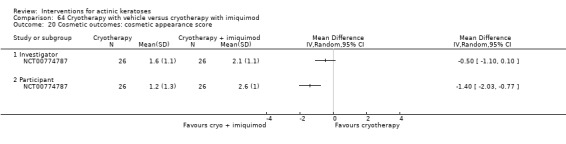
Comparison 64 Cryotherapy with vehicle versus cryotherapy with imiquimod, Outcome 20 Cosmetic outcomes: cosmetic appearance score.
65.
Cryotherapy with imiquimod versus cryotherapy with vehicle
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Participant complete clearance of all lesions | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 5.04 [1.37, 18.51] |
| 1.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 2.48 [0.70, 8.76] |
| 1.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 9.12 [3.36, 24.79] |
| 2 Mean percentage of reduction in all lesion counts | 2 | 301 | Mean Difference (IV, Random, 95% CI) | 23.69 [1.34, 46.03] |
| 2.1 5% imquimod | 1 | 54 | Mean Difference (IV, Random, 95% CI) | 11.20 [‐4.13, 26.53] |
| 2.2 3.75% imiquimod | 1 | 247 | Mean Difference (IV, Random, 95% CI) | 34.10 [26.82, 41.38] |
| 3 Withdrawal due to adverse events | 2 | 312 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.33, 3.56] |
| 3.1 5% imiquimod | 1 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 0.34 [0.01, 8.13] |
| 3.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 1.30 [0.36, 4.73] |
| 4 Skin irritation | 2 | 311 | Risk Ratio (M‐H, Random, 95% CI) | 2.55 [0.65, 10.04] |
| 4.1 5% imiquimod | 1 | 64 | Risk Ratio (M‐H, Random, 95% CI) | 1.60 [0.50, 5.13] |
| 4.2 3.75% imiquimod | 1 | 247 | Risk Ratio (M‐H, Random, 95% CI) | 6.72 [0.84, 53.83] |
66.
(3% diclofenac in 2.5% hyaluronic acid + ALA‐red light PDT) versus (2.5% hyaluronic acid + ALA‐red light PDT)
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global Improvement Indices (‐2 to 4) at 6 months | Other data | No numeric data | ||
| 2 Mean reduction of lesion counts | Other data | No numeric data |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | This was a randomised, active‐controlled, double‐blind, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 1% colchicine cream twice daily for 10 days + 10 more days if weak response (N = 8 participants) Control intervention B: 0.5% colchicine cream twice daily for 10 days + 10 more days if weak response (N = 8 participants) |
|
| Outcomes |
Outcomes of the trial 1) Complete healing (= participant complete clearance) 2) Reduction rate in number of actinic keratoses (= lesion complete response) at 1 month 3) Mean reduction of lesion counts at 1 month 4) Number of participants treated (pooled data) with strong, weak, or no inflammatory reaction 5) Minor adverse events (qualitative) 6) Number of participants with decreased infiltration and disappearance of crust (cosmetic) at 1 month 7) Clinical laboratory tests 8) Relapse at 6 months Efficacy Methods: quantitative assessment by counting visible and palpable actinic keratoses in each test area Time points: at baseline; end of treatment; 1, 2, and 6 months post‐treatment Safety Methods: 1. clinical examination, 2. routine laboratory tests (complete blood cell counts, urinalysis, and fasting chemistry) Time points: 1. each study visit (clinical exam), 2. before and after treatment (laboratory tests) |
|
| Funding | The drug was provided by Dr. F Frik Drug Company. | |
| Notes | Thick surface scales were removed by 10% salicylic acid 2 days before treatment. There were severe inflammation reactions in the majority of participants (11/16). In cases of inflammation, a weak antiseptic or antibiotic ointment was applied. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 200): "Patients were randomly assigned to treatment with 0.5% colchicine cream or 1% colchicine cream." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Both the investigators and the participants were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The investigators were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | An intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported even if there was no difference between treatment groups. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions | A 1‐month run‐in was performed during which participants used a placebo formulation (hydrophilic ointment) twice daily on both right and left forearms. Intervention A: 2‐(Difluoromethyl)‐dl‐ornithine (DFMO) twice daily for 6 months (N = 48 participants) Control intervention B: placebo twice daily for 6 months (N = 48 participants) |
|
| Outcomes |
Primary outcomes of the trial 1) Mean numbers of lesions at baseline and 6 months (the mean reduction in lesion counts was calculated) 2) Percentage reduction in the number of actinic keratoses 3) Skin concentrations of drug and products due to its mechanism of action at 6 months Secondary outcomes of the trial 1) Tolerance (qualitative) 2) Compliance Efficacy Methods: quantitative assessment by circling and counting of individual lesions on each arm by a dermatologist and photography using a Nikon N5005 camera with a 60‐mm Micor Nikkor lens, SB‐21 Macro Speedlight, and Kodachrome ASA 64 film Time points: at baseline and end of treatment Safety Methods: 1. assessment of clinical toxicity frequency and severity [scale 0 (none) to 3 (severe)] by the study dermatologist, 2. complete blood counts and serum chemistry panels (SMA20s) Time points: 1. before the first application of the placebo ointment, at randomisation, and at each monthly visit (toxicity), 2. run‐in and at the end of the study (laboratory tests) |
|
| Funding | This study was supported by USPHS Grant PO1 CA27502. | |
| Notes | There was no evidence of systemic toxicity. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page1282): "Before randomisation, participants were stratified on the basis of gender and numbers of actinic keratoses on the forearms. Participants were then randomly assigned, in a double‐blind fashion, to treatment with hydrophilic DFMO ointment on the right versus the left forearm and placebo hydrophilic ointment on the contralateral forearm twice daily for 6 months." Comment: Stratification was used for randomisation sequence generation. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 6 dropouts (the reasons were reported) Control ‐ B: 6 dropouts (the reasons were reported) Comment: The associated risk with PP analysis is unclear because the same number of participants were lost in both treatment groups. |
| Selective reporting (reporting bias) | High risk | The percentage reduction in lesion counts was given only for the DFMO‐treated group. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: isotretinoin 0.1% cream twice daily for 24 weeks (N = 50 participants?) Control intervention B: placebo cream twice daily for 24 weeks (N = 50 participants?) |
|
| Outcomes |
Outcomes of the trial 1) Investigators' evaluation of global therapeutic response (= global improvement indice‐completely cleared) 2) Mean number of actinic keratosis lesions over time by anatomical area 3) Mean reduction of lesion counts by anatomical area at end of treatment 4) Number of participants with severe, moderate, mild, or no local irritation on the face (= skin irritation) 5) Minor adverse events (qualitative) 6) Serious adverse events (including basal and squamous cell carcinoma) 7) Clinical laboratory tests Efficacy Methods: quantitative assessment by lesion counting and photography Time points: 1. at baseline and every 4 weeks (counting), 2. at baseline, week 12, and end of treatment (photography) Definitions for the global evaluation: 1. worsening (increase in lesions in treated area), 2. partial response (between 30% < 100% reduction in the number of lesions), and 3. complete response (total clearing) Safety Methods: 1. local tolerability was scored (absent to severe) by investigator, 2. clinical evaluation and reported adverse events, 3. routine laboratory tests Time points: 1. at each visit (tolerability and adverse events), 2. before and after treatment (laboratory tests) |
|
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 448): "Patients were randomly assigned to treatment with 0.1% isotretinoin or a color‐matched vehicle." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | 2 independent investigators counted lesions. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Modified intention‐to‐treat (ITT) analysis was used (i.e. participants with at least 1 postbaseline assessment were included in the analysis, N = 93), but the number of participants lost to follow up was higher than 20%. The numbers used for analysis were unclear. 1 participant in the isotretinoin group was missing in the data for skin irritation. Intervention ‐ A: 11 dropouts (the reasons were reported) Control ‐ B: 10 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | A poor efficiency was reported. |
| Other bias | High risk | The partial response criteria was very large (30% to 100%). Baseline mean withinparticipant differences in lesion number on the face was significantly different between treatment and control groups (P = 0.04). |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: December 2003 End date: December 2004 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod once per day 3 times per week, 4 weeks on, 4 weeks off (repeated if not cleared) (N = 129 participants) Control intervention B: vehicle, once per day 3 times per week, 4 weeks on, 4 weeks off (repeated if not cleared) (N = 130 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates at week 8 (1 treatment) and at week 16 (1or 2 treatments) Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates at week 16 2) Lesion complete response rates at weeks 8 and 16 Other outcomes of the trial 1) Participants experiencing at least 1 adverse event 2) Local skin reactions 3) Minor adverse events 4) Serious adverse events Efficacy Methods: 1. quantitative assessment using lesion counting and mapping with the use of a clear plastic template and photography, 2. histological confirmation using biopsy of a target lesion site Time points: 1. at week 1, week 2, end of treatment (EOT) (week 4 for 1 treatment, week 12 for 2 treatments), and the 4‐week post‐treatment visit (week 8 for 1 treatment, week 16 form 2 treatments), 2. pretreatment and 8‐week post‐treatment visit (week 12 for 1 treatment, week 20 for 2 treatments) (biopsy) Safety Methods: 1. assessment of the presence and intensity of specific local skin reactions by the investigator and rating on a scale [0 (none) to 3 (severe)], 2. safety evaluations including clinical laboratory tests (haematology, blood chemistry, and urinalysis) and urine pregnancy tests for women of child‐bearing potential, 3. physical examination including vital sign measurements, adverse events, and monitoring of concomitant medications Time points: 1. each visit (local skin reactions, adverse events, and medication monitoring), 2. pre‐study and poststudy (safety evaluations), 3. pre‐study, week 4 and week 12 visits (physical exam) |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | Rest periods were allowed in case of local skin reaction or treatment site adverse events. There were significant differences between imiquimod and vehicle groups in term of numbers and intensities of local skin reactions. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 134): "Eligible patients were randomised to either imiquimod 5% cream or vehicle cream in a 1:1 ratio." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blind and used 2 independent blinded dermatologists for histological evaluation. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used, and all subjects were accounted for. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 3 dropouts (the reasons were not all reported) |
| Selective reporting (reporting bias) | High risk | The participant complete clearance for the face and scalp was reported for the imiquimod group but not the vehicle group. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, double‐dummy, vehicle‐controlled, parallel‐group study. Start date: September 2006 End date: June 2007 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: once daily 0.025% ingenol mebutate gel (PEP005) for 3 days (N = 50 participants) B: once daily 0.05% ingenol mebutate gel (PEP005) for 2 days (N = 55 participants) C: once daily 0.05% ingenol mebutate gel (PEP005) for 3 days (N = 57 participants) Control intervention D: once daily vehicle for 3 days (N = 60 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Partial clearance rate (= participant partial clearance) Secondary outcomes of the trial 1) Complete clearance rate (= participant complete clearance for all lesions) 2) Baseline clearance rate (= participant complete clearance for target lesions) 3) Median percentage reduction of target lesions Other outcomes of the trial 1) Application site reactions 2) Local skin reactions overtime (pooled ingenol mebutate data and no data for vehicle) 3) Global severity rating of local reactions 4) Minor adverse events 5) Treatment‐related adverse events 6) Serious adverse events 7) Clinical laboratory tests 8) Cosmetic outcomes: pigmentation and scarring (pooled data) 9) Participants' satisfaction Efficacy Methods: quantitative assessment using counting of clinically‐visible lesions in the selected treatment area (including baseline and new lesions) Time points: at day 57 (end‐of‐study visit) Definitions: 1. partial clearance rate (proportion of participants with > 75% reduction in the number of lesions identified at baseline), 2. complete clearance rate (proportion of participants with no clinically‐visible lesions in the selected treatment area ‐ lesions present at baseline or emergent during the study period), 3. baseline clearance rate (proportion of participants with 100% reduction in the number of lesions identified at baseline), and 4. percentage reduction of the number of lesions (number of lesions present in the treatment area at baseline minus the number of lesions present at the end of the study divided by the number of lesions present at baseline) Safety Methods: 1. assessment of any local skin reactions and global severity rating, and monitoring of adverse events by a qualified dermatologist, 2. clinical laboratory tests Time points: 1. at day 3, follow‐up visits on days 8, 15, 29, and 57 (end‐of‐study visit); 2. at screening visit and on day 8 (laboratory tests) Cosmetic Methods: questionnaire with a 7‐point Likert scale, in which a score of 1 is very negative, 4 is neutral, and 7 is very positive |
|
| Funding | This study was supported by Peplin Ltd. | |
| Notes | Participants' satisfaction (P = 0.0005) and overall satisfaction (P < 0.001) were higher in the treatments groups compared with vehicle. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Each centre was allocated an initial block of 4 randomisation numbers and enrolled participants were assigned a participant number in ascending order. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The centre personnel and the participants were blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The investigator was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Modified ITT analysis was used (i.e. at least 1 dose and 1 postbaseline assessment). Intervention ‐ A: 0 dropouts, B: 1 dropout, C: 0 dropouts Control ‐ D: 0 dropouts |
| Selective reporting (reporting bias) | High risk | Data were pooled for safety and cosmetic outcomes. Based on the protocol NCT00375739, safety was supposed to be the primary outcome. However, efficacy data were presented first. |
| Other bias | Unclear risk | ‐ |
| Methods | The was a single‐centre, randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% 5‐fluorouracil twice daily on both arms for 2 to 4 weeks and 0.05% tretinoin nightly on 1 randomised arm for 2 to 4 weeks (N = 20 participants) Control intervention B: 5% 5‐fluorouracil twice daily on both arms for 2 to 4 weeks and placebo nightly on 1 randomised arm for 2 to 4 weeks (N = 20 participants) |
|
| Outcomes |
Outcomes of the trial 1) Mean number of actinic keratosis lesions at baseline and 3 months (mean reduction of lesion counts was calculated) 2) Relative irritation (skin irritation) Efficacy Methods: quantitative assessment using counting of residual actinic keratoses and biopsy of doubtful lesions Time points: at baseline and week 12 |
|
| Funding | The treatments were provided by Ortho Pharmaceutical Corporation and Hoffman‐LaRoche, Inc. | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 550): "Tretinoin 0.05% cream (RETIN‐A Cream, Ortho Pharmaceutical Corp., Raritan, NJ, U.S.A.) and a placebo cream, Eucerin (Beiersdorf Inc., Norwalk, CT, U.S.A.) were supplied to each patient in unmarked jars labelled only with the randomly assigned side to which the medication was to be applied." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It was unclear if the only lost participant was included or not in the analysis. Intraindividual study: Intervention ‐ A: 1 dropout (the reasons were reported) Control ‐ B: 1 dropout (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Based on the text, the data should have been presented as percentage of reduction in lesion count, but only the absolute counts at baseline and 3 months were presented. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: January 2002 End date: August 2002 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: imiquimod 5% cream once per day, 3 times per week for 3 weeks on, 4 weeks off (repeat once if 75% of lesions hadn't cleared) Control intervention B: placebo once per day, 3 times per week for 3 weeks on, 4 weeks off (repeat once if 75% of lesions hadn't cleared) |
|
| Outcomes |
Primary outcome of the trial 1) Participant partial (> 75%) clearance rates at 14 weeks Other outcomes of the trial 1) Mean number of actinic keratosis lesions overtime (graphical representation) 2) Participant complete clearance 3) Local skin reactions Efficacy Methods: quantitative assessment using counting of the number of lesions in the treatment area by the same investigator and photography Time points: at each weekly visit Safety Methods: reporting of local skin reactions [severity on a 0 (none) to 3 (severe) scale] and adverse events Time points: at each weekly visit |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | All adverse effects were gone at follow‐up. An increase in the number of lesions during treatment was observed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 252): "Randomisation codes were prepared using permuted blocks of four and stratifying by study centre." |
| Allocation concealment (selection bias) | Low risk | Quote (page 252): "Randomisation codes were concealed within opaque, sealed envelopes." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind, and randomisation codes were not revealed until all final assessments were completed. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was double‐blind, and randomisation codes were not revealed until all final assessments were completed. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol analysis was used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 1 dropout (the reasons were reported) 5 participants with protocol violation (4 imiquimod, 1 placebo) were excluded, and 1 with protocol violation was included (imiquimod not cured). |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, intraindividual study. Start date: July 2001 End date: March 2002 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention
A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 17 participants) Control intervention B: placebo‐PDT (N = 17 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage Cream concentration (%): ‐‐ Application of cream: 1 mm thick to lesional field and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: visible non‐coherent light Light source: Waldmann PDT 1200 Wavelength (nm): 600‐730 Energy fluence (J/cm²): 75 inten (mW/cm²): 80 Exposure time: ‐‐ Others: Each participant received 1 g paracetamol orally 1 hour before illumination. Additionally, a fan was used to cool the treated area and to reduce discomfort during illumination. |
|
| Outcomes |
Outcomes of the trial 1) Complete response rates of the lesional area (= participant complete clearance) at 16 weeks after 2nd treatment 2) Reduction in the number of lesions (= lesion complete response) at 16 weeks after 2nd treatment 3) Minor adverse events (qualitative) 4) Discomfort on a visual analogue scale (VAS) Efficacy Methods: quantitative assessment by inspection, photography, and palpation of the lesional area Time points: at 1, 4, 8, and 16 weeks after the 2nd treatment Definitions: 1. complete response (complete clinical regression of all lesions within the treated area), 2. partial response (incomplete reduction in size or number of the lesions within the treated area) Safety Methods: reporting of adverse events including the local phototoxicity reactions Time points: before and after illumination, and at 1, 4, 8, and 16 weeks after the 2nd treatment |
|
| Funding | ‐ | |
| Notes | There was no quantification of adverse events, but discomfort was reported higher for MAL than placebo. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 197): "Two lesional areas within a patient, measuring a maximum of 4 X 4 cm, were randomised to receive 2 consecutive treatments of topical PDT 1 week apart using either MAL or placebo cream." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was double‐blind, but because discomfort was higher with MAL than placebo cream, the blinding could have been broken. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study was double‐blind, but because discomfort was higher with MAL than placebo cream, the blinding could have been broken. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Low risk | The same data were reported in the abstract and published paper. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, intraindividual study. Start date: 2003 End date: 2004 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 3% diclofenac/2.5% hyaluronic acid twice daily for 90 days (N = 20 participants, 32 lesions) Control intervention B: 2.5% hyaluronic acid twice daily for 90 days (N = 20 participants, 32 lesions) |
|
| Outcomes |
Outcomes of the trial 1) Lesion complete response rates 2) Reduction in lesion size 3) Number of participants experiencing irritation (skin irritation) Efficacy Time points: at the end of treatment Definitions: 1. partial response (any reduction in the lesion size compared to baseline), 2. complete response (complete disappearance of the lesion) |
|
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 347): "Sixty‐four lesions of actinic keratosis in 20 patients were evaluated, 32 for active treatment and another 32 lesions with relatively similar characteristics but on the opposite side, as controls. Lesions were randomised to receive either 3% diclofenac in 2.5% hyaluronic acid gel or placebo (the inactive gel vehicle, hyaluronic acid only) 0.5 g twice daily in each 5 cm² treatment area for 90 days." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | The presentation of the data was confusing. The wording in the manuscript for the efficacy analysis created confusion between 'lesion complete response' and 'participant complete clearance', but based on the numbers and the percentages given, the outcome was lesion complete response. |
| Methods | This was a single‐centre, randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 12.5% DL‐α‐tocopherol (vitamin E) on right or left arm twice daily for 6 months (N = 48 participants) Control intervention B: placebo on right or left arm twice daily for 6 months (N = 48 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Biochemical and immunological outcomes Secondary outcome of the trial 1) Mean reduction of lesion counts Other outcome of the trial 1) Number of reports of symptoms (redness, itchiness, burning, dryness) Efficacy Methods: 1. quantitative assessment using circling of lesions and photographs, 2. shave biopsies by physician Time points: before and at the end of treatment Safety Methods: 1. physical exams, 2. clinical staff inquired about adverse events (severity, date of onset, duration, and date of resolution) Time points: 1. before treatment and at the end of treatment (physical exams), 2. at monthly visits (adverse events) |
|
| Funding | This study was supported by NIH grants CA‐27502 and CA‐23074. | |
| Notes | This was a phase IIb study. Vitamin E was well tolerated, i.e. only 14 reports for moderate and severe symptoms, which were similar for treatment and placebo. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A progressive randomisation program was used to make sure that allocation did not vary by gender, age, or actinic keratosis lesions (stratification). |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 6 dropouts (the reasons were reported) Control ‐ B: 6 dropouts (the reasons were reported) Comment: The associated risk with PP analysis is unclear because the same number of participants were lost in both treatment groups. |
| Selective reporting (reporting bias) | Low risk | No statistically different results were reported. |
| Other bias | High risk | Unprecise evaluation: 8 participants at baseline and 6 at the end of treatment had lesions too numerous to count and a number of 78 was used for analysis. |
| Methods | This was a multicentre, randomised, double‐blind, open, placebo‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 88 participants) Control interventions B: placebo ‐PDT: Placebo (N = 23 participants) C: cryotherapy: no prior preparation, variable liquid nitrogen spray unit, 1 to 2 mm rim of frozen tissue beyond marked outline, a single timed freeze‐thaw cycle; mean diameter < 10 mm = mean freeze time of 12 + 13 seconds, 10 to 20 mm = 16 + 15 seconds, > 20 mm = 26 + 11 seconds (N = 89 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage Cream concentration (%): 16 Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 50 to 250 Exposure time: 10 minutes |
|
| Outcomes |
Outcomes of the trial 1) Lesion complete response rates at 3 months 2) Participants experiencing at least 1 adverse event 3) Local skin/adverse reactions 4) Minor adverse events (given only for MAL‐PDT) 5) Cosmetic outcomes: overall and individual lesions at 3 months (MAL‐PDT vs cryotherapy) 6) Participant satisfaction Efficacy Methods: quantitative assessment using mapping with acetate sheets, marking of lesions and anatomical landmarks and Polaroid photography Time points: at 3 months after the beginning of treatment Definitions for lesion response: 1. complete response (complete disappearance of the lesion, both visually and by palpation), 2. non‐complete response (incomplete disappearance of the lesion) Safety Methods: adverse events reported by the participant or elicited through open (non‐leading) questioning by the investigator Time points: before, during, and after treatment; at 2 weeks by telephone contact; and at a final examination 3 months after treatment Cosmetic Methods: 1. overall cosmetic outcome of completely cleared participants (by investigator and participant), 2. individual lesion cosmetic outcome for completely cleared lesions (hypopigmentation, hyperpigmentation, scar formation and tissue defect rated as none, slight, or obvious) Time points: at 3 months after the beginning of treatment Definitions for overall outcome: 1. excellent (no scarring, atrophy or induration, and no or slight occurrence of redness or change in pigmentation compared with adjacent skin), 2. good (no scarring, atrophy or induration but moderate redness or change in pigmentation compared with adjacent skin), 3. fair (slight to moderate occurrence of scarring, atrophy, or induration), 4. poor (extensive occurrence of scarring, atrophy, or induration) |
|
| Funding | This study was supported by Photocure ASA, Olso, Norway. | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was performed per participant for each treatment option (first: PDT or cryo, second: MAL or placebo) and stratified by centre. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes were used to conceal the allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was double‐blind for the comparison between MAL‐PDT and placebo‐PDT and open for the comparison between MAL‐PDT and cryotherapy. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study was double‐blind for the comparison between MAL‐PDT and placebo‐PDT and open for the comparison between MAL‐PDT and cryotherapy. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Both intention‐to‐treat (ITT) and per‐protocol (PP) analyses were used, but only values for PP were presented and the authors only mentioned that results were similar for ITT analysis. Intervention ‐ A: 11 dropouts Control ‐ B: 4 dropouts, C: 3 dropouts Thus, there was less lost in cryotherapy (3.4%) than placebo‐PDT (17%) and MAL‐PDT (12.5%). |
| Selective reporting (reporting bias) | High risk | Types of adverse events were not reported separately for PDT and cryotherapy treatments, but more adverse events were reported for PDT than cryotherapy, and more for MAL‐PDT than placebo‐PDT. Risk of bias for more than 2 groups: cosmetic outcomes were not presented for the placebo‐PDT treatment group, and satisfaction was reported only for PDT participants. Similar data were presented in abstract form and published paper. |
| Other bias | High risk | There was a difference in baseline; the placebo PDT group included a greater proportion of men, and slightly more participants with skin type I and fewer with skin type 2. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: 1994 End date: 1995 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 0.25 g of 3% diclofenac in 2.5% hyaluronic acid gel twice daily for 12 weeks (N = 73 participants) Control intervention B: 2.5% hyaluronic acid gel alone twice daily for 12 weeks (N = 77 participants) |
|
| Outcomes |
Outcomes of the trial 1) Mean reduction of lesion counts at end of treatment and at 30 days post‐treatment 2) Participant complete resolution rates (= participant complete clearance) at end of treatment and at 30 days post‐treatment 3) Participant with 50% or greater reduction rates at end of treatment and at 30 days post‐treatment 4) Minor adverse events 5) Serious adverse events 6) Clinical laboratory tests 7) Compliance Efficacy Methods: quantitative assessment using lesion counting by a single doctor in each centre throughout the entire study Time points: at baseline, end of treatment (12 weeks), and at 16 weeks Definitions: 1. complete clearance (proportion of participants with complete resolution of lesions), 2. partial clearance (proportion of participants with a > 50% reduction in lesions) Safety Methods: 1. medical history and physical examination (baseline only), 2. haematology and biochemistry testing, 3. adverse events were recorded in the case report form [type (serious or non‐serious), onset date, severity (mild, moderate or severe), duration, any action taken, assumed relationship to treatment and outcome] Time points: at baseline, week 12 , and week 16 |
|
| Funding | This study was supported by Hyal Pharmaceutical Corporation. | |
| Notes | The safety assessment showed no difference between groups. A high compliance was observed for both groups. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 40):"They were randomly allocated to either active treatment (N = 73) or placebo (N = 77)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Intention‐to‐treat (ITT) analysis was used and 1 withdrawn participant was accounted for in the wrong treatment group (see below). Intervention ‐ A: 23 dropouts stated but 22 based on the details of the reasons Control ‐ B: 12 dropouts stated but 13 based on the details of the reasons. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: April 2000 End date: December 2000 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: 5% imiquimod (2 times/week) for 8 weeks (N = 31 participants) B: 5% imiquimod (3 times/week) for 8 weeks (N = 29 participants) C: 5% imiquimod (5 times/week) for 8 weeks (N = 30 participants) D: 5% imiquimod (7 times/week) for 8 weeks (N = 30 participants) Control intervention E: vehicle (2, 3, 5, 7 times/week) for 8 weeks (N = 29 participants, 7 to 8/dosing regimen pooled together) |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates at week 16 Secondary outcome of the trial 1) Participant partial (> 75%) clearance rates at week 16 Other outcomes of the trial 1) Application site reactions 2) Local skin reactions 3) Treatment‐related adverse events 4) Serious adverse events 5) Clinical laboratory tests 6) Compliance 7) Rest periods Efficacy Methods: 1. quantitative assessment using lesion counts within the target area performed by a qualified dermatologist using a transparent plastic template to track lesions, 2. qualitative assessment using lesion descriptions by investigator, i.e. degree of hyperkeratosis, size and confluence of the lesions, and degree of solar damage between lesions Time points: at baseline, week 4, and end of study (week 16) Definitions: 1. complete clearance rate (proportion of subjects at end of study with no lesions in the treatment area), 2. partial clearance rate (proportion of subjects at their last study visit with at least 75% reduction in lesions in the treatment area) Safety Methods: 1. recording of vital signs and adverse events, 2. assessment of defined local skin reactions (erythema, oedema, induration, vesicles, erosion, excoriation ⁄flaking, scabbing), 3. photography, 4. physical examination, 5. haematology and chemistry tests and pregnancy testing (adverse events were coded and summarised by body system and preferred term using a modified World Health Organization Adverse Reactions Terminology dictionary) Time points: 1. at weeks 1, 2, 3, 4, 6, 8 (end of treatment), 12, and 16 (8 weeks post‐treatment, end of study), 2. at baseline, end of treatment, and end of study (physical exam and laboratory tests) Definitions for grading: 1. mild (subject is aware of the signs and symptoms, but the signs and symptoms are easily tolerated), 2. moderate (signs and symptoms are sufficient to restrict, but not prevent, usual daily activity for the subject), and 3. severe (signs and symptoms are such that the subject is unable to perform usual daily activity) |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | The number of local skin reactions and adverse events increased with dosing frequency. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was double‐blind for intervention versus control but not for the frequency of application. 4 groups of vehicle were used to match the number of application and conceal treatment allocation, but there was not use of the vehicle to have all groups applying cream for 7 days/week, e.g. tubes labelled for each day of the week (with intervention or control) to conceal frequency allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was double‐blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 4 dropouts, B: 2 dropouts, C: 9 dropouts, D: 12 dropouts Control ‐ E: 1 dropout The imiquimod 5X/week and 7X/week groups lost 30% and 40% of participants, respectively. |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported even if efficacy was low. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: January 2008 End date: July 2008 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: 3.75% imiquimod, once daily for 3 weeks on, 3 weeks off, 3 weeks on (N = 162 participants) B: 2.5% imiquimod, once daily for 3 weeks on, 3 weeks off, 3 weeks on (N = 164 participants) Control intervention C: placebo, once daily for 3 weeks on, 3 weeks off, 3 weeks on (N = 164 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates at week 17 Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates at week 17 2) Median percentage of changes in lesion counts 3) Local skin reactions Other outcomes of the trial 1) Participants experiencing at least 1 adverse event 2) Application site reactions (including irritation) 3) Minor adverse events 4) Treatment‐related adverse events 5) Serious adverse events 6) Clinical laboratory tests 7) Investigator global integrated photodamage (IGIP‐cosmetic outcome) 8) Temporary treatment interruption Efficacy Methods: quantitative assessment using counting of all visible or palpable lesions ‐ baseline or new ‐ in the treatment area by the investigator Time points: baseline; at weeks 1, 2, 3 (end of cycle 1), 6 (beginning of cycle 2), 7, 8, 9 (end of cycle 2), 13, and 17 (end of study) (subjects who discontinued the study prematurely were requested to return for the end‐of‐study visit) Definitions: 1. complete clearance rate (proportion of subjects at the end‐of‐study visit with a count of zero lesions in the treatment area), 2. partial clearance rates (proportion of subjects with 75% or greater reduction in lesion count in the treatment area at the end‐of‐study visit as compared with baseline), and 3. percentage of changes in lesion count (per cent change in lesion number at the end‐of‐study visit as compared with baseline) Safety Methods: 1. measurement of vital signs, 2. recording of adverse events, 3. investigator assessment of local skin reactions (erythema, edema, weeping/exudate, flaking/scaling/dryness, scabbing/crusting, and erosion/ulceration) graded as none, mild, moderate, or severe, 4. hematology, serum chemistry, urinalyses, and urine pregnancy tests (treatment‐emergent adverse events were summarised for each treatment group by preferred term, intensity, and investigator assessment of relationship to study cream. The local skin reactions were summarised by the most intense score for each reaction and by the sum score at each visit and over the course of the study) Time points: 1. baseline, at weeks 1, 2, 3 (end of cycle 1), 6 (beginning of cycle 2), 7, 8, 9 (end of cycle 2), 13, and 17 (end of study), 2. pre‐study visit and end‐of‐study visit (laboratory tests) Cosmetic Methods: qualitative and quantitative assessment (IGIP score) Time points: at end‐of‐study visit Definition: IGIP score (overall assessment of the subject's photodamage change from baseline in the treatment area including an integrated assessment of fine wrinkling, coarse wrinkling, mottled pigmentation, roughness, sallowness, skin laxity, and telangiectasias) [the details on score were not provided, but both numerical results (score with standard deviation) and the number of participants with "significantly or much improved' cosmetic outcome were presented] |
|
| Funding | This study was supported by Graceway Pharmaceuticals. | |
| Notes | Data from 2 studies were pooled together. Temporary dosing interruptions could have been instructed by the investigator to manage local skin reactions and adverse events. 96% of subjects were compliant with dosing. A sample size calculation was provided. There was a follow‐up study published (Hanke 2011). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 575): "Eligible subjects were randomised to placebo, imiquimod 2.5%, or imiquimod 3.75% cream in a 1:1:1 treatment allocation." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was double‐blind, but authors mentioned that local effects of imiquimod may have led to investigator and subject bias. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study was double‐blind, but authors mentioned that local effects of imiquimod may have led to investigator and subject bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 10 dropouts, B: 7 dropouts Control ‐ C: 10 dropouts |
| Selective reporting (reporting bias) | Unclear risk | All outcomes from the protocol NCT00603798 were reported, but additional outcomes were also presented in the published paper (e.g. cosmetic). |
| Other bias | Unclear risk | Data for safety were reported differently in the published and the study results section of the protocol NCT00603798 in clinicaltrials.gov. |
| Methods | This was a single‐centre, randomised, active‐controlled, parallel‐group study. Start date: October 1, 2000 End date: October 30, 2002 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 2 passes of carbon dioxide laser resurfacing (N = 8 participants) Control interventions B: 30% trichloroacetic acid peel (N = 10 participants) C: 5% fluorouracil twice daily for 3 weeks (N = 9 participants) D: not randomised control group without treatment (data not presented and not included in our review) (N = 5 participants, 2 participants not included and reasons were given) |
|
| Outcomes |
Outcomes of the trial 1) Mean number of actinic keratosis lesions at baseline and 3 months (transformed to mean reduction in lesion counts) 2) Mean percentage of reduction of lesion counts at 3 months 3) Incidence of new non‐melanoma skin cancer for 5 years (4 groups) 4) Minor adverse events (qualitative) Efficacy Methods: quantitative assessment using the number and locations of existing lesions charted on a diagram of the head (at each visit, any lesions suggestive of basal or squamous cell carcinoma were biopsied. The VA Palo Alto Health Care System (VAPAHCS) medical and pathologic records were reviewed for each participant through June 30, 2005, to evaluate for any subsequent development of skin cancer in treated areas) Time points: at enrolment and every 3 months for a minimum of 24 months (at the end of the 24‐month study, participants continued routine general dermatology clinic surveillance) Definitions for rates of cancer formation: 1. cancer incidence rates (ratio of the total number of cancers to the total number of participant‐years followed in each group), 2. number of days from baseline/treatment to diagnosis of the first non‐melanoma skin cancer Safety Methods: monitoring for any adverse events Time points: at every 3 months for a minimum of 24 months |
|
| Funding | ‐ | |
| Notes | Every 3 months, new or remaining lesions were treated with cryosurgery. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 977): "Patients were prospectively randomised to 1 of 3 treatment arms..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was open because physically different treatments were used. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of assessor was not stated and physically different treatments were used. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The type of analysis was unclear. Intervention ‐ A: 2 dropouts (the reasons were reported) Controls ‐ B: 0 dropouts, C: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blinded, placebo‐controlled, parallel‐group study. Start date: March 2006, End date: December 2007 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 3 to 8 self‐adhesive patches of PD P506A (aminolevulinic acid ‐ ALA)‐photodynamic therapy (PDT) (N = 69 participants) Control intervention B: 3 to 8 self‐adhesive patches of placebo‐PDT (N = 34 participants) Characteristics of PDT intervention: Type of treatment: individual lesions Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: no Cream concentration (%): patches containing 8 mg Application of cream: self‐adhesive patch Incubation with cream: 4 hours Type of light: red light LED Light source: Aktilite CL 128 or Omnilux Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
|
| Outcomes |
Primary outcome of the trial 1) Complete clinical clearance rates on lesion basis (= lesion complete response) at 12 weeks post‐treatment Secondary outcomes of the trial 1) Participant complete clearance rates at 12 weeks post‐treatment 2) Adverse reactions at the treatment site (= application site reactions) during and the day after the treatment 3) Local skin/adverse reactions (presented for ALA‐PDT only) 4) Serious adverse events 5) Treatment‐related adverse events 6) Participant and investigator cosmetic outcomes of cleared lesions 7) Participant satisfaction Efficacy Methods: clinical diagnosis being regarded as usual procedure in dermatological practice Time points: at 12 weeks post‐treatment Definitions: complete clinical clearance of a lesion (no visual evidence of persisting lesion on treated surface; no evidence of adherent scaling plaques on treated skin surface when palpated; lesions no longer perceptible to touch; and slight pink or red foci might be visible at lesion sides) Safety Methods: 1. recording of local reactions by clinical staff, 2. a diary for the documentation of local reactions by participant during the 4 weeks after therapy, 3. blood samples for monitoring hepatic aminotransferases (alanine aminotransferase and aspartate aminotransferase) and ɣ‐glutamyltransferase, 4. documentation of adverse events Time points: 1. during patch application, illumination, and thereafter (local reactions), 2. before and day of treatment (blood tests), 3. each study visit (adverse events) Cosmetic Methods: 1. participants' and investigators' assessment of the cosmetic outcome of cleared lesions ('excellent', 'good', 'fair', or 'poor'), 2. participants' overall satisfaction with the cosmetic outcome ('very satisfied', 'satisfied', 'poorly satisfied', 'not satisfied') Time points: at 12 weeks post‐treatment |
|
| Funding | This study was supported by Photonamic GmbH & Co. | |
| Notes | The manuscript included 2 independent phase III studies (AK03 and AK04). This study was AK03. Adverse events were given for individual studies and pooled for ALA‐PDT, but pooled only for placebo‐PDT. Thus, pooled data were used for analysis (under Hauschild 2009a). A follow‐up study was published (Szeimies 2010a). Data for intention‐to‐treat analysis were used for the meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratification was performed by centre. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was double‐blind, and treatment was performed by a second investigator to guarantee an observer‐blinded status. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Modified ITT analysis was used (4 participants were not included, and the criteria were not specified but the numbers correspond to the following: ALA‐PDT: 1 missed control visit, 1 curettage before the study, 1 stop of illumination, and placebo‐PDT: 1 consent withdrawn). Intervention ‐ A: 17 dropouts (the reasons were reported) Control ‐ B: 8 dropouts (the reasons were reported) 24% of participants were lost before the end of study, but similar percentages were lost for both treatment arms. |
| Selective reporting (reporting bias) | High risk | Details on investigator cosmetic outcomes and adverse events for placebo group were not given. Outcomes in protocol (NCT00308854) were all presented in published paper. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, open, parallel‐group study. Start date: March 2006 End date: November 2007 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 4 to 8 self‐adhesive patches of PD P506A (aminolevulinic acid ‐ALA)‐ photodynamic therapy (PDT) (N = 148 participants) Control interventions B: 4 to 8 self‐adhesive patches of placebo‐PDT (N = 49 participants) C: cryosurgery: nozzles of size C, 1 cycle and freeze time between 5 and 10 seconds (N = 149 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: no Cream concentration (%): patches containing 8 mg Application of cream: self‐adhesive patch Incubation with cream: 4 hours Type of light: red light LED Light source: Aktilite CL 128 or Omnilux Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
|
| Outcomes |
Primary outcome of the trial 1) Complete clinical clearance rates on lesion basis (= lesion complete response) at 12 weeks post‐treatment Secondary outcomes of the trial 1) Participant complete clearance rates at 12 weeks post‐treatment 2) Adverse reactions at the treatment site (= application site reactions) during and the day after the treatment 3) Local skin/adverse reactions (presented for ALA‐PDT only) 4) Serious adverse events 5) Treatment‐related adverse events 6) Participant and investigator cosmetic outcomes of cleared lesions 7) Participant satisfaction Efficacy Methods: clinical diagnosis being regarded as usual procedure in dermatological practice Time points: at 12 weeks post‐treatment Definitions: complete clinical clearance of a lesion (no visual evidence of persisting lesion on treated surface; no evidence of adherent scaling plaques on treated skin surface when palpated; lesions no longer perceptible to touch; and slight pink or red foci might be visible at lesion sides) Safety Methods: 1. recording of local reactions by clinical staff, 2. a diary for the documentation of local reactions by participant during the 4 weeks post‐treatment, 3. blood samples for monitoring hepatic aminotransferases (alanine aminotransferase and aspartate aminotransferase) and ɣ‐glutamyltransferase, 4. documentation of adverse events Time points: 1. during patch application, illumination, and thereafter for PDT, and during the spraying procedure and thereafter for cryotherapy (local reactions), 2. before and day of treatment (blood tests), 3. each study visit (adverse events) Cosmetic Methods: participants' and investigators' assessment of the cosmetic outcome of cleared lesions ('excellent', 'good', 'fair', or 'poor') Time points: at 12 weeks post‐treatment |
|
| Funding | This study was supported by Photonamic GmbH & Co. | |
| Notes | The manuscript included 2 independent phase III studies (AK03 and AK04). This study was AK04. Adverse events were given for individual studies and pooled for ALA‐PDT, but only pooled for placebo‐PDT. Thus, pooled data were used for analysis for ALA‐PDT vs placebo‐PDT (under Hauschild 2009a). A follow‐up study was published (Szeimies 2010a). Data for intention‐to‐treat analysis were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratification was performed by centre. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was open because the treatments were physically distinct. Similar patches were used for ALA‐PDTand placebo‐PDT, but no concealment was possible for the physically distinct treatments, i.e. PDT versus cryotherapy. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study was open. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 19 dropouts (the reasons were reported) Controls ‐ B: 6 dropouts (the reasons were reported), C: 23 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Details on investigator cosmetic outcomes and adverse events for placebo group were not given. Outcomes in protocol (NCT00308867) were all presented in published paper. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, assessor‐blinded, active‐controlled, parallel group study. Start date: January 2005 End date: July 2005 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: 1 hour of PD P506A (aminolevulinic acid (ALA) self‐adhesive patch)‐photodynamic therapy (PDT) (N = 38 participants) B: 2 hours of PD P506A‐PDT (N = 34 participants) C: 4 hours of PD P506A‐PDT (N = 34 participants) Control intervention D: 0.5 hour of PD P506A‐PDT (N = 34 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: no Cream concentration (%): patches containing 8 mg Application of cream: self‐adhesive patch Incubation with cream: 0.5 to 4 hours Type of light: red light Light source: Aktilite CL 128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
|
| Outcomes |
Primary outcome of the trial 1) Lesion complete response rates at 4 and 8 weeks Secondary outcomes of the trial 1) Participant complete clearance rates at 4 and 8 weeks 2) Local skin reactions presented graphically for 3 periods (during ALA patch application, during illumination, and after illumination) as well as by severity of the reactions 3) Treatment‐related adverse events (minor adverse events) 4) Minor adverse events (pooled) 5) Serious adverse events 6) Clinical laboratory tests 7) New actinic keratoses Efficacy Methods: clinical diagnosis, the usual procedure in dermatological practice Time points: at 4 and 8 weeks after PDT Definitions: complete clinical clearance of a lesion (no visual evidence of persisting lesions on treated surface, no evidence of adherent scaling plaques on treated skin surface when palpated, lesions no longer perceptible to touch, slight pink or red foci might be visible at lesion sides) Safety Methods: 1. inspection of study lesions for tolerability, 2. recording of local reactions and adverse events (local reactions were always assumed to be related to study therapy. For adverse events, the investigator judged the relation to the study therapy) Time points: 1. at 1 day and 1 week after PDT (tolerability), 2. entire study duration (local reactions and adverse events) |
|
| Funding | This study was supported by Photoamic. | |
| Notes | Percentages of participants with local skin reactions were given graphically. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 118): "Patients were randomly allocated to treatment." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was no stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | To keep the assessor blinded, 1 investigator performed the evaluation, and another investigator administered the treatments. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used. Lost participants were mentioned (9) but not by treatment group and the reasons were not given. |
| Selective reporting (reporting bias) | High risk | Adverse events were not always clearly reported by group, and the number of participants included in the safety analysis was not clear. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, active‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: betulin‐based oleogel applied twice daily (N = 15) B: combination therapy with initial cryotherapy followed by betulin‐based oleogel twice daily (N = 15) Control intervention C: cryotherapy in the form of a spray coat method with liquid nitrogen (20 to 40 seconds) (N = 15) |
|
| Outcomes |
Outcomes of the trial 1) Complete clearing (= participant complete clearance) rates at 3 months 2) Therapy responders with > 75% of clearing of the lesions (= participant partial clearance) rates at 3 months 3) Histological analysis of biopsies before treatment and at the end of treatment 4) Minor adverse events (qualitative) Efficacy Methods: 1. quantitative assessment using documentation of visual and photographic evaluation in the case report forms, 2. punch biopsies from 4 participants out of the oleogel group, 2 participants out of the cryotherapy group and 2 participants out of the combination therapy group for evaluation of the degree of dysplasia, number of dyskeratoses and thickness of epidermis and stratum corneum Time points: 1. at 1, 2, and 3 months after the beginning of treatment, 2. before treatment and at the end of treatment (biopsy) Definitions: 1. responders [participants with complete (100 %) and with extensive (> 75 %) total clearing of the lesions], and 2. non‐responders (participants with disappearance of < 75 % of the lesions) Safety Methods: assessment of the subjective parameters itching and stinging by a questionnaire and grading as follows: 0 = absent, 1 = mild, 2 = moderate, and 3 = severe |
|
| Funding | This study was supported by Birken GmbH. | |
| Notes | This study was a Phase II pilot study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation plan was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This study was open. This study did not use placebo cream to conceal the allocation to cryotherapy only versus cryotherapy with betulin‐based oleogel. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This study was open. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 1 dropout (the reason was reported), B: 1 dropout (the reason was reported) Control ‐ C: 1 dropout (the reason was reported) Comment: The effect of PP analysis is difficult to assess because the same number of participants was lost in each treatment group. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, assessor‐blinded, vehicle‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 36 participants) Control intervention B: vehicle‐PDT (N = 36 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 (if complete response not achieved) Interval between treatments: 8 weeks Preparation of lesions: ‐‐ Cream concentration (%): 20% Application of cream: 3 applications onto lesion and a rim of 2 to 4 mm, air dry between applications Incubation with cream: 14 to 18 hours Type of light: blue light Light source: DUSA BLU‐417 Wavelength (nm): 417 Energy fluence (J/cm²): 2, 5, or 10 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
|
| Outcomes |
Outcomes of the trial 1) Clinical response of actinic keratosis lesions including completely cleared (= lesion complete response) rates for individual (at 8 weeks) and all (at 8 and 16 weeks) light doses 2) Number of participants with 0, 1, 2 (all) cleared lesions (= participant complete clearance) for individual (at 8 weeks) and all [at 8 (ALA and placebo) and 16 (ALA only) weeks] light doses 3) Application site reactions during (illumination) and after treatment reported per lesions 4) Clinical laboratory tests 5) Changes in pigmentation (cosmetic) per lesions 6) PpIX fluorescence Efficacy Time points: at baseline; immediately after PDT; at 24 and 72 hours; and at weeks 1, 4, 8, 9 (retreated participants), 12, and 16 Definitions: 1. complete response (completely cleared with no evidence of adherent scale on the surface of the treated skin when palpated), 2. partial response (≥ 50% reduction in lesion size), and 3. no response (< 50% reduction in lesion size) Safety Methods: 1. evaluation of objective changes in erythema, oedema, wheal, vesiculation, ulceration, haemorrhage, and necrosis on a graded scale (0: none; 1: minimal; 2: moderate; 3: severe), 2. subjective assessment of participant discomfort from pain, burning/stinging, and itching was graded (0: none; 1: minimal; 2: moderate; 3: severe), 3. standard hematologic and biochemical laboratory parameters, 4. reporting of adverse events Time points: 1. at baseline; immediately after PDT; at 24 hours; at 72 hours; and at weeks 1, 4, 8, 9 (retreated participants), 12, and 16, 2. at baseline and again at 1 week post‐treatment (laboratory tests) |
|
| Funding | This study was supported by DUSA pharmaceuticals, Inc. | |
| Notes | Clinical response was dependant upon dose of light administered (5 or 10 J/cm² were more effective than 2 J/cm²). Visual detection of PpIX confirmed absence of cross contamination of treatment and placebo creams. Grade 1 (30/39 = 77%) lesions had better response than grade 2 (16/31 = 52%). PpIX fluorescence significantly correlated with clinical response (P < 0.001). Only data from 8 week's visit was used, because data from 16 weeks included participants with or without additional treatment. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 97): "Each patient had a minimum of 4 target lesions with 2 lesions being randomised to ALA solution and 2 to vehicle." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | A non‐blinded investigator performed treatments. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Different investigators were involved for the treatment and the analysis. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intraindividual study: Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, open (treatment duration), vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: 0.5% 5‐fluorouracil, once daily for 1 week (N = 47 participants) B: 0.5% 5‐fluorouracil, once daily for 2 weeks (N = 46 participants) C: 0.5% 5‐fluorouracil, once daily for 4 weeks (N = 45 participants) Control intervention D: Vehicle, once daily for 1, 2, or 4 weeks (pooling not clear ‐ see page 336 of the study) (N = 69 participants) |
|
| Outcomes |
Primary outcomes of the trial 1) Physician Global Assessment of Improvement (PGAI = Global improvement indices) 2) Per cent reduction of lesions (= mean percentage of reduction in lesion counts) 3) Absolute mean reduction in lesion counts Other outcomes of the trial 1) Proportion of participants achieving total clearance (= participant complete clearance) 2) Skin irritation (percentages of participants, severity, overtime) 3) Application and local skin reactions and minor adverse events (pooled data from 2 studies included in Carac product insert, i.e. Jorizzo 2002 and Weiss 2002) 4) Serious adverse events Efficacy Methods: 1. quantitative assessment using lesion counting, 2. qualitative assessment (PGAI mean score, +5 = total clearance and ‐4 = much worse) Time points: at baseline and 4 weeks post‐treatment Safety Methods: 1. adverse events (including details of facial irritation): maximum severity, symptoms, onset and overall duration, post‐treatment duration, and summary by visit, 2. facial irritation graded as 0 = none, 1 = mild, 2 = moderate, or 3 = severe (to ensure that all facial irritation was recorded, the last observed value for facial irritation was carried forward if a participant discontinued treatment for any reason) |
|
| Funding | This study was supported by Dermik Laboratories. | |
| Notes | Data from this study were included in the Carac product insert. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 336): "Of the 207 randomised participants,..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This study was double‐blind (treatment vs placebo) and open (treatment duration). No placebo cream was used to conceal the treatment duration. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This study was double‐blind (treatment vs placebo) and open (treatment duration). No placebo cream was used to conceal the treatment duration. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | It was unclear which type of analysis was used. Intervention ‐ A: 2 dropouts, B: 1 dropout, C: 1 dropout Control ‐ D: 0 dropouts (the reasons were not reported) |
| Selective reporting (reporting bias) | High risk | Values for absolute reductions in lesion numbers, standard deviations on mean percentages, and PGAI were not given. Details on local skin reactions and adverse events, other than facial irritation, was not reported in the published version of the study. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel group study. Start date: October 2001 End date: February 2002 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: topical 0.5% 5‐fluorouracil, once daily for 7 days. At 4 weeks post‐treatment, residual lesions treated with cryotherapy. (N = 72 participants) Control intervention B: vehicle, once daily for 7 days. At 4 weeks post‐treatment, residual lesions treated with cryotherapy (N = 72 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Absolute and per cent of mean reduction in lesion counts at 4 weeks (topical only) and 6 months (topical + cryotherapy) Other outcomes of the trial 1) Participant complete clearance rates at 4 weeks and 6 months 2) Application site reactions (also reported in Jorizzo 2006) 3) Eye irritation (= minor adverse events) Efficacy Methods: quantitative assessment using the counting of visible or palpable lesions, or both, by the same evaluator Time points: before initial treatment and at the 4‐week and 6‐month follow‐up visits Safety Methods: 1. recording of severe application site reactions (erythema, edema, dryness, pain, erosion, burning, and pruritus), 2. eye irritation (burning, sensitivity, itching, stinging, and watering), 3. any other adverse events Time points: at each visit |
|
| Funding | This study was supported by Dermik Laboratories. | |
| Notes | This study (interim analysis) is part of a 3‐cycle study published in Jorizzo 2006, which was also included in this review. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. All study personnel, and participants were blinded to actual treatment assignment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. Investigators were blinded to actual treatment assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Modified Intention‐to‐treat (ITT) analysis was used (i.e. participants at 4 week evaluation based on information, efficacy: 142, safety: 143‐received one treatment). Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 7 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 3 topical/cryosurgery cycles: topical 0.5% 5‐fluorouracil, once daily for 7 days. At 4 weeks post‐treatment, residual lesions were treated with cryosurgery (N = 72 participants) Control intervention B: 3 topical/cryosurgery cycles: topical vehicle, once daily for 7 days. At 4 weeks post‐treatment, residual lesions were treated with cryosurgery (N = 70 participants) |
|
| Outcomes |
Primary outcomes of the trial 1) Mean lesion counts at baseline and different treatment cycles (transformed to mean reduction in lesion counts) 2) Mean percentage of reduction in lesion counts 3) Participant complete clearance rates at 3 topical/cryosurgery cycles (before cryosurgery) 4) New involvement of actinic keratosis lesions Other outcomes of the trial 1) Application site reactions (severe) 2) Treatment‐related adverse events (= minor adverse events) 3) Serious adverse events including basal and squamous cell carcinoma Efficacy Methods: quantitative assessment using the counting of visible or palpable actinic keratoses, or both, by the same evaluator Time points: before initial treatment and at the follow‐up visits Definitions: 1. actinic keratosis reduction (number of lesions present at the 4‐week follow‐up visit for each topical/cryosurgery cycle minus the number of baseline lesions for that cycle), 2. clearance (complete lack of lesions in the treatment area at the 4‐week follow‐up visit for each topical/cryosurgery cycle), 3. new involvement [presence of new lesions in the treatment area at the start of topical/cryosurgery cycle 2 (week 26) or 3 (week 52)] Safety Methods: 1. monitoring the incidence, onset, duration, and severity of adverse events observed, 2. participants reporting the occurrence of any adverse event, and study personnel questioned participants during study visits to monitor safety, 3. all adverse events were categorised by the investigator as serious or not Time points: from time of enrolment to the end of the follow‐up period Definitons: serious adverse events [adverse events resulting in death, life threatening; required inpatient hospitalisation or prolongation of existing hospitalisation, resulted in persistent or significant disability or incapacity, those described as important medical events (e.g. diagnosis cancer during the course of treatment)] |
|
| Funding | This study was supported by Dermik Laboratories. | |
| Notes | More participants in the vehicle group had cryosurgery. New actinic keratosis lesions were observed over time in both groups, but a lower percentage of participants in the 5‐fluorouracil group than the vehicle group was obtained. The number of participants based on ITT analyses were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. All study personnel and participants were blinded to the actual treatment assignment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. Investigators were blinded to the actual treatment assignment. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was stated. Intervention ‐ A: 5 dropouts (the reasons were reported) Control ‐ B: 12 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Standard deviations on percentage of mean reduction in lesion counts were not reported. |
| Other bias | High risk | There was some inconsistency in the numbers in the manuscript as well as with the previous paper, Jorizzo 2004, for efficacy outcomes. |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod, once per day, 3 days per week for 4 weeks on, 4 weeks off, 1 or 2 courses (N = 123 participants) Control intervention B: vehicle: once per day, 3 days per week for 4 weeks on, 4 weeks off, 1 or 2 courses (N = 123 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Recurrence at 1 year Other outcomes of the trial 1) Participant complete and partial (> 75%) clearance rates after course 1 or overall 2) Individual lesion clearance (= lesion complete response) rates after course 1 or overall 3) Minor adverse events (qualitative) 4) Clinical laboratory tests Efficacy Methods: quantitative assessment using lesion counting Time points: at week 8, 16, and 1 year follow‐up (relapse for participants achieving complete clearance) Definitions: 1. complete clearance rate (proportion of participants who cleared all lesions in the treatment area), 2. partial clearance rate (proportion of participants with at least a 75% reduction in baseline lesions) Safety Methods: 1. monitoring for adverse events and local skin reactions, 2. clinical laboratory tests (hematology and chemistry blood tests, and urinalysis), 3. culture of suggested skin infections |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | 1‐year recurrence rate of 39% and 57% were respectively found for imiquimod and vehicle. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 266): "Randomised patients applied imiquimod or vehicle cream (randomised 1:1)..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Authors reported that blinded investigators may have been biased toward participants treated with imiquimod identified by treatment site reactions. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 4 dropouts (the reasons were not reported) Control ‐ B: 3 dropouts (the reasons were not reported) |
| Selective reporting (reporting bias) | High risk | Statistically significantly more application site reactions, local skin reactions, and adverse events for imiquimod were reported but not all the numbers supporting it were reported. |
| Other bias | High risk | There was no demographic description. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel group study. Start date: May 2009 End date: February 2010 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: cryotherapy followed by 3.75% imiquimod, daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 126 participants) B: no cryotherapy followed by 3.75% imiquimod, daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 126 participants) Control interventions C: cryotherapy followed by placebo, daily for 2 weeks on, 2 weeks off, 2 weeks on (N =121 participants) D: no cryotherapy followed by placebo, daily for 2 weeks on, 2 weeks off, 2 weeks on (N =121 participants) Randomisation was performed for imiquimod or placebo treatment, but the method used to select which lesions were treated with or without cryotherapy was not specified. At least 5 lesions were not treated with cryosurgery, and 5 to 14 actinic keratoses were treated with cryosurgery. |
|
| Outcomes |
Primary outcome of the trial 1) Mean and median per cent changes from baseline for all lesions (= mean percentage of reduction in lesion counts) at week 26 Secondary outcomes of the trial 1) Participant complete clearance rates for all lesions at week 26 2) Local skin reactions (severe) Other outcomes of the trial 1) Application site reactions including irritation 2) Treatment‐related adverse events (= minor adverse events) 3) Minor adverse events 4) Serious adverse events including basal and squamous cell carcinoma 5) Cosmetic outcomes (photodamage) 6) Rest periods 7) Participant satisfaction Efficacy Methods: quantitative assessment using lesion counting and mapping on a facial diagram, including lesions that were initially treated with cryosurgery Time points: 1. at each visit (counting), 2. baseline and end of study at week 26 (mapping) Safety Methods: investigator assessment of local skin reactions (erythema, oedema, weeping/exudate, flaking/scaling/dryness, scabbing/crusting, and erosion/ulceration) graded as none, mild, moderate, or severe and summarised by the most intense score for each reaction, 2. recording of adverse events coded using MedORA® (Medical Dictionary for Regulatory Activities), Version 12.0 (treatment‐emergent AEs were summarised for each treatment group by preferred term, intensity, and investigator assessment of relationship to study cream), 3. serious adverse events and discontinuations due to adverse events Time points: at each clinic visit Cosmetic Methods: assessment of facial photodamage by the investigator using previously published 5‐point scales that rated fine lines, mottled pigmentation, tactile roughness, sallowness, and global photoageing Time points: at baseline (prior to cryosurgery) and at weeks 10, 14, 20, and 26/end of study |
|
| Funding | This study was supported by Graceway Pharmaceuticals, LLC. | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated allocation sequence was generated. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was double‐blind for imiquimod versus placebo (subject, caregiver, investigator). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study was double‐blind for imiquimod versus placebo (outcomes assessor). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A & B: 14 dropouts (the reasons were reported) Controls ‐ C & D: 10 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | All outcomes from the protocol were reported, and the data were consistent in conference abstract, published paper, and the study results section of the protocol (NCT00894647) in clinicaltrials.gov. Additonal outcomes were reported in the published report. The data for "no cryotherapy" was not given for all outcomes. Cosmetic outcomes were reported separately for each criteria but were not reported as a global assessment. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, placebo‐controlled, active‐controlled, assessor‐blinded, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: 0.1% adapalene gel, once daily for 4 weeks; twice daily from 4 weeks to 36 weeks (N = 30 participants) B: 0.3% adapalene gel, once daily for 4 weeks; twice daily from 4 weeks to 36 weeks (N = 30 participants) Control intervention C: placebo, once daily for 4 weeks; twice daily from 4 weeks to 36 weeks (N = 30 participants) |
|
| Outcomes |
Primary outcomes of the trial 1) Mean reduction/changes of lesion counts 2) Morphological changes of target lesions Secondary outcomes of the trial 1) Physician global assessment improvement (PGAI) from worse to clear (= Global Improvement Indices) 2) Histological analysis of biopsies before and after treatment Other outcomes of the trial 1) Tolerability (= local skin reactions) 2) Minor adverse events 3) Serious adverse events 4) Photoaging characteristic improvement (cosmetic outcome) Efficacy Methods: 1. quantitative assessment using total lesion counts, 2. assessment of morphologic changes in target actinic keratoses (induration, scaling, and erythema evaluated on a scale of 0 [none] to 3 [severe]), 3. qualitative assessment of improvement (PGAI) as clear, marked, moderate, slight, no change, or worse, 4. biopsy specimens evaluated in a blinded manner by a board‐certified dermatopathologist at 1 centre (University of Michigan, Ann Arbor, Michigan) Time points: at week 2, 4, 12, 18, 24, 30, and 36 Cosmetic Methods: standardised photographs were taken of 45 participants by a professional photographer at 1 centre (University of Michigan). [The photographs were evaluated retrospectively in a randomised, blinded fashion to assess the effects of the 3 treatments on photoageing characteristics (mottled hyperpigmentation, fine wrinkles, coarse wrinkles, rosy glow‐healthy pink complexion, and global photoageing severity). Each parameter was graded for improvement on a scale of 0 to 6. If there was no difference or worsening between before‐ and after‐treatment photographs, then a score of 0 was given] Time points: at baseline and after 3, 6, and 9 months of treatment |
|
| Funding | This study was supported by Galderma Corporation, Texas, US. | |
| Notes | Histology on 36 participants showed no significant difference between treatment groups. There was no follow‐up period. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation sequence in blocks of 9 using a unique 4‐digit number was generated by a computer program. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 2 dropouts (the reasons were not reported), B: 3 dropouts (the reasons were reported) Control ‐ C: 2 dropouts (the reasons were not completely reported) |
| Selective reporting (reporting bias) | High risk | Authors pooled together selected PGAI data, i.e. for clear, marked, moderate (but not slight) improvement to reach statistically significant difference. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, open, active‐controlled, intraindividual study. Start date: January 2005 End date: February 2006 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 121 participants) Control intervention B: cryotherapy: double freeze/thaw (1 to 2 mm frozen rim outside marked outline of lesion), 20 seconds (1 or 2 treatments with a 12‐week interval) (N = 121 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 12 weeks Preparation of lesions: gentle scraping Cream concentration (%): 16 % Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: 3 hours Type of light: red light LED Light source: Aktilite CL128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
|
| Outcomes |
Primary outcome of the trial 1) Lesion complete response rates for baseline lesions at week 24 Secondary outcomes of the trial 1) Cosmetic outcome assessed by investigator and participants 2) Participant preference Other outcomes of the trial 1) Mean percentage of reduction in lesion counts 2) Treatment‐related adverse events (= minor adverse events) 3) Participants reporting at least one adverse event 4) Minor adverse events 5) Serious adverse events including squamous cell carcinoma 6) Observation of Bowen's disease and new actinic keratoses Efficacy Methods: quantitative assessment using lesion counting [efficacy evaluations only included lesions present at baseline (i.e. lesions appearing after baseline, if any, were to be reported as an adverse event)] Time points: at baseline and weeks 12 and 24 Definitions for lesion response: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance) Safety Methods: spontaneous reporting of adverse events by the participant or elicited following non‐leading questioning (severity, duration and need for additional therapy) Time points: at each follow‐up visit Definitions for adverse events: 1. phototoxic reaction (any observed erythema, oedema, itching, pain, etc), 2. cryotherapy reaction (any observed blisters, infection, etc) Cosmetic Methods: 1. investigator assessment of cosmetic outcome for all lesions with complete response, 2. participant assessment of cosmetic outcome Time points: at week 24 Definitions for investigator assessment: 1. excellent (only slight occurrence of redness or change in pigmentation), 2. good (moderate redness or change in pigmentation), 3. fair (slight to moderate scarring, atrophy or induration), 4. poor (extensive scarring, atrophy, or induration) Definitions for participant assessment on a 5‐point scale: –2 (cryotherapy a lot better than MALPDT) to 2 (MAL‐PDT a lot better than cryotherapy) |
|
| Funding | This study was supported by Galderma. | |
| Notes | A participant questionnaire showed that participants preferred MAL‐PDT over cryotherapy for all questions except for effectiveness of treatment. A sample size calculation was provided. Intention‐to‐treat values were used for meta‐analyses. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 995) : "At the baseline visit, eligible patients received treatment with PDT using MAL... and conventional cryotherapy, randomly allocated to alternate sides of the body." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | There was no blinding. This study was open because physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | There was no blinding. This study was open because physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Both per‐protocol (PP) and intention‐to‐treat (ITT) analyses were used. Intraindividual study: Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) Comment: ITT and PP populations were respectively 121 and 106. Thus, 11 participants were not accounted for, and it was not always clear which analysis type was used for the different outcomes reported. |
| Selective reporting (reporting bias) | High risk | The standard deviation associated with the mean percentage of reduction in lesion counts were not provided. |
| Other bias | High risk | Participant's assessment of cosmetic outcomes has negative value if cryotherapy is better and positive value if MAL‐PDT is better. This could influence the participant perception. |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: August 2001 End date: August 2002 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod applied 3 times per week for 16 weeks (N = 242 participants) Control intervention B: vehicle cream applied 3 times per week for 16 weeks (N = 250 participants). Applied to entire treatment area at same time of day (before sleeping). Cream to remain in place for approximately 8 hours. Rest periods allowed at discretion of investigator, but did not alter length of 16‐week treatment |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates for all lesions at 8 weeks post‐treatment Secondary outcome of the trial 1) Participant partial (> 75%) clearance rates for all lesions at 8 weeks post‐treatment Other outcomes of the trial 1) Median percentage reduction of baseline lesions at 8 weeks post‐treatment 2) Participants experiencing at least 1 adverse event 3) Application site reactions 4) Local skin reactions 5) Frequency and severity of adverse events (= minor adverse events) 6) Serious adverse events 7) Rest periods 8) Skin quality (cosmetic outcome) Efficacy Methods: quantitative assessment using lesion counting (baseline and new lesions) Time points: at weeks 4, 8, and 16, and post‐treatment week 8 Definitions: 1. complete clearance rate (proportion of participants at the 8‐week post‐treatment visit with no clinically‐visible lesions in the treatment area), 2. partial clearance rate (proportion of participants at the 8‐week post‐treatment visit with at least a 75% reduction in the number of baseline lesions in the treatment area) Safety Methods: 1. reviewing concomitant medication use, 2. assessing the incidence and severity of adverse events spontaneously reported, 3. assessing of local skin reactions (erythema, edema, erosion/ulceration, scabbing/crusting, weeping/exudation, vesicles, and flaking/scaling/dryness) rated as 0 = none, 1= mild, 2 = moderate, and 3 = severe by investigator (reactions within the treatment area that were not assessed as local skin reactions were reported as adverse events) Time points: at weeks 1, 2, 4, 6, 8, 10, 12, 14, and 16, and post‐treatment weeks 4 and 8 Cosmetic Methods: investigator‐performed (visual, clinical, and tactile examinations) skin quality assessments including skin surface (roughness/dryness/scaliness), hyperpigmentation, hypopigmentation, mottled or irregular pigmentation (hyperpigmentation and hypopigmentation), degree of scarring, and degree of atrophy area rated as 0 = none; 1 = mild; 2 = moderate; and 3 = severe Time points: at the treatment initiation and the 8‐week post‐treatment visit |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | 2 phase III studies were included. Clearance rates increased with intensity of erythema. Increase in lesion counts (new or subclinical lesions) was higher in imiquimod group at any point during the treatment period. Long‐term clinical outcomes were presented in Lee 2005. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation was achieved by a computer‐generated randomisation schedule. |
| Allocation concealment (selection bias) | Low risk | Participants were assigned the next sequential participant study number and the corresponding study cream. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 15 dropouts (the reasons were reported) Control ‐ B: 15 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Skin quality rating was not reported for placebo. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, open‐label, active‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 3% diclofenac sodium in 2.5% hyaluronic acid, once daily for 12 weeks (N = 24 participants) Control intervention B: 5% imiquimod, 3 times/week for 12 weeks (N = 25 participants) |
|
| Outcomes |
Outcomes of the trial 1) Investigator (IGII) and participant (PGII) global improvement indices at the end of treatment 2) Lesion severity index 3) Local skin reactions 4) Participants experiencing at least 1 treatment‐related adverse event 5) Clinical laboratory tests Efficacy Methods: 1. quantitative assessment using lesion counting, 2. severity of actinic keratoses at baseline, 3. qualitative assessment (GII) by the investigator and participant Time points: at baseline and monthly up to 1 year follow‐up Definitions for global improvement indices on a 7‐point scale: ‐2 (significantly worse), ‐1 (slightly worse), 0 (no change), 1 (slightly improved), 2 (moderately improved), 3 (significantly improved), and 4 (completely improved) Definitions for baseline severity index: 0 (no lesions visible), 1 (clearly visible lesions), 2 (many visible, small, moderately‐thick lesions, or a few large, thick, rough scaly lesions), 3 (many thick, hypertrophic lesions, which are clearly visible and palpable with well‐defined borders) Safety Methods: 1. assessment of tolerability by investigator (erythema, itching, dry skin, and scaling), 2. clinical examination, 3. reporting of adverse events, 4. routine laboratory tests (complete blood cell counts, urine analysis, and fasting chemistry) Time points: 1. monthly up to 1 year follow‐up, 2. before and after treatment (laboratory tests) |
|
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 159): "Patients were randomly assigned to treatment with DFS or IMI." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was open. The difference in dosing regimen frequency of the 2 topical treatments was not concealed by the use of double‐dummy technique. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study was open. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropout Control ‐ B: 0 dropout |
| Selective reporting (reporting bias) | High risk | Participant partial (> 75%) clearance was mentioned, but no data were reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, active‐controlled, parallel‐group study. Start date: August 2004 End date: February 2005 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 0.25 g of 5% imiquimod cream 3 times per week for 8 hours each over a span of 4 weeks, 1 or 2 treatments with a 4‐week rest period (N = 26 participants) Control interventions B: 5% 5‐fluorouracil cream twice daily for 4 weeks (N= 24 participants) C: cryosurgery using bursts of 20 to 40 seconds, 1 or 2 treatments with a 2‐week rest period (N = 25 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates at test of cure and 12 months after the end of treatment Other outcomes of the trial 1) Participant histological clearance rates at test of cure 2) Negative predictive value, i.e. ratio between histological and clinical clearance 3) Serious adverse events 4) Global cosmetic outcome assessments by participant and investigator (presented graphically) 5) Skin quality 6) Recurrence (individual lesion and field) at 12 months after the end of treatment Efficacy Methods: 1. quantitative assessment using precise documentation of location of each lesion on body grid charts with raster and photographs, 2. a 4 mm punch biopsy specimen obtained from 1 of the selected lesions and evaluated independently by 2 expert dermatopathologists Time points: at baseline, test of cure (6, 4, and 8 weeks after last treatment for cryotherapy, 5‐fluorouracil and imiquimod, respectively), and 1 year after the end of treatment (recurrence) Definitions: 1. complete clearance (absence of clinically detectable lesions in treated skin regions), 2. recurrence (for cryotherapy: recurrence of initially cleared lesions determined and expressed as percentage of participants with lesion recurrence in relation to all treated participants; for 5‐fluorouracil and imiquimod: total recurrences within the cleared cancer field determined and expressed as percentage of participants with field recurrence in relation to all treated participants, new lesions considered; missing values counted as recurrence) Cosmetic Methods: global assessment by investigator and participant based on the amount of scarring, atrophy, or indurations and on pigment change within the treatment area by comparison to adjacent, untreated skin Time points: at test of cure Definitions: 1. excellent (treated skin was indistinguishable from normal skin), 2. good (moderate redness or change in pigmentation), 3. fair (moderate redness or change in pigmentation and slight to moderate scarring, atrophy, or induration), 4. poor (extensive scarring, atrophy, or indurations) |
|
| Funding | ‐ | |
| Notes | 1 week of rest period for imiquimod and 5‐fluorouracil during treatment in case of acute inflammation was allowed. The data on non‐recurrence at 1 year follow‐up were as follows: 86% (19/22) for imiquimod, 57% (13/23) for 5‐fluorouracil, and 41% (7/17) for cryotherapy. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 35): "Each patient was randomly assigned to one of [the] equally sized treatment groups (cryosurgery, 5‐FU, or IMIQ) by 'on‐call randomisation' provided by a specialised external company." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | Randomisation involving "on call" randomisation by an external company was used to conceal the allocation. (See previous quote.) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was not stated, but the study compared physically distinct interventions and topical treatments with different application regimens. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | There was no statement about assessor‐blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts at test of cure Control ‐ B: 0 dropouts at test of cure, C: 0 dropouts at test of cure |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, active‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 10% masoprocol applied topically twice daily for 4 weeks (N = 27 participants) Control intervention B: 5% 5‐fluorouracil applied topically twice daily for 4 weeks (N = 30 participants) |
|
| Outcomes |
Outcomes of the trial 1) Investigator global assessment (= global improvement indices) at week 8 2) Absolute and per cent of mean reduction in lesion counts 3) Number of events for adverse events and their severity 4) Percentage of participants experiencing adverse events represented graphically in function of the event severity (= minor adverse events) 5) Percentage of participants who discontinued treatment 6) Mean max pain score Efficacy Methods: 1. quantitative assessment using lesions counting and rating, 2. qualitative assessment (global assessment) Time points: at baseline; days 7, 14, 21, and 28 (the last day of treatment); at day 42 and day 56; and at 1 year and 2 years (recurrence) Definitions for lesion rating: 1. mild (thin actinic keratoses, visible and palpable), 2. moderate (moderately thick actinic keratoses, easily seen and palpated), 3. severe (thick and florid actinic keratoses with distinct borders) Definitions for Global assessment: 1. cured (clear of palpable lesions, slight residual erythema remaining), 2. marked improvement (majority of lesions absent and scales of remaining lesions barely palpable), 3. moderate improvement (many lesions absent and scales decreased in thickness), 4. slight improvement (some lesions cleared, some decreased in scale, but many lesions remain), 5. no change (slightly worse, more or rougher larger lesions remain), 6. much worse (significantly more lesions or majority of lesions rougher, larger, or both) Safety Methods: 1. recording of all adverse experiences noted by participants and questioning of participants; 2. evaluation by investigator on key adverse reactions based on the appearance of the lesions, i.e. degree of erythema, necrosis, ulceration, and erosion in the lesions, and the degree of erythema and contact dermatitis in tissue surrounding and scored as 0 = none, 1 = mild, 2 = moderate, or 3 = severe; 3. intensity of the pain rated by participant on a 9‐point scale ranging from 0 = no pain to 8 = severe pain Time points: at each visit |
|
| Funding | This study was supported by Reed & Carnrick Pharmaceuticals, A Division of Block Drug Company, Inc. | |
| Notes | The numbers for per‐protocol analysis were used for meta‐analyses of mean reduction in lesion counts, and intention‐to‐treat numbers were used for meta‐analysis of global improvement indices. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 162): "Patients were randomised in a double‐blind fashion to twice‐daily treatment with either 10% masoprocol in an emollient cream base or 5% 5‐fluorouracil in a vanishing cream base." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded, and treatments were given with the same application regiment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol analysis was used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | A significantly higher percentage (65.5%) of participants treated with 5‐fluorouracil failed to complete 28 days of treatment than participants treated with masoprocol (16%). |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: September 2001 End date: August 2002 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod cream, once per day, twice weekly for 16 weeks or less (N = 215 participants) Control intervention B: vehicle, once per day, twice weekly for 16 weeks or less (N = 221 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates (all lesions) at 8 weeks post‐treatment Secondary outcome of the trial 1) Participant partial (> 75% of baseline lesions) clearance rates at 8 weeks post‐treatment Other outcomes of the trial 1) Median per cent reduction in baseline lesions at 8 weeks post‐treatment 2) Clinical laboratory tests 3) Participants experiencing at least 1 adverse event 4) Application site reactions 5) Local skin reaction: severe erythema, flaking/scaling/dryness, scabbing/crusting 6) Serious adverse events 7) Skin quality (cosmetic) 8) Increase in the number of lesions during the study Efficacy Methods: quantitative assessment using clinical counting Time points: at baseline; weeks 1, 2, 4, 6, 8, 10, 12, 16 (end of treatment), 20, and 24 Definitions: 1. complete clearance rate (proportion of participants at the 8‐week post‐treatment visit with a count of 0 clinically‐visible lesions in the treatment area), 2. partial clearance rate (proportion of participants at the 8‐week post‐treatment visit with at least a 75% reduction in the number of lesions counted at baseline in the treatment area) Safety Methods: 1. clinical laboratory tests [hematology (haemoglobin, hematocrit, reel blood cell, white blood cell, and platelet counts), serum chemistry (random glucose, blood urea nitrogen, creatinine, total bilirubin, serum glutamic oxaloacetic transaminase, serum glutamic pyruvic transaminase, lactate dehydrogenase, alkaline phosphatase, potassium, sodium, calcium, chloride, total protein, albumin, phosphorous, and cholesterol) and urine analysis for colour/appearance, specific gravity, pH, protein, glucose, and ketones and a microscopic examination]; 2. vital sign measurements and physical examinations; 3. photography; 4. recording of adverse events; 5. assessment of local skin reactions (erythema, oedema, erosion/ulceration, scabbing/crusting, weeping/exudate, vesicles, or flaking/scaling/dryness) rated by a study investigator on a scale of 0 to 3, where 0 = none, 1 = mild, 2 = moderate, and 3 = severe; and 6. recording of concomitant medication use Time points: 1. at each visit, 2. pre‐study visit and end of treatment at week 16 (physical exam and laboratory tests) Definitions for spontaneous participant‐reported adverse events: 1. mild (participant was aware of the signs and symptoms, but the signs and symptoms were easily tolerated), 2. moderate (the signs and symptoms were sufficient to restrict, but not prevent, usual daily activity), and 3. severe (the participant was unable to perform usual daily activity) Cosmetic Methods: assessment of skin quality by visual, clinical, and tactile examinations of the treatment area by investigator [skin surface, hyperpigmentation, hypopigmentation, mottled or irregular pigmentation (both hyperpigmentation and hypopigmentation), degree of scarring, and atrophy on a scale of 0 to 3, where 0 = none, 1 = mild, 2 = moderate, and 3 = severe] Time points: at treatment initiation and 8‐week post‐treatment visit |
|
| Funding | This study was supported by 3M Pharmaceuticals, St Paul, Minnesota. | |
| Notes | An increase in lesion counts was observed during treatment. A sample size calculation was provided. Pooled data from 2 phase III studies were presented in Aldara product insert. Long‐term clinical outcomes were presented in Lee 2005. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 9 dropouts (the reasons were not all reported) Control ‐ B: 11 dropouts (the reasons were not all reported) |
| Selective reporting (reporting bias) | High risk | As a similar study (Szeimies 2004) was also supported by Graceway/3M Pharmaceuticals, not all skin quality outcomes were reported. All outcomes presented in the product insert were reported in the published paper. Another Graceway clinical trial on the arms and hands (NCT00115154) was not published or included in the product insert. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, assessor‐blinded, active‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 0.5% 5‐fluorouracil on either side of face, once daily for 4 weeks. Sunscreen/moisturiser was provided when needed (N = 21 participants). Control intervention B: 5% 5‐fluorouracil on either side of face, twice daily for 4 weeks. Sunscreen/moisturiser was provided when needed (N = 21 participants). |
|
| Outcomes |
Primary outcomes of the trial 1) Absolute and per cent of mean reduction in lesion counts at week 8 2) Total clearance (= participant complete clearance) rates at week 8 Other outcomes of the trial 1) Facial irritation (= skin irritation) 2) Eye irritation (= minor adverse events) 3) Serious adverse events including basal cell carcinoma 4) Participant preference Efficacy Methods: quantitative assessment using counting of palpable or visible (to the unaided eye) lesions by a designated blinded evaluator Time points: during screening period and at 4 weeks post‐treatment Safety Methods: 1. participant‐reported adverse events, 2. photography and evaluation of facial irritation (erythema, edema, dryness, pain, erosion, burning, pruritus, and other signs) on a scale from 0 to 3 (0 = none, 1 = mild, 2 = moderate, 3 = severe) by the same blinded individual and recording of days of onset and resolution (adverse events that occurred on the head were included in the assessment of facial irritation.) Time points: at baseline and weekly throughout the 4‐week treatment (twice weekly for facial irritation) and the post‐treatment periods |
|
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation list was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Participants and clinic staff were not blinded to the difference in dosing regimens (once versus twice daily). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was evaluator blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | Only estimates on clearance rates were provided, i.e. exact values were not given and standard deviations for absolute and per cent mean values were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: September 1994 End date: January 1996 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 3% diclofenac in 2.5% hyaluronic acid gel, applied to single keratosis twice daily for 8 to 24 weeks. Sunscreen was applied after morning application (N = 65 participants). Control intervention B: 2.5% hyaluronic acid gel alone, applied to single keratosis twice daily for 8 to 24 weeks. Sunscreen was applied after morning application (N = 65 participants). |
|
| Outcomes |
Primary outcome of the trial 1) Response to treatment (including participant complete response) rates at end of treatment Other outcomes of the trial 1) Local adverse reactions 2) Serious adverse events Safety Methods: diary recording of any adverse effects and any change in use of concomitant medications Time points: at 8, 16, and 24 weeks |
|
| Funding | This study was supported by Hyal Pharmaceutical Australia Ltd. | |
| Notes | There was no follow‐up period. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A permuted block randomisation design of size 10 was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The participants, the investigator, and the data managers were kept "blind" as to the treatment until all assessments and data entry had been completed. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The participants, the investigator, and the data managers were kept "blind" as to the treatment until all assessments and data entry had been completed. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 31 participants did not complete the 24‐week treatment (the reasons were reported) Control ‐ B: 16 participants did not complete the 24‐week treatment (the reasons were reported), and 29 completed. There was no information given for 20 participants. There were also inconsistency between data presented in a table and the description in the text. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | End of treatment varied as participants ceased treatment at varying times. 34% (diclofenac) and 20% (hyaluronic acid) of participants ceased the treatment before 8 weeks. Between 8 and 16 weeks, 11% (diclofenac) and 5% (hyaluronic acid) of participants ceased the treatment. Between 16 and 24 weeks, 55% (diclofenac) and 75% (hyaluronic acid) of participants ceased the treatment. |
| Methods | This was a randomised, double‐blind, active‐controlled, intraindividual study. Start date: January 1988 End date: June 1988 |
|
| Participants |
Inclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 0.05% (0.5 g) Ro14‐9706 cream (arotinoid methyl sulfone) applied to 1 side of the face twice daily (N = 26 participants) Control intervention B: 0.05% tretinoin cream applied to 1 side of the face twice daily (N = 26 participants) |
|
| Outcomes |
Outcomes of the trial 1) Participant overall response (= global improvement indices) rates at 16 weeks 2) Mean per cent decrease in the number of lesions (= mean percentage of reduction in lesion counts) at 16 weeks 3) Clinical laboratory tests 4) Tolerability (erythema and scaling) scoring 5) Increase of lesions during treatment 6) Rest periods Efficacy Methods: 1. quantitative assessment using lesion counting by 2 independent investigators, 2. qualitative assessment using photography of the treated areas Time points: 1. at the beginning of the study and weekly intervals (counting), 2. at baseline and after 4, 8, 12, and 16 weeks (photography) Definitions: healed or completely cleared lesion (the site had been replaced by normal, smooth, hypopigmented or hyperpigmented skin, and lesion not palpable) Definitions for overall response: 1. worsening (increase in the number of lesions), 2. no response (no change or less than 50% reduction in the total number of lesions), 3. partial response (reduction greater than 50%, but less than 100%, in the number of lesions), 4. complete response (total clearing of lesions) Safety Methods: 1. assessment of local tolerability (erythema and scaling) on a scale (in case of severe reactions, therapy was interrupted until the inflammation had disappeared), 2. routine laboratory (hematologic and biochemical) tests Time points: 1. weekly, 2. before and after treatment (laboratory tests) Definitions for tolerability scale: 0 (none), 1 (mild, minimal), 2 (moderate, more intense), and 3 (severe, very intense erythema, and scaling with exudation) |
|
| Funding | This study was supported by La Roche Ltd. | |
| Notes | Ro 14‐9706 had better tolerability. An initial increase in the number of visible actinic keratoses with tretinoin was observed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 448): "The study was randomised, double‐blind, with each agent applied to opposite sides of the patient's face at a 0.05% concentration, for a period of 16 weeks." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The assessment was performed by 2 independent investigators. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was not reported) Control ‐ B: 1 dropout (the reason was not reported) |
| Selective reporting (reporting bias) | Low risk | Negative (tolerability) data for the sponsored product were reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single centre, randomised, double‐blind, active‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 16 participants) Control intervention B: methyl aminolevulinate (MAL)‐PDT (N = 16 participants) There was a 2‐week interval between the 2 treatments (right versus left scalp). Characteristics of PDT intervention: Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: hyperkeratotic lesions were treated with white paraffin gel to remove any keratotic debris Cream concentration (%): 20% Application of cream: visible layer Incubation with cream: occlusive dressing over cream for 3 (MAL) or 5 (ALA) hours Type of light: red light Light source: Waldmann PDT lamp MSR 1200 Wavelength (nm): 580‐740 Energy fluence (J/cm²): 50 Intensities (mW/cm²): 50 Exposure time: 16 minutes 40 seconds |
|
| Outcomes |
Outcomes of the trial 1) Field complete clearance (= participant complete clearance) rates at 1 month post‐treatment 2) Mean number of lesions at baseline and at 1 month post‐treatment 3) Mean reduction in lesion counts at 1 month post‐treatment 4) Minor adverse events (qualitative) 5) Visual analogue score (VAS) for pain 6) Duration of discomfort 7) Participant preference Efficacy Methods: 1. grading of PpIX fluorescence on a scale of 1 to 3 using a Wood's light (1 = light ⁄ pale; 2 = moderate; and 3 = strong), 2. clinical response (clear, improved, or no response, and number of residual palpable lesions) assessed by an investigator not involved in safety assessment Time points: 1. before treatment (fluorescence), 2. at baseline and 1 month post‐treatment Safety Methods: 1. assessment of pain using a VAS (1 to 100 mm) (If treatment had to be discontinued because of pain, the timing of this was recorded), 2. documentation of adverse effects, and 3. assessment of erythema and erosions by 1 investigator Time points: 1. at 3, 6, 12, and 16 minutes during treatment (pain), 2. 4 days after their first and second treatments (erythema and erosion) |
|
| Funding | ‐ | |
| Notes | ALA‐PDT was more painful than MAL‐PDT. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 88): "Patients were randomised so that half would receive ALA and half MAL as their first split scalp treatment." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 88): "Both patients and investigators remained blinded until study completion." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessment was performed by a second investigator. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol (PP) analysis. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | Wood's light was used to look at PpIX fluorescence after cream incubation, but the results were not mentioned. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: March 2008 End date: Not available |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 1% nicotinamide, twice daily for 6 months (information from the protocol) (N = 13 participants) Control intervention B: placebo, twice daily for 6 months (information from the protocol) (N = 17 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Mean percentage of reduction in lesion counts from baseline at 3 and 6 months Secondary outcome of the trial 1) Total count for appearance of new/subclinical lesions at 3 months (protocol) Other outcome of the trial 1) Serious adverse events including basal cell carcinoma and squamous cell carcinoma Efficacy Methods: quantitative assessment using counting of lesions by a single observer and photography Safety Methods: reporting of all adverse events |
|
| Funding | This study was supported by Cancer Council NSW, the Dermatology Research Foundation and Epiderm. | |
| Notes | This was a pilot study. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 1138): "Participants were randomised (unstratified, size six randomised block)." |
| Allocation concealment (selection bias) | Low risk | Allocation was concealed within sealed opaque envelopes (protocol). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 1138): "Patients and observers (F.M., M.V.) remained blinded until all patients had completed the study." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote (page 1138): "Patients and observers (F.M., M.V.) remained blinded until all patients had completed the study." |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It was unclear if intention‐to‐treat (ITT) or per‐protocol (PP) analysis was used, but ITT was stated in the protocol. Intervention ‐ A: 0 dropouts Control ‐ B: 2 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Appearance of new/subclinical lesions was not reported, but this outcome was included in the protocol ACTRN12607000428460 at anzctr.org.au. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, cross‐over study (2‐part study). The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: etretinate, 75 mg/day (25 mg tablet 3 times daily) for 2 months (N = 25 participants) Control intervention B: placebo. 3 times daily for 2 months (N = 25 participants) Then treatment changes for 2 more months (placebo group gets etretinate, and vice versa) |
|
| Outcomes |
Outcomes of the trial Part 1 1) Complete remission (= participant complete clearance) rates 2) Partial remission (50% size reduction of 75% of lesions) rates 3) Clinical laboratory tests Part 2 (alternate therapy given to each group) 1) Complete remission 2) Partial remission rates 3) Clinical laboratory tests 4) Minor adverse events (for the 2 phases) Efficacy Methods: quantitative assessment using direct measurement and photography of the lesions Time points: every month Safety Methods: 1. haematological and biochemical screen, 2. measurement of plasma vitamin A |
|
| Funding | ‐ | |
| Notes | 17 participants required dosage reduction due to toxicity. Response was maintained when dosage was reduced. Vitamin A type unwanted effects (dry mouth, skin rash, desquamation, etc) were observed. Only part 1 data has been included in this review. Data for intention‐to‐treat analysis was used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (pages 364 to 365): "Each treatment was given for 2 months and the order of administration was randomised." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 3 dropouts (the reasons were not reported) Control ‐ B: 2 dropouts (the reasons were not reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, open‐label, active‐controlled, intraindividual study. Start date: March 2004 End date: April 2005 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: methyl aminolevulinate (MAL)‐photodynamic (PDT) (N = 119 participants) Control intervention B: cryotherapy: double freeze‐thaw (16 seconds total) using liquid nitrogen spray, lesions with non‐complete response were retreated at 12 weeks 1 (assessment at 12 weeks) or 2 treatments (assessment at 24 weeks) (N = 119 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 12 weeks Preparation of lesions: gentle scraping Cream concentration (%): 16% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light LED Light source: AktiliteCL 128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: 8 to 10 minutes |
|
| Outcomes |
Primary outcomes of the trial 1) Lesion complete response rates of baseline lesions only at 24 weeks 2) Participant preference Secondary outcomes of the trial 1) Lesion complete response rates of baseline lesions only at 12 weeks 2) Cosmetic outcomes by investigator at 12 and 24 weeks 3) Investigator preference Other outcomes of the trial 1) Mean per cent reduction in lesion counts from baseline at 12 and 24 weeks 2) Skin‐related adverse events 3) Skin discomfort and pain after first or second treatments on a visual analogue scale (VAS) Efficacy Methods: quantitative assessment using lesion counting [efficacy evaluations included only lesions present at baseline (i.e. lesions appearing after baseline, if any, were to be reported as adverse events)] Time points: at baseline, and at weeks 12 and 24 Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance) Safety Methods: 1. immediate evaluation of skin discomfort by participant after each procedure using a VAS of 0 (no discomfort) to 10 (worst possible skin discomfort), 2. adverse events reported spontaneously by the participant or elicited following non‐leading questioning (severity, duration, and need for additional therapy) (If pain was the only reaction and concomitant treatment was not needed, it was not reported as an adverse event, as it was already recorded as skin discomfort) Time points: at each visit (adverse events) Definitions: 1. phototoxic reaction (any observed erythema, oedema, itching, etc), 2. cryotherapy reaction (any observed blisters, infection, etc) Cosmetic Methods: assessment of the overall cosmetic outcome Time points: at weeks 12 and 24 Definitions: 1. excellent (only slight occurrence of redness or change in pigmentation), 2. good (moderate redness or change in pigmentation), 3. fair (slight to moderate scarring, atrophy, or induration), 4. poor (extensive scarring, atrophy, or induration) |
|
| Funding | This study was supported by Galderma France. | |
| Notes | The treatments were comparable in terms of efficacy; however, participants significantly preferred MAL‐PDT over cryotherapy. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1030): "At baseline visit, eligible subjects received treatment with PDT using MAL... and conventional
cryotherapy, randomly allocated to alternate sides of the
face/scalp." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This study was open because 2 physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This study was open because 2 physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Per‐protocl (PP) and intention‐to‐treat (ITT) analyses were used. Intraindividual study: Intervention ‐ A: 6 dropouts (the reasons were reported) Control ‐ B: 6 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Standard deviations for the mean percentages of reduction in lesion counts were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, assessor‐blinded, active‐controlled, intraindividual study. Start date: October 2008 End date: September 2009 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: cryotherapy followed by 5% imiquimod 3 times per week for 4 weeks (N = 27 participants) Control intervention B: cryotherapy (N = 27 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Mean percentage of reduction in lesion counts at 4 to 8 weeks post‐treatment Secondary outcome of the trial 1) Cosmetic appearance scores by participant and investigator at 4 to 8 weeks post‐treatment Other outcomes of the trial 1) Participant complete clearance rates at 4 to 8 weeks post‐treatment (posthoc analysis) 2) Local skin reactions (severity scores) 3) Minor adverse events (pooled) 4) Serious adverse events Efficacy Methods: quantitative assessment using counting of all (baseline and new) actinic keratoses in each respective treatment area Time points: at baseline and 4 to 8 weeks post‐treatment Definitions: 1. per cent change = [(actinic keratoses count at 4 to 8 weeks post‐treatment)‐(actinic keratoses count at baseline)]/(actinic keratoses count at baseline)]*100%, 2. complete clearance (actinic keratosis count of 0) Safety Methods: 1. mean maximum postbaseline intensity of investigator‐assessed local skin reactions (erythema, edema, weeping/exudate, flaking/scaling/dryness, scabbing/crusting, erosion/ulceration) scored as 0 = none, 1 = mild, 2 = moderate, 3 = severe per treatment area; 2. serious adverse events; 3. adverse events collected by non‐systematic assessment Time points: postbaseline to the end of study Cosmetic Methods: Cosmetic appearance score based on comparison to appearance at baseline by investigator and participant Time points: at 4 to 8 weeks post‐treatment Definitions for 7‐point scale: +3 (treatment area is much better appearing), +2 (treatment area is moderately better appearing), + 1 (treatment area is slightly better appearing), 0 (treatment area appears same), ‐1 (treatment area is slightly worse appearing), ‐2 (treatment area is moderately worse appearing), and ‐3 (treatment area is much worse appearing) |
|
| Funding | ‐ | |
| Notes | Local skin reactions were reported as scores, but their values were similar between the imiquimod‐treated and topical untreated sides. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1 of study data document): "Study design: Allocation: Randomised..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This study was single‐blinded (assessor). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was single‐blinded (assessor). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis for participant complete clearance and per‐protocol (PP) analysis for other analyses were used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | A "favourable" efficacy outcome was analysed posthoc, whereas the prespecified outcome was not favourable. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: June 2008 End date: May 2009 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod (Taro), once per day, twice weekly for 16 weeks (N = 183 participants) Control intervention B: vehicle, once per day, twice weekly for 16 weeks (N = 30 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Number of participants with 100% clearance of lesions (= participant complete clearance) at week 24 Secondary outcome of the trial 1) Participants experiencing at least 1 adverse event Other outcomes of the trial 1) Application site reactions including irritation 2) Minor adverse events 3) Serious adverse events including squamous cell carcinoma Efficacy Methods: quantitative assessment using lesion counting Time points: at baseline and week 24 Definitions: the participant is 100% clear of lesions (all lesions that were identified at baseline are no longer present, and there are no new lesions) Safety Methods: reporting of adverse events and serious adverse events termed from Medical Dictionary for Regulatory Activities (MedDRA) Time points: at each follow‐up visit |
|
| Funding | This study was supported by Taro Pharmaceuticals USA. | |
| Notes | This was an equivalence study and was divided into 2 studies for our review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1 of study data document): "Study design: Allocation: Randomised..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded (subject, caregiver, investigator). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded (outcomes assessor). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Per‐protocol (PP) and modified (no postbaseline assessment but received treatment) intention‐to‐treat ( ITT) analyses were used. Intervention ‐ A: 31 dropouts (the reasons were reported) Control ‐ B: 7 dropouts of the entire group, i.e. 60 participants (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: June 2008 End date: May 2009 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod (Aldara), once per day, twice weekly for 16 weeks (N = 179 participants) Control intervention B: vehicle, once per day, twice weekly for 16 weeks (N = 30 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Number of participants with 100% clearance of lesions (= participant complete clearance) at week 24 Secondary outcome of the trial 1) Participants experiencing at least 1 adverse event Other outcomes of the trial 1) Application site reactions including irritation 2) Minor adverse events 3) Serious adverse events including squamous cell carcinoma Efficacy Methods: quantitative assessment using lesion counting Time points: at baseline and week 24 Definitions: the participant is 100% clear of lesions (all lesions that were identified at baseline are no longer present, and there are no new lesions) Safety Methods: reporting of adverse events and serious adverse events termed from Medical Dictionary for Regulatory Activities (MedDRA) Time points: at each follow‐up visit |
|
| Funding | This study was supported by Taro Pharmaceuticals USA. | |
| Notes | This was an equivalence study and was divided into 2 studies for our review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1 of study data document): "Study design: Allocation: Randomised..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded (subject, caregiver, investigator). |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded (outcomes assessor). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Per‐protocol (PP) and modified (no postbaseline assessment but received treatment) Intention‐to‐treat (ITT) analyses were used. Intervention ‐ A: 29 dropouts (the reasons were reported) Control ‐ B: 7 dropouts of the entire group, i.e. 60 participants (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 10% masoprocol cream applied twice daily for 14 to 28 days (N = 131 participants) Control intervention B: placebo applied twice daily for 14 to 28 days (N = 45 participants) |
|
| Outcomes | 1) Investigator global assessment (global improvement indices‐cured) at 1 month post‐treatment 2) Mean reduction in lesion counts 3) Median percentage reduction in lesion counts between baseline and 1 month post‐treatment 4) Skin irritation Efficacy Methods: 1. quantitative assessment by counting all actinic keratoses in the test area, 2. qualitative assessment (global assessment) Time points: at baseline and 1 month post‐treatment Definitions: evaluable (participant who completed at least 14 days of therapy and returned for the follow‐up visit 1 month after the drug was stopped) Definitions for global assessment: 1. cured (clear of palpable lesions, slight residual erythema may remain), 2. marked improvement (majority of lesions absent and scales of remaining lesions are barely palpable), 3. moderate improvement (many lesions are now absent and scales have decreased in thickness), 4. slight improvement (some lesions cleared, some decreased in scale, but many lesions remain), 5. no change, 6. slightly worse (more or rougher, larger lesions, or both, remain), and 7. much worse (significantly more lesions or majority of lesions rougher, larger, or both) Safety Methods: clinical assessment and participant history Time points: at each visit (weekly during treatment) |
|
| Funding | This study was supported by Chemex Pharmaceuticals, Denver. | |
| Notes | Inflammatory response was not essential for therapeutic activity of masoprocol. The percentage of reduction in lesion counts did not correlate with baseline lesion severity. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 739): "Patients were randomly assigned to treatment with topical masoprocol or vehicle in a 3:1 ratio, respectively." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used, and there was a difference in the percentages of participant lost between treatment (14%) and placebo (9%). Intervention ‐ A: 18 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | 25 participants stopped treatment between 14 & 28 days due to adverse reactions, but it was stated that their results were comparable to those who completed the full 28 days of treatment. |
| Methods | This was a single‐centre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod, applied to 5 lesions, once per day 3 times per week for up to 16 weeks, biopsy of 1 lesion after 2 weeks of treatment (N = 12 participants) Control intervention B: vehicle, applied to 5 lesions, once per day 3 times per week for up to 16 weeks, biopsy of 1 lesion after 2 weeks of treatment (N = 6 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Immunological outcome Other outcomes of the trial 1) Participant complete clearance rates at the end of treatment 2) Clearance rates 3) Percentage lesion reduction (proportion of baseline lesions cleared at end of treatment = lesion complete response) rates 4) Clinical laboratory tests 5) Application site reactions 6) Local skin reactions 7) Minor adverse events 8) Serious adverse events Efficacy Time points: at the end‐of‐treatment visit Definitions: 1. clearance (clinical resolution of > 50% of the 4 treated, non‐biopsied lesions), 2. complete clearance rate (proportion of participants with 100% clinical clearance of treated lesions) Safety Methods: 1. laboratory tests, 2. recording of local skin reactions and adverse events Time points: pre‐study and end of treatment (laboratory tests) |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a phase I study, mainly on cutaneous immune response (biomarker changes). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation schedule was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: February 2006 End date: January 2007 |
|
| Participants |
Inclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod, once per day, 3 times per week, 4 weeks on, 4 weeks off, 4 weeks on (N = 9 participants) Control intervention B: vehicle, once per day, 3 times per week, 4 weeks on, 4 weeks off, 4 weeks on (N = 3 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Comparison of evaluation techniques Secondary outcome of the trial 1) Histological clearance (confirmation) Other outcomes of the trial 1) Mean lesion counts at baseline and week 20 (transformed to mean reduction in lesion counts) 2) New/sub‐clinical lesions during the study 3) Minor adverse events (pooled) Efficacy Methods: quantitative assessment using clinical counting Time points: at baseline and weeks 4, 8, 12, and 20 Safety Methods: 1. general physical examination, 2. recording of any adverse events Time points: 1. at the start and end of the study (physical exam), 2. at each visit (adverse events) |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a pilot study. Cross polarised light photography, fluorescence diagnostic, and clinical lesion counting were used for efficacy analysis, but only data obtained with clinical counting were used for analyses because it was the technique used in the other studies. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 641): "Eligible patients were randomised in a 3:1 ratio (active:vehicle) to either imiquimod or vehicle cream." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Low risk | All outcomes in the protocol (NCT00294320) were reported in the published study. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, double‐blind, active‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% 5‐fluorouracil twice daily for 4 to 7 weeks followed by chlorhexidine cream (N = 27 participants) Control intervention B: Er:YAG laser resurfacing with oral prophylactic antibiotics and antivirals, Erbium mode: 7 to 28 J/cm², 10 to 12 pulses per second with 50% CO₂ from 2 to 4 W (N = 28 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Recurrence rates according to clinical evaluation at 12 months post‐treatment Secondary outcomes of the trial 1) Recurrence rates according to clinical evaluation at 3 and 6 months post‐treatment 2) Recurrence rates according to histological evaluation at 3 months and at time of recurrence Other outcomes of the trial 1) Mean reduction in lesion counts at 3, 6, and 12 months post‐treatment 2) Mean per cent lesions cleared (= mean percentage of reduction in lesion counts) at 6 and 12 months post‐treatment 3) Skin irritation 4) Minor adverse events after treatment, at 3, 6, and 12 months post‐treatment 5) Cosmetic outcome: proportion of participants with improvement of surface with actinic damage 6) Cosmetic outcome: proportion of participants with decrease in photoageing score 7) Cosmetic outcome: number of participants with changes in pigmentation or scarring Efficacy Methods: 1. quantitative assessment using clinical evaluation including the number of lesions and the surface of actinic damage (0% to 25%, 25% to 50%, 50% to 75%, and 75% to 100%) performed by 2 investigators, 2. histopathological evaluation (3 months after treatment and evaluation of recurrence) Time points: at baseline; at 3 days (laser only); at 1 (laser only), 2, and 4 weeks; and at 3, 6, and 12 months post‐treatment Safety Methods: evaluation of adverse effects Time points: at day 3 (laser only), at weeks 1 (laser only), 2 and 4 , and at 3, 6, and 12 months post‐treatment Cosmetic Methods: photoageing score (simplified form of the Glogau score) performed by 2 investigators Time points: at baseline; at day 3 (laser only); at weeks 1 (laser only), 2, and 4; and at 3, 6, and 12 months post‐treatment |
|
| Funding | ‐ | |
| Notes | There were significantly less recurrences in the laser group than 5‐fluorouracil group. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer program, Sampsize 2.0, was used to generate the allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | The standard deviations associated with mean values were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: MAL‐photodynamic therapy (PDT) (N = 42 participants) Control intervention B: placebo‐PDT (N = 38 participants) Characteristics of PDT intervention: Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage and gentle scraping Cream concentration (%): 16% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: ‐‐ Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 50 to 200 Exposure time: 8 min |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates at 3 months after last treatment Other outcomes of the trial 1) Lesion complete response rates at 3 months after last treatment 2) Local adverse events 3) Minor adverse events 4) Serious adverse events 5) Cosmetic outcome with MAL‐PDT in completely cleared participants: physician assessment, participant assessment 6) Participants' satisfaction Efficacy Methods: quantitative assessment using inspection, photography, and palpation of each lesion by the same investigator at each centre Time points: at baseline and at 3 months after the second PDT Definitions: complete response (complete disappearance of the lesion) Safety Methods: 1. recording of local skin reactions, phototoxicity reactions, or both, by study‐centre personnel who were not involved in evaluation of the participant, 2. adverse events reported spontaneously by the participant or elicited after non‐leading questioning (severity, duration, and need for additional therapy) and rated (the clinician assessed the causal relationship of the event to the study treatment as related, uncertain, or not related) Time points: at baseline, during, and immediately after PDT; at week 2; and at 3 months after the second PDT treatment Definitions for adverse events (any unfavourable and unintended sign, symptom, or disease) rating: 1. mild (the event was transient and easily tolerated), 2. moderate (the event caused the participant discomfort and interrupted usual activities), and 3. severe (the event caused considerable interference with usual activities and may have been incapacitating or life‐threatening) Cosmetic Methods: assessment of overall cosmetic outcome in participants with complete response in all lesions by both the investigator and participant using a 4‐point rating scale Definitions for the 4‐point scale: 1. excellent (no scarring, atrophy, or induration, and no or slight occurrence of redness or change in pigmentation compared with adjacent skin), 2. good (no scarring, atrophy, or induration, but moderate redness or change in pigmentation compared with adjacent skin), 3. fair (slight to moderate occurrence of scarring, atrophy, or induration), 4. poor (extensive occurrence of scarring, atrophy, or induration) |
|
| Funding | This study was supported by PhotoCure ASA. | |
| Notes | The response rate was similar for mild and moderate lesions. Most pain and erythema was gone within 24 hours. Data for intention‐to‐treat analysis were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Prerandomised numbers assigned to participants at screening visit and stratified per centre were used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 3 dropouts (the reasons were reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | High risk | The cosmetic outcomes were reported for MAL‐PDT participants only. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: January 2006 End date: December 2006 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: MAL‐photodynamic therapy (PDT) (N = 49 participants) Control intervention B: placebo‐PDT (N = 47 participants) Characteristics of PDT intervention: Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage and gentle scraping Cream concentration (%): 16.8% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light LED Light source: Aktilite CL 128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: 8 min |
|
| Outcomes |
Primary outcome of the trial 1) Complete participant response rates (= participant complete clearance) at 3 months post‐treatment Secondary outcomes of the trial 1) Complete lesion response rates (= lesion complete response) at 3 months post‐treatment 2) Application site and local adverse reactions (in general and severe) Other outcomes of the trial 1) Serious adverse events 2) New lesions during the study Efficacy Methods: clinical assessment by inspection, palpation, and characterisation of lesions (in accordance with Olsen 1991) by the same blinded investigator Time points: at baseline and 3 months post‐treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion) (non‐completely responding lesions were treated at the discretion of the investigator) Safety Methods: adverse events reported spontaneously or elicited by non‐leading questioning (severity, localisation, duration, and need for additional treatment) (the clinician assessed the causal relationship of the event to the study treatment as related, uncertain, or not related.) Time points: after lesion preparation before cream application, at the end of the 3‐hour cream application and after illumination during each treatment session, and at 2 weeks and 3 months post‐treatment Definitions for the severity of the adverse events: 1. mild (transient and easily tolerated), 2. moderate (caused the participant's discomfort and interrupted usual activities), 3. severe (caused considerable interference with usual activities and may have been incapacitating or life‐threatening) |
|
| Funding | This study was supported by PhotoCure ASA. | |
| Notes | Within the MAL‐PDT group, complete lesion response rates were slightly higher in grade‐1 than grade‐2 lesions (89% vs 80%), and in lesions on the scalp than on the face (93% vs 87%). Larger lesions (diameter > 20 mm) had lower complete response rates than smaller lesions (74% vs 86% to 90%). At 3 months post‐treatment, 31% (15/49) of participants treated with MAL had new lesions compared to 26% (12/47) of participants treated with vehicle. A sample size calculation was provided. This study is study #1 in the Metvixia product insert 2008. The studies included in the Metvixia product insert were changed between 2004 (PhotoCure) and 2008 (Galderma), which correspond to the use of different types of light. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Prerandomised numbers assigned to participants at screening visit and stratified per centre were used for allocation generation. |
| Allocation concealment (selection bias) | Low risk | Randomisation sequence was prepared by sponsor. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was evaluator‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropout Control ‐ B: 0 dropout |
| Selective reporting (reporting bias) | Low risk | All outcomes from the protocol (NCT00306800) and protocol mistakes were presented. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single centre, randomised, active‐controlled, intraindividual study. Start date: May 2004 End date: August 2005 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 4 participants) Control intervention B: 5‐fluorouracil twice daily for 3 weeks (N = 4 participants) Characteristics of PDT intervention: Type of treatment: field‐directed treatment Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: scales removed by curettage Cream concentration (%): 16% Application of cream: 1 mm thick onto area Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Paterson PDT Omnilux Wavelength (nm): 633 + 15 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 80 Exposure time: 8 min |
|
| Outcomes |
Outcomes of the trial 1) Complete resolution of lesional area (= participant complete clearance) rates at 1, 3, and 6 months 2) Overall reduction in lesional area 3) Local skin reactions (pooled for carcinomas in situ and actinic keratoses) 4) Minor adverse events (pooled for carcinomas in situ and actinic keratoses) 5) Cosmestic outcomes by participant and investigator (pooled for carcinomas in situ and actinic keratoses) 6) Treatment‐associated pain score 7) Participant's preference Efficacy Methods: quantitative assessment using photographic mapping, tracing of the clinical margins of each lesional area onto transparencies and calculating the surface area by overlaying on 1 mm squared graph paper Time points: before treatment, at 1, 3, and 6 months post‐treatment Definitions: 1. complete response (complete clinical resolution of the treated lesion), 2. partial response (at least a 30% reduction in the surface area of the lesion after treatment based upon the European Organisation for Research and Treatment of Cancer (EORTC) guidelines for the evaluation of tumour treatment response), 3. non‐responders (lesions that failed to meet the criteria for partial response) Safety Methods: 1. participants kept a record of pain and erythema using a 4‐point scale (0 = none, 1 = mild, 2 = moderate, and 3 = severe); 2. documentation of other local skin reactions, such as pruritus, erosions, ulceration, crusting, skin infection, and scarring Time points: daily Cosmetic Methods: cosmetic scoring by clinician and participant Time points: at the 6‐month assessment Definitions: 1. poor (extensive scarring, atrophy, or induration), 2. moderate (slight to moderate occurrence of scarring), 3. good (no scarring, atrophy or induration, but moderate redness or pigmentation change compared with adjacent skin), 4. excellent (no scarring, atrophy, or induration, and no or slight occurrence of redness or pigmentation change compared with adjacent skin) |
|
| Funding | The light source was provided by Omnilux. | |
| Notes | This study with immunocompromised participants included participants with actinic keratoses (N = 4) or carcinoma in situ (N = 5). Separated data were presented for efficacy but not for local skin reactions, adverse events, and cosmetic outcomes. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 321): "Each patient was randomly assigned to apply topical 5‐FU cream to 1 lesional area twice daily for 3 weeks and to
receive topical PDT twice at a 1‐week interval to the other lesional area." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This study was open‐label. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote (page 322): "Assessments were not blinded." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used for the review based on the individual data presented in a table. Intraindividual study: Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, vehicle‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% Imiquimod, 3 times per week for 8 weeks or less (clearance achieved) (N = 22 participants) Control intervention B: vehicle, 3 times per week for 8 weeks or less (clearance achieved) (N = 22 participants) |
|
| Outcomes |
Outcomes of the trial 1) Mean lesion counts (transformed to mean reduction of lesion counts) at baseline and week 16 2) Changes in lesion size 3) Participants experiencing at least 1 adverse event (pooled) 4) Local adverse reactions (pooled) 5) Rest periods Efficacy Methods: quantitative assessment using lesion counting and photography (only participants who completed the 8‐week course of treatment with imiquimod were assessed for changes in lesion size) Time points: at baseline; at weeks 2, 4, 6, 8, and 16 Safety Methods: 1. monitoring of concomitant medications, and 2. recording of the type and severity (mild, moderate, or severe) of local adverse reactions (erythema, itching, scabbing) Time points: at baseline; at weeks 2, 4, 6, and 8, or until total clearance of lesions |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | If needed, a rest period of 3 weeks was allowed and the dosing frequency reduced to 2 times per week. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 554): "Application sites were randomised at the
time of treatment." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | This was not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis with 23% of lost participants was used. Intraindividual study: Intervention ‐ A: 5 dropouts (the reasons were reported) Control ‐ B: 5 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | The adverse events were not reported separately for the 2 treatments. The standard deviations associated with mean values were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 88 participants) Control intervention B: placebo‐PDT (N = 23 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: debridement Cream concentration (%): 16.8% Application of cream: onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 2.5 to 4 hours Type of light: red light Light source: CureLight BroadBand Model CureLight 01 Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
|
| Outcomes |
Outcomes of the trial 1) Complete responders (= participant complete clearance) rates at 3 months 2) Participant partial (> 75%) clearance rates at 3 months 3) Lesion complete response rates at 3 months 4) Local adverse reactions 5) Cosmetic outcomes: hyperpigmentation Efficacy Time points: at 3 months after the second treatment session Definitions: 1. cleared lesion (not visible and not palpable), 2. complete responder (participant with all treated lesions cleared) |
|
| Funding | This study was supported by Photocure ASA. | |
| Notes | Participants with > 4 lesions had lower success rates than those with < 4 lesions when treated with MAL‐PDT. Lesions that were slightly palpable, i.e. grade 1, had a better success rate than lesions that were visible and palpable, i.e. grade 2. Local skin reactions and cosmetic outcomes from Photocure ASA Australian and US studies were combined. The studies included in the Metvixia product insert were changed between 2004 (PhotoCure) and 2008 (Galderma), which correspond to the use of different types of light. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 2): "These trials were not identical; however, both were randomised, multicenter, and double‐blinded with patients randomised to Metvixia‐PDT and Vehicle‐PDT study arms that required 2 treatment sessions (7 days apart)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | This was not stated, and the information provided did not allow to make any conclusion. There was no information provided on study dropouts. |
| Selective reporting (reporting bias) | High risk | The adverse events were reported for the whole study, i.e. not separated for MAL and vehicle. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 42 participants) Control intervention B: placebo‐PDT (N = 38 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: debridement Cream concentration (%): 16.8% Application of cream: onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 2.5 to 4 hours Type of light: red light Light source: CureLight BroadBand Model CureLight 01 Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
|
| Outcomes |
Outcomes of the trial 1) Complete responders (= participant complete clearance) rates at 3 months 2) Participant partial (> 75%) clearance rates at 3 months 3) Lesion complete response rates at 3 months 4) Local adverse reactions 5) Cosmetic outcomes: hyperpigmentation Efficacy Time points: at 3 months after the second treatment session Definitions: 1. cleared lesion (not visible and not palpable), 2. complete responder (participant with all treated lesions cleared) |
|
| Funding | This study was supported by Photocure ASA. | |
| Notes | Lesions that were slightly palpable, i.e. grade 1, had a better success rate than lesions that were visible and palpable, i.e. grade 2. Local skin/adverse reactions and cosmetic outcomes from Photocure ASA Australian and US studies were combined. The studies included in the Metvixia product insert were changed between 2004 (PhotoCure) and 2008 (Galderma), which correspond to the use of different types of light. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 2): "These trials were not identical; however, both were randomised, multicenter, and double‐blinded with patients randomised to Metvixia‐PDT and Vehicle‐PDT study arms that required 2 treatment sessions (7 days apart)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | This was not stated and the information provided did not allow to make any conclusion. There was no information provided on study dropouts. |
| Selective reporting (reporting bias) | High risk | The adverse events were reported for the whole study, i.e. not separated for MAL and vehicle. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, assessor‐blinded, vehicle‐controlled, parallel group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 181 participants) Control intervention B: placebo‐PDT (N = 62 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 8 weeks Preparation of lesions: ‐‐ Cream concentration (%): 20% Application of cream: ‐‐ Incubation with cream: 14 to 18 hours Type of light: blue light Light source: Blu‐U Wavelength (nm): 417 + 5 Energy fluence (J/cm²): 10 Intensities (mW/cm²): 10 Exposure time: 1000 seconds (16 minutes) |
|
| Outcomes |
Outcomes of the trial Assessments at 8 weeks (1 treatment) and 12 weeks (1 or 2 treatments): 1) Clearing of individual lesions (= lesion complete response) rates 2) Percentage of participants who experienced 100% clearance of all target lesions (= participant complete clearance) 3) Percentage of participants who experienced 75% or greater clearance of all target lesions (= participant partial clearance) 4) Clinical laboratory tests 5) Application site reactions 6) Local skin reactions by location, i.e. face or scalp 7) Treatment‐related adverse events (= minor adverse events) given for ALA treatment only 8) Minor adverse events in general and by location, i.e. face or scalp 9) Serious adverse events 10) Changes in pigmentation reported per lesion Efficacy Methods: assessments performed by a blinded investigator Time points: at weeks 4, 8, and 12 Safety Methods: 1. laboratory tests, 2. assessment of phototoxic effects such as erythema, edema, stinging or burning, by an unblinded investigator, 3. assessment of adverse events Time points: 1. at baseline and 24 hours after initial light treatment and at week 8 and 24 hours after retreatment (laboratory tests), 2. every visit (phototoxic effect), 3. before, during, and after treatment and at each visit during the study period (adverse events) Cosmetic Methods: assessment of changes in pigmentation by an unblinded investigator Time points: at every visit |
|
| Funding | This study was supported by DUSA Pharmaceuticals. | |
| Notes | Pooled results from 2 independent and identical phase III clinical trials also presented in the Levulan kerastick product insert. Recurrence was presented in a follow‐up paper (Fowler 2002). Data for intention‐to‐treat analysis were used for the meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The method used for allocation generation is unclear, but it was performed separately for each centre. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote (page 42): "Drug application and activation, light treatment, and all safety evaluations were performed by an unblinded investigator..." |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This study was assessor‐blinded for efficacy, but not for safety. Safety and efficacy assessments were performed by different investigators. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 7 dropouts (the reasons were reported) Control ‐ B: 3 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | The Levulan kerastick product insert mentioned 7 clinical trials. Efficacy data from only 2 randomised phase III (Piacquadio 2004) and open studies were presented. Only safety data from the 2 phase III studies were presented. Levulan kerastick reported efficacy outcomes separately for the face and scalp, but Piacquadio 2004 did not. Piacquadio 2004 reported adverse events for ALA‐PDT, whereas Levulan kerastick reported them for both ALA‐PDT and placebo‐PDT. |
| Other bias | High risk | Data between Piacquadio 2004 (PP) and the Levulan kerastick (ITT?) product insert was not always the same. Data from Piacquadio 2004 was used for analyses. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: 1995 End date: 1996 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: topical 3% diclofenac in 2.5% hyaluronic acid gel, twice daily for 30 days (0.5 g/treatment with 6 hours between applications) (N = 49 participants) B: topical 3% diclofenac in 2.5% hyaluronic acid gel, twice daily for 60 days (0.5 g/treatment with 6 hours between applications) (N = 48 participants) Control interventions C: topical 2.5% hyaluronic acid gel, twice daily for 30 days (0.5 g/treatment with 6 hours between applications) (N = 49 participants) D: topical 2.5% hyaluronic acid gel, twice daily for 60 days (0.5 g/treatment with 6 hours between applications) (N = 49 participants) |
|
| Outcomes |
Primary outcomes of the trial 1) Investigator global improvement indices (IGII) 2) Participant global improvement indices (PGII) 3) Number of participants with complete target lesion clearance at day 60 (target lesion number score = TLNS) 4) Number of participants with complete lesion clearance including new lesions (cumulative lesion number score = CLNS) (TLNS and CLNS transformed to participant complete clearance) 5) Mean numbers of target lesions at baseline and follow‐up (transformed to mean reduction in lesion counts). 6) Total thickness score (TTS) Other outcomes of the trial 1) Clinical laboratory tests 2) Application site reactions 3) Minor adverse events 4) Serious adverse events (including basal and squamous cell carcinoma) Efficacy Methods: 1. quantitative assessment using outlining and counting of lesions on a 5 cm² transparent grid, 2. TTS determined by palpation and visual assessments, 3. qualitatively assessments of overall improvements by investigator and participant (IGII and PGII), 4. severity of the lesions (mild, moderate, and severe) Time points: at baseline (visit 2, day 1 of treatment) and subsequent visits Definitions for TTS: R (lesion resolved completely), 0 (lesion visible, but not palpable), 1 (lesion visible and palpable), 2 (lesion raised with visible scaling), 3 (lesion hyperkeratotic and > 1 mm in thickness) Definitions for the 7‐point scale for IGII and PGII: ‐2 (significantly worse), ‐1 (slightly worse), 0 (no change), 1 (slightly improved), 2 (moderately improved), 3 (significantly improved), 4 (completely improved) Safety Methods: 1. participant‐recorded concomitant medications taken and side‐effects experienced, 2. review of adverse events and photography, 3. clinical laboratory analyses (standard haematological, biochemical parameters, assessment of electrolytes and urinalysis), 4. serology (antidiclofenac antibodies) Time points: 1. daily (participant record), 2. at each visit (adverse events review), 3. at screening and end of treatment or onset of reaction (serology) |
|
| Funding | This study was supported by Hyal Pharmaceutical Corporation. | |
| Notes | A sample size calculation was provided. Data were reported in the Solaraze gel product insert. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 95): "This randomised, double‐blind, placebo‐controlled, parallel‐group trial was conducted between 1995 and 1996 at 6 Canadian centres." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 3 dropouts, B: 5 dropouts (the reasons were reported) Control ‐ C: 2 dropouts, D: 1 dropout (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | Statistically significant and non‐significant outcomes were reported. |
| Other bias | High risk | There were different safety data between Rivers 2002 and the Solaraze product insert. |
| Methods | This was a single‐centre, randomised, assessor‐blinded, placebo‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: calcipotriol (vitamin D), twice daily for 12 weeks on right/left side (N = 9 participants) Control intervention B: placebo twice daily for 12 weeks on right/left side (N = 9 participants) |
|
| Outcomes |
Outcomes of the trial 1) Mean numbers of lesions at baseline and week 12 (transformed to mean reduction in lesion counts) 2) Mean diameter of target lesion at baseline and week 12 3) Local skin reactions graded on scale 4) Minor adverse events at week 1 and 12 5) Total score for cosmetic appearance of a target lesion (1 target lesion per treatment side) Efficacy Methods: quantitative assessment using lesion counting and recording of diameters of the target lesions Time points: at baseline and weeks 3, 6, 9, and 12 of therapy Safety Methods: side‐effects, such as erythema, dryness, burning sensation, and pruritus, graded from 0 to 3 (0 = none, 1 = mild, 2 = moderate, and 3 = severe) Time points: at baseline and weeks 3, 6, 9, and 12 of therapy Cosmetic Methods: total scores of the target lesions = the sum of erythema, desquamation, and induration scored between 0 to 3 Time points: at baseline and weeks 3, 6, 9, and 12 of therapy |
|
| Funding | ‐ | |
| Notes | Similar number of adverse events reports between to treatment and placebo were reported. Neutrogena sunscreen was used. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate allocation generation was achieved with a random digits table. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was assessor‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used, and there was missing information about participants lost after the baseline evaluation. |
| Selective reporting (reporting bias) | High risk | The severity of local skin reactions was not reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) followed by imiquimod once per day, twice per week for 16 weeks (N = 25 participants) Control intervention B: ALA‐PDT followed by vehicle once per day, twice per week for 16 weeks (N = 25 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 2 Interval between treatments: 4 weeks Preparation of lesions: microdermabrasion Cream concentration (%): 20% Application of cream: ‐‐ Incubation with cream: 1 hour Type of light: blue light Light source: Blu‐U 4170 Wavelength (nm): (417) Energy fluence (J/cm²): ‐‐ Intensities (mW/cm²): ‐‐ Exposure time: 8 min |
|
| Outcomes |
Primary outcome of the trial 1) Median and mean per cent reduction of the number of lesions at baseline and month 12 (= mean percentage of reduction in lesion counts) Secondary outcome of the trial 1) Severe local skin reactions (pooled) Other outcomes of the trial 1) Median and mean lesion counts at baseline and month 12 (converted to mean reduction of lesion counts) 2) Participant complete clearance rates 3) Treatment‐related adverse events (= minor adverse events) 4) Rest periods Efficacy Methods: quantitative assessment using lesion counting and mapping by marking their locations on clear acetate templates using permanent marker Time points: at baseline and months 1, 2, 3, 4, 6, and 12 of the study Safety Methods: 1. incidence of severe local skin reactions (erythema, edema, erosion/ulceration, scabbing/crusting, weeping/exudates, vesicles, and flaking/scaling/dryness) associated with imiquimod treatment and compare the incidence to those reported in imiquimod studies in which PDT pretreatment was not utilised, 2. assessment of local skin reactions types on a 4‐point scale (0 = none, 1 = mild, 2 = moderate, 3 = severe) by the investigator Time points: at months 2, 3, 4, and 6 |
|
| Funding | This study was supported by 3M Pharmaceuticals and Graceway Pharmaceuticals. | |
| Notes | The data were changed for intention‐to‐treat (ITT) analysis for meta‐analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A previous generated list using a random number generator was used. |
| Allocation concealment (selection bias) | Low risk | Sequential assignment upon participation enrolment was used. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported, and the lost was before treatment) Control ‐ B: 1 dropout (the reason was reported, and the lost was before treatment) |
| Selective reporting (reporting bias) | High risk | The number of participants with severe local skin reactions was not given separately for the 2 treatments. The standard deviations associated with mean values were not given. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: March 2005 End date: September 2005 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: 0.0025% ingenol mebutate, once per day at days 1 & 2 or days 1 & 8 (N = 15 participants) B: 0.01% ingenol mebutate, once per day at days 1 & 2 or days 1 & 8 (N = 16 participants) C: 0.05% ingenol mebutate, once per day at days 1 & 2 or days 1 & 8 (N = 15 participants) Control intervention D: vehicle, once per day at days 1 & 2 or days 1 & 8 (N = 12 participants) |
|
| Outcomes |
Primary outcomes of the trial 1) Application site reactions (pain) 2) Local skin responses (= local skin reactions) 3) Treatment‐related adverse events (= minor adverse events) 4) Serious adverse events 5) Clinical laboratory tests Secondary outcomes of the trial 1) Lesion complete response rates at 85 days (target lesions) 2) Lesion complete and marked clinical clearance rates at 85 days 3) Participant partial (> 80%) clearance rates at 85 days [included in participant partial (> 75%) clearance] 4) Participant histological clearance rates at 85 days Other outcomes of the trial 1) Cosmetic outcomes: changes in pigmentation Efficacy Methods: 1. clinical evaluation of each lesion by the investigator, 2. histological evaluation by a central blinded dermatopathologist based on a repeat biopsy of the lesion biopsied prior to treatment. Time points: at day 85 (end of study) Definitions: 1. complete clearance (no evidence of residual disease), 2. marked clearance (50% to 90% improvement), 3. slight clearance (10% to 50% improvement), 4. unchanged (10%), and 5. worsened (clinically‐observable growth) Safety Methods: 1. vital signs, 2. physical examinations, 3. laboratory tests (haematology, serum chemistry, liver function tests, and urinalysis), 4. recording of local skin reactions (itching, erythema,oedema, erosion/ulceration, scabbing/crusting, weeping/exudates, vesicles, flaking/scaling/dryness) and abnormal skin proliferation (treatment was withheld if a severe local skin reaction occurred prior to the second scheduled dose) Time points: 1. at each visit (vital signs), 2. at screening and final visit (day 85) (physical exam), 3. at screening, last day of treatment, and day 85 (or early exit) (laboratory tests) Cosmetic Methods: recording of hypopigmentation, hyperpigmentation and scarring Time points: at day 85 Definitions for local skin reaction rating: 1. mild (easily tolerated), 2. moderate (associated with discomfort sufficient to interfere with usual activities), and 3. severe (incapacitating with inability to work or perform usual activities). |
|
| Funding | This study was supported by Peplin Ltd. | |
| Notes | This was a phase IIa study. Application was done only on predetermined 5 lesions with 2 template diameters. There were 2 application regimens, i.e. days 1 & 2 or days 1 & 8, but no differences were detected and results were pooled together. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 17): "Randomisation was performed by an independent clinical research organisation and identical packaging was used to maintain blinding of both investigator and patients regarding allocation to active or vehicle gel." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | Randomisation sequence was generated by an independent company (see previous quote in random sequence generation). |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Modified intention‐to‐treat (ITT) analysis was used (participants with no treatment were excluded). Intervention ‐ A: 0 dropouts, B: 1 dropout (the reason was reported), C: 0 dropouts Control ‐ D: 0 dropout |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported based on protocol NCT00107965, as well as the mistakes made in the dose application schedule. |
| Other bias | High risk | There were higher percentages of women and scalp lesions in the vehicle group at baseline. |
| Methods | This was a randomised, active‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: aminolevulinic acid (ALA)‐blue light photodynamic therapy (PDT) (N = 12 participants) B: ALA‐pulsed dye laser (PDL) PDT (N = 12 participants) Control intervention C: 0.5% 5‐fluorouracil once or twice daily for 4 weeks (N = 12 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 2 Interval between treatments: 4 weeks Preparation of lesions: ‐‐ Cream concentration (%): 20% Application of cream: ‐‐ Incubation with cream: 1 hour Type of light: blue light or pulsed dye laser Light source: Blu‐U Photodynamic Therapy Illuminator Wavelength (nm): 595 (PDL) Energy fluence (J/cm²): 7.5 (PDL) Intensities (mW/cm²): ‐‐ Exposure time: 1000 sec (16 min), 10 ms (PDL) |
|
| Outcomes |
Outcomes of the trial 1) 100% lesions cleared (= participant complete response) at 4 weeks post‐treatment 2) > 75% lesions cleared (= participant partial clearance) at 4 weeks post‐treatment 3) Tolerability, i.e. grading of local skin reactions 4) Photoageing Efficacy Methods: 1. grading of target lesions on a 4‐point scale (from resolved to very thick, markedly keratotic, or both), 2. high magnification digital photography of lesions identified with 3 black ink dots and small adhesive label Time points: at baseline and the end of treatment, 2 weeks and 4 weeks post‐treatment Definitions: therapeutic success (sustained clearance of 75% or more of target lesions) Safety Methods: grading of erythema, edema, crusting/erosions, and stinging/ burning Time points: immediately after PDT treatments Definitions for erythema score: 0 (none), 1 (minimal, scant rare erythema), 2 (mild, easily‐seen erythema up to ⅓ of the treated area), 3 (moderate, easily‐seen erythema involving between ⅓ to ⅔ of the treated area), 4 (severe, easily‐seen erythema involving over ⅔ of the treated area) Definitions for oedema score: 0 (none), 1 (minimal, scant rare oedema), 2 (mild, easily‐seen oedema, minimally palpable, involving up to ⅓ of the treated area), 3 (moderate, easily‐seen oedema and typically palpable involving between ⅓ to ⅔ of the treated area), 4 (severe, easily‐seen oedema, indurated in some areas involving over ⅔ of the treated area) Definitions for crusts and erosions score: 0 (none), 1 (rare, a few 3 mm or smaller areas), 2 (mild, up to 12 lesions 3 mm or less, areas readily seen), 3 (moderate), 4 (severe) Definitions for stinging/burning score: 0 (none), 1 (minimal), 2 (moderate), 3 (severe) Cosmetic Methods: 1. global response, 2. assessment of tactile roughness by lightly palpitating by stroking gently with the index finger and molted hyperpigmentation (including area involved, the colour intensity, and the evenness of pigment distribution) Definitions for global response: 0 (complete response = complete resolution of photodamage), 1 (almost complete response = very significant improvement in photodamage, approximately 90% improvement), 2 (marked response = significant improvement in photodamage, approximately 75% improvement), 3 (moderate response = intermediate improvement in photodamage, approximately 50% improvement), 4 (slight response = some improvement in photodamage), 5 (no response), 6 (condition worsened) Definitions for tactile roughness grading: 0 (skin is very smooth), 1 (skin is smooth with very occasional rough area), 2 (mild roughness), 3 (moderate roughness), 4 (severe roughness) Definitions for molted hyperpigmentation grading: 0 (evenly pigmented skin), 1 (light hyperpigmentation involving small areas), 2 (moderate hyperpigmentation involving small areas, light hyperpigmentation involving moderate areas), 3 (moderate hyperpigmentation involving moderate sized areas, light hyperpigmentation involving large areas, small areas of heavy hyperpigmentation), 4 (heavy hyperpigmentation) |
|
| Funding | This study was supported by DUSA Laboratories. | |
| Notes | PDT treatments were better tolerated than 5‐fluorouracil. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 630): "Caucasian patients with a minimum of 4 nonhyperkeratotic AK of either the face or scalp were recruited and
randomised to 3 treatment groups." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The blinding was not stated, but 2 physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The blinding was not stated, but 2 physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A & B: 0 dropouts Control ‐ C: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | The percentages of participants reporting adverse events were not given except for stinging. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 3% diclofenac in hyaluronic acid for 90 days (N = 53 participants) Control intervention B: hyaluronic acid for 90 days (N = 55 participants) |
|
| Outcomes |
Outcomes of the trial 1) Complete clearing of lesions (= participant complete clearance) rates at 30 days post‐treatment 2) Application site reactions (for the 3 studies included in insert, i.e. Rivers 2002; Solaraze study 2; Wolf 2001) reported as incidences (i.e. number of events, not number of participants) 3) Minor adverse events (for the 3 studies included in insert, i.e. Rivers 2002; Solaraze study 2; Wolf 2001) reported as incidences (i.e. number of events, not number of participants) |
|
| Funding | This study was supported by Nycomed US Inc. | |
| Notes | This study was included in the product package insert as study 2. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation was not stated in the product insert, but the other 2 studies included were randomised. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The blinding was not stated in the product insert, but the other 2 studies were double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The blinding was not stated in the product insert, but the other 2 studies were double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The type of analysis was not stated, but the 2 other studies had intention‐to‐treat (ITT) analysis. |
| Selective reporting (reporting bias) | High risk | This was the only study of 3 presented in the Solaraze product insert that was not published and with no significant difference for participant complete clearance. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, active‐controlled, intraindividual study. Start date: September 2007 End date: July 2008 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 30 participants) Control intervention B: 5% imiquimod once per day, 3 times per week for 4 weeks on, 4 weeks off (N = 30 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 15 days Preparation of lesions: crust removed by curettage Cream concentration (%): 20% Application of cream: onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 4 hours Type of light: red light Light source: Waldmann PDT 1200 Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 75 Exposure time: ‐‐ |
|
| Outcomes |
Outcomes of the trial 1) Lesion complete response rates at 1 and 6 months 2) Application site reactions 3) Local skin reactions 4) Investigator‐assessed cosmetic outcome 5) Participant's preference Efficacy Methods: quantitative assessment using counting and recording of lesions by the same examiners Time points: at baseline and 1 and 6 months post‐treatment Definitions: 1. clinical lesion response (complete response = complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion). Safety Methods: recording of adverse events (severity, duration, and need for additional therapy) Time points: at each visit Cosmetic Methods: assessment by investigators based on the amount of scarring, atrophy, induration, erythema, and pigment change within the treated area in comparison with adjacent, untreated skin Time points: at month 6 post‐treatment Definitions: 1. excellent (no erythema, change in pigmentation, scarring, atrophy, or induration), 2. good (slight to moderate erythema or change in pigmentation, but no scarring, atrophy, or induration), 3. fair (slight scarring, atrophy, or induration), 4. poor (moderate to extensive scarring, atrophy, or induration) |
|
| Funding | ‐ | |
| Notes | There was a difference in lesion complete response between treatments for grade II lesions, i.e. 57.8% for PDT and 37% for imiquimod, but not for grade I lesions (71% to 72%). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1062): "Eligible patients received PDT treatment and treatment with imiquimod 5% cream randomly allocated to alternate upper extremities." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The blinding was not stated, but 2 physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The blinding was not stated, but 2 physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 2 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | All outcomes were reported, i.e. significantly different or not. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod cream, 3 times (or less based on adverse events) per week for a maximum of 12 weeks (N = 25 participants) Control intervention B: placebo, 3 times (or less based on adverse events) per week for a maximum of 12 weeks (N = 11 participants) |
|
| Outcomes |
Outcomes of the trial 1) Participant complete clearance rates at 14 weeks. 2) Participant partial clearance rates 3) Local skin reactions (graphical representation) 4) Minor adverse events (graphical representation) 5) Recurrence 6) Compliance 7) Rest periods Efficacy Methods: 1. clinical evaluation, 2. biopsy (histology) assessed by the same dermatopathologist Time points: at baseline and week 14 after treatment initiation Definitions: 1. complete clearance (complete clinical clearance confirmed histologically), 2. partial clearance (the clearance of 1 or more lesions treated with imiquimod) Safety Methods: 1. vital signs recording; 2. photography; 3. assessment and recording of local and systemic adverse or abnormal effects; 4. recording of the incidence and severity of erythema, edema, induration, vesicles, erosion, ulceration, excoriation or flaking, and scabbing on a scale of 1 (mild) to 3 (severe) Time points: at each visit (at weeks 2, 3, 6, 9, and 12) |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | The recurrence rate at 1 year was 10% (2/25) for participants treated with imiquimod. A sample size calculation was based on rate of spontaneous healing of actinic keratoses lesions. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 1500): "Each patient was randomly assigned a number that was paired with a box containing 12 sachets." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | The key to codes were held by the pharmaceutical company. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: January 2008 End date: June 2008 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: 3.75% imiquimod, once daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 160 participants) B: 2.5% imiquimod, once daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 160 participants) Control intervention C: placebo, once daily for 2 weeks on, 2 weeks off, 2 weeks on (N = 159 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates at week 14 Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates at week 14 2) Median percentage of reduction in lesion counts 3) Local skin reactions Other outcomes of the trial 1) Participants experiencing at least 1 adverse event 2) Application site reactions including irritation 3) Treatment‐related adverse events (= minor adverse events) 4) Serious adverse events 5) Clinical laboratory tests 6) Investigator global integrated photodamage (IGIP‐cosmetic outcome) 7) Number of participants with the different cosmetic outcomes 8) Rest periods Efficacy Methods: quantitative assessment by counting all of the visible or palpable lesions (baseline or new) in the treatment area by the investigator Time points: at each visit Definitions: 1. complete clearance rate (proportion of participants at the end‐of‐study visit with a count of 0 lesions in the treatment area), 2. partial clearance rates (proportion of participants with 75% or more reduction in lesion count in the treatment area at the end‐of‐study visit as compared with baseline), 3. per cent change (changes in lesion number at the end‐of‐study visit as compared with baseline) Safety Methods: 1. measurement of vital signs; 2. recording and coding (Medical Dictionary for Regulatory Activities) of adverse events; 3. investigator assessment of local skin reactions (erythema, edema, weeping/exudate, flaking/scaling/dryness, scabbing/crusting, and erosion/ulceration) graded as none, mild, moderate, or severe; 4. laboratory tests (hematology, serum chemistry, and urinalyses) (treatment‐emergent adverse events were summarised for each treatment group by preferred term, intensity, and investigator assessment of relationship to study cream) Time points: 1. at each visit, 2. pre‐study visit and end‐of‐study visit (laboratory tests) Cosmetic Methods: an overall assessment (IGIP score) of the participant's photodamage change from baseline in the treatment area (including an integrated assessment of fine wrinkling, coarse wrinkling, mottled pigmentation, roughness, shallowness, skin laxity, and telangiectasias) rated on a 7‐point symmetric scale, ranging from significantly improved = +3 to significantly worse = ‐3 Time points: at end‐of‐study visit |
|
| Funding | This study was supported by Graceway Pharmaceuticals LLC. | |
| Notes | Data from 2 studies were pooled together. Temporary dosing interruptions could have been instructed by the investigator to manage local skin reactions and adverse events. A sample size calculation was provided. A follow‐up study was published (Hanke 2011). | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 584): "Eligible patients were centrally randomised to placebo, imiquimod 2.5%, or imiquimod 3.75% cream in a 1:1:1 treatment allocation." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation was used. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This study was double‐blinded (subject, caregiver, investigator), but the authors mentioned that adverse events could be an issue for the concealment of the assigned treatment in some participants. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This study was double‐blinded (outcomes assessor), but authors mentioned that adverse events could be an issue for the concealment of the assigned treatment in some participants. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 11 dropouts (the reasons were reported), B: 6 dropouts (the reasons were reported) Control ‐ C: 9 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | All outcomes from the protocol (NCT00605176) were reported. |
| Other bias | Unclear risk | Data for safety were reported differently in the published study, and the data results linked to the protocol. |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel‐group study. Start date: August 2008 End date: February 2009 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 0.05% ingenol mebutate for 2 days (N = 117 participants) Control intervention B: vehicle for 2 days (N = 118 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates at day 57 Secondary outcome of the trial 1) Participant partial (percentage criteria was not specified) clearance rates at day 57 Other outcomes of the trial 1) Median percentage reduction in lesion counts 2) Local skin reactions (qualitative) 3) Treatment‐related adverse events (qualitative) 4) Serious adverse events 5) Pigmentation changes (cosmetic) 6) Compliance Efficacy Time points: on days 3, 8, 15, 29, and 57 Safety Methods: 1. assessment of the incidence rate of adverse events, serious adverse events, and local skin responses; 2. grading of local skin responses Time points: on days 3, 8, 15, 29, and 57 Cosmetic Methods: assessment of pigmentation and scarring Time points: on days 3, 8, 15, 29, and 57 |
|
| Funding | This study was supported by Peplin Ltd. | |
| Notes | This study report was a conference abstract and was included in the following study awaiting classification Lebwohl 2012. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page AB2): "A total of 255 patients were randomised to treatment with 0.05'X, ingenol mebutate gel or vehicle." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The type of analysis was not stated. Only 1 dropout due to adverse events was reported, but the treatment group was not specified. |
| Selective reporting (reporting bias) | Unclear risk | All outcomes in the protocol (NCT00742391) were reported in the published study. Another similar study (NCT00942604) has not been published yet. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, open, active‐controlled, parallel‐group study. Start date: April 1999 End date: November 1999 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) (N = 102 participants) Control intervention B: cryotherapy: prior skin preparation, variable liquid nitrogen spray unit, 1 to 2 mm rim of frozen tissue beyond marked outline, 2 freeze‐thaw cycles in the same session; mean freeze time of 24 + 18 seconds, (N = 100 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: once for face and scalp, twice for others (8% of lesions) Interval between treatments: 1 week Preparation of lesions: crusts removed by curettage Cream concentration (%): 16% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: ‐‐ Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 70 to 200 Exposure time: 10 min |
|
| Outcomes |
Outcomes of the trial 1) Lesion complete response rates at 3 months post‐treatment 2) Skin irritation 3) Local adverse reactions 4) Investigator's and participant's cosmetic outcomes in participants with > 75% reduction of total lesions: number of participants (excellent and good pooled together) 5) Participants' satisfaction Efficacy Time points: at 3 months after the initial treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion) Safety Methods: recording of adverse events (including the local phototoxicity due to PDT) Time points: before and after illumination, after 2 weeks by telephone contact, and after a final examination 3 months post‐treatment Cosmetic Methods: assessment and grading of overall cosmetic outcome Time points: at 3 months after the initial treatment Definitions: 1. excellent (only slight occurrence of redness or change in pigmentation), 2. good (moderate redness or change in pigmentation), 3. fair (slight to moderate scarring, atrophy, or induration), and 4. poor (extensive scarring, atrophy, or induration) |
|
| Funding | This study was supported by Photocure ASA. | |
| Notes | Higher response rates were obtained with thin lesions. High participant satisfaction was obtained with MAL‐PDT. 43% of participants treated with MAL‐PDT reported local adverse events compared to 26% treated with placebo. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate allocation sequence was generated by stratification with respect to the number of lesions. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This study was open because 2 physically distinct treatments were compared. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This study was open because 2 physically distinct treatments were compared. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There was discrepancy in the text (415) and table (384) for number of lesions in the PDT group. Per‐protocol (PP) analysis was used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 5 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Overall cosmetic outcomes were pooled together. Satisfaction was reported for PDT participants only. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, vehicle‐controlled, parallel group study. Start date: January 2002 End date: March 2003 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: imiquimod 5% cream, once per day, 3 days per week for 16 weeks or less (N = 147 participants) Control intervention B: vehicle, once per day, 3 days per week for 16 weeks or less (N = 139 participants) |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates at 8 weeks post‐treatment Secondary outcome of the trial 1) Participant partial (> 75%) clearance rates at 8 weeks post‐treatment Other outcomes of the trial 1) Histological clearance at 8 weeks post‐treatment 2) Clinical laboratory tests 3) Application site reactions (including irritation) 4) Local skin reactions 5) Minor adverse events 6) Serious adverse events 7) Skin quality (cosmetic) Efficacy Methods: 1. quantitative assessment using clinical counting and recording the number lesions present in the treatment area, 2. by the histologic result from the biopsy specimen of a predefined lesion biopsy site Time points: 1. at weeks 1, 2, 4, 8, 12, 16 (end of treatment), and 24 (8 weeks post‐treatment); 2. at the 8‐week post‐treatment visit (biopsy) Definitions: 1. complete clearance rate (proportion of participants at the 8‐week post‐treatment visit with no evidence of lesion on the histology result of the post‐treatment target lesion biopsy site and no clinically‐visible lesions in the remainder of the treatment area), 2. partial clearance rate (proportion of participants at the 8‐week post‐treatment visit with at least 75% reduction in the number of lesions counted at baseline in the treatment area) Safety Methods: 1. photography of the treatment area; 2. reviewing adverse events and local skin reactions (erythema, edema, erosion/ulceration, scabbing/crusting, weeping/exudate, vesicles, or flaking/scaling/dryness) rated on a scale of 0 (none) to 3 (severe) and concomitant medication use; 3. clinical laboratory tests (hematology, serum chemistry, urinalyses, and pregnancy test) Time points: 1. at weeks 1, 2, 4, 8, 12, 16 (end of treatment), and 24 (8 weeks post‐treatment), 2. pre‐study and end‐of‐study visits (laboratory tests) Cosmetic Methods: visual, clinical, and tactile examinations of skin quality within the treatment area by investigator [skin surface (roughness/dryness/scaliness), hyperpigmentation, hypopigmentation, mottled or irregular pigmentation (both hyperpigmentation and hypopigmentation), degree of scarring, and atrophy] on a scale of 0 (none) to 3 (severe) Time points: at the treatment initiation and 8‐week post‐treatment visits |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a phase III study. A high rate of agreement was observed between clinical and histologic lesion clearances. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | An adequate method of randomisation was achieved by the use of a computer‐generated randomisation schedule. |
| Allocation concealment (selection bias) | Low risk | Adequate allocation concealment was achieved by sealed, tamper‐proof envelopes containing a number allocated to each participant. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Intention‐to‐treat (ITT) analysis was used, but some lost to follow up participants were missing for the description. Intervention ‐ A: 10 dropouts (the reasons were not all reported) Control ‐ B: 18 dropouts (the reasons were not all reported) |
| Selective reporting (reporting bias) | High risk | As a similar study (Lebwohl 2004) was also supported by 3M Pharmaceuticals not all skin quality outcomes were reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, active‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: 0.03% resiquimod, once per day, 3 days per week, 4 week on, 8 weeks off, 1or 2 courses (N = 31 participants) B: 0.06% resiquimod, once per day, 3 days per week, 4 week on, 8 weeks off, 1or 2 courses (N = 32 participants) C: 0.1% resiquimod, once per day, 3 days per week, 4 week on, 8 weeks off, 1or 2 courses (N = 34 participants) Control intervention D: 0.01 % resiquimod, once per day, 3 days per week, 4 week on, 8 weeks off, 1or 2 courses (N = 35 participants) The gel application was done using a dosing paper template. |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates after 1 to 2 treatment courses (week 24) Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates after 1 to 2 treatment courses 2) Participant complete clearance rates after 1 course only (week 12) . Other outcomes of the trial 1) Application site reactions 2) Severe local skin reactions 3) Treatment‐related adverse events (minor adverse events) 4) Serious adverse events 5) Clinical laboratory tests 6) Compliance Efficacy Methods: quantitative assessment using lesion counting and mapping with a transparent plastic template by a qualified dermatologist Time points: at baseline; weeks 2, 4, 8, and 12 for course 1, and if applicable, at weeks 14, 16, 20, and 24 for course 2 Definitions: 1. overall complete clearance rate (proportion of participants at the end of course 1 (week 12) or course 2 (week 24) with no lesions in the treatment area), 2. partial clearance rate (proportion of participants at their last study visit with at least 75% reduction in the number of lesions in the treatment area) Safety Methods: 1. recording of adverse events, 2. assessment of local skin reactions (erythema, oedema, erosion ⁄ulceration, weeping ⁄exudate, flaking ⁄scaling ⁄dryness, and scabbing ⁄crusting), 3. photographs of treatment area, 4. laboratory tests (haematology, biochemistry, urine analysis, and where applicable, pregnancy tests), 5. vital signs measurements and physical examination, and if appropriate, skin cultures (suspected infection) or skin biopsy (lesion suspicious for malignancy) Time points: at weeks 2, 4, 8, and 12 for course 1, and if applicable, at weeks 14, 16, 20, and 24 for course 2 |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a phase II study. Serious adverse events and local skin reactions were more frequent with higher doses, and there was lowest compliance in the 0.1% group. A sample size calculation was provided. Intention‐to‐treat data were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Pre‐assigned numbering with 1:1:1:1 randomisation with a block size 4 was used for allocation generation. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was not stated. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | This was not stated. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) and per‐protocol (P) analyses were used. Intervention ‐ A: 5 dropouts (the reasons were reported), B: 10 dropouts (the reasons were reported), C: 14 dropouts (the reasons were reported) Control ‐ D: 2 dropouts (the reasons were reported) Dropping rates were higher for higher doses, i.e. 6%, 16%, 31%, and 41% for 0.01, 0.03, 0.06, and 0.1% resiquimod groups. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | High risk | The values for overall partial (> 75%) clearance were lower than overall complete clearance. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: March 2006 End date: January 2007 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: MAL‐photodynamic therapy (PDT) (N = 57 participants) Control intervention B: placebo‐PDT (N = 58 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 2 Interval between treatments: 1 week Preparation of lesions: crusts and scales removed by curettage Cream concentration (%): 16% Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light LED Light source: Aktilite CL 128 Wavelength (nm): 630 Energy fluence (J/cm²): 37 Intensities (mW/cm²): 56 to 83 Exposure time: 9 min |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete response (= participant complete clearance) rates at 3 months after last treatment Secondary outcomes of the trial 1) Lesion complete response rates at 3 months post‐treatment 2) Treatment site reactions (= application site reactions) reported by events (i.e. not per participants) 3) Local skin reactions (in general and severe) Other outcomes of the trial 1) Participants experiencing at least one adverse event 2) Treatment‐related adverse events (= minor adverse events) reported by events (i.e. not per participants) 3) Minor adverse events 4) Serious adverse events including squamous cell carcinoma 5) Percentage of participants with new lesions Efficacy Methods: 1. quantitative assessment using inspection, palpation, and characterisation of lesions (Olsen 1991) by the same investigator, who was not involved in the treatment procedure, 2. documentation of any lesions not present at baseline (lesions with a non‐complete response were treated at the discretion of the investigator) Time points: at baseline and 3 months post‐treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion), 3. participant complete response (all participants in whom 100% of lesions had responded completely 3 months post‐treatment) Safety Methods: 1. assessment of tolerability, 2. recording of adverse events (severity, localisation, duration, and need for additional treatment) (the clinician assessed the causal relationship of the event to the study treatment as related, uncertain, or not related) Time points: after lesion preparation before cream application, at the end of the 3‐hour cream application, after illumination during each treatment session, and at 2 weeks and 3 months post‐treatment |
|
| Funding | This study was supported by Photocure ASA. | |
| Notes | A sample size calculation was provided. This study was study #2 in the Metvixia product insert 2008. The studies included in the Metvixia product insert were changed between 2004 (PhotoCure) and 2008 (Galderma), which correspond to the use of different types of light. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A computer‐generated randomisation scheme was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | A clinician not involved in treatment procedure assessed response. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Low risk | All outcomes were presented based on the protocol (NCT00304239), and protocol mistakes were acknowledged. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: ALA‐photodynamic therapy (PDT) (N = 81 participants) Control intervention B: placebo‐PDT (N = 41 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 12 weeks Preparation of lesions: crusts removed by curettage, roughening, and alcohol wiping Cream concentration (%): BF‐200 gel Application of cream: air dry for 10 min Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Aktilite CL 128 or PhotoDyn 750 Wavelength (nm): 590‐670 (Aktilite), 595‐1400 (PhotoDyn) Energy fluence (J/cm²): 37 (Aktilite), 170 (PhotoDyn) Intensities (mW/cm²): 50‐70 (Aktilite), 196 (PhotoDyn) Exposure time: 15 minutes (PhotoDyn) |
|
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance at 12 and 24 weeks Secondary outcome of the trial 1) Lesion complete response at 12 and 24 weeks Other outcomes of the trial 1) Local skin reactions for first and second treatment by light sources and in general 2) Cosmetic outcomes: general Efficacy Methods: 1. quantitative assessment of lesion clearance by visual inspection and by palpation by an investigator not involved in treatment and safety evaluation, 2. histological assessment using biopsy of a lesion defined and marked before the PDT treatment Time points: 1. at baseline; 3, and 12 weeks post‐treatment, 2. end‐of‐study visit (biopsy) Definitions: participant complete clearance (all lesions were considered to be cleared both by the clinical and histological assessment) Safety Methods: 1. recording of adverse effects, 2. documentation of local adverse reactions (pain, itching, burning, erythema, oedema, and induration) at the application site and rated as mild, moderate, and severe by the assessing physician or reporting participants, 3. serious adverse events Time points: 1. at 1 week after PDT (by phone) and 3 weeks, 2. during and after PDT (local adverse reactions) Cosmetic Methods: 1. general cosmetic outcome assessed by the investigator as very good, good, unsatisfactory, and impaired; 2. assessment of skin quality Time points: at 12 weeks post‐treatment |
|
| Funding | This study was supported by Biofrontera Bioscience GmbH. | |
| Notes | Pain, itching, and burning were reported separately for 1st and 2nd treatment, anatomical area, and light sources. In general, more symptoms were reported for Aktilite CL128 than PhotoDyn 750. A sample size calculation was provided. Data with intention‐to‐treat analyses were used for meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | A validated SAS programme based on the random number function RANUNI and random blocks for 6 participants was used. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Treatment and safety assessment were performed by 1 investigator and efficacy assessment by another. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) and per‐protocol (PP) analyses were used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 4 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | All outcomes were reported, but little information was given on adverse events and cosmetic outcomes. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, parallel‐group study (part 1). Start date: April 2005 End date: December 2006 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions | Part 1: Intervention A: cryotherapy: 3 to 5 second freeze cycle followed (2 weeks after) by 5% imiquimod cream applied twice weekly for 8 weeks (N = 33 participants) Control intervention B: cryotherapy: 3 to 5 second freeze cycle followed (2 weeks after) by vehicle cream for 8 weeks (N = 32 participants) Part 2: Participants with residual actinic keratosis lesions were offered cryotherapy and open‐label imiquimod twice weekly for 8 weeks. |
|
| Outcomes |
Primary outcomes of the trial (protocol) 1) Recurrence rate 2) Time to recurrence of lesions Secondary outcomes of the trial (protocol) 1) Time to reach treatment success 2) Proportion of participants completely clear [= participant complete clearance rates for target, subclinical and total lesions at week 22 (i.e. part 1 only)] 3) Participant improvement assessment Other outcomes of the trial 1) Clearance rates of target lesion (= lesion complete response) 2) Skin irritation 3) Treatment‐related adverse events (= minor adverse events) 4) Serious adverse events 5) New actinic keratoses (subclinical) during the study Efficacy Methods: quantitative assessment using lesion mapping on a transparent overlay map Time points: at baseline, at week 22 after cryotherapy (end of part 1) Definitions: 1. target lesions (those within a designated 50 cm² treatment field established at baseline), 2. subclinical lesions (those within the designated treatment field unapparent at baseline), 3. total lesions (the sum of target and subclinical lesions) Safety Methods: monitoring of the frequency and duration of adverse events (local and systemic) Time points: at every study visit |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | An increase in subclinical actinic keratoses was observed within the first 3 weeks of imiquimod treatment with a subsequent progressive reduction there after, but this was not observed with vehicle. This was a 2‐part study: part 1, included in the meta‐analyses, is randomised, double‐blind, and controlled, but part 2 is an optional open study not included in this review. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 2): "The initial randomised double‐blind phase in which subjects were allocated to either vehicle or imiquimod 5% cream twice weekly for 8 weeks was followed by an optional open‐label phase..." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded for part I. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded for part I. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | High risk | Some outcomes (recurrence rate, time to recurrence, time to reach success, participant improvement assessment) in the protocol (NCT00110682) were not presented in the published study. |
| Other bias | High risk | There were differences between the protocol and the published report. The primary and secondary outcomes in the published report were different than the protocol. The protocol did not mention the second part of the study. |
| Methods | This was a multicentre, randomised, assessor‐blinded, active‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% 5‐fluorouracil twice daily for 2 to 4 weeks (N = 20 participants) Control intervention B: 5% imiquimod twice weekly for 16 weeks (N = 19 participants) |
|
| Outcomes |
Outcomes of the trial 1) Physician global assessment as scores (presented in a graph) 2) Lesion counts at baseline, during and after treatment (= lesion complete response) 3) Participant complete and partial (> 66%) clearance 4) Mean percentage of reduction in lesion counts 5) Physician's grading of erythema (scores represented in a graph) 6) Local skin reactions (qualitative) 7) Participant's perception of efficacy 8) New/subclinical lesions Efficacy Methods: 1. quantitative assessment using lesion counting, 2. qualitative assessment using physician's global assessment and participant's perception of efficacy Time points: at baseline and weeks 4, 8, 12, 16, and 24 Definitions for physician's global assessment and the participant's perception of efficacy scales: 1 (very effective), 2 (moderately effective), 3 (slightly effective), 4 (not effective at all) Safety Methods: 1. physician's assessment of erythema on a 0 = none to 3 = severe scale, 2. participant perception of discomfort associated with the treatment. Time points: at baseline and weeks 4, 8, 12, 16, and 24 Definitions for participant's perception of discomfort scale: 1 (very painful), 2 (moderately painful), 3 (slightly painful), 4 (not painful at all) |
|
| Funding | This study was supported by Valeant Pharmaceuticals International. | |
| Notes | Treatment with 5‐fluorouracil, but not imiquimod, uncovered and treated subclinical lesions. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 145): "Patients were randomly assigned to receive one of the following treatments to be applied in a thin layer to completely cover each affected cosmetic unit:.." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The 2 distinct dosing schedules were not concealed with a double‐dummy technique. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The assessor was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat was used. Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 2 dropouts (the reasons were provided) |
| Selective reporting (reporting bias) | High risk | Values for participant’s perception of efficacy were not presented. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, open, active‐controlled, parallel‐group study. Start date: January 2002 End date: October 2002 |
|
| Participants |
Inclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: methyl aminolevulinate (MAL)‐photodynamic therapy (PDT) once (N = 105 participants) Control intervention B: MAL‐PDT twice with a 1 week interval (N = 106 participants) There was possible retreatment for single treatment group (not included in meta‐analysis). Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 1 week Preparation of lesions: crusts removed by curettage and gentle scrapping Cream concentration (%): 16 Application of cream: 1 mm thick onto lesion and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Aktilite CL 16 Wavelength (nm): 590‐670 Energy fluence (J/cm²): 37 Intensities (mW/cm²): 750 to 2050 Exposure time: 8 min |
|
| Outcomes |
Outcomes of the trial 1) Lesion complete response at 3 months post‐treatment 2) Participant complete response (= participant complete clearance) 3) Participants experiencing at least 1 adverse event 4) Local adverse events 5) Lesion cosmetic outcomes Efficacy Methods: assessment by investigator Time points: at 3 months post‐treatment Definitions: 1. lesion complete response (complete disappearance of the lesion), 2. lesion non‐complete response (incomplete disappearance of the lesion) Safety Methods: recording of adverse events, including local phototoxicity reactions that normally occur after PDT, and rating as mild, moderate, or severe (the clinician assessed the causal relationship of any adverse events to the study treatment as related, uncertain, or not related) Time points: before and after illumination, and at 3 months post‐ treatment Cosmetic Methods: assessment of hypopigmentation, hyperpigmentation, scar formation, and tissue defect by their rating as none, slight, or obvious for each lesion that had responded completely Time points: at 3 months post‐treatment |
|
| Funding | This study was supported by PhotoCure ASA. | |
| Notes | A sample size calculation was provided. Data for intention‐to‐treat analysis were used for the meta‐analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 425): "The randomisation was performed after the patient was included in the study." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | Sealed envelopes were used to conceal the allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This study was open. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | This study was open. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 0 dropouts Control ‐ B: 6 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Low risk | The results were similar to previously reported data in an abstract. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single ‐centre, randomised, placebo‐controlled, parallel‐group study. Start date: September 1991 End date: March 1992 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: sunscreen SPF 17 (8% 2‐ethyl‐hexyl p‐methoxycinnamate/2% 4‐tert‐butyl‐4‐methoxy‐4‐dibenzoylmethane), as needed daily for 7 months (N = 221 evaluable participants) Control intervention B: placebo, as needed daily for 7 months (N = 210 evaluable participants) |
|
| Outcomes |
Outcomes of the trial 1) Mean reduction/increase in lesion counts at 7 months (= mean reduction in lesion counts) 2) New lesions 3) Number and per cent of baseline lesion remitting (= lesion complete response) 4) Compliance Efficacy Methods: quantitative assessment using recording of lesions Time points: at baseline and 7 months (end of the trial) Definitions: incident lesions (the number of new lesions appearing during the study) Safety Methods: recording of any untoward reactions to the creams |
|
| Funding | This study was supported by grants from the Victorian Health Promotion Foundation, Melbourne; the Skin and Cancer Foundation, Sydney; the Skin and Psoriasis Foundation, Melbourne; the Uoyd Williams Trust, Maryborough; the Sydney Melanoma Foundation; and the Australasian College of Dermatologists. | |
| Notes | A sex‐based difference in the change in the number of lesions was noted. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation was achieved by stratification according to sex and self‐rated skin. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This was not stated. Base cream (vehicle) and sunscreen cream had the same consistency by adding 10% mineral oil to the vehicle. Participant blinding concealment was tested by a question at the end of study, and answers were not significant for both treatment arms. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | This was not stated. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The type of analysis was unclear. The initial number of participants randomised and the number of dropouts (157) were given for the 2 groups together. The reasons for withdrawal were provided in a table, but because 1 participant could have more than 1 reason, it was impossible to determine how many participants withdrew in each treatment group. |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: β‐1,3‐D‐glucan, twice daily for 7 days (N = 20 participants) Control intervention B: placebo, twice daily for 7 days (N = 20 participants) |
|
| Outcomes |
Outcomes of the trial 1) Mean lesion counts (converted to mean reduction of lesion counts) 2) Tolerability (= local skin/adverse reactions) 3) Minor adverse events Efficacy Methods: quantitative assessment using lesion counting by the same investigator Time points: at baseline, and weeks 1, 4, and 8 Safety Methods: 1. grading of erythema and burning/stinging as absent, mild, moderate, or severe, 2. participant‐reported adverse events and concomitant medication use Time points: at baseline and weeks 1, 4, and 8 |
|
| Funding | ‐ | |
| Notes | This was a pilot study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 137): "...the respective arms to which each preparation was applied were determined by randomisation. Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | An unclear type of analysis was used. Intraindividual study: Intervention ‐ A: 0 dropouts Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | High risk | All outcomes were presented even if efficacy was higher for placebo. The standard deviations associated with mean values were not given. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: November 2002 End date: September 2005 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod, 3 times per week for 16 weeks (N = 29 participants) Control intervention B: vehicle, 3 times per week for 16 weeks (N = 14 participants) |
|
| Outcomes |
Primary outcomes of the trial 1) Safety of the graft (rejection) 2) Application site reactions (imiquimod only) 3) Skin irritation (qualitative) 4) Minor adverse events (imiquimod only) 5) Serious adverse events 6) Clinical laboratory tests Secondary outcome of the trial 1) Participant complete or partial (> 75%) clearance Other outcomes of the trial 1) Lesion complete response 2) Skin quality (cosmetic) Efficacy Methods: 1. quantitative assessment using clinical counting of visible lesions in the treatment area, 2. biopsy of a lesion mapped at baseline (week 24) Time points: at weeks 7, 12, and 16 (treatment period) and weeks 19 and 24 (post‐treatment) Safety Methods: 1. monitoring of safety of the graft by an independent and blinded safety committee; monitoring of transplant rejection status, laboratory results, adverse events, local skin reactions, vital signs measurements, and the dosage of immunosuppressive medications, changes in haematology and serum chemistry (specifically: levels of serum creatinine, C‐reactive protein, and proteinuria for renal transplant recipients; levels of gamma glutamyl‐transpeptidase, glutamic‐pyruvic transaminase, glutamic‐oxalacetic transaminase, and bilirubin for liver transplant recipients; GOT and GPT, white cell blood count, serum creatinine, haemoglobin, and signs of heart failure for heart transplant recipients), 2. assessment of local skin reactions (erythema, oedema, erosion/ulceration, scabbing/crusting, weeping/exudate, vesicles, and flaking/scaling/dryness) Time points: at weeks 1, 2, 3, 5, 7, 9, 12, and 16 during the treatment period, and weeks 19 and 24 post‐treatment Cosmetic Methods: assessment of skin quality of the treatment area (skin surface, hyperpigmentation, hypopigmentation, the degree of scarring, and any atrophy) Time points: at the 8‐week post‐treatment visit (week 24) |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | The participants were organ transplant recipients, i.e. immunocompromised participants. There was no graft rejection during the study. There was an increase in the number of lesions in the vehicle group only. Clinical clearance was confirmed histologically. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 3): "Baseline data were collected, and the patients were randomised to study drug." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 4 dropouts (the reasons were reported) Control ‐ B: 6 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Little was reported on skin quality outcomes. A lot of outcomes were reported for the imiquimod‐treated group only. All outcomes from the protocol (NCT00189267) were reported in the published study. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 3% diclofenac in 2.5% hyaluronic acid, twice daily for 16 weeks (N = 24 participants) Control intervention B: vehicle, 2.5% hyaluronic acid, twice daily for 16 weeks (N = 8 participants) |
|
| Outcomes |
Outcomes of the trial 1) Participant complete clearance at 20 weeks and 24 months (recurrence) 2) Participant partial (> 75%) clearance at 20 weeks and 24 months (recurrence) 3) Average percentage reduction of lesions at 20 weeks (= mean percentage of lesion counts) 4) Safety of the graft (rejection) 5) Minor adverse events (qualitative) 6) Skin irritation (tolerability, presented graphically) 7) Clinical laboratory tests 8) Cosmetic outcomes 9) Skin quality (cosmetic) at 20 weeks 10) 24‐month follow up for development of new lesions and invasive squamous cell carcinoma Efficacy Methods: 1. quantitative assessment using clinical counting of visible lesions supported by a transparent grid, 2. 3 to 4 mm punch biopsy of a target lesion mapped at the initiation visit Time points: at baseline, at each visit (weeks 4, 8, 12, 16) and at the post‐treatment visit (week 20) Definitions: 1. complete clearance rate (proportion of participants at the 4‐week post‐treatment visit who had no evidence of lesions on the histology results of a target biopsy lesion site and no clinically‐visible lesions in the remainder of the treatment area), 2. partial clearance rate (proportion of participants at the 4‐week post‐treatment visit who obtained at least 75% reduction in the number of lesions counted at baseline in the treatment area), 3. clearance rate of individual lesions (percentage reduction of lesions from baseline to the 4‐weeks post‐treatment visit) Safety Methods: monitoring of transplant rejection status, laboratory results, adverse events, local skin reactions, vital signs measurements, and the dosage of immunosuppressive medications; clinical laboratory analyses: serum levels of immunosuppressive medication; levels in serum creatinine, C‐reactive protein, and proteinuria for renal transplant recipients; gamma glutamyltranspeptidase, glutamic‐pyruvic transaminase, glutamicoxalacetic transaminase, and bilirubin for liver transplant recipients; GOT and GPT, white cell blood count, serum creatinine, hemoglobin, and signs of heart failure for heart transplant recipients) Time points: at each visit (weeks 4, 8, 12, 16) and at the post‐treatment visit (week 20) Cosmetic Methods: assessment of skin quality by investigator based on skin surface, hyperpigmentation, hypopigmentation, the degree of scarring, and any atrophy Time points: at the post‐treatment visit (week 20) |
|
| Funding | This study was supported by Shire Pharmaceuticals. | |
| Notes | New actinic keratoses developed at an average of 9.3 months, but no invasive squamous cell carcinoma developed within a period of 24 months. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page): "Patients with one of three organ transplant types (kidney, liver, heart) were randomised 3:1 (active:vehicle) in this vehicle‐controlled, double‐blind, parallel group design." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 2 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | There was discrepancy in the number of participants completely cleared between the abstract and published report. The lowest number in the published report was used for meta‐analysis. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, vehicle‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 3% diclofenac in 2.5% hyaluronic acid gel twice daily for 4 weeks, 2 weeks off followed by aminolevulinic acid (ALA)‐photodynamic therapy (PDT) (N = 10 participants) Control intervention B: 2.5% hyaluronic acid gel twice daily for 4 weeks, 2 weeks off followed by ALA‐PDT (N = 10 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: no Cream concentration (%): ‐‐ Application of cream: ‐‐ Incubation with cream: occlusive dressing over cream for 4 hours Type of light: red light Light source: Omnilux Wavelength (nm): 633 Energy fluence (J/cm²): 80 Intensities (mW/cm²): ‐‐ Exposure time: 16 minutes (fractions) |
|
| Outcomes |
Outcomes of the trial 1) Mean reduction of lesion counts in a 5 X 5 cm area 2) Participant and investigator global improvement indices (GIIs) expressed as scores 3) Total thickness scores 4) Pain score 5) Severe local adverse reactions (qualitative) Efficacy Methods: 1. quantitative assessment using lesions counting in a 5 X 5 cm area, 2. assessment of lesion thickness visually and by palpation and scored on a 1 to 4 scale, 3. qualitative assessment of global improvement by the investigator and the participant (an independent dermatologist evaluated the efficacy by using photographs) Time points: at baseline, at 6 weeks and 6 months after PDT, and 8 of 10 participants were examined 12 months post‐treatment. Definitions: 1. total lesion score (number of lesions counted in 5 × 5 cm area), 2. total thickness score (sum of the thickness scores for individual lesions) Definitions for thickness score: 1 (lesion visible, but not palpable), 2 (lesion visible and palpable), 3 (elevated and keratotic lesion), 4 (hyperkeratotic lesion > 1 mm in thickness) Definitions for GIIs: –2 (significantly worse), –1 (slightly worse), 0 (no change), 1 (some improvement), 2 (moderate improvement), 3 (significant improvement), 4 (complete remission) Safety Methods: 1. scoring of pain, 2. participant‐recorded side‐effects in daily diary (number and severity) Time points: 1. during PDT (pain), 2. at each visit (side‐effects) Definitions for pain score: 0 (painless), 1 (mild pain), 2 (moderate pain), 3 (severe pain), 4 (unbearable pain) |
|
| Funding | ‐ | |
| Notes | This was a pilot study. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 260): "The pharmacist of the hospital randomly assigned the vehicle to 1 hand and the active drug to the other hand on each patient." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Low risk | A third party (the pharmacist) randomly assigned the vehicle to 1 hand. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | An independent dermatologist evaluated the efficacy. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Modified intention‐to‐treat (ITT, exclusion of only 1 participant who withdrew before PDT) was used. Intraindividual study: Intervention ‐ A: 2 dropouts (the reasons were reported) Control ‐ B: 2 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | High risk | Only mean values were reported i.e. the associated standard deviations were not provided. |
| Other bias | Unclear risk | ‐ |
| Methods | Randomised, double‐blind, active‐controlled, parallel‐group study The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions | 2 subgroups: with and without cooling spray Intervention A: methyl aminolevulinate (MAL)‐ visible + water‐filtered infrared A (VIS + wIRA) PDT (N = 40 participants) Control intervention B: MAL‐light‐emitting diode (LED) red light PDT (N = 40 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 or 2 Interval between treatments: 3 months Preparation of lesions: gentle removal of scales Cream concentration (%): 16 Application of cream: 1 mm thick to lesion area and 5 mm of surrounding normal tissue Incubation with cream: occlusive dressing over cream for 3 hours Type of light: visible + water‐filtered infrared A (VIS + wIRA) or LED red light Light source: Hydrosun type 505 Broadband with 7‐mm water cuvette and orange filter OG590 (VIS + wIRA), Aktilite CL 128 (red light) Wavelength (nm): 580‐1400 (VIS + wIRA), 630 (red light) Energy fluence (J/cm²): 240 including 60 VIS (VIS + wIRA), 37 (red light) Intensities (mW/cm²): 200 including 50 VIS (VIS + wIRA), 75 (red light) Exposure time: 20 minutes (VIS+ wIRA), 8 minutes (red light) |
|
| Outcomes |
Outcomes of the trial 1) Participant complete and partial (> 75%) clearance at 3 (1 treatment), 6 (1 or 2 treatments), and 12 (1 or 2 treatments) months 2) Efficacy on a visual assessment scale (VAS) 3) Pain on a VAS (first outcome presented) 4) Local skin reactions 5) Serious adverse events 6) Satisfaction and quality of life on a VAS 7) Number of spray cooling and illumination interruptions Efficacy Methods: 1. documentation of the global aspect of total actinic keratosis area by physicians, 2. rating of efficacy on a VAS [‐50 mm (extreme worsening), 0 mm (unchanged), +50 mm (extreme improvement)], 3. rating of efficacy on a five‐point scale‐rated variable 'percentage of the cleared area in relation to the initial total actinic keratosis area' (100% clearance, > 75% of the total area cleared, > 50% of the total area cleared, > 25% of the total area cleared, no relevant part of the area cleared) Time points: before PDT; at 2 weeks; and at 3, 6, and 12 months after the first PDT Safety Methods: 1. evaluation of the extent of erythema, scaling, crusts, indurations, erosions, ulcerations, and oedema on a VAS [0 (non‐existent) to 100 mm (extremely high)] by physicians, 2. evaluation of the intensity of pain, side‐effects on a VAS [0 (none) to 100 mm (extremely high)] by participants Time points: 1. before PDT; 2 weeks; and 3, 6, and 12 months after the first PDT, 2. 2, 4, 6, 8, 10, 13, 15, 20, 22, and 25 minutes after the start of PDT (pain) Cosmetic Methods: 1. evaluation of the extent of skin atrophy, scar formation, and pigmentation on a visual analogue scale (VAS) [0 (non‐existent) to 100 mm (extremely high)] by physicians, 2. assessment of cosmetic appearance by physicians and participants [VAS: 0 (extremely bad) to 100 mm (extremely good)] Time points: before treatment; at 2 weeks; and at 3, 6, and 12 months after the first PDT |
|
| Funding | This study was supported by Erwin Braun Foundation. | |
| Notes | Efficacy was lower in participants receiving cooling spray. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 608): "The patient number was randomly assigned to either group 1 (VIS + wIRA PDT, n = 40 patients) or group 2 (red light PDT, n = 40 patients, Table 1)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was a double‐blinded. Quote (page 609): "Both patients and investigators remained blinded until study completion." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The assessor was not involved in treatment. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used. Intervention ‐ A: 1 dropout (the reasons were reported) Control ‐ B: 3 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind (treatment vs placebo),open (treatment duration), vehicle‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Interventions A: 0.5% fluorouracil cream (microsphere) applied once daily to affected areas for 1 week with 4 week follow‐up (N = 38 participants) B: 0.5% fluorouracil cream (microsphere) applied once daily to affected areas for 2 weeks with 4 week follow‐up (N = 41 participants) C: 0.5% fluorouracil cream (microsphere) applied once daily to affected areas for 4 weeks with 4 week follow‐up (N = 40 participants) Control intervention D: vehicle applied once daily to affected areas for 1, 2, or 4 weeks with 4‐week follow‐up (N = 58 participants) |
|
| Outcomes |
Outcomes of the trial 1) Physician global assessment of improvement (PGAI = global improvement indices) 2) Proportion of participants achieving total clearance (= participant complete clearance) 3) Per cent reduction of lesions (= mean percentage of reduction in lesion counts) 4) Mean number of lesions at baseline and end of study (transformed to absolute mean reduction in lesion counts) 5) Median number of lesions at baseline and end of study 6) Skin irritation (number of participants, severity, overtime) 7) Serious adverse events Efficacy Methods: 1. quantitative assessment using lesion counting, 2. qualitative assessment using PGAI (+5 = total clearance and ‐4 = much worse) reported as mean score Time points: at baseline and 4 weeks post‐treatment Safety Methods: 1. monitoring of adverse events (onset, duration, severity, and frequency), 2. separate recording for adverse events affecting the facial skin and scalp, 3. monitoring of facial irritation including maximum severity (0 = none, 1 = mild, 2 = moderate, or 3 = severe), symptoms (edema, erythema, dryness, erosion, pain, burning), onset, overall duration, and post‐treatment duration Time points: during treatment: days 1 and 8 (1‐week groups); days 1, 8, and 15 (2‐week groups); or days 1, 8, 15, and 29 (4‐week groups); post‐treatment: weekly visits and a final evaluation 4 weeks after completing or discontinuing treatment |
|
| Funding | ‐ | |
| Notes | ‐ | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 23): "Patients were randomised to receive 0.5% fluorouracil cream or vehicle control for 1, 2, or 4 weeks (Figure 1)." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This study was double‐blinded (treatment vs placebo) and open (treatment duration). Indeed, placebo cream was not used to conceal allocation to 1, 2 , or 4 weeks. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Different assessment time points were used for 1‐, 2‐, or 4‐week groups. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Intention‐to‐treat (ITT) analysis was used. Intervention ‐ A: 1 dropout (the reason was not reported), B: 1 dropout (the reason was reported), C: 4 dropouts (the reasons were reported) Control ‐ D: 1 dropout (the reason was not reported) |
| Selective reporting (reporting bias) | High risk | The standard deviations associated with the mean number of lesions and percentages were not reported. Adverse events were not reported in this study, but they were reported in a similar study included (Jorizzo 2002). |
| Other bias | High risk | There was slight difference (P = 0.048) in the women ratio [more in 4‐week group (C) and less in placebo group (D)] at baseline. |
| Methods | This was a single‐centre, randomised, assessor‐blind, active‐controlled, intraindividual study. Start date: May 2006 End date: February 2007 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: methyl aminolevulinate (MAL)‐red light photodynamic therapy (PDT) (N = 30 participants) Control intervention B: MAL‐daylight PDT (N = 30 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: crusts and hyperkeratoses removed Cream concentration (%): 16.8 Application of cream: 1 g applied to lesion area Incubation with cream: occlusive dressing over cream for 0.5 hour (daylight) and 3 hours (red light) Type of light: LED red light or daylight Light source: Aktilite CL 128 (red light), sun (daylight) Wavelength (nm): 575‐670 (red light), 290‐670 (daylight) Energy fluence (J/cm²): 37 (red light), 11.7‐65.9 (mean = 43.2, measured with a dosimeter) Intensities (mW/cm²): ‐‐ Exposure time: 2.5 hours (daylight) |
|
| Outcomes |
Outcomes of the trial 1) Mean reduction in lesion counts at 3 months post‐treatment 2) Local adverse events 3) Participant's pain scores 4) Participant's satisfaction Efficacy Methods: quantitative assessment using counting, grading (Olsen 1991), mapping, and photography of lesions Time points: before treatment and at 3 months post‐treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion) Safety Methods: 1. scoring (0 = no pain to 10 = worst imaginable pain) of the pain in the 2 treated areas by participants, 2. evaluation of adverse events (erythema, crusting or pain) Time points: 1. during daylight exposure, during red LED light illumination, and after treatment (pain), 2. at 1 to 3 days after PDT (adverse events) |
|
| Funding | This study was supported by The Eva and Henry Frænkels Memorial Foundation. | |
| Notes | PpIX fluorescence measured before, during, and after treatments showed less fluorescence associated with daylight. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation sequence generation was achieved by drawing lots. |
| Allocation concealment (selection bias) | Low risk | Opaque sealed envelopes were used to conceal allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The 2 treatments were physically distinct. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The evaluator was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Per‐protocol (PP) analysis was used. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | Unclear risk | No outcomes were specified in the protocol (NCT00432224). |
| Other bias | Unclear risk | ‐ |
| Methods | This was a randomised, double‐blind, active‐controlled, intraindividual study. Start date: June 2007 End date: December 2007 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 16% methyl aminolevulinate (MAL)‐daylight photodynamic therapy (PDT) (N = 30 participants) Control intervention B: 8% MAL ‐daylight PDT (N = 30 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: sunscreen SPF20, crusts and hyperkeratoses removed Cream concentration (%):8 or 16 Application of cream: 1 g applied to lesion area Incubation with cream: all day Type of light: daylight Light source: sun (daylight) Wavelength (nm): 290‐670 (daylight) Energy fluence (J/cm²): measured with dosimeter Intensities (mW/cm²): ‐‐ Exposure time: all day |
|
| Outcomes |
Outcomes of the trial 1) Mean reduction in lesion counts 2) Total lesion count (lesion complete response) 3) Pain scores 4) Erythma scale and per cent by measurements before and after treatment with skin reflectance meter 5) Participant's preference Efficacy Methods: quantitative assessment using counting, grading (Olsen 1991), mapping, and photography of lesions Time points: before treatment and at 3 months post‐treatment Definitions: 1. complete response (complete disappearance of the lesion), 2. non‐complete response (incomplete disappearance of the lesion) Safety Methods: 1. scoring (0 = no pain to 10 = worst imaginable pain) of the pain in the 2 treated areas by participants in a diary every hour, 2. evaluation of adverse events (erythema, crusting, or pain) visually on a 4‐point scale by a dermatologist, 3. quantitative evaluation of erythema [erythema percentage measured with a skin reflectance meter (Optimize Scientific; Chromo Light Aps,Skodsborg, Denmark)] Time points: 1. during daylight exposure, during red LED light illumination, and after PDT (pain), 2. 1 to 3 days after PDT(adverse events), 3. before curettage and on the day after PDT (erythema evaluation) Definitions for visual evaluation of erythema: 1. 0 (no visible erythema), 2. (+) (just perceptible erythema), 3. + (uniform erythema), 4. ++ (bright red erythema and induration) |
|
| Funding | ‐ | |
| Notes | PpIX fluorescence was measured using fluorescence camera, and there was no difference between the 2 cream concentrations. There was a correlation with light exposure/dose and response rate. Pain increased with light exposure. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Adequate randomisation sequence was achieved by drawing lots. |
| Allocation concealment (selection bias) | Low risk | Opaque sealed envelopes were used to conceal allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 1309): "The evaluating dermatologist and participants were blinded to the concentrations of creams." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote (page 1309): "The evaluating dermatologist and participants were blinded to the concentrations of creams." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The type of data analysis was unclear, but only 1 participant was lost to follow up. Intraindividual study: Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 1 dropout (the reason was reported) |
| Selective reporting (reporting bias) | High risk | The number of participants and average time spent outside were different between the abstract and published report. There was a little confusion regarding the type of efficacy outcome reported in the abstract. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, active‐controlled, parallel‐group study. Start date: June 2008 End date: January 2009 |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 2h MAL‐1.5h daylight photodynamic therapy (PDT) (N = 58 participants) Control intervention B: 3h MAL‐2.5h daylight PDT (N = 62 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: sunscreen SPF20, crusts and hyperkeratoses removed Cream concentration (%): 16 Application of cream: thick layer applied to lesion area Incubation with cream: 2 or 3 hours (0.5 hour without light exposure) Type of light: daylight Light source: sun (daylight) Wavelength (nm): 290‐670 (daylight) Energy fluence (J/cm²): 8.6 (1.5 hours), 10.2 (2.5 hours) Intensities (mW/cm²): ‐‐ Exposure time: 131 + 37 min (1.5 hours) or 187 + 52 min (2.5 hours) |
|
| Outcomes |
Primary outcomes of the trial 1) Mean reduction in lesion counts at 3 months post‐treatment 2) Mean percentage of reduction in lesion counts at 3 months post‐treatment [which correspond to the primary and only outcome "response rate" in the protocol NCT00711178 based on the published report (see efficacy definitions below)] Other outcomes of the trial 1) Pain scores 2) Local adverse reactions: erythema and pustular eruptions (pooled data) 3) Participants' satisfaction 4) New actinic keratosis lesions Efficacy Methods: quantitative assessment using lesion grading and counting using a template Time points: at baseline and 3 months post‐treatment Definitions: 1. lesion response rate (number of completely responding lesions divided by the number of lesions treated within the individual participants), 2. complete response (complete disappearance of the lesion, visually and by palpation ‐ mild erythema might remain), 3. non‐complete response (incomplete disappearance of the lesion, visually and by palpation) Safety Methods: 1. participant‐recorded pain score (0 = no pain and 10 = worst imaginable pain) in diary, 2. erythema and pustular eruption rating by investigators (other adverse events were recorded if the investigators considered these to be related to treatment) Time points: 1. every half hour during the day of treatment and 4 time‐points the following day (pain), 2. at 2 days after PDT (erythema and pustules) Definitions for erythema rating: 1. none (no redness), 2. mild (visibly pink colour), 3. moderate (red colour), 4. severe (dark red purple) Definitions for pustular eruptions rating: 1. none (no pustules), 2. mild (few pustules), 3. moderate (several pustules), 4. severe (severe pustular eruption with yellow crusting) |
|
| Funding | This study was supported by Department of dermatology, Bisebjerg Hospital, Copenhagen. | |
| Notes | The effective daylight dose was measured with electronic wristband dosimeter. The weather conditions were monitored every half hour in diary. An increase in pain and erythema was associated with higher effective light dose. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 2): "The randomisation procedure was performed by a computer‐generated sequence blocked by centre." |
| Allocation concealment (selection bias) | Low risk | Quote (page 2): "Allocations were contained in opaque, sequentially numbered, sealed envelopes and were concealed from assessors throughout the study." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was not stated, but the participants were exposed to light for different periods of time. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was assessor‐blinded (based on the protocol NCT00711178, clinicaltrials.gov). |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Intention‐to‐treat (ITT) analysis was used for primary outcomes, and per‐protocol (PP) analysis was used for secondary outcomes. Intervention ‐ A: 1 dropout (the reason was reported) Control ‐ B: 0 dropouts |
| Selective reporting (reporting bias) | Unclear risk | All primary outcomes were reported based on the protocol NCT00711178. Additional secondary outcomes, which were not included in our review, were also reported. |
| Other bias | Unclear risk | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 3% diclofenac gel in 2.5% hyaluronic acid gel, 0.5 g twice daily for 90 days (N = 58 participants based on safety data) Control intervention B: 2.5% hyaluronic acid gel only, 0.5 g twice daily for 90 days (N = 59 participants based on safety data) |
|
| Outcomes |
Primary outcomes of the trial 1) Participant Global Improvement Indices = 4 2) Investigator Global Improvement Indices = 4 3) Participants with target lesion number score = 0 at 30 days post‐treatment (= participant complete clearance) 4) Participants with cumulative lesion number score = 0 at 30 days post‐treatment (= participant complete clearance) Other outcomes of the trial 1) Participants experiencing at least 1 adverse event 2) Application site reactions 3) Minor adverse events 4) Serious adverse events 5) Clinical laboratory tests Efficacy Methods: 1. quantitative assessment using lesion counting, 2. qualitative assessments (IGII and PGII) Time points: at each visit Definitions: 1. target lesion number score (number of lesions identified in the designated treatment blocks at baseline), 2. cumulative lesion number score (number of lesions identified ‐target or new‐in the designated treatment blocks) Definitions for IGII and PGII 7‐point scale: + 2 (significantly worse), +1 (slightly worse), 0 (no change), 1 (slightly improved), 2 (moderately improved), 3 (significantly improved), and 4 (completely improved) Safety Methods: 1. participant‐recorded concomitant medications and adverse events in diary; 2. assessment of adverse events for duration, intensity, and causality by physician; 3. standard laboratory analyses [hematology, biochemistry, urinalysis, and serology (antidiclofenac)] Time points: at screening and end of treatment (laboratory tests) |
|
| Funding | This study was supported by Hyal Pharmaceutical Co. | |
| Notes | A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 710): "One week after the Screening Visit, patients were randomised to receive either the active treatment, 3% diclofenac in 2.5% hyaluronan gel (SolarazeTM Bioglan) or placebo, which consisted of the inactive gel vehicle, hyaluronan only." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Intention‐to‐treat (ITT) analysis was used, but the initial randomised numbers for each group were not given explicitly. Intervention ‐ A: 14 dropouts (the reasons were reported) Control ‐ B: 8 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
| Methods | This was a single‐centre, randomised, double‐blind, placebo‐controlled, intraindividual study. The start and end dates were not specified. |
|
| Participants |
Inclusion criteria of the trial
Demographics
|
|
| Interventions |
Intervention A: 5% imiquimod, once weekly for 24 weeks (N = 20 participants) Control intervention B: placebo, once weekly for 24 weeks (N = 20 participants) |
|
| Outcomes |
Outcomes of the trial 1) Investigator assessment scale (= global improvement indice) 2) Total lesion number score 3) Local skin reactions (qualitative) 4) Serious adverse events 5) Skin irritation score (graph) Efficacy Methods: 1. qualitative assessment with an investigator assessment scale (7‐point), and 2. quantitative assessment with lesion counting Time points: every 4 weeks during the treatment period and at 4 weeks post‐treatment Definitions for the investigator assessment scale: ‐2 (much worse), ‐1 (slightly worse), 0 (no change), 1 (mild improvement), 2 (moderate improvement), 3 (marked improvement), 4 (cured) Definitions: total lesion number score (total number of lesions present in the target area was determined for each side, 0 = 0 lesion, 1 = 1 to 3, 2 = 4 to 6, 3 = > 6 lesions) Safety Methods: 1. monitoring the occurrence of local adverse events and systemic adverse events, 2. rating of skin irritation by participants on a 6‐point scale (there was no objective measure of local side‐effects) Time points: at each visit Definitions for skin irritation scale: 0 (no irritation), 1 (trace irritation), 2 (mild irritation) 3 (moderate irritation), 4 (marked irritation), 5 (severe irritation) |
|
| Funding | This study was supported by 3M Pharmaceuticals. | |
| Notes | This was a pilot study. A sample size calculation was provided. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page): "Enrolled patients were randomised, 1:1, to apply imiquimod to a 20‐cm²area on the right or left side." Comment: Insufficient detail was reported about the method used to generate this allocation sequence. |
| Allocation concealment (selection bias) | Unclear risk | This was not stated. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This study was double‐blinded. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | This study was double‐blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Per‐protocol (PP) analysis was used with 25% of subjects lost. Intraindividual study: Intervention ‐ A: 5 dropouts (the reasons were reported) Control ‐ B: 5 dropouts (the reasons were reported) |
| Selective reporting (reporting bias) | Unclear risk | ‐ |
| Other bias | Unclear risk | ‐ |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alberts 2004 | We assessed actinic damage in general, and the study was not specific to actinic keratoses. Only 60% of participants had clinically‐evaluable actinic keratoses on the forearms. |
| Alexiades‐Armenakas 2003 | The study did not randomise all the participants. |
| Apalla 2010a | This was a conference abstract without numerical values for efficacy. |
| Apalla 2010b | The study did not meet the outcome requirements of the review. |
| Apalla 2010c | The efficacy of the intervention was on new actinic keratosis lesions (prevention), not on baseline lesions. |
| Babilas 2006 | The study was not randomised. Predefined sides were treated with 2 different light sources for PDT, i.e. always the same side for 1 treatment. |
| Babilas 2007 | The study did not meet the outcome requirements of the review. |
| Babilas 2008 | The study did not meet the outcome requirements of the review. |
| Bartels 2009 | The study did not meet the outcome requirements of the review. |
| Berlin 2008 | The study was not randomised; the participants were allocated based on participant and physician judgement. |
| Biecha‐Thalharnmer 2003 | The study did not meet the outcome requirements of the review. |
| Braathen 2009 | The study did not meet the outcome requirements of the review. |
| Breza 1976 | The study did not present efficacy results numerically. |
| de Sévaux 2003 | The study did not present efficacy results numerically; they were given in graphical form. |
| Dermik 2003 | The study did not clearly present efficacy measures. The data were presented in graph form with no quantitative numbers. |
| Dirschka 2010 | The study was randomised (90‐ versus 180‐day treatments), but only partial data at 3 months were presented based on the conference abstract. And only data from the 90‐day group were presented in the peer‐reviewed paper, i.e. there was no comparison. |
| DUSA 2009 | This trial was terminated due to "Orphan Drug Designation for this indication not granted". |
| Edwards 1986 | The type of interventions were not covered in this review, i.e. injection in participant lesion. |
| Elmets 2010 | This study was on the prevention of actinic keratosis lesions. |
| Epstein 2006 | The study did not meet the outcome requirements of the review. |
| Ericson 2004 | The study did not meet the outcome requirements of the review. |
| Fowler 2002 | This was a follow‐up study of an included study. |
| Gold 2006 | It was not clear if the study was randomised. |
| Goldman 2003 | The randomisation was based on participant preference, and there was no efficacy comparison between the 2 treatments. |
| Green 1998 | Out of 80 participants, only 4 had actinic keratosis lesions, and mean lesions counts were reported for all subjects. |
| Griffin 1991 | The randomisation was not mentioned. Little information was given. |
| Grimaître 2000 | The randomisation was not mentioned. |
| Gupta 2004 | The study did not meet the outcome requirements of the review. |
| Hanke 2011 | This was a follow‐up study for Hanke 2010; Swanson 2010a. |
| Humphreys 1996 | Actinic keratosis lesions were not distinguished from lentigines for efficacy data. |
| Jury 2005 | The criteria for outcomes was not met, i.e. median lesion counts were provided instead of mean lesion counts. |
| Kurwa 1999 | This study did not meet the outcome requirements of the review; it presented results as the mean reduction in lesion area. |
| Marrero 1998 | There was an inadequate method of randomisation: Every other participant was given a combination treatment on the left side of the face and a monotherapy treatment on the right. All other participants were given the opposite treatment. |
| Morales 2010 | This study did not meet the outcome requirements of the review. |
| Naylor 1995 | This study was on the prevention of actinic keratosis lesions formation, not an intervention to cure. |
| NCT00005097 | The trial was terminated because of the low conditional power for a positive study. |
| Puizina‐Ivic 2008a | This study did not meet the outcome requirements of the review. |
| Radakovic‐Fijan 2005 | This study did not meet the outcome requirements of the review. |
| Robins 2002a | 50% of participants were lost to follow up. No statistics were reported. There was too much variability within and between groups as far as following instructions for application. There was no initial counting of lesions. |
| Rosen 2010 | There was not enough information in this conference abstract of a phase II trial to be able to use the data. |
| Shuttleworth 1989 | The type of interventions were not covered in this review, i.e. injection in participant lesion, and it was unclear if the study was randomised |
| Simmonds 1973 | There were no numerical results. |
| Smith 2006 | This study did not meet the outcome requirements of the review. |
| Sotiriou 2011 | This study did not meet the outcome requirements of the review. |
| Spencer 2010 | No enough numerical information was provided, e.g. number of participants in each of the 8 treatment groups. |
| Stockfleth 2004 | This was a long‐term follow‐up to a previous study: Stockfleth 2002 |
| Szeimies 2010a | This was a follow‐up study of Hauschild 2009a; Hauschild 2009b; and Hauschild 2009c. |
| Touma 2004 | The study did not present efficacy results numerically; they were given in graph form. |
| Tsoukas 2010 | It was unclear if the study was randomised based on this conference abstract. |
| Valeant 2004 | The randomisation was not mentioned. |
| Vbeam 2005 | This trial was terminated due low accrual. |
| Weinstock 2010 | The study did not present numerical data. |
| Wennberg 2008 | This study was on prevention of new lesions and mixed lesion types, i.e. not only actinic keratoses. |
| Wulf 2006 | This study does not provide efficacy data on intervention. The primary outcome was mean time to occurrence of first new lesion. |
| Yamauchi 2002 | There was not enough information. Results taken from 2 studies and combined, i.e. no direct comparison. |
Characteristics of studies awaiting assessment [ordered by study ID]
| Methods | This was a single‐centre, randomised, open, assessor‐blinded, placebo‐ and active‐controlled, parallel‐group study. The start and end dates were not specified. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: 3% diclofenac in 2.5% hyaluronic acid, twice daily for 12 weeks (N = 21 participants) Control interventions B: 5% imiquimod, twice per week for 16 weeks (N = 20 participants) C: placebo (Ultrabase cream; Schering Alman, Istanbul, Turkey), twice daily for 12 weeks (N = 20 participants) |
| Outcomes |
Outcomes of the trial 1) Total thickness score (TTS) 2) Patient global improvement index (PGII) 3) Complete clearance (= lesion complete response) 4) Local skin reactions Efficacy Methods: quantitative assessment using a scale and photography of lesions Time points: before treatment and at every 4 weeks up to 24 weeks Definitions: 1. TTS scale, 0 = complete clearance, 1 = lesion visible but not palpable, 2 = lesion visible and palpable, 3 = lesion raised with visible scaling, 4 = lesion hyperkeratotic and > 1 mm in thickness; 2. PGII is a self‐report scale, and participants evaluated themselves according to a 7‐point scale (0 = significantly worse, 1 = slightly worse, 2 = no change, 3 = slightly improved, 4 = moderately improved, 5 = significantly improved, 6 = completely improved) Safety Methods: evaluation of local skin reactions (erythema, oedema, erosion ⁄ ulceration, scabbing ⁄ crusting, weeping⁄ exudates, vesicles, and scaling ⁄ dryness) on a 0 to 3 scale Time points: at every 4 weeks up to 24 weeks Definitions: 0 = none, 1 = mild, 2 = moderate, 3 =severe |
| Notes | ‐ |
| Methods | This was a single‐centre, randomised, open, assessor‐blinded, intraindividual study. Start date: February 2009 End date: May 2010 |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Interventions A: 50 mW/cm² ALA‐red light photodynamic therapy (PDT) (N = 50 participants) B: 75 mW/cm² ALA‐red light PDT (N = 50 participants) Control intervention C: 25 mW/cm² ALA‐red light PDT (N = 50 participants) Characteristics of PDT intervention Type of treatment: individual lesion Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: ‐‐ Cream concentration (%): 20 Application of cream: ‐‐ Incubation with cream: 4 hours with occlusion Type of light: red Light source: Waldmann PDT 1200 (non‐coherent light) Wavelength (nm): 570‐670 Energy fluence (J/cm²): 75 Intensities (mW/cm²): 25‐75 Exposure time:‐‐ At the 3‐month follow‐up visit, lesions without a complete response were treated with surgical techniques (cryosurgery or excision). |
| Outcomes |
Primary outcomes of the trial 1) Pain on a visual analogue scale (VAS) during illumination Secondary outcomes of the trial 1) Lesion complete response at 3 and 12 months post‐treatment 2) Adverse events (qualitative) Efficacy Methods: 1. quantitative assessment using lesion counting, Time points: at baseline; and at 3, 6, and 12 months post‐treatment Definitions: 1. complete response (CR): complete absence of any clinical sign indicative of actinic keratoses, 2. non‐complete response (non‐CR): remaining clinical signs indicative of actinic keratoses Safety Methods: recording of adverse events Time points: from 5‐ALA application time point until the end‐of‐study |
| Notes | ‐ |
| Methods | This was a randomised, double‐blind, placebo‐controlled, parallel‐group study. Start date: January 2010 End date: March 2011 |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: cryotherapy followed by Acnalene 0.1% gel at day 10, twice daily for 3 months Control intervention B: cryotherapy followed by placebo at day 10, twice daily for 3 months |
| Outcomes |
Primary outcomes of the trial 1) Changes in the average number of actinic keratosis lesions (= mean reduction in lesion counts) 2) Mean frequency of reduction (= mean percentage of reduction in lesion counts) 3) Clinical outcome based on change in lesion number [including 'recovery rate of over 75%' (= participant partial (> 75%) clearance)] Secondary outcomes of the trial 1) Adverse events 2) Cosmetic outcomes Efficacy Methods: quantitative assessment by lesion counting and photography Time points: at baseline and monthly after the beginning of the topical treatment Definitions: 1. full recovery (greater than or equal to 75% reduction in the number of lesions), 2. relative recovery (30 to 75 per cent reduction in the number of lesions), 3. no response (no reduction or [greater than or equal to] 30% reduction in the number of lesions), 4. worsening (an increase in the number of lesions) Safety Methods: clinical examination and questioning of participants Cosmetic Methods: assessment of changes in pigmentation and scar formation |
| Notes | This study corresponded to the protocol IRCT201010104901N1. |
| Methods | This was a single‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group study. The start and end dates were not specified. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: administration of 500 mg nicotinamide tablet, twice daily for 4 months Control intervention B: placebo tablet made of lactose. The dose, frequency, and duration of treatment are the same as for the nicotinamide group (i.e. 500 mg twice a day for 4 months) |
| Outcomes |
Primary outcomes of the trial 1) Reduction in total actinic keratosis count at 4 months compared with baseline count Secondary outcomes of the trial 1) Reduction in site‐specific (i.e. face, arms, scalp) actinic keratosis count at 2 and 4 months compared with baseline count 2) Reduction in skin cancers (posthoc) Efficacy Methods: quantitative assessment by blinded clinical examination by a medically‐qualified observer Time points: at baseline, and 2 and 4 months |
| Notes | ‐ |
| Methods | Randomised, open, active‐controlled, intraindividual study The start and end dates were not specified. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: topical anaesthetic with occlusion (lidocaine 5% and prilocaine 5%) was applied for 2 hours before cryopeeling (on a field) followed by treatment of individual lesions with liquid nitrogen (LN) (N = 16 participants) Control intervention B: topical anaesthetic with occlusion (lidocaine 5% and prilocaine 5%) was applied for 2 hours before cryopeeling (on a field) followed by treatment of individual lesions with dimethyl ether, propane, and isobutane gases in a portable system (PS) (N = 16 participants) Topical Vaseline was used in the postoperative period to moisturise the skin and reduce the discomfort of healing. |
| Outcomes |
Outcomes of the trial 1) Percentage of lesions completely healed (= lesion complete response) at 2 months post‐treatment 2) Pain on the visual analogue scale (VAS) from zero (no discomfort) to 10 (worst discomfort possible) 3) Global preference of physician and participant based on a 5‐point scale, which ranged from ‐2 (right much better than left) to +2 (left much better than right) 4) Cosmetic outcomes Efficacy Methods: quantitative assessment using marking of the lesions with acetate sheets and permanent‐ink pen, standardised photographic documentation, and counting Time points: at baseline and days 7, 14, 21, 3, and 60 Definitions: lesions that had healed completely (showed no sign of a previous lesion) Cosmetic Methods: 3 blinded dermatologists evaluated how much the appearance of the skin had improved following a standardised scale based on comparison with pictures taken at baseline Time points: at 60 days Definitions: 0 (no improvement), 1 (a little better), 2 (much better) |
| Notes | ‐ |
| Methods | This was a multicentre, randomised, assessor‐blind, placebo‐ and active‐controlled, parallel‐group study. The start and end dates were not specified. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: BF‐200 (10%) ALA gel‐photodynamic therapy (PDT) (N = 248 participants) Control interventions B: 16% MAL cream ‐PDT (N = 247 participants) C: placebo gel‐PDT (N = 76 participants) Characteristics of PDT intervention Type of treatment: individual lesions Number of treatments: 1 or 2 Interval between treatments: 12 weeks Preparation of lesions: mild curettage, roughening and alcohol wiping Cream concentration (%): ‐‐ Application of cream: ‐‐ Incubation with cream: occlusive dressing over cream for 3 hours Type of light: red light Light source: Aktilite CL 128, PhotoDyn 750/505, Omnilux PDT, Waldmann PDT 1200L Wavelength (nm): 630 (Aktilite, Omnilux), 580‐1400 (PhotoDyn), 600‐750 (Waldmann) Energy fluence (J/cm²): 37 (Aktilite, Omnilux), 170 (PhotoDyn), 100 (Waldmann) Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance at 12 weeks after the first and last PDT Secondary outcome of the trial 1) Lesion complete response at 12 weeks after the last PDT Other outcomes of the trial 1) Local skin reactions at application sites before and after PDT 2) Adverse events 3) Serious adverse events 3) Pain on a visual analogue scale (VAS: 0 represents no pain, 1 to 3 are interpreted as 'mild', 4 to 7 as 'moderate', and 8 to 10 as 'severe' pain) during PDT 4) Cosmetic outcomes: general Outcomes were stratified for different light sources, mild and moderate lesions, and lesions on the face and scalp. Efficacy Methods: quantitative assessment of lesion clearance by visual inspection and by palpation by an investigator not involved in treatment and safety evaluation Time points: at baseline, and 3 and 12 weeks post‐treatment Definitions: participant complete clearance (all lesions were considered to be cleared both by the clinical assessment) Safety Methods: 1. recording of adverse effects, 2. documentation of local adverse reactions (pain, itching, burning, erythema, oedema, and induration) at the application site and rated as mild, moderate, and severe by the assessing physician or reporting participants, 3. serious adverse events Time points: 1. at 1 week after PDT (by phone) and 3 weeks, 2. during and after PDT (local adverse reactions), 3. throughout the study (serious adverse events) Cosmetic Methods: 1. general cosmetic outcome assessed by the investigator as very good, good, unsatisfactory, and impaired; 2. assessment of skin quality Time points: at 12 weeks post‐treatment |
| Notes | This was a confirmatory phase III study. |
| Methods | This was a multicentre, randomised, assessor‐blind, intraindividual study. The start and end dates were not specified. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: pretreatment with tazarotene gel (0.1 %) twice daily for 1 week + ALA‐blue light photodynamic therapy (PDT) on the entire treatment area, e.g. extensor surface of the hand or forearm between the elbow and the base of the fingers, which includes target area (N = 10 participants) Control intervention B: no pretreatment + ALA‐blue light PDT on the entire treatment area (N = 10 participants) Characteristics of PDT intervention Type of treatment: field‐directed Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: ‐‐ Cream concentration (%): 20 Application of cream: first applied to individual lesions and then to entire treatment area Incubation with cream: 1 hour, with occlusion on the control side Type of light: blue light Light source: BLU‐U Wavelength (nm): ‐‐ Energy fluence (J/cm²): 10 Intensities (mW/cm²): 10 Exposure time: 16 minutes 40 seconds |
| Outcomes |
Outcomes of the trial 1) Lesion counts on the target and entire treatment areas after pretreatment and 8 weeks after PDT compared to baseline 2) Median per cent reduction in lesion counts in the target and entire treatment areas after pretreatment and 8 weeks after PDT 3) Participants with different percentage reduction in lesion counts in the target and entire treatment areas (100% = participant complete clearance) after pretreatment (target area only) and 8 weeks after PDT 4) Investigator global assessment (IGA) after pretreatment and 8 weeks after PDT 5) Tolerance (pigmentary changes, erythema, edema, stinging/burning, scaling/dryness, and oozing/vesiculation) at baseline, after pretreatment, after PD, and 8 weeks after PDT 6) Participant satisfaction on 5‐point scale in which 0 = poor and 4 = excellent Efficacy Methods: 1. quantitative assessment by lesion counting, 2. qualitative assessment by investigator (IGA) on a 5‐point scale Time points: at baseline, after pretreatment, and 8 weeks after PDT Definitions: IGA: 0 = clear and 5 = very severe Safety Methods: 1. postinflammatory hyperpigmentation, erythema, scaling & dryness, edema, and oozing/crusting/vesiculation were assessed using a 5‐point ordinal scale (0 = none to 4 = severe), 2. participants rated stinging and burning on a 4‐point scale (0 = none and 3 = severe), 3. adverse events were recorded Time points: at each visit and phone call 24 to 48 hours after PDT |
| Notes | ‐ |
| Methods | This was a randomised, parallel‐group study. The start and end dates were not specified. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Interventions A: 20 J/cm² ALA‐Intense Pulsed Light (IPL) PDT (N = 4 participants) B: 25 J/cm² ALA‐IPL PDT (N = 4 participants) C: 40 J/cm² (2 passes of 20 J/cm²) ALA‐IPL PDT (N = 5 participants) D: 50 J/cm² (2 passes of 25 J/cm²) ALA‐IPL PDT (N = 5 participants) Control intervention E: IPL PDT (N = 3 participants) Characteristics of PDT intervention Type of treatment: ‐‐ Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: ‐‐ Cream concentration (%): 20 Application of cream: twice Incubation with cream: 2 hours Type of light: IPL Light source: ‐‐ Wavelength (nm): ‐‐ Energy fluence (J/cm²): ‐‐ Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
| Outcomes |
Outcomes of the trial 1) Global response to treatment (actinic keratoses and photodamage) was determined by a 7‐point scale (= grade) 2) Mean clearance rates for actinic keratoses only (= difference between mean grades before PDT and 8 weeks after PDT) 3) Tolerability (adverse reactions) at 48 hours after PDT 4) Participant discomfort during PDT Efficacy Methods: 1. quantitative assessment of 5 previously identified lesions (non‐hyperkeratotic, < 1 cm in diameter, dry, rough, yellowish, with scales) marked in each participant, numbered from 1 to 5, and documented by the FotoFindermediscope system (photography); 2. qualitative assessment (global response) on a 7‐point scale Time points: at baseline, and 48 hours and 8 weeks after PDT Definitions: 7‐point scale: 0 = complete response, 1 = 90% improvement, 2 = 75% improvement, 3 = 50% improvement, 4 = 10% improvement, 5 = no improvement, and 6 = condition worsened Safety Methods: 1. erythema, edema, crusts, and erosions were graded on a 5‐point scale (0= none to 4= severe), 2. burning or stinging during treatment was graded on a 4‐point scale (0 = none to 3 = severe). Time points: at 48 hours after PDT |
| Notes | The study included participants with actinic keratoses, photodamage, or both. |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group studies (4 studies). Start date: September 2008 End date: October 2009 |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: face or scalp: 0.015% PEP005 gel, once daily for 3 days (N = 277 participants) trunk or extremities: 0.05% PEP005 gel, once daily for 2 days (N = 226 participants) Control intervention B: vehicle gel, once daily for 2 or 3 days (N = 502 participants) |
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates at day 57 Secondary outcomes of the trial 1) Participant partial (> 75%) clearance rates at day 57 2) Median percentage changes from baseline in total number of lesions at day 57 and 12 months follow‐up (posthoc) Other outcomes of the trial 1) New actinic keratosis lesions or recurrence at 12 months follow‐up for participants complete cleared in 3 of the studies (posthoc) 2) Local skin reactions on a 5‐point scale (individual scores and time course of the mean composite score) 3) Application‐site adverse reactions 4) Adverse events 5) Serious adverse events 6) Pigmentation changes and scarring (cosmetic) Efficacy Methods: quantitative assessment by a study investigator who examined the selected treatment area in person Time points: at baseline, and day 57 and 12 months follow‐up Safety Methods: 1. assessment of the incidence rate of adverse events, serious adverse events, and local skin responses; 2. grading of local skin responses with photographic guides to ensure uniform reporting for the following: erythema, flaking or scaling, crusting, swelling, vesiculation or pustulation, and erosion or ulceration Time points: on days 3 (trunk or extremities), 4 (face or scalp), 8, 15, 29, and 57 Definitions: composite local‐skin response score (sum of the 6 individual scores that were reported at each study visit for each participant ‐ maximum composite score = 24) Cosmetic Methods: assessment of pigmentation and scarring Time points: on days 3, 8, 15, 29, and 57 |
| Notes | This included 4 studies: NCT00742391 (= Swanson 2010b included in analyses), NCT0091551, NTC00916006, and NCT00942604. |
| Methods | This was a randomised, active‐controlled, parallel‐group study. Start date: January 2009 The end date was not specified. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: MAL‐red light PDT followed 1 month later by 5% imiquimod, 3 times per week for 4 weeks (N = 32 participants) Control interventions B: MAL‐red light PDT (N = 40 participants) C: 5% imiquimod, 3 times per week for 4 weeks (N = 33 participants) Characteristics of PDT intervention Type of treatment: field‐directed Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: curettage of the most hyperkeratotic lesions Cream concentration (%): 16 Application of cream: whole treatment area Incubation with cream: 3 hours with occlusion Type of light: red light Light source: Aktilite CL 1 Wavelength (nm): ‐‐ Energy fluence (J/cm²): 37 Intensities (mW/cm²): ‐‐ Exposure time: 8 minutes |
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rates (clinical) at 1 month post‐treatment Other outcomes of the trial 1) Participant partial (> 75%) clearance rates (clinical) at 1 month post‐treatment 2) Clinicopathologic response (= clinical complete clearance and histological clearance) at 1 month post‐treatment 3) Local skin reactions for imiquimod treatment at week 4 4) Tolerance (comfort, discomfort, pain, local skin reaction, side‐effects, waiting time, and duration of treatment) evaluated on an analogue scale of 0 (well tolerated) to 10 (very poorly tolerated) after PD, imiquimod treatment, or both 5) Participant satisfaction (benefit, improvement achieved, side‐effects, and tolerance) on an analogue scale of 0 (very dissatisfied) to 10 (very satisfied) at 1 month post‐treatment Efficacy Methods: 1. quantitative assessment by visual examination and palpation, 2. histological evaluation (biopsy) on 2 prespecified lesions identified by photography: lesion 1 before treatment and lesion 2 after treatment Time points: at baseline, and at 1 month post‐treatment Definitions: 1. complete clinical clearance (total absence of actinic keratoses or lesions clinically suspected of being actinic keratoses), 2. clinicopathologic response (complete clinical response and absence of actinic keratoses in the biopsy specimen of lesion 2), 3. absence of actinic keratoses (normalisation of the stratum corneum with no parakeratosis and normal maturation of epidermal keratinocytes with no atypical keratinocytes) Safety Methods: evaluation of the intensity of the local reaction (mild, moderate, or severe) Time points: at week 4 of treatment Definitions: 1. mild local reaction (occasional appearance of limited edema and mild erythema in the treatment area), 2. moderate local reaction (erythema, edema, ulceration, and flaking in at least 50% of the treatment area), 3. severe local reaction (erythema, edema, ulceration, and crusts occupying almost all of the treatment area and even extending beyond the treatment area) |
| Notes | ‐ |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group study. Start date: 2008 End date: 2009 |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: 0.5% 5‐FU in combination with 10% salicylic acid (SA) solution (LAS41005), once daily for up to 12 weeks (N = 98 participants) Control interventions B: 5‐FU/SA vehicle, once daily for up to 12 weeks (N = 187 participants) C: 3% diclofenac in hyaluronic acid (HA)(LAS106521), twice daily for up to 12 weeks (N = 185 participants) If severe side‐effects occurred, frequency of drug application could be reduced to 3 times per week (5‐FU ⁄SA and vehicle) or to once daily (diclofenac/HA). |
| Outcomes |
Primary outcome of the trial 1) Histological clearance rate of 1 predefined lesion Secondary outcomes of the trial 1) Lesion counts at baseline, and at week 12 (end of treatment) and 20 (8 weeks post‐treatment) 2) Mean total lesion area at baseline and week 20 (8 weeks post‐treatment) 3) Participant complete clearance (clinical) at week 20 4) Investigator's and participant's global assessment at weeks 6, 12, and 20 5) Treatment‐emergent adverse events (including application site reactions) 6) Tolerance (inflammation and burning) 7) Serious adverse events Efficacy Methods: 1. quantitative assessment by visual inspection of the treatment area and determination of the number and size of lesions, 2. biopsy of 1 representative target lesion performed by 3 mm punch biopsy and a second predefined clinically identical lesion for the biopsy at the post‐treatment visit evaluated by an independent and blinded dermatopathologist. Biopsy sites were selected on the basis of clinical appearance (clinical grade, area, and size). Photodocumentation and grid documentation were performed for the purposes of lesion identification. No skin tattoos were used, and no photodocumentation was performed during interim visits, 3. qualitative assessment by physician and participant ranging from 'very good' to 'none' Time points: at weeks 2, 4, 6, 10, 12 (end of treatment), and 20 (8 weeks post‐treatment) Safety Methods: participant‐reported adverse events Time points: at each visit |
| Notes | This corresponded to protocol NCT00987246 and EudraCT No. 2007‐003889‐18. |
| Methods | This was a randomised, assessor‐blinded, active‐controlled, intraindividual study. Start date: December 2008 End date: May 2009 |
| Participants |
Inclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: MAL‐2.5h low‐intensity artificial daylight photodynamic therapy (PDT) (N = 20 participants) Control intervention B: MAL‐red light‐emitting diode (LED) PDT (N = 20 participants) Characteristics of PDT intervention Type of treatment: field‐directed treatment (100 cm²) Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: scales and hyperkeratoses removed with a curette Cream concentration (%): 16 Application of cream: ‐‐ Incubation with cream: 3 hours (under occlusion for 0.5 hours for daylight and 3 hours for red LED) Type of light: artificial daylight, red light Light source: Xenon H4 light bulbs (daylight) Wavelength (nm): ‐‐ Energy fluence (J/cm²): 37 (red LED) Intensities (mW/cm²): ‐‐ Exposure time: 2.5 hours for artificial daylight |
| Outcomes |
Primary outcome of the trial 1) Lesion complete response Other outcomes of the trial 1) Mean reduction in lesion counts at 3 months post‐treatment 2) Participant complete response (= participant complete clearance) at 3 months post‐treatment 3) Pain scores (0 = no pain and 10 = worst imaginable pain) every half hour during 'daylight' exposure and every 1.5 minutes during red LED treatment 4) New actinic keratosis lesions 5) PpIX fluorescence Efficacy Methods: quantitative assessment using lesion grading and counting using a template Time points: at baseline and 3 months post‐treatment Definitions: complete response (complete disappearance of the lesion, visually and by palpation ‐ mild erythema might remain) |
| Notes | ‐ |
Characteristics of ongoing studies [ordered by study ID]
| Trial name or title | Randomised, double‐blind, placebo‐controlled study to assess efficacy of oral nicotinamide (500 mg daily) in the treatment and prevention of actinic keratoses |
| Methods | This is a randomised, double‐blind, placebo‐controlled, parallel‐group. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: oral nicotinamide 500 mg daily for 4 months Control intervention B: placebo tablets (lactose tablets identical in appearance and size to nicotinamide tablets but without active ingredient) daily for 4 months |
| Outcomes |
Primary outcome of the trial 1) Reduction in total actinic keratosis count at 2 and 4 months from baseline |
| Starting date | August 2010 |
| Contact information | A/Prof Diona Damian (diona.damian@sswahs.nsw.gov.au) Dermatology Gloucester House Level 3 Royal Prince Alfred Hospital Missenden Rd Camperdown 2050 Australia |
| NCT | ‐ |
| Notes | This study is ongoing and the last update was in August 2010. (April 2012) |
| Trial name or title | Vehicle‐Controlled, Double‐Blind Study to Assess the Safety and Efficacy of Imiquimod 5% Cream for the Treatment of Actinic Keratosis on the Upper Extremities |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: imiquimod 5% cream, once per day, 2 days per week for 16 weeks Control intervention B: vehicle cream, once per day, 2 days per week for 16 weeks |
| Outcomes |
Primary outcome of the trial 1) Efficacy of imiquimod 5% cream compared to vehicle cream Secondary outcome of the trial 1) Safety of treatment with imiquimod 5% cream |
| Starting date | May 2005 |
| Contact information | Graceway Pharmaceuticals, LLC |
| NCT | NCT00115154 |
| Notes | This study has been completed and the last update was in February 2007 (April 2012). |
| Trial name or title | Comparison of the Efficacy and Tolerability of Solaraze for 3 vs. 6 Months in Patients With Mild to Moderate Actinic Keratosis Located at the Face and Head |
| Methods | This is a multicentre, randomised, open, active‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: diclofenac, twice daily for 3 months Control intervention B: diclofenac, twice daily for 6 months |
| Outcomes |
Primary outcome of the trial 1) Histologically controlled complete clearance of the actinic keratosis at 6 weeks post‐treatment |
| Starting date | June 2005 |
| Contact information | Claus Garbe, MD Skin Cancer Program, Department of Dermatology, University Hospital Tübingen |
| NCT | NCT00204542 |
| Notes | This study has been completed in December 2010, and the last update was in August 2011 (April 2012). |
| Trial name or title | A Multicentre, Randomised Study of Photodynamic Therapy(PDT) With Metvix® 160 mg/g Cream in Immuno‐compromised Patients With Non‐melanoma Skin Cancer |
| Methods | This was a multicentre, randomised, open, active‐controlled, intraindividual study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: the treatment area on a randomised side (5 x 10 cm²) will be treated at baseline and at visits at 3, 9, and 15 months. At baseline the area will be treated with fractionated Metvix® PDT treatment consisting of 2 treatments 1 week apart and at visits at 3, 9, and 15 months with single Metvix® PDT treatment Control intervention B: in the contralateral control area (5 x 10 cm²), new and recurrent lesions and lesions in non‐complete response will be treated with lesion‐specific treatment at the discretion of the investigator at each study visit |
| Outcomes |
Primary outcomes of the trial 1) Occurrence of new lesions (actinic keratosis, basal cell carcinoma, squamous cell carcinoma and warts) at 3, 9, 15, 21, and 27 months after first treatment 2) Number of actinic keratosis lesions that show complete response at 3, 9, 15, 21, and 27 months after first treatment Secondary outcomes of the trial 1) Number of BCC lesions that show complete response in the treated area with the contralateral control area at 3, 9, 15, 21, and 27 months after first treatment 2) Number of recurrent lesions at 9, 15, 21, and 27 months after first treatment 3) Assess the cosmetic outcome at 3, 9, 15, 21, and 17 months after first treatment 4) Investigate product safety in this participant population at 3, 9, 15, 21, and 27 months after first treatment |
| Starting date | July 2003 |
| Contact information | Ann‐Marie Wennberg, MD, PhD (PI) Sahlgrenska University Hospital Gothenburg, Sweden |
| NCT | NCT00472459 |
| Notes | This study has been completed in July 2006, and the last update was in September 2010 (April 2012). |
| Trial name or title | Phase 2a Randomized, Placebo‐Controlled, Double‐Blind Trial of Topical Perillyl Alcohol in Sun Damaged Skin |
| Methods | This is a single‐centre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Disease characteristics
Exclusion criteria of the trial
Demographics
|
| Interventions |
Interventions A: perillyl alcohol (POH) cream (0.3%) applied topically to each dorsal forearm twice daily for 3 months in the absence of unacceptable toxicity B: perillyl alcohol (POH) cream (0.76%) applied topically to each dorsal forearm twice daily for 3 months in the absence of unacceptable toxicity Control intervention C: placebo cream applied topically to each dorsal forearm twice daily for 3 months in the absence of unacceptable toxicity |
| Outcomes |
Primary outcome of the trial 1) To determine if topical administration of perillyl alcohol (POH) cream can reverse actinic damage as evidenced by normalisation of quantitative skin histopathology scores in skin tissue biopsy samples from participants with moderate to severe sun damage Secondary outcomes of the trial 1) To determine if topically‐administered POH results in significant alterations in surrogate end point biomarkers of epidermal cell proliferation, including optical coherence tomography, p53 expression, c‐Fos expression, and apoptosis (as measured by activated caspase‐3 expression) 2)To determine if topically‐administered POH results in normalisation of nuclear chromatin patterns in skin biopsy tissue from these participants, as determined by karyometric analysis 3) To determine if topical POH can be administered safely to the forearms of these participants |
| Starting date | May 2004 |
| Contact information | Steve Stratton, MD (study chair) University of Arizona |
| NCT | NCT00608634 |
| Notes | The status of this study is unknown, and the last update was in September 2010 (April 2012). |
| Trial name or title | A Randomized Right/Left Clinical Trial to Evaluate the Use of Biafine Cream Versus Standard Care in Subjects With Actinic Keratosis Post Cryotherapy |
| Methods | This is a single‐centre, randomised, assessor‐blinded, active‐controlled, intraindividual study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: cryotherapy followed by Biafine cream Control intervention B: cryotherapy followed by standard care |
| Outcomes |
Primary outcome of the trial 1) The change of the target lesions from baseline to end of treatment in the IGA at 4 weeks |
| Starting date | October 2006 |
| Contact information | Steve Feldman, MD, PhD (PI) Wake Forest University |
| NCT | NCT00695578 |
| Notes | This study has been completed in February 2008, and the last update was in February 2009 (April 2012). |
| Trial name or title | A Multicenter, Randomized, Double‐blind, Vehicle‐controlled, Dose‐ranging Study to Evaluate the Safety and Efficacy of 0.005%, 0.01% and 0.015% PEP005 Topical Gel When Used to Treat Actinic Keratoses on the Head (Face or Scalp) |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Interventions A: PEP005 topical gel 0.005%, 2‐day treatment B: PEP005 topical gel 0.01%, 2‐day treatment C: PEP005 topical gel 0.015%, 2‐day treatment D: PEP005 topical gel 0.005%, 3‐day treatment E: PEP005 topical gel 0.01%, 3‐day treatment F: PEP005 topical gel 0.015%, 3‐day treatment Control interventions G: vehicle gel, 2‐day treatment H: vehicle gel, 3‐day treatment |
| Outcomes |
Primary outcome of the trial 1) Safety and toleration (incidence of adverse events, serious adverse events, and skin responses) at 57 days Secondary outcome of the trial 1) Efficacy (clearance of actinic keratosis lesions) at 57 days |
| Starting date | June 2008 |
| Contact information | Peplin |
| NCT | NCT00700063 |
| Notes | This study has been completed in October 2008, and the last update was in July 2010 (April 2012). Incomplete data were published in abstract form in an excluded study (Spencer 2010). |
| Trial name or title | A Randomized Controlled Paired Comparison of Photo‐therapy With a Topical Retinoid Cream Pretreatment Versus PDT Alone for Actinic Keratoses |
| Methods | This is a single‐centre, randomised, double‐blind, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: topical retinoid, for 4 weeks followed by blue‐light photodynamic therapy with photosensitising agent (at week 4) Control intervention B: blue‐light photodynamic therapy with photosensitising agent at week 4 |
| Outcomes |
Primary outcomes of the trial 1) Live blinded rater and blinded photo rater analysis of areas at week 0 and week 6 for erythema, edema, crusting, ulceration, palpability, and the need to cease/delay treatment 2) Overall response in reduction of number of AKs at 6 weeks Secondary outcomes of the trial 1) Participants will assess pain, burning, and itching on a scale of 0 to 3 at week 0, during retinoid treatment, during phototherapy, 1 day after, and at week 6 2) Principal investigator will evaluate adverse events at week 6 |
| Starting date | August 2008 |
| Contact information | Murad Alam, MD (PI) Department of Dermatology Northwestern University |
| NCT | NCT00756288 |
| Notes | This study is ongoing and the last update was in April 2011 (April 2012). |
| Trial name or title | Randomized, Multicenter, Double Blind Study to Compare the Efficacy and Tolerability of Oleogel‐S‐10 for 3 Month Versus Placebo Only in Patients With Mild to Moderate Actinic Keratoses Located at the Face and Head Oleogel‐S‐10 in Actinic Keratoses Trial |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Interventions A: oleogel‐S‐10 for 3 months once a day (N = 54 participants) B: oleogel‐S‐10 for 3 months twice a day (N = 54 participants) Control interventions C: placebo (petroleum jelly) for 3 months once a day (N = 27 participants) D: placebo (petroleum jelly) for 3 months twice a day (N = 27 participants) |
| Outcomes |
Primary outcome of the trial 1) Objective response of the marker actinic keratosis, defined as histologically complete or partial clearance (partial clearance = down‐grading in Cockerell‐classification) assessed at 18 weeks. The marker actinic keratosis is defined as an initially‐selected lesion within the target area that will be used for final biopsy Secondary outcomes of the trial 1) Histologically‐controlled complete clearance, 2) Histologically‐controlled down staging 75% clearance rate 3) Dose response relationship 4)Time to clinically complete response 5) Tolerability Assessment at 18 weeks |
| Starting date | October 2008 |
| Contact information | Birken GmbH Principal investigator Claus Garbe, Prof. Dr, Universitätshautklinik Tübingen |
| NCT | NCT00786994 |
| Notes | This study has been completed in November 2010 and the last update was in January 2012 (April 2012). |
| Trial name or title | A Multicenter, Double‐Blind, Vehical‐Controlled Study Comparing Imiquimod Cream, 5% (Apotex Inc.) to Aldara™ Cream, 5%(3M Pharmaceutials, U.S.) and Aldara™ Cream, 5%(3M Pharmaceuticals, Canada) in the Treatments of Actinic Keratosis |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Interventions A: Apotex, 5% imiquimod applied as a thin layer to target area once a day, 2 days each week, 16 weeks B: Aldara US, 5% imiquimod applied as a thin layer to target area once a day, 2 days each week, 16 weeks C: Aldara Canada, 5% imiquimod applied as a thin layer to target area once a day, 2 days each week, 16 weeks Control intervention D: vehicle applied as a thin layer to target area once a day, 2 days each week, 16 weeks |
| Outcomes |
Primary outcomes of the trial 1) The primary objectives are to establish the therapeutic equivalence of imiquimod cream 5%, manufactured by Apotex Inc. and 2 Aldara (imiquimod) creams, manufactured by 3M (US & Canada) at 24 weeks 2) Superiority over vehicle in the treatment of AK at 24 weeks Secondary outcome of the trial 1) The secondary objective is to compare the safety profiles of the 3 creams at 24 weeks |
| Starting date | February 2008 |
| Contact information | William Brooks (study director) Apotex Inc |
| NCT | NCT00859105 |
| Notes | This study has been completed in November 2008, and the last update was in March 2009 (April 2012). |
| Trial name or title | A Multicenter, Double‐Blind, Randomized, Parallel Group, Vehicle‐Controlled Study to Determine the Clinical Equivalence of a Generic Imiquimod Cream, 5% and Aldara™ Cream in Subjects With Actinic Keratosis |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: generic 5% topical cream dispensed in individual 0.25 g sachets applied twice a week for 16 weeks Control interventions B: Aldara 5% topical cream dispensed in individual 0.25 g sachets applied twice a week for 16 weeks C: topical cream vehicle matching generic imiquimod dispensed in individual 0.25 g sachets applied twice a week for 16 weeks |
| Outcomes |
Primary outcome of the trial 1) Proportion of participants in each treatment group with complete clearance of actinic keratosis lesions at 8 weeks post‐treatment (week 24, test of cure/TOC) visit Secondary outcomes of the trial 1) The partial clearance rates, defined as the proportion of subjects with at least a 75% reduction in the number of lesions counted at baseline 2) Proportion of participants with complete clearance of lesions at week 16 (end of treatment) and week 24 (TOC) |
| Starting date | May 2008 |
| Contact information | Christine M. Winslow, Ph.D. (study director) Actavis Mid‐Atlantic LLC |
| NCT | NCT00948428 |
| Notes | This study has been completed in April 2009, and last update was in August 2010 (April 2012). |
| Trial name or title | Double‐blind, Randomized, Multi‐centre Phase II Study to Evaluate the Efficacy and Safety of Topically Applied LAS41007 Once Daily and LAS41007 Twice Daily Versus LAS106521 Gel Twice Daily in the Treatment of Actinic Keratosis Grade I to II |
| Methods | This is a multicentre, randomised, double‐blind, active‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Interventions A: LAS41007 once daily B: LAS41007 twice daily Control intervention C: LAS106521 (3% diclofenac in hyaluronic acid) |
| Outcomes |
Primary outcomes of the trial 1) Histological clearance of 1 pre‐selected target lesion at day 120 2) Complete clinical clearance of all target lesions in the treatment areas at day 120 Secondary outcome of the trial 1) Physician's Global Tolerability Assessment (PGT) at day 120 |
| Starting date | August 2009 |
| Contact information | Christoph Willers, MD, MBA Almirall Hermal GmbH |
| NCT | NCT00991861 |
| Notes | This study has been completed in February 2010, and the last update was in July 2010 (April 2012). |
| Trial name or title | An Exploratory, Open‐label Study of Sequential Field‐directed Treatment of Actinic Keratoses of the Face With Imiquimod 3.75% Cream Followed by Photodynamic Therapy |
| Methods | This was a multicentre, randomised, open, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: imiquimod 3.75% cream, up to 2 packets, applied topically daily for 2 2‐week cycles separated by a no‐treatment interval of 2 weeks, followed 4 weeks later by photodynamic therapy with 20% aminolevulinic acid and blue light exposure of the entire face Control intervention B: Imiquimod 3.75% cream, up to 2 packets, applied topically daily for 2 2‐week cycles separated by a no‐treatment interval of 2 weeks, followed by observation |
| Outcomes |
Primary outcomes of the trial 1) Actinic keratosis count at week 18 2) The per cent change in actinic keratosis count as compared to the baseline lesion count Secondary outcomes of the trial 1) Complete clearance at week 18 2) The proportion of participants with complete clearance of actinic keratoses in the treatment area (entire face) 3) Cosmetic appearance at week 18 4) Change in investigator and participant scores of cosmetic appearance of the treatment area (entire face) |
| Starting date | September 2010 |
| Contact information | Julie Biron jbiron@goldskincare.com |
| NCT | NCT01203878 |
| Notes | This study is currently recruiting, and the last update was in July 2011 (April 2012). |
| Trial name or title | An Investigator‐Initiated Study to Assess the Safety and Efficacy of Imiquimod 3.75% Cream When Used After Cryotherapy in the Treatment of Hypertrophic Actinic Keratoses (AK) on Dorsal Hands and Forearms |
| Methods | This is a single‐centre, randomised, assessor‐blinded, intraindividual study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics:
|
| Interventions |
Intervention A: cryotherapy + imiquimod 3.75% daily for 2 weeks on, 2 weeks off, 2 weeks on on one arm Control intervention B: cryotherapy alone on the other arm |
| Outcomes |
Primary outcomes of the trial 1) Clearance of actinic keratoses assessed at 14 weeks 2) Actinic keratosis lesion count 3) Photography Secondary outcome of the trial 1) Local skin reactions assessed at 14 weeks |
| Starting date | October 2010 |
| Contact information | Gary S Goldenberg, MD (garygoldenbergmd@gmail.com) Giselle Singer, BS (giselle.singer@mssm.edu) |
| NCT | NCT01229319 |
| Notes | This study is currently recruiting, and the last update was in June 2011 (April 2012). |
| Trial name or title | Conventional Versus Fractional CO2 Laser Assisted Photodynamic Therapy for Basal Call Carcinomas and Actinic Keratoses |
| Methods | This is a single‐centre, randomised, assessor‐blinded, active‐controlled, intraindividual. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: fractional CO₂ laser assisted photodynamic therapy (PDT) pretreatment with fractional CO₂ laser before methyl‐aminolevulinate (MAL)‐red light PDT (37 J/cm²) Control intervention B: conventional photodynamic therapy using methyl‐aminolevulinate (MAL) and red light (37 J/cm²) |
| Outcomes |
Primary outcomes of the trial 1) Treatment response at 3 months, clinical evaluation by a blinded physician 2) Reoccurrence at 12 months, clinical evaluation by a blinded physician 3) Treatment response at 12 months Secondary outcomes of the trial 1) Pain during treatment, participant score (VAS 0 to 10) 2) Adverse effects at 12 months 3) Scaring, hyper‐ and hypopigmentation 4) Fluorescence at 3 hours, MAL uptake 5) Cosmetic result at 12 months, 4‐point scale |
| Starting date | October 2010 |
| Contact information | Christina S Haak, MD (christinahaak@dadlnet.dk) Katrine Togsverd‐Bo, MD (KTOG0001@regionh.bbh.dk) |
| NCT | NCT01260987 |
| Notes | This study is currently recruiting, and the last update was June 2011 (April 2012). |
| Trial name or title | Double‐blind, Randomized, Vehicle‐ and Comparator‐controlled, Multi‐center Trial to Evaluate the Efficacy and Safety of LAS41007 in the Treatment of Actinic Keratosis |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: LAS41007, twice daily, once in the morning and once in the evening. Per application, not more than 1.5 g of the immunologically mediated photodermatoses (IMP) should be applied, which is sufficient to cover a total area of 75 cm² (corresponding to 3 single TAs, each with a size of 25 cm² ) in maximum. The IMPs will be applied for 90 days in maximum Control interventions B: LASW1510, twice daily, once in the morning and once in the evening. Per application, not more than 1.5 g of the IMP should be applied, which is sufficient to cover a total area of 75 cm² (corresponding to 3 single TAs, each with a size of 25 cm²) in maximum. The IMPs will be applied for 90 days in maximum C: vehicle, twice daily, once in the morning and once in the evening. Per application, not more than 1.5 g of the IMP should be applied, which is sufficient to cover a total area of 75 cm² (corresponding to 3 single TAs, each with a size of 25 cm²) in maximum. The IMPs will be applied for 90 days in maximum |
| Outcomes |
Primary outcomes of the trial 1) Superiority of LAS41007 compared to vehicle at day 1 2) Superiority of LAS41007 compared to LASW1510 assessed by histology to evaluate the histological clearance of one pre‐selected target lesion 3) Superiority of LAS41007 compared to vehicle at day 150 4) Superiority of LAS41007 compared toLASW1510 each assessed by histology to evaluate the histological clearance of one pre‐selected target lesion Secondary outcomes of the trial 1) Superiority of LAS41007 compared to vehicle at day 1 2) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 1 3) Superiority of LAS41007 compared to vehicle at day 21 4) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 21 5) Superiority of LAS41007 compared to vehicle at day 56 6) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 56 7) Superiority of LAS41007 compared to vehicle at day 90 8) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 90 9) Superiority of LAS41007 compared to vehicle at day 150 10) Improved clinical efficacy of LAS41007 compared to LASW1510 with respect to clinical efficacy at day 150 |
| Starting date | November 2010 |
| Contact information | Sven Silberborth, PhD (sven.silberborth@almirall.com) |
| NCT | NCT01265602 |
| Notes | This study is currently recruiting, and the last update was in December 2010 (April 2012). |
| Trial name or title | Phase 3 Study of Brand Generic and Placebo in Treatment of Actinic Keratosis |
| Methods | This is a randomised, double‐blind, placebo‐ and active‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: generic 0.5% 5‐fluorouracil, once daily (duration of treatment was not specified) Control interventions B: carac 0.5% 5‐fluorouracil, once daily (duration of treatment was not specified) C: placebo, once daily (duration of treatment was not specified) |
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance at 6 weeks |
| Starting date | September 2010 |
| Contact information | ‐ |
| NCT | NCT01354717 |
| Notes | This study has been completed in March 2011, and the last update was in JUne 2011 (April 2012). |
| Trial name or title | Prospective Comparator Controlled Randomized Exploratory Study on the Efficacy of LAS 41005 Compared to Cryotherapy in Subjects With Hyperkeratotic Actinic Keratosis |
| Methods | This is a multicentre, randomised, open, active‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: LAS41005 (0.5% 5‐fluorouracil/ 10% salicylic acid) once daily (number of weeks was not specified) Control intervention B: cryotherapy 1 to 2 times during the treatment time |
| Outcomes |
Primary outcome of the trial 1) Histological clearance of 1 predefined target lesion at 8 weeks after the end of treatment with LAS41005 or 14 weeks after first cryotherapy Secondary outcomes of the trial 1) Participant complete clearance rates at days 21, 42, and 98 2) Participant partial (% was not specified) clearance rates at days 21, 42, and 98 |
| Starting date | April 2011 |
| Contact information | Rosario Rodríguez Almirall, S.A. |
| NCT | NCT01358851 |
| Notes | This study is ongoing, and the last update was January 2012 (April 2012). |
| Trial name or title | A Double‐blind, Randomized, Placebo‐controlled, 2‐way Crossover Study to Assess the Potential Effect of Topically Applied Imiquimod Cream on Atrial Ectopy in Patients With Actinic Keratosis |
| Methods | This is a single‐centre, randomised, double‐blind, placebo‐controlled, cross‐over study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: 3.75% imiquimod cream, daily for 2 weeks Control intervention B: placebo cream, daily for 2 weeks |
| Outcomes |
Primary outcome of the trial 1) Change in 24‐hour supraventricular beat count at day 14 Secondary outcomes of the trial 1) Change in 24‐hour supraventricular premature couplet and run counts and atrial fibrillation (per cent time) at day 14 2) Change in 24‐hour mean heart rate at day 14 3) Change in 24‐hour ventricular premature beat count, ventricular premature couplet, and run counts at day 14 |
| Starting date | July 2011 |
| Contact information | Irma Benavides (ibenavides@cnsmail.com) |
| NCT | NCT01413763 |
| Notes | This study is recruiting, and the last update was in August 2011 (April 2012). |
| Trial name or title | Long‐term Effects of Aldara® 5% Cream and Solaraze® 3% Gel in the Treatment of Actinic Keratoses on the Face or Scalp With Respect to the Risk of Progression to In‐situ and Invasive Squamous Cell Carcinoma |
| Methods | This is a multicentre, randomised, open, active‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: 5% imiquimod, 3 times per week for 4 weeks on, 4 weeks off, once or twice Control intervention B: 3% diclofenac in hyaluronic acid, twice daily for 12 weeks |
| Outcomes |
Primary outcome of the trial 1) Long‐term outcome (3 years) with respect to the risk of progression to SCC (in situ, invasive, or both) Secondary outcomes of the trial 1) Recurrence rate at 3 years post‐treatment 2) Time to recurrence at 3 years post‐treatment 3) Need of rescue treatment at 3 years post‐treatment 4) Cosmetic outcome at 3 years post‐treatment |
| Starting date | October 2011 |
| Contact information | Dr Ursula Petzold MEDA Pharma GmbH & Co. KG |
| NCT | NCT01453179 |
| Notes | This study is ongoing, and the last update was in March 2012 (April 2012). |
| Trial name or title | A Phase II Study of Photodynamic Therapy With LEVULAN® Topical Solution + Blue Light Versus LEVULAN® Topical Solution Vehicle + Blue Light for the Treatment of Actinic Keratoses on the Upper Extremities |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: 3 hours 20% ALA‐blue light PDT Control intervention B: 3 hours vehicle‐blue light PDT |
| Outcomes |
Primary outcome of the trial 1) Clearance rate for baseline grade I/II lesions at week 12 Secondary outcomes of the trial 1) Clearance rate for all baseline lesions at weeks 8 and 12 2) Clearance rate for baseline grade I/II lesions at week 8 3) Per cent change in total actinic keratoses at weeks 8 and 12 4) Participant complete clearance at weeks 8 and 12 5) Participant complete clearance excluding grade III lesions at weeks 8 and 12 6) Participant partial (> 75%) clearance at weeks 8 and 12 7) Participant satisfaction score 8) Changes in pigmentation (hypo and hyper) at 2, 8, and 12 weeks after PDT 9) Local skin reactions during PDT (stinging/burning); 5 minutes after PDT (erythema, edema); and 2, 8, and 12 weeks after PDT (erythema, edema, stinging/burning, scaling/dryness, oozing/vesiculation/crusting) |
| Starting date | November 2011 |
| Contact information | Jim Berg (jberg@therapeuticsinc.com) Dan Piacquadio, MD (danp@therapeuticsinc.com) |
| NCT | NCT01458587 |
| Notes | This study is recruiting, and the last update was in November 2011 (April 2012). |
| Trial name or title | Evaluation of the Formulation of 5‐aminolevulinic Acid With Dimethylsulfoxide in Photodynamic Therapy for Treatment of Actinic Keratosis |
| Methods | This is a single‐centre, randomised, open, active‐controlled, intraindividual study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: 4 hours 20% ALA‐red light PDT, once or twice with a 3‐month interval Control intervention B: cryotherapy, once or twice with a 3‐month interval |
| Outcomes |
Primary outcome of the trial 1) Changes in lesion area at 0, 3, and 6 months Secondary outcomes of the trial 1) Pain on a visual analogue scale (VAS) and a graduated scale at the time 0 and 15 minutes after each intervention 2) Cosmetic outcome evaluated subjectively by the participant and investigator (awful, bad, regular, good, excellent) and objectively by the investigator (presence or absence of 1 or more of these criteria: hypochromia, hyperpigmentation, hyperemia, scar) |
| Starting date | November 2010 |
| Contact information | Catarina Robert, MD René AC Vieira, PHD Fundação Pio XII ‐ Hospital de Câncer de Barretos |
| NCT | NCT01459393 |
| Notes | This study is ongoing, and the last update was in October 2011 (April 2012). |
| Trial name or title | IIntra‐individual Comparison of Efficacy and Safety of Metvix® Natural Daylight Photodynamic Therapy Versus Conventional Metvix® Photodynamic Therapy in Subject With Mild Actinic Keratoses |
| Methods | This is a multicentre, randomised, assessor‐blind, active‐controlled, intraindividual study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A:16% MAL‐daylight photodynamic therapy (PDT), once or twice with a 12‐week interval Control intervention B: 16% MAL‐conventional PDT, once or twice with a 12‐week interval |
| Outcomes |
Primary outcome of the trial 1) Per cent change from baseline in total number of treated mild lesions per side at week 12 2) Participant assessment of maximal pain on a scale from 0 (no pain) to 10 (extreme pain) per side at baseline |
| Starting date | March 2012 |
| Contact information | Catherine Bosc (catherine.bosc@galderma.com) |
| NCT | NCT01475071 |
| Notes | This study is recruiting, and the last update was in March 2012 (April 2012). |
| Trial name or title | A Phase II Study of Photodynamic Therapy With LEVULAN® Topical Solution + Blue Light Versus LEVULAN® Topical Solution Vehicle + Blue Light Using Spot and Broad Area Application and Incubation Times of 1, 2 and 3 Hours for the Treatment of Multiple Actinic Keratoses on the Face or Scalp |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Interventions A: field application 1 hours 20% ALA‐blue light photodynamic therapy (PDT) B: field application 2 hours 20% ALA‐blue light PDT C: field application 3 hours 20% ALA‐blue light PDT D: Individual lesion application 2 hours 20% ALA‐blue light PDT Control intervention E: field/individual application 1 to 3 hours 20% ALA‐blue light PDT |
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rate at week 12 Secondary outcomes of the trial 1) Participant complete clearance rate at weeks 4, 8, and 24 2) Participant partial (> 75%) clearance rate at weeks 4, 8, 12, and 24 3) Per cent change in lesion number at weeks 4, 8, 12, and 24 4) Participant satisfaction score on a 0 to 3 scale 5) Changes in pigmentation (hyper‐ and hypo‐) at 24 to 48 hours after PDT, and at weeks 2, 4, 8, 12, and 24 6) Local skin reactions during PDT (stinging/burning); at 5 minutes after PDT (erythema, edema); at 24 to 48 hours after PDT; and weeks 2, 4, 8, 12, and 24 (erythema, edema, stinging/burning, scaling/dryness, oozing/vesiculation/crusting) |
| Starting date | December 2011 |
| Contact information | Jim Berg (jberg@therapeuticsinc.com) Dan Piacquadio, MD (danp@therapeuticsinc.com) |
| NCT | NCT01475955 |
| Notes | This study is recruiting, and the last update was in March 2012 (April 2012). |
| Trial name or title | Prospective, Single‐center, Investigator‐blinded, Randomized, Half‐side, Comparative Study of Photodynamic Therapy vs. CO2 Laser Therapy in Treatment of Actinic Keratoses |
| Methods | This is a single‐centre, randomised, assessor‐blind, active‐controlled, intraindividual study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: CO₂ laser therapy Control intervention B: 4h ALA‐ red light photodynamic therapy (PDT) |
| Outcomes |
Primary outcome of the trial 1) Number of actinic keratoses at 3 months post‐treatment Secondary outcomes of the trial 1) Histologic features at 1 month post‐treatment 2) Epidermal thickness in optical coherence tomography at 1 month post‐treatment |
| Starting date | March 2011 |
| Contact information | Dr. Nina Scola, Consultant in Ruhr University of Bochum, Ruhr University of Bochum |
| NCT | NCT01481155 |
| Notes | This study is ongoing, and the last update was in December 2011 (April 2012). |
| Trial name or title | A Randomized, Double‐Blind, Parallel, Vehicle‐Controlled Phase III Trial to Assess the Efficacy and Safety of Topical SR‐T100 Gel in the Treatment of Patients With Actinic Keratosis |
| Methods | This is a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: SR‐T100 gel, once daily with an occlusive dressing for 16 weeks Control intervention B: vehicle gel, once daily with an occlusive dressing for 16 weeks |
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance at 8 weeks post‐treatment (week 24) Secondary outcomes of the trial 1) Lesion size reduction at 8 weeks post‐treatment (week 24) 2) Participant partial (> 75%) clearance at 8 weeks post‐treatment (week 24) 3)Tolerance |
| Starting date | October 2011 |
| Contact information | Kou‐Wha Kuo, Ph.D (kwkuo@geherbs.com.tw) Tony Chiu, B.S (tonychiu@geherbs.com.tw) |
| NCT | NCT01493921 |
| Notes | This study is recruiting, and the last update in March 2012 (April 2012). |
| Trial name or title | A Multicenter, Randomized, Double‐Blind, Vehicle‐Controlled, Parallel Group Comparison Study to Determine the Therapeutic Equivalence of Generic Imiquimod Cream, 3.75% and Zyclara™ (Imiquimod) Cream, 3.75% in Subjects With Actinic Keratoses |
| Methods | This was a multicentre, randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: generic 3.75% imiquimod, once daily for 2 weeks on, 2 weeks off, 2 weeks on Control interventions B: placebo, once daily for 2 weeks on, 2 weeks off, 2 weeks on C: Zyclara 3.75% imiquimod, once daily for 2 weeks on, 2 weeks off, 2 weeks on |
| Outcomes |
Primary outcomes of the trial 1) Participant complete clearance rate at 8 weeks post‐treatment (week 14) 2) Compliance 3) Severity and frequency of adverse events 4) Severity and frequency of local skin reactions Secondary outcome of the trial 1) Participant partial (> 75%) clearance rate at 8 weeks post‐treatment (week 14) 2) Per cent change in the lesion number at 8 weeks post‐treatment (week 14) |
| Starting date | February 2011 |
| Contact information | Daniel Piacquadio, M.D Therapeutics, Inc |
| NCT | NCT01502020 |
| Notes | This study has been completed in November 2011, and the last update was in December 2011 (Apil 2012). |
| Trial name or title | A Double‐blind, Randomized, Vehicle‐controlled, Parallel‐group, Phase II Dose‐ranging Study to Evaluate the Efficacy and Safety of SR‐T100 Gel in Patients With Actinic Keratosis (AK) on the Head (Face and/or Scalp) |
| Methods | This is a randomised, double‐blind, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Interventions A: SR‐T100 with 1.0% of SM in Solanum undatum plant extract for 16 weeks B: SR‐T100 with 2.3% of SM in Solanum undatum plant extract for 16 weeks Control intervention B: placebo for 16 weeks |
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance rate at 8 weeks post‐treatment (week 24) Secondary outcome of the trial 1) Participant partial (> 75%) clearance rate at 8 weeks post‐treatment (week 24) |
| Starting date | ‐ |
| Contact information | Dr Kou‐Wha Kuo (kwkuo@geherbs.com.tw) |
| NCT | NCT01516515 |
| Notes | This study is not yet recruiting, and the last update was January 2012 (April 2012). |
| Trial name or title | Combination Therapy With 5‐Fluorouracil and Photodynamic Therapy for the Treatment of Post‐transplant Premalignant Skin Disease |
| Methods | This is a single‐centre, randomised, open, parallel‐group (1 arm with immunocompetent controls and 1 arm with immunosuppressed organ transplant recipients), intraindividual (1 side with topical and PDT and 1 side with only PDT) study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: 5% 5‐fluorouracil once daily for 6 days followed by 3 hours MAL‐red light phototherapy (PDT) in immunocompetent and immunosuppressed participants Control intervention B: 3 hours MAL‐red light PDT in immunocompetent and immunosuppressed participants |
| Outcomes |
Primary outcome of the trial 1) Accumulation of PpIX at 3 hours after MAL application Secondary outcomes of the trial 1) Rate of lesion clearance at day 14 and at 3 months 2) Rate of development of new AK at months 3, 6 9, and 12 |
| Starting date | September 2011 |
| Contact information | Margo Riha, BSN, RN (riham@ccf.org) Sara Lohser, MD (lohsers@ccf.org) |
| NCT | NCT01525329 |
| Notes | This study is recruiting, and the last update was in February 2012 (April 2012). |
| Trial name or title | Topical Imiquimod 5% Cream Therapy Versus Photodynamic Therapy With Methyl‐aminolaevulinate 16% Cream of Actinic Keratoses in Organ Transplant Recipients |
| Methods | This is a single‐centre, randomised, open, active‐controlled, intraindividual study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: 3 hours 16% MAL‐red light photodynamic therapy (PDT) (70 J/cm²), twice with a 2 week interval Control intervention B: 5% imiquimod, 3 times per week for 4 weeks |
| Outcomes |
Primary outcome of the trial 1) Lesion complete response rate at 4 weeks post‐treatment Secondary outcomes of the trial 1) Lesion complete response rate at 6 and 12 months post‐treatment 2) Global reduction in the area of specific PpIX fluorescence at 1, 6, and 12 months post‐treatment 3) Global participant's satisfaction on a 10 cm visual analogue scale (VAS, 0 means extremely unsatisfied, 1 to 3 means unsatisfied, 5 to 7 means moderately satisfied, 8 to 10 means highly satisfied) at 3, 6, and 12 months post‐treatment |
| Starting date | April 2012 |
| Contact information | Stanislava Tzaneva, Doz. Dr (stanislava.tzaneva@meduniwien.ac.at) Alexandra Geusau, Prof. Dr (alexandra.geusau@meduniwien.ac.at) |
| NCT | NCT01538901 |
| Notes | This study is not yet recruiting, and the last update was in February 2012 (April 2012). |
| Trial name or title | Clinical Effect of Photodynamic Treatment When Treating Actinic Keratoses With Different Light Doses |
| Methods | This is a single‐centre, randomised, participant‐blinded, active‐controlled, intraindividual study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: 20% ALA‐red light photodynamic therapy (PDT) (70 J/cm²), twice with a 2‐week interval Control intervention B: 20% ALA‐red light PDT (100 J/cm²), twice with a 2‐week interval |
| Outcomes |
Primary outcome of the trial 1) Relapse of clinically cleared actinic keratosis, evaluation by two investigators for clinical/histological relapse at 1, 3, 6, 12, and 24 months post‐treatment Secondary outcome of the trial 1) Pain during treatment, participant scoring on a visual analogue scale (VAS) |
| Starting date | April 2010 |
| Contact information | Evelina Buinauskaite, MD Skaidra Valiukeviciene, Prof. Lithuanian University of Health Sciences, Medical Academy, Department of Skin and Venereal Diseases |
| NCT | NCT01541228 |
| Notes | This study is ongoing, and the last update was in February 2012 (April 2012). |
| Trial name or title | A Sequential Treatment Regimen of Cryotherapy and Picato® for the Treatment of Actinic Keratosis on the Face and Scalp |
| Methods | This is a multicentre, randomised, double‐blinded, placebo‐controlled, parallel‐group study. |
| Participants |
Inclusion criteria of the trial
Exclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: cryotherapy followed by 0.015% PEP005 (ingenol mebutate) gel (field) daily for 3 consecutive days Control intervention B: cryotherapy followed by vehicle gel (field) daily for 3 consecutive days |
| Outcomes |
Primary outcome of the trial 1) Participant complete clearance at week 11 Secondary outcomes of the trial 1) Per cent reduction from baseline in number of lesions at week 11 2) Participant complete clearance for 12 months (recurrence) 3) Per cent reduction from baseline in number of lesions at week 11 through to month 12 4) Participant partial (> 75%) clearance at week 11 4) Participant partial (> 75%) clearance at week 11 through to month 12 |
| Starting date | March 2012 |
| Contact information | Birgitte Vestbjerg (birgitte.vestbjerg@leo‐pharma.com) |
| NCT | NCT01541553 |
| Notes | This study is recruiting, and the last update was in March 2012 (April 2012). |
| Trial name or title | Temperature modulated photodynamic therapy for the treatment of actinic keratoses on the extremities |
| Methods | This is a randomised, blinded, active‐controlled, intraindividual study. |
| Participants |
Inclusion criteria of the trial
Demographics
|
| Interventions |
Intervention A: ALA + heat followed by blue light photodynamic therapy (PDT) (N= 20 participants) Control interventions B: ALA + no heat followed by blue light PDT (N = 20 participants) Characteristics of PDT intervention Type of treatment: ‐‐ Number of treatments: 1 Interval between treatments: ‐‐ Preparation of lesions: ‐‐ Cream concentration (%): 20 Application of cream: ‐‐ Incubation with cream: 1 hour under occlusion Type of light: blue light Light source: ‐‐ Wavelength (nm): 417 nm Energy fluence (J/cm²): 10 Intensities (mW/cm²): ‐‐ Exposure time: ‐‐ |
| Outcomes |
Outcomes of the trial 1) Clearance rates 2) Tolerability during and following treatment on a 4‐point scale Efficacy Methods: quantitative assessment by lesion counting performed by an unblinded investigator and photographs evaluation by a blinded investigator Time points: at baseline, and 2 and 6 months post‐treatment |
| Starting date | ‐ |
| Contact information | ‐ |
| NCT | ‐ |
| Notes | Abstract from 31st Annual Conference of the American Society for Laser Medicine and Surgery, ASLMS 2011 |
Differences between protocol and review
Authorship: Since the publication of the Cochrane Protocol in 2009, the review has been updated by new authors.
Background: This was updated as the protocol was published in 2003. The 'Disease definition' section was modified to emphasise the relationship between actinic keratosis and squamous cell carcinoma, and previous information about the differential diagnosis was moved to the 'Clinical features' section. The 'Epidemiology and causes' section title was changed to 'Pathogenesis and epidemiology' to reflect better the order of the information presented. Finally, a 'How the intervention might work' section was added to follow the recommendation in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Methods:
Criteria for considering studies for this review:
Types of interventions: The sentence on the comparators accepted was slightly modified to include any variation of the treatment (duration, concentration, etc) because several studies investigated the influences of these parameters on primary and secondary outcomes.
Types of outcome measures: The rationale for selecting only per participant outcomes (i.e. randomisation per participant and not per lesion) was presented, as well as a description on the general method used to evaluate efficacy in the included studies.
Changes in the primary outcomes:
a) To make a distinction between "Global degree of improvement in symptoms and/or signs as rated by participant or medical practitioner" (subjective) and "Participant complete clearance" (objective), subjective assessment was added before the outcome description. Only global improvement indices for completely improved or cleared were considered for inclusion in meta‐analysis.
b) "Lesion clearance rate of 100% and 75%" were changed for "participant complete (100%) or partial (> 75%) clearance" to make a distinction between "lesion complete response" expressed per lesion, which is not included in our review, and the number of participants with the percentage of lesions cleared. "Participant complete clearance" and "Participant partial (> 75%) clearance" were presented separately.
c) The outcomes reported in actinic keratoses studies that could be reported as "Improvement in quality of life" were cosmetic outcomes, which were reported separately as secondary outcomes. Thus, 'Improvement in quality of life' was removed from our primary outcomes.
The objective assessment, "Mean reduction in lesion counts" expressed as absolute values or percentages was included in the review because this per participant outcome was often reported, and it was the only efficacy outcome that could be analysed by meta‐analysis for intraindividual studies. Thus, all primary outcomes became efficacy outcomes.
Changes in the secondary outcomes:
(a) "Severe adverse events, i.e. severe enough to require withdrawal of treatment" was changed to "Withdrawal due to adverse events" to avoid problems with the interpretation of data.
(c) "Minor patient‐reported adverse events, not sufficient to require cessation of treatment, excluding skin irritation" was replaced by "Minor adverse events excluding skin irritation" because the source, reported by participant or investigator, was generally not specified.
To fulfil the requirement of a 'per participant' outcome for meta‐analysis, all safety outcomes were the number of participants experiencing events in general or a specific event, and cosmetic outcomes were the number of participants with the different cosmetic measurement.
Search methods for identification of studies:
Electronic searches: We updated all searches in March 2011, and the LILACs database was added, as well as different online ongoing trial registers.
Unpublished literature: We searched additional companies, as well as the FDA website, for clinical trials in the product insert.
Conference proceedings: We searched conference proceedings from additional associations.
Data collection and analysis:
Selection of studies/Data extraction and management:
Several collaborators contributed to this review over the years, and they shared different responsibilities in the study selection, data extraction, and data analysis. During a global revision of the manuscript undertaken in March 2011, the different roles were redistributed to the current authors, and they may differ from the protocol.
Assessment of risk of bias in included studies:
Cochrane methodology was changed for assessment of methodological quality. Thus, the quality of the data was assessed with two new tools: 1) the 'Risk of bias' tables included in RevMan 5.1, and 2) the quality of evidence by the GRADEpro software.
Analysis:
The section of the protocol on planed data analysis has been divided into the following sections: measures of treatment effect, unit of analysis issues, assessment of heterogeneity, data synthesis, and subgroup analysis and investigation of heterogeneity.
In 'Measures of treatment effect', the original protocol stated that the results will be expressed as odds ratios for dichotomous outcomes. However, the results were presented as risk ratios because they are easier to interpret.
In 'Unit of analysis issues', a strategy for analysis and reporting of data from intraindividual studies and studies with multiple treatments was added.
In 'Assessment of heterogeneity', a cut‐off value of the I² statistic as a measure of heterogeneity was added.
In 'Data synthesis', no meta‐analysis method was specified in the protocol, but a random‐effects model was prespecified for all analyses.
In 'Subgroup analysis and investigation of heterogeneity', subgroup analysis in the protocol was referred to the different types of interventions (i.e. topical, oral, mechanical, etc), which were analysed in separate comparisons. Subgroup analysis in the review referred to subgroup analysis within a comparison.
Because of the large number of randomised studies included in this review, non‐randomised controlled studies were not listed as mentioned in the protocol.
Contributions of authors
We indicate below the contributions made by the reviewers.
Link with editorial base and coordinate contributions from co‐reviewers (AG)
Draft protocol (AG, RW, with contributions from all)
Run search (AG, WB, and MP)
Identify relevant titles and abstracts from searches, i.e. broad screen (MP and WB)
Obtain full text copies of studies (WB and MP)
Select trials to include (WB, MP, and AG as arbitrator when necessary)
Extract data from trials (WB and MP)
Enter data into RevMan (MP)
Carry out the analysis (MP and EV)
Interpret the analysis (MP)
Draft final review (MP, WB, and AG)
Update the review (MP)
Declarations of interest
Dr Aditya Gupta, lead author of this review, participated in a clinical trial sponsored by DUSA in 2004, which was excluded from the review because it did not meet the inclusion criteria.
Dr Stephen Keohane, clinical referee for this review, states: "I have been paid for lectures and advisory boards by Galderma, Almirall, Intendis, Inc., Shire, and Bayer."
Edited (no change to conclusions)
References
References to studies included in this review
Akar 2001 {published data only}
- Akar A, Tastan HB, Erbil H, Arca E, Kurumlu Z, Gur AR. Efficacy and safety assessment of 0.5% and 1% colchicine cream in the treatment of actinic keratoses. Journal of Dermatological Treatment 2001;12(4):199‐203. [PUBMED: 12241628] [DOI] [PubMed] [Google Scholar]
Alberts 2000 {published data only}
- Alberts DS, Dorr RT, Einspahr JG, Aickin M, Saboda K, Xu MJ, et al. Chemoprevention of human actinic keratoses by topical 2‐(difluoromethyl)‐dl‐ornithine. Cancer Epidemiology, Biomarkers & Prevention 2000;9(12):1281‐6. [PUBMED: 11142412] [PubMed] [Google Scholar]
- Einspahr JG, Nelson MA, Saboda K, Warneke J, Bowden GT, Alberts DS. Modulation of biologic endpoints by topical difluoromethylornithine (DFMO), in subjects at high‐risk for nonmelanoma skin cancer. Clinical Cancer Research 2002;8(1):149‐55. [PUBMED: 11801552 ] [PubMed] [Google Scholar]
Alirezai 1994 {published data only}
- Alirezai M, Dupuy P, Amblard P, Kalis B, Souteyrand P, Frappaz A, et al. Clinical evaluation of topical isotretinoin in the treatment of actinic keratoses. Journal of the American Academy of Dermatology 1994;30(3):447‐51. [PUBMED: 8113458] [DOI] [PubMed] [Google Scholar]
Alomar 2007 {published data only}
- Alomar A, Bichel J, McRae S. Vehicle‐controlled, randomized, double‐blind study to assess safety and efficacy of imiquimod 5% cream applied once daily 3 days per week in one or two courses of treatment of actinic keratoses on the head. British Journal of Dermatology 2007;157(1):133‐41. [PUBMED: 17501955 ] [DOI] [PubMed] [Google Scholar]
Anderson 2009 {published data only}
- Anderson L, Schmieder GJ, Werschler WP, Tschen EH, Ling MR, Stough DB, et al. Randomized, double‐blind, double‐dummy, vehicle controlled study of ingenol mebutate gel 0.025% and 0.05% for actinic keratosis. Journal of the American Academy of Dermatology 2009;60(6):934‐43. [PUBMED: 19467365 ] [DOI] [PubMed] [Google Scholar]
- NCT00375739. Study to Determine the Safety of PEP005 0.025% and 0.05% Topical Gel in Patients With Actinic Keratoses. clinicaltrials.gov/ct2/show/NCT00375739 (accessed 14 March 2011).
Bercovitch 1987 {published data only}
- Bercovitch L. Topical chemotherapy of actinic keratoses of the upper extremity with tretinoin and 5‐fluorouracil: a double‐blind controlled study. British Journal of Dermatology 1987;116(4):549‐52. [PUBMED: 3555597] [DOI] [PubMed] [Google Scholar]
Chen 2003 {published data only}
- Chen K, Yap LM, Marks R, Shumack S. Short‐course therapy imiquimod 5% cream for solar keratoses: A randomized controlled trial. Australasian Journal of Dermatology 2003;44(4):250‐5. [PUBMED: 14616490] [DOI] [PubMed] [Google Scholar]
Dragieva 2004a {published and unpublished data}
- Dragieva G, Prinz B, Hafner J, Dummer R, Burg G, Binswanger U, et al. Topical photodynamic therapy with MAL in the treatment of actinic keratoses in transplant recipients. [Abstract PD‐16] The 85th BAD Annual Meeting 5‐8th July 2005, Glasgow, UK. British Journal of Dermatology 2005;153(Suppl 1):97. [Google Scholar]
- Dregieva G, Prinz B, Hafner J, Dummer R, Burg G, Binswanger U, et al. A randomized controlled clinical trial of topical photodynamic therapy with methyl aminolaevulinate in the treatment of actinic keratoses in transplant recipients. British Journal of Dermatology 2004;151(1):196‐200. [PUBMED: 15270891] [DOI] [PubMed] [Google Scholar]
Fariba 2006 {published data only}
- Fariba I, Ali A, Hossein SA, Atefeh S, Atarzadeh Behbahan SA. Efficacy of 3% diclofenac gel for the treatment of actinic keratoses: a randomized, double‐blind, placebo controlled study. Indian Journal of Dermatology, Venereology & Leprology 2006;72(5):346‐9. [PUBMED: 17050927] [DOI] [PubMed] [Google Scholar]
Foote 2009 {published data only}
- Foote JA, Ranger‐Moore JR, Einspahr JG, Saboda K, Kenyon J, Warneke J, et al. Chemoprevention of human actinic keratoses by topical DL‐α‐tocopherol. Cancer Prevention Research 2009;2(4):394‐400. [PUBMED: 19336724] [DOI] [PMC free article] [PubMed] [Google Scholar]
Freeman 2003 {published and unpublished data}
- Foley P, Freeman M, Vinciullo C, Spelman L, Murrell D, Weightman W, et al. Photodynamic therapy using methyl aminolaevulinate with cryotherapy in actinic keratosis: an Australian study. [Abstract P‐31] The 85th BAD Annual Meeting 5‐8th July 2005, Glasgow, UK. British Journal of Dermatology 2005; Vol. 153, issue Suppl 1:29.
- Freeman M, Vinciullo C, Francis D, Spelman L, Nguyen R, Fergin P, et al. A comparison of photodynamic therapy using topical methyl aminolevulinate (Metvix) with single cycle cryotherapy in patients with actinic keratosis: a prospective, randomized study. Journal of Dermatological Treatment 2003;14(2):99‐106. [PUBMED: 12775317] [DOI] [PubMed] [Google Scholar]
Gebauer 2003 {published data only}
- Gebauer K, Brown P, Varigos G. Topical diclofenac in hyaluronan gel for the treatment of solar keratoses. Australasian Journal of Dermatology 2003;44(1):40‐3. [PUBMED: 12581080 ] [DOI] [PubMed] [Google Scholar]
- Paquet M. Confirmation of ITT analysis for efficacy [personal communication]. Email to: Dr Gebauer 27 March 2011.
Gebauer 2009 {published data only}
- Gebauer K, Shumack S, Cowen P S. Effect of dosing frequency on the safety and efficacy of imiquimod 5% cream for treatment of actinic keratosis on the forearms and hands: a phase II, randomized placebo‐controlled trial. British Journal of Dermatology 2009;161(4):897‐903. [PUBMED: 19545297] [DOI] [PubMed] [Google Scholar]
Hanke 2010 {published and unpublished data}
- Hanke CW, Beer KR, Stockfleth E, Wu J, Rosen T, Levy S. Imiquimod 2.5% and 3.75% for the treatment of actinic keratoses: results of two placebo‐controlled studies of daily application to the face and balding scalp for two 3‐week cycles. Journal of the American Academy of Dermatology 2010;62(4):573‐81. [PUBMED: 20133012] [DOI] [PubMed] [Google Scholar]
- NCT00603798. Safety and Effectiveness Study of Imiquimod Creams for the Treatment of Actinic Keratoses (actinic keratoses). clinicaltrials.gov/ct2/show/NCT00603798 (accessed 14 March 2011).
Hantash 2006 {published data only}
- Hantash BM, Stewart DB, Cooper ZA, Rehmus WE, Koch RJ, Swetter SM. Facial resurfacing for nonmelanoma skin cancer prophylaxis. Archives of Dermatology 2006;142(8):976‐82. [PUBMED: 16924046 ] [DOI] [PubMed] [Google Scholar]
Hauschild 2009a {published data only}
- Hauschild A, Stockfleth E, Popp G, Borrosch F, Bruning H, Dominicus R, et al. Optimization of photodynamic therapy with a novel self‐adhesive 5‐aminolaevulinic acid patch: results of two randomized controlled phase III studies. British Journal of Dermatology 2009;160(5):1066‐74. [PUBMED: 19222455] [DOI] [PubMed] [Google Scholar]
- NCT00308854. PD P 506 A‐PDT Versus Placebo‐PDT for the treatment of actinic keratose. clinicaltrials.gov/ct2/show/NCT00308854 (accessed 14 March 2011).
Hauschild 2009b {published data only}
- Hauschild A, Stockfleth E, Popp G, Borrosch F, Bruning H, Dominicus R, et al. Optimization of photodynamic therapy with a novel self‐adhesive 5‐aminolaevulinic acid patch: results of two randomized controlled phase III studies. British Journal of Dermatology 2009;160(5):1066‐74. [PUBMED: 19222455] [DOI] [PubMed] [Google Scholar]
- NCT00308867. PD P 506 A‐PDT Versus Placebo‐PDT and Cryosurgery for the Treatment of actinic keratose. clinicaltrials.gov/ct2/show/NCT00308867 (accessed 14 March 2011).
Hauschild 2009c {published data only}
- Hauschild A, Popp G, Stockfleth E, Meyer K G, Imberger D, Mohr P, et al. Effective photodynamic therapy of actinic keratoses on the head and face with a novel, self‐adhesive 5‐aminolaevulinic acid patch. Experimental Dermatology 2009;18(2):116‐21. [PUBMED: 18643849] [DOI] [PubMed] [Google Scholar]
Huyke 2009 {published data only}
- Huyke C, Reuter J, Rodig M, Kersten A, Laszczyk M, Scheffler A, et al. Treatment of actinic keratoses with a novel betulin‐based oleogel. A prospective, randomized, active‐controlled pilot study. Journal Der Deutschen Dermatologischen Gesellschaft 2009;7(2):128‐133. [PUBMED: 18808378] [DOI] [PubMed] [Google Scholar]
Jeffes 2001 {published data only}
- Jeffes E, McCullough JL, Weinstein GD, Kaplan R, Glazer SD, Taylor JR. Photodynamic therapy of actinic keratoses with topical aminolevulinic acid hydrochloride and fluorescent blue light. Journal of the American Academy of Dermatology 2001;45(1):96‐104. [PUBMED: 11423841] [DOI] [PubMed] [Google Scholar]
Jorizzo 2002 {published and unpublished data}
- Dermik Laboratories. Carac product insert. //products.sanofi.us/carac/carac.html (accessed 24 March 2011).
- Jorizzo J. Topical treatment of actinic keratosis with fluorouracil: is irritation associated with efficacy?. Journal of Drugs in Dermatology 2004;3(1):21‐6. [PUBMED: 14964743] [PubMed] [Google Scholar]
- Jorizzo J, Stewart D, Bucko A, Davis SA, Espy P, Hino P, et al. Randomized trial evaluating a new 0.5% fluorouracil formulation demonstrates efficacy after 1‐, 2‐, or 4‐week treatment in patients with actinic keratosis. Cutis 2002;70(6):335‐9. [PUBMED: 12502122] [PubMed] [Google Scholar]
Jorizzo 2004 {published data only}
- Jorizzo J, Weiss J, Furst K, VandePol C, Levy S. Effect of a 1‐week treatment with 0.5% topical fluorouracil on occurrence of actinic keratosis after cryosurgery: A randomized, vehicle‐controlled clinical trial. Archives of Dermatology 2004;140:813‐6. [PUBMED: 15262691] [DOI] [PubMed] [Google Scholar]
Jorizzo 2006 {published data only}
- Jorizzo J, Weiss J, Vamvakias G. One‐week treatment with 0.5% fluorouracil cream prior to cryosurgery in patients with actinic keratoses: a double‐blind, vehicle‐controlled, long‐term study. Journal of Drugs in Dermatology 2006;5(2):133‐9. [PUBMED: 16485881 ] [PubMed] [Google Scholar]
Jorizzo 2007 {published data only}
- Jorizzo J, Dinehart S, Matheson R, Moore JK, Ling M, Fox TL, et al. Vehicle‐controlled, double‐blind, randomized study of imiquimod 5% cream applied 3 days per week in one or two courses of treatment for actinic keratoses on the head. Journal of the American Academy of Dermatology 2007;57(2):265‐8. [PUBMED: 17512087] [DOI] [PubMed] [Google Scholar]
Jorizzo 2010 {published and unpublished data}
- Jorizzo JL, Markowitz O, Lebwohl MG, Bourcier M, Kulp J, Meng TC, et al. A randomized, double‐blinded, placebo‐controlled, multicenter, efficacy and safety study of 3.75% imiquimod cream following cryosurgery for the treatment of actinic keratoses. Journal of Drugs in Dermatology 2010;9(9):1101‐8. [PUBMED: 20865842] [PubMed] [Google Scholar]
- Lee J, Jorizzo J, Lebwohl M, Markowitz O, Levy S. Cryosurgery followed by imiquimod 3.75% to treat actinic keratosis. 69th Annual Meeting of the American Academy of Dermatology New Orleans, LA United States, February 2011. Journal of the American Academy of Dermatology 2011; Vol. 64, issue 2 Suppl 1:AB2.
- NCT00894647. Safety and effectiveness study of actinic keratosis treatment following cryosurgery. clinicaltrials.gov/ct2/show/NCT00894647 (accessed 14 March 2011).
Kang 2003 {published data only}
- Kang S, Goldfarb MT, Weiss JS, Metz RD, Hamilton TA, Voorhees JJ, et al. Assessment of adapalene gel for the treatment of actinic keratoses and lentigines: a randomized trial. Journal of the American Academy of Dermatology 2003;49(1):83‐90. [PUBMED: 12833014 ] [DOI] [PubMed] [Google Scholar]
Kaufmann 2008 {published data only}
- Kaufmann R, Spelman L, Weightman W, Reifenberger J, Szeimies R M, Verhaeghe E, et al. Multicentre intraindividual randomized trial of topical methyl aminolaevulinate‐photodynamic therapy vs. cryotherapy for multiple actinic keratoses on the extremities. British Journal of Dermatology 2008;158(5):994‐9. [PUBMED: 18341663] [DOI] [PubMed] [Google Scholar]
Korman 2005 {published data only}
- Korman N, Moy R, Ling M, Matheson R, Smith S, McKane S, et al. Dosing with 5% imiquimod cream 3 times per week for the treatment of actinic keratosis: Results of two phase 3, randomized, double‐blind, parallel‐group, vehicle‐controlled trials. Archives of Dermatology 2005;141(4):467‐73. [PUBMED: 15837864 ] [DOI] [PubMed] [Google Scholar]
Kose 2008 {published data only}
- Kose O, Koc E, Erbil AH, Caliskan E, Kurumlu Z. Comparison of the efficacy and tolerability of 3% diclofenac sodium gel and 5% imiquimod cream in the treatment of actinic keratosis. Journal of Dermatological Treatment 2008;19(3):159‐63. [PUBMED: 18569272 ] [DOI] [PubMed] [Google Scholar]
Krawtchenko 2007 {published data only}
- Krawtchenko N, Roewert‐Huber J, Ulrich M, Mann I, Sterry W, Stockfleth E. A randomized study of topical 5% imiquimod vs. topical 5‐fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1‐year follow‐up. British Journal of Dermatology 2007;157(Suppl 2):34‐40. [PUBMED: 18067630] [DOI] [PubMed] [Google Scholar]
Kulp‐Shorten 1993 {published data only}
- Kulp‐Shorten C, Konnikov N, Callen J. Comparative Evaluation of the efficacy and Safety of Masoprocol and 5‐Fluorouracil Cream for the Treatment of Multiple Actinic Keratoses of the Head and Neck. Journal of Geriatric Dermatology 1993;1(4):161‐8. [Google Scholar]
Lebwohl 2004 {published and unpublished data}
- Graceway Pharmaceuticals. Aldara product monograph. //www.gracewaypharma.com/sites/default/files/products/aldara_ppi.pdf (accessed 24 March 2011).
- Lebwohl M, Dinehart S, Whiting D, Lee PK, Tawfik N, Jorizzo J, et al. Imiquimod 5% cream for the treatment of actinic keratosis: results from two phase III, randomized, double‐blind, parallel group, vehicle‐controlled trials. Journal of the American Academy of Dermatology 2004;50(5):714‐21. [PUBMED: 15097955] [DOI] [PubMed] [Google Scholar]
Loven 2002 {published data only}
- Loven K, Stein L, Furst K, Levy S. Evaluation of the efficacy and tolerability of 0.5% fluorouracil cream and 5% fluorouracil cream applied to each side of the face in patients with actinic keratosis. Clinical Therapeutics 2002;24(6):990‐1000. [PUBMED: 12117087] [DOI] [PubMed] [Google Scholar]
McEwan 1997 {published data only}
- McEwan LE, Smith JG. Topical diclofenac/hyaluronic acid gel in the treatment of solar keratoses. Australasian Journal of Dermatology 1997;38(4):187‐9. [PUBMED: 9431711] [DOI] [PubMed] [Google Scholar]
Misiewicz 1991 {published data only}
- Misiewicz J, Sendagorta E, Golebiowska A, Lorenc B, Czarnetzki BM, Jablonska S. Topical treatment of multiple actinic keratoses of the face with aretinoid methyl sulfone (Ro 14‐9706) cream versus tretinoin cream: a double‐blind, active‐controlled study. Journal of the American Academy Dermatology 1991;24(3):448‐51. [PUBMED: 2061443] [DOI] [PubMed] [Google Scholar]
Moloney 2007 {published data only}
- Moloney FJ, Collins P. Randomized, double‐blind, prospective study to compare topical 5‐aminolaevulinic acid methylester with topical 5‐aminolaevulinic acid photodynamic therapy for extensive scalp actinic keratosis. British Journal of Dermatology 2007;157(1):87‐91. [PUBMED: 17501954 ] [DOI] [PubMed] [Google Scholar]
Moloney 2010 {published data only}
- ACTRN12609000490279. Randomized, double‐blind, placebo controlled study to assess efficacy of topical nicotinamide in the treatment and prevention of actinic keratosis.. anzctr.org.au/trial_view.aspx?ID=83879 (accessed 14 March 2011).
- Moloney F, Vestergaard M, Radojkovic B, Damian D. Randomized, double‐blinded, placebo controlled study to assess the effect of topical 1% nicotinamide on actinic keratoses. British Journal of Dermatology 2010; Vol. 162, issue 5:1138‐9. [PUBMED: 20199551] [DOI] [PubMed]
Moriarty 1982 {published data only}
- Moriarty M, Dunn J, Darragh A, Lambe R, Brick I. Etretinate in treatment of actinic keratosis: a double‐blind crossover study. Lancet 1982;1(8268):364‐5. [PUBMED: 6120350] [DOI] [PubMed] [Google Scholar]
Morton 2006 {published data only}
- Morton C, Campbell S, Gupta G, Keohane S, Lear J, Zaki I, et al. Intraparticipant, right‐left comparison of topical methyl aminolaevulinate‐photodynamic therapy and cryotherapy in subjects with actinic keratoses: a multicentre, randomized controlled study. British Journal of Dermatology 2006;155(5):1029‐36. [PUBMED: 17034536] [DOI] [PubMed] [Google Scholar]
NCT00774787 {unpublished data only}
- NCT00774787. Topical imiquimod cream in combination with cryotherapy for the treatment of actinic keratoses. http://www.clinicaltrials.gov/ct2/show/NCT00774787 (accessed 14 March 2011).
NCT00828568 Aldara {unpublished data only}
- NCT00828568. A Therapeutic Equivalence Study of Two Imiquimod Cream 5% Treatments for Patients With Actinic Keratosis. http://www.clinicaltrials.gov/ct2/show/NCT00828568 (accesssed 14 March 2011).
NCT00828568 Taro {unpublished data only}
- NCT00828568. A Therapeutic Equivalence Study of Two Imiquimod Cream 5% Treatments for Patients With Actinic Keratosis. clinicaltrials.gov/ct2/show/NCT00828568 (accessed 14 March 2011).
Olsen 1991 {published data only}
- Olsen EA, Abernethy ML, Kulp‐Shorten C, Callen JP, Glazer SD, Huntley A, et al. A double‐blind, vehicle‐controlled study evaluating masoprocol cream in the treatment of actinic keratoses on the head and neck. Journal of the American Academy of Dermatology 1991;24(5 Pt 1):738‐43. [PUBMED: 1869646] [DOI] [PubMed] [Google Scholar]
Ooi 2006 {published data only}
- Ooi T, Barnetson RS, Zhuang L, McKane S, Lee JH, Slade HB, et al. Imiquimod‐induced regression of actinic keratosis is associated with infiltration by T lymphocytes and dendritic cells: a randomized controlled trial. British Journal of Dermatology 2006;154(1):72‐8. [PUBMED: 16403097] [DOI] [PubMed] [Google Scholar]
Ortonne 2010 {published data only}
- NCT00294320. Evaluation of Two Different Non‐Invasive Techniques to Monitor the Clearance of Actinic Keratosis Lesions. clinicaltrials.gov/ct2/show/NCT00294320 (accessed 14 March 2011).
- Ortonne JP, Gupta G, Ortonne N, Duteil L, Queille C, Mallefet P. Effectiveness of cross polarized light and fluorescence diagnosis for detection of sub‐clinical and clinical actinic keratosis during imiquimod treatment. Experimental Dermatology 2010;19(7):641‐7. [PUBMED: 20201959] [DOI] [PubMed] [Google Scholar]
Ostertag 2006 {published data only}
- Ostertag JU, Quaedvlieg PJF, Geer S, Nelemans P, Christianen ME, Neumann MH, et al. A clinical comparison and long‐term follow‐up of topical 5‐fluorouracil versus laser resurfacing in the treatment of widespread actinic keratoses. Lasers in Surgery & Medicine 2006; Vol. 38, issue 8:731‐9. [PUBMED: 16912977] [DOI] [PubMed]
Pariser 2003 {published data only}
- Pariser DM, Lowe NJ, Stewart DM, Jarratt MT, Lucky AW, Pariser RJ, et al. Photodynamic therapy with topical methyl aminolevulinate for actinic keratosis: results of a prospective randomized multicenter trial. Journal of the American Academy of Dermatology 2003;48(2):227‐32. [PUBMED: 12582393] [DOI] [PubMed] [Google Scholar]
Pariser 2008 {published and unpublished data}
- Galderma. Metvixia product label. http://metvixia.com/pi/MetvixiaPI.pdf (accessed on 26 April 2011).
- NCT00306800. Metvix PDT Versus Vehicle PDT With Aktilite CL128 Lamp in Patients With Actinic Keratosis on the Face and Scalp. clinicaltrials.gov/ct2/show/NCT00306800 (accessed 14 March 2011). [NCT00306800]
- Pariser D, Loss R, Jarratt M, Abramovits W, Spencer J, Geronemus R, et al. Topical methyl‐aminolevulinate photodynamic therapy using red light‐emitting diode light for treatment of multiple actinic keratoses: a randomized, double‐blind, placebo‐controlled study. Journal of the American Academy of Dermatology 2008;59(4):569‐76. [PUBMED: 18707799 ] [DOI] [PubMed] [Google Scholar]
Perrett 2007 {published data only}
- Perrett CM, McGregor JM, Warwick J, Karran P, Leigh IM, Proby CM, et al. Treatment of post‐transplant premalignant skin disease: a randomized intrapatient active‐controlled study of 5‐fluorouracil cream and topical photodynamic therapy. British Journal of Dermatology 2007;156(2):320‐8. [PUBMED: 17223873 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Persaud 2002 {published data only}
- Persaud AN, Shamuelova E, Sherer D, Lou W, Singer G, Cervera C, et al. Clinical effect of imiquimod 5% cream in the treatment of actinic keratosis. Journal of the American Academy of Dermatology 2002;47(4):553‐6. [PUBMED: 12271300] [DOI] [PubMed] [Google Scholar]
Photocure‐Australian 2004 {unpublished data only}
- Photocure ASA. Metvixia cream, 16.8%, product insert. Data on file 2004.
Photocure‐US 2004 {unpublished data only}
- Photocure ASA. Metvixia cream 16.8%, product insert. Data on file 2004.
Piacquadio 2004 {published and unpublished data}
- DUSA. Levulan Kerastick (aminolevulinic acid HCl) for topical solution, 20%. Product monograph. accessdata.fda.gov/drugsatfda_docs/label/2010/020965s007lbl.pdf (accessed 5 April 2011).
- Fowler JF, Zax RH. Aminolevulinic acid hydrochloride with photodynamic therapy: efficacy outcomes and recurrence 4 years after treatment. Cutis 2002;69(6 Suppl):2‐7. [PUBMED: 12095067 ] [PubMed] [Google Scholar]
- Piacquadio DJ, Chen DM, Farber HF, Fowler JF, Glazer SD, Goodman JJ, et al. Photodynamic therapy with aminolevulinic acid topical solution and visible blue light in the treatment of multiple actinic keratoses of the face and scalp: investigator‐blinded, phase 3, multicenter trials. Archives of Dermatology 2004;140(1):41‐6. [PUBMED: 14732659] [DOI] [PubMed] [Google Scholar]
Rivers 2002 {published and unpublished data}
- Solaraze gel: Diclofenac Sodium 3% ‐ package insert. www.solaraze.com/solaraze/pdf/solaraze_pi.pdf (accessed January 2012).
- Rivers JK, Arlette J, Shear N, Guenther L, Carey W, Poulin Y. Topical treatment of actinic keratoses with 3.0% diclofenac in 2.5% hyaluronoan gel. British Journal of Dermatology 2002;146(1):94‐100. [PUBMED: 11841372] [DOI] [PubMed] [Google Scholar]
Seckin 2009 {published data only}
- Seckin D, Cerman AA, Yildiz A, Ergun T. Can topical calcipotriol be a treatment alternative in actinic keratoses? A preliminary report. Journal of Drugs in Dermatology 2009;8(5):451‐4. [PUBMED: 19537367] [PubMed] [Google Scholar]
Shaffelburg 2009 {published data only}
- Shaffelburg M. Treatment of actinic keratoses with sequential use of photodynamic therapy; and imiquimod 5% cream. Journal of Drugs in Dermatology 2009;8(1):35‐9. [PUBMED: 19180894] [PubMed] [Google Scholar]
Siller 2009 {published data only}
- NCT00107965. Study to Determine the Safety of Two Applications of PEP005 Topical Gel to Actinic Keratoses. clinicaltrials.gov/ct2/show/NCT00107965 (accessed 14 March 2011).
- Siller G, Gebauer K, Welburn P, Katsamas J, Ogbourne SM. PEP005 (ingenol mebutate) gel, a novel agent for the treatment of actinic keratosis: results of a randomized, double‐blind, vehicle‐controlled, multicentre, phase IIa study. Australasian Journal of Dermatology 2009;50(1):16‐22. [PUBMED: 19178487 ] [DOI] [PubMed] [Google Scholar]
Smith 2003 {published data only}
- Smith S, Piacquadio D, Morhenn V, Atkin D, Fitzpatrick R. Short incubation PDT versus 5‐FU in treating actinic keratosis. Journal of Drugs in Dermatology 2003;2(6):629‐35. [PUBMED: 14711141] [PubMed] [Google Scholar]
Solaraze study 2 {unpublished data only}
- Solaraze gel: Diclofenac Sodium 3% ‐ package insert. https://www.solaraze.com/solaraze/pdf/solaraze_pi.pdf (accessed 24 March 2011).
Sotiriou 2009 {published data only}
- Sotiriou E, Apalla Z, Maliamani F, Zaparas N, Panagiotidou D, Ioannides D. Intraparticipant, right‐left comparison of topical 5‐aminolevulinic acid photodynamic therapy vs. 5% imiquimod cream for actinic keratoses on the upper extremities. Journal of the European Academy of Dermatology & Venereology 2009;23(9):1061‐5. [PUBMED: 19470041 ] [DOI] [PubMed] [Google Scholar]
Stockfleth 2002 {published data only}
- Stockfleth E, Meyer T, Benninghoff B, Salasche S, Paradoulos L, Ulrich C, et al. A randomized, double‐blind, vehicle‐controlled study to asses 5% imiquimod cream for the treatment of multiple actinic keratoses. Archives of Dermatology 2002;138(11):1498‐502. [PUBMED: 12437457] [DOI] [PubMed] [Google Scholar]
Swanson 2010a {published and unpublished data}
- Graceway Pharmaceuticals. Zyclara product monograph. http://www.gracewaypharma.com/sites/default/files/products/Zyclara%20Full%20PI.pdf (accessed 26 April 2011).
- NCT00605176. Safety and Effectiveness Study of Imiquimod Creams for Treatment of Actinic Keratoses (actinic keratoses). //clinicaltrials.gov/ct2/show/NCT00605176 (accessed 14 March 2011).
- Swanson N, Abramovits W, Berman B, Kulp J, Rigel DS, Levy S. Imiquimod 2.5% and 3.75% for the treatment of actinic keratoses: results of two placebo‐controlled studies of daily application to the face and balding scalp for two 2‐week cycles. Journal of the American Academy of Dermatology 2010;62(4):582‐90. [PUBMED: 20133013] [DOI] [PubMed] [Google Scholar]
Swanson 2010b {published and unpublished data}
- Lebwohl M, Swanson N, Anderson LL, Melgaard A, Xu Z, Berman B. Ingenol mebutate gel for actinic keratosis. New England Journal of Medicine 2012;366(11):1010‐19. [PUBMED: 22417254] [DOI] [PubMed] [Google Scholar]
- NCT00742391. A Multicenter Study to Evaluate the Efficacy and Safety of PEP005 (Ingenol Mebutate) Gel When Used to Treat Actinic Keratoses (actinic keratoses) on the Non Head Locations. //clinicaltrials.gov/ct2/show/NCT00742391 (accessed 14 March 2011).
- Swanson N. Multicenter, randomized, parallel‐group, double‐blind, vehicle‐controlled study to evaluate the efficacy and safety of PEP005 (ingenol mebutate) gel, 0.05% in patients with actinic keratoses on nonhead locations. Journal of the American Academy of Dermatology 2010; Vol. 62, issue 3 Suppl 1:AB2.
Szeimies 2002 {published data only}
- Szeimies RM, Karrer S, Radakovic‐Fijan S, Tanew A, Calzavara‐Pinton PG, Zane C, et al. Photodynamic therapy using topical methyl 5‐aminolevulinate compared with cryotherapy for actinic keratosis: a prospective, randomized study. Journal of the American Academy of Dermatology 2002;47(2):258‐62. [PUBMED: 12140473] [PubMed] [Google Scholar]
Szeimies 2004 {published data only}
- Szeimies RM, Gerritsen MJ, Gupta G, Ortonne JP, Serresi S, Bichel J, et al. Imiquimod 5% cream for the treatment of actinic keratosis: Results from a phase III, randomized, double‐blind, vehicle‐controlled, clinical trial with histology. Journal of the American Academy Dermatology 2004;51(4):547‐55. [PUBMED: 15389189] [DOI] [PubMed] [Google Scholar]
Szeimies 2008 {published data only}
- Szeimies RM, Bichel J, Ortonne JP, Stockfleth E, Lee J, Meng TC. A phase II dose‐ranging study of topical resiquimod to treat actinic keratosis. British Journal of Dermatology 2008;159(1):205‐10. [PUBMED: 18476957] [DOI] [PubMed] [Google Scholar]
Szeimies 2009 {published and unpublished data}
- Galderma. Metvixia product label. http://metvixia.com/pi/MetvixiaPI.pdf (accessed 26 April 2011).
- NCT00304239. Metvix PDT Versus Vehicle PDT With Aktilite CL128 Lamp in Patients With Actinic Keratosis on the Face and Scalp. clinicaltrials.gov/ct2/show/NCT00304239 (accessed 14 March 2011).
- Szeimies R, Matheson RT, Davis SA, Bhatia AC, Frambach Y, Klovekorn W, et al. Topical methyl aminolevulinate photodynamic therapy using red light‐emitting diode light for multiple actinic keratoses: a randomized study. Dermatologic Surgery 2009;35(4):586‐592. [PUBMED: 19309347] [DOI] [PubMed] [Google Scholar]
Szeimies 2010b {published data only}
- Szeimies RM, Radny P, Sebastian M, Borrosch F, Dirschka T, Krahn‐Senftleben G, et al. Photodynamic therapy with BF‐200 ALA for the treatment of actinic keratosis: results of a prospective, randomized, double‐blind, placebo‐controlled phase III study. British Journal of Dermatology 2010;163(2):386‐94. [PUBMED: 20518784] [DOI] [PubMed] [Google Scholar]
Tan 2007 {published data only}
- NCT00110682. Study of Imiquimod 5% Cream in Addition to Cryotherapy in the Management of Actinic Keratoses. http://clinicaltrials.gov/ct2/show/NCT00110682 (accessed 14 March 2011).
- Tan JK, Thomas DR, Poulin Y, Maddin F, Tang J. Efficacy of imiquimod as an adjunct to cryotherapy for actinic keratoses. Journal of Cutaneous Medicine and Surgery 2007;11(5):195‐201. [PUBMED: 18042331] [DOI] [PubMed] [Google Scholar]
Tanghetti 2007 {published and unpublished data}
- Paquet M. Tanghetti and Werschler 2007. Email to: E A Tanghetti 12 April 2011.
- Tanghetti E, Werschler P. Comparison of 5% 5‐fluorouracil cream and 5% imiquimod cream in the management of actinic keratoses on the face. Journal of Drugs in Dermatology 2007;6(2):144‐7. [PUBMED: 17373172] [PubMed] [Google Scholar]
Tarstedt 2005 {published and unpublished data}
- Tarstedt M, Rosdahl I, Berne B, Svanberg K, Wennberg AM. A Randomized Multicenter Study to Compare Two Treatment Regimens of Topical Methyl Aminolevulinate (Metvix)‐PDT in Actinic Keratosis of the Face and Scalp. Acta Dermato‐Venereologica 2005;85(5):424‐428. [PUBMED: 16159735] [DOI] [PubMed] [Google Scholar]
- Tarstedt M, Wennberg A‐M, Rosdahl I, Persson BE, Berne B, Bojs G, et al. A comparison of two treatment regimes using photodynamic therapy with Metvix in actinic keratosis [Abstract FC8‐3] [The 12th Congress of the European Academy of Dermatology and Venerology. Barcelona, Spain 15‐18th October 2003]. Journal of the European Academy of Dermatology & Venerology 2003;17(Suppl 3):126. [Google Scholar]
Thompson 1993 {published data only}
- Thompson SC, Jolley D, Marks R. Reduction of solar keratoses by regular sunscreen use. New England Journal of Medicine 1993;329(16):1147‐51. [PUBMED: 8377777] [DOI] [PubMed] [Google Scholar]
Tong 1996 {published data only}
- Tong DW, Barnetson RS. Beta‐1,3‐D‐glucan gel in the treatment of solar keratoses. Australasian Journal of Dermatology 1996;37(3):137‐8. [PUBMED: 8771866] [DOI] [PubMed] [Google Scholar]
Ulrich 2007 {published data only}
- NCT00189267. A Study to Evaluate the Effectiveness and Safety of Multiple Applications of Imiquimod 5% Cream for the Treatment of Actinic Keratoses in Organ Transplant Recipients. clinicaltrials.gov/ct2/show/NCT00189267 (accessed 14 March 2011).
- Ulrich C, Bichel J, Euvrard S, Guidi B, Proby CM, Kerkhof PC, et al. Topical immunomodulation under systemic immunosuppression: results of a multicentre, randomized, placebo‐controlled safety and efficacy study of imiquimod 5% cream for the treatment of actinic keratoses in kidney, heart, and liver transplant patients. British Journal of Dermatology 2007;157(Suppl 2):25‐31. [PUBMED: 18067628] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ulrich 2010 {published and unpublished data}
- Ulrich C. The efficacy and safety of diclofenac 3% gel treatment of multiple actinic keratoses in organ transplant patients: A double‐blind, randomized, vehicle‐controlled study. Journal of the American Academy of Dermatology 2010; Vol. 62, issue 3 Suppl 1:AB107.
- Ulrich C, Johannsen A, Rowert‐Huber J, Ulrich M, Sterry W, Stockfleth E. Results of a randomized, placebo‐controlled safety and efficacy study of topical diclofenac 3% gel in organ transplant patients with multiple actinic keratoses. European Journal of Dermatology 2010;20(4):482‐8. [PUBMED: 20507841] [DOI] [PubMed] [Google Scholar]
Van der Geer 2009 {published data only}
- Geer S, Krekels GA. Treatment of actinic keratoses on the dorsum of the hands: ALA‐PDT versus diclofenac 3% gel followed by ALA‐PDT. A placebo‐controlled, double‐blind, pilot study. Journal of Dermatological Treatment 2009;20(5):259‐65. [PUBMED: 19370466] [DOI] [PubMed] [Google Scholar]
von Felbert 2010 {published data only}
- Felbert V, Hoffmann G, Hoff‐Lesch S, Abuzahra F, Renn C N, Braathen L R, et al. Photodynamic therapy of multiple actinic keratoses: reduced pain through use of visible light plus water‐filtered infrared A compared with light from light‐emitting diodes. British Journal of Dermatology 2010;163(3):607‐15. [PUBMED: 20426780] [DOI] [PubMed] [Google Scholar]
Weiss 2002 {published and unpublished data}
- Carac® Cream, 0.5% ‐ product insert. http://products.sanofi.us/carac/carac.html (accessed January 2012).
- Jorizzo J. Topical treatment of actinic keratosis with fluorouracil: is irritation associated with efficacy?. Journal of Drugs in Dermatology 2004;3(1):21‐6. [PUBMED: 14964743] [PubMed] [Google Scholar]
- Weiss J, Menter A, Hevia O, Jones T, Ling M, Rist T, et al. Effective Treatment of Actinic Keratosis With 0.5% Fluorouracil Cream for 1, 2 or 4 Weeks. Cutis 2002;70(2 Suppl):22‐9. [PUBMED: 12353677] [PubMed] [Google Scholar]
Wiegell 2008 {published data only}
- NCT00432224. Treatment of Actinic Keratoses With Photodynamic Therapy Using Sunlight Versus Red Light. clinicaltrials.gov/ct2/show/NCT00432224 (accessed 14 March 2011).
- Wiegell SR, Haedersdal M, Philipsen PA, Eriksen P, Enk CD, Wulf HC. Continuous activation of PpIX by daylight is as effective as and less painful than conventional photodynamic therapy for actinic keratoses; a randomized, controlled, single‐blinded study. British Journal of Dermatology 2008;158(4):740‐6. [PUBMED: 18294318] [DOI] [PubMed] [Google Scholar]
Wiegell 2009 {published data only}
- Wiegell SR, Haedersdal M, Eriksen P, Wulf HC. Photodynamic therapy of actinic keratoses with 8% and 16% methyl aminolaevulinate and home‐based daylight exposure: a double‐blinded randomized clinical trial. British Journal of Dermatology 2009;160(6):1308‐14. [PUBMED: 19416257] [DOI] [PubMed] [Google Scholar]
- Wiegell SR, Hædersdal M, Wulf H. Photodynamic therapy of actinic keratoses with two different concentrations of methylaminolevulinate and home‐based daylight exposure – a randomized, controlled, double blinded study. Journal of Investigative Dermatology 2008;128(S1):S204. [Google Scholar]
Wiegell 2011a {published data only}
- NCT00711178. A Randomized Multicenter Study of Daylight Mediated Photodynamic Therapy (PDT). clinicaltrials.gov/ct2/show/NCT00711178 (accessed 14 March 2011).
- Wiegell SR, Fabricius S, Stender IM, Berne B, Kroon S, Andersen BL, et al. A randomized, multicentre study of directed daylight exposure times of 1½ vs. 2½h in daylight‐mediated photodynamic therapy with methyl aminolaevulinate in patients with multiple thin actinic keratoses of the face and scalp.. British Journal of Dermatology 2011;164(5):1083‐90. [PUBMED: 21219287] [DOI] [PubMed] [Google Scholar]
Wolf 2001 {published and unpublished data}
- Solaraze gel: Diclofenac Sodium 3% ‐ package insert. Doak Dermatologics.
- Wolf JE, Taylor JR, Tschen E, Kang S. Topical 3.0% diclofenac in 2.5% hyaluronan gel in the treatment of actinic keratoses. International Journal of Dermatology 2001;40(11):709‐13. [PUBMED: 11737438] [DOI] [PubMed] [Google Scholar]
Zeichner 2009 {published data only}
- Zeichner JA, Stern DW, Uliasz A, Itenberg S, Lebwohl M. Placebo‐controlled, double‐blind, randomized pilot study of imiquimod 5% cream applied once per week for 6 months for the treatment of actinic keratoses. Journal of the American Academy of Dermatology 2009;60(1):59‐62. [PUBMED: 18937999 ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Alberts 2004 {published data only}
- Alberts D, Ranger‐Moore J, Einsphahr J, Saboda K, Bozzo P, Liu Y, et al. Safety and efficacy of dose‐intensive oral vitamin A in subjects with sun‐damaged skin. Clinical Cancer Research 2004;10(6):1875‐80. [PUBMED: 15041701] [DOI] [PubMed] [Google Scholar]
Alexiades‐Armenakas 2003 {published data only}
- Alexiades‐Armenakas MR, Geronemus RG. Laser‐mediated photodynamic therapy of actinic keratoses. Archives of Dermatology 2003;139(10):1313‐20. [PUBMED: 14568836] [DOI] [PubMed] [Google Scholar]
Apalla 2010a {unpublished data only}
- Apalla Z, Panagiotidou D, Ioannides D, Sotiriou E. Does the fluence rate influence clinical outcome and pain experienced by the patients with actinic keratosis treated with PDT?. Melanoma Research 2010;20(e‐suppl):e73. [Google Scholar]
Apalla 2010b {unpublished data only}
- Apalla Z, Panagiotidou D, Ioannides D, Sotiriou E. Which light dose is the optimal for treating actinic keratosis with PDT? An intraindividual randomized active‐controlled study. Abstracts of the 6th Congress of the European Association of Dermatologic Oncology [P70]. Melanoma Research 2010;20(e‐Suppl):e73‐4. [Google Scholar]
Apalla 2010c {published data only}
- Apalla Z, Sotiriou E, Chovarda E, Lefaki I, Devliotou‐Panagiotidou D, Ioannides D. Skin cancer: preventive photodynamic therapy in patients with face and scalp cancerization. A randomized placebo‐controlled study. British Journal of Dermatology 2010;162(1):171‐5. [PUBMED: 19863513] [DOI] [PubMed] [Google Scholar]
Babilas 2006 {published data only}
- Babilas P, Kohl E, Maisch T, Backer H, Grob B, Branzan AL, et al. In vitro and in vivo comparison of two different light sources for topical photodynamic therapy. British Journal of Dermatology 2006;154(4):712‐718. [PUBMED: 16536815 ] [DOI] [PubMed] [Google Scholar]
Babilas 2007 {published data only}
- Babilas P, Knobler R, Hummel S, Gottschaller C, Maisch T, Koller M, et al. Variable pulsed light is less painful than light‐emitting diodes for topical photodynamic therapy of actinic keratosis: a prospective randomized controlled trial. British Journal of Dermatology 2007;157(1):111‐7. [PUBMED: 17542980 ] [DOI] [PubMed] [Google Scholar]
Babilas 2008 {published data only}
- Babilas P, Travnik R, Werner A, Landthaler M, Szeimies RM. Split‐face‐study using two different light sources for topical PDT of actinic keratoses:non‐inferiority of the LED system. Journal der Deutschen Dermatologischen Gesellschaft 2008;6(1):25‐32. [PUBMED: 17995967 ] [DOI] [PubMed] [Google Scholar]
Bartels 2009 {published data only}
- Bartels P, Yozwiak M, Einspahr J, Saboda K, Liu Y, Brooks C, et al. Chemopreventive efficacy of topical difluoromethylornithine and/or triamcinolone in the treatment of actinic keratoses analyzed by karyometry. Analytical & Quantitative Cytology & Histology 2009;31(6):355‐66. [PUBMED: 20698351 ] [PMC free article] [PubMed] [Google Scholar]
Berlin 2008 {published data only}
- Berlin JM, Rigel DS. Diclofenac sodium 3% gel in the treatment of actinic keratoses postcryosurgery. Journal of Drugs in Dermatology 2008;7(7):669‐73. [PUBMED: 18664159] [PubMed] [Google Scholar]
Biecha‐Thalharnmer 2003 {unpublished data only}
- Biecha‐Thalharnmer U, Honigsmann H, Tanew A. Comparison of incoherent versus pulsed monochromatic light for photodynamic therapy of actinic keratoses. [Abstract FC13‐3]. The 12th Congress of the European Academy of Dermatology and Venereology. Barcelona, Spain 15‐18th October 2003. Journal of the European Academy of Dermatology & Venereology 2003;17(Suppl 3):143. [Google Scholar]
Braathen 2009 {published data only}
- Braathen LR, Paredes BE, Saksela O, Gardio K, Morken T, Florich KW, et al. Short incubation with methyl aminolevulinate for photodynamic therapy of actinic keratoses. European Academy of Dermatology & Venereology 2009;23(5):550‐555. [PUBMED: 19415804] [DOI] [PubMed] [Google Scholar]
Breza 1976 {published data only}
- Breza T, Taylor JR, Eaglestein WH. Noninflammatory destruction of actinic keratoses by fluorouracil. Archives of Dermatology 1976;112(9):1256‐8. [PUBMED: 999302] [PubMed] [Google Scholar]
Dermik 2003 {published data only}
- Dermik. Product monograph: Carac Cream 0.5% (fluorouracil cream). carac.info/hcp/efficacy.aspx (accessed 26 April 2011).
de Sévaux 2003 {published data only}
- Sévaux R, Smit J, Jong E, Kerkhof P, Hoitsma A. Acitretin treatment of premalignant and malignant skin disorders in renal transplant recipients: Clinical effects of a randomized trial comparing two doses of acitretin. Journal of the American Academy of Dermatology 2003;49:407‐12. [PUBMED: 12963902] [DOI] [PubMed] [Google Scholar]
Dirschka 2010 {published and unpublished data}
- Dirschka T, Bierhoff E, Pflugfelder A, Garbe C. 3.0% diclofenac in 2.5% hyaluronic acid inverts the process of cancerous transformation in actinic keratoses and maintains therapeutic response by prolongation of treatment. Melanoma Research 2010;20(e‐suppl):e39. [Google Scholar]
- Dirschka T, Bierhoff E, Pflugfelder A, Garbe C. Topical 3.0% diclofenac in 2.5% hyaluronic acid gel induces regression of cancerous transformation in actinic keratoses. Journal of the European Academy of Dermatology & Venereology 2010;24(3):258‐63. [PUBMED: 19709346] [DOI] [PubMed] [Google Scholar]
DUSA 2009 {unpublished data only}
- NCT00865878. ALA‐PDT Versus Vehicle PDT for Treatment of AK and Reduction of New NMSC in Solid Organ Transplant Recipients. clinicaltrials.gov/ct2/show/NCT00865878 (accessed 14 March 2011).
Edwards 1986 {published data only}
- Edwards L, Levine N, Weidner M, Piepkorn M, Smiles K. Effect of intralesional interferon on actinic keratoses. Archives of Dermatology 1986;122(7):779‐82. [PUBMED: 3524471 ] [PubMed] [Google Scholar]
Elmets 2010 {published data only}
- Elmets CA, Viner JL, Pentland AP, Cantrell W, Lin HY, Bailey H, et al. Chemoprevention of nonmelanoma skin cancer with celecoxib: a randomized, double‐blind, placebo‐controlled trial. Journal of the National Cancer Institute 2010;102(24):1835‐44. [PUBMED: 21115882] [DOI] [PMC free article] [PubMed] [Google Scholar]
Epstein 2006 {published data only}
- Epstein E. Twice daily vs. four times daily 5‐fluorouracil therapy for actinic keratoses: a split face study. British Journal of Dermatology 2006;154(4):794‐5. [PUBMED: 16536841] [DOI] [PubMed] [Google Scholar]
Ericson 2004 {published data only}
- Ericson M, Sandburg C, Stenquist B, Gudmundson F, Karlsson M, Ros AM, et al. Photodynamic therapy of actinic keratosis at varying fluence rates: assessment of photobleaching, pain and primary clinical outcome. British Journal of Dermatology 2004;151(6):1204‐12. [PUBMED: 15606516] [DOI] [PubMed] [Google Scholar]
Fowler 2002 {published data only}
- Fowler JF, Zax RH. Aminolevulinic acid hydrochloride with photodynamic therapy: efficacy outcomes and recurrence 4 years after treatment. Cutis 2002;69(6):2‐7. [PUBMED: 12095067] [PubMed] [Google Scholar]
Gold 2006 {published data only}
- Gold MH, Bradshaw VL, Boring MM, Bridges TM, Biron JA. Split‐face comparison of photodynamic therapy with 5‐aminolevulinic acid and intense pulsed light versus intense pulsed light alone for photodamage. Dermatologic Surgery 2006;32(6):795‐801. [PUBMED: 16792644 ] [DOI] [PubMed] [Google Scholar]
Goldman 2003 {published data only}
- Goldman MP, Atkin DH. ALA‐PDT in the treatment of actinic keratosis: spot versus confluent therapy. Journal of Cosmetic & Laser Therapy 2003;5(2):107‐10. [PUBMED: 12850802] [DOI] [PubMed] [Google Scholar]
Green 1998 {published data only}
- Green C, Orchard G, Cerio R, Hawk JL. A clinicopathological study of the effects of topical retinyl propionate cream in skin photoageing. Clinical & Experimental Dermatology 1998;23(4):162‐7. [PUBMED: 9894360] [DOI] [PubMed] [Google Scholar]
Griffin 1991 {published data only}
- Griffin TD, Scott EJ. Use of pyruvic acid in the treatment of actinic keratoses: a clinical and histopathological study. Cutis 1991;47(5):325‐9. [PUBMED: 2070654] [PubMed] [Google Scholar]
Grimaître 2000 {published data only}
- Grimaitre M, Etienne A, Fathi M, Piletta PA, Saurat JH. Topical colchicine therapy for actinic keratoses. Dermatology 2000;200(4):346‐8. [PUBMED: 10894974] [DOI] [PubMed] [Google Scholar]
Gupta 2004 {unpublished data only}
- Gupta AK. Single‐blind, randomized controlled trial to evaluate the efficacy and safety of photodynamic therapy using aminolevulinic acid versus 5‐fluorouracil in the management of actinic keratosis. Journal of the American Academy of Dermatology 2004;50(S3):P129. [Google Scholar]
Hanke 2011 {published data only}
- Hanke CW, Swanson N, Bruce S, Berman B, Kulp J, Levy S. Complete clearance is sustained for at least 12 months after treatment of actinic keratoses of the face or balding scalp via daily dosing with imiquimod 3.75% or2.5% cream. Journal of Drugs in Dermatology 2011;10(2):165‐70. [PUBMED: 21283921] [PubMed] [Google Scholar]
Humphreys 1996 {published data only}
- Humphreys TR, Werth V, Dzubow L, Kligman A. Treatment of photodamaged skin with trichloroacetic acid and topical tretinoin. Journal of the American Academy of Dermatology 1996;34(4):638‐44. [PUBMED: 8601654 ] [DOI] [PubMed] [Google Scholar]
Jury 2005 {published data only}
- Jury CS, Ramraka‐Jones VS, Gudi V, Herd RM. A randomized trial of topical 5% 5‐fluorouracil (Efudix cream) in the treatment of actinic keratoses comparing daily with weekly treatment. British Journal of Dermatology 2005;153(4):808‐810. [PUBMED: 16181465 ] [DOI] [PubMed] [Google Scholar]
Kurwa 1999 {published data only}
- Kurwa HA, Yong‐Gee SA, Seed PT, Markey AC, Barlow RJ. A randomized paired comparison of photodynamic therapy and topical 5‐fluorouracil in the treatment of actinic keratoses. Journal of the American Academy of Dermatology 1999;41(3 Pt 1):414‐8. [PUBMED: 10459115] [DOI] [PubMed] [Google Scholar]
Marrero 1998 {published data only}
- Marrero GM, Katz BE. The new fluor‐hydroxy pulse peel: a combination of 5‐fluorouracil and glycolic acid. Dermatological Surgery 1998;24(9):973‐8. [PUBMED: 9754085 ] [PubMed] [Google Scholar]
Morales 2010 {published data only}
- Morales Toquero A, Ocampo Candiani J, Gomez Flores M, Gonzalez Gonzalez SE, Eguia Rodriguez R, Mendez Olvera NP, et al. Clinical and histological evaluation of 5% imiquimod cream vs 5% 5‐fluorouracil ointment in patients with actinic keratosis on the face [Evaluación clínica e histológica de imiquimod a 5% en crema vs5‐fluorouracilo a 5% en ungüento en pacientes con queratosisactínicas en la cara]. Dermatologia Revista Mexicana 2010;54(6):326‐31. [Google Scholar]
Naylor 1995 {published data only}
- Naylor MF, Boyd A, Smith DW, Cameron GS, Hubbard D, Neldner KH. High sun protection factor sunscreens in the suppression of actinic neoplasia. Archives of Dermatology 1995;131(2):170‐5. [PUBMED: 7857113] [PubMed] [Google Scholar]
NCT00005097 {unpublished data only}
- NCT00005097. Green Tea Extract in Treating Patients With Actinic Keratosis. clinicaltrials.gov/ct2/show/NCT00005097 (accessed 14 March 2011).
Puizina‐Ivic 2008a {published data only}
- Puizina‐Ivic N, Zorc H, Vanjaka‐Rogosic L, Miric L, Persin A. Fractionated illumination improves the outcome in the treatment of precancerous lesions with photodynamic therapy. Collegium Antropologicum. 2008;32(Suppl 2):67‐73. [PUBMED: 19138010] [PubMed] [Google Scholar]
Radakovic‐Fijan 2005 {published data only}
- Radakovic‐Fijan S, Blecha‐Thalhammer U, Kittler H, Honigsmann H, Tanew A. Efficacy of 3 different light doses in the treatment of actinic keratoses with 5‐aminolevulinic acid photodynamic therapy: a randomized, observer‐blinded, intrapatient, comparison study. Journal of the American Academy of Dermatology 2005;53:823‐7. [PUBMED: 16243131 ] [DOI] [PubMed] [Google Scholar]
Robins 2002a {published data only}
- Robins P. Pulse therapy with 5‐FU in eradication actinic keratoses with less than recommended dosage. Journal of Drugs in Dermatology 2002;1(1):25‐30. [PUBMED: 12847751] [PubMed] [Google Scholar]
Rosen 2010 {unpublished data only}
- Rosen R. Clinical development of ingenol mebutate (PEP005) gel for actinic keratosis. 43rd Annual Scientific Meeting of the Australasian College of Dermatologists Darwin, NT Australia. Australasian Journal of Dermatology 2010; Vol. 51, issue Suppl 1:A5.
Shuttleworth 1989 {published data only}
- Shuttleworth D, Marks R. A comparison of the effects of intralesional interferon a‐2b and topical 5% 5‐fluorouracil cream in the treatment of solar keratoses and Bowen's disease. Journal of Dermatological Treatment 1989;1(2):65‐8. [Google Scholar]
Simmonds 1973 {published data only}
- Simmonds WL. Double‐blind investigation comparing a 1%‐vs‐5% 5‐fluorouracil topical cream in patients with multiple actinic keratoses. Cutis 1973;12(4):615‐8. [Google Scholar]
Smith 2006 {published data only}
- Smith SR, Morhenn VB, Piacquadio DJ. Bilateral comparison of the efficacy and tolerability of 3% diclofenac sodium gel and 5% 5‐fluorouracil cream in the treatment of actinic keratoses of the face and scalp. Journal of Drugs in Dermatology 2006;5(2):156‐9. [PUBMED: 16485883] [PubMed] [Google Scholar]
Sotiriou 2011 {published data only}
- Sotiriou E, Apalla Z, Chovarda E, Goussi C, Trigoni A, Ioannides D. Single vs. fractionated photodynamic therapy for face and scalp actinic keratoses: a randomized, intraindividual comparison trial with 12‐month follow‐up. Journal of the European Academy of Dermatology & Venereology 2011 Mar 2 [Epub ahead of print]. [PUBMED: 21366709] [DOI] [PubMed]
Spencer 2010 {published data only}
- Spencer J. Multicenter, randomized, double‐blind, vehicle‐controlled, dose‐ranging study to evaluate the efficacy and safety of PEP005 (ingenol mebutate) gel 0.005%, 0.01%, and 0.015% when used to treat actinic keratoses on the head. Journal of the American Academy of Dermatology 2010; Vol. 62, issue 3 Suppl 1:AB105.
Stockfleth 2004 {published data only}
- Stockfleth E, Christophers E, Benninghoff B, Sterry W. Low incidence of new actinic keratoses after topical 5% imiquimod cream treatment: a long‐term follow‐up study. Archives of Dermatology 2004;140(12):1542. [PUBMED: 15611446 ] [DOI] [PubMed] [Google Scholar]
Szeimies 2010a {published data only}
- Szeimies RM, Stockfleth E, Popp G, Borrosch F, Bruning H, Dominicus R, et al. Long‐term follow‐up of photodynamic therapy with a self‐adhesive 5‐aminolaevulinic acid patch: 12 months data. British Journal of Dermatology 2010;162(2):410‐4. [PUBMED: 19804593] [DOI] [PubMed] [Google Scholar]
Touma 2004 {published data only}
- Paquet M. Touma et al. 2004 [personal communication]. Email to: BA Gilchrest 29 March 2011.
- Touma D, Yaar M, Whitehead S, Konnikov N, Gilchrest B. A Trial of Short Incubation, Broad‐Area Photodynamic Therapy for Facial Actinic Keratoses and Diffuse Photodamage. Archives of Dermatology 2004;140(1):33‐40. [PUBMED: 14732657] [DOI] [PubMed] [Google Scholar]
Tsoukas 2010 {unpublished data only}
- Tsoukas M. Treatment of actinic keratosis with 5‐ALA PDT versus topical imiquimod in healthy and immunosuppressed patients. Melanoma Research 2010;20(e‐suppl):e39‐40. [Google Scholar]
Valeant 2004 {published data only}
- Valeant. Efudex topical solutions and cream (fluorouracil). Product monograph. valeant.com/fileRepository/products/PI/Efudex‐40_Cream_5_Solution_2‐5_PI_Apr04.pdf (accessed 26 April 2011).
Vbeam 2005 {unpublished data only}
- NCT00524485. Photodynamic Therapy Using Topical Aminolevulinic Acid in Treating Patients With Actinic Keratosis. clinicaltrials.gov/ct2/show/NCT00524485 (accessed 14 March 2011).
Weinstock 2010 {unpublished data only}
- Weinstock MA, Bingham SF. High‐dose topical tretinoin for reducing multiplicity of actinic keratoses. 2010 Annual Meeting of the Society for Investigative Dermatology Atlanta, GA United States. Journal of Investigative Dermatology 2010; Vol. Suppl 1, issue 130:S63.
Wennberg 2008 {published data only}
- Wennberg AM, Stenquist B, Stockfleth E, Keohane S, Lear JT, Jemec G, et al. Photodynamic therapy with methyl aminolevulinate for prevention of new skin lesions in transplant recipients: a randomized study. Transplantation 2008;86(3):423‐9. [PUBMED: 18698246] [DOI] [PubMed] [Google Scholar]
Wulf 2006 {published data only}
- Wulf HC, Pavel S, Stender I, Bakker‐Wensveen CA. Topical photodynamic therapy for prevention of new skin lesions in renal transplant recipients. Acta Dermato‐Venereologica 2006;86(1):25‐8. [PUBMED: 16585985] [DOI] [PubMed] [Google Scholar]
Yamauchi 2002 {published data only}
- Yamauchi PS, Lowe NJ, Lask GP, Patnaik R, Moore D, Foley P. Methyl aminolevulinate and photodynamic therapy in the treatment of actinic keratosis. [Abstract 804]. The 63rd annual meeting of the Society for Investigative Dermatology, 15‐18 May, Los Angeles, USA. Journal of Investigative Dermatology 2002; Vol. 119, issue 1:341.
References to studies awaiting assessment
Akarsu 2011 {published data only}
- Akarsu S, Aktan S, Atahan A, Koc P, Ozkan S. Comparison of topical 3% diclofenac sodium gel and 5% imiquimod cream for the treatment of actinic keratoses. Clinical & Experimental Dermatology 2011;36(5):479‐84. [PUBMED: 21418281] [DOI] [PubMed] [Google Scholar]
Apalla 2011 {published data only}
- Apalla Z, Sotiriou E, Panagiotidou D, Lefaki I, Goussi C, Ioannides D. The impact of different fluence rates on pain and clinical outcome in patients with actinic keratoses treated with photodynamic therapy. Photodermatology, Photoimmunology & Photomedicine 2011;27(4):181‐5. [PUBMED: 21729165] [DOI] [PubMed] [Google Scholar]
Azimi 2012 {published data only}
- Azimi H, Zadeh MG, Jaberian M, Omid M. Comparison of the efficacy of cryotherapy and 0.1% Acnalen gel vs. cryotherapy and placebo in the treatment of actinic keratoses. Pakistan Journal of Medical Sciences 2012;28(1):54‐7. [Google Scholar]
- IRCT201010104901N1. Comparison of the efficacy of cryotherapy and 0.1 % acnalen gel vs. cryotherapy alone in the treatment of actinic keratoses: a randomized, double‐blind clinical trial. irct.ir/searchresult.php?id=4901&number=1 (accessed 14 March 2011).
Damian 2011 {unpublished data only}
- ACTRN12609000490279. Randomized, double‐blind, placebo controlled study to assess efficacy of oral nicotinamide in the treatment and prevention of actinic keratoses. anzctr.org.au/trial_view.aspx?ID=83879 (accessed 14 March 2011).
- Damian D, Surjana D, Martin A, Halliday G. Oral nicotinamide for skin cancer prevention. 41st Annual Meeting of the European Society for Dermatological Research, ESDR 2011 Barcelona Spain. 7‐10 September 2011. Journal of Investigative Dermatology 2011;131(Suppl 2):S99. [PubMed] [Google Scholar]
Deonizio 2011 {published data only}
- Deonizio JM, Mulinari‐Brenner FA. Cryopeeling for treatment of photodamage and actinic keratosis: liquid nitrogen versus portable system. Anais Brasileiros De Dermatologia 2011;86(3):440‐4. [PUBMED: 21738958] [DOI] [PubMed] [Google Scholar]
Dirschka 2012 {published data only}
- Dirschka T, Radny P, Dominicus R, Mensing H, Bruning H, Jenne L, et al. Photodynamic therapy with BF‐200 ALA for the treatment of actinic keratosis: Results of a multicentre, randomized, observer‐blind phase III study in comparison with a registered methyl‐5‐aminolaevulinate cream and placebo. British Journal of Dermatology 2012;166(1):137‐46. [PUBMED: 21910711] [DOI] [PubMed] [Google Scholar]
Galitzer 2011 {published data only}
- Galitzer BI. Effect of retinoid pretreatment on outcomes of patients treated by photodynamic therapy for actinic keratosis of the hand and forearm. Journal of Drugs in Dermatology 2011;10(10):1124‐32. [PUBMED: 21968662] [PubMed] [Google Scholar]
Haddad 2011 {published data only}
- Haddad A, Santos ID, Gragnani A, Ferreira LM. The effect of increasing fluence on the treatment of actinic keratosis and photodamage by photodynamic therapy with 5‐aminolevulinic acid and intense pulsed light. Photomedicine & Laser Surgery 2011;29(6):427‐32. [PUBMED: 21631378] [DOI] [PubMed] [Google Scholar]
Lebwohl 2012 {unpublished data only}
- Anderson L, Melgaard A, Schmeider G, Xu Z. Two‐day topical treatment with ingenol mebutate gel, 0.05% for actinic keratoses on the trunk and extremities: Analysis of data pooled from two trials. 70th Annual Meeting of the American Academy of Dermatology San Diego, CA United States, 16‐20 March 2012. Journal of the American Academy of Dermatology 2012;66(4 Suppl):AB159. [Google Scholar]
- Berman B, Melgaard A, Marmur E, Larsson T. Three‐day topical treatment with ingenol mebutate gel, 0.015% for actinic keratoses on the face and scalp: analysis of data pooled from two trials. 70th Annual Meeting of the American Academy of Dermatology San Diego, CA United States, 16‐20 March 2012. Journal of the American Academy of Dermatology 2012;66(4 Suppl):AB3. [Google Scholar]
- Lebwohl M, Melgaard A, Kobayashi K, Swanson N. Local skin responses associated with ingenol mebutate gel for the treatment of actinic keratosis: Two analyses of pooled data, 70th Annual Meeting of the American Academy of Dermatology San Diego, CA United States, 16‐20 March 2012. Journal of the American Academy of Dermatology 2012;66(4 Suppl):AB153. [Google Scholar]
- Lebwohl M, Swanson N, Anderson LL, Melgaard A, Xu Z, Berman B. Ingenol mebutate gel for actinic keratosis. New England Journal of Medicine 2012;366(11):1010‐9. [PUBMED: 22417254] [DOI] [PubMed] [Google Scholar]
- Lebwohl M, Swanson N, Anderson LL, Melgaard A, Xu Z, Berman B. Ingenol mebutate gel for actinic keratosis‐online supplement. New England Journal of Medicine 2012; Vol. 366, issue 11. [PUBMED: 22417254] [DOI] [PubMed]
- Leo Pharma. Picato product label information. http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Label_ApprovalHistory#labelinfo (accessed 5 April 2012).
- NCT00915551. A multi‐center study to evaluate the efficacy and safety of PEP005 (ingenol mebutate) gel, when used to treat actinic keratoses on the head (face or scalp). clinicaltrials.gov/ct2/show/NCT00915551 (accessed 14 March 2011).
- NCT00916006. A multi‐center study to evaluate the efficacy and safety of PEP005 (ingenol mebutate) gel, when used to treat actinic keratoses on the head (face or scalp). clinicaltrials.gov/ct2/show/NCT00916006 (accessed 14 March 2011).
- NCT00942604. A multicenter study to evaluate the efficacy and safety of PEP005 (ingenol mebutate) gel when used to treat actinic keratoses (AKs) on the non head locations. clinicaltrials.gov/ct2/show/NCT00942604 (accessed 14 March 2011).
Serra‐Guillen 2012 {published data only}
- Serra‐Guillen C, Nagore E, Hueso L, Traves V, Messeguer F, Sanmartin O, et al. A randomized pilot comparative study of topical methyl aminolevulinate photodynamic therapy versus imiquimod 5% versus sequential application of both therapies in immunocompetent patients with actinic keratosis: Clinical and histologic outcomes. Journal of the American Academy of Dermatology 2012;66(4):e131‐7. [PUBMED: 22226430] [DOI] [PubMed] [Google Scholar]
Stockfleth 2011 {published data only}
- NCT00987246. Study on the efficacy of LAS41005 in the treatment of actinic keratosis. clinicaltrials.gov/ct2/show/NCT00987246ls.gov (accessed 14 March 2011).
- Stockfleth E, Kerl H, Zwingers T, Willers C. Low‐dose 5‐fluorouracil in combination with salicylic acid as a new lesion‐directed option to treat topically actinic keratoses: histological and clinical study results. British Journal of Dermatology 2011;165(5):1101‐8. [PUBMED: 21517801] [DOI] [PubMed] [Google Scholar]
Wiegell 2011b {published data only}
- Wiegell SR, Heydenreich J, Fabricius S, Wulf HC. Continuous ultra‐low‐intensity artificial daylight is not as effective as red LED light in photodynamic therapy of multiple actinic keratoses. Photodermatology, Photoimmunology & Photomedicine 2011;27(6):280‐5. [PUBMED: 22092730] [DOI] [PubMed] [Google Scholar]
- Willey A. Temperature‐modulated photodynamic therapy for the treatment of actinic keratoses on the extremities. 70th Annual Meeting of the American Academy of Dermatology San Diego, CA United States, 16‐20 March 2012. Journal of the American Academy of Dermatology 2012;66(4 Suppl):AB177. [Google Scholar]
References to ongoing studies
ACTRN12610000689077 {unpublished data only}
- ACTRN12610000689077. Effect of nicotinamide versus placebo on numbers of actinic keratoses. anzctr.org.au/trial_view.aspx?ID=335868 (accessed 14 March 2011).
NCT00115154 {unpublished data only}
- NCT00115154. Study to assess the safety and efficacy of imiquimod 5% cream for the treatment of actinic keratosis on the arms and hands. clinicaltrials.gov/ct2/show/NCT00115154 (accessed 14 March 2011).
NCT00204542 {unpublished data only}
- NCT00204542. Comparison of the efficacy and tolerability of Solaraze for 3 versus 6 months in patients with mild to moderate actinic keratosis located on the face and head. clinicaltrials.gov/ct2/show/NCT00204542 (accessed 14 March 2011).
NCT00472459 {unpublished data only}
- NCT00472459. PDT with Metvix® 160 mg/g cream in organ transplant recipients with non‐melanoma skin cancer. clinicaltrials.gov/ct2/show/NCT00472459 (accessed 14 March 2011).
NCT00608634 {unpublished data only}
- NCT00608634. Topical perillyl alcohol in treating patients with sun damaged skin and actinic keratoses. clinicaltrials.gov/ct2/show/NCT00608634 (accessed 14 March 2011).
NCT00695578 {unpublished data only}
- NCT00695578. Clinical trial to evaluate biafine cream versus standard care in subjects with actinic keratosis post cryotherapy. clinicaltrials.gov/ct2/show/NCT00695578 (accessed 14 March 2011).
NCT00700063 {unpublished data only}
- NCT00700063. A multicenter study to evaluate the safety and efficacy of PEP005 topical gel when used to treat actinic keratoses on the head (face or scalp). clinicaltrials.gov/ct2/show/NCT00700063 (accessed 14 March 2011).
NCT00756288 {unpublished data only}
- NCT00756288. Photo‐therapy with a topical retinoid versus photo‐therapy alone for actinic keratoses. clinicaltrials.gov/ct2/show/NCT00756288 (accessed 14 March 2011).
NCT00786994 {unpublished data only}
- NCT00786994. The efficacy and tolerability of oleogel‐S‐10 in patients with actinic keratoses. clinicaltrials.gov/ct2/show/NCT00786994 (accessed 14 March 2011).
NCT00859105 {unpublished data only}
- NCT00859105. A randomized, double‐blind, parallel‐group, vehicle‐controlled therapeutic equivalence study of three Imiquimod cream 5% treatments for patients with actinic keratosis. clinicaltrials.gov/ct2/show/NCT00859105 (accessed 14 March 2011).
NCT00948428 {unpublished data only}
- NCT00948428. Bioequivalence of generic imiquimod cream, 5% when compared to Aldara™ (imiquimod) cream, 5% in the treatment of actinic keratosis. clinicaltrials.gov/ct2/show/NCT00948428 (accessed 14 March 2011).
NCT00991861 {unpublished data only}
- NCT00991861. Efficacy and safety study of LAS41007 in the treatment of actinic keratosis. clinicaltrials.gov/ct2/show/NCT00991861 (accessed 14 March 2011).
NCT01203878 {unpublished data only}
- NCT01203878. Treatment of actinic keratoses of the face with imiquimod 3.75% cream followed by photodynamic therapy. clinicaltrials.gov/ct2/show/NCT01203878 (accessed 14 March 2011).
NCT01229319 {unpublished data only}
- NCT01229319. Imiquimod 3.75% cream in combination with cryotherapy in the treatment of hypertrophic actinic keratoses. clinicaltrials.gov/ct2/show/NCT01229319 (accessed 14 March 2011).
NCT01260987 {unpublished data only}
- NCT01260987. Fractional CO2 laser assisted photodynamic therapy. clinicaltrials.gov/ct2/show/NCT01260987 (accessed 14 March 2011).
NCT01265602 {unpublished data only}
- NCT01265602. Double‐blind, randomized, vehicle‐ and comparator‐controlled, multi‐center trial to evaluate the efficacy and safety of LAS41007 in the treatment of actinic keratosis. clinicaltrials.gov/ct2/show/NCT01265602 (accessed 14 March 2011).
NCT01354717 {unpublished data only}
- NCT01354717. Bioequivalence study of generic fluorouracil 0.5% cream and 0.5% Carac® and placebo. clinicaltrials.gov/ct2/results?term=NCT01354717 (accessed 5 April 2012).
NCT01358851 {unpublished data only}
- NCT01358851. LAS41005 in hyperkeratotic actinic keratosis. clinicaltrials.gov/ct2/results?term=NCT01358851 (accessed 5 April 2012).
NCT01413763 {unpublished data only}
- NCT01413763. Potential effect of topical imiquimod on atrial ectopy in patients with actinic aeratosis. clinicaltrials.gov/ct2/results?term=NCT01413763 (accessed 5 April 2012).
NCT01453179 {unpublished data only}
- NCT01453179. Long‐term effects of imiquimod and diclofenac in actinic keratoses (LEIDA 2). clinicaltrials.gov/ct2/results?term=NCT01453179 (accessed 5 April 2012).
NCT01458587 {unpublished data only}
- NCT01458587. Levulan PDT versus vehicle for extremity actinic keratoses (AK). clinicaltrials.gov/ct2/results?term=NCT01458587 (accessed 5 April 2012).
NCT01459393 {unpublished data only}
- NCT01459393. Comparison between 5‐aminolevulinic acid photodynamic therapy versus cryotherapy for actinic keratosis treatment. clinicaltrials.gov/ct2/results?term=NCT01459393 (accessed 5 April 2012).
NCT01475071 {unpublished data only}
- NCT01475071. Intra‐individual comparison of efficacy and safety of Metvix® natural daylight photodynamic therapy versus conventional Metvix® photodynamic therapy in subject with mild actinic keratoses (CoMet). clinicaltrials.gov/ct2/results?term=NCT01475071 (accessed 5 April 2012).
NCT01475955 {unpublished data only}
- NCT01475955. Short‐incubation Levulan photodynamic therapy versus vehicle for face/scalp actinic keratosis (AK). clinicaltrials.gov/ct2/results?term=NCT01475955 (accessed 5 April 2012).
NCT01481155 {unpublished data only}
- NCT01481155. Comparative study of photodynamic therapy vs. CO2 laser therapy in treatment of actinic keratoses. clinicaltrials.gov/ct2/results?term=NCT01481155 (accessed 5 April 2012).
NCT01493921 {unpublished data only}
- NCT01493921. Efficacy study & safety evaluation of SR‐T100 gel in actinic keratosis treatment. clinicaltrials.gov/ct2/results?term=NCT01493921 (accessed 5 April 2012).
NCT01502020 {unpublished data only}
- NCT01502020. A bioequivalence study with clinical endpoints comparing generic imiquimod cream, 3.75% and Zyclara™ (imiquimod) cream, 3.75% in subjects with actinic keratoses. clinicaltrials.gov/ct2/results?term=NCT01502020 (accessed 5 April 2012).
NCT01516515 {unpublished data only}
- NCT01516515. Efficacy and safety phase II dose‐ranging study of SR‐T100 to treat actinic keratosis. clinicaltrials.gov/ct2/results?term=NCT01516515 (accessed 5 April 2012).
NCT01525329 {unpublished data only}
- NCT01525329. Combination therapy with 5‐FU and PDT for the treatment of post‐transplant premalignant skin disease. clinicaltrials.gov/ct2/results?term=NCT01525329 (accessed 5 April 2012).
NCT01538901 {unpublished data only}
- NCT01538901. Imiquimod versus photodynamic therapy of actinic keratoses in organ transplant recipients. clinicaltrials.gov/ct2/results?term=NCT01538901 (accessed 5 April 2012).
NCT01541228 {unpublished data only}
- NCT01541228. Clinical effect of photodynamic treatment when treating actinic keratoses with different light doses. clinicaltrials.gov/ct2/results?term=NCT01541228 (accessed 5 April 2012).
NCT01541553 {unpublished data only}
- NCT01541553. A sequential treatment regimen of cryotherapy and Picato® for the treatment of actinic keratosis on the face and scalp. clinicaltrials.gov/ct2/results?term=NCT01541553 (accessed 5 April 2012).
Willey 2011 {unpublished data only}
- Willey A, Sakamoto F. Temperature modulated photodynamic therapy for the treatment of actinic keratoses on the extremities. 31st Annual Conference of the American Society for Laser Medicine and Surgery, ASLMS 2011 Grapevine, TX United States, 30/3‐3/4 2011. Lasers in Surgery & Medicine 2011;43(S23):957‐958. [Google Scholar]
Additional references
Adamson 2002
- Adamson DJ, Frew D, Tatoud R, Wolf CR, Palmer CN. Diclofenac antagonizes peroxisome proliferator‐activated receptor‐gamma signaling. Molecular Pharmacology 2002;61(1):7‐12. [DOI] [PubMed] [Google Scholar]
Alam 1995
- Alam CA, Seed MP, Willoughby DA. Angiostasis and vascular regression in chronic granulomatous inflammation induced by diclofenac in combination with hyaluronan in mice. Journal of Pharmacy & Phamacology 1995;47(5):407‐11. [DOI] [PubMed] [Google Scholar]
Amini 2010
- Amini S, Viera MH, Valins W, Berman B. Nonsurgical innovations in the treatment of nonmelanoma skin cancer. The Journal of Clinical & Aesthetic Dermatology 2010;3(6):20‐34. [PMC free article] [PubMed] [Google Scholar]
Askew 2009
- Askew DA, Mickan SM, Soyer HP, Wilkinson D. Effectiveness of 5‐fluorouracil treatment for actinic keratosis‐‐a systematic review of randomized controlled trials. International Journal of Dermatology 2009;48(5):453‐63. [PUBMED: 19416373] [DOI] [PubMed] [Google Scholar]
Berman 2006
- Berman B, Villa AM, Ramirez CC. Mechanisms of action of new treatment modalities for actinic keratosis. Journal of Drugs in Dermatology 2006;5(2):167‐73. [PUBMED: 16485885] [PubMed] [Google Scholar]
Brown 2005
- Brown MB, Jones SA. Hyaluronic acid: a unique topical vehicle for the localized delivery of drugs to the skin. Journal of the European Academy of Dermatology & Venereology 2005;19:308‐18. [PUBMED: 15857456] [DOI] [PubMed] [Google Scholar]
Callen 1997
- Callen JP, Bickers DR, Moy RL. Actinic keratoses. Journal of the American Academy of Dermatology 1997;36(4):650‐3. [DOI] [PubMed] [Google Scholar]
Chakrabarty 2004
- Chakrabarty A, Geisse JK. Medical therapies for non‐melanoma skin cancer. Clinics in Dermatology 2004;22(3):183‐8. [DOI] [PubMed] [Google Scholar]
Cockerell 2000
- Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma ("actinic keratosis"). Journal of the American Academy of Dermatology 2000;42:S11‐17. [PUBMED: 10607351] [DOI] [PubMed] [Google Scholar]
Correale 2002
- Correale CE. Actinic keratoses: new treatment options. Advances in Dermatology 2002;18:339‐55. [PubMed] [Google Scholar]
Dinehart 2000
- Dinehart SM. The treatment of actinic keratoses. Journal of the American Academy of Dermatology 2000;42(1 Pt 2):25‐8. [DOI] [PubMed] [Google Scholar]
Eaglstein 1970
- Eaglstein WH, Weinstein GD, Frost P. Fluorouracil: mechanism of action in human skin and actinic keratoses. Effect on DNA synthesis in vivo. Archives of Dermatology 1970;101(2):132‐9. [PUBMED: 5413250] [PubMed] [Google Scholar]
Engel 1988
- Engel A, Johnson M, Haynes SG. Health effects of sunlight exposure in the United States. Results from the first National Health and Nutrition Examination Survey, 1971‐1974. Archives of Dermatology 1988;124(1):72‐9. [PUBMED: 3257372] [PubMed] [Google Scholar]
Fayter 2010
- Fayter D, Corbett M, Heirs M, Fox D, Eastwood A. A systematic review of photodynamic therapy in the treatment of pre‐cancerous skin conditions, Barrett's oesophagus and cancers of the biliary tract, brain, head and neck, lung, oesophagus and skin. Health Technology Assessment 2010;14(37):1‐288. [PUBMED: 20663420] [DOI] [PubMed] [Google Scholar]
Feldman 2011
- Feldman SR, Fleischer AB. Progression of actinic keratosis to squamous cell carcinoma revisited: clinical and treatment implications. Cutis 2011;87:201‐7. [PUBMED: 21644496] [PubMed] [Google Scholar]
Fink‐Puches 1997
- Fink‐Puches R, Hofer A, Smolle J, Kerl H, Wolf P. Primary clinical responses and long‐term follow‐up of solar keratoses treated with topically applied 5‐ALA and irradiation by different wavebands of light. Journal of Photochemistry & Photobiology 1997;41(1‐2):145‐51. [DOI] [PubMed] [Google Scholar]
Frost 2000
- Frost C, Williams G, Green A. High incidence and regression rates of solar keratoses in a Queensland community. Journal of Investigative Dermatology 2000;115(2):273‐7. [DOI] [PubMed] [Google Scholar]
Fulton 1999
- Fulton JE, Rahimi AD, Helton P, Dahlberg K, Kelly AG. Disappointing results following resurfacing of facial skin with CO2 lasers for prophylaxis of keratoses and cancers. Dermatologic Surgery 1999;25(9):729‐732. [PUBMED: 10491067] [DOI] [PubMed] [Google Scholar]
Glogau 2000
- Glogau RG. The risk of progression to invasive disease. Journal of the American Academy of Dermatology 2000;42(1 Pt 2):23‐4. [DOI] [PubMed] [Google Scholar]
Gold 2008
- Gold MH. Continuing medical education article‐skin treatment: Photodynamic therapy: indications and treatment. Aesthetic Surgery Journal 2008;28(5):545‐52. [DOI] [PubMed] [Google Scholar]
Goldberg 2010
- Goldberg LH, Kaplan B, Vergilis‐Kalner I, Landau J. Liquid nitrogen: temperature control in the treatment of actinic keratosis. Dermatologic Surgery 2010;36(12):1956‐61. [DOI] [PubMed] [Google Scholar]
Hadley 2006
- Hadley G, Derry S, Moore RA. Imiquimod for actinic keratosis: systematic review and meta‐analysis. Journal of Investigative Dermatology 2006;126(6):1251‐55. [DOI] [PubMed] [Google Scholar]
Hemmi 2002
- Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small anti‐viral compounds activate immune cells via the TLR7 MyD88‐dependent signaling pathway. Nature Immunology 2002;3(2):196‐200. [PUBMED: 11812998] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration. Available from www.cochrane‐handbook.org.
Holmes 2007
- Holmes C, Foley P, Freeman M, Chong AH. Solar keratosis: epidemiology, pathogenesis, presentation and treatment. Australasian Journal of Dermatology 2007;48(2):67‐76. [PUBMED: 17535191] [DOI] [PubMed] [Google Scholar]
Ibrahim 2009
- Ibrahim SF, Brown MD. Actinic keratoses. A comprehensive update. Journal of Clinical & Aesthetic Dermatology 2009;2(7):43‐8. [PUBMED: 20729970] [PMC free article] [PubMed] [Google Scholar]
Jeffes 2000
- Jeffes EW, Tang EH. Actinic keratoses. Current treatment options. American Journal of Clinical Dermatology 2000;1(3):167‐79. [DOI] [PubMed] [Google Scholar]
Juarranz 2008
- Juarranz A, Jaen P, Sanz‐Rodriguez F, Cuevas J, Gonzalez S. Photodynamic therapy of cancer. Basic principles and applications. Clinical & Translational Oncology 2008;10(3):148‐54. [DOI] [PubMed] [Google Scholar]
Juzeniene 2007
- Juzeniene A, Peng Q, Moan J. Milestones in the development of photodynamic therapy and fluorescence diagnosis. Photochemical & Photobiological Sciences 2007;6(12):1234‐45. [DOI] [PubMed] [Google Scholar]
Kaur 2010
- Kaur RR, Alikhan A, Maibach HI. Comparison of topical 5‐fluorouracil formulations in actinic keratosis treatment. Journal of Dermatological Treatment 2010;21(5):267‐71. [PUBMED: 19878034] [DOI] [PubMed] [Google Scholar]
Lebwohl 2003
- Lebwohl M. Actinic keratosis: epidemiology and progression to squamous cell carcinoma. British Journal of Dermatology 2003;149(Suppl 66):31‐33. [PUBMED: 14616345] [DOI] [PubMed] [Google Scholar]
Lee 2005
- Lee PK, Harwell WB, Loven KH, Phillips TJ, Whiting DA, Andres KL, et al. Long‐term clinical outcomes following treatment of actinic keratosis with imiquimod 5% cream. Dermatologic Surgery 2005;31(6):659‐63. [PUBMED: 15996416] [DOI] [PubMed] [Google Scholar]
Lu 1995
- Lu X, Xie W, Reed D, Bradshaw WS, Simmons DL. Nonsteroidal anti‐inflammatory drugs cause apoptosis and induce cyclooxygenases in chicken embryo fibroblasts. Proceedings of the National Academy of Sciences of the United States of America 1995;92(17):7961‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marks 1993
- Marks VJ. Actinic keratosis: a premalignant skin lesion. Otolaryngologic Clinics of North America 1993;26(1):23‐35. [PUBMED: 8433840] [PubMed] [Google Scholar]
Martin 2011
- Martin GM. Impact of interval and combination therapies on the management of actinic keratosis: review and clinical considerations. Journal of Dermatological Treatment 2011;22:288‐97. [PUBMED: 20528483] [DOI] [PubMed] [Google Scholar]
Moan 1991
- Moan J, Berg K. The photodegradation of porphyrins in cells can be used to estimate the lifetime of singlet oxygen. Photochemistry & Photobiology 1991;53(4):549‐53. [DOI] [PubMed] [Google Scholar]
Moy 2000
- Moy RL. Clinical presentation of actinic keratoses and squamous cell carcinoma. Journal of the American Academy of Dermatology 2000;42(1 Pt 2):8‐10. [DOI] [PubMed] [Google Scholar]
Nelson 1994
- Nelson MA, Einspahr JG, Alberts DS, Balfour CA, Wymer JA, Welch KL, et al. Analysis of the p53 gene in human precancerous actinic keratosis lesions and squamous cell cancers. Cancer Letters 1994;85(1):23‐9. [DOI] [PubMed] [Google Scholar]
NHANES I
- Center for Disease Control and Prevention/ National Center for Health Statistics. National Health and Nutrition Examination Survay (NHANES I). http://www.cdc.gov/nchs/nhanes/nhanesi.htm (accessed 24 August 2010).
Ramsay 2003
- Ramsay HM, Fryer AA, Hawley CM, Smith AG, Nicol DL, Harden PN. Factors associated with nonmelanoma skin cancer following renal transplantation in Quenssland Australia. Journal of the American Academy of Dermatology 2003;49(3):397‐406. [PUBMED: 12963901] [DOI] [PubMed] [Google Scholar]
Rigel 2008
- Rigel DS, Cockerell CJ, Carucci J, Wharton J. Actinic keratosis, basal cell carcinoma and squamous cell carcinoma. In: Bolognia JL, Jorizzo JL, Rapini RP editor(s). Dermatology. 2nd Edition. Vol. 2, Mosby Elsevier, 2008:1641‐59. [ISBN 9781416029991] [Google Scholar]
Rivers 2004
- Rivers JK. Topical 3% diclofenac in 2.5% hyaluronan gel for the treatment of actinic keratoses. Skin Therapy Letters 2004;9(1):1‐3. [PubMed] [Google Scholar]
Robins 2002b
- Robins P, Gupta AK. The use of topical fluorouracil to treat actinic keratosis. Cutis 2002;70(2 Suppl):4‐7. [PUBMED: 12353679] [PubMed] [Google Scholar]
Roewert‐Huber 2007
- Roewert‐Huber J, Stockfleth E, Kerl H. Pathology and pathobiology of actinic (solar) keratosis ‐ an update. British Journal of Dermatology 2007;157(Suppl 2):18‐20. [PUBMED: 18067626] [DOI] [PubMed] [Google Scholar]
Salasche 2000
- Salasche SJ. Epidemiology of actinic keratoses and squamous cell carcinoma. Journal of the American Academy of Dermatology 2000;42(1 Pt 2):4‐7. [DOI] [PubMed] [Google Scholar]
Schwartz 1997
- Schwartz RA. The actinic keratosis: a perspective and update. Dermatologic Surgery 1997;23(11):1009‐19. [PUBMED: 9391557] [DOI] [PubMed] [Google Scholar]
Seed 1997
- Seed MP, Brown JR, Freemantle CN, Papworth JL, Colville‐Nash PR, Willis D, et al. The inhibition of colon‐26 adenocarcinoma development and angiogenesis by topical diclofenac in 2.5% hyaluronan. Cancer Research 1997;57(9):1625‐29. [PubMed] [Google Scholar]
Stanley 1999
- Stanley MA. Mechanism of action of imiquimod. Papillomavirus Rep 1999;10(2):23‐9. [Google Scholar]
Thai 2004
- Thai KE, Fergin P, Freeman M, Vinciullo C, Francis D, Spelman L, et al . A prospective study of the use of cryosurgery for the treatment of actinic keratoses. International Journal of Dermatology 2004;43(9):687‐92. [DOI] [PubMed] [Google Scholar]
Vatve 2007
- Vatve M, Ortonne J‐P, Birch‐Machin MA, Gupta G. Management of field change in actinic keratosis. British Journal of Dermatology 2007;157(Suppl 2):21‐24. [PUBMED: 18067627] [DOI] [PubMed] [Google Scholar]
Vidal 2006
- Vidal D. Topical imiquimod: mechanism of action and clinical applications. Mini‐Reviews in Medicinal Chemistry 2006;6(5):499‐503. [DOI] [PubMed] [Google Scholar]
Warino 2006
- Warino L, Tusa M, Camacho F, Teuschler H, Fleischer AB Jr, Feldman SR. Frequency and cost of actinic keratosis treatment. Dermatologic Surgery 2006;32(8):1045‐49. [PUBMED: 16918567] [DOI] [PubMed] [Google Scholar]
Wilkerson 1984
- Wilkerson MG, Goldberg LH, Rapini RP. Actinic keratoses. American Family Physician 1984;30(1):103‐8. [PubMed] [Google Scholar]