

Restriction factors provide the first line of defense against retrovirus infection by posing several blocks to the viral replication cycle. SERINC5 is a novel restriction factor that strongly blocks HIV-1 entry, although it is counteracted by Nef. Currently, it is still unclear how HIV-1 entry is blocked by SERINC5. Notably, this entry block is dependent on viral Env proteins. Laboratory-adapted HIV-1 strains are sensitive, whereas primary isolates are highly resistant to SERINC5. Env proteins mediate virus entry via extensive conformational rearrangements from a closed ground state to a CD4-bound open state. We detected Env-Env associations and Env-SERINC5 interactions in live cells by a novel bimolecular fluorescence assay. We demonstrate that CD4 expression increases the Env sensitivity to SERINC5 and allows SERINC5 to dissociate the Env complex, suggesting that SERINC5 restriction is dependent on Env conformation. Our results provide new insights into the poorly defined Env-dependent SERINC5 antiviral mechanism.
KEYWORDS: CD4, entry, Env, HIV-1, Nef, restriction factor, SERINC5
ABSTRACT
Serine incorporator 5 (SERINC5) is a recently identified restriction factor that strongly blocks HIV-1 entry but is counteracted by Nef. Notably, tier 1 HIV-1 Env proteins are sensitive to SERINC5, whereas the majority of tier 2/3 Env proteins are resistant to SERINC5, when viruses are produced from CD4-negative cells and tested by a single-round replication assay. Here, we investigated the Env-dependent SERINC5 antiviral mechanism by comparing tier 1 NL Env with tier 3 AD8 Env proteins. We found that when NL and AD8 viruses were inoculated into CD4+ T cells and human peripheral blood mononuclear cells (PBMCs), the propagation of the two viruses was restricted to a similar level when Nef was not expressed. Using a bimolecular fluorescence complementation (BiFC) assay, we detected Env-Env association and Env-SERINC5 interactions. A much greater level of NL Env-SERINC5 interactions was detected than was AD8 Env-SERINC5 interactions, which was further validated by immunoprecipitation assays. In addition, SERINC5 dissociated the NL Env trimeric complex more effectively than the AD8 Env trimeric complex when CD4 was not expressed. However, when CD4 was expressed, SERINC5 became more capable of interacting with AD8 Env and dissociating its trimeric complex. Moreover, AD8 and several other tier 2/3 viruses produced in the presence of CD4 became sensitive to SERINC5 when measured by the single-round replication assay. Because tier 1 and tier 2/3 Env trimers have open and closed conformations, respectively, and CD4 opens the closed conformation, we conclude that SERINC5 selectively dissociates Env trimers with an open conformation to restrict HIV-1 replication.
IMPORTANCE Restriction factors provide the first line of defense against retrovirus infection by posing several blocks to the viral replication cycle. SERINC5 is a novel restriction factor that strongly blocks HIV-1 entry, although it is counteracted by Nef. Currently, it is still unclear how HIV-1 entry is blocked by SERINC5. Notably, this entry block is dependent on viral Env proteins. Laboratory-adapted HIV-1 strains are sensitive, whereas primary isolates are highly resistant to SERINC5. Env proteins mediate virus entry via extensive conformational rearrangements from a closed ground state to a CD4-bound open state. We detected Env-Env associations and Env-SERINC5 interactions in live cells by a novel bimolecular fluorescence assay. We demonstrate that CD4 expression increases the Env sensitivity to SERINC5 and allows SERINC5 to dissociate the Env complex, suggesting that SERINC5 restriction is dependent on Env conformation. Our results provide new insights into the poorly defined Env-dependent SERINC5 antiviral mechanism.
INTRODUCTION
The serine incorporator (SERINC) family has five members that are type III membrane proteins with 9 to 11 transmembrane domains (1). SERINC5 (Ser5) is a novel restriction factor that strongly inhibits HIV-1 replication during virus entry (2, 3). Although SERINC proteins share 31 to 58% sequence homology, only Ser5 and Ser3 have antiviral activity, albeit Ser3 activity is weak. Five Ser5 alternatively spliced isoforms are expressed in humans, but only the longest isoform is stably expressed and exhibits antiviral activity (4). Ser5 was initially identified as the counteractive target of HIV-1 Nef that increases viral infectivity (2, 3). Nef antagonism plays an important role in the prevalence of primate lentiviruses in their hosts (5). In addition to Nef, Ser5 is antagonized by murine leukemia virus (MLV) glycosylated Gag (glycoGag) (2, 3) and equine infectious anemia virus (EIAV) S2 proteins (6, 7). We recently reported that Nef, glycoGag, and S2 proteins all target Ser5 to the endosome/lysosome pathways for degradation (8–10). Thus, Ser5 is an important restriction factor for a wide range of retroviruses.
It is still unclear how Ser5 blocks virus entry. From its potential role in phospholipid metabolism on the plasma membrane, it was postulated that Ser5 modifies the lipid composition of the viral membrane to inhibit virus fusion. However, recent studies strongly argue against this model (6, 11, 12). Nevertheless, Ser5 inhibits both virus and cell fusion at the stage of fusion pore formation (11). In addition, Ser5 renders HIV-1 more sensitive to neutralizing antibodies (11, 13–15), suggesting that Ser5 modifies Env conformation likely by directly targeting the Env trimers.
Notably, Ser5 inhibitory activity is dependent on Env glycoproteins in a strain-specific manner when viruses are produced from CD4-negative cells and viral infectivity is measured by a single-round replication assay. Based on neutralization sensitivity, HIV-1 isolates are divided into three distinct tiers (16). Laboratory-adapted viruses that are sensitive to neutralizing antibodies belong to tier 1A/1B, whereas the primary circulating strains that are resistant to neutralizing antibodies belong to tier 2/3. Interestingly, tier 1 strains, such as NL, HXB2, SF162, and 89.2, are sensitive, whereas the majority of tier 2/3 primary isolates, such as AD8 and JRFL, are resistant to the Ser5 restriction (3, 11, 13). Thus, there is a concordance between Env sensitivities to neutralization and Ser5 restriction.
To explore the Env-dependent Ser5 antiviral mechanism, we compared the tier 1 NL Env and tier 3 AD8 Env sensitivities to Ser5 in the presence and absence of CD4 using viral and biochemical assays. We demonstrate that CD4 expression and Env conformation are critical for Ser5 restriction of HIV-1.
RESULTS
Comparison of NL and AD8 virus restriction by Ser5.
The HIV-1 CCR5-tropic AD8 strain belongs to tier 3 and is highly resistant to HIV-1 neutralizing antibodies (17). Like HIV-1 from a number of other tier 2/3 strains, AD8 viruses are also highly resistant to Ser5 when viruses are produced in the absence of CD4 and tested by a single-round HIV-1 replication assay (13). To confirm Ser5 resistance, wild-type (WT) and Nef-deficient (ΔN) HIV-1 viruses expressing AD8 and NL Env proteins were produced from 293T cells in the presence of increasing amounts of pBJ5-Ser5 vector. Viral infectivity was measured via single-round infection of TZM-bI cells. NL-ΔN infectivity was strongly reduced by Ser5 in a dose-dependent manner, whereas NL-WT infectivity was less effectively reduced due to antagonism of Ser5 by Nef (Fig. 1A). To understand whether Ser5 affects viral protein expression, NL-WT and NL-ΔN viruses were produced in the presence of increasing amounts of pCMV6-Ser5 vector, and viral protein expression was analyzed by Western blotting. Compared to the pBJ5 vector that has a weaker simian virus 40-human T cell leukemia virus (SV40-HTLV) hybrid promoter, pCMV6 vector has a very strong human cytomegalovirus (CMV) immediate early enhancer and promoter. Thus, to be consistent with the levels of Ser5 expression from pBJ5, ∼20-fold less pCMV6 was used to express Ser5, as we did previously (4). It was found that Ser5 did not reduce steady-state expression of any viral proteins, indicating that the inhibition was not due to a decrease in Env expression (Fig. 1B). In contrast, Ser5 reduced neither AD8-WT nor AD8-ΔN viral infectivity significantly. Thus, AD8 Env confers strong HIV resistance to Ser5, as does Nef under this experimental setting.
FIG 1.

NL and AD8 virus restriction by Ser5. (A) 293T cells were transfected with 2 μg pH22, pH22ΔN, pH-AD8, or pH-AD8ΔN in the presence of indicated amounts of pBJ5-Ser5-HA. An equal amount of viruses was used to infect TZM-bI cells, and viral infectivity was determined. The relative infectivity is shown, with the WT infectivity in the absence of Ser5 set as 100%. (B) 293T cells were transfected with 2 μg pNL4-3 or pNLΔN in the presence of increasing amounts of pCMV6-Ser5-FLAG. Viral proteins were detected by polyclonal human anti-HIV-1, and Ser5 was detected by anti-FLAG. Sizes of protein markers are indicated on the right. (C) A total of 2 × 105 J-LTRG-R5 and PHA-activated human PBMCs from three healthy donors were seeded into individual wells of a 96-well plate in 200 μl medium. Cells were inoculated with WT or ΔN mutant NL and AD8 viruses (10 ng p24Gag antigen). Half of the medium was exchanged every 2 days with fresh medium, and p24Gag antigen in the supernatants was analyzed by ELISA. (D) Cells expressing high levels of CD4 (CD4high) were isolated from J-TAg and J-TAg-KO cells that express low levels of CD4 (CD4low) and analyzed by flow cytometry. CD4high J-TAg and J-TAg-KO cells were infected with WT and ΔN mutant NL viruses, and viral growth curves were determined by p24Gag ELISA as in panel C. (A, C, and D) Error bars represent standard deviation (SDs) of the results from three independent experiments. ***, P < 0.001.
Next, we compared NL and AD8 virus replication in Jurkat cells and human peripheral blood mononuclear cells (PBMCs). Jurkat cells were chosen because of their high levels of Ser5 expression (2). To allow AD8 virus replication, a Jurkat cell line that naturally expresses CCR5 (J-LTRG-R5) was used (18). These cells were inoculated with WT and ΔN NL or AD8 viruses, and viral propagation was detected by p24Gag enzyme-linked immunosorbent assay (ELISA). Compared to their WT viruses, the replication of both NL-ΔN and AD8-ΔN viruses was suppressed to similar levels in J-LTRG-R5 and PBMCs (Fig. 1C). These results demonstrate that NL and AD8 viruses are restricted similarly when Nef is not expressed, indicating that NL and AD8 Env proteins are equally sensitive to restriction in these CD4+ cells.
It was reported that CD4 could inhibit HIV-1 replication in primary cells (19–21), likely by targeting glycoprotein 120 (gp120) (22, 23). We reported that Nef uses a similar mechanism to downregulate Ser5 and CD4 (10). To distinguish the roles of CD4 and Ser5 in HIV-1 restriction, we compared NL-WT and NL-ΔN virus replication in the Jurkat cell line J-TAg and its Ser5/Ser3 knockout (KO) cell line J-TAg-KO. We could not test AD8 viruses in these cells due to their lack of CCR5 expression. Because J-TAg cells express low levels of CD4, populations of cells expressing high levels of CD4 (CD4high) were isolated from J-TAg and J-TAg-KO cells by flow cytometry to ensure sufficient levels of CD4 expression for infection (Fig. 1D). NL-WT and NL-ΔN showed similar levels of replication in J-TAg-KO cells, while NL-ΔN replication was suppressed in J-TAg cells. Thus, Ser5, but not CD4, appears to be responsible for HIV-1 restriction in these CD4high J-TAg cells. Collectively, these results suggest that NL and AD8 viruses were very likely restricted by Ser5 in J-LTRG-R5 cells and human PBMCs.
Detection of Env-Env association by BiFC.
The HIV-1 Env is a trimer of gp120-gp41 heterodimers. In order to elucidate the Ser5 antiviral mechanism, we adopted a bimolecular fluorescence complementation (BiFC) assay to detect Env-Env association in live cells (24). We previously detected the specific Nef-Ser5, glycoGag-Ser5, and S2-Ser5 interactions using this assay (8–10). A hemagglutinin (HA)-tagged VN or FLAG-tagged VC gene fragment was linked to the last codon of NL and AD8 gp160, and these new gene fragments were inserted into a proviral vector, pH22, by replacing the entire gp160 and 5′ portion of nef (Fig. 2A). Thus, these new vectors express NL-VN, NL-VC, AD8-VN, or AD8-VC Env fusion proteins and all other HIV-1 proteins except Nef. When these vectors were transfected into 293T cells, similar levels of Env (gp160 and gp41) and Gag (p24 and p55) proteins were detected by Western blotting (Fig. 2B). Specific Env fusion protein expression was also confirmed by anti-HA or anti-FLAG. In addition, all these Env fusion proteins showed levels of surface gp120 expression similar to those of their WT counterparts. Thus, NL-VN, NL-VC, AD8-VN, and AD8-VC Env fusion proteins are properly expressed and processed from the pH22 vector.
FIG 2.

Detection of Env-Env association by BiFC. (A) A schematic of Env BiFC fusion proteins is presented. NL and AD8 Env proteins were fused to the fluorescent protein Venus N-terminal region from residues 2 to 173 (VN) that has an HA tag, or its C-terminal region from residues 154 to 238 (VC) that has a FLAG tag. These fusion fragments were inserted into pH22 proviral vector by replacing gp160 and partial nef. LTR, long terminal repeat. (B) These vectors were transfected into 293T cells, and the total Env and Gag expression levels were detected by Western blotting. Cell surface expression of gp120 was detected by human anti-HIV-1 broadly neutralizing antibody 35O22. Ctrl, control. (C) J-TAg cells were spinoculated with VSV-G pseudotyped viruses expressing NL-VN, NL-VC, AD8-VN, or AD8-VC, as indicated. After 48 h, cells were permeabilized and stained with Pacific Blue-conjugated anti-HA and APC-conjugated anti-FLAG antibodies. Fluorescent signals from these infected cells were detected by flow cytometry. (D) The NL-VN- and NL-VC-coinfected cells were observed by confocal microscopy. Two different levels of magnification were used to visualize single cells (scale bar = 5 μm) and multiple cells (scale bar = 10 μm).
Next, vesicular stomatitis virus G (VSV-G) pseudotyped viruses expressing NL-VN, AD8-VN, NL-VC, or AD8-VC fusion proteins were generated. J-TAg cells were infected with these viruses individually or pairwise and analyzed by flow cytometry. The expression of these Env fusion proteins was specifically detected by blue fluorescent anti-HA or red fluorescent anti-FLAG (Fig. 2C). These experiments also demonstrated that similar levels of NL-VN, AD8-VN, NL-VC, or AD8-VC were expressed in these cells. In addition, BiFC signals were only detected from pairwise infections, and importantly, the levels of BiFC-positive populations (11.5 to 13.4%) corresponded to the levels of the dual-positive populations (11.5 to 12.7%). Furthermore, the expression of NL-VN and NL-VC in cells was specifically detected by confocal microscopy and found to overlap the BiFC signals (Fig. 2D). Thus, we established a BiFC assay to detect Env-Env associations.
Detection of Env-Ser5 interaction by BiFC.
We created vectors expressing Ser5-VN and Ser5-VC fusion proteins in our previous studies (9, 10). Because Ser2 does not have any antiviral activity (4), we additionally created vectors expressing Ser2-VN and Ser2-VC fusion proteins as controls. The various expression vectors were transfected into 293T cells, and specific Ser5-VN, Ser5-VC, Ser2-VN, and Ser2-VC expression was confirmed by Western blotting using anti-HA and anti-FLAG (Fig. 3A). Notably, Ser2 fusion proteins were expressed at much higher levels than Ser5 fusion proteins. When their expression levels were titrated by flow cytometry, we found that Ser2 fusion proteins were expressed at ∼20-fold higher levels than were Ser5 fusion proteins (Fig. 3B). The expression levels of Ser2 and Ser5 fusion proteins were then normalized by reducing the amount of the Ser2 expression vectors during transfection, and similar levels of their expression were confirmed by Western blotting (Fig. 3C). When Ser2 and Ser5 fusion proteins were expressed with NL-VN or NL-VC under such conditions, the Ser5-NL Env pairs produced almost 4-fold more BiFC-positive cells than did the Ser2/NL Env pairs. These results suggest that Env proteins likely selectively interact with Ser5.
FIG 3.

Detection of Env-Ser5 interaction by BiFC. (A) 293T cells were transfected with 1 μg Ser2-VN, Ser2-VC, Ser5-VN, and Ser5-VC expression vectors, and their expression was determined by Western blotting using anti-HA and anti-FLAG. (B) Indicated amounts of Ser2-VN, Ser2-VC, Ser5-VN, and Ser5-VC expression vectors were transfected into 293T cells. After permeabilization, Ser2- and Ser5-VN were stained with Pacific Blue-conjugated anti-HA, and Ser2-VC and Ser5-VC were stained with APC-conjugated anti-FLAG. Fluorescent signals were detected by flow cytometry. (C) Indicated amounts of Ser2, Ser5, and NL Env BiFC fusion expression vectors were transfected into 293T cells individually or pairwise. Their BiFC fluorescent green signals were analyzed by flow cytometry. The levels of Env interaction with Ser2 and Ser5 were then calculated and presented as relative values, with the levels of the Ser5-VN/NL-VC pair set as 100%. The levels of Ser2-VN, Ser2-VC, Ser5-VN, and Ser5-VC expression were determined by Western blotting. Error bars represent SDs of the results from three independent experiments. (D) Indicated Ser5-Env BiFC pairs were expressed in 293T cells. Cells were stained with Alexa Fluor-647-conjugated anti-HA or Pacific Blue-conjugated anti-FLAG. Fluorescent signals were visualized by confocal microscopy (scale bar = 5 μm). (E) Indicated Env-Env and Ser5-Env BiFC pairs were expressed with a mCherry fusion protein expressing the N-terminal 14 residues of the ER protein calnexin (CNX). Their colocalization was determined by confocal microscopy (scale bar = 5 μm). DAPI, 4′,6-diamidino-2-phenylindole.
The Ser5-VN/NL-VC and NL-VN/Ser5-VC BiFC signals were further analyzed by confocal microscopy after immunofluorescent staining. These signals colocalized with their corresponding fusion proteins, which were distributed on the plasma membrane and in the cytoplasm (Fig. 3D). To understand where the Ser5-Env interaction occurs, we expressed NL-VN/NL-VC and Ser5-VN/NL-VC pairs with a fluorescent endoplasmic reticulum (ER) marker, CNX-N-14. As expected, the Env-Env complex colocalized with this marker, confirming that Env is oligomerized in the ER (Fig. 3E). The Ser5-Env complex also colocalized with this marker, suggesting that the Ser5-Env interaction could also occur in the ER. Thus, we established a BIFC assay to detect Env-Ser5 interaction.
Comparison of NL and AD8 Env interaction with Ser5.
Ser5-VN and Ser5-VC fusion proteins were expressed with NL- and AD8-VN or VC fusion proteins in 293T cells, and their interactions were compared by flow cytometry. Both Ser5-VN/NL-VC and NL-VN/Ser5-VC pairs produced almost 2-fold more BiFC-positive cells than did the Ser5-VN/AD8-VC and AD8-VN/Ser5-VC pairs (Fig. 4A). Because NL-VN, NL-VC, AD8-VN, and AD8-VC were expressed at a similar level (Fig. 2B and C), these results suggest that Ser5 likely interacts with NL Env to a greater extent than it does with AD8 Env.
FIG 4.

NL and AD8 Env interaction with Ser5. (A) Indicated Ser5-Env BiFC pairs were expressed in 293T cells and analyzed by flow cytometry. The NL and AD8 Env interactions with Ser5 were then calculated and presented as relative values, with the levels of the Ser5-VN/NL-VC pair set as 100%. (B) 293T cells were transfected with 1 μg pH22ΔN or pH-AD8ΔN in the presence of 25 ng pCMV6-Ser2-FLAG or pCMV6-Ser5-FLAG. Proteins were pulled down by anti-FLAG and analyzed by Western blotting. Pulled-down Env proteins were quantified by ImageJ, and their relative values are shown on the bottom, with the levels of NL Env set as 100%. (A and B) Error bars represent SDs of the results from three independent experiments. **, P < 0.01.
To validate this conclusion, we set up an immunoprecipitation (IP) assay to further compare these interactions. NL and AD8 Env proteins were expressed with FLAG-tagged Ser2 or Ser5, and proteins were pulled down by anti-FLAG. Env (gp160 and gp41) proteins were expressed at comparable levels, and Ser2 was expressed at higher levels than was Ser5 in the cell lysate (Fig. 4B). In addition, Ser2 and Ser5 were pulled down at comparable levels. Both Ser2 and Ser5 in these pulldown samples showed a molecular mass higher than 150 kDa, which was caused by a boiling procedure during IP that triggered aggressive Ser2 and Ser5 protein aggregation. Nonetheless, Ser5 pulled down ∼3-fold more NL than AD8 Env proteins, and Ser2 barely pulled down any Env proteins. Thus, results from this IP experiment support those from the BiFC measurement. Collectively, these results demonstrate that Ser5 but not Ser2 interacts with Env, and importantly, that the Ser5-NL Env interaction occurs to a much greater extent than the Ser5-AD8 Env interaction.
Dissociation of NL but not AD8 Env trimeric complex by Ser5.
To understand whether Ser5 affects Env-Env associations, we compared NL Env-Env and AD8 Env-Env associations in J-TAg and J-TAg-KO cells. Initially, we compared HIV-1 receptor expression levels in J-TAg and J-TAg-KO cells with J-LTRG-R5 cells. J-LTRG-R5 cells expressed not only high levels of CCR5, but also much higher levels of CXCR4 and CD4 than did J-TAg and J-TAg-KO cells (Fig. 5A). Next, we compared the infectivity of NL-ΔN or AD8-ΔN viruses released from J-TAg and J-TAg-KO cells. NL viruses were at least 20-fold more infectious when produced from J-TAg-KO than from J-TAg cells, and AD8 viruses were equally infectious from the two cell lines (Fig. 5B). Thus, we confirmed that Ser5 does not restrict AD8 viruses in these J-TAg cells as reported previously (13).
FIG 5.

Dissociation of NL Env trimers by Ser5. (A) CCR5, CXCR4, and CD4 expression on the surface of J-TAg, J-TAg-KO, and J-LTRG-R5 cells was determined by flow cytometry. (B) J-TAg and J-TAg-KO cells were infected with VSV-G pseudotyped NL-VN/NL-VC or AD8-VN/AD8-VC viruses by spinoculation. After normalization by p24Gag ELISA, the infectivity of released virions was determined in TZM-bI cells. Infectivity was presented as relative values, with the ΔN mutant viral infectivity in J-TAg-KO set as 100%. (C) J-TAg, J-TAg-KO, and J-TAg-KO-Ser5 cells were spinoculated with VSV-G pseudotyped viruses expressing NL-VN/NL-VC or AD8-VN/AD8-VC, and BiFC signals were detected by flow cytometry. Levels of Env-Env association were calculated and presented as relative values, with the levels of Env in J-TAg-KO set as 100%. (B and C) Error bars represent SDs of the results from three independent experiments. *, P < 0.05; **, P < 0.01.
Next, J-TAg and J-TAg-KO cells were infected with VSV-G pseudotyped NL-VN/NL-VC or AD8-VN/AD8-VC viruses, and Env-Env association was determined by flow cytometry. Notably, the NL Env-Env BiFC-positive signals were strongly reduced from 39.5% in J-TAg-KO cells to 15.9% in J-TAg cells, whereas the levels of AD8 Env-Env BiFC-positive signals were similar in the two cell lines (Fig. 5C). Because J-TAg-KO cells lack both Ser3 and Ser5 genes, we also did this experiment in J-TAg-KO-Ser5 cells where the Ser5 gene was reconstituted (4). The levels of NL Env-Env BiFC-positive signals were 24.1% in J-TAg-KO-Ser5 cells, and the levels of AD8 Env-Env BiFC-positive signals in this cell line were similar to those in J-TAg and J-TAg-KO cells. These results suggest that Ser5 likely selectively dissociates the NL but not the AD8 Env trimeric complex in these Jurkat cells expressing low levels of CD4. In addition, it cannot be excluded that the Ser3 activity in J-TAg cells contributed to NL restriction.
Increased sensitivity of tier 2/3 viruses to Ser5 mediated by CD4.
To confirm the Ser5 effect on Env-Env association, we measured the association in the presence of Ser5 in 293T cells. NL- or AD8-VN/VC pairs were expressed with increasing Ser5 or Ser2 expression, and Env-Env associations were analyzed by flow cytometry. Ser5 reduced NL Env-Env associations in a dose-dependent manner but had little effect on AD8 Env-Env association; Ser2 did not have any effect (Fig. 6, left). These results confirm Ser5 activity and AD8 Env resistance to this activity.
FIG 6.

Dissociation of AD8 Env trimers by Ser5 in the presence of CD4. NL and AD8 Env BiFC fusion proteins were expressed pairwise in 293T cells with increasing amounts of pBJ5-Ser5-HA or pBJ6-Ser2-HA in the presence or absence of 1 μg pCMV6-CD4-FLAG. Levels of Env-Env association were detected by flow cytometry and calculated, with the levels in the absence of SERINC proteins set as 100%. Error bars represent SDs of the results from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We observed that AD8 virus restriction differed between the J-TAg and J-LTRG-R5 cell lines. AD8 viral replication was restricted in J-LTRG-R5 cells (Fig. 1C), but its Env-Env association was not restricted in J-TAg cells (Fig. 5B and C). Because J-LTRG-R5 cells express much higher levels of CD4 than J-TAg cells (Fig. 5A), we wondered whether the restriction is dependent on the level of CD4 expression. Thus, we again tested the effect of Ser5 on Env-Env association in 293T cells but in the presence of CD4 expression. We found that Ser5 reduced not only NL, but also AD8 Env-Env association to similar levels in a dose-dependent manner, and that Ser2 did not have any effect (Fig. 6, right). Thus, CD4 expression conferred AD8 Env sensitivity to Ser5. In addition, CD4 alone could also reduce the Env-Env association likely caused by CD4-Env interactions.
To confirm the CD4 effect on Env sensitivity to Ser5, we tested how CD4 affects the Ser5 interaction with AD8 Env. Ser5 and AD8 Env BiFC pairs were expressed in the presence of increasing amounts of a CD4 expression vector (0, 0.125, 0.25, 0.5, 1 μg) in 293T cells, and the Ser5-Env interaction was analyzed by flow cytometry. Initially, we compared the CD4 levels in our 293T cells to those in primary T cells by flow cytometry. CD4 was expressed at much higher levels in primary T cells than in 293T cells (Fig. 7A), indicating that the amount of CD4 used in our experiment did not exceed physiological levels. Under this experimental setting, CD4 increased the AD8 Env-Ser5 interaction up to 4-fold in a dose-dependent manner (Fig. 7B). Thus, CD4 enhances the AD8 Env-Ser5 interaction, supporting the hypothesis that CD4 increases AD8 virus sensitivity to Ser5.
FIG 7.

Increase in AD8 Env-Ser5 interaction by CD4. (A) 293T cells were transfected with indicated amounts of pCMV6-CD4-FLAG and stained with APC-conjugated anti-human CD4. The levels of CD4 surface expression were analyzed by flow cytometry. In addition, human PBMCs were stained with Pacific Blue-conjugated anti-human CD3 and APC-conjugated anti-human CD4. The CD3+ cell population was gated, and their CD4 surface expression was analyzed by flow cytometry. (B) AD8 Env and Ser5 BiFC pairs were expressed with increasing amounts of pCMV6-CD4-FLAG in 293T cells. Levels of Env-Ser5 interaction were detected by flow cytometry and calculated, with the levels in the presence of 1 μg pCMV6-CD4-FLAG set as 100%. Error bars represent SDs of the results from three independent experiments.
To further confirm the CD4 effect on Env sensitivity to Ser5, we tested if Ser5 affects AD8 viral infectivity when viruses are produced in the presence of CD4 using the single-round replication assay. Ser5 reduced both NL and AD8 viral infectivity to similar levels in a dose-dependent manner (Fig. 8A). We also tested several other Ser5-resistant CCR5-tropic HIV-1 strains derived from tier 2/3 transmitted founder viruses, including YU2, THRO, WITO, and RHPA (13). Although they were resistant to Ser5 when produced in the absence of CD4, they all became sensitive to Ser5 when produced in the presence of CD4 (Fig. 8B). Collectively, these results demonstrate that CD4 expression in viral producer cells increases Env sensitivity to Ser5.
FIG 8.
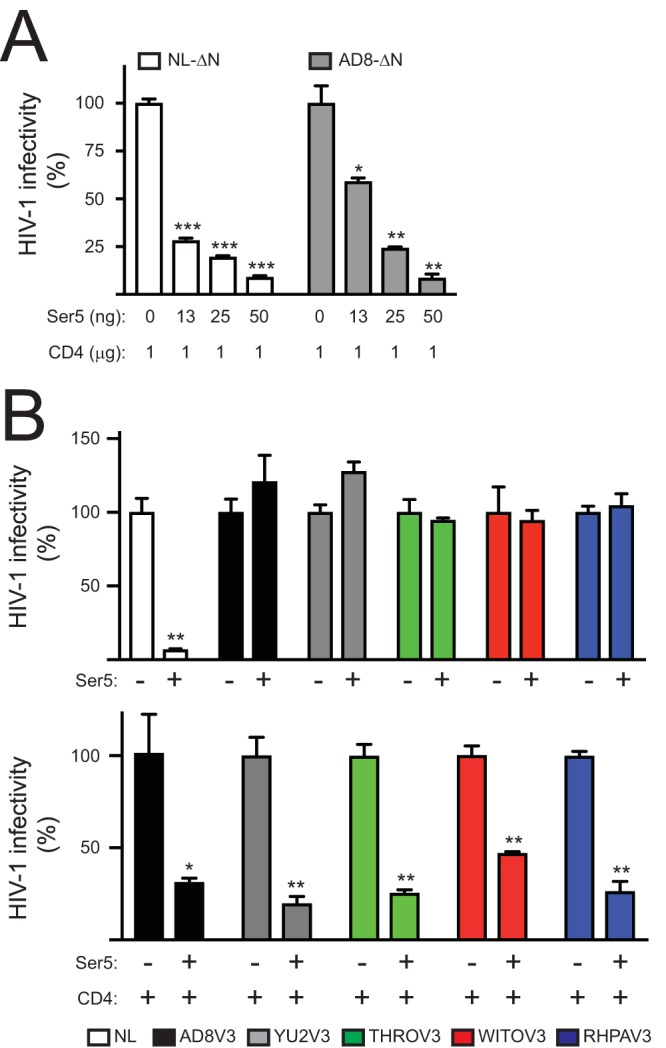
Inhibition of tier 2/3 virus replication by Ser5 in the presence of CD4. (A) NL-ΔN and AD8-ΔN viruses were produced from 293T cells in the presence of indicated amounts of pCMV6-CD4-FLAG and pCMV6-Ser5-FLAG. Viral infectivity was determined in TZM-bI cells and presented as relative values, with the infectivity of viruses produced in the absence of Ser5 set as 100%. (B) Indicated viruses were produced from 293T cells in the presence or absence of 50 ng pCMV6-Ser5-FLAG with or without 1 μg pCMV6-CD4-FLAG. Viral infectivity was determined in TZM-bI cells and presented as relative values, with the infectivity in the absence of Ser5 set as 100%. Error bars indicate SDs of the results from three experiments.
Ser5 restriction of another HIV-1 CCR5-tropic primary strain, BaL.
We sought to further confirm our findings by examining whether another CCR5-tropic virus, BaL, is resistant to Ser5. When NL-ΔN and BaL-ΔN viruses were produced in the presence of Ser5, the NL infectivity was strongly reduced by Ser5 in a dose-dependent manner, whereas BaL infectivity was not (Fig. 9A). In addition, when human primary T cells were infected with NL-WT, NL-ΔN, BaL-WT, and BaL-ΔN viruses, NL-ΔN and BaL-ΔN viral replication were suppressed to a similar level compared to their WT viruses (Fig. 9B). When the infectivities of virions released from these primary cells were compared by a single-round replication assay, both NL-WT and BaL-WT viruses showed a >10-fold higher infectivity than their ΔN mutant counterparts (Fig. 9C). Thus, like AD8 viruses, BaL viruses are resistant to Ser5 when produced in CD4-negative cells and tested by the single-round replication assay, but they likely become sensitive to Ser5 when propagated in CD4+ cells.
FIG 9.

Restriction of BaL viruses by Ser5. (A) NL-ΔN and BaL-ΔN viruses were produced from 293T cells in the presence of increasing amounts of pBJ-iHA-Ser5. Viral infectivity was analyzed after infection of TZM-bI cells. (B) PHA-activated primary T cells purified from 3 healthy donors were infected with R9 (NL-WT), R9ΔN (NL-ΔN), R9BaL (BaL-WT), or R9BaLΔN (BaL-ΔN). Viral growths were determined by p24Gag ELISA as in Fig. 1C. (C) Virus supernatants were collected during high virus production of infection in panel B, and viral infectivity was determined by infection of P4 or P4-CCR5 cells followed by staining cultures with X-Gal. Infectivity values were calculated as the ratio of the number of blue-stained foci per well to the p24Gag content of the inoculum. Error bars indicate SDs of the results from three independent experiments.
DISCUSSION
We investigated the Env-dependent Ser5 antiviral mechanism by comparing the tier 1 NL with tier 3 AD8 Env proteins. Env proteins from tier 2/3 isolates have been suggested to antagonize Ser5 more efficiently than Nef by an independent mechanism (13). However, if this is the case, it is unclear why Nef is required to counteract Ser5 during HIV-1 infection. In this paper, we demonstrate that these primary strains become sensitive to Ser5 when they are produced in the presence of CD4. In addition, AD8 and BaL viruses are restricted as efficiently as NL viruses in CD4+ T cells in the absence of Nef, which is consistent with a previous report that Nef strongly promoted a large number of CCR5-tropic HIV-1 primary isolates to replicate in PBMCs (25). Upon binding to CD4, trimers of the gp120/gp41 heterodimer undergo profound conformational alterations (26, 27) via transition from a prefusion closed conformation to a CD4-bound, open conformation that is ready for coreceptor binding and membrane fusion (28, 29). However, even in the absence of CD4, native tier 1 Env trimers predominantly adopt the open conformation, while the native tier 2/3 Env trimers retain the closed conformation (29, 30). Thus, the Env resistance to Ser5 is likely caused by the refractory closed conformation of Env trimers rather than an antagonistic mechanism.
We established a BiFC assay to detect Env-Env associations and Ser5-Env interactions. BiFC assays have become very popular to study protein interactions and report the subcellular localization of the observed protein complex. However, the generation of BiFC signals is dependent on spatial proximity of two proteins, not necessarily a direct interaction between them. Thus, we conducted several experiments to validate our results. First, we validated the suitability of our expression vectors. The specific Env BiFC fusion protein expression and their proper processing into gp120 and gp41 were detected by Western blotting, and their surface expression was detected by flow cytometry. These experiments also showed that all these Env fusion proteins were expressed at similar levels. The generation of the BiFC signals from these Env fusion proteins was shown by both flow cytometry and confocal microscopy. Second, we included Ser2 as a negative control. After validation of the specific Ser2 and Ser5 BiFC fusion expression and normalization of their expression to similar levels, we demonstrate that Ser5 interacts with Env to a much greater extent than does Ser2. Third, we verified the specificity of Ser5-Env interaction independently by IP. From these experiments, we conclude that in the absence of CD4, Ser5 interacts with NL Env to a greater extent than with AD8 Env, and Ser5 selectively dissociates the NL Env trimeric complex. Because we showed that Ser5 did not affect Env expression, NL Env dissociation is not caused by a reduction in Env expression.
CD4 expression is a critical determinant for Env sensitivity to Ser5. Unlike previous reports, our results suggest that CD4 likely does not play an important role in viral restriction. Instead, CD4 likely makes tier 2/3 viruses become sensitive to Ser5. In the presence of CD4, Ser5 showed increased capacity to interact with AD8 Env and dissociate AD8 Env proteins when measured by BiFC, and tier 2/3 viruses thus became sensitive to Ser5 restriction when measured by a single-round viral replication assay. Thus, CD4 likely increases Env sensitivity to Ser5 by opening the closed Env conformation.
HIV-1 has evolved two independent mechanisms to downregulate CD4 at posttranscriptional levels. CD4 is downregulated by Nef via the lysosomal pathway, and it is also downregulated by Vpu via the ER-associated protein degradation (ERAD) pathway (31). CD4 downregulation prevents HIV-1 superinfection (32) and facilitates virus release (22, 23), resulting in controlled and productive infection. Because CD4-Env interaction could occur cotranslationally, Vpu and Nef likely do not prevent the Env conformational shift induced by CD4 in infected cells. Nonetheless, our findings suggest that HIV-1 should gain another benefit from low levels of CD4 expression, which is to escape from Ser5 restriction. This may contribute to the importance of relatively low CD4-expressing cells, such as macrophages, to HIV-1 infection.
Our results have important implications toward understanding the poorly defined Env-dependent Ser5 antiviral mechanism. We hypothesize that during natural HIV-1 infection, CD4 binds to HIV-1 Env proteins cotranslationally, resulting in opening the closed Env conformation in the ER. Subsequently, Ser5 interacts with Env trimers with an open conformation and dissociates the Env trimeric complex. Env dissociation may sufficiently alter Env conformation, or even promote Env monomerization, to the extent of jeopardizing Env function and blocking viral entry. Recently, Pizzato and colleagues speculated that Ser5 likely impairs the functional clustering of the Env trimers, which is consistent with our hypothesis (33). Thus, more effort is needed to further understand the interplay between CD4, Env, and Ser5 in HIV-1 infection.
MATERIALS AND METHODS
Cells.
Human 293T cells were obtained from the ATCC. Human Jurkat-TAg cell line and its ser3-ser5 double-knockout cell line Jurkat-TAg-KO-(1) were provided by Heinrich Göttlinger (3). The Jurkat-TAg-KO-Ser5 cell line was created by stable transduction of the Jurkat-TAg-KO cells with pMSCV-Ser5-FLAG retroviral vector (4). TZM-bI and Jurkat LTR-GFP CCR5+ (J-LTRG-R5) cells were obtained from the NIH AIDS Reagent Program. P4 and P4-CCR5 cells were provided by Chris Aiken (34).
Human peripheral blood mononuclear cells (PBMCs) were purchased from Gulf Coast Regional Blood Center. Heparinized human blood was also obtained from healthy adult volunteers according to protocols approved by the institutional review board of Vanderbilt University Medical School. CD3+ T cells were selected from PBMCs by rosetting with 2-aminoethylisothiouronium bromide (AET; Sigma)-treated sheep red blood cells (SRBC; Cocalico Biologicals, Reamstown, PA). SRBC-T rosettes were removed by Ficoll-Paque density gradient sedimentation. High-purity CD3+ T cells were obtained by separation of the E-rosette-positive population from SRBC by hypotonic lysis using distilled water. PBMCs and purified CD3+ T cells were activated with 3 μg/ml phytohemagglutinin (PHA; Sigma) and cultured in complete RPMI 1640 medium containing 10% fetal bovine serum (FBS) with 40 U/ml recombinant human interleukin-2 (rhIL-2) (NIH AIDS Reagent Program).
Plasmids.
Infectious HIV-1 proviral vectors pNL4-3 and pNLΔN were described before (35). pNL4-3-derived infectious HIV-1 proviral vectors pH22 and pH22ΔN were described previously (36). pNL(AD8) was provided by Eric Freed (NCI, Frederick, MD) (37). pH-AD8, which expresses the KpnI/BsmI fragment of the AD8 Env gene in pNL(AD8) was described previously (38). pH-AD8ΔN was created by introducing a frameshift mutation at the XhoI site in pH-AD8. Infectious HIV-1 proviral vectors R9, R9ΔN, R9BaL, and R9BaLΔN were provided by Chris Aiken (34). R9 is a chimera created by replacing the BssHII-BamHI fragment of R7 that encodes HXB2 with the corresponding fragment from pNL4-3 (39). pCMV6-Ser5-FLAG and pCMV6-Ser2-FLAG were described before (4). pCMV6-CD4-FLAG was described before (10). The pBJ5-Ser5-HA that has a C-terminal HA and pBJ5-iHA-Ser5 that has an HA tag inserted between residues 290 and 291 of Ser5 was provided by Heinrich Göttlinger (3). The pBJ6-Ser2-HA and pBJ6-Ser5-HA that have a C-terminal HA were provided by Massimo Pizzato (2). pNL(YU2V3), pNL(THROV3), pNL(AD8V3), pNL(WITOV3), and pNL(RHPAV3) were provided by Chen Liang (13). The mCherry-Calnexin-N-14 expression vector was obtained from Michael Davidson via Addgene (no. 55005).
pcDNA3.1-Ser5-VN-HA and pcDNA3.1-Ser5-FLAG-VC were described previously (10). pcDNA3.1-Ser2-VN-HA and pcDNA3.1-Ser2-FLAG-VC were created by swapping the XhoI/BspEI fragment in pcDNA3.1-Ser5-VN-HA and pcDNA3.1-Ser5-FLAG-VC with that from pBJ6-Ser2-HA. To construct HIV-1 proviral vectors expressing Env-VN and Env-VC fusions, a bridge vector, pH22-NcoI/XhoI, was created by introducing an NcoI-EcoRI-XhoI linker immediately after the gp41 gene in the pH22 vector after BamHI/XhoI digestion. pH-NL-VN-HA and pH-NL-FLAG-VC were created by cloning NL-Env-VN-HA and NL Env-FLAG-VC into the bridge vector after NcoI/XhoI digestion. pH-AD8-VN-HA and pH-AD8-FLAG-VC were created by swapping the SalI/BamHI fragment in pH-NL-VN-HA and pH-NL-V5-VC with that from pH-AD8. A more detailed protocol for these cloning experiments is available upon request.
Immunoprecipitation.
To detect Ser5-Env interaction, 293T cells were cultured in 6-well plates and transfected with 1 μg pH22 or pH-AD8, plus 25 ng pCMV6-Ser5 or pCMV6-Ser2 using polyethyleneimine (PEI) as the transfection reagent. Proteins were pulled down by an anti-FLAG M2 antibody (Sigma) using the Pierce Classic IP kit (Thermo Scientific), following the manufacturer’s manual, and analyzed by Western blotting.
Single-round HIV-1 infection.
To determine Ser5 antiviral activity from producer cells, viruses were produced from 293T cells after transfection with WT or ΔN mutant HIV-1 proviral and Ser5 expression vector using PEI. After 48 h, viruses were collected from culture supernatants and quantified by p24Gag ELISA, as reported previously (40). Equal amounts of viruses were inoculated into the HIV-1 firefly luciferase reporter cell line TZM-bI in 96-well plates. After 48 h, cells were lysed and analyzed using the Firefly luciferase assay kit 2.0 (Biotium).
Viral infectivity was also measured by the multinuclear activation of a galactosidase indicator (MAGI) assay, as described before (41). Briefly, P4 or P4-CCR5 cells (20,000) were plated 1 day prior to infection in 48-well plates. After overnight growth, the medium was removed, and the cells were infected in triplicate using 10-fold serial dilutions of virus stocks (0.125-ml volumes). Two days after infection, cells were fixed and stained with 5-bromo-4-chloro-3-indolyl-galactopyranoside (X-Gal), and the number of infectious units was determined by counting the blue-stained foci under the light microscope. Wells containing between 30 and 1,000 infectious units were used for counting; within this range, the assay was linear and consistent with values obtained for virus dilutions. Uninfected wells consistently exhibited fewer than 5 blue cells per well. All values shown were the results of triplicate infections, which typically varied by less than 20%. Infectivity values were calculated by dividing the number of infectious units per well by the p24 content of the inoculum.
Measurement of HIV-1 propagation in T cells and PBMCs.
Jurkat cells and PHA-activated PBMCs were plated at 2 × 105 cells per well in 200 μl culture medium in 96-well plates. Aliquots of HIV-1 stocks containing 10 ng of p24Gag antigen were used to infect each well. Supernatants (100 μl) from each well were withdrawn daily or every other day and replaced with fresh medium. The p24Gag concentrations in the supernatants were determined by ELISA.
Confocal microscopy analysis.
Forty-eight hours after transfection, 293T cells were collected and washed twice with phosphate-buffered saline (PBS). Cells were treated with fixation/permeabilization solution (BD Biosciences) for 30 min at room temperature. After being washed 3 times with PBS, cells were incubated with Alexa Fluor-647 anti-HA (BioLegend) and Pacific Blue anti-FLAG (Cell Signaling) at a 1:500 dilution at 4°C for 1 h. After being washed 3 times with PBS, 5 × 105 cells were collected on a slide at 1,000 rpm for 3 min by cytospin. After being dried by air, cells were mounted by ProLong gold antifade mountant (Thermo Fisher) and observed with an Olympus spectral FV1000 microscope (Japan).
Flow cytometry.
Cells were washed with PBS twice and incubated with primary antibodies at 4°C for 1 h. After being washed three times, cells were incubated with secondary antibodies at 4°C for 30 min. After further washing and fixation with formaldehyde, stained cells were analyzed by a BD LSR II flow cytometer. Phycoerythrin (PE)-conjugated mouse anti-human CCR5 was purchased from BD Pharmingen, antigen-presenting cell (APC)-conjugated rat anti-human CD4 and Pacific Blue-conjugated mouse anti-human CD3 were purchased from BioLegend, PE-conjugated goat anti-human IgG was purchased from Santa Cruz Biotechnology, and primary mouse anti-human CXCR4 and HIV-1 broadly neutralizing antibody 35O22 were obtained from the NIH AIDS Reagent Program. To detect BiFC signals, 293T cells were transfected with vectors expressing BiFC fusion proteins. Alternatively, Jurkat cells were spinoculated with VSV-G pseudotyped viruses expressing NL-VN, NL-VC, AD8-VN, or AD8-VC, as indicated in Fig. 2C and Fig. 4A. After 48 h, cells were analyzed using the fluorescein isothiocyanate (FITC) channel. In some experiments, cells were permeabilized and stained with Pacific Blue-conjugated anti-HA and APC-conjugated anti-FLAG and then analyzed.
Western blotting.
Cells were lysed with 1% NP-40 in PBS containing protease inhibitor cocktail (Sigma). After the removal of nuclei by low-speed centrifugation, samples were resolved by SDS-PAGE gels. After transferring to an Immun-Blot polyvinylidene difluoride (PVDF) membrane (Bio-Rad), proteins were incubated with primary and secondary antibodies. The anti-Gag (catalog no. 1513) and anti-gp41 (catalog no. 526) antibodies were obtained from the NIH AIDS Reagent Program. A human polyclonal anti-HIV-1 antibody was described previously (38). Mouse anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) was purchased from Proteintech. Mouse anti-actin was purchased from Santa Cruz. Horseradish peroxidase (HRP)-conjugated anti-human, anti-rabbit, or anti-mouse immunoglobulin G secondary antibodies were purchased from Pierce. HRP-conjugated anti-FLAG was purchased from Sigma. The enhanced chemiluminescence detection kit was purchased from Amersham Bioscience.
Statistics.
Statistical tests were performed using Microsoft Excel. The significance of differences between samples was assessed using an unpaired two-tailed Student t test. The variance was estimated by calculating the standard deviation (SD) and is represented by error bars. Experiments were repeated as described in the figure legends, with a representative experiment being shown. Statistical significance was denoted as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001.
ACKNOWLEDGMENTS
We thank Thomas E. Smithgall for critical comments on the manuscript. We thank Henrich Göttlinger, Massimo Pizzato, Chris Aiken, Eric Freed, Chen Liang, and Michael Davidson as well as the NIH AIDS Reagent Program for providing various reagents.
Y.-H.Z. is supported by grants (AI120189, AI122863, and AI138707) from the National Institutes of Health.
REFERENCES
- 1.Inuzuka M, Hayakawa M, Ingi T. 2005. Serinc, an activity-regulated protein family, incorporates serine into membrane lipid synthesis. J Biol Chem 280:35776–35783. doi: 10.1074/jbc.M505712200. [DOI] [PubMed] [Google Scholar]
- 2.Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, Santoni FA, Pizzato M. 2015. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526:212–217. doi: 10.1038/nature15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Usami Y, Wu Y, Gottlinger HG. 2015. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526:218–223. doi: 10.1038/nature15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X, Zhou T, Yang J, Lin Y, Shi J, Zhang X, Frabutt DA, Zeng X, Li S, Venta PJ, Zheng YH. 2017. Identification of SERINC5-001 as the predominant spliced isoform for HIV-1 restriction. J Virol 91:e00137-17. doi: 10.1128/JVI.00137-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heigele A, Kmiec D, Regensburger K, Langer S, Peiffer L, Sturzel CM, Sauter D, Peeters M, Pizzato M, Learn GH, Hahn BH, Kirchhoff F. 2016. The potency of Nef-mediated SERINC5 antagonism correlates with the prevalence of primate lentiviruses in the wild. Cell Host Microbe 20:381–391. doi: 10.1016/j.chom.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahi YS, Zhang S, Thappeta Y, Denman A, Feizpour A, Gummuluru S, Reinhard B, Muriaux D, Fivash MJ, Rein A. 2016. Functional interplay between murine leukemia virus glycogag, Serinc5, and surface glycoprotein governs virus entry, with opposite effects on gammaretroviral and ebolavirus glycoproteins. mBio 7:e01985-16. doi: 10.1128/mBio.01985-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chande A, Cuccurullo EC, Rosa A, Ziglio S, Carpenter S, Pizzato M. 2016. S2 from equine infectious anemia virus is an infectivity factor which counteracts the retroviral inhibitors SERINC5 and SERINC3. Proc Natl Acad Sci U S A 113:13197–13202. doi: 10.1073/pnas.1612044113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmad I, Li S, Li R, Chai Q, Zhang L, Wang B, Yu C, Zheng YH. 2019. The retroviral accessory proteins S2, Nef, and glycoMA use similar mechanisms for antagonizing the host restriction factor SERINC5. J Biol Chem 294:7013–7024. doi: 10.1074/jbc.RA119.007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li S, Ahmad I, Shi J, Wang B, Yu C, Zhang L, Zheng YH. 2018. Murine leukemia virus glycosylated Gag reduces murine SERINC5 protein expression at steady-state levels via endosome/lysosome pathway to counteract the SERINC5 antiretroviral activity. J Virol 93:e01651-18. doi: 10.1128/JVI.01651-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi J, Xiong R, Zhou T, Su P, Zhang X, Qiu X, Li H, Li S, Yu C, Wang B, Ding C, Smithgall TE, Zheng YH. 2018. HIV-1 Nef antagonizes SERINC5 restriction by downregulation of SERINC5 via the endosome/lysosome system. J Virol 92:e00196-18. doi: 10.1128/JVI.00196-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sood C, Marin M, Chande A, Pizzato M, Melikyan GB. 2017. SERINC5 protein inhibits HIV-1 fusion pore formation by promoting functional inactivation of envelope glycoproteins. J Biol Chem 292:6014–6026. doi: 10.1074/jbc.M117.777714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trautz B, Wiedemann H, Luchtenborg C, Pierini V, Kranich J, Glass B, Krausslich HG, Brocker T, Pizzato M, Ruggieri A, Brugger B, Fackler OT. 2017. The host-cell restriction factor SERINC5 restricts HIV-1 infectivity without altering the lipid composition and organization of viral particles. J Biol Chem 292:13702–13713. doi: 10.1074/jbc.M117.797332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beitari S, Ding S, Pan Q, Finzi A, Liang C. 2017. Effect of HIV-1 Env on SERINC5 antagonism. J Virol 91:e02214-16. doi: 10.1128/JVI.02214-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lai RP, Yan J, Heeney J, McClure MO, Gottlinger H, Luban J, Pizzato M. 2011. Nef decreases HIV-1 sensitivity to neutralizing antibodies that target the membrane-proximal external region of TMgp41. PLoS Pathog 7:e1002442. doi: 10.1371/journal.ppat.1002442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Usami Y, Gottlinger H. 2013. HIV-1 Nef responsiveness is determined by Env variable regions involved in trimer association and correlates with neutralization sensitivity. Cell Rep 5:802–812. doi: 10.1016/j.celrep.2013.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seaman MS, Janes H, Hawkins N, Grandpre LE, Devoy C, Giri A, Coffey RT, Harris L, Wood B, Daniels MG, Bhattacharya T, Lapedes A, Polonis VR, McCutchan FE, Gilbert PB, Self SG, Korber BT, Montefiori DC, Mascola JR. 2010. Tiered categorization of a diverse panel of HIV-1 Env pseudoviruses for assessment of neutralizing antibodies. J Virol 84:1439–1452. doi: 10.1128/JVI.02108-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gautam R, Nishimura Y, Lee WR, Donau O, Buckler-White A, Shingai M, Sadjadpour R, Schmidt SD, LaBranche CC, Keele BF, Montefiori D, Mascola JR, Martin MA. 2012. Pathogenicity and mucosal transmissibility of the R5-tropic simian/human immunodeficiency virus SHIV(AD8) in rhesus macaques: implications for use in vaccine studies. J Virol 86:8516–8526. doi: 10.1128/JVI.00644-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ochsenbauer-Jambor C, Jones J, Heil M, Zammit KP, Kutsch O. 2006. T-cell line for HIV drug screening using EGFP as a quantitative marker of HIV-1 replication. Biotechniques 40:91–100. doi: 10.2144/000112072. [DOI] [PubMed] [Google Scholar]
- 19.Arganaraz ER, Schindler M, Kirchhoff F, Cortes MJ, Lama J. 2003. Enhanced CD4 down-modulation by late stage HIV-1 nef alleles is associated with increased Env incorporation and viral replication. J Biol Chem 278:33912–33919. doi: 10.1074/jbc.M303679200. [DOI] [PubMed] [Google Scholar]
- 20.Glushakova S, Munch J, Carl S, Greenough TC, Sullivan JL, Margolis L, Kirchhoff F. 2001. CD4 down-modulation by human immunodeficiency virus type 1 Nef correlates with the efficiency of viral replication and with CD4+ T-cell depletion in human lymphoid tissue ex vivo. J Virol 75:10113–10117. doi: 10.1128/JVI.75.21.10113-10117.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lundquist CA, Tobiume M, Zhou J, Unutmaz D, Aiken C. 2002. Nef-mediated downregulation of CD4 enhances human immunodeficiency virus type 1 replication in primary T lymphocytes. J Virol 76:4625–4633. doi: 10.1128/JVI.76.9.4625-4633.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lama J, Mangasarian A, Trono D. 1999. Cell-surface expression of CD4 reduces HIV-1 infectivity by blocking Env incorporation in a Nef- and Vpu-inhibitable manner. Curr Biol 9:622–631. doi: 10.1016/S0960-9822(99)80284-X. [DOI] [PubMed] [Google Scholar]
- 23.Ross TM, Oran AE, Cullen BR. 1999. Inhibition of HIV-1 progeny virion release by cell-surface CD4 is relieved by expression of the viral Nef protein. Curr Biol 9:613–621. doi: 10.1016/S0960-9822(99)80283-8. [DOI] [PubMed] [Google Scholar]
- 24.Kerppola TK. 2008. Bimolecular fluorescence complementation (BiFC) analysis as a probe of protein interactions in living cells. Annu Rev Biophys 37:465–487. doi: 10.1146/annurev.biophys.37.032807.125842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papkalla A, Munch J, Otto C, Kirchhoff F. 2002. Nef enhances human immunodeficiency virus type 1 infectivity and replication independently of viral coreceptor tropism. J Virol 76:8455–8459. doi: 10.1128/JVI.76.16.8455-8459.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu J, Bartesaghi A, Borgnia MJ, Sapiro G, Subramaniam S. 2008. Molecular architecture of native HIV-1 gp120 trimers. Nature 455:109–113. doi: 10.1038/nature07159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pancera M, Zhou T, Druz A, Georgiev IS, Soto C, Gorman J, Huang J, Acharya P, Chuang GY, Ofek G, Stewart-Jones GB, Stuckey J, Bailer RT, Joyce MG, Louder MK, Tumba N, Yang Y, Zhang B, Cohen MS, Haynes BF, Mascola JR, Morris L, Munro JB, Blanchard SC, Mothes W, Connors M, Kwong PD. 2014. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 514:455–461. doi: 10.1038/nature13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guttman M, Cupo A, Julien JP, Sanders RW, Wilson IA, Moore JP, Lee KK. 2015. Antibody potency relates to the ability to recognize the closed, pre-fusion form of HIV Env. Nat Commun 6:6144. doi: 10.1038/ncomms7144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munro JB, Gorman J, Ma X, Zhou Z, Arthos J, Burton DR, Koff WC, Courter JR, Smith AB III, Kwong PD, Blanchard SC, Mothes W. 2014. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 346:759–763. doi: 10.1126/science.1254426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munro JB, Mothes W. 2015. Structure and dynamics of the native HIV-1 Env trimer. J Virol 89:5752–5755. doi: 10.1128/JVI.03187-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sugden SM, Bego MG, Pham TN, Cohen EA. 2016. Remodeling of the host cell plasma membrane by HIV-1 Nef and Vpu: a strategy to ensure viral fitness and persistence. Viruses 8:67. doi: 10.3390/v8030067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Little SJ, Riggs NL, Chowers MY, Fitch NJ, Richman DD, Spina CA, Guatelli JC. 1994. Cell surface CD4 downregulation and resistance to superinfection induced by a defective provirus of HIV-1. Virology 205:578–582. doi: 10.1006/viro.1994.1683. [DOI] [PubMed] [Google Scholar]
- 33.Firrito C, Bertelli C, Vanzo T, Chande A, Pizzato M. 2018. SERINC5 as a new restriction factor for human immunodeficiency virus and murine leukemia virus. Annu Rev Virol 5:323–340. doi: 10.1146/annurev-virology-092917-043308. [DOI] [PubMed] [Google Scholar]
- 34.Lundquist CA, Zhou J, Aiken C. 2004. Nef stimulates human immunodeficiency virus type 1 replication in primary T cells by enhancing virion-associated gp120 levels: coreceptor-dependent requirement for Nef in viral replication. J Virol 78:6287–6296. doi: 10.1128/JVI.78.12.6287-6296.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng YH, Plemenitas A, Linnemann T, Fackler OT, Peterlin BM. 2001. Nef increases infectivity of HIV via lipid rafts. Curr Biol 11:875–879. doi: 10.1016/S0960-9822(01)00237-8. [DOI] [PubMed] [Google Scholar]
- 36.Zheng YH, Plemenitas A, Fielding CJ, Peterlin BM. 2003. Nef increases the synthesis of and transports cholesterol to lipid rafts and HIV-1 progeny virions. Proc Natl Acad Sci U S A 100:8460–8465. doi: 10.1073/pnas.1437453100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freed EO, Englund G, Martin MA. 1995. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J Virol 69:3949–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang X, Zhou T, Frabutt DA, Zheng YH. 2016. HIV-1 Vpr increases Env expression by preventing Env from endoplasmic reticulum-associated protein degradation (ERAD). Virology 496:194–202. doi: 10.1016/j.virol.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gallay P, Swingler S, Aiken C, Trono D. 1995. HIV-1 infection of nondividing cells: C-terminal tyrosine phosphorylation of the viral matrix protein is a key regulator. Cell 80:379–388. doi: 10.1016/0092-8674(95)90488-3. [DOI] [PubMed] [Google Scholar]
- 40.Wehrly K, Chesebro B. 1997. p24 antigen capture assay for quantification of human immunodeficiency virus using readily available inexpensive reagents. Methods 12:288–293. doi: 10.1006/meth.1997.0481. [DOI] [PubMed] [Google Scholar]
- 41.Kimpton J, Emerman M. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J Virol 66:2232–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]