

Worldwide, more than 70 million people are infected with hepatitis C virus (HCV), which is a leading cause of hepatocellular carcinoma and liver transplantation. Despite the development of potent direct acting antivirals (DAAs) for HCV treatment, a vaccine is urgently needed due to the high cost of treatment and the possibility of reinfection after cure. Induction of multiple broadly neutralizing antibodies (bNAbs) that target distinct epitopes on the HCV envelope proteins is one approach to vaccine development. However, antigenic sites targeted by bNAbs in individuals with spontaneous control of HCV have not been fully defined. In this study, we characterize 13 monoclonal antibodies (MAbs) from a single person who cleared an HCV infection without treatment, and we identify 3 new sites targeted by neutralizing antibodies. The sites targeted by these MAbs could inform HCV vaccine development.
KEYWORDS: broadly neutralizing antibodies, epitope, Flaviviridae, hepatitis C virus, hepatitis C virus clearance, humoral immunity, neutralizing antibodies
ABSTRACT
Increasing evidence indicates that broadly neutralizing antibodies (bNAbs) play an important role in immune-mediated control of hepatitis C virus (HCV) infection, but the relative contribution of neutralizing antibodies targeting antigenic sites across the HCV envelope (E1 and E2) proteins is unclear. Here, we isolated thirteen E1E2-specific monoclonal antibodies (MAbs) from B cells of a single HCV-infected individual who cleared one genotype 1a infection and then became persistently infected with a second genotype 1a strain. These MAbs bound six distinct discontinuous antigenic sites on the E1 protein, the E2 protein, or the E1E2 heterodimer. Three antigenic sites, designated AS108, AS112 (an N-terminal E1 site), and AS146, were distinct from previously described antigenic regions (ARs) 1 to 5 and E1 sites. Antibodies targeting four sites (AR3, AR4-5, AS108, and AS146) were broadly neutralizing. These MAbs also displayed distinct patterns of relative neutralizing potency (i.e., neutralization profiles) across a panel of diverse HCV strains, which led to complementary neutralizing breadth when they were tested in combination. Overall, this study demonstrates that HCV bNAb epitopes are not restricted to previously described antigenic sites, expanding the number of sites that could be targeted for vaccine development.
IMPORTANCE Worldwide, more than 70 million people are infected with hepatitis C virus (HCV), which is a leading cause of hepatocellular carcinoma and liver transplantation. Despite the development of potent direct acting antivirals (DAAs) for HCV treatment, a vaccine is urgently needed due to the high cost of treatment and the possibility of reinfection after cure. Induction of multiple broadly neutralizing antibodies (bNAbs) that target distinct epitopes on the HCV envelope proteins is one approach to vaccine development. However, antigenic sites targeted by bNAbs in individuals with spontaneous control of HCV have not been fully defined. In this study, we characterize 13 monoclonal antibodies (MAbs) from a single person who cleared an HCV infection without treatment, and we identify 3 new sites targeted by neutralizing antibodies. The sites targeted by these MAbs could inform HCV vaccine development.
INTRODUCTION
Worldwide, more than 70 million people are infected with hepatitis C virus (HCV), which can lead to hepatocellular carcinoma and end-stage liver disease (1). Despite the development of potent direct acting antivirals (DAAs) for HCV treatment, the majority of persons with HCV are unaware of their infection, so they do not seek treatment and may continue to infect others (2). The high cost of treatment also limits access for many infected individuals, and those who are treated remain at risk for reinfection after cure (3–6). Few countries are on pace to achieve the goal of the World Health Organization (WHO) to eliminate HCV as a public health problem by 2030, since nearly 60% of surveyed countries had more infections than cures in 2016 (7, 8). An effective preventative vaccine would play a major role in controlling the HCV pandemic.
There is evidence from natural HCV infection that a vaccine could be effective. Approximately 25% of acutely HCV-infected individuals clear their infections without treatment (9–11), and those who clear one infection clear subsequent infections more than 80% of the time, demonstrating that a protective anti-HCV immune response can develop naturally (12). We and others have shown that spontaneous clearance of HCV infection is associated with early development of broadly neutralizing antibodies (bNAbs) (10, 13), and we also demonstrated a mechanism by which bNAbs mediated spontaneous clearance of infection by two human subjects (14), suggesting that identification or design of antigens capable of inducing bNAbs is a reasonable approach to vaccine development. Therefore, clear definition of the epitopes targeted by bNAbs from individuals with clearance of infection is critical to inform vaccine design.
The majority of HCV envelope protein-specific human monoclonal antibodies (MAbs) were isolated from individuals with persistent HCV infection (15–25). Based upon E1E2 competition-binding experiments and alanine-scanning mutagenesis E1E2-binding experiments with these MAbs, commonly targeted antigenic sites on the E1 and E2 proteins were designated domains A–E or antigenic regions (ARs) 1–5 (reviewed in references 26–28). Previously described MAbs targeting AR1/domain C binding residues in the central beta sandwich (amino acids [aa] 486 to 566) and back layer (aa 597 to 645) of E2 and are weakly or nonneutralizing. MAbs targeting AR2 have narrow neutralizing breadth and also bind at the back layer of E2. MAbs targeting AR3/domain B are often broadly neutralizing, targeting epitopes spanning the E2 front layer (aa 424 to 459) and CD81 binding loop (aa 519 to 535) (26, 29). Prototypical AR4 and AR5 MAbs, designated AR4A and AR5A, are also broadly neutralizing. These MAbs bind to epitopes that are still poorly defined, but both MAbs appear to bind near the C terminus of E2, and each requires E1 and E2 in complex for binding (29). To date, few E1-specific MAbs have been isolated from infected humans. One example, designated IGH526, bound near the C terminus of E1 and demonstrated relatively narrow neutralizing breadth (30).
Recently, we and others isolated bNAbs from the B cells of individuals who cleared their HCV infections without treatment (31, 32). However, MAb isolation has been attempted from very few individuals who cleared HCV infection. Therefore, it remains unclear whether particular antigenic sites are consistently targeted by bNAbs in these individuals and whether all commonly targeted antigenic sites have been identified.
In this study, we isolated 13 HCV envelope protein-specific MAbs from B cells of a single HCV-infected individual who cleared 1 genotype 1a infection and then became persistently infected with a second genotype 1a strain. These MAbs bound six distinct antigenic sites on the E2 protein, the E1E2 heterodimer, or at a discontinuous epitope near the N terminus of E1. Three of these sites, including the N-terminal E1 site, were distinct from previously described AR1–5 and E1 antigenic sites. Notably, MAbs targeting four of these antigenic sites were broadly neutralizing. These MAbs also displayed distinct patterns of relative neutralizing potency (i.e., neutralization profiles) across a panel of diverse HCV strains, which led to complementary neutralizing breadth when they were tested in combination.
RESULTS
Subject C18 spontaneously cleared one HCV infection and then became persistently reinfected with a second HCV strain.
At enrollment in the Baltimore Before and After Acute Study of Hepatitis (BBAASH), subject C18 was HCV seronegative and HCV RNA negative (Fig. 1). He became infected with a genotype 1a HCV strain 8 months after enrollment. He experienced an unusual 70-day period of undetectable viremia beginning approximately 100 days after infection, followed by a return of detectable viremia with a virus that was likely the same as the initial infecting strain since both were genetically very similar, based on sequencing of core and E1 genes (nucleotide divergence, <0.01) (33). He ultimately cleared this first infection without treatment 522 days after initial infection. Following a period of aviremia that lasted 637 days, subject C18 was reinfected with a second genetically distinct genotype 1a strain (nucleotide divergence from first infecting strain, 0.11) and remained persistently infected for over 4 years of subsequent follow-up.
FIG 1.
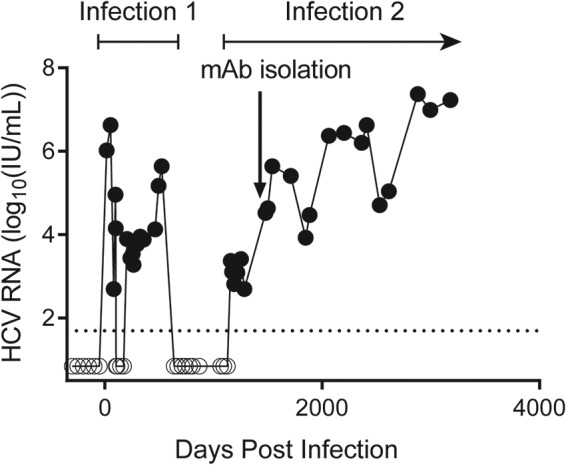
Subject C18 spontaneously cleared one HCV infection and then became persistently infected with a second HCV strain. The limit of detection (LOD) of the viral load assay (50 international units [IU]/ml) is indicated by a dashed line. Values below the LOD are set at 25 IU/ml and marked with open circles. The arrow at 1,483 days postinfection indicates the time point at which MAbs were isolated from circulating B cells.
C18 MAbs targeted six distinct antigenic sites based on hierarchical clustering analysis.
We isolated 13 HCV envelope protein-specific MAbs from subject C18 B cells 1,483 days after initial infection, which was 642 days after onset of the second, persistent infection (Fig. 1). We performed binding assays using a comprehensive alanine mutant library spanning the envelope proteins (E1 and E2) of genotype 1a strain H77 HCV to identify E1E2 residues important for binding of these MAbs. For reference, we also reanalyzed previously published alanine-scanning E1E2-binding results of 13 well-characterized MAbs targeting multiple distinct antigenic sites on the E1 protein (IGH526), the E2 protein (MAbs targeting AR1, AR2, and AR3), or the E1E2 heterodimer (MAbs targeting AR4 and AR5) (29, 31). Alanine-scanning E1E2-binding assays for these reference MAbs and C18 MAbs were performed using the same methods in the same laboratory. Probable binding residues identified in this manner may contribute directly to the MAb-binding epitope or they may be important for the local structure of that site.
We grouped MAbs with shared binding residues using hierarchical clustering analysis based on binding of these MAbs across the library of alanine mutant E1E2 proteins, as previously described (26) (Fig. 2). The number of MAb clusters in this hierarchical analysis indicated that C18 MAbs targeted six distinct antigenic sites. Some C18 MAbs clustered with reference MAbs in this analysis and others did not. The largest cluster included reference bNAbs AR3A–AR3D, HEPC3, HEPC74, and HEPC43, which were previously mapped to the AR3 antigenic site (31) and C18 MAbs HEPC122, HEPC151-1, HEPC153, and HEPC154. A second cluster included reference bNAbs AR4A and AR5A as well as two C18 MAbs, HEPC111 and HEPC130. It is reasonable that reference bNAbs AR4A and AR5A clustered in this analysis, although they did not compete in E1E2 competition-binding experiments in a prior study (18) since both require E1 and E2 in complex for binding and both are thought to bind near the C terminus of E2. Given this clustering, we consider AR4 and AR5 a single antigenic region, AR4-5, for the remainder of this report. A third cluster included reference MAb AR1A and C18 MAb HEPC167. C18 MAbs HEPC108, HEPC132, HEPC158, and HEPC151-2 formed a cluster that excluded all reference MAbs, indicating that they bind to a distinct antigenic site (hereafter designated AS108). Similarly, C18 MAbs HEPC112 and HEPC146 did not cluster with each other or any reference MAbs, indicating that each also binds to a distinct antigenic site (hereafter designated AS112 and AS146, respectively). Taken together, these data indicate that the epitopes of the C18 MAbs involve six distinct antigenic sites, including previously described AR1, AR3, and AR4-5 antigenic sites as well as non-AR1–5 antigenic sites designated AS108, AS112, and AS146.
FIG 2.

C18 MAbs targeted six distinct antigenic sites. Hierarchical clustering of 13 C18 MAbs (in bold type) and 13 reference MAbs based on Ward’s minimal variance analysis of MAb binding to a library of strain H77 E1E2 proteins with single alanine mutations spanning the full E1E2 sequence, depicted as an unrooted tree. Bootstrap values greater than 80 are shown. MAbs that clustered together with bootstrap values of >90 were highlighted in the same color and given a label indicating a previously described (AR1, AR2, AR3, and AR4-5) or novel (AS108, AS112, and AS146) antigenic site.
To identify probable binding residues of each C18 MAb and each reference MAb, we ranked E1E2 residues from greatest to least effect of alanine mutation on binding of each MAb. Since all C18 MAbs bound native but not denatured E1E2 proteins in an enzyme-linked immunosorbent assay (ELISA), indicating that they bind to conformational epitopes (Fig. 3), residues at which alanine mutation reduced binding of all E1 and E1E2-specific MAbs, or all E2 and E1E2-specific MAbs, were excluded as potential binding residues under the assumption that they reduced MAb binding by globally disrupting protein folding (29). For each MAb, after exclusion of these global misfolding positions, up to 15 residues were selected at which alanine mutation produced the greatest reduction in binding and also reduced binding by at least 50% relative to wild-type protein binding (Table 1). We then identified the reference MAb which shared the most probable binding residues with each C18 MAb (Table 2). Similarity of C18 MAbs to reference MAbs ranged from 14 of 15 probable binding residues shared (C18 MAb HEPC111 and reference MAb AR5A) down to 2 of 15 probable binding residues shared (C18 MAb HEPC112 and reference MAb IGH526).
FIG 3.
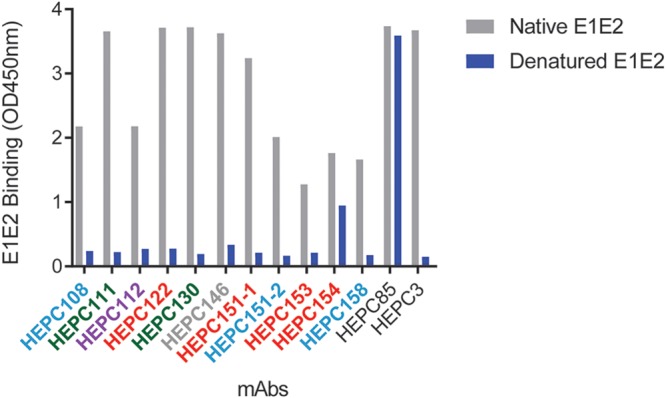
Conformational E1E2 binding. Binding of C18 MAbs to native and denatured E1E2 proteins. HEPC85 and HEPC3 bind to linear and conformational epitopes, respectively, and are shown as controls (31).
TABLE 1.
Probable E1E2-binding residues for each C18 MAb and reference MAb
| Antigenic site | C18 MAb | Probable binding residues | Reference MAb | Probable binding residues |
|---|---|---|---|---|
| AR1 | HEPC-167 | R543 P544 P545 L546 G547 N548 W549 Y594 C597 G598 G635 | AR1A | Y485 P498 G517 T519 N532 T534 P544 P545 G547 N548 W549 C597 G635 W646 D653 |
| AR3 | HEPC-122 | I422 T425 N428 C429 L441 H445 C503 T518 D520 G523 W529 G530 D535 W616 C620 | AR3A | T425 N428 C429 G436 W437 L438 G440 L441 F442 Y485 C503 W529 G530 D535 W616 |
| HEPC-151-1 | T425 L427 N428 C429 W437 A499 C503 V515 T518 D520 G523 W529 G530 D535 | AR3B | T425 L427 N428 C429 W437 G440 L441 F442 C503 D520 G523 W529 G530 D535 W616 | |
| HEPC-153 | T425 L427 N428 C429 W437 L441 F442 C503 T519 D520 G523 Y527 W529 D535 W616 | AR3C | T425 N428 C429 W437 L438 G440 L441 F442 Y443 A499 C503 W529 G530 D535 W616 | |
| HEPC-154 | L356 N428 C429 W437 F442 A499 K500 C503 V515 G523 G530 D535 P605 G635 T647 | AR3D | T425 L427 N428 C429 G436 W437 G440 L441 F442 C503 T518 D520 G530 D535 W616 | |
| HEPC3 | T425 L427 N428 G436 W437 L438 L441 F442 C503 G517 T518 D520 W529 G530 D535 | |||
| HEPC74 | S424 T425 L427 N428 C429 G436 W437 A499 C503 T518 T519 D520 G523 G530 D535 | |||
| HEPC43 | S424 T425 L427 N428 C429 W437 L441 A499 C503 T518 T519 D520 G523 G530 D535 | |||
| AR4-5 | HEPC-111 | Y201 N205 C486 R543 C569 G573 C585 F586 Y594 C597 G635 C652 R657 D658 C677 | AR4A | Y201 N205 C486 W487 R543 P545 C569 C585 Y594 C597 G635 C652 R657 C677 D698 |
| HEPC-130 | Y201 N205 C486 W487 R543 P544 P545 C569 G573 C585 Y594 C597 G635 C652 C677 | AR5A | Y201 N205 C486 W487 R543 C569 G573 C585 Y594 C597 G635 C652 R657 D658 C677 | |
| AS108 | HEPC-108 | I472 Y474 R543 P544 P545 L546 G547 N548 W549 C569 C585 Y594 C597 G598 G635 | ||
| HEPC-151-2 | I472 Y474 R543 P544 P545 G547 N548 W549 C569 G573 T593 Y594 C597 G598 G635 | |||
| HEPC-132 | I472 C486 R543 P544 P545 W549 C569 C581 C585 H589 E591 Y594 R596 C597 G635 | |||
| HEPC-158 | I472 Y474 C486 R543 P544 P545 L546 G547 N548 W549 C569 Y594 C597 G598 G635 | |||
| AS146 | HEPC-146 | D321 A330 G517 D520 W529 T534 D535 W549 | ||
| AS112 | HEPC-112 | E215 E232 G233 V246 R249 K252 R259 R260 D263 R297 W299 G354 Y361 F378 D382 | ||
| E1 C terminus | IGH526 | H316 M324 M323 Q336 W320 D321 C652 D653 T329 Y361 E215 G315 K588 N325 G573 | ||
| HVR1 | HEPC98 | L402 P405 K408 | ||
| AR2 | AR2A | S557 L615 F627 T647 |
TABLE 2.
Reference MAbs sharing the greatest number of probable binding residues with each C18 MAb
| C18 MAb | Reference MAb(s) | No. of shared residues involved in epitope formation |
|---|---|---|
| HEPC153 | AR3B | 13 |
| HEPC151-1 | HEPC43, HEPC74 | 12 |
| HEPC122 | AR3B | 11 |
| HEPC154 | AR3B | 8 |
| HEPC111 | AR5A | 14 |
| HEPC130 | AR4A, AR5A | 13 |
| HEPC167 | AR1A | 7 |
| HEPC108 | AR1A, AR4A | 7 |
| HEPC158 | AR4A | 7 |
| HEPC151-2 | AR4A, AR5A | 6 |
| HEPC132 | AR4A | 8 |
| HEPC146 | HEPC3 | 4 |
| HEPC112 | IGH526 | 2 |
For each identified antigenic site, probable binding residues of one C18 MAb representative of each antigenic site and its most closely related reference MAb or MAbs were mapped onto the HCV E2 ectodomain structure from Flyak et al. (34) (Fig. 4). First, we mapped probable binding residues of C18 MAbs that clustered with reference MAbs in the hierarchical analysis. As expected, based on clustering with AR3-site MAbs, C18 MAb HEPC153 probable binding residues fell in the CD81 binding loop and the front layer of E2, which was comparable to AR3B, the reference MAb with which it shared the most probable binding residues (Fig. 4A). C18 MAb HEPC111 probable binding residues fell in both E1 and E2 proteins (Fig. 4B), which was expected based upon hierarchical clustering of this MAb with AR4A and AR5A reference MAbs, which are known to require complexed E1 and E2 for binding (18). Most HEPC167 probable binding residues fell in the central beta sandwich (aa 486 to 566) and back layer (aa 597 to 645) of E2, and 7 of 11 were shared by reference MAb AR1A (Fig. 4C).
FIG 4.

Mapping of probable binding residues of each MAb onto the E2 ectodomain crystal structure. One C18 MAb representative of each antigenic cluster in Fig. 2 is shown, with the reference MAb or MAbs with which is shares the most probable binding residues, namely, (A) HEPC153, (B) HEPC111, (C) HEPC167, (D) HEPC108, (E) HEPC146, and (F) HEPC112. For each MAb, up to 15 probable E2-binding residues are shown, marked with colored spheres on the HCV E2 structure (Flyak et al., PDB 6MEI) (34). Probable E1-binding residues are shown in tabular form. These 15 E1E2 binding residues were also subdivided into tertiles of greatest effect (magenta), middle effect (orange), and least effect (yellow) of mutation to alanine on MAb binding. E2 structural components were highlighted in PyMOL, including the AS412 site (purple), front layer (cyan), VR2 (brown), CD81 binding loop (blue), beta sandwich (red), VR3 and post-VR3 (gray), and back layer (green). Dashed line indicates the stalk region of E2 not present in the ectodomain structure. MAb names are color-coded according to hierarchical clustering in Fig. 2.
Next, we mapped probable binding residues of MAbs that bound AS108, AS146, and AS112. HEPC108 probable binding residue fell in the beta sandwich, VR2, VR3, and back layer of E2. Seven of 15 HEPC108 probable binding residues were also binding residues for reference MAb AR1A, and 7 of 15 HEPC108 binding residues were binding residues for reference MAb AR4A, indicating that AS108 overlaps the AR1 and AR4-5 sites (Fig. 4D). HEPC146, which did not cluster with any other MAbs in Fig. 2, demonstrated an unusual binding pattern with probable binding residues in the CD81 binding loop and the central beta sheet of E2, and two probable binding residues in E1 (Fig. 4E). This MAb shared four of its eight probable binding residues with HEPC3, its most closely related reference MAb. Finally, HEPC112, which also did not cluster with any other MAbs in Fig. 2, had probable binding residues falling only in E1 (Fig. 4F). Only 2 of 15 HEPC112 probable E1-binding residues were also binding residues for IGH526, an E1-specific reference MAb, suggesting that HEPC112 binds to a novel antigenic site on E1 (AS112).
Taken together, these data confirm that C18 MAbs bound six distinct antigenic sites, including previously described AR1, AR3, and AR4-5 antigenic sites as well as non-AR1–5 antigenic sites on E1 (AS112) and E2 (AS108 and AS146). These results also demonstrate overlap between probable binding residues of MAbs targeting the AS108, AR1, and AR4-5 antigenic sites and between probable binding residues of MAbs targeting the AR3 and AS146 antigenic sites.
C18 MAbs formed five groups based on E1E2 competition binding.
To assess relationships among MAbs by a different method, we performed pairwise E1E2 competition-binding experiments with 12 C18 MAbs, grouping MAbs that were mutually inhibitory, i.e., binding of each MAb inhibited each other MAb in the group and was inhibited by each other MAb in the group (Fig. 5). C18 MAb HEPC132 was not included in this analysis due to poor ELISA binding. Competition-binding results were largely concordant with hierarchical clustering analysis in Fig. 2. Four C18 MAbs from the AR3 cluster were mutually inhibitory (HEPC151-1, HEPC122, HEPC154, and HEPC153), as were three C18 MAbs from the AS108 cluster (HEPC108, HEPC158, and HEPC151-2), and two C18 MAbs from the AR4-5 cluster (HEPC111 and HEPC130). HEPC146 and HEPC112 each displayed a unique pattern of inhibition by the panel of blocking antibodies, confirming that each binds to a distinct antigenic site. Although HEPC167 fell in the AR1 rather than the AS108 hierarchical cluster in Fig. 2, it grouped with AS108 MAbs in the binding competition analysis, which is likely explained by binding residues shared between AS108 MAbs and HEPC167 (i.e., 11 of 15 HEPC108 probable binding residues were shared by HEPC167) (Fig. 4C and D). Notably, AS108 MAbs did not compete with HEPC111 or HEPC130 (AR4-5-site MAbs) despite sharing multiple probable binding residues with these MAbs (i.e., 6 of 15 HEPC108 probable binding residues were shared by HEPC111), indicating that these binding residues shared between AS108 and AR4-5 MAbs do not have a large energetic effect on MAb binding. Alternatively, these shared probable binding residues could be sites that influence E2 conformation rather than MAb contact residues. Taken together, these data demonstrate that with the exception of competition between AS108 MAbs and HEPC167 (an AR1-site MAb), binding competition results were concordant with hierarchical clustering analysis.
FIG 5.
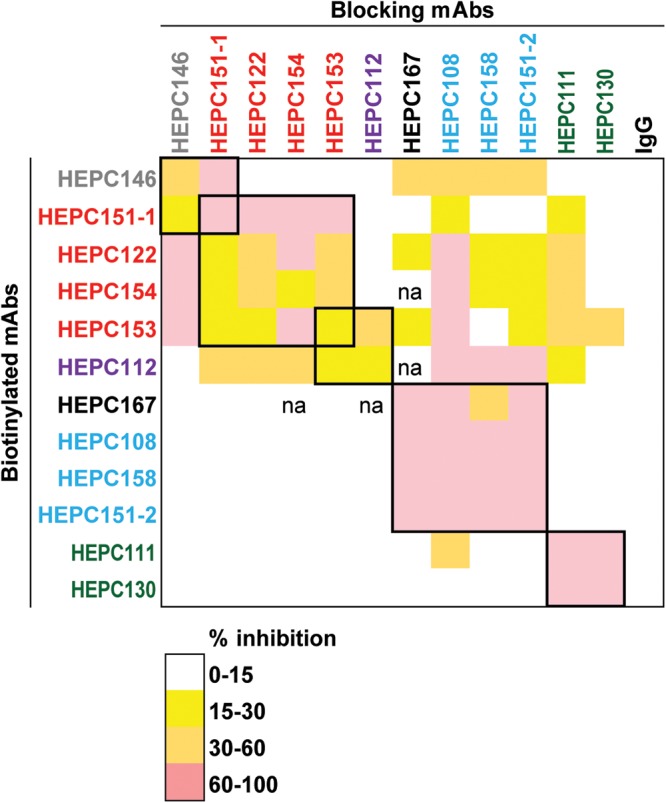
C18 MAbs formed five groups based on E1E2 competition binding. Percent inhibition of binding of 2 μg/ml or 0.4 μg/ml of biotinylated MAbs to E1E2 proteins in an ELISA in the presence of blocking MAbs at a concentration of 20 μg/ml. Squares at the intersection between blocking and biotinylated MAbs are color coded by the degree of inhibition. Groups of MAbs that were mutually inhibitory (i.e., binding of each MAb inhibited each other MAb in the group and was inhibited by each other MAb in the group) are marked with thick black boxes. Values are averages of three independent experiments, each performed in duplicate. MAb names are color coded according to hierarchical clustering in Fig. 2 Data not available is marked “na.”
MAbs targeting four antigenic sites were broadly neutralizing.
We used a diverse panel of 19 genotype 1a and 1b HCV pseudoparticles (HCVpps) as well as genotypes 2 to 6 of cell culture-produced HCV (HCVcc) to measure the neutralizing breadth of C18 MAbs. The HCVpp panel comprises 94% of the E1E2 amino acid polymorphisms present at greater than 5% frequency in a reference panel of 643 genotype 1 HCV isolates from GenBank. Using ≥50% neutralization as a threshold, the neutralizing breadth of the MAbs at a 50-μg/ml concentration ranged from 5 of 6 genotypes, including 17 of 19 genotype 1 HCVpps, neutralized by HEPC153 to 0 HCVccs or HCVpps neutralized by HEPC132 (Fig. 6). Overall, 8 of 13 C18 MAbs were broadly neutralizing, defined as greater than 50% neutralization of strains from at least 2 genotypes, and more than half of the genotype 1 HCVpp panel. These bNAbs targeted AR3 (HEPC153, HEPC151-1, HEPC154, and HEPC122), AR4-5 (HEPC111 and HEPC130), AS108 (HEPC108), and AS146 (HEPC146).
FIG 6.

MAbs targeting four antigenic sites were broadly neutralizing. Neutralizing breadth of the C18 MAbs was measured using a diverse panel of genotype 1a and 1b HCV pseudoparticles (HCVpps) and genotype 2–6 HCVccs. Percent neutralization of each strain by each MAb at a 50-μg/ml concentration is shown, with HCVpp or HCVcc on the y axis and MAbs arranged from greatest to least neutralizing breadth on the x axis. HCVpp values are averages of two independent experiments, each performed in duplicate. HCVcc values are from a single experiment performed in duplicate. MAb names are color coded according to hierarchical clustering in Fig. 2.
In many cases, the neutralizing breadth of C18 MAbs was consistent with the previously described neutralizing breadth of closely related reference MAbs. Two of the three most broadly neutralizing C18 MAbs (HEPC153 and HEPC151-1) bound at AR3, the target of many previously described bNAbs (19). In addition to these AR3-site MAbs, HEPC111 was also broadly neutralizing (17 of 24 strains [4 of 6 genotypes] neutralized), which was similar to the previously described neutralizing breadth of closely related reference MAb AR4A (12 of 19 genotype 1 HCVpps were neutralized by AR4A in reference 31). HEPC167, which clustered with the weakly neutralizing reference MAb AR1A in the binding analysis, also demonstrated poor neutralizing breadth against the HCVpp panel (2 of 24 strains [1 of 6 genotypes] neutralized).
The neutralizing breadth of AS108 MAbs varied widely. HEPC108 was broadly neutralizing (19 of 24 strains [5 of 6 genotypes] neutralized) despite sharing probable binding residues with weakly neutralizing reference MAb AR1A and weakly neutralizing C18 MAb HEPC167. Furthermore, HEPC132, which also bound at AS108 and shared 10 of 15 HEPC108 probable binding residues, neutralized 0 of 24 strains, further demonstrating that the neutralizing breadth of MAbs is not determined solely by the antigenic site targeted. HEPC112, which binds a novel site in E1 (AS112), neutralized 7 of 24 strains (1 of 6 genotypes), which did not meet our threshold of broad neutralization. Taken together, these results demonstrate that C18 MAbs targeting known antigenic sites (AR3 and AR4-5) as well as non-AR1–5 antigenic sites (AS108 and AS146) were broadly neutralizing.
bNAbs targeting multiple antigenic sites were encoded by IgHV1-69.
We sequenced the heavy and light chain variable gene sequences of each of the MAbs (Table 3). As we and others have previously observed (19, 31, 32, 35), multiple AR3-site MAbs (HEPC122, HEPC151-1, and HEPC153) were encoded by the same antibody heavy chain variable gene segment, VH1-69. Of note, one AR4-5-site MAb (HEPC111) and one AS108-site MAb (HEPC108) also used VH1-69. Collectively, these data indicate that VH1-69 usage favors broad neutralization and binding of HCV across multiple distinct antigenic sites. Of note, we also found that HEPC151-2 and HEPC158, which were biologically cloned from different B cells using limiting dilution and flow sorting, displayed identical heavy chain and light chain-variable gene sequences, indicating that this clonotype was relatively frequent among HCV-specific B cells in this subject. As we have previously observed, all MAbs, including bNAbs, were encoded by antibody genes with relatively sparse somatic mutations, ranging from 87% to 94% identity to their germ line heavy chain variable heavy (VH) gene sequences and 89% to 98% identity to their germ line light chain variable light (VL) gene sequences, indicating that extensive somatic hypermutation was not necessary for acquisition of broad neutralizing activity.
TABLE 3.
Germ line origin genes and variable region analysis of subject C18 MAbs
| Antigenic site | MAb | Heavy chain variable gene sequence |
Light chain variable sequence |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| V gene and allele | V region nucleotide homology to V-gene(%) | D gene and allele | J gene and allele | CDR3 amino acids | CDR3 length (aa)a | V gene and allele | V region nucleotide homology to V gene (%) | J gene and allele | CDR3 amino acids | CDR3 length (aa) | ||
| AR1 | HEPC-167 | IGHV1-18*04 | 91.7 | IGHD3-16*01 | IGHJ4*03 | ARDSVTGGMDV | 11 | IGLV3-25*03 | 92.8 | IGLJ3*02 | QFADISGTYRRV | 12 |
| AR3 | HEPC-122 | IGHV1-69*17 | 93.8 | IGHD3-3*01 | IGHJ4*02 | ATFDYDFYSGAPSRSYFVH | 19 | IGKV3-20*01 | 97.2 | IGKJ4*01 | QQYGSSPT | 8 |
| HEPC-151-1 | IGHV1-69*01 | 87.9 | IGHD3-10*01 | IGHJ6*02 | ARDGGGHSGLGRFYDLNGMDV | 21 | IGKV3-15*01 | 95.3 | IGKJ2*01 | QQYDNWPPRYT | 11 | |
| HEPC-153 | IGHV1-69*01 | 92.7 | IGHD1-20*01 | IGHJ5*02 | VREMRYNWNPGRSSWFDP | 18 | IGKV3-11*01 | 93.6 | IGKJ3*01 | QQRSIRPPS | 9 | |
| HEPC-154 | IGHV3-53*01 | 88.8 | IGHD3-16*01 | IGHJ3*02 | ARCFAGLMAMDI | 12 | IGKV2-28*01 | 97.3 | IGKJ3*01 | MQGLQIPPT | 9 | |
| AR4-5 | HEPC-111 | IGHV1-69*01 | 91.7 | IGHD3-3*01 | IGHJ3*01 | ARSEDVLSGYYSVSIDAFNV | 20 | IGLV1-51*01 | 94.0 | IGLJ2*01 | ATWDRSLSIAV | 11 |
| HEPC-130 | IGHV3-30*03 | 87.2 | IGHD2-2*01 | IGHJ6*02 | ARNSVPATMTSYYFYALDL | 19 | IGKV1-39*01 | 88.9 | IGKJ1*01 | LQTYDTVWT | 9 | |
| AS108 | HEPC-108 | IGHV1-69*06 | 94.1 | IGHD6-19*01 | IGHJ4*02 | ARASIVNISGFVRFRFDS | 18 | IGKV1-33*01 | 95.7 | IGKJ2*01 | QHYDDVPPVYT | 11 |
| HEPC-151-2 | IGHV1-18*04 | 89.6 | IGHD3-22*01 | IGHJ3*02 | ARHDSSGYEALDI | 13 | IGLV3-19*01 | 97.9 | IGLJ1*01 | NSRDTSPYHHFV | 12 | |
| HEPC-132 | IGHV4-31*03 | 91.8 | IGHD3-3*01 | IGHJ6*02 | ARDGADGFWSGHNFGMDA | 18 | IGKV3-11*01 | 93.6 | IGKJ4*01 | QQRSAWPRT | 9 | |
| HEPC-158 | IGHV1-18*04 | 89.6 | IGHD3-22*01 | IGHJ3*02 | ARHDSSGYEALDI | 13 | IGLV3-19*01 | 97.9 | IGLJ1*01 | NSRDTSPYHHFV | 12 | |
| AS146 | HEPC-146 | IGHV4-61*01 | 92.1 | IGHD5-24*01 | IGHJ1*01 | ARTSQMTTLLL | 11 | IGLV2-8*01 | 94.4 | IGLJ3*02 | SSYVGNDNLL | 10 |
| AS112 | HEPC-112 | IGHV3-48*03 | 92.4 | IGHD3-3*01 | IGHJ4*02 | VRDGREFWGGSRIPFDH | 17 | IGKV3-20*01 | 95.0 | IGKJ2*01 | QQYATTPDT | 9 |
aa, amino acid.
Polymorphisms outside MAb binding sites influence bNAb resistance.
To further investigate the wide variation in neutralization sensitivity of HCVpp to C18 MAbs, we analyzed the conservation of probable binding residues of three MAbs (HEPC111, HEPC153, and HEPC151-1) across all strains in the HCVpp panel (Fig. 7). Conservation or variation at probable binding residues did not predict neutralization sensitivity or resistance of HCVpp strains. For example, sensitivity of HCVpp strains to HEPC111 varied more than 30-fold despite 100% conservation of HEPC111 probable binding residues across all strains. Similarly, two strains with 100% conservation of HEPC151-1 probable binding residues (1a31 and 1a129) differed by more than 100-fold in sensitivity to HEPC151-1 neutralization. For HEPC153, a strain that was highly sensitive to neutralization (1b09) had a substitution relative to the H77 sequence (W437F) at a HEPC153 probable binding residue, but a strain that was more than 100-fold more resistant to HEPC153 (1a31) did not have any substitutions relative to H77 at probable HEPC153 binding residues. Taken together, these results demonstrate that conservation or lack of conservation of probable binding residues did not predict neutralization sensitivity or resistance of HCVpp strains. These results may indicate that not all critical binding residues were identified by our alanine-scanning analysis. Alternatively, these results might be explained by allosteric effects of polymorphisms distant from binding epitopes. Prior studies have demonstrated that polymorphisms in hypervariable region 1 (HVR1) or in the central beta sheet of E2 can exert allosteric effects that influence neutralization sensitivity of HCV isolates. (36–38).
FIG 7.

Polymorphisms outside MAb binding sites influence bNAb resistance. Amino acids present at probable binding residues of HEPC111 (A), HEPC153 (B), and HEPC151-1 (C) across all strains in the HCVpp panel. Homology to the strain H77 sequence is indicated with a dot. Percent neutralization of each HCVpp strain by 50 μg/ml of each MAb is shown. Values are averages of two independent experiments, each performed in duplicate. For each MAb, full neutralization curves were also measured in duplicate with 5-fold serial dilutions of MAb starting at 100 μg/ml for one sensitive and two more resistant HCVpp strains. Error bars indicate standard deviations. MAb names are color coded according to hierarchical clustering in Fig. 2.
C18 MAbs formed three functional groups based on neutralization profiles.
In addition to providing a measure of neutralizing breadth, the pattern of relative neutralizing potency across a diverse panel of HCVpps (i.e., the neutralization profile) is unique to each MAb and can be used to identify functional relationships among MAbs (36, 37). We have previously demonstrated that polymorphisms distant from binding epitopes can have significant effects on neutralization sensitivity, so functional relationships among MAbs are not always predicted by binding to shared or distinct antigenic sites (36, 37). We measured pairwise Pearson correlations between neutralization profiles of C18 MAbs and previously published neutralization profiles of reference MAbs (31, 36) to identify MAbs with the most closely related neutralization profiles (Fig. 8). As an internal control, we also repeated neutralization testing of reference MAb HEPC74 in parallel with testing of C18 MAbs. As expected, the neutralization profile of this repeat test of HEPC74 (HEPC74-test) correlated more closely with the previously published HEPC74 neutralization profile (HEPC74-reference) than with any other reference MAb, demonstrating that neutralization profiles of individual MAbs are distinct and reproduceable across independent experiments (Fig. 8A). C18 MAbs fell into three functional groups, which were designated AR3-like, AR1-like, and AR5-like based upon the reference MAb to which they were most functionally similar (Fig. 8B).
FIG 8.

C18 MAbs formed three functional groups based on neutralization profiles. Neutralization of 18 HCVpps by 50 μg/ml of each MAb was measured in duplicate, generating a neutralization profile for each MAb (i.e., a ranking of HCVpp strains by relative neutralization sensitivity). Pearson’s correlations were measured between these C18 MAb neutralization profiles and neutralization profiles of reference MAbs. (A) Neutralization testing of reference MAb HEPC74 was performed in parallel with testing of C18 MAbs. Correlation was measured between the neutralization profile of this repeat test of HEPC74 (HEPC74-test) and the previously published HEPC74 neutralization profile (HEPC74-reference, left) or an unrelated MAb (AR5A-reference, right). Each point indicates a single HCVpp, with rank of neutralization sensitivity to one MAb on the x axis and another MAb on the y axis. (B) Pearson r values of pairwise correlations between neutralization profiles of each C18 MAb (y axis) and each reference MAb (x axis). Only r values that are statistically significant (P < 0.05) are shown, and darker green color indicates a stronger positive correlation. The highest r value for each C18 MAb is boxed and in bold type. C18 MAbs clustered into three functional groups, namely, AR3-like, AR1-like, and AR5-like.
In many cases, neutralization profiles of C18 MAbs correlated best with neutralization profiles of reference MAbs that bound to the same antigenic site. As shown in Fig. 8B, neutralization profiles of C18 MAbs HEPC153, HEPC122, and HEPC154, which each target the AR3 antigenic site, showed the greatest correlation with reference MAbs AR3B, HEPC43, and AR3A, respectively, which are also AR3-site MAbs. Neutralization profiles of HEPC111 and HEPC130, which bind at the AR4-5 antigenic site, each showed the greatest correlation with reference MAb AR5A, which also binds at this site. Similarly, the neutralization profile of HEPC167, which binds at the AR1 antigenic site, correlated best with the neutralization profile of the AR1A reference MAb.
In other cases, neutralization profiles of C18 MAbs could not be predicted from the antigenic site targeted. Unexpectedly, the neutralization profile of HEPC151-1, which bound at the AR3 site, correlated best with reference MAb AR1A. The neutralization profile of HEPC108, which bound the AS108 site, correlated best with the neutralization profile of AR3A. Neutralization profiles of HEPC158 and HEPC151-2, two other MAbs binding at AS108, correlated best with the neutralization profile of reference MAb AR1A, as did the neutralization profile of HEPC112, the MAb which bound exclusively to residues on the E1 protein. HEPC146, which bound at the CD81 binding loop of E2, correlated best with reference MAb AR5A. Taken together, these results demonstrate that C18 MAbs fell into AR3-like, AR1-like, and AR5-like functional groups. In many but not all cases, neutralization profiles of C18 MAbs were predicted by the antigenic site targeted. These results highlight the functional significance of small variations in binding residues targeted, as well as the important but still poorly understood role that polymorphisms distant from MAb binding sites play in bNAb resistance.
C18 MAbs displayed complementary neutralizing breadth.
Since C18 MAbs fell into three functional groups based on their neutralization profiles, we hypothesized that these MAbs might display complementation of neutralizing breadth, i.e., that HCV strains resistant to one MAb might be sensitive to another, leading to improved neutralizing breadth of the MAbs in combination. It should be noted that this was not an analysis of synergy or cooperativity between MAbs but rather a comparison of neutralizing breadth of individual MAbs versus combinations when the total MAb concentration was held constant. We assessed for complementation using two different combinations of three MAbs. For each combination, one MAb was selected from the AR3-like, AR1-like, and AR5-like functional groups. Individual MAbs were tested at 50 μg/ml for neutralization of the panel of 19 HCVpps and 5 HCVccs (24 strains total), and each combination was tested at a 50-μg/ml total antibody concentration (i.e., 16.7 μg/ml of each MAb), with neutralization defined as ≥50% inhibition. As shown in Fig. 9A, HEPC146, HEPC153, and HEPC151-1 individually neutralized 15, 19, and 19 strains, respectively, whereas the combination neutralized 22 of 24 strains, confirming that the individual MAbs were complementary. Results with the second MAb combination were similar (Fig. 9B). HEPC111, HEPC153, and HEPC158 individually neutralized 16, 19, and 2 HCVpps, respectively. Despite the poor neutralizing activity of HEPC158, these three MAbs in combination neutralized 21 of 24 strains, which was more than any individual MAb. Notably, strain 1a129 HCVpp (genotype 1a) and strain S52 HCVcc (genotype 3a) were resistant to neutralization by both MAb combinations, and these strains were also resistant to neutralization by every individual C18 MAb (Fig. 9 and 6), making it unlikely that they would be effectively neutralized by any combination of C18 MAbs. Together, these results confirm that MAbs with distinct neutralization profiles arising in the same individual can display complementary neutralizing breadth but that relatively rare HCV strains demonstrate resistance to multiple individual bNAbs as well as bNAb combinations.
FIG 9.
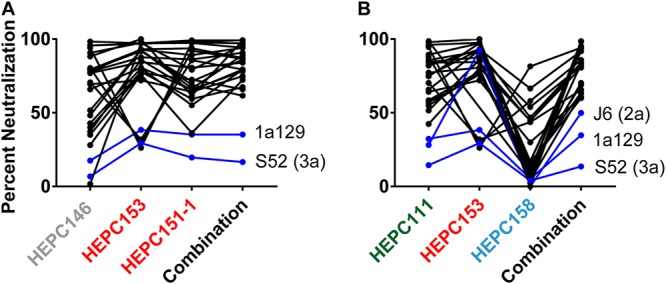
C18 MAbs displayed complementary neutralizing breadth. C18 MAbs were tested for neutralization of the HCVpp and HCVcc panels individually at 50 μg/ml or in two different 3-MAb combinations at 50-μg/ml total antibody concentration (i.e., 16.7 μg/ml of each MAb), namely, (A) HEPC146, HEPC153, and HEPC151-1; and (B) HEPC111, HEPC153, and HEPC158. Lines connect neutralization of each HCV strain by each individual MAb and the 3-MAb combination. Blue lines indicate strains not neutralized >50% by the MAb combination. MAb names are color coded according to hierarchical clustering in Fig. 2. Percent neutralization values are the average of duplicate measurements.
DISCUSSION
In this study, we isolated 13 E1E2-specific MAbs from B cells of a single HCV-infected donor who cleared 1 genotype 1a infection and then became persistently infected with a second genotype 1a strain. Based on analysis of probable binding residues, these MAbs bound six distinct antigenic sites on the E1 protein, the E2 protein, or the E1E2 heterodimer, including three sites (AS108, AS112, and AS146) that are distinct from previously described AR1–5 and E1 sites. Notably, MAbs targeting four antigenic sites (AR3, AR4-5, AS108, and AS146) were broadly neutralizing. These MAbs also displayed distinct patterns of relative neutralizing potency (i.e., neutralization profiles) across a panel of diverse HCV strains, which led to complementary neutralizing breadth when they were tested in combination.
These data support prior studies showing that human MAbs targeting the AR3, AR4, and AR5 antigenic sites are often broadly neutralizing (18, 19) and that bNAbs targeting the AR3 site develop commonly in individuals who clear HCV infection (31, 32). In contrast with prior studies, we found that some MAbs targeting non-AR3 and non-AR4-5 antigenic sites also displayed broad neutralizing activity. The identification here of three non-AR1–5 antigenic sites targeted by neutralizing antibodies (AS108, AS112, and AS146) suggests that MAb isolation from additional human donors could continue to identify additional bNAb epitopes.
The E1-specific MAb HEPC112 characterized in this study is of particular interest since few human MAbs targeting the E1 protein have previously been described. Prior to this study, two regions of E1 were identified as MAb targets. IGH526 and the related MAb IGH505, which have neutralizing activity across multiple HCV strains, bind predominantly to a linear epitope spanning amino acids 313 to 327 near the C terminus of E1 (30, 39). A weakly neutralizing MAb designated H-111 binds to a linear epitope spanning amino acids 192 to 211 at the N terminus of E1 (40). In contrast, HEPC112 appears to bind a distinct, discontinuous site predominantly spanning amino acids 215 to 299. Although the neutralizing breadth of HEPC112 was relatively narrow, isolation of this conformation-sensitive MAb with cross-reactivity across multiple heterologous HCV strains suggests that E1 could be a useful target for vaccine development.
In addition to the notable neutralizing breadth of individual MAbs, we also found that bNAbs arising in subject C18 displayed complementary neutralizing breadth. It is possible that this enhanced neutralizing breadth due to the expression of multiple bNAbs in combination explains the ability of subject C18 to clear one genotype 1a infection. However, the timing of development of these MAbs remains unclear. In addition, one strain in the heterologous HCVpp panel (1a129) and one HCVcc strain (S52) were resistant to neutralization by all individual MAbs and both MAb combinations tested. These holes in the neutralizing repertoire of subject C18, represented by strain 1a129 and S52 resistance, might explain the persistence of this subject’s second infection. Further studies using autologous viral envelopes will be necessary to test this hypothesis.
This study has important implications for HCV vaccine development. The identification of probable binding residues that were targeted by multiple bNAbs, as well as probable binding residues shared by bNAbs and weakly neutralizing MAbs, could guide the engineering of vaccine antigens to favor the induction of antibodies against broadly neutralizing epitopes, while limiting the induction of antibodies targeting nonneutralizing domains. This study also suggests that simultaneous induction of bNAbs targeting multiple E1E2 domains may be advantageous, so incorporation into a vaccine of full-length E1E2 might provide an opportunity for induction of multiple, complementary bNAbs.
The alanine scanning and binding competition assays used to identify antigenic sites targeted by C18 MAbs each have limitations. Since most MAbs in this study bind to conformational epitopes, positions at which mutation to alanine reduced binding could be either energetically important MAb-E1E2 contact residues or they could be positions that influence MAb binding allosterically by altering protein conformation. This is a particularly complex problem for AR4-5-like MAbs, which require both E1 and E2 in a native complex for binding. We controlled for protein folding effects by discounting “global misfolding positions” that eliminated binding of all conformational E1 and E1E2-specific or all conformational E2 and E1E2-specific MAbs since these mutations likely reduced binding by causing misfolding of the E1 or E2 proteins. Nevertheless, some positions identified as probable binding residues may not be true epitope residues. Binding competition assays have limitations as well. It is well established that some MAbs binding to adjacent but nonoverlapping epitopes can compete for E1E2 binding (22, 41–43). Conversely, HEPC74 and AR3C, two MAbs now structurally proven to bind largely overlapping epitopes (34), displayed minimal binding competition in a prior study (31). Given these limitations, fine resolution of epitopes targeted by these MAbs may ultimately require generation of MAb-E1E2 crystal structures.
An additional limitation of this study was the lack of reference MAbs binding to domains A–E. It is possible that AS108, AS146, and AS112 are more closely related to these domains than to AR1–5. However, domains A–E fall in E2 only, so they are not likely to be closely related to AS112, which falls in E1. Based on previously published critical binding residues (26), domain C MAb CBH-7 shares some but not all critical binding residues with AR1 and AS108 MAbs. However, unlike weakly neutralizing CBH-7 and AR1A, some AS108 MAbs were broadly neutralizing. Further studies are needed to better define the differences between the overlapping AS108/AR1/domain C regions of the E2 protein.
In summary, we isolated 13 HCV envelope protein-specific MAbs from B cells of a single HCV-infected individual who cleared 1 genotype 1a infection and then became persistently infected with a second genotype 1a strain. These MAbs bound six distinct antigenic sites on the E2 protein, the E1E2 heterodimer, or at a discontinuous site near the N terminus of E1. Three of these sites (AS108, AS112, and AS146) were distinct from previously described AR1–5 and E1 sites. Notably, MAbs targeting four antigenic sites were broadly neutralizing (AR3, AR4-5, AS108, and AS146), and MAbs also displayed complementary neutralizing breadth when they were tested in combination. Overall, this study demonstrates that HCV bNAb epitopes are not restricted to previously described antigenic sites, expanding the number of sites that could be targeted for vaccine development.
MATERIALS AND METHODS
Study subjects.
Peripheral blood mononuclear cells (PBMCs) were obtained from the BBAASH cohort (44). The study was approved by the Institutional Review Board of Johns Hopkins Hospital, and informed consent was obtained from all study participants.
Cell lines.
HEK293T cells and Hep3B cells were obtained from American Type Culture Collection (ATCC).
HCV viral load and serology testing.
HCV viral loads (IU/ml) were quantified after RNA extraction with the use of commercial real-time reagents (Abbot HCV real-time assay) migrated onto a research-based real-time PCR platform (Roche 480 LightCycler). HCV seropositivity was determined using the Ortho HCV version 3.0 ELISA test system (Ortho Clinical Diagnostics).
C18 MAb isolation.
Human hybridomas were generated as described previously (31). In brief, cryopreserved PBMC samples from subject C18 were transformed with EBV, CpG, and additional supplements. After 7 days, cells from each well of the 384-well culture plates were expanded into four 96-well culture plates using cell culture medium containing irradiated heterologous human PBMCs and incubated for an additional 4 days. B cell culture supernatants were screened in ELISAs with E1E2-transfected cell lysates and in neutralization assays with HCVpp using autologous (strain 1a115) or heterologous (strain 1b38) HCV E1E2. Cells from wells with supernatants reacting with HCV antigens were fused with HMMA2.5 myeloma cells using an electrofusion technique. After fusion, hybridoma cell lines were cloned by limited dilutions and single-cell fluorescence-activated cell sorting and expanded in postfusion medium as previously described. HiTrap protein G or HiTrap MabSelect Sure columns were used to purify HCV-specific antibodies from filtered cell culture supernatants.
Alanine scanning and identification of probable binding residues.
Alanine scanning mutagenesis was performed using a comprehensive HCV E1E2 mutation library (genotype 1a, strain H77) in which every residue of the E1E2 proteins was individually mutated to alanine. E1E2 alanine residues were mutated to serine (45). Cells expressing HCV E1E2 mutants were fixed, permeabilized, and immunostained with the indicated antibodies in duplicate. Mean cellular fluorescence was detected using an Intellicyt flow cytometer. To identify probable binding residues for each MAb, E1E2 residues were ranked from greatest to least effect of alanine mutation on MAb binding. Global misfolding positions, defined as residues at which alanine mutation reduced binding of all E1 and E1E2-specific MAbs, or all E2 and E1E2-specific MAbs to <75% of binding to wild type (121 of 555 E1E2 residues), were excluded as potential binding residues. For each MAb, after exclusion of global misfolding positions, up to 15 mutations with greatest effect on binding that also reduced binding by at least 50% relative to wild-type protein binding were selected as probable binding residues. The maximum number of probable binding residues was chosen based crystal structures of antibody/antigen complexes, demonstrating that the average epitope includes 15 ± 4 contact residues (46).
Hierarchical clustering of MAbs based on relative binding across the E1E2 alanine scanning library.
Hierarchical clustering analysis of alanine-scanning E1E2-binding values for each MAb was performed using code in R developed by Pierce et al. (26). Prior to analysis, 0% binding values were converted to 1%, and all values were log10 transformed. Global misfolding positions were excluded. Ward’s minimum variance method in the hclust R package was used, and an unrooted clustering tree was created with the ape R library. Bootstrap values for each cluster were generated from 10,000 replicates using the approximately unbiased method in the pvclust R package (47).
E1E2 ELISA.
ELISAs were performed as previously described (31). 293T cells were transfected with strain 1a31 or 1a53 E1E2 expression plasmids and then lysed and used to coat ELISA plates. The E1E2 strain was chosen for each competition-binding experiment to ensure adequate binding of all biotinylated MAbs. HEPC122 and HEPC153 were tested using strain 1a53, and all other MAbs were tested using strain 1a31. Blocking MAbs were added at 20 μg/ml, and biotinylated testing MAbs were added at either 0.4 μg/ml (HEPC122 and HEPC153) or at 2 μg/ml (all other C18 MAbs). For denatured protein ELISA, E1E2 was boiled for 5 min in Tris-buffered saline (TBS)-10% fetal calf serum (FCS) containing 1.0% sodium dodecyl sulfate and 50 mM dithiothreitol prior to addition to Galanthus nivalis (GNA)-lectin-coated ELISA plates.
HCVpp production and neutralization.
HCVpps were produced by lipofectamine-mediated transfection of HCV E1E2, pNL4-3.Luc.R-E, and pAdVantage (Promega) plasmids into HEK293T cells as previously described (11, 36, 48). Only HCVpps with an average relative light unit (RLU) value >10× mock HCVpp values were used. Neutralization assays were performed as described previously with MAbs at a 50-μg/ml concentration with the exception of HEPC141 (40 μg/ml), or with serial 5-fold dilutions of MAbs starting at 100-μg/ml concentration (49). Fifty percent inhibitory concentrations (IC50) were calculated from neutralization curves using nonlinear regression in Prism v7 (GraphPad Software).
HCVcc neutralization assays.
Chimeric genotype 2–6 HCVcc constructs (50, 51) were a gift of Jens Bukh, (Copenhagen University Hospital, Copenhagen, Denmark). HCVcc neutralization assays were performed as described elsewhere (52). Briefly, human hepatoma Huh7.5.1 cells (a gift of Charles Rice, The Rockefeller University, New York City, New York, USA) were maintained in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and nonessential amino acids. A total of 8,000 Huh7.5.1 cells per well were plated in flat-bottom 96-well tissue culture plates and incubated overnight at 37°C. The following day, HCVccs were mixed with MAb (50 μg/ml) then incubated at 37°C for 1 h. Medium was removed from the cells and replaced with 50 μl of the HCVcc/antibody mixture. The plates were placed in a CO2 incubator at 37°C overnight, after which the HCVccs were removed and replaced with 100 μl of Huh7.5.1 medium and incubated for 48 hours at 37°C. After 48 hours, the medium was removed and cells were fixed and stained with primary anti-NS5A antibody 9E10 (a gift from Charles Rice, The Rockefeller University, New York City, New York, USA) at a 1:2,000 dilution for 1 hour at room temperature and with secondary antibody Dylight 488-conjugated goat anti-mouse IgG (Abcam) at a 1:500 dilution for 1 hour at room temperature. Images were acquired and spot-forming units were counted in the presence of MAb (HCVccSFUtest) or media only (HCVccSFUcontrol) using an AID iSpot reader spectrum operating AID ELISpot reader v7.0. Percent neutralization was calculated as 100% × (1−[HCVccSFUtest/HCVccSFUcontrol]).
Antibody variable region gene sequencing.
Antibody heavy chain- and light chain-variable region genes were sequenced from antigen-specific hybridoma lines that had been cloned biologically from flow cytometry. Total RNA was extracted using the RNeasy mini kit (Qiagen). A modified 5′ rapid amplification of cDNA ends (RACE) approach was utilized similar to reference 53. Briefly, 5-μl total RNA was mixed with cDNA synthesis primer mix (10 μM each) and incubated for 2 min at 70°C and then at 42°C to anneal the synthesis primers (1 to 3 min). After incubation, a mix containing 5× first-strand buffer (Clontech), dithiothreitol (DTT; 20 mM), 5′ template switch oligonucleotide (10 μM), deoxynucleoside triphosphate (dNTP) solution (10 mM each), and 10× SMARTScribe reverse transcriptase (Clontech) was added to the primer-annealed total RNA reaction and incubated for 60 min at 42°C. The first-strand synthesis reaction was purified using the Ampure size select magnetic bead kit at a ratio of 0.6× (Beckman Coulter). Following, a single PCR amplification reaction mixture containing 5-μl first-strand cDNA, 2× Q5 high fidelity mastermix (New England BioLabs [NEB]), dNTP (10 mM each), forward universal primer (10 μM), and reverse primer mix (0.2 μM each in heavy chain mix, 0.2 μM each in light chain mix) were subjected to thermal cycling with the following conditions: initial denaturation for 1 min 30 s followed by 30 cycles of denaturation at 98°C for 10 s, annealing at 60°C for 20 s, and extension at 72°C for 40 s, followed by a final extension step at 72°C for 4 min. The first PCR was purified using the Ampure size select magnetic bead kit at a ratio of 0.6× (Beckman Coulter). Amplicon libraries were then prepared according to the Pacific Biosciences multiplex SMRT sequencing protocol and sequenced on a Pacific Biosciences Sequel platform. Raw sequencing data were demultiplexed, and circular consensus sequences (CCS) were determined using the Pacific Biosciences SMRT analysis tool suite. The identities of gene segments and mutations from germ lines were determined by alignment using ImMunoGenetics database (54).
HCV sequence analysis.
Core-E1 nucleotide sequences (366 bp) were amplified from longitudinal plasma samples, and divergence calculated using the Tamura-Nei model in Mega v7.0.21, as previously described (33). E1E2 sequences were aligned using Muscle and trimmed in Bioedit to include only MAb probable binding residues.
Correlation of MAb neutralization profiles.
A neutralization profile was generated for each MAb by ranking each of the 19 HCVpps by greatest neutralization to least neutralization, as previously described (36). Pairwise Pearson correlations were calculated between these neutralization profiles in Prism v7 (GraphPad Software). Neutralization of strain 1a116 HCVpp was not measured in neutralization assays with C18 MAbs used to generate neutralization profiles, so these data were omitted from reference MAb neutralization profiles prior to correlation with C18 MAbs.
Combination neutralization assays.
MAbs were tested individually at a total concentration of 50 μg/ml against each HCVpp or HCVcc in the panel. Neutralization by each combination of MAbs was simultaneously tested at a total concentration of 50 μg/ml (16.7 μg/ml of each MAb).
Accession number(s).
The GenBank accession numbers of E1E2 clones used to generate HCVpp are KJ187972 to KJ187978, KJ187980 to KJ187985, KJ187987 to KJ187990, KM660628, and KM660629).
ACKNOWLEDGMENTS
We thank Brian G. Pierce for the use of his R code as well as Alexander Gooden and Jordan Salas for technical support.
This research was supported by National Institutes of Health grants R01AI127469 (to J.R.B. and J.E.C.) and U19 AI088791 (to J.R.B.) and NIH contract HHSN272201400058C (to B.J.D.). A.I.F. is a Cancer Research Institute Irvington Fellow supported by the Cancer Research Institute. Content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
REFERENCES
- 1.World Health Organization. 2017. Global hepatitis report, 2017. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 2.Gravitz L. 2011. Introduction: a smouldering public-health crisis. Nature 474:S2–S4. doi: 10.1038/474S2a. [DOI] [PubMed] [Google Scholar]
- 3.Midgard H, Bjøro B, Mæland A, Konopski Z, Kileng H, Damås JK, Paulsen J, Heggelund L, Sandvei PK, Ringstad JO, Karlsen LN, Stene-Johansen K, Pettersson JH-O, Dorenberg DH, Dalgard O. 2016. Hepatitis C reinfection after sustained virological response. J Hepatol 64:1020–1026. doi: 10.1016/j.jhep.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Martin TC, Martin NK, Hickman M, Vickerman P, Page EE, Everett R, Gazzard BG, Nelson M. 2013. Hepatitis C virus reinfection incidence and treatment outcome among HIV-positive MSM. AIDS 27:2551–2557. doi: 10.1097/QAD.0b013e32836381cc. [DOI] [PubMed] [Google Scholar]
- 5.Pineda JA, Núñez-Torres R, Téllez F, Mancebo M, García F, Merchante N, Pérez-Pérez M, Neukam K, Macías J, Real LM. 2015. Hepatitis C virus reinfection after sustained virological response in HIV-infected patients with chronic hepatitis C. J Infect 71:571–577. doi: 10.1016/j.jinf.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Martinello M, Grebely J, Petoumenos K, Gane E, Hellard M, Shaw D, Sasadeusz J, Applegate TL, Dore GJ, Matthews GV. 2017. HCV reinfection incidence among individuals treated for recent infection. J Viral Hepat 24:359–370. doi: 10.1111/jvh.12666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hill AM, Nath S, Simmons B. 2017. The road to elimination of hepatitis C: analysis of cures versus new infections in 91 countries. J Virus Erad 3:117–123. [PMC free article] [PubMed] [Google Scholar]
- 8.Reference deleted.
- 9.Osburn WO, Snider AE, Wells BL, Latanich R, Bailey JR, Thomas DL, Cox AL, Ray SC. 2014. Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology 59:2140–2151. doi: 10.1002/hep.27013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pestka JM, Zeisel MB, Blaser E, Schurmann P, Bartosch B, Cosset FL, Patel AH, Meisel H, Baumert J, Viazov S, Rispeter K, Blum HE, Roggendorf M, Baumert TF. 2007. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc Natl Acad Sci U S A 104:6025–6030. doi: 10.1073/pnas.0607026104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, Feinstone SM, Alter H, Rice CM, McKeating JA. 2004. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci U S A 101:10149–10154. doi: 10.1073/pnas.0403519101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osburn WO, Fisher BE, Dowd KA, Urban G, Liu L, Ray SC, Thomas DL, Cox AL. 2010. Spontaneous control of primary hepatitis C virus infection and immunity against persistent reinfection. Gastroenterology 138:315–324. doi: 10.1053/j.gastro.2009.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raghuraman S, Park H, Osburn WO, Winkelstein E, Edlin BR, Rehermann B. 2012. Spontaneous clearance of chronic hepatitis C virus infection is associated with appearance of neutralizing antibodies and reversal of T-cell exhaustion. J Infect Dis 205:763–771. doi: 10.1093/infdis/jir835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kinchen VJ, Zahid MN, Flyak AI, Soliman MG, Learn GH, Wang S, Davidson E, Doranz BJ, Ray SC, Cox AL, Crowe JE Jr, Bjorkman PJ, Shaw GM, Bailey JR. 2018. Broadly neutralizing antibody mediated clearance of human hepatitis C virus infection. Cell Host Microbe 24:717–730.e715. doi: 10.1016/j.chom.2018.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Keck Z-Y, Saha A, Xia J, Wang Y, Lau P, Krey T, Rey FA, Foung SK. 2011. Mapping a region of hepatitis C virus E2 that is responsible for escape from neutralizing antibodies and a core CD81-binding region that does not tolerate neutralization escape mutations. J Virol 85:10451–10463. doi: 10.1128/JVI.05259-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Keck Z-Y, Li SH, Xia J, von Hahn T, Balfe P, McKeating JA, Witteveldt J, Patel AH, Alter H, Rice CM, Foung SK. 2009. Mutations in hepatitis C virus E2 located outside the CD81 binding sites lead to escape from broadly neutralizing antibodies but compromise virus infectivity. J Virol 83:6149–6160. doi: 10.1128/JVI.00248-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kong L, Giang E, Robbins JB, Stanfield RL, Burton DR, Wilson IA, Law M. 2012. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc Natl Acad Sci U S A 109:9499–9504. doi: 10.1073/pnas.1202924109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giang E, Dorner M, Prentoe JC, Dreux M, Evans MJ, Bukh J, Rice CM, Ploss A, Burton DR, Law M. 2012. Human broadly neutralizing antibodies to the envelope glycoprotein complex of hepatitis C virus. Proc Natl Acad Sci U S A 109:6205–6210. doi: 10.1073/pnas.1114927109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Law M, Maruyama T, Lewis J, Giang E, Tarr AW, Stamataki Z, Gastaminza P, Chisari FV, Jones IM, Fox RI, Ball JK, McKeating JA, Kneteman NM, Burton DR. 2008. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat Med 14:25–27. doi: 10.1038/nm1698. [DOI] [PubMed] [Google Scholar]
- 20.Hadlock KG, Lanford RE, Perkins S, Rowe J, Yang Q, Levy S, Pileri P, Abrignani S, Foung SK. 2000. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J Virol 74:10407–10416. doi: 10.1128/JVI.74.22.10407-10416.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keck Z-Y, Olson O, Gal-Tanamy M, Xia J, Patel AH, Dreux M, Cosset F-L, Lemon SM, Foung SK. 2008. A point mutation leading to hepatitis C virus escape from neutralization by a monoclonal antibody to a conserved conformational epitope. J Virol 82:6067–6072. doi: 10.1128/JVI.00252-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keck Z, Wang W, Wang Y, Lau P, Carlsen TH, Prentoe J, Xia J, Patel AH, Bukh J, Foung SK. 2013. Cooperativity in virus neutralization by human monoclonal antibodies to two adjacent regions located at the amino terminus of hepatitis C virus E2 glycoprotein. J Virol 87:37–51. doi: 10.1128/JVI.01941-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krey T, Meola A, Keck ZY, Damier-Piolle L, Foung SK, Rey FA. 2013. Structural basis of HCV neutralization by human monoclonal antibodies resistant to viral neutralization escape. PLoS Pathog 9:e1003364. doi: 10.1371/journal.ppat.1003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansson DX, Voisset C, Tarr AW, Aung M, Ball JK, Dubuisson J, Persson MA. 2007. Human combinatorial libraries yield rare antibodies that broadly neutralize hepatitis C virus. Proc Natl Acad Sci U S A 104:16269–16274. doi: 10.1073/pnas.0705522104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keck Z-Y, Xia J, Wang Y, Wang W, Krey T, Prentoe J, Carlsen T, Li AY-J, Patel AH, Lemon SM, Bukh J, Rey FA, Foung SK. 2012. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog 8:e1002653. doi: 10.1371/journal.ppat.1002653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pierce BG, Keck ZY, Lau P, Fauvelle C, Gowthaman R, Baumert TF, Fuerst TR, Mariuzza RA, Foung SK. 2016. Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: implications for vaccine design. Proc Natl Acad Sci U S A 113:E6946–E6954. doi: 10.1073/pnas.1614942113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinchen VJ, Cox AL, Bailey JR. 2018. Can broadly neutralizing monoclonal antibodies lead to a hepatitis C virus vaccine? Trends Microbiol 26:854–864. doi: 10.1016/j.tim.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong L, Jackson KN, Wilson IA, Law M. 2015. Capitalizing on knowledge of hepatitis C virus neutralizing epitopes for rational vaccine design. Curr Opin Virol 11:148–157. doi: 10.1016/j.coviro.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gopal R, Jackson K, Tzarum N, Kong L, Ettenger A, Guest J, Pfaff JM, Barnes T, Honda A, Giang E, Davidson E, Wilson IA, Doranz BJ, Law M. 2017. Probing the antigenicity of hepatitis C virus envelope glycoprotein complex by high-throughput mutagenesis. PLoS Pathog 13:e1006735. doi: 10.1371/journal.ppat.1006735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meunier J-C, Russell RS, Goossens V, Priem S, Walter H, Depla E, Union A, Faulk KN, Bukh J, Emerson SU, Purcell RH. 2008. Isolation and characterization of broadly neutralizing human monoclonal antibodies to the e1 glycoprotein of hepatitis C virus. J Virol 82:966–973. doi: 10.1128/JVI.01872-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bailey JR, Flyak AI, Cohen VJ, Li H, Wasilewski LN, Snider AE, Wang S, Learn GH, Kose N, Loerinc L, Lampley R, Cox AL, Pfaff JM, Doranz BJ, Shaw GM, Ray SC, Crowe JE Jr. 2017. Broadly neutralizing antibodies with few somatic mutations and hepatitis C virus clearance. JCI Insight 2:92872. doi: 10.1172/jci.insight.92872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merat SJ, Molenkamp R, Wagner K, Koekkoek SM, van de Berg D, Yasuda E, Bohne M, Claassen YB, Grady BP, Prins M, Bakker AQ, de Jong MD, Spits H, Schinkel J, Beaumont T. 2016. Hepatitis C virus broadly neutralizing monoclonal antibodies isolated 25 years after spontaneous clearance. PLoS One 11:e0165047. doi: 10.1371/journal.pone.0165047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rose R, Lamers SL, Massaccesi G, Osburn W, Ray SC, Thomas DL, Cox AL, Laeyendecker O. 2018. Complex patterns of hepatitis-C virus longitudinal clustering in a high-risk population. Infect Genet Evol 58:77–82. doi: 10.1016/j.meegid.2017.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flyak AI, Ruiz S, Colbert MD, Luong T, Crowe JE Jr, Bailey JR, Bjorkman PJ. 2018. HCV broadly neutralizing antibodies use a CDRH3 disulfide motif to recognize an E2 glycoprotein site that can be targeted for vaccine design. Cell Host Microbe 24:703–716.e703. doi: 10.1016/j.chom.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan CH, Hadlock KG, Foung SK, Levy S. 2001. V(H)1-69 gene is preferentially used by hepatitis C virus-associated B cell lymphomas and by normal B cells responding to the E2 viral antigen. Blood 97:1023–1026. doi: 10.1182/blood.V97.4.1023. [DOI] [PubMed] [Google Scholar]
- 36.Bailey JR, Wasilewski LN, Snider AE, El-Diwany R, Osburn WO, Keck Z, Foung SK, Ray SC. 2015. Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. J Clin Invest 125:437–447. doi: 10.1172/JCI78794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Diwany R, Cohen VJ, Mankowski MC, Wasilewski LN, Brady JK, Snider AE, Osburn WO, Murrell B, Ray SC, Bailey JR. 2017. Extra-epitopic hepatitis C virus polymorphisms confer resistance to broadly neutralizing antibodies by modulating binding to scavenger receptor B1. PLoS Pathog 13:e1006235. doi: 10.1371/journal.ppat.1006235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prentoe J, Velazquez-Moctezuma R, Foung SK, Law M, Bukh J. 2016. Hypervariable region 1 shielding of hepatitis C virus is a main contributor to genotypic differences in neutralization sensitivity. Hepatology 64:1881–1892. doi: 10.1002/hep.28705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong L, Kadam RU, Giang E, Ruwona TB, Nieusma T, Culhane JC, Stanfield RL, Dawson PE, Wilson IA, Law M. 2015. Structure of hepatitis C virus envelope glycoprotein E1 antigenic site 314-324 in complex with antibody IGH526. J Mol Biol 427:2617–2628. doi: 10.1016/j.jmb.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Keck Z-Y, Sung VM, Perkins S, Rowe J, Paul S, Liang TJ, Lai MM, Foung SK. 2004. Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. J Virol 78:7257–7263. doi: 10.1128/JVI.78.13.7257-7263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang P, Zhong L, Struble EB, Watanabe H, Kachko A, Mihalik K, Virata-Theimer ML, Alter HJ, Feinstone S, Major M. 2009. Depletion of interfering antibodies in chronic hepatitis C patients and vaccinated chimpanzees reveals broad cross-genotype neutralizing activity. Proc Natl Acad Sci U S A 106:7537–7541. doi: 10.1073/pnas.0902749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kachko A, Frey SE, Sirota L, Ray R, Wells F, Zubkova I, Zhang P, Major ME. 2015. Antibodies to an interfering epitope in hepatitis C virus E2 can mask vaccine-induced neutralizing activity. Hepatology 62:1670–1682. doi: 10.1002/hep.28108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Keck Z-Y, Girard-Blanc C, Wang W, Lau P, Zuiani A, Rey FA, Krey T, Diamond MS, Foung SK. 2016. Antibody response to hypervariable region 1 interferes with broadly neutralizing antibodies to hepatitis C virus. J Virol 90:3112–3122. doi: 10.1128/JVI.02458-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cox AL, Netski DM, Mosbruger T, Sherman SG, Strathdee S, Ompad D, Vlahov D, Chien D, Shyamala V, Ray SC, Thomas DL. 2005. Prospective evaluation of community-acquired acute-phase hepatitis C virus infection. Clin Infect Dis 40:951–958. doi: 10.1086/428578. [DOI] [PubMed] [Google Scholar]
- 45.Davidson E, Doranz BJ. 2014. A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology 143:13–20. doi: 10.1111/imm.12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kringelum JV, Nielsen M, Padkjær SB, Lund O. 2013. Structural analysis of B-cell epitopes in antibody:protein complexes. Mol Immunol 53:24–34. doi: 10.1016/j.molimm.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki R, Shimodaira H. 2006. Pvclust: an R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22:1540–1542. doi: 10.1093/bioinformatics/btl117. [DOI] [PubMed] [Google Scholar]
- 48.Hsu M, Zhang J, Flint M, Logvinoff C, Cheng-Mayer C, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins mediate pH-dependent cell entry of pseudotyped retroviral particles. Proc Natl Acad Sci U S A 100:7271–7276. doi: 10.1073/pnas.0832180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dowd KA, Netski DM, Wang XH, Cox AL, Ray SC. 2009. Selection pressure from neutralizing antibodies drives sequence evolution during acute infection with hepatitis C virus. Gastroenterology 136:2377–2386. doi: 10.1053/j.gastro.2009.02.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gottwein JM, Scheel TK, Jensen TB, Lademann JB, Prentoe JC, Knudsen ML, Hoegh AM, Bukh J. 2009. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 49:364–377. doi: 10.1002/hep.22673. [DOI] [PubMed] [Google Scholar]
- 51.Scheel TK, Gottwein JM, Jensen TB, Prentoe JC, Hoegh AM, Alter HJ, Eugen-Olsen J, Bukh J. 2008. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc Natl Acad Sci U S A 105:997–1002. doi: 10.1073/pnas.0711044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wasilewski LN, El-Diwany R, Munshaw S, Snider AE, Brady JK, Osburn WO, Ray SC, Bailey JR. 2016. A hepatitis C virus envelope polymorphism confers resistance to neutralization by polyclonal sera and broadly neutralizing monoclonal antibodies. J Virol 90:3773–3782. doi: 10.1128/JVI.02837-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turchaninova MA, Davydov A, Britanova OV, Shugay M, Bikos V, Egorov ES, Kirgizova VI, Merzlyak EM, Staroverov DB, Bolotin DA, Mamedov IZ, Izraelson M, Logacheva MD, Kladova O, Plevova K, Pospisilova S, Chudakov DM. 2016. High-quality full-length immunoglobulin profiling with unique molecular barcoding. Nat Protoc 11:1599–1616. doi: 10.1038/nprot.2016.093. [DOI] [PubMed] [Google Scholar]
- 54.Brochet X, Lefranc MP, Giudicelli V. 2008. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res 36:W503–W508. doi: 10.1093/nar/gkn316. [DOI] [PMC free article] [PubMed] [Google Scholar]