

Abstract
A 73-year-old man developed diplopia after the administration of pembrolizumab for lung adenocarcinoma. He had ptosis and external ophthalmoplegia without general muscle weakness. Serum CK levels were elevated. Although autoantibodies to acetylcholine receptor and muscle-specific kinase, the edrophonium test, and the repetitive nerve stimulation test were all negative, anti-titin autoantibody was positive, leading to the diagnosis of myasthenia gravis (MG). Muscle pathology showed necrotizing myopathy with tubular aggregates. Unlike previously reported cases of pembrolizumab-associated MG, the present case showed ocular MG. This is the first case of pembrolizumab-associated MG with anti-titin antibody, as well as the first case with tubular aggregates.
Keywords: pembrolizumab, ocular myasthenia gravis, anti-titin antibody, necrotizing myopathy, tubular aggregates
Introduction
Pembrolizumab is an anti-programmed cell death 1 (PD-1) monoclonal antibody that is used for therapy against lung cancer. Increasing evidence suggests that it can cause myasthenia gravis (MG) and necrotizing myopathy, although the detailed clinicopathological features are still unclear (1).
Case Report
A 73-year-old man with hypertension and mitral valve regurgitation with no relevant family history of neuromuscular disorders was found to have a lung nodule on a regular medical checkup in 2016. Subsequently, a diagnosis of lung adenocarcinoma with brain and bone metastases was made by pulmonologists. The tumor proportion score of the programmed cell death-ligand 1 (PD-L1) expression was 85%, so he was started on pembrolizumab in July 2017 (day 1). On day 23, he was found to have diplopia, and his CK level had increased from 55 to 600 U/L. He was admitted to our hospital on day 30 with stable vital signs.
On a physical examination, he had diplopia and ptosis in the left eye with daily fluctuation. He had no easy fatigability or weakness in the limbs and trunk. On laboratory testing, his creatine kinase (CK) level was 7,311 U/L, aldolase 16.5 IU/L, aspartate aminotransferase (AST) 172 U/L, alanine aminotransferase (ALT) 74 U/L, lactate dehydrogenase (LDH) 631 U/L, creatinine 1.17 mg/dL, C-reactive protein (CRP) 0.68 mg/dL, erythrocyte sedimentation rate (ESR) 34 mm/h, and D-dimer 1.4 μg/mL. His thyroid function was within the normal range [thyroid stimulating hormone (TSH) 1.75 μIU/mL, FT3 2.61 pg/mL, FT4 1.05 ng/dL]. Rheumatoid factor, antinuclear antibody, anti-double stranded DNA (dsDNA) antibody, anti-ribonucleoprotein (RNP) antibody, anti-histidyl transfer RNA synthetase (Jo-1) antibody, anti-aminoacyl transfer RNA synthetase (ARS) antibody, anti-mitochondrial M2 (M2) antibody, anti-signal recognition particle (SRP) antibody, and anti-3-hydroxy-3-methylglutary-coenzyme A reductase (HMGCR) antibody were all negative. Anti-acetylcholine receptor (AChR) antibody, anti-muscle-specific kinase (MuSK) antibody, and anti-voltage-gated potassium channel Kv1.4 antibody were also negative, but anti-titin antibody was positive, leading to the diagnosis of MG, although both the repetitive nerve stimulation test (right accessory nerve, axillary nerve, median nerve, and ulnar nerve) and the edrophonium test were negative. The ice pack test was not evaluated.
Arterial blood gas analyses and spirometry showed no evidence of respiratory insufficiency. Echocardiography showed a good ejection fraction (70%) and no myocarditis.On electromyography of the right deltoid, biceps brachii, and iliopsoas, fibrillation potentials were seen only in the biceps muscle. Low-amplitude and short-duration motor unit potentials were recorded in all muscles, indicating myogenic changes. Magnetic resonance imaging of the thigh muscles showed no evidence of myopathy. A muscle biopsy from the left biceps brachii showed scattered necrotic and regenerating muscle fibers with minimal reactive mononuclear cell infiltration (Fig. 1A, B). Tubular aggregates were seen in some fibers (Fig. 1C, D). On immunohistochemistry, major histocompatibility complex (MHC)-I was mildly expressed in fibers in some areas (Fig. 1E), and membrane attack complex (MAC) was deposited on the sarcolemma of some non-necrotic fibers, in addition to the cytoplasm of necrotic fibers (Fig. 1F).
Figure 1.
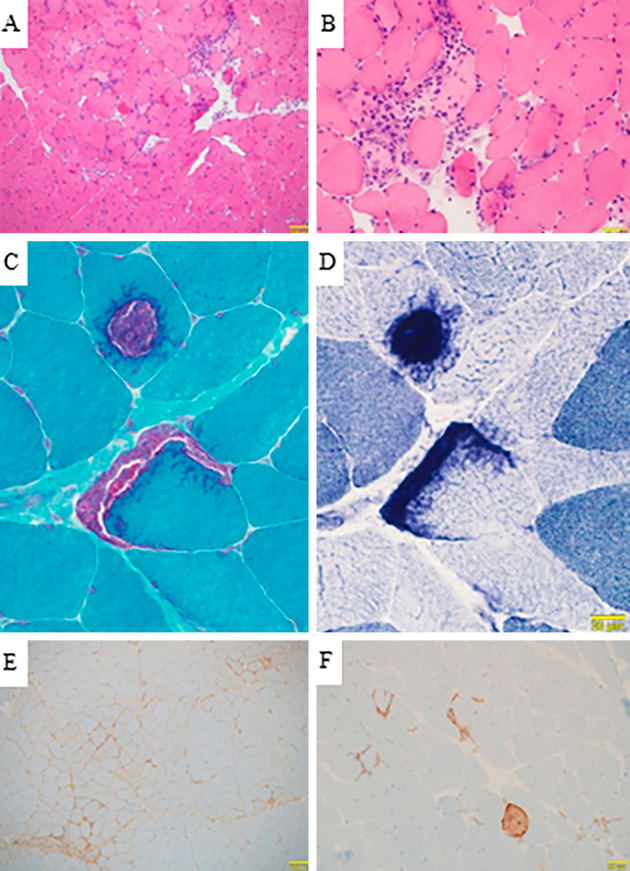
Pathological features of necrotizing myopathy. A, B: Hematoxylin and Eosin staining demonstrates necrosis and regeneration of muscle fibers and necrotizing myopathy with inflammatory cell infiltration only around necrotic fibers. A: scale bar 100 μm, B: scale bar 50 μm. C: Gomori trichrome staining, D: dihydronicotinamide adenine dinucleotide (NADH) staining. Tubular aggregates can be seen. C, D: scale bar 20 μm. E: Major histocompatibility complex (MHC)-I staining demonstrates light staining of muscle fibers. Scale bar 100 μm. F: Membrane attack complex (MAC) staining demonstrates the deposition of necrotic fibers, with light deposition of non-necrotic fibers. Scale bar 50 μm.
Based on the above results, a diagnosis of ocular MG (Myasthenia Gravis Foundation of America I) with anti-titin antibody and necrotizing myopathy with tubular aggregates was made. The Quantitative MG score was 6 points (ptosis and diplopia: 3 and 3 points, respectively). The patient was given an ascending-dose regimen of prednisolone that increased by 5 mg every 5 days to a total of 20 mg. He initially had exacerbation of ptosis and opthalmoplegia with diplopia. The CK level showed a trend toward reduction, but it remained high. Therefore, steroid pulse therapy (methylprednisolone 1 g/day 3 times) was given. The CK level then normalized promptly, and his symptoms improved gradually. All abnormal neurological findings disappeared four months after the start of steroid therapy (Fig. 2).
Figure 2.

Clinical course. The patient showed elevation of CK to 600 U/L at 23 days after and 7,311 U/L at 30 days after the administration of pembrolizumab, at which point he became aware of diplopia and ptosis. Various investigations were negative, and myasthenia gravis was diagnosed based on his symptoms that co-existed with myopathy. A left biceps muscle biopsy was performed. The patient received an ascending-dose regimen of prednisolone that increased by 5 mg every 5 days to a total of 20 mg. After steroid therapy, he developed exacerbations of ptosis and diplopia, and extraocular muscle weakness appeared. This was considered initial worsening. The CK level showed a trend toward reduction, but it remained high, so steroid pulse therapy (methylprednisolone 1 g/day 3 times) was given. The CK level then decreased to the normal range, and his symptoms improved. After four months, ptosis disappeared, and extraocular muscle weakness improved.
Discussion
Immunotherapy is a recently established treatment method for lung cancer. Representative agents include antibodies targeting immune checkpoints, such as PD-1, PD-L1, and cytotoxic T-lymphocyte associated antigen-4 (CTLA-4). Pembrolizumab and nivolumab are anti-PD-1 monoclonal antibodies. These agents connect with PD-1 expressed on activated T cells and exert their anticancer activity by eliminating the suppression of activated T cells (1). These immune checkpoint inhibitors are reported to cause immune-related adverse events (irAEs), including MG and myopathy (2), although the pathomechanism is still unclear.
Although the previously reported cases of anti-PD-1 monoclonal antibody-associated MG (anti-PD-1-associated MG) showed a relatively severe generalized type (3), the present case showed ocular MG without generalized symptoms. There have been four cases of anti-PD-1-associated MG that presented with the ocular type (Table) (3-5). Even if patients show ocular MG, there is the possibility of general inflammation, such as the elevation of the serum CK level and myopathy on electromyography and a muscle biopsy.
Table.
Cases of Ocular-type Anti-PD-1-associated MG.
| Case/age (y)/sex | Cancer | PD-1 | Onset | Diplopia/ ptosis |
AChR/ MuSK/titin |
CK U/L | Treatment | Outcome | Reference number |
|---|---|---|---|---|---|---|---|---|---|
| 1/65/M | NSCLC | Nivolumab | 25 days | +/+ | -/-/ND | ND | ChEI | CR | 4 |
| 2/81/M | Melanoma | Pembrolizumab | 58 days | +/+ | -/ND/ND | ND | PRED | CR | 5 |
| 3/74/F | Melanoma | Nivolumab | 28 days | +/+ | 4.0/ND/ND | 654 | ChEI, PRED | PR | 3 |
| 4/57/M | NSCLC | Nivolumab | 58 days | +/- | 0.5/ND/ND | 57 | ChEI, PRED | MM | 3 |
| 5/73/M | NSCLC | Pembrolizumab | 23 days | +/+ | -/-/+ | 7,311 | PRED | CR | Present |
NSCLC: non-small-cell lung cancer, ChEI: cholinesterase inhibitor, PRED: prednisolone, CR: complete response, MM: minimal manifestation, PR: partial response, N/D: no data, Onset: the time between the appearance of MG symptoms and the administration of anti-PD-1 monoclonal antibody
There are some cases of co-existing anti-striated muscle antibody (anti-titin, anti-ryanodine, anti-Kv1.4 antibody) and anti-AChR antibody in idiopathic MG with myopathy and myocarditis (6). In the present case, anti-AChR antibody was negative, but anti-titin antibody was positive. Apparently, anti-striated muscle antibody was positive in previously reported cases of anti-PD-1-associated MG (7-9), but the details were not described. To our knowledge, this is the first case of anti-PD-1-associated MG with anti-titin antibody. In idiopathic MG, 80% of patients have anti-AChR antibody, and 10% have anti-MuSK antibody (10). In anti-PD-1-associated MG, however, the positive rate for anti-AChR antibody differs widely, from 20% (9) to 73% (11). Anti-MuSK antibody has never been reported in a case of anti-PD-1-associated MG.
There have been several cases of anti-PD-1 monoclonal antibody-associated myopathy in which a muscle biopsy was performed. The reported pathological changes have varied, including necrotizing myopathy, nonspecific myopathy, and myopathy with complement-mediated microvasculopathy (2,9). Serum CK is not elevated in all cases, but it is elevated in patients with necrotizing myopathy, as in the present case. This is the first case of anti-PD-1 monoclonal-antibody-associated myopathy that showed tubular aggregates on muscle pathology. Tubular aggregates are found in cases of periodic paralysis, congenital myasthenia, and myopathy due to abnormal store-operated Ca2+ channels (12), but the present case had neither clinical manifestations nor a family history suggesting any of these conditions. The relationship between tubular aggregates and anti-PD-1 monoclonal antibody therefore remains unclear.
Conclusion
We herein report a case of anti-PD-1-associated MG. Unlike other cases with anti-PD-1-associated MG, the present case showed ocular MG without limb or trunk weakness, although the serum CK level was high, and necrotic and regenerating fibers were seen in the muscle. Interestingly, anti-titin antibody was positive, and tubular aggregates were seen on muscle pathology, although the associations of these findings with PD-1 are still unclear. Similar cases will need to be accumulated in order to better understand this relationship.
The authors state that they have no Conflict of Interest (COI).
References
- 1. Miyauchi E, Inoue A. Immune checkpoint therapy for non-small-cell lung cancer. Gan To Kagaku Ryoho 43: 666-671, 2016(in Japanese, Abstract in English). [PubMed] [Google Scholar]
- 2. Kao JC, Liao B, Markovic SN, et al. . Neurological complications associated with anti-programmed death 1 (PD-1) antibodies. JAMA Neurol 74: 1216-1222, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Suzuki S, Ishikawa N, Konoeda F, et al. . Nivolumab-related myasthenia gravis with myositis and myocarditis in Japan. Neurology 89: 1127-1134, 2017. [DOI] [PubMed] [Google Scholar]
- 4. Polat P, Donofrio PD. Myasthenia gravis induced by nivolumab therapy in a patient with non-small-cell lung cancer. Muscle Nerve 54: 507, 2016. [DOI] [PubMed] [Google Scholar]
- 5. Nguyen BH, Kuo J, Budiman A, Christie H, Ali S. Two cases of clinical myasthenia gravis associated with pembrolizumab use in responding melanoma patients. Melanoma Res 27: 152-154, 2017. [DOI] [PubMed] [Google Scholar]
- 6. Suzuki S, Utsugisawa K, Nagane Y, Suzuki N. Three types of striational antibodies in myasthenia gravis. Autoimmune Dis, 2011(Epub ahead of print). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bilen MA, Subudhi SK, Gao J, Tannir NM, Tu SM, Sharma P. Acute rhabdomyolysis with severe polymyositis following ipilimumab-nivolumab treatment in a cancer patient with elevated anti-striated muscle antibody. J Immunother Cancer 4: 36, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Norwood TG, Westbrook BC, Johnson DB, et al. . Smoldering myocarditis following immune checkpoint blockade. J Immunother Cancer 5: 91, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liewluck T, Kao JC, Mauermann ML. PD-1 inhibitor-associated myopathies: emerging immune-mediated myopathies. J Immunother 41: 208-211, 2018. [DOI] [PubMed] [Google Scholar]
- 10. Hurst RL, Gooch CL. Muscle-specific receptor tyrosine kinase (MuSK) myasthenia gravis. Curr Neurol Neurosci Rep 16: 61, 2016. [DOI] [PubMed] [Google Scholar]
- 11. Gonzalez NL, Puwanant A, Lu A, Marks SM, Živković SA. Myasthenia triggered by immune checkpoint inhibitors: new case and literature review. Neuromuscul Disord 27: 266-268, 2017. [DOI] [PubMed] [Google Scholar]
- 12. Kim NR, Suh YL. Tubular aggregate myopathy: a case report. J Korean Med Sci 18: 135-140, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]