

Virus capsids assemble spontaneously with surprisingly high fidelity. This requires strict geometry and a narrow range of association energies for these protein-protein interactions. It was hypothesized that requiring subunits to undergo a conformational change to become assembly active could regulate assembly by creating an energetic barrier and attenuating association. We found that woodchuck hepatitis virus capsid protein undergoes structural transitions between its dimeric and its 120-dimer capsid states. It is likely that the closely related hepatitis B virus capsid protein undergoes similar structural changes, which has implications for drug design. Regulation of assembly by structural transition may be a common mechanism for many viruses.
KEYWORDS: HBV, WHV, self-assembly
ABSTRACT
Hepadnaviruses are hepatotropic enveloped DNA viruses with an icosahedral capsid. Hepatitis B virus (HBV) causes chronic infection in an estimated 240 million people; woodchuck hepatitis virus (WHV), an HBV homologue, has been an important model system for drug development. The dimeric capsid protein (Cp) has multiple functions during the viral life cycle and thus has become an important target for a new generation of antivirals. Purified HBV and WHV Cp spontaneously assemble into 120-dimer capsids. Though they have 65% identity, WHV Cp has error-prone assembly with stronger protein-protein association. We have taken advantage of the differences in assemblies to investigate the basis of assembly regulation. We determined the structures of the WHV capsid to 4.5-Å resolution by cryo-electron microscopy (cryo-EM) and of the WHV Cp dimer to 2.9-Å resolution by crystallography and examined the biophysical properties of the dimer. We found, in dimer, that the subdomain that makes protein-protein interactions is partially disordered and rotated 21° from its position in capsid. This subdomain is susceptible to proteolysis, consistent with local disorder. WHV assembly shows similar susceptibility to HBV antiviral molecules, suggesting that HBV assembly follows similar transitions. These data show that there is an entropic cost for assembly that is compensated for by the energetic gain of burying hydrophobic interprotein contacts. We propose a series of stages in assembly that incorporate a disorder-to-order transition and structural shifts. We suggest that a cascade of structural changes may be a common mechanism for regulating high-fidelity capsid assembly in HBV and other viruses.
IMPORTANCE Virus capsids assemble spontaneously with surprisingly high fidelity. This requires strict geometry and a narrow range of association energies for these protein-protein interactions. It was hypothesized that requiring subunits to undergo a conformational change to become assembly active could regulate assembly by creating an energetic barrier and attenuating association. We found that woodchuck hepatitis virus capsid protein undergoes structural transitions between its dimeric and its 120-dimer capsid states. It is likely that the closely related hepatitis B virus capsid protein undergoes similar structural changes, which has implications for drug design. Regulation of assembly by structural transition may be a common mechanism for many viruses.
INTRODUCTION
Hepatitis B virus (HBV) is a widespread virus that causes chronic infection in around 257 million people. Annually, more than 800,000 deaths result from chronic HBV and its complications, notably liver failure, cirrhosis, and hepatocellular carcinoma (1, 2). Woodchuck hepatitis virus (WHV), an HBV homolog, causes symptoms similar to those of HBV (3) and has been a critical animal model, contributing to our understanding of HBV and development of antivirals (4, 5).
WHV and HBV have similar architectures; an envelope surrounds an icosahedral capsid, which contains the partially double-stranded circular DNA genome (4). The HBV core protein (Cp), a small 183-residue homodimer, plays multiple roles (6). Cp dimers specifically assemble around a complex of viral RNA and polymerase. This compartment provides the environment for reverse transcription, has signal for nuclear import, has signals for binding viral envelop protein, and releasees mature genomes into the nucleus (7). Cp dimers are also found in the nucleus associated with viral genomes (8). The unique characteristics and multiple functions make Cp an important target for developing direct-acting antivirals (6).
HBV capsids have 120 Cp dimers arranged in T=4 icosahedral symmetry (9, 10). Cp is comprised of a 149-residue assembly domain (human Cp149, or hCp149) and a nucleic acid binding domain. The assembly domain has a stable chassis domain linked to subdomains by glycine/proline hinges; two of these subdomains are the spike tips (the external-most part of the four-helix bundle that forms the dimer interface) and helix 5, which makes the protein-protein interactions required for capsid formation (Fig. 1a) (11, 12). In vitro, HBV Cp capsid assembly is a reaction with high fidelity (13, 14) that can be initiated and manipulated by pH, ionic strength, and temperature using the N-terminal assembly domain of Cp, hCp149 (13). Hypothetically misassembled capsids will lead to failure of the HBV life cycle. In vitro studies have provided understanding of the biology of capsid assembly in vivo, contributing to development of antivirals that misregulate or misdirect capsid assembly. The Cp of WHV shares 63% sequence identity with HBV Cp (15). However, the assembly domain of WHV, wCp149, has low assembly fidelity (15, 16). Kukreja et al. and Pierson et al. have shown that wCp149 assembles much faster than hCp149, with stronger protein-protein interactions, frequently leading to irregular and oversized capsids (15, 16).
FIG 1.

WHV and HBV have similar assembly domains. (a) A capsid protein dimer is represented using pipes and planks with the two monomers shown in different shades of gray. The positions of key residues discussed in text, Y132 and G111, are marked by spheres. The first residue, Met1, of one monomer and the end residue solved in crystal structure, Ser141, of the other monomer are also indicated. (b) Sequence alignment shows that wCp149 and hCp149 have 63% sequence identity. The sequence is correlated with α-helixes (cyan), numbered as in panel a.
Here, to identify the structural basis for differences between wCp149 and hCp149, we determined the wCp149 dimer structure using the assembly-incompetent mutant (11) wCp149-Y132A (wY132A) and the WHV capsid structure. In the WHV dimer assembly domain, the C-terminal region, consisting of residues 110 to 140, that is responsible for interdimer interaction is shifted by 21° from its location in capsids and is partially disordered. Proteolysis studies confirmed that WHV dimer has a more flexible C terminus than HBV. Our findings suggest that the entropy of the C-terminal region plays an important role in regulating assembly.
(This article was submitted to an online preprint archive [17].)
RESULTS
wY132A shows unique crystal packing.
Kukreja et al. showed that wCp149 and hCp149 have distinctly different assembly behaviors (15) despite 63% sequence identity (Fig. 1b). To understand the basis for those differences, we sought to determine the structure of wCp149 free dimer to compare it with that of the hCp149 dimer. Because wild-type wCp149 spontaneously self-assembles, we used an assembly-incompetent mutant, wCp149-Y132A (wY132A), that was designed based on previous work with assembly-incompetent human Cp149-Y132A (hY132A) (11, 18). The Y132A mutation is on an exposed loop of human capsid protein where it is expected to eliminate a substantial fraction of surface that is buried during assembly but have little effect on protein folding (Fig. 1a). The crystal structure of hY132A is similar to that of hCp149 in a capsid; it can coassemble with hCp149 but does not assemble on its own (11, 12). wY132A purification followed the same protocol developed for hY132A and, like hY132A, remained dimeric under various assembly conditions, based on size exclusion chromatography (SEC) and light scattering. wY132A did not crystalize under the conditions used to determine the structure of hY132A (11, 12). After exhaustive screening, we identified and optimized a novel condition for obtaining well-ordered crystals: 13% polyethylene glycol 400, 240 mM KCl, and 50 mM morpholineethanesulfonic acid (MES), pH 5.8.
The crystal structure of wY132A was determined to 2.9-Å resolution and had two wY132A dimers, AB and CD, in the crystallographic asymmetric unit (ASU) (Fig. 2a) in a novel arrangement of dimers. To appreciate the significance of the crystal packing for wY132A, it must be recognized that in crystals of hY132A, the protein is always arranged in sheets with triangular repeats (Fig. 2b); this trigonal arrangement of dimers is based on interdimer contacts that are similar to the interactions found in capsids and involve many of the same protein-protein contacts (11, 12).
FIG 2.
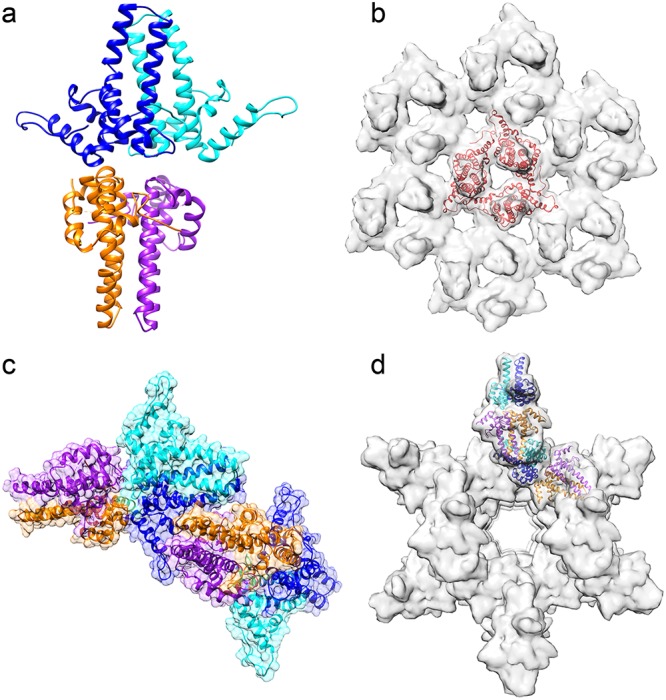
Unlike hY132A crystal packing, wY132A crystal packs differently. (a) The asymmetric unit (ASU) of the wY132A crystal structure has two dimers, AB (blue and cyan) and CD (orange and purple). (b) Crystal packing of hY132A (PDB accession number 4BMG) adopts capsid-like interactions to form trimers of dimers (red triangle). (c) Unlike hY132A, the crystallographic ASU of wY132A has two dimers in a base-to-base arrangement. (d) By crystallographic symmetry, the wY132A repeats the ASU to form a cylinder in the crystal. In this packing arrangement, the region around residue 132 for the CD dimer does not contact other proteins.
In wY132A crystals, interactions between dimers involve contacts not found in capsids (Fig. 2a and c). The two dimers, AB and CD, from the wY132A ASU are stacked base to base (Fig. 2a), where the base normally forms the lumen of a capsid. ASUs in the crystal are arranged so that the top of the four α-helix bundle of the CD dimer is wedged into the gap formed by α-helix 5 and the four-α-helix bundle of the AB dimer from an adjacent ASU (Fig. 1a and 2c). Clusters resulting from such interactions are arranged to form a hollow cylinder (Fig. 2d). Crystallization conditions did not cause wY132A to form cylinders in solution, based on transmission electron microscopy (TEM), suggesting that such interaction and structure are specific for crystal packing.
wY132A dimers adopt a single conformation but with some important differences.
Though the two dimers in the crystallographic asymmetric unit have very different environments, they have nearly the same dimer structures, except for some missing density in the CD dimer (Fig. 3a and b). In overlays, all monomers in the wY132A asymmetric unit had one conformation: aligned models of A, B, C, and D showed little deviation (Fig. 3c and d). The root mean square deviations (RMSD) of the alignments were 0.367 Å (AB), 0.671 Å (AC), and 0.534 Å (AD). In contrast, a T=4 HBV capsid has two asymmetric dimers resulting in four unique monomer structures, and the hY132A crystal structure (PDB accession number 3KXS) (11) has three asymmetric dimers, resulting in two classes of monomer structures. The wY132A dimers have a nearly 2-fold symmetry, off by only 0.65°; AB and CD dimers of wY132A aligned with little difference in Cα positions over both subunits and both dimers (Fig. 3).
FIG 3.

All subunits from the wY132A crystal structure asymmetric unit adopt one conformation. Structure alignment of the AB dimer and CD dimer (PDB accession code 6ECS) shows that the two dimers have similar tertiary structures (shown in side view in panel a and top view in panel b). Overlapping all subunits (A, B, C, and D) shows that all subunits from the asymmetric unit exhibit a single conformation (shown in side view in panel c and top view in panel d). (e) To quantitively compare four subunits, the Cα displacements of chain B, C, and D relative to chain A were calculated and plotted against residue number. Most of the residues have displacements of less than 1 Å, indicating that all four subunits have a similar tertiary structures. The exceptions to this generalization are the spike tips and the C terminal regions, including where electron density starts to degrade.
Despite the overall conformational similarity, the AB and CD dimers display an important difference in their density maps (Fig. 4). At 1.5-σ contour level, the electron density map of the AB dimer is continuous, with no breaks (Fig. 4a and b). However, at the same contour level, the map for the CD dimer has missing density, corresponding to residues 126 to 136, a loop structure linking α-helix 5 to the C-terminal extended structure of the assembly domain (Fig. 4c and d). This disorder has never been observed in hCp149 capsid or hY132A dimer structures. The ordered density in the loop of the AB dimer may be related to packing interactions with neighboring CD dimers. The equivalent loop in CD dimers is surrounded by solvent. The missing density of the loop in CD dimers leads us to suggest that the loop is similarly disordered when the dimer is in solution. Though this region is not involved in crystallographic contacts for CD dimers, it plays a critical role in the dimer-dimer interactions involved in building capsids (9, 10). The disorder of the sequence from residue 126 to 136 is consistent with the proteolytic sensitivity of Arg127 in human HBV Cp149 (19).
FIG 4.

wY132A dimers have different features. 2Fo − Fc (where Fo and Fc are the observed and calculated structure factor amplitudes, respectively) electron density map of the wY132A crystal structure at 1.5-σ contour level with Cα chains and a subunit comparison among asymmetric units. The A subunit is shown in blue, the B subunit is in cyan, the C subunit is in orange, and the D subunit is in purple. The side view (a) and top view (b) of the AB dimer show that electron density covers the entire backbone. In comparison to views of the AB dimer, the side view (c) and top view (d) of the CD dimer show missing electron density at the loop, from residue 126 to 136, where dimers make contacts during assembly.
wY132A and hY132A have substantive structural and biochemical differences.
Human HBV dimers have a central core with mobile peripheral subdomains (9, 11). This also seems to be the case with wY132A dimers (Fig. 3a and 4a) but with differences. The AB dimer of wY132A was superimposed with the EF dimer (PDB accession no. 3KXS), a 2.25-Å resolution structure of hY132A (Fig. 5). There was a large difference in Cα positions at the spike tips connecting α-helix 3 and α-helix 4a, a notably flexible region (6). The view of the overlay from what would be the capsid interior also showed that α-helix 5 of wY132A is rotated about 8.5° away from hY132A, using residue Gly111 as the hinge residue (Fig. 5b). With this position for α-helix 5, wY132A could not pack into trimers of dimers, as seen in the crystallographic packing of hY132A (Fig. 2b) (11). Comparisons of the two aligned structures showed that the Cα positions for the spike tips (residues 60 to 100) and the α-helix 5 interdimer interaction region were displaced up to 8 Å whereas core regions were displaced by less than 1 Å (Fig. 5c).
FIG 5.

wY132A AB dimer has a different tertiary structure than hY132A. (a and b) wY132A AB dimer (PDB accession number 6ECS) is overlaid with hY132A EF dimer (PDB accession number 3KXS; labeled in yellow). The top half of the four α-helix bundles and C termini show a distinct structure shift, which is quantified by plots of Cα displacements against residue numbers (c). (d) PONDR VL-XT predicts that hY132A and wY132A have similar disorder patterns, especially at the C termini where missing density is observed (Fig. 4c and d).
To test if the sequence of the disordered loop in wY132A, residues 126 to 136, and the equivalent region of hY132A had a predilection for disorder, we used PONDR (http://www.pondr.com/) to analyze both proteins. Given the 63% sequence identity (Fig. 1b), it is not surprising that the two proteins led to similar predictions (Fig. 5d). Both sequences had notable peaks in the predicted disorder scores at the loop formed by residues 126 to 136 as well as in a region from residues 35 to 50 that interacts with helix 5, which had been suggested as a fulcrum that modulated α-helix 5 mobility (11).
To investigate whether disorder in the loop from residues 126 to 136 of wY132A corresponded to increased protein flexibility compared to that of hY132A, we tested their susceptibilities to proteolysis. In hCp149, it was discovered that Arg127, though part of α-helix 5, was the first site cleaved by trypsin (19). In the current study, we observed that wY132A was digested by trypsin much faster than hY132A (Fig. 6a). As expected from previous work (19), cleavage of both proteins was at Arg127, as confirmed by mass spectrometry. In the earlier study, we determined that unfolding, or “opening,” of the Arg127 site was the rate-limiting factor for proteolysis. The faster digestion of wY132A suggests that residues 126 to 136 are more disordered. To test if the higher cleavage rate of wY132A indicates that the protein is partially unfolded in solution and has a correspondingly larger hydrodynamic radius, we used size exclusion chromatography (SEC). Paradoxically, wY132A eluted later than hY132A, suggesting that wY132A has a more compact Stokes radius (Fig. 6b).
FIG 6.
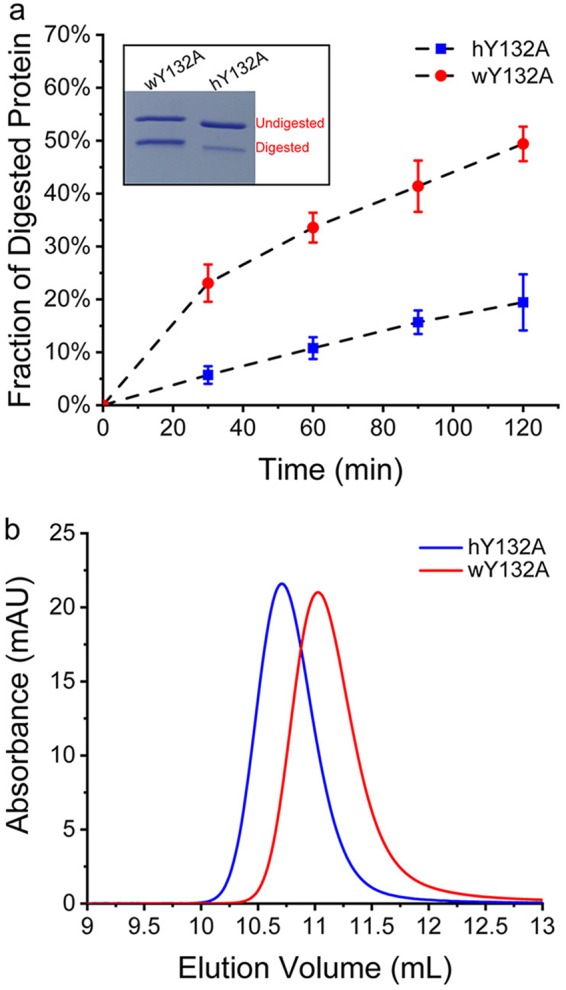
hY132A and wY132A have different rates of proteolysis in a region where the sequence is identical. (a) Proteolysis of hY132A and wY132A by trypsin was quantified by SDS-PAGE and showed that wY132A is digested much faster than hY132A. The inset is an example of SDS-PAGE of trypsin digestion. (b) An SEC experiment using hY132A and wY132A with high salt (1 M NaCl) shows that wY132A elutes later than hY132A, indicating that wY132A has a smaller Stokes radius and a more compact form. AU, arbitrary units.
wCp149 from dimer to capsid: assembly is more thermodynamically favored than that of hCp149.
Assembly of hCp149 and wCp149 is a temperature-dependent and entropy-driven reaction, consistent with the observed burial of hydrophobic surface at the dimer-dimer contacts (6). However, wCp149 dimers have stronger association energies, and wCp149 assembly is favored in a broad range of temperatures (15). To quantify assembly of wCp149, we used very low ionic strength (50 mM NaCl and 10 mM HEPES, pH 7.5) at different temperatures, conditions where hCp149 showed no assembly (Fig. 7a). We note that under these assembly conditions, wCp149 has a linear van’t Hoff plot, which suggested that wCp149 assembly has a heat capacity close to zero (Fig. 7b). This is different from the positive heat capacity previously obtained using higher ionic strength (15).
FIG 7.
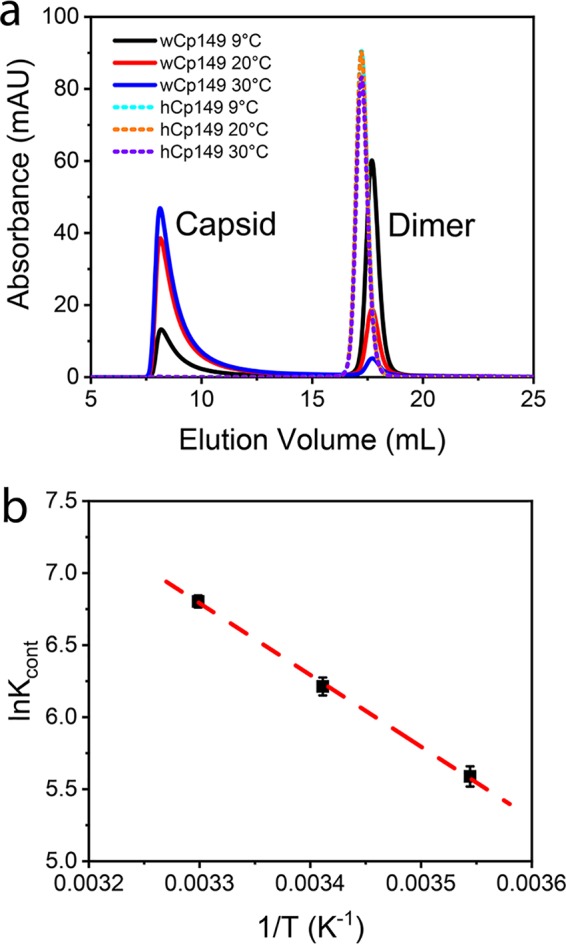
wCp149 assembly is thermodynamically favored. (a) Assemblies of 10 μM hCp149 and wCp149 were compared. Assembly was initiated by addition of 50 mM NaCl. wCp149 shows increasing assembly as the temperature increased. At all temperatures tested, hCp149 showed no assembly with this relatively low ionic strength. (b) A van’t Hoff plot of wCp149 assembly shows that wCp149 assembly at low-ionic strength has a linear temperature (T) dependence. The values for Kcont (the equilibrium constant [K] per pairwise interaction) were derived from the SEC-measured equilibrium concentrations of capsid and dimer, as previously described (15).
wY132A needs conformational changes to form capsid.
Superposition of wY132A AB dimer with AB and CD dimers of HBV capsid (PDB accession no. 1QGT) revealed a critical structure difference in the position of α-helix 5, which rotated 21° from the dimer in capsid form (Fig. 8a). Even though AB and CD dimers from a HBV capsid are asymmetric and not identical, the differences are in the same regions as those of wY132A. In addition to the highly variable spike tips (residues 80 to 90), Cα positions for the region containing residues 120 to 140 move up to 12 Å.
FIG 8.

α-Helix 5 modulates the capsid assembly process by changing its conformations. (a) Alignment of wY132A (red), hY132A (green; PDB accession number 3KXS), and dimer in WHV capsid (blue) shows that the three dimers have different positions for residues from α-helix 5 to the C termini. wY132A needs to shift 8.5° to match the conformation of hY132A and 21° to match wCp149. (b) A hypothesized scheme of the assembly process that progresses through the different dimer conformations (structures are labeled using the same colors used in panel a). We propose the following sequence of events. Assembly starts with free dimers (wY132A; red). Initiation of assembly induces a 8.5° shift of α-helix 5 to generate nuclei (based on hY132A, trimer of dimers) (11, 13). As assembly proceeds, the α-helix of dimers keep rotating to 21°, where dimers have a conformation that corresponds to those in a capsid (WHV capsid).
To test whether WHV core proteins have the same conformation in the capsid form or if the dimers change their conformations when they assemble, we determined the cryo-electron microscopy (cryo-EM) structure of WHV capsid to 4.5-Å resolution (Fig. 9a and b). Comparison between WHV and HBV capsid density maps suggests that they are virtually identical (Fig. 9a), which was confirmed by structural alignment between the asymmetric dimers of WHV and HBV capsids (Fig. 9c). This observation led us to infer that wCp149 dimers undergo structural changes during assembly. Furthermore, the structural similarity of WHV and HBV suggests that WHV is a suitable model system for testing and development of capsid-directed antivirals.
FIG 9.

WHV and HBV have similar capsid density maps. (a) A close view of the overlay of the cryo-EM density map of the WHV capsid (EMD-9031; blue) with a calculated HBV capsid density map (PDB accession number 1QGT; yellow) shows similar structures. The HBV capsid density map was calculated to 4.5 Å based on the crystal structure of HBV capsid (PDB accession number 1QGT) using the molmap function in the UCSF Chimera software program. The position of one asymmetric dimer (ASU) of each capsid is boxed by a red rhombus. (b) Fourier shell correlation (FSC) for the WHV capsid reconstruction. The dashed line indicates a correlation of 0.143. (c) Top view of the overlay between the WHV (blue, cyan, orange, and purple) and HBV (dark gray) ASUs with the same arrangement as shown in panel a. AB and CD shows similar dimer and capsid structures.
To test this hypothesis, we further compared the wY132A crystallographic dimer model to the cryo-EM WHV capsid density using a quasi-6-fold hexamer extracted from the WHV capsid to avoid biased interpretation. Six wY132A dimers were fit as rigid bodies into the extracted capsid density, a process that was dominated by the successful match of the four-helix bundle at the dimer interface (Fig. 10). The majority of the wY132A molecular model matches the density map except the C terminus from helix 5 to the end of the visible protein density, residues 112 to 141 (Fig. 10a). Dimers from the wY132A crystal structure, with alanines at residue 132 reverted to tyrosines, were refined to density using molecular dynamics flexible fitting (MDFF) (20) and PHENIX real-space refinement with icosahedral symmetry constraints (21). The resulting models fit into the hexamer density (Fig. 10b). These models are nearly identical to the corresponding dimers from human HBV capsid (Fig. 9c) (9). Nevertheless, comparing the wY132A dimers and dimers from the refined WHV capsid structure shows major changes at the regions following Gly111 and modest changes at the spike tip and α-helix 2.
FIG 10.
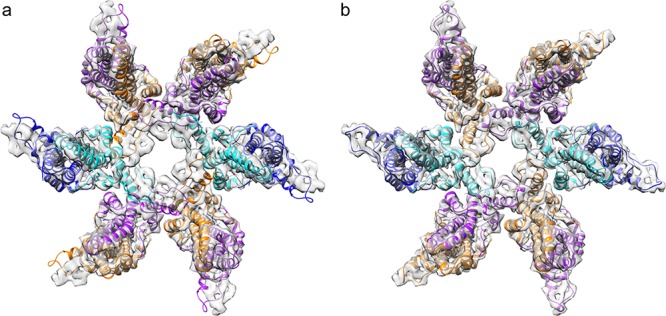
wY132A AB dimers did not fit into WHV capsid density. (a) wY132A AB dimers were rigid-body fitted into WHV hexamer density. The crystal structures and the capsid density map match each other well, except for the C-terminal subdomain starting at helix 5. The models are out of density and clash with adjacent subunits. (b) The same capsid hexamer density map is well occupied by the refined molecular models of WHV capsid dimers (PDB accession number 6EDJ).
wCp149 assembly can be distorted by assembly-directed antivirals.
WHV and human HBV have similar capsid structures (Fig. 9a and c), supporting the feasibility of WHV as a model system for testing and developing assembly-directed antivirals. To test this hypothesis, wCp149 assembly was observed with or without two antiviral molecules, HAP12 and HAP13 (22), and their assembly products were observed using TEM. Under conditions where wCp149 showed modest assembly, HAP12 significantly increased wCp149 assembly, and HAP13 showed a similar assembly effect but to a lesser extent (Fig. 11a). wCp149 mainly assembles into T=4 capsids at physiological ionic strength (150 mM NaCl, pH 7.5) (Fig. 11b). With the addition of HAP12, wCp149 assembles into products with larger, aberrant complexes, often with large openings (Fig. 11c and e). Consistent with its effect on assembly kinetics, HAP13 led to aberrant assembly products, but defects were not as gross as with HAP12 (Fig. 11d and f). These observations for wCp149 parallel our previous results with HBV Cp149 (22), supporting the utility of using WHV as a tool for testing and developing assembly-directed antivirals.
FIG 11.

wCp149 assembly is distorted by antiviral molecules, HAP12 and HAP13. (a) Under conditions where 4 μM wCp149 has slight assembly, 4 μM HAP12 and HAP13 increased wCp149 assembly. (b) In the absence of HAPs, most of the wCp149 assembly products are icosahedral T=4 capsids. With the addition of 4 μM HAP12 (c) or HAP13 (d), wCp149 assembles into a mixture of oversized products with few normal-sized capsids. Increasing the concentration of the HAP molecules to 10 μM (e, HAP12; f, HAP13) showed more dramatic effects on the assembly products. These negative-stain micrographs were recorded at ×50,000 magnification.
DISCUSSION
To investigate the basis of regulated HBV capsid assembly, we have determined the structures of wY132A and a WHV capsid. The wY132A structure is nearly symmetric, and the dimers are not involved in capsid-like interactions (seen in hY132A structures [11, 12]). Observed wY132A symmetry is consistent with nuclear magnetic resonance (NMR) analysis of hCp149, albeit the NMR study was at pH 9.5 (23). Without the constraint of quaternary structure in the crystallographic packing, the subdomain comprised by α-helix 5 and the following residues pivot around Gly111, shifting 8.5° away from its position in hY132A, which is constrained by a trigonal crystallographic lattice, and 21° from hCp149 in a T=4 capsid (Fig. 8a). Though the two wY132A dimers in the ASU are nearly identical, the CD dimer has missing density for residues 126 to 136, whereas those residues are ordered in the AB dimer (Fig. 4). The missing density indicates that in the WHV dimer, this disordered loop fits a large ensemble of conformations. The disordered region matches PONDR predictions (Fig. 5d) and the previously observed susceptibility of Arg127 to proteolysis (19). In this study, we observed a much higher digestion rate for wY132A (Fig. 6a), suggesting that in WHV dimers, this loop is more dynamic. Structurally disordered regions have been proposed to possess functional roles, becoming ordered upon interaction with a target molecule or protein (24, 25). In WHV Cp, the disordered loop becomes ordered by interaction with another dimer during capsid assembly. Although there is an entropic cost for ordering the loop, it is apparently offset by the energetic advantage of assembly. Indeed, the position of helix 5 may also be extremely mobile until pinned down by assembly, at an entropic cost. We suggest that wY132A structure, with its symmetry, shifted α-helix 5, and that the disordered loop may be an instructive representative of free dimer. We cannot exclude the possibility that the wCp149-Y132A structure is unlike structures seen in solution.
In previous work, we observed that the association energy of assembly was less than predicted based on structure and suggested that the difference was the energetic cost of a conformational change relating to assembly (11, 13). This hypothesis is qualitatively consistent with the shift of α-helix 5 (residues 111 to 128) and ordering of the associated loop (residues 126 to 136). Similarly, molecular dynamics simulations show that even in a stable HBV capsid, dimers change conformations constantly (26). This leads us to propose two models for regulation of capsid assembly.
The first model of assembly is based on sequential conformational change. We have observed three distinct conformations for dimer: (i) the partially disordered form shown here for wY132A, (ii) planar sheets of triangles found in hY132A crystals, and (iii) capsids where dimer-dimer interactions conform to a capsid’s radius of curvature. We suggest that the three conformations correspond to free dimer, early assembly intermediates before curvature is locked in, and capsid-like assembly intermediates and products. Assembly factors (i.e., temperature, ionic strength, and pH) trigger a conformational change by rotating α-helix 5 by 8.5° so that free dimers can associate into triangles to form nuclei and small oligomers. As the assembly products grow, the planar sheet curves, forcing further rotation of α-helix 5 to 21°, which then leads to T=4 icosahedral capsids (Fig. 8).
The second model posits that assembly is regulated entropically (24). In the entropic model, dimers have an ensemble of conformations that change in response to solution conditions. For free dimers, this presumably includes equilibration between ordered and disordered states for the loop from residues 126 to 136. As stated by Hilser and Thompson, the effect of allosteric coupling between two disordered regions will accelerate the conformational changes and dimer-dimer interactions because binding of one site of the disordered region will cooperatively turn the other disordered region into its ordered form and increase the binding affinity to new incoming subunits (24). Therefore, the energetic cost of ordering the first few subunits provides a basis for the nucleation phase. The allosteric effect of subsequent assembly suggests an element of cooperativity (Fig. 8), an idea originally described as autostery by Caspar (27). Importantly, these models are not mutually exclusive.
Both models provide explanations for normal and abnormal assembly reactions. Because of the presence of different conformations under different assembly stages, there must be an orchestrated assembly process to adjust the protein conformations to produce high-fidelity capsids. Conversely, random shifts of the position of α-helix 5 will almost certainly lead to incorrect dimer-dimer geometry, eventually forming an abnormal capsid. This effect has been demonstrated with assembly induced by small molecules that bind to a pocket formed by α-helix 5, residues 136 to 140, and α-helix 2 (10).
The structural similarities of WHV and HBV capsids suggest that human HBV undergoes the same transitions observed for WHV. These similarities extend to the WHV and HBV responses to two core protein-directed antiviral compounds, HAP12 and HAP13 (Fig. 11). With human HBV, HAP12 had a much greater effect than HAP13 on assembly and antiviral activity (22, 28). Similarly, HAP12 had a stronger effect on wCp149 than HAP13 though both led hCp149 and wCp149 assembly to form complexes that were oversized, elongated, and even broken (Fig. 11). Of note, the effect of HAP13 on WHV is attenuated compared to what that on HBV (see Fig. 5 in Ruan et al. [28]). We suggest that this difference arises because wCp149 assembly association energy is stronger than that of hCp149 (Fig. 8); similarly, it was observed that core protein mutations that stabilize HBV capsids confer greater resistance against the weaker HAPs (28). This set of observations is also consistent with the hypothesis that, in the presence of assembly-directed antivirals, there is competition between normal and aberrant assembly (41).
In conclusion, we observed that dimers have different conformations at different assembly stages. Loss of disorder in the loop at residues 126 to 136, as well as conformational shift of α-helix 5, suggests that there is an entropic cost for assembly in addition to the entropic gain from burial of hydrophobic surface. We propose that regulating conformation, or an ensemble of conformations, can trigger assembly and control the cascade of assembly reactions that produce uniform capsids.
MATERIALS AND METHODS
Cloning of wCp149-Y132A (wY132A) and protein purification.
The woodchuck hepatitis virus (WHV) capsid protein assembly domain (wCp149) was expressed and purified as described previously (15). The expression plasmid for wCp149, pET11c-wCp149, was mutated to wCp149-Y132A (wY132A) using QuikChange mutagenesis (Stratagene). wY132A and its human homolog hCp149-Y132A (hY132A) were expressed and purified using the same previously described protocol (11, 18).
Protein crystallization.
wY132A was dialyzed into 50 mM Tris, pH 9, and concentrated to 17.5 mg/ml using an Amicon Ultra 30-kDa centrifugal filter. A crystallization screening of a variety of conditions gave a promising hit with 12% polyethylene glycol 400, 400 mM KCl, and 50 mM HEPES, pH 7.5, at room temperature. Crystallization conditions were refined to 13 to 16% polyethylene glycol 400, 220 to 320 mM KCl, and 50 mM MES, pH 5.8, in a sitting-drop format. The crystal used for solving the wY132A structure grew in 13% polyethylene glycol 400, 240 mM KCl, and 50 mM MES, pH 5.8. For data collection, crystals were cryoprotected by briefly soaking in 25% glycerol, 15% polyethylene glycol 400, 260 mM KCl, and 50 mM MES, pH 5.5.
wY132A structure determination and refinement.
Diffraction data were collected at the Advanced Light Source (ALS), beamline 4.2.2. Data were processed using the XDS package (XDS, XSCALE, and XDSCONV) (29). The high-resolution cutoff was set to 2.9 Å to ensure that values of I/σ and the Pearson correlation coefficient (CC) of two half data sets (CC1/2) in the highest resolution bin (3.0 to 2.9 Å) were higher than 1 and 0.5, respectively (30). A molecular replacement density map was calculated using a dimer from the hY132A (PDB accession no. 3KXS) (11) as the phasing model. Iterative refinement and manual model building were accomplished with programs in the Phenix suite (31) and Coot (32). TLS (translation/libration/screw) refinement notably improved the R factors. Throughout the refinement process, 7% of the reflections were used for Rfree to avoid structure overfitting. For the final model, Rwork was 19.8%, and Rfree was 22.8% (Table 1).
TABLE 1.
Data collection and refinement statistics of wCp149-Y132A
| Parameter | Value for the parametera |
|---|---|
| PDB accession code | 6ECS |
| Wavelength (Å) | 1 |
| Resolution range (Å) | 63.09– 2.9 (3.004–2.9) |
| Space group | P3221 |
| Unit cell parameters | |
| a, b, c (Å) | 116.41, 116.41, 161.76 |
| α, β, γ (°) | 90, 90, 120 |
| Total no. of reflections | 312,472 (27,830) |
| No. of unique reflections | 28,665 (2,817) |
| Multiplicity | 10.9 (9.9) |
| Completeness (%) | 99.93 (99.93) |
| Mean I/sigma(I) | 19.47 (1.28) |
| Wilson B factor (A2) | 78.95 |
| Rmerge | 0.1083 (1.638) |
| Rmeas | 0.1136 (1.728) |
| Rpim | 0.0342 (0.5449) |
| CC1/2 | 0.999 (0.589) |
| CC* | 1 (0.861) |
| No. of reflections used in refinement | 28,656 (2,818) |
| No. of reflections used for Rfree | 1,993 (197) |
| Rwork | 0.1984 (0.3492) |
| Rfree | 0.2278 (0.3843) |
| CCwork | 0.968 (0.696) |
| CCfree | 0.964 (0.620) |
| No. of non-hydrogen atoms | 4,393 |
| No. of protein residues | 542 |
| RMSD | |
| Bond length (Å) | 0.007 |
| Bond angles (°) | 0.86 |
| Ramachandran plot | |
| Favored (%) | 96.23 |
| Allowed (%) | 3.77 |
| Outlier (%) | 0.00 |
| Rotamer outliers (%) | 0.00 |
| Clashscore | 9.71 |
| Avg B-factor (Å2) | 84.23 |
| Number of TLS groups | 23 |
Statistics for the highest-resolution shell are shown in parentheses.
Proteolysis reactions.
hY132A and wY132A were first dialyzed into 50 mM HEPES, pH 7.5, at 4°C. Sequencing-grade modified trypsin (Promega) was stored as an inactive stock in 50 mM acetic acid at a concentration of ∼4 μM. Each proteolysis reaction mixture consisted of 10 μM hY132A or wY132A, 0.2 μM trypsin, and 50 mM HEPES, pH 7.5, in a total volume of 200 μl. At time points 0, 30, 60, 90 and 120 min, 10 μl of each reaction product was mixed with 10 μl of denaturing loading buffer and boiled for 7 min.
Solution characterization of dimers and dimer assembly.
Dimer hydrodynamic radii were compared by size exclusion chromatography (SEC) using a Superdex 75 10/300 GL column (GE Healthcare) equilibrated with 1 M NaCl and 50 mM HEPES, pH 7.5. hY132A and wY132A were dialyzed into 50 mM HEPES, pH 7.5, at 4°C and adjusted to 5 μM. Proteins were mixed 1:1 (vol/vol) with 2 M NaCl and 50 mM HEPES, pH 7.5, and incubated overnight at room temperature.
To characterize the thermodynamics of wCp149 assembly, assembly reaction products were equilibrated, and the amounts of capsid and dimer were determined by SEC. As previously published (15), 10 μM wCp149 was mixed with 50 mM NaCl and 10 mM HEPES, pH 7.5, and incubated for 48 h at temperatures of 9°C, 20°C, and 30°C using a Superose 6 10/300 GL column.
wCp149 assembly kinetics were characterized using 90° light scattering as described previously (13). For control reactions, 4 μM wCp149 was mixed with an equal volume of 300 mM NaCl and 10 mM HEPES, pH 7.5, giving 2 μM wCp149, 150 mM NaCl, and 10 mM HEPES, pH 7.5. For reaction mixtures containing antivirals, 8 μM or 20 μM HAP12 or HAP13 in 1% dimethyl sulfoxide (DMSO) was mixed with the salt solution (300 mM NaCl, 10 mM HEPES, pH 7.5); then, equal volumes of protein and salt were mixed, giving reaction mixtures with 4 μM or 10 μM HAP12 or HAP13. To visualize assembly products by TEM, 4 μl of each assembly reaction product was applied to a glow-discharged carbon film with a 300-mesh Cu grid and stained with 2% uranyl acetate. Micrographs were collected using a JEOL JEM 1400Plus transmission electron microscope equipped with a 4,000-by-4,000 pixel charge-coupled-device (CCD) camera.
Cryo-EM structure determination of WHV capsids.
wCp149 was dialyzed into 50 mM HEPES, pH 7.5, at 4°C, and 25 μM wCp149 was assembled by mixing at 1:1 (vol/vol) with 300 mM NaCl, 50 mM HEPES, pH 7.5, and incubated overnight. The wCp149 assembly mixture was loaded onto a 10 to 40% (wt/vol) continuous sucrose gradient containing 300 mM NaCl and 50 mM HEPES, pH 7.5, and centrifuged at 285,000 × g for 5 h in a SW40 swinging bucket rotor. wCp149 capsids, visible as well-defined bands, were extracted from the gradient tube, dialyzed into 300 mM NaCl and 50 mM HEPES, pH 7.5, and concentrated to 15 mg/ml.
Preparation of wCp149 capsids for cryo-EM and image analysis followed previously described methods (10). Briefly, 4 μl of purified wCp149 capsids was applied to a glow-discharged UltrAuFoil R2/2 holey gold film grid and vitrified using a FEI Vitrobot (Mark III). Grids were imaged using an FEI Titan Krios microscope operated at 300 kV. Low-dose (∼30 e−/Å2) cryo-EM images, at a nominal magnification of ×81,000 (0.86 Å per pixel), were collected on a Gatan K2 Summit detector in superresolution mode. Leginon automated data collection software (33) was used to collect 251 images. A total of 25,800 particles were extracted using e2boxer_old.py from EMAN2 (34). Contrast transfer function parameters were estimated using ctffind4 (35). All particles were subjected to reference-free two-dimensional (2D) classification using RELION (version 2.1) (36). Particles in classes that showed blurred density were discarded, leaving 23,700 particles. A low-resolution initial model was built de novo using EMAN2. Briefly, the data set was subjected to reference-free 2D classification in EMAN2. A collection of 41 classes having good signal-to-noise ratios and well-defined characteristics was used to build the initial model using e2initialmodel.py. This model was used as a starting model with the RELION 3D auto-refine procedure. During 3D refinement, an initial angular sampling of 7.5° was selected, and icosahedral symmetry was imposed. The parameters were chosen based on earlier success in near-atomic resolution of HBV capsid structure determination (10). The data set was split into two parts prior to refinement, which was performed iteratively until the correlation between the two halves of the data set converged. The resolution cutoff was determined using the gold standard Fourier shell correlation of 0.143 (Fig. 9). A final 4.5-Å electron density map was obtained from 7,911 particles. Density was sharpened with a negative B factor of −214, obtained using the Guinier fitting procedure from RELION (42). The final density map of WHV capsid was deposited in the EMDB database as EMD-9031.
Capsid model determination.
To model the WHV capsid structure, the crystal structure of wY132A with Y132A reverted to Y132 was rigid-body docked into density corresponding to a dimer extracted from the WHV capsid map. Using molecular dynamic flexible fitting (MDFF) implemented in VMD and NAMD (20, 37, 38), the model was fit into the capsid density. A WHV capsid structure was generated using the fitted wY132A structure, and this was rigid-body fitted into the WHV capsid density, applying symmetry restraints (39). Molecular coordinates from the final frame of MDFF were extracted and refined using the PHENIX real-space refinement function with imposed icosahedral symmetry (21) (Table 2). All structures and maps were analyzed and displayed using UCSF Chimera (40).
TABLE 2.
Data collection and refinement statistics of wCp149 capsid
| Parameter | Value for the parameter |
|---|---|
| Data collection | |
| Microscope | FEI Titan Krios |
| Voltage (kV) | 300 |
| Detector | Gatan K2 Summit |
| Dose (e−/Å2) | 30 |
| Pixel size (Å) | 0.86 |
| Defocus range (μm) | 0.739–3.888 |
| Reconstruction (RELION) | |
| No. of micrographs | 251 |
| No. of particles | 7911 |
| Symmetry | Icosahedron |
| Sharpening B-factor (Å2) | −214 |
| Final resolution (Å) | 4.52 |
| EMDB accession code | EMD-9031 |
| Model refinement (PHENIX) | |
| CCvolume | 0.855 |
| CCmasked | 0.858 |
| RMSD bond length (Å) | 0.006 |
| RMSD bond angles (°) | 0.871 |
| Ramachandran plot | |
| Favored (%) | 95.46 |
| Allowed (%) | 4.54 |
| Outliers (%) | 0.00 |
| Clashscore | 3.28 |
| Avg B-factor (Å2) | 133.17 |
| PDB code | 6EDJ |
Data availability.
The final structure coordinates of wY132A were deposited in the Protein Data Bank (PDB) under accession number 6ECS. WHV capsid coordinates were deposited in the PDB under accession number 6EDJ.
ACKNOWLEDGMENTS
This research used resources of the Advanced Light Source, which is a Department of Energy Office of Science User Facility under contract no. DE-AC02-05CH11231. This work was supported by NIH grant R01-AI118933 to A.Z. and the Indiana Clinical and Translational Sciences Institute.
We thank Jay Nix for his assistance during X-ray data collection at beamline 4.2.2. We also acknowledge the Purdue cryo-EM facility, Thomas Klose, and Valerie Bowman for assistance collecting cryo-EM images. We also acknowledge Alex Kukreja for his preliminary experiments with WHV and antiviral molecules.
A.Z. has an interest in biotechnology companies that are developing capsid protein-directed antivirals.
REFERENCES
- 1.Gish RG, Given BD, Lai CL, Locarnini SA, Lau JY, Lewis DL, Schluep T. 2015. Chronic hepatitis B: virology, natural history, current management and a glimpse at future opportunities. Antiviral Res 121:47–58. doi: 10.1016/j.antiviral.2015.06.008. [DOI] [PubMed] [Google Scholar]
- 2.Zhao Z, Zlotnick A. 2018. Virus assembly antagonists from redesigned antibodies. Structure 26:1297–1299. doi: 10.1016/j.str.2018.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Summers J, Smolec JM, Snyder R. 1978. A virus similar to human hepatitis B virus associated with hepatitis and hepatoma in woodchucks. Proc Natl Acad Sci U S A 75:4533–4537. doi: 10.1073/pnas.75.9.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tennant BC, Gerin JL. 2001. The woodchuck model of hepatitis B virus infection. ILAR J 42:89–102. doi: 10.1093/ilar.42.2.89. [DOI] [PubMed] [Google Scholar]
- 5.Menne S, Cote PJ. 2007. The woodchuck as an animal model for pathogenesis and therapy of chronic hepatitis B virus infection. World J Gastroenterol 13:104–124. doi: 10.3748/wjg.v13.i1.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venkatakrishnan B, Zlotnick A. 2016. The structural biology of hepatitis B virus: form and function. Annu Rev Virol 3:429–451. doi: 10.1146/annurev-virology-110615-042238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kann M, Schmitz A, Rabe B. 2007. Intracellular transport of hepatitis B virus. World J Gastroenterol 13:39–47. doi: 10.3748/wjg.v13.i1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qian G, Jin F, Chang L, Yang Y, Peng H, Duan C. 2012. NIRF, a novel ubiquitin ligase, interacts with hepatitis B virus core protein and promotes its degradation. Biotechnol Lett 34:29–36. doi: 10.1007/s10529-011-0751-0. [DOI] [PubMed] [Google Scholar]
- 9.Wynne SA, Crowther RA, Leslie A. 1999. The crystal structure of the human hepatitis B virus capsid. Mol Cell 3:771–780. doi: 10.1016/S1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- 10.Schlicksup CJ, Wang JC, Francis S, Venkatakrishnan B, Turner WW, VanNieuwenhze M, Zlotnick A. 2018. Hepatitis B virus core protein allosteric modulators can distort and disrupt intact capsids. Elife 7:e31473. doi: 10.7554/eLife.31473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Packianathan C, Katen SP, Dann CE 3rd, Zlotnick A. 2010. Conformational changes in the hepatitis B virus core protein are consistent with a role for allostery in virus assembly. J Virol 84:1607–1615. doi: 10.1128/JVI.02033-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alexander CG, Jurgens MC, Shepherd DA, Freund SM, Ashcroft AE, Ferguson N. 2013. Thermodynamic origins of protein folding, allostery, and capsid formation in the human hepatitis B virus core protein. Proc Natl Acad Sci U S A 110:E2782–91. doi: 10.1073/pnas.1308846110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zlotnick A, Johnson JM, Wingfield PW, Stahl SJ, Endres D. 1999. A theoretical model successfully identifies features of hepatitis B virus capsid assembly. Biochemistry 38:14644–14652. doi: 10.1021/bi991611a. [DOI] [PubMed] [Google Scholar]
- 14.Lutomski CA, Lyktey NA, Zhao Z, Pierson EE, Zlotnick A, Jarrold MF. 2017. Hepatitis B virus capsid completion occurs through error correction. J Am Chem Soc 139:16932–16938. doi: 10.1021/jacs.7b09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kukreja AA, Wang JC, Pierson E, Keifer DZ, Selzer L, Tan Z, Dragnea B, Jarrold MF, Zlotnick A. 2014. Structurally similar woodchuck and human hepadnavirus core proteins have distinctly different temperature dependences of assembly. J Virol 88:14105–14115. doi: 10.1128/JVI.01840-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pierson EE, Keifer DZ, Kukreja AA, Wang JC, Zlotnick A, Jarrold MF. 2016. Charge detection mass spectrometry identifies preferred non-icosahedral polymorphs in the self-assembly of woodchuck hepatitis virus capsids. J Mol Biol 428:292–300. doi: 10.1016/j.jmb.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Z, Wang JC-Y, Gonzalez-Gutierrez G, Venkatakrishnan B, Zlotnick A. 2019. Structural differences between the woodchuck hepatitis virus core protein in dimer and capsid states indicate entropic and conformational regulation of assembly. bioRxiv 10.1101/512319. [DOI] [PMC free article] [PubMed]
- 18.Bourne CR, Katen SP, Fulz MR, Packianathan C, Zlotnick A. 2009. A mutant hepatitis B virus core protein mimics inhibitors of icosahedral capsid self-assembly. Biochemistry 48:1736–1742. doi: 10.1021/bi801814y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hilmer JK, Zlotnick A, Bothner B. 2008. Conformational equilibria and rates of localized motion within hepatitis B virus capsids. J Mol Biol 375:581–594. doi: 10.1016/j.jmb.2007.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. 2008. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure 16:673–683. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. 2012. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bourne C, Lee S, Venkataiah B, Lee A, Korba B, Finn MG, Zlotnick A. 2008. Small-molecule effectors of hepatitis B virus capsid assembly give insight into virus life cycle. J Virol 82:10262–10270. doi: 10.1128/JVI.01360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freund SMV, Johnson CM, Jaulent AM, Ferguson N. 2008. Moving towards high-resolution descriptions of the molecular interactions and structural rearrangements of the human hepatitis B core protein. J Mol Biol 384:1301–1313. doi: 10.1016/j.jmb.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 24.Hilser VJ, Thompson EB. 2007. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci U S A 104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Motlagh HN, Wrabl JO, Li J, Hilser VJ. 2014. The ensemble nature of allostery. Nature 508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hadden JA, Perilla JR, Schlicksup CJ, Venkatakrishnan B, Zlotnick A, Schulten K. 2018. All-atom molecular dynamics of the HBV capsid reveals insights into biological function and cryo-EM resolution limits. Elife 7:e32478. doi: 10.7554/eLife.32478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caspar DL. 1980. Movement and self-control in protein assemblies. Quasi-equivalence revisited. Biophys J 32:103–138. doi: 10.1016/S0006-3495(80)84929-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruan L, Hadden JA, Zlotnick A. 2018. Assembly properties of hepatitis B virus core protein mutants correlate with their resistance to assembly-directed antivirals. J Virol 92:e01082-18. doi: 10.1128/JVI.01082-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kabsch W. 2010. XDS. Acta Crystallogr D Biol Crystallogr 66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karplus PA, Diederichs K. 2015. Assessing and maximizing data quality in macromolecular crystallography. Curr Opin Struct Biol 34:60–68. doi: 10.1016/j.sbi.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. 2010. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. 2005. Automated molecular microscopy: the new Leginon system. J Struct Biol 151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 34.Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. 2007. EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Rohou A, Grigorieff N. 2015. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192:216–221. doi: 10.1016/j.jsb.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimanius D, Forsberg BO, Scheres SH, Lindahl E. 2016. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 5:e18722. doi: 10.7554/eLife.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38, 27-8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 38.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chan KY, Gumbart J, McGreevy R, Watermeyer JM, Sewell BT, Schulten K. 2011. Symmetry-restrained flexible fitting for symmetric EM maps. Structure 19:1211–1218. doi: 10.1016/j.str.2011.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 41.Kondylis P, Schlicksup CJ, Brunk NE, Zhou J, Zlotnick A, Jacobson SC. 2018. Competition between normative and drug-induced virus self-assembly observed with single-particle methods. J Am Chem Soc 141:1251–1260. doi: 10.1021/jacs.8b10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rosenthal PB, Henderson R. 2003. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol 333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The final structure coordinates of wY132A were deposited in the Protein Data Bank (PDB) under accession number 6ECS. WHV capsid coordinates were deposited in the PDB under accession number 6EDJ.