

The scaffolding proteins of several envelope viruses required for virion assembly form high-order lattice structures. However, information on the significance of their lattice formation in infected cells is limited. Herpesviruses acquire envelopes twice during their viral replication. The first envelop acquisition (primary envelopment) is one of the steps in the vesicle-mediated nucleocytoplasmic transport of nascent nucleocapsids, which is unique in biology. HSV-1 NEC, thought to be conserved in all members of the Herpesviridae family, is critical for primary envelopment and was shown to form a hexagonal lattice structure. Here, we investigated the significance of the interhexamer contact site for hexagonal lattice formation of the NEC in HSV-1-infected cells and present evidence suggesting that the lattice formation of the NEC hexamer has an important role in HSV-1 replication by regulating primary envelopment. Our results provide insights into the mechanisms of the envelopment of herpesviruses and other envelope viruses.
KEYWORDS: UL31, UL34, herpes simplex virus, nuclear membrane
ABSTRACT
During the nuclear export of nascent nucleocapsids of herpes simplex virus 1 (HSV-1), the nucleocapsids acquire a primary envelope by budding through the inner nuclear membrane into the perinuclear space between the inner and outer nuclear membranes. This unique budding process, termed primary envelopment, is initiated by the nuclear egress complex (NEC), composed of the HSV-1 UL31 and UL34 proteins. Earlier biochemical approaches have shown that the NEC has an intrinsic ability to vesiculate membranes through the formation of a hexagonal lattice structure. The significance of intrahexamer interactions of the NEC in the primary envelopment of HSV-1-infected cells has been reported. In contrast, the contribution of lattice formation of the NEC hexamer to primary envelopment in HSV-1-infected cells remains to be elucidated. Therefore, we constructed and characterized a recombinant HSV-1 strain carrying an amino acid substitution in a UL31 residue that is an interhexamer contact site for the lattice formation of the NEC hexamer. This mutation was reported to destabilize the interhexamer interactions of the HSV-1 NEC. Here, we demonstrate that the mutation causes the aberrant accumulation of nucleocapsids in the nucleus and reduces viral replication in Vero and HeLa cells. Thus, the ability of HSV-1 to form the hexagonal lattice structure of the NEC was linked to an increase in primary envelopment and viral replication. Our results suggest that the lattice formation of the NEC hexamer has an important role in HSV-1 replication by regulating primary envelopment.
IMPORTANCE The scaffolding proteins of several envelope viruses required for virion assembly form high-order lattice structures. However, information on the significance of their lattice formation in infected cells is limited. Herpesviruses acquire envelopes twice during their viral replication. The first envelop acquisition (primary envelopment) is one of the steps in the vesicle-mediated nucleocytoplasmic transport of nascent nucleocapsids, which is unique in biology. HSV-1 NEC, thought to be conserved in all members of the Herpesviridae family, is critical for primary envelopment and was shown to form a hexagonal lattice structure. Here, we investigated the significance of the interhexamer contact site for hexagonal lattice formation of the NEC in HSV-1-infected cells and present evidence suggesting that the lattice formation of the NEC hexamer has an important role in HSV-1 replication by regulating primary envelopment. Our results provide insights into the mechanisms of the envelopment of herpesviruses and other envelope viruses.
INTRODUCTION
Herpes simplex virus 1 (HSV-1), a member of the Alphaherpesvirinae subfamily of the Herpesviridae family, causes a variety of diseases in humans, including mucocutaneous diseases, keratitis, skin diseases, and encephalitis (1). Herpesviruses, including HSV-1, replicate their genomes and package the nascent progeny viral genomes into capsids in the nucleus, which are then translocated to the cytoplasm, where they acquire final envelopes (2, 3). During the nuclear export of the nascent nucleocapsids, these viruses utilize a unique nuclear egress mechanism termed vesicle-mediated nucleocytoplasmic transport. Thus, nucleocapsids acquire a primary envelope by budding through the inner nuclear membrane (INM) into the perinuclear space between the INM and the outer nuclear membrane (ONM) (primary envelopment), and enveloped nucleocapsids in the perinuclear space then fuse with the ONM to release nucleocapsids into the cytoplasm (deenvelopment) (2, 3). HSV-1 UL31 and UL34, both of which are thought to be conserved in all members in the Herpesviridae family, form a heterodimeric complex in HSV-1-infected cells. UL34 is anchored to the INM by a carboxyl-terminal transmembrane domain (4, 5), whereas UL31 localizes to the INM through interactions with UL34 (5). It was reported that the deletion of either UL31 or UL34 caused the mislocalization of other components in the complex, the aberrant accumulation of nucleocapsids in the nucleus, and significantly reduced viral replication (5). The complex, therefore, has a critical role in the nuclear egress of nucleocapsids and is termed the nuclear egress complex (NEC). For primary envelopment, nucleocapsids need to bypass the nuclear lamina to engage the INM, which is then deformed to wrap around the nucleocapsid. Vesiculation is finalized by the abscission of the INM (2, 3, 6). HSV-1 NEC and/or its homolog(s) in other herpesvirus(es) was reported to have multiple roles in these steps during primary envelopment, including the recruitment of nucleocapsids to the INM, deformation of the INM, and recruitment of host cellular factors, such as members of the protein kinase C (PKC) family and components of the ESCRT-III machinery, which are thought to dissolve the nuclear lamina by the phosphorylation of lamin proteins (7) and to mediate abscission of the INM (8), respectively (6, 9, 10).
The NECs of herpesviruses are considered to have an intrinsic ability to vesiculate membranes, based on observations that the ectopic expression of NECs in cells was sufficient to drive the vesiculation of the INM in the absence of any other viral proteins (11–13) and that the purified NECs themselves vesiculated synthetic liposomes in an in vitro budding assay (14, 15). The structure of herpesvirus NEC has been reported (16–20), and the recently solved structures of the NECs of HSV-1 and other herpesviruses (16–18) have shown that, in crystals, the HSV-1 NEC forms a hexagonal lattice resembling a honeycomb (Fig. 1) (16) and that similar hexagonal coats were observed on the inner surface of budding vesicles of the HSV-1 NEC in vitro as well as in cells expressing the NEC of another alphaherpesvirus, pseudorabies virus (PRV) (14, 21). Notably, alanine substitutions for aspartic and glutamic acids at residues 35 and 37 (D35A/E37A) in UL34, located in a hexameric interface of the HSV-1 NEC, blocked the formation of the hexagonal coats on membranes and abolished budding in vitro (Fig. 1) (14, 16). In agreement with this observation, these mutations also blocked primary envelopment in HSV-1-infected cells in a dominant negative manner (22). These findings suggested a crucial role for the hexagonal formation of the NEC in primary envelopment. In contrast, mutations in the interhexamer contacts (L142E, D286R, and E153R in UL31) designed to destabilize the interhexamer interactions of the HSV-1 NEC (Fig. 1) moderately reduced in vitro budding to 48% to 69% of the wild-type level (16), only implying the importance of the lattice formation of the NEC hexamer in budding in vitro. Furthermore, the significance of the lattice formation of the NEC hexamer in HSV-1-infected cells remains to be addressed. This study clarified these unsolved issues by investigating the effects of mutations in the interhexamer contacts of the NEC (L142E, D286R, and E153R in UL31) in HSV-1-infected cells.
FIG 1.

Mutagenesis design in UL31 based on the crystal structure of the HSV-1 NEC. (A) In the HSV-1 NEC structure (16), UL34 is shown in gray and UL31 is shown in green. The locations of amino acids mutated in this study are indicated in blue (L142, E153, and D286) or magenta (V247). Molecular graphics and analyses were performed with the PyMOL molecular graphics system, version 2.0.6 (Schrödinger, LLC). (B) Predicted model for hexagonal lattice formation of NEC from biochemical analysis of purified HSV-1 NEC (16). UL31 and UL34 form a stable NEC heterodimer. Individual NEC heterodimers then assemble into hexameric rings. Finally, the NEC hexamers are linked to each other and form a hexagonal lattice to vesiculate the membrane. The UL34-D25A/E37A and UL31-V247F mutants are expected to be defective for NEC hexamer formation. The UL31-L142E, -E153R, and -D286R mutants are expected to be defective for interhexamer interactions and the lattice formation of the hexamer of the NEC.
RESULTS
Effect of mutations in contacts between hexamers of HSV-1 NEC (L142E, D286R, and E153R in UL31) on complementation of a UL31-null mutant virus.
In an earlier study (16), mutations (L142E, D286R, and E153R) in UL31 were designed to destabilize the interhexamer interactions of NEC by disrupting salt bridges (E153R and D286R in UL31) or hydrophobic interactions (L142E in UL31) (Fig. 1). To investigate the effect of these mutations on the functionality of UL31 in HSV-1 replication, we tested the ability of expression plasmids encoding the wild type or UL31 mutants to complement the replication of a UL31-null mutant virus, YK720 (ΔUL31) (Fig. 2) (23). Vero cells transfected with an empty expression plasmid or expression plasmids for either the wild type or UL31 mutants were superinfected with YK720 (ΔUL31). At 24 h after infection, total virus from cell culture supernatants and infected cells was harvested and titrated on UL31-expressing cells. As shown in Fig. 3A, ectopic expression of wild-type UL31 in YK720 (ΔUL31)-infected cells increased progeny virus yields by 39.1-fold. In contrast, although the ectopic expression of UL31 mutants carrying either L142E, D286R, or E153R (UL31L142E, UL31D286R, or UL31E153R, respectively) in these infected cells moderately but consistently increased progeny virus yields (5.5- to 9.6-fold), the progeny virus yields of the UL31 mutants were significantly lower (4.1- to 7.1-fold) than those of wild-type UL31. The levels of accumulation of the wild-type and mutant UL31 proteins were comparable when these UL31 proteins were ectopically expressed in Vero cells (Fig. 3B). These results suggest that the UL31 amino acid residues (L142, D286, and E153) on the interhexamer contacts of the HSV-1 NEC are required for the proper functionality of UL31 during HSV-1 replication. Because the effects of the three mutations in UL31 in these assays were comparable, we therefore focused on one of the mutations, D286R in UL31, for further experiments.
FIG 2.
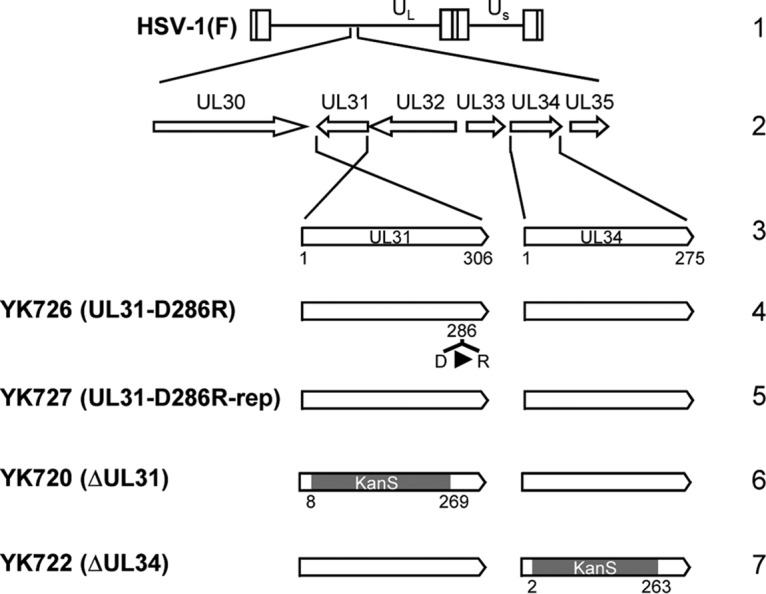
Schematic diagrams of the genomic structure of wild-type HSV-1(F) and the relevant domains of the recombinant viruses used in this study. Line 1, the wild-type HSV-1(F) genome (UL and US, unique long and unique short regions, respectively); line 2, domains of the UL30 to UL35 genes; line 3, domains of the UL31 and UL34 genes; lines 4, 5, and 6, recombinant viruses with mutations in the UL31 gene; line 7, recombinant virus with a mutation in the UL34 gene.
FIG 3.
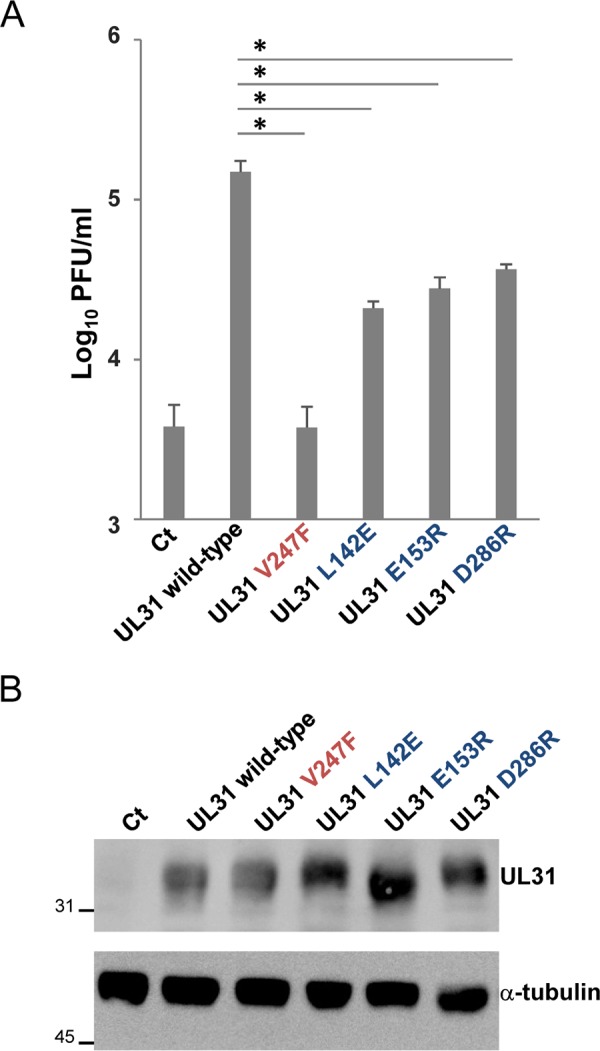
Complementation of the replication of the UL31-null mutant virus YK720 (ΔUL31) by expression plasmids for UL31 mutants. (A) Vero cells were transfected with a series of UL31-expressing plasmids or the control plasmid [pcDNA3.1/myc-His(−)A] for 4 h and then superinfected with YK720 (ΔUL31) at an MOI of 3 for 24 h. The progeny virus was collected and titrated on UL31-CV-1 cells. Data are the mean ± standard error from 4 independent experiments. *, P < 0.0001 (Tukey’s test). (B) Vero cells were transfected with the indicated plasmids for 28 h. The cells were analyzed by immunoblotting with anti-UL31 and anti-α-tubulin antibodies. The numbers to the left of the gels are molecular masses (in kilodaltons). Ct, control.
We also tested the effect of the V247F mutation in UL31 in these assays. Similar to UL34 D35/E37, UL31 V247 is located in a hexameric interface of the HSV-1 NEC, and therefore, an amino acid substitution at this residue was designed to disrupt the intrahexamer interactions of the HSV-1 NEC (Fig. 1) (16). Although the mutation was reported to significantly reduce budding in vitro (16), the effect of the mutation in the context of HSV-1 infection has not been tested. In agreement with an earlier report (22) showing that the UL34 mutant carrying the D35A/E37A mutations barely complemented the defect of virus production of a UL34-null mutant virus, the ectopic expression of the UL31 mutant carrying the V247F mutation was unable to increase virus progeny yields in YK720 (ΔUL31)-infected cells (Fig. 3A). This mutation in UL31 had no effect on the accumulation of UL31 proteins when ectopically expressed in Vero cells (Fig. 3B). These observations were in agreement with those of previous studies (16, 21), where hexamer formation of the NEC had a critical role in HSV-1-infected cells by regulating primary envelopment.
Construction and characterization of a recombinant virus harboring the D286R mutation in UL31.
To investigate the effects of the interhexamer contact site of the NEC in HSV-1-infected cells in more detail, we constructed a recombinant virus, YK726 (UL31-D286R), carrying the D286R mutation in UL31 and its repaired virus, YK727 (UL31-D286R-repair) (Fig. 2). As shown in Fig. 4, Vero or HeLa cells infected with YK726 (UL31-D286R) at a multiplicity of infection (MOI) of 3 for 18 h or an MOI of 10 for 24 h accumulated UL31 proteins and other HSV-1 proteins, UL34, ICP4, and ICP8, at levels similar to those for cells infected with wild-type HSV-1(F) or YK727 (UL31-D286R-repair). These results indicate that the D286R mutation in UL31 has no obvious effect on the accumulation of NEC components or other HSV-1 proteins (ICP4 and ICP8) in HSV-1-infected cells. We also examined the effect of the D286R mutation in UL31 on its interaction with UL34. As shown in Fig. 5, anti-UL34 antibody coprecipitated UL31 D286R with UL34 from lysates of Vero cells infected with YK726 (UL31-D286R) at an MOI of 3 for 18 h as efficiently as it coprecipitated wild-type UL31 with UL34 from cells infected with wild-type HSV-1(F). In these studies, the same antibody also precipitated UL47, which was reported to interact with UL34 and UL31 in HSV-1-infected cells (24), from the lysates of cells infected with YK726 (UL31-D286R) as efficiently as the antibody precipitated this HSV-1 protein from the lysates of cells infected with wild-type HSV-1(F) but barely precipitated UL31 from the lysates of cells infected with YK722 (ΔUL34) (Fig. 5). The anti-UL50 antibody did not precipitate UL31, UL34, or UL47 from the lysates of cells infected with wild-type HSV-1(F), and both the UL34 and UL50 antibodies did not precipitate UL31, UL34, or UL47 from the lysates of mock-infected cells (Fig. 5). These results suggest that the D286R mutation in UL31 does not affect NEC formation in HSV-1-infected cells and are in agreement with the findings of a previous study reporting that the mutation has no effect on dimerization between UL31 and UL34 in vitro (16).
FIG 4.

Effects of the D286R mutation in UL31 on the accumulation of viral proteins in HSV-1-infected cells. Vero (A) or HeLa (B) cells were mock infected or infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 3 for 18 h or at an MOI of 10 for 24 h, respectively. The infected cells were analyzed by immunoblotting with the indicated antibodies. The numbers to the left of the gels are molecular masses (in kilodaltons).
FIG 5.
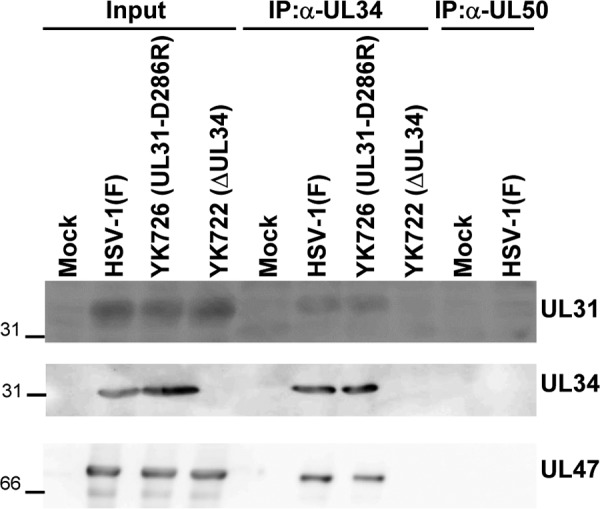
Effects of the D286R mutation in UL31 on its interaction with UL34 in HSV-1-infected cells. Vero cells were mock infected or infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK722 (ΔUL34) at an MOI of 3 for 18 h, harvested, immunoprecipitated (IP) with anti-UL34 or anti-UL50 antibody, and analyzed by immunoblotting with anti-UL34, anti-UL31, and anti-UL47 antibodies. The numbers to the left of the gels are molecular masses (in kilodaltons).
Effect of the D286R mutation in UL31 on HSV-1 replication.
We next examined progeny virus yields at various times after infection in Vero or HeLa cells infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 3 or 0.01 or at an MOI of 10 or 0.05, respectively. In agreement with the complementation assays (Fig. 3), YK726 (UL31-D286R) growth was less than that of wild-type HSV-1(F) and YK727 (UL31-D286R-repair) in Vero and HeLa cells infected at all the MOIs tested (Fig. 6 and 7). The progeny virus yields of YK726 (UL31-D286R) in Vero cells at an MOI of 3 at 24 h after infection and at an MOI of 0.01 at 48 h after infection were significantly less than those of wild-type HSV-1(F) and YK727 (UL31-D286R-repair) (7.5- and 6.9-fold, respectively, at 24 h after infection and 12.2- and 8.8-fold, respectively, at 48 h after infection) (Fig. 6 and Tables 1 and 2). Similarly, in HeLa cells, the progeny virus yields of YK726 (UL31-D286R) at an MOI of 10 at 36 h postinfection and at an MOI of 0.05 at 72 h postinfection were significantly less than those of wild-type HSV-1(F) and YK727 (UL31-D286R-repair) (7.4- and 15.1-fold, respectively, at 24 h postinfection and 34.5- and 53.4-fold, respectively, at 72 h postinfection) (Fig. 7 and Tables 3 and 4). To compare the effects of the UL31-D286R mutation with those of the UL31-null mutation on progeny viral yields in Vero and HeLa cells, these cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), YK727 (UL31-D286R-repair), or YK720 (ΔUL31), and progeny virus yields were determined. As shown in Fig. 8A and B, the progeny virus yields of YK726 (UL31-D286R) in Vero cells at an MOI of 3 at 24 h postinfection and at an MOI of 0.01 at 48 h postinfection were significantly higher than those of YK720 (ΔUL31) (185.7-fold at 24 h after infection and 314.2-fold at 48 h after infection, respectively) (Fig. 8A and B). Similarly, the progeny virus yields of YK726 (UL31-D286R) in HeLa cells at an MOI of 10 at 36 h postinfection and at an MOI of 0.05 at 72 h postinfection were significantly higher than those of YK720 (ΔUL31) (300.0-fold at 24 h postinfection and 189.4-fold at 72 h postinfection, respectively) (Fig. 8C and D). These results indicate that UL31 residue D286 is required for efficient HSV-1 replication in both Vero and HeLa cells.
FIG 6.

Effects of the D286R mutation in UL31 on HSV-1 growth in Vero cells. Vero cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 3 (A) or 0.01 (B), harvested at the indicated times after infection, and assayed on Vero cells. Each data point is the mean ± standard error from 3 independent experiments. *, P < 0.05 (Tukey’s test) for YK726 (UL31-D286R) versus HSV-1(F) and YK726 (UL31-D286R) versus YK727 (UL31-D286R-repair).
FIG 7.

Effects of the D286R mutation in UL31 on viral growth in HeLa cells. HeLa cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 10 (A) or 0.05 (B), harvested at the indicated times postinfection, and assayed on Vero cells. Each data point is the mean ± standard error from 3 independent experiments. *, P < 0.05 (Tukey’s test) for YK726 (UL31-D286R) versus HSV-1(F) and YK726 (UL31-D286R) versus YK727 (UL31-D286R-repair).
TABLE 1.
Effects of the D286R mutation in UL31 on HSV-1 growth in Vero cells when infected at an MOI of 3
| Virus | Expt no. and statistic | Viral yield (PFU/ml) |
|||
|---|---|---|---|---|---|
| 4 hpia | 10 hpi | 14 hpi | 24 hpi | ||
| HSV-1(F) | 1 | 1.0 × 104 | 4.0 × 106 | 1.4 × 107 | 2.5 × 107 |
| 2 | 2.5 × 104 | 5.0 × 106 | 9.0 × 106 | 1.5 × 107 | |
| 3 | 3.5 × 104 | 4.0 × 106 | 1.5 × 107 | 2.0 × 107 | |
| Mean | 2.3 × 104 | 4.3 × 106 | 1.3 × 107 | 2.0 × 107 | |
| SE | 7.3 × 103 | 3.3 × 105 | 1.8 × 106 | 2.9 × 106 | |
| YK726 (UL31-D286R) | 1 | 1.0 × 104 | 1.1 × 106 | 2.5 × 106 | 3.0 × 106 |
| 2 | 3.0 × 104 | 5.0 × 105 | 3.0 × 106 | 2.0 × 106 | |
| 3 | 4.0 × 104 | 1.3 × 106 | 2.5 × 106 | 3.0 × 106 | |
| Mean | 2.7 × 104 | 9.3 × 105 | 2.7 × 106 | 2.7 × 106 | |
| SE | 8.8 × 103 | 2.2 × 105 | 1.7 × 105 | 3.3 × 105 | |
| YK727 (UL31-D286R-rep) | 1 | 1.5 × 104 | 4.5 × 106 | 1.3 × 107 | 2.0 × 107 |
| 2 | 1.5 × 104 | 4.0 × 106 | 1.7 × 107 | 1.5 × 107 | |
| 3 | 2.0 × 104 | 6.5 × 106 | 1.4 × 107 | 2.0 × 107 | |
| Mean | 1.7 × 104 | 5.0 × 106 | 1.5 × 107 | 1.8 × 107 | |
| SE | 1.7 × 103 | 7.6 × 105 | 1.0 × 106 | 1.7 × 106 | |
hpi, hours postinfection.
TABLE 2.
Effects of the D286R mutation in UL31 on HSV-1 growth in Vero cells when infected at an MOI of 0.01
| Virus | Expt no. and statistic | Viral yields (PFU/ml) |
|||
|---|---|---|---|---|---|
| 10 hpia | 24 hpi | 36 hpi | 48 hpi | ||
| HSV-1(F) | 1 | 3.0 × 102 | 9.0 × 105 | 1.2 × 107 | 2.5 × 107 |
| 2 | 6.0 × 102 | 8.0 × 105 | 1.5 × 107 | 2.5 × 107 | |
| 3 | 1.5 × 102 | 1.2 × 106 | 9.5 × 106 | 3.0 × 107 | |
| Mean | 3.5 × 102 | 9.5 × 105 | 1.2 × 107 | 2.7 × 107 | |
| SE | 1.3 × 102 | 1.0 × 105 | 1.6 × 106 | 1.5 × 106 | |
| YK726 (UL31-D286R) | 1 | 3.0 × 101 | 2.5 × 104 | 2.5 × 105 | 3.0 × 106 |
| 2 | 2.0 × 101 | 2.5 × 104 | 8.0 × 105 | 1.5 × 106 | |
| 3 | 2.0 × 101 | 4.0 × 104 | 5.0 × 105 | 2.0 × 106 | |
| Mean | 2.3 × 101 | 3.0 × 104 | 5.2 × 105 | 2.2 × 106 | |
| SE | 3.3 × 100 | 5.0 × 103 | 1.6 × 105 | 4.4 × 105 | |
| YK727 (UL31-D286R-rep) | 1 | 7.0 × 102 | 1.4 × 106 | 1.6 × 107 | 1.8 × 107 |
| 2 | 8.0 × 102 | 7.0 × 105 | 8.5 × 106 | 2.7 × 107 | |
| 3 | 4.5 × 102 | 2.2 × 106 | 2.1 × 107 | 1.4 × 107 | |
| Mean | 6.5 × 102 | 1.4 × 106 | 1.5 × 107 | 1.9 × 107 | |
| SE | 1.0 × 102 | 4.2 × 105 | 3.5 × 106 | 3.8 × 106 | |
hpi, hours postinfection.
TABLE 3.
Effects of the D286R mutation in UL31 on HSV-1 growth in HeLa cells when infected at an MOI of 10
| Virus | Expt no. and statistic | Viral yield (PFU/ml) |
|||
|---|---|---|---|---|---|
| 6 hpia | 12 hpi | 24 hpi | 36 hpi | ||
| HSV-1(F) | 1 | 9.0 × 103 | 3.0 × 105 | 1.4 × 107 | 8.5 × 106 |
| 2 | 2.5 × 104 | 9.5 × 105 | 4.5 × 106 | 1.1 × 107 | |
| 3 | 2.0 × 104 | 3.5 × 105 | 2.5 × 106 | 1.4 × 107 | |
| Mean | 1.8 × 104 | 5.3 × 105 | 6.8 × 106 | 1.1 × 107 | |
| SE | 4.7 × 103 | 2.1 × 105 | 3.4 × 106 | 1.6 × 106 | |
| YK726 (UL31-D286R) | 1 | 2.3 × 104 | 3.5 × 104 | 8.0 × 105 | 2.0 × 106 |
| 2 | 3.0 × 104 | 6.0 × 104 | 6.0 × 105 | 1.3 × 106 | |
| 3 | 3.5 × 104 | 5.0 × 104 | 3.5 × 105 | 1.2 × 106 | |
| Mean | 2.9 × 104 | 4.8 × 104 | 5.8 × 105 | 1.5 × 106 | |
| SE | 3.6 × 103 | 7.3 × 103 | 1.3 × 105 | 2.6 × 105 | |
| YK727 (UL31-D286R-rep) | 1 | 3.0 × 104 | 2.1 × 106 | 1.0 × 107 | 2.8 × 107 |
| 2 | 1.7 × 104 | 3.5 × 105 | 7.0 × 106 | 1.6 × 107 | |
| 3 | 3.5 × 104 | 7.5 × 105 | 3.0 × 106 | 2.3 × 107 | |
| Mean | 2.7 × 104 | 1.1 × 106 | 6.7 × 106 | 2.2 × 107 | |
| SE | 5.5 × 103 | 5.1 × 105 | 2.0 × 106 | 3.5 × 106 | |
hpi, hours postinfection.
TABLE 4.
Effects of the D286R mutation in UL31 on HSV-1 growth in HeLa cells when infected at an MOI of 0.05
| Virus | Expt no. and statistic | Viral yields (PFU/ml) |
|||
|---|---|---|---|---|---|
| 12 hpia | 24 hpi | 48 hpi | 72 hpi | ||
| HSV-1(F) | 1 | 3.0 × 102 | 1.0 × 104 | 1.4 × 105 | 3.0 × 105 |
| 2 | 3.0 × 102 | 8.0 × 103 | 7.0 × 104 | 4.0 × 105 | |
| 3 | 8.0 × 102 | 2.0 × 104 | 1.5 × 105 | 3.0 × 105 | |
| Mean | 4.7 × 102 | 1.3 × 104 | 1.2 × 105 | 3.3 × 105 | |
| SE | 1.7 × 102 | 3.7 × 103 | 2.5 × 104 | 3.3 × 104 | |
| YK726 (UL31-D286R) | 1 | 5.0 × 101 | 2.0 × 103 | 3.5 × 104 | 8.0 × 103 |
| 2 | 3.5 × 102 | 2.0 × 103 | 1.5 × 103 | 1.0 × 103 | |
| 3 | 2.0 × 102 | 1.5 × 103 | 3.5 × 103 | 2.0 × 104 | |
| Mean | 2.0 × 102 | 1.8 × 103 | 1.3 × 104 | 9.7 × 103 | |
| SE | 8.7 × 101 | 1.7 × 102 | 1.1 × 104 | 5.5 × 103 | |
| YK727 (UL31-D286R-rep) | 1 | 4.0 × 102 | 8.0 × 103 | 1.5 × 105 | 7.0 × 105 |
| 2 | 6.0 × 102 | 1.5 × 104 | 5.5 × 104 | 5.0 × 105 | |
| 3 | 3.0 × 102 | 3.5 × 104 | 3.0 × 105 | 3.5 × 105 | |
| Mean | 4.3 × 102 | 1.9 × 104 | 1.7 × 105 | 5.2 × 105 | |
| SE | 8.8 × 101 | 8.1 × 103 | 7.1 × 104 | 1.0 × 105 | |
hpi, hours postinfection.
FIG 8.
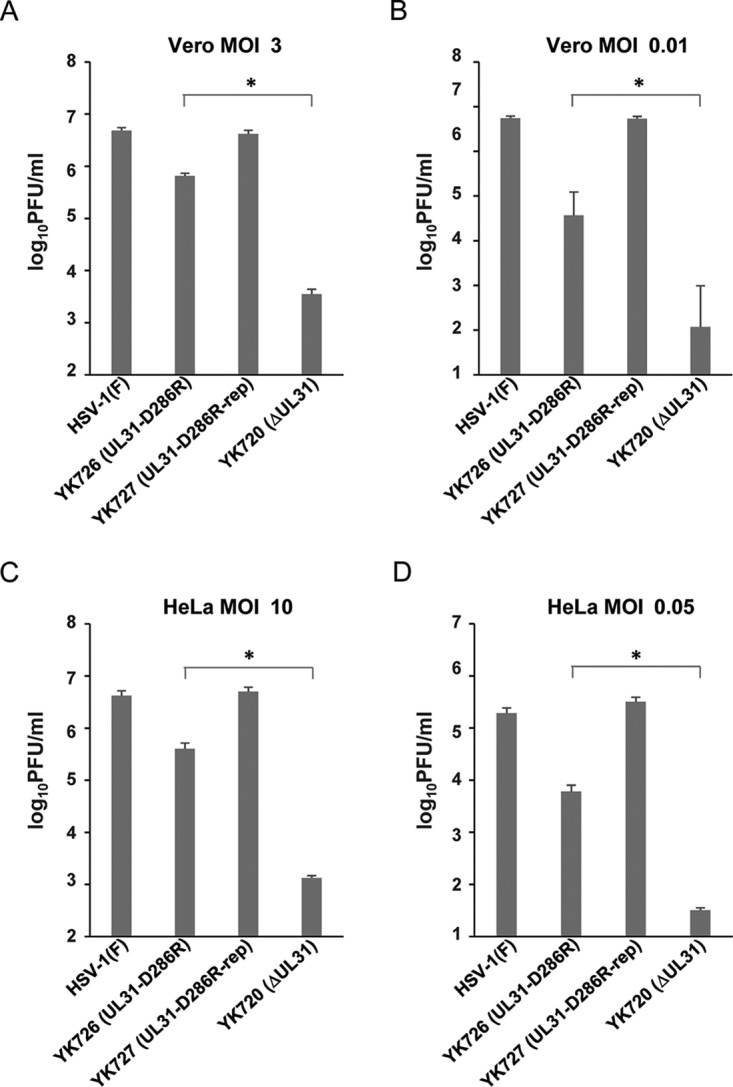
Comparison of effects of the D286R and null mutation in UL31 on viral growth in Vero and HeLa cells. (A and B) Vero cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), YK727 (UL31-D286R-repair), or YK720 (ΔUL31) at an MOI of 3 (A) or 0.01 (B) and harvested at 24 h (A) or 48 h (B) after infection. (C and D) HeLa cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), YK727 (UL31-D286R-repair), or YK720 (ΔUL31) at an MOI of 10 (C) or 0.05 (D) and harvested at 36 h (C) or 72 h (D) after infection. Cells and supernatants were titrated in UL31-CV-1 cells. Each data point is the mean ± standard error from 3 (A and B) or 4 (C and D) independent experiments. *, P < 0.05 (the unpaired Student's t test).
Effect of the D286R mutation in UL31 on localization of the NEC in HSV-1-infected cells.
To investigate the effect of the interhexamer contact site of the NEC on localization of the NEC in HSV-1-infected cells, Vero and HeLa cells infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 3 for 18 h or an MOI of 10 for 24 h, respectively, were examined by confocal microscopy. As reported previously (5), UL31 and UL34 were smoothly distributed and colocalized around the nuclear rim, and furthermore, UL34 was colocalized with lamin A/C, a marker for the inner nuclear membrane (8, 25) in Vero and HeLa cells infected with wild-type HSV-1(F) or YK727 (UL31-D286R-repair) (Fig. 9 and 10).
FIG 9.
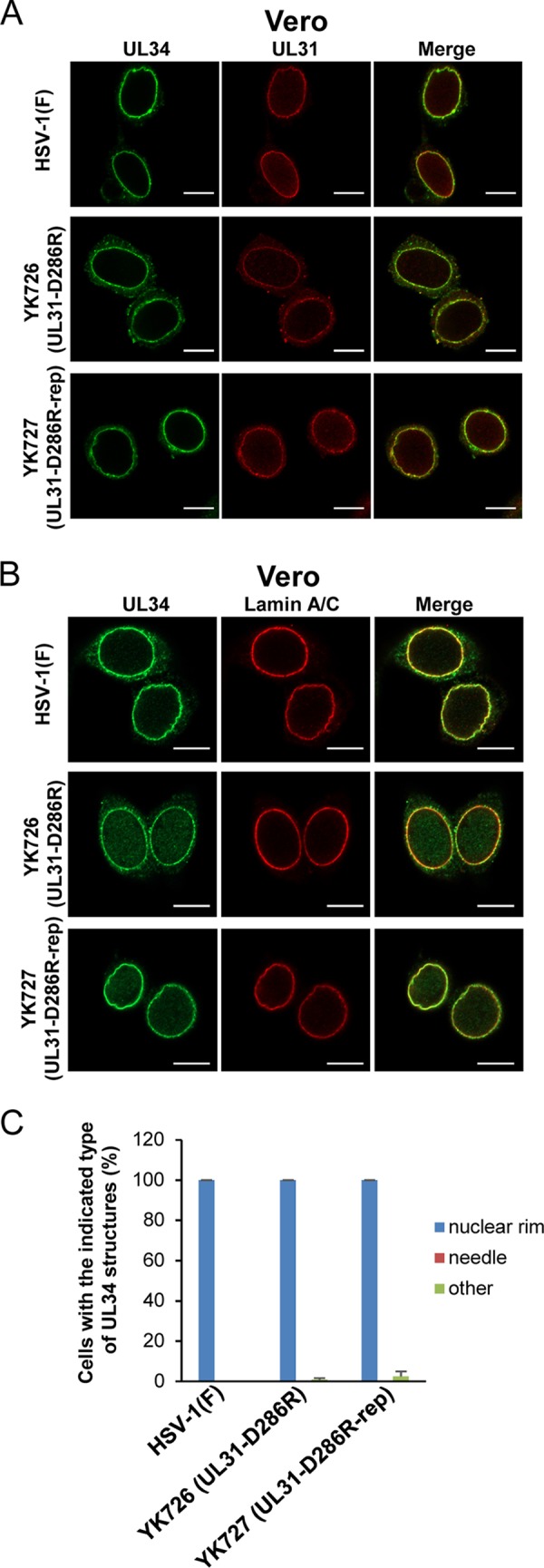
Effects of the D286R mutation in UL31 on the subcellular localization of UL31 and UL34 in HSV-1-infected Vero cells. (A and B) Vero cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 3, fixed at 18 h postinfection, permeabilized, stained with anti-UL31 and anti-UL34 antibodies (A) or anti-UL34 and lamin A/C antibodies (B), and examined by confocal microscopy. Bars, 10 μm. (C) Percentage of cells (of 30 to 40 cells in each experiment) showing a nuclear rim localization of UL34 (nuclear rim), needle-like structures of UL34 (needle), and other punctate structures of UL34 (other) in the experiment whose results are presented in panel B. Data are shown as the mean ± SEM from 3 independent experiments.
FIG 10.
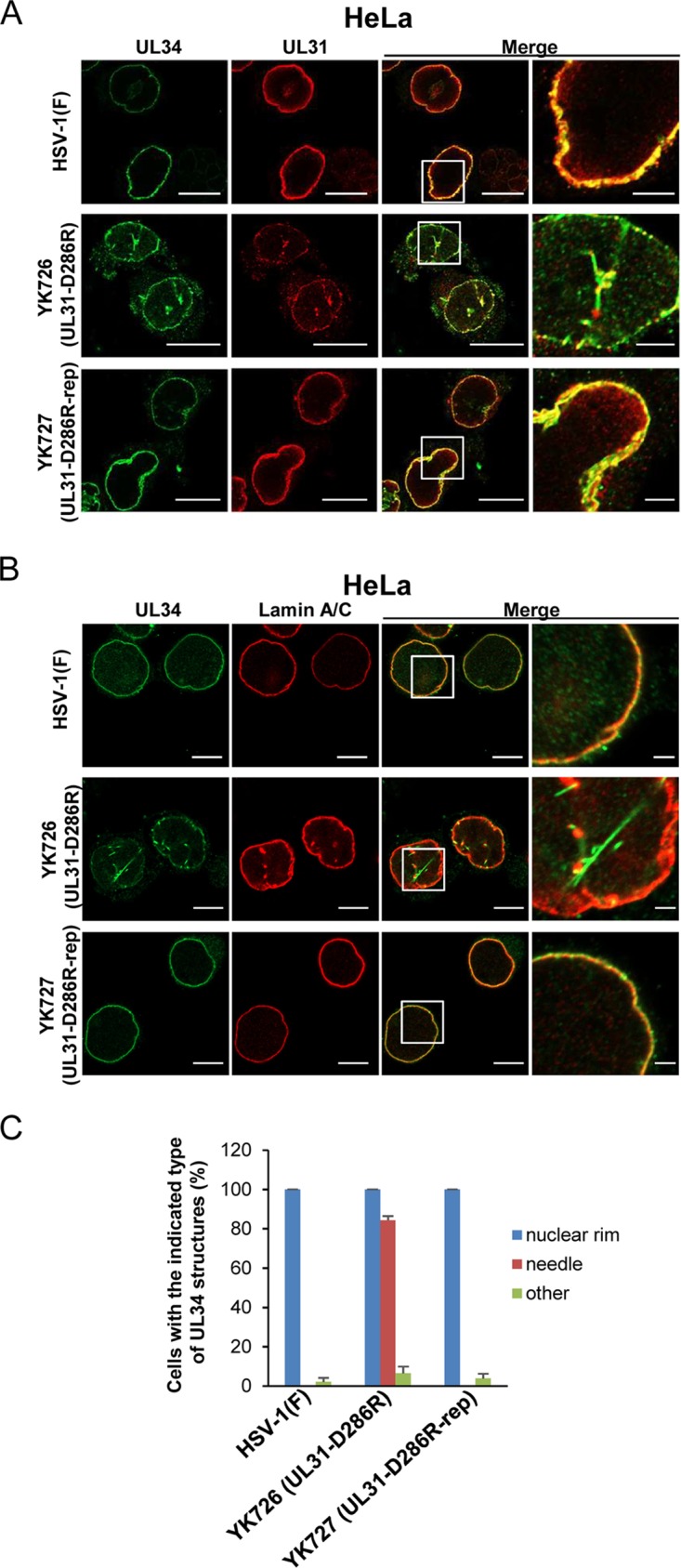
Effects of the D286R mutation in UL31 on the subcellular localization of UL31 and UL34 in HSV-1-infected HeLa cells. (A and B) HeLa cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 10, fixed at 24 h postinfection, permeabilized, stained with anti-UL31 and anti-UL34 antibodies (A) or anti-UL34 and lamin A/C antibodies (B), and examined by confocal microscopy. Each image in the far-right columns is the magnified image of the boxed area in the image to its left. Bars, 10 μm (first to third columns) and 2 μm (fourth column). (C) Percentage of cells (of 30 to 40 cells in each experiment) showing a nuclear rim localization of UL34 (nuclear rim), needle-like structures of UL34 (needle), and other punctate structures of UL34 (other) in the experiment whose results are presented in panel B. Data are shown as the mean ± SEM from 3 independent experiments.
In Vero cells infected with YK726 (UL31-D286R), the patterns of localization of UL31 and UL34 could not be differentiated from those in cells infected with wild-type HSV-1(F) or YK727 (UL31-D286R-repair) (Fig. 9). In contrast, in the majority (84.3%) of YK726 (UL31-D286R)-infected HeLa cells, aberrant needle-like and discrete punctate structures were observed at the nuclear rim and nucleus, where UL31 and UL34 were colocalized (Fig. 10A and C). The needle-like structures appeared to extend from the nuclear rim (Fig. 10). In most of these infected cells, lamin A/C was mislocalized to the punctate structures at the nuclear rim and the nucleus, but the needle-like structures of lamin A/C were barely detectable (Fig. 10B). However, the punctate structures of lamin A/C appeared to partially colocalize at either end of the needle-like structures of UL34 (Fig. 10B). The needle-like structures of UL34 were barely observed in Vero cells infected with YK726 (UL31-D286R) and in HeLa cells infected with wild-type HSV-1(F) or YK727 (UL31-D286R-repair) (Fig. 9C and 10C). These results indicate that the D286R mutation in UL31 induces the aberrant localization of the NEC as well as lamin A/C in HSV-1-infected cells in a cell type-dependent manner.
Effect of the D286R mutation in UL31 on HSV-1 morphogenesis.
To determine the step(s) at which the interhexamer contact site of the NEC acts during HSV-1 replication, we investigated viral morphogenesis by quantitating the number of virus particles at different morphogenetic stages by electron microscopy of Vero or HeLa cells infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 5 for 18 h or an MOI of 10 for 24 h, respectively. As shown in Table 5, capsids were markedly accumulated in the nucleus of Vero cells infected with YK726 (UL31-D286R). Thus, while 55.6% or 58.2% of virus particles were detected in the nucleus in Vero cells infected with wild-type HSV-1(F) or YK727 (UL31-D286R-repair), respectively, the fraction of virus particles that were capsids in the nucleus of cells infected with YK726 (UL31-D286R) was increased, and almost all (96.5%) virus particles were capsids in the nucleus in these infected cells. In contrast, in Vero cells infected with wild-type HSV-1(F) or YK727 (UL31-D286R-repair), 6.2% or 6.0%, respectively, of virus particles were primary enveloped virions in the perinuclear space (Table 5), but cells infected with YK726 (UL31-D286R) had no detectable primary enveloped virions in the perinuclear space (Table 5). Furthermore, whereas 38.2% or 35.9% of virus particles were in the cytoplasm and extracellular space in Vero cells infected with wild-type HSV-1(F) or YK727 (UL31-D286R-repair), respectively, the fraction of virus particles in the cytoplasm and extracellular space decreased to 3.5% in cells infected with YK726 (UL31-D286R) (Table 5). Similar results were also obtained with infected HeLa cells (Table 6). These results indicate that the D286R mutation in UL31 results in an increase in the fraction of capsids in the nucleus and a decrease in the fractions of virus particles in the perinuclear space, cytoplasm, and extracellular space.
TABLE 5.
Effect of the UL31-D286R mutation on the distribution of virus particles in infected Vero cells
| Virus | Mean % of virus particles in each morphogenetic stage ± SEa |
Total no. of particles/total no. of cells counted | ||||
|---|---|---|---|---|---|---|
| Nucleocapsids in nucleus | Enveloped virions in perinuclear space | Nucleocapsids in cytoplasm | Enveloped virions in cytoplasm | Extracellular enveloped virions | ||
| HSV-1(F) | 55.6 ± 9.0 (633) | 6.2 ± 2.4 (71) | 12.0 ± 4.3 (136) | 3.1 ± 0.8 (35) | 23.1 ± 3.5 (263) | 1,138/12 |
| YK726 (UL31-D286R) | 96.5 ± 18.3 (626) | 0 (0) | 1.5 ± 0.6 (10) | 0.6 ± 0.3 (4) | 1.4 ± 0.6 (9) | 649/12 |
| YK727 (UL31-D286R-repair) | 58.2 ± 8.8 (899) | 6.0 ± 2.3 (92) | 9.1 ± 2.6 (140) | 3.2 ± 0.9 (50) | 23.6 ± 3.0 (364) | 1,545/12 |
Numbers in parentheses are the numbers of virus particles.
TABLE 6.
Effect of UL31-D286R mutation on the distribution of virus particles in infected HeLa cells
| Virus | Mean % of virus particles in each morphogenetic stage ± SEa |
Total no. of particles/total no. of cells counted | ||||
|---|---|---|---|---|---|---|
| Nucleocapsids in nucleus | Enveloped virions in perinuclear space | Nucleocapsids in cytoplasm | Enveloped virions in cytoplasm | Extracellular enveloped virions | ||
| HSV-1(F) | 54.7 ± 10.5 (690) | 4.3 ± 2.0 (54) | 14.0 ± 3.6 (177) | 4.4 ± 1.5 (56) | 22.6 ± 4.3 (285) | 1,262/12 |
| YK726 (UL31-D286R) | 92.7 ± 18.8 (758) | 0 (0) | 4.8 ± 1.5 (39) | 0.5 ± 0.3 (4) | 2.1 ± 1.1 (17) | 818/12 |
| YK727 (UL31-D286R-repair) | 52.6 ± 12.1 (687) | 4.7 ± 2.3 (62) | 10.4 ± 2.4 (136) | 4.1 ± 1.6 (53) | 28.2 ± 7.6 (368) | 1,306/12 |
Numbers in parentheses are the numbers of virus particles.
Regarding Vero and HeLa cells infected with YK726 (UL31-D286R), although the capsids closely apposed at the INM were easily observed, enveloped and even partially enveloped virions in the perinuclear space were not detectable (Fig. 11 and 12), as shown in Tables 5 and 6. Notably, multiple capsids that accumulated in close association with the INM, accompanied by a large curvature of the nuclear membrane (NM), were observed in YK726 (UL31-D286R)-infected Vero cells but not in YK726 (UL31-D286R)-infected HeLa cells (Fig. 11 and 12). In contrast, rod-like structures were observed in the nucleus of YK726 (UL31-D286R)-infected HeLa cells but not in that of YK726 (UL31-D286R)-infected Vero cells (Fig. 11 and 12), in agreement with the cell type-specific mislocalization of the NEC and lamin A/C observed in these infected cells by confocal microscopy. The rod-like structures consisted of two straight membranes, which appeared to be derived from the INM, were coated with electron-dense materials, and contained some capsids (Fig. 12). The curvature of the NM associated with capsids and the membranous rod-like structures specifically were observed in YK726 (UL31-D286R)-infected Vero and HeLa cells, respectively, but were not detectable in these cells infected with wild-type HSV-1(F) (Fig. 11 and 12). These results indicate that the D286R mutation in UL31 had different effects on the integrity of the INM in HSV-1-infected cells in a cell type-dependent manner.
FIG 11.

Effects of the D286R mutation in UL31 on HSV-1 nuclear egress in Vero cells. Vero cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 3, fixed at 18 h postinfection, and examined by transmission electron microscopy. N, nucleus; C, cytoplasm; NM, nuclear membrane. Bars, 500 nm.
FIG 12.

Effects of the D286R mutation in UL31 on HSV-1 nuclear egress in HeLa cells. HeLa cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 10, fixed at 24 h postinfection, and examined by transmission electron microscopy. Arrowheads indicate membranous structures in the nucleus. N, nucleus; C, cytoplasm; NM, nuclear membrane. Bars, 500 nm.
DISCUSSION
The accumulation of nuclear capsids and the lack of primary enveloped virions in the perinuclear space due to the D286R mutation in UL31 assessed by quantitative electron microscopic analysis may reflect an imbalance between the rate of virion delivery into the perinuclear space and the rate of egress from this region. It seems likely that the D286R mutation in UL31 significantly decreased the rate of viral egress from the nucleoplasm but had no effect on the rate of egress from the perinuclear space. Based on these results, UL31 D286 appeared to be required for efficient primary envelopment during HSV-1 nuclear egress. Supporting this hypothesis, it was reported that viruses with mutations in the viral and cellular regulatory proteins for HSV-1 primary envelopment exhibited similar phenotypes in HSV-1 virion morphogenesis (23, 24, 26, 27). As described above, UL31 D286 is one of the interhexamer contacts in the NEC, and the D286R mutation in UL31 was predicted to destabilize the interhexamer interactions of the NEC based on its solved structure (16). Therefore, taken together with the observation here that the mutation significantly impaired viral replication in cell cultures, the current study findings suggest that the lattice formation of the NEC hexamer has an important role in HSV-1 replication by regulating primary envelopment, probably by promoting vesiculation of the INM, as previously proposed by in vitro budding analyses (14, 16, 21). This hypothesis is consistent with a model proposed for the virion assembly of human immunodeficiency virus (HIV), where the lattice formation of the hexamer of Gag proteins induces vesiculation of the membrane during virion assembly at the plasma membrane, based on in vitro analyses and analyses of infected cells similar to those performed in this and previous HSV-1 studies (28–30). Furthermore, it was reported that the scaffolding proteins required for virion assembly of several other envelope viruses, including the measles virus matrix, vaccinia virus D13, and influenza virus M1 proteins, form lattice structures in infected cells and/or in vitro (31–34). However, we cannot completely eliminate the possibility that the D286R mutation in UL31 has no effect on the formation of hexagonal lattice structures of the NEC in HSV-1-infected cells, unlike the observations in the in vitro budding analyses (14, 16, 21).
Of note, the D286R mutation in UL31 might not perturb the interhexamer interaction of the NEC but, rather, might cause the misfolding of UL31 to prevent the functional consequences of the NEC. Although we could not eliminate this possibility completely, it is unlikely, because UL31 D286 is a solvent-exposed residue, based on the solved structure of the HSV-1 NEC, which minimizes the chance of misfolding when mutated (16). Furthermore, the NEC carrying the D286R mutation in UL31 (NEC/UL31D286R) appeared to retain multiple properties of the wild-type NEC in HSV-1-infected cells. Thus, we present evidence in this study showing that the UL31 D286 mutant forms a complex with UL34 and UL47 as efficiently as wild-type UL31 does in HSV-1-infected Vero cells (Fig. 5) and that the NEC/UL31D286R is recruited normally to the NM in HSV-1-infected Vero and HeLa cells, although the mutant is additionally localized to the aberrant rod-like structures in the nucleus of infected HeLa cells (Fig. 9 and 10). In addition, electron micrographs of Vero and HeLa cells infected with YK726 (UL31-D286R) showed that multiple capsids were closely associated with the INM (Fig. 11 and 12), suggesting that the NEC/UL31D286R might recruit capsids to the INM in HSV-1-infected cells, which is an important function of the NEC. However, since we have not investigated the status of nuclear lamina in cells infected with YK726 (UL31-D286R), we could not completely exclude the possibility that a defect(s) in lamina disruption accounts for the phenotype in these infected cells.
To our surprise, the D286R mutation in UL31 reduced HSV-1 replication only modestly. This may be due to the incomplete abrogation of NEC lattice formation by the mutation. In agreement with this possibility, the reduction in progeny virus yield in cells infected by HSV-1 carrying the D286R mutation in UL31 was significantly less than that in cells infected by HSV-1 carrying the UL31-null mutation. Alternatively, a compensatory mutation(s) outside of UL31 gene might have a role(s) to play in diminishing the severity of the growth defect observed for the UL31 mutant HSV-1, as previously reported (35–37). We verified that the sequences of the genes encoding UL31 and those encoding HSV-1 proteins interacting with UL31, including UL34, UL47, ICP22, and Us3 in the YK726 (UL31-D286R) genome, were identical to those in the parental strain genome, except for the introduced mutation of interest (D286R in UL31). However, we could not completely eliminate the possibility that a secondary mutation(s) outside these genes might have an effect on the phenotypes of YK726 (UL31-D286R) in infected cells.
Of note, we observed cell type-specific effects of the D286R mutation in UL31 on the NM and localization of the NEC in this study. By electron microscopy, we observed rod-like structures consisting of two straight membranes derived from the INM and containing capsids between the membranes in the nucleus of HeLa cells infected with YK726 (UL31-D286R) but not in infected Vero cells. By confocal microscopy, we observed needle-like structures of the NEC, which appeared to be associated with lamin A/C at either end of the nucleus in HeLa cells infected with YK726 (UL31-D286R) but not in infected Vero cells. These observations suggest that the rod-like and needle-like structures of the NEC, observed by electron microscopy and confocal microscopy, respectively, were identical and that these aberrant membranous structures were therefore induced by the NEC/UL31D286R. Furthermore, by electron microscopy the NM curvature associated with multiple capsids was observed in Vero cells infected with YK726 (UL31-D286R) but not in infected HeLa cells. Because wild-type NEC is thought to have an intrinsic ability to deform the INM for primary envelopment, as described above, a mutant NEC might be unable to deform INMs correctly, thereby inducing aberrant rod-like structures or NM curvature as well as preventing primary envelopment. In a previous study (16), the purified NEC/UL31D286R could produce intraluminal vesicles within giant unilamellar vesicles in vitro, but the number of them was 69% of that of wild-type NEC. As that study did not report what happened within giant unilamellar vesicles, it would be interesting to analyze whether the mutant NEC produced needle-like structures in vitro, as observed in HSV-1-infected cells.
At present, why the effects of the mutant NEC on the NM differ in Vero and HeLa cells remains unknown. One possible explanation is that HSV-1-infected Vero and HeLa cells may differ regarding the plasticity of the NM and/or the rigidity of the nuclear lamina, which closely interacts with the INM and establishes mechanical support for the nucleus, including the NM (38). In agreement with this, it was reported that HSV-1 infection regulated lamin A/C expression in a manner dependent on the cell type (39, 40). Furthermore, it was shown that HSV-1 infection induced the hyperphosphorylation of emerin, an INM protein that has an important role in NM integrity, and that the levels of the HSV-1-induced phosphorylation of emerin were different between cell types (41). The phosphorylation of emerin was important for the regulation of NM integrity (42–44). Another possible explanation is that the activities of cellular factors involved in primary envelopment differ between cell types. It was reported that the Drosophila cellular large ribonucleoprotein complex utilizes vesicle-mediated nucleocytoplasmic transport (45), indicating that it can be performed solely by the intrinsic cellular machinery, and that herpesviruses may expropriate this transport mechanism. Indeed, several cellular proteins, including PKCs, p32, the CD98 heavy chain, β1 integrin, and ESCRT-III proteins, which promote the nuclear egress of HSV-1 nucleocapsids, have been reported (8, 26, 27, 46, 47). Among the cellular proteins, p32 was reported to function in HSV-1 primary envelopment in a manner dependent on cell type (27, 46, 48). Moreover, cell type-dependent cellular activities that compensate for the functions of the NEC have been suggested (49, 50). Thus, an UL34-null mutant PRV serially passaged in rabbit kidney cells grew as efficiently as wild-type PRV in these cells but not in rat neurons (49). Similarly, an UL31-null mutant HSV-1 produced virus titers in rabbit skin cells only 10- to 50-fold lower than those of a virus harboring a restored UL31 gene, whereas in Vero cells it produced virus titers more than 2,000-fold lower (50). Notably, in rabbit skin cells infected with UL31-null mutant HSV-1 but not in infected Vero cells, UL34 was localized at the NM, as observed with UL34 in wild-type HSV-1-infected cells, suggesting a cell type-specific activity that allows the correct localization of UL34 to the NM (50).
MATERIALS AND METHODS
Cells and viruses.
Vero, HeLa, UL31-CV-1, UL34-Vero, and rabbit skin cells were described previously (50–52). Wild-type HSV-1(F), UL31-null mutant virus YK720 (ΔUL31), and UL34-null mutant virus YK723 (ΔUL34) (Fig. 2) were described previously (23, 51, 52). YK720 (ΔUL31) and YK723 (ΔUL34) were propagated and titrated in UL31-CV-1 and UL34-Vero cells, respectively.
Plasmids.
The plasmids pcDNA3.1/myc-His(−)A (Invitrogen, Carlsbad, CA) and pcDNA3.1-UL31, used as a control and for the expression of HSV-1 UL31, respectively, were described previously (52). Plasmids pcDNA3.1-UL31-L142E, -E153R, -V247F, and -D286R were constructed by replacing UL31 codon L142, E153, V247, or D286 in pcDNA3.1-UL31 with glutamic acid, arginine, phenylalanine, or arginine, respectively, as previously described (53).
Complementation analysis.
Vero cells were transfected with a series of UL31 expression plasmids or control plasmid pcDNA3.1/myc-His(−)A using the Lipofectamine 2000 reagent (Invitrogen) for 4 h. These cells were superinfected with YK720 (ΔUL31) at an MOI of 3 for 24 h. Then, these cells and their supernatants were harvested and the progeny virus was titrated on UL31-CV-1 cells.
Mutagenesis of viral genomes and generation of recombinant HSV-1.
Recombinant virus YK726 (UL31-D286R) carrying the D286R mutation in UL31 (Fig. 2) was generated by the two-step Red-mediated mutagenesis procedure (54–56) using Escherichia coli GS1783 containing pYEbac102Cre and the following primers: 5′-GTGTCGGCCGCAGACATTTATTGTAAAATGAGGGACATCAGCTTCAGGGGGGGGCTCATGAGGATGACGACGATAAGTAGGG-3′ and 5′-TAGAGCCTTTGATACTCTAGCATGAGCCCCCCCCTGAAGCTGATGTCCCTCATTTTACAACAACCAATTAACCAATTCTGATTAG-3′. Recombinant virus YK727, in which the UL31-D286R mutation in YK726 was repaired (Fig. 2), was generated as described above, except that the following primers were used: 5′-GTGTCGGCCGCAGACATTTATTGTAAAATGAGGGACATCAGCTTCGACGGGGGGCTCATGAGGATGACGACGATAAGTAGGG-3′ and 5′-TAGAGCCTTTGATACTCTAGCATGAGCCCCCCGTCGAAGCTGATGTCCCTCATTTTACAACAACCAATTAACCAATTCTGATTAG-3′. To verify the sequences of the gene encoding UL31 and those encoding UL34, Us3, UL47, and ICP22, all of which were reported to interact with UL31 (5, 23, 24, 57), Vero cells were infected with YK726 (UL31-D286R) or its parental virus, YK304 (wild-type), at an MOI of 5, harvested at 24 h postinfection, and lysed in 500 μl lysis buffer (10 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1.5 mM MgCl2, 0.1% Nonidet P-40 [NP-40]). After a brief centrifugation, β-mercaptoethanol and EDTA were added to 400 μl of each supernatant to final concentrations of 50 mM and 1 mM, respectively. DNA was extracted with phenol-chloroform and precipitated with ethanol. The sequences of the genes encoding UL31, UL34, UL47, ICP22, and Us3 were determined. The sequences of these genes in the YK726 (UL31-D286R) genome were identical to the sequences of these genes in the YK304 (parental strain) genome, except for the introduced mutation of interest (D286R in UL31).
Antibodies.
Mouse monoclonal antibodies to α-tubulin (catalog number DM1A; Sigma), lamin A/C (catalog number 636; Santa Cruz Biotechnology), ICP4 (catalog number 58S; ATCC), and ICP8 (catalog number 10A3; Millipore) were used in this study. Mouse polyclonal antibody to UL31 and rabbit polyclonal antibodies to UL31, UL34, UL47, and UL50 were described previously (24, 58).
Immunoblotting.
Vero or HeLa cells were mock infected or infected with viruses at an MOI of 3 for 18 h or at an MOI of 10 for 24 h, respectively. The infected cells were analyzed by immunoblotting as previous reported (56, 59).
Immunofluorescence.
Vero or HeLa cells were infected with viruses at an MOI of 3 for 18 h or at an MOI of 10 for 24 h, respectively. The cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.1% Triton X-100 for 10 min, and blocked with phosphate-buffered saline containing 10% human serum for 30 min. These cells were reacted with the antibodies indicated above for 1 h at room temperature, followed by reaction with secondary antibodies conjugated to Alexa Fluor (Invitrogen) for 1 h at room temperature. Then, the cells were examined with a Zeiss LSM800 microscope (Zeiss).
Immunoprecipitation.
Vero cells were mock infected or infected with viruses at an MOI of 3 for 18 h. These cells were harvested and lysed with 0.5% NP-40 buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.5% NP-40) containing a protease inhibitor cocktail (Nacalai Tesque). After centrifugation, the supernatants were reacted with protein A-Sepharose beads (GE Healthcare Life Sciences) for 10 min at 4°C. Then, protein A-Sepharose beads were removed by centrifugation, and the supernatants were reacted with the same amount of anti-UL34 antibody or anti-UL50 antibody as a control for 30 min on ice. These samples were reacted with protein A-Sepharose beads with rotation for 2 h at 4°C. The precipitates were collected by a brief centrifugation, washed extensively with 0.1% NP-40 buffer, and analyzed by immunoblotting.
Electron microscopy.
Vero or HeLa cells were infected with wild-type HSV-1(F), YK726 (UL31-D286R), or YK727 (UL31-D286R-repair) at an MOI of 3 for 18 h or an MOI of 10 for 24 h, respectively. At the indicated time for each experiment, the cells were fixed with 2% paraformaldehyde and 1% glutaraldehyde overnight at 4°C, postfixed with 2% osmium tetroxide on ice for 2 h, washed with distilled water, dehydrated with an ethanol gradient series, incubated in propylene oxide, and embedded in an Epon 812 resin mixture. Then, these samples were sectioned on grids, stained with 2% uranyl acetate and Reynold's lead citrate, and examined by transmission electron microscopy as described previously (60). Representative cells were randomly chosen, and the number of virus particles at different morphogenetic stages was quantitated in Vero or HeLa cells.
ACKNOWLEDGMENTS
We thank Risa Abe and Hiroshi Sagara for their excellent technical assistance.
This study was supported by Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS), grants for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology of Japan (16H06433, 16H06429, 16K21723, 19H05286, and 19H05417), contract research funds from the Program of Japan Initiative for the Global Research Network on Infectious Diseases (J-GRID) (JP18fm0108006) and the Research Program on Emerging and Re-emerging Infectious Diseases (19fk018105h0001) from the Japan Agency for Medical Research and Development (AMED), a grant for the Joint Research Project of the Institute of Medical Science, the University of Tokyo, grants from the Takeda Science Foundation, GSK Japan Research grant 2017, and a grant from the Uehara Memorial Foundation.
REFERENCES
- 1.Roizman B, Knipe DM, Whitley RJ. 2013. Herpes simplex viruses, p 1823–1897. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed Lippincott-Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 2.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 3.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 4.Shiba C, Daikoku T, Goshima F, Takakuwa H, Yamauchi Y, Koiwai O, Nishiyama Y. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J Gen Virol 81:2397–2405. doi: 10.1099/0022-1317-81-10-2397. [DOI] [PubMed] [Google Scholar]
- 5.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hellberg T, Paßvogel L, Schulz KS, Klupp BG, Mettenleiter TC. 2016. Nuclear egress of herpesviruses: the prototypic vesicular nucleocytoplasmic transport. Adv Virus Res 94:81–140. doi: 10.1016/bs.aivir.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 7.Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80:494–504. doi: 10.1128/JVI.80.1.494-504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arii J, Watanabe M, Maeda F, Tokai-Nishizumi N, Chihara T, Miura M, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2018. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat Commun 9:3379. doi: 10.1038/s41467-018-05889-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marschall M, Muller YA, Diewald B, Sticht H, Milbradt J. 2017. The human cytomegalovirus nuclear egress complex unites multiple functions: recruitment of effectors, nuclear envelope rearrangement, and docking to nuclear capsids. Rev Med Virol 27:e1934. doi: 10.1002/rmv.1934. [DOI] [PubMed] [Google Scholar]
- 10.Lv Y, Zhou S, Gao S, Deng H. 2018. Remodeling of host membranes during herpesvirus assembly and egress. Protein Cell 10:315–326. doi: 10.1007/s13238-018-0577-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc Natl Acad Sci U S A 104:7241–7246. doi: 10.1073/pnas.0701757104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luitweiler EM, Henson BW, Pryce EN, Patel V, Coombs G, McCaffery JM, Desai PJ. 2013. Interactions of the Kaposi's sarcoma-associated herpesvirus nuclear egress complex: ORF69 is a potent factor for remodeling cellular membranes. J Virol 87:3915–3929. doi: 10.1128/JVI.03418-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J. 2012. Reconstitution of the Kaposi's sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J Virol 86:594–598. doi: 10.1128/JVI.05988-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. doi: 10.1038/ncomms5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lorenz M, Vollmer B, Unsay JD, Klupp BG, Garcia-Saez AJ, Mettenleiter TC, Antonin W. 2015. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J Biol Chem 290:6962–6974. doi: 10.1074/jbc.M114.627521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bigalke JM, Heldwein EE. 2015. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J 34:2921–2936. doi: 10.15252/embj.201592359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zeev-Ben-Mordehai T, Weberruß M, Lorenz M, Cheleski J, Hellberg T, Whittle C, El Omari K, Vasishtan D, Dent KC, Harlos K, Franzke K, Hagen C, Klupp BG, Antonin W, Mettenleiter TC, Grünewald K. 2015. Crystal structure of the herpesvirus nuclear egress complex provides insights into inner nuclear membrane remodeling. Cell Rep 13:2645–2652. doi: 10.1016/j.celrep.2015.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lye MF, Sharma M, El Omari K, Filman DJ, Schuermann JP, Hogle JM, Coen DM. 2015. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J 34:2937–2952. doi: 10.15252/embj.201592651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leigh KE, Sharma M, Mansueto MS, Boeszoermenyi A, Filman DJ, Hogle JM, Wagner G, Coen DM, Arthanari H. 2015. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc Natl Acad Sci U S A 112:9010–9015. doi: 10.1073/pnas.1511140112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walzer SA, Egerer-Sieber C, Sticht H, Sevvana M, Hohl K, Milbradt J, Muller YA, Marschall M. 2015. Crystal structure of the human cytomegalovirus pUL50-pUL53 core nuclear egress complex provides insight into a unique assembly scaffold for virus-host protein interactions. J Biol Chem 290:27452–27458. doi: 10.1074/jbc.C115.686527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hagen C, Dent KC, Zeev-Ben-Mordehai T, Grange M, Bosse JB, Whittle C, Klupp BG, Siebert CA, Vasishtan D, Bäuerlein FJB, Cheleski J, Werner S, Guttmann P, Rehbein S, Henzler K, Demmerle J, Adler B, Koszinowski U, Schermelleh L, Schneider G, Enquist LW, Plitzko JM, Mettenleiter TC, Grünewald K. 2015. Structural basis of vesicle formation at the inner nuclear membrane. Cell 163:1692–1701. doi: 10.1016/j.cell.2015.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roller RJ, Bjerke SL, Haugo AC, Hanson S. 2010. Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J Virol 84:3921–3934. doi: 10.1128/JVI.01638-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, Arii J, Kato A, Kawaguchi Y. 2014. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol 88:7445–7454. doi: 10.1128/JVI.01057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88:4657–4667. doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reynolds AE, Liang L, Baines JD. 2004. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes U(L)31 and U(L)34. J Virol 78:5564–5575. doi: 10.1128/JVI.78.11.5564-5575.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leach NR, Roller RJ. 2010. Significance of host cell kinases in herpes simplex virus type 1 egress and lamin-associated protein disassembly from the nuclear lamina. Virology 406:127–137. doi: 10.1016/j.virol.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Yang Y, Wu S, Pan S, Zhou C, Ma Y, Ru Y, Dong S, He B, Zhang C, Cao Y. 2014. p32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J Biol Chem 289:35795–35805. doi: 10.1074/jbc.M114.603845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carlson LA, Bai Y, Keane SC, Doudna JA, Hurley JH. 2016. Reconstitution of selective HIV-1 RNA packaging in vitro by membrane-bound Gag assemblies. Elife 5:e14663. doi: 10.7554/eLife.14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schur FK, Hagen WJ, Rumlova M, Ruml T, Muller B, Krausslich HG, Briggs JA. 2015. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 517:505–508. doi: 10.1038/nature13838. [DOI] [PubMed] [Google Scholar]
- 30.von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J Virol 77:5439–5450. doi: 10.1128/JVI.77.9.5439-5450.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szajner P, Weisberg AS, Lebowitz J, Heuser J, Moss B. 2005. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J Cell Biol 170:971–981. doi: 10.1083/jcb.200504026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hyun JK, Accurso C, Hijnen M, Schult P, Pettikiriarachchi A, Mitra AK, Coulibaly F. 2011. Membrane remodeling by the double-barrel scaffolding protein of poxvirus. PLoS Pathog 7:e1002239. doi: 10.1371/journal.ppat.1002239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Calder LJ, Wasilewski S, Berriman JA, Rosenthal PB. 2010. Structural organization of a filamentous influenza A virus. Proc Natl Acad Sci U S A 107:10685–10690. doi: 10.1073/pnas.1002123107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ke Z, Strauss JD, Hampton CM, Brindley MA, Dillard RS, Leon F, Lamb KM, Plemper RK, Wright ER. 2018. Promotion of virus assembly and organization by the measles virus matrix protein. Nat Commun 9:1736. doi: 10.1038/s41467-018-04058-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vu A, Poyzer C, Roller R. 2016. Extragenic suppression of a mutation in herpes simplex virus 1 UL34 that affects lamina disruption and nuclear egress. J Virol 90:10738–10751. doi: 10.1128/JVI.01544-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roller RJ, Haugo AC, Kopping NJ. 2011. Intragenic and extragenic suppression of a mutation in herpes simplex virus 1 UL34 that affects both nuclear envelope targeting and membrane budding. J Virol 85:11615–11625. doi: 10.1128/JVI.05730-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haugo AC, Szpara ML, Parsons L, Enquist LW, Roller RJ. 2011. Herpes simplex virus 1 pUL34 plays a critical role in cell-to-cell spread of virus in addition to its role in virus replication. J Virol 85:7203–7215. doi: 10.1128/JVI.00262-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prokocimer M, Davidovich M, Nissim-Rafinia M, Wiesel-Motiuk N, Bar DZ, Barkan R, Meshorer E, Gruenbaum Y. 2009. Nuclear lamins: key regulators of nuclear structure and activities. J Cell Mol Med 13:1059–1085. doi: 10.1111/j.1582-4934.2008.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simpson-Holley M, Baines J, Roller R, Knipe DM. 2004. Herpes simplex virus 1 U(L)31 and U(L)34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J Virol 78:5591–5600. doi: 10.1128/JVI.78.11.5591-5600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scott ES, O'Hare P. 2001. Fate of the inner nuclear membrane protein lamin B receptor and nuclear lamins in herpes simplex virus type 1 infection. J Virol 75:8818–8830. doi: 10.1128/JVI.75.18.8818-8830.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leach N, Bjerke SL, Christensen DK, Bouchard JM, Mou F, Park R, Baines J, Haraguchi T, Roller RJ. 2007. Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J Virol 81:10792–10803. doi: 10.1128/JVI.00196-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ellis JA, Craxton M, Yates JR, Kendrick-Jones J. 1998. Aberrant intracellular targeting and cell cycle-dependent phosphorylation of emerin contribute to the Emery-Dreifuss muscular dystrophy phenotype. J Cell Sci 111:781–792. [DOI] [PubMed] [Google Scholar]
- 43.Hirano Y, Segawa M, Ouchi FS, Yamakawa Y, Furukawa K, Takeyasu K, Horigome T. 2005. Dissociation of emerin from barrier-to-autointegration factor is regulated through mitotic phosphorylation of emerin in a Xenopus egg cell-free system. J Biol Chem 280:39925–39933. doi: 10.1074/jbc.M503214200. [DOI] [PubMed] [Google Scholar]
- 44.Lammerding J, Hsiao J, Schulze PC, Kozlov S, Stewart CL, Lee RT. 2005. Abnormal nuclear shape and impaired mechanotransduction in emerin-deficient cells. J Cell Biol 170:781–791. doi: 10.1083/jcb.200502148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Moore MJ, Budnik V. 2012. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149:832–846. doi: 10.1016/j.cell.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Z, Kato A, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 2015. Role of host cell p32 in herpes simplex virus 1 de-envelopment during viral nuclear egress. J Virol 89:8982–8998. doi: 10.1128/JVI.01220-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hirohata Y, Arii J, Liu Z, Shindo K, Oyama M, Kozuka-Hata H, Sagara H, Kato A, Kawaguchi Y. 2015. Herpes simplex virus 1 recruits CD98 heavy chain and beta1 integrin to the nuclear membrane for viral de-envelopment. J Virol 89:7799–7812. doi: 10.1128/JVI.00741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu S, Pan S, Zhang L, Baines J, Roller R, Ames J, Yang M, Wang J, Chen D, Liu Y, Zhang C, Cao Y, He B. 2016. Herpes simplex virus 1 induces phosphorylation and reorganization of lamin A/C through the gamma134.5 protein that facilitates nuclear egress. J Virol 90:10414–10422. doi: 10.1128/JVI.01392-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klupp BG, Granzow H, Mettenleiter TC. 2011. Nuclear envelope breakdown can substitute for primary envelopment-mediated nuclear egress of herpesviruses. J Virol 85:8285–8292. doi: 10.1128/JVI.00741-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang L, Tanaka M, Kawaguchi Y, Baines JD. 2004. Cell lines that support replication of a novel herpes simplex virus 1 UL31 deletion mutant can properly target UL34 protein to the nuclear rim in the absence of UL31. Virology 329:68–76. doi: 10.1016/j.virol.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 51.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maeda F, Arii J, Hirohata Y, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2017. Herpes simplex virus 1 UL34 protein regulates the global architecture of the endoplasmic reticulum in infected cells. J Virol 91:e00271-17. doi: 10.1128/JVI.00271-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Arii J, Shindo K, Koyanagi N, Kato A, Kawaguchi Y. 2016. Multiple roles of the cytoplasmic domain of herpes simplex virus 1 envelope glycoprotein D in infected cells. J Virol 90:10170–10181. doi: 10.1128/JVI.01396-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kato A, Oda S, Watanabe M, Oyama M, Kozuka-Hata H, Koyanagi N, Maruzuru Y, Arii J, Kawaguchi Y. 2018. Roles of the phosphorylation of herpes simplex virus 1 UL51 at a specific site in viral replication and pathogenicity. J Virol 92:e01035-18. doi: 10.1128/JVI.01035-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jarosinski KW, Margulis NG, Kamil JP, Spatz SJ, Nair VK, Osterrieder N. 2007. Horizontal transmission of Marek's disease virus requires US2, the UL13 protein kinase, and gC. J Virol 81:10575–10587. doi: 10.1128/JVI.01065-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kato A, Tsuda S, Liu Z, Kozuka-Hata H, Oyama M, Kawaguchi Y. 2014. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J Virol 88:655–666. doi: 10.1128/JVI.02710-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fujii H, Kato A, Mugitani M, Kashima Y, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 2014. The UL12 protein of herpes simplex virus 1 is regulated by tyrosine phosphorylation. J Virol 88:10624–10634. doi: 10.1128/JVI.01634-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83:11624–11634. doi: 10.1128/JVI.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]