

Abstract
Anthrax lethal toxin (LT) is known to induce NLRP1B inflammasome activation and pyroptotic cell death in macrophages from certain mouse strains in its metalloprotease activity‐dependent manner, but the underlying mechanism is unknown. Here, we establish a simple but robust cell system bearing dual‐fluorescence reporters for LT‐induced ASC specks formation and pyroptotic lysis. A genome‐wide siRNA screen and a CRISPR‐Cas9 knockout screen were applied to this system for identifying genes involved in LT‐induced inflammasome activation. UBR2, an E3 ubiquitin ligase of the N‐end rule degradation pathway, was found to be required for LT‐induced NLRP1B inflammasome activation. LT is known to cleave NLRP1B after Lys44. The cleaved NLRP1B, bearing an N‐terminal leucine, was targeted by UBR2‐mediated ubiquitination and degradation. UBR2 partnered with an E2 ubiquitin‐conjugating enzyme UBE2O in this process. NLRP1B underwent constitutive autocleavage before the C‐terminal CARD domain. UBR2‐mediated degradation of LT‐cleaved NLRP1B thus triggered release of the noncovalent‐bound CARD domain for subsequent caspase‐1 activation. Our study illustrates a unique mode of inflammasome activation in cytosolic defense against bacterial insults.
Keywords: anthrax lethal toxin, N‐end rule pathway, NLRP1B inflammasome, UBR2
Subject Categories: Immunology; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Vertebrates have evolved both innate and adaptive immune systems to fight against infection, disease, and other unwanted biological invasions. The innate immune system is the first defense line; it recruits various classes of pattern‐recognition receptors (PRRs) to coordinate rapid and versatile responses to these challenges. PRRs, including Toll‐like receptors (TLRs), nucleotide‐binding oligomerization domain‐like receptors (NLRs), and RIG‐I‐like receptors (RLRs), sense pathogen‐associated molecular patterns (PAMPs) or damage‐associated molecular patterns (DAMPs), resulting in the release of pleiotropic effector molecules, reactive oxygen or nitrogen intermediates, costimulatory molecules, chemokines, cytokines, etc. Different stimuli are recognized by specific PRRs that, under certain situations, signals the formation of a large protein platform known as inflammasome for activating caspase‐1 (Martinon et al, 2002). To date, there are five well‐studied canonical inflammasomes—distinguished based on their particular PRRs—including NLRP1, NLRP3, NAIP‐NLRC4, AIM2, and Pyrin inflammasomes (Broz & Dixit, 2016; Zhao & Shao, 2016). Inflammasome‐activated caspase‐1 proteolytically processes pro‐IL‐1β and pro‐IL‐18, which function as “alarm” cytokines to stimulate immune responses to eliminate potential threats. Caspase‐1 also cleaves and activates the pore‐forming protein gasdermin D (GSDMD), causing pyroptotic cell death and thereby massive release of cellular contents (Kayagaki et al, 2015; Shi et al, 2015; Ding et al, 2016).
NLRP1 was the first NLR protein proposed to form an inflammasome complex (Martinon et al, 2002). Humans have one NLRP1 gene, while the C57BL/6 mouse genome bears three polymorphic paralogs, Nlrp1a, 1b, and 1c (Boyden & Dietrich, 2006). In addition to the NOD and LRR domains, typical of the NLR family, NLRP1 has two additional domains at the C‐terminus: a FIIND (function to find domain) and a CARD domain (Tschopp et al, 2003); mouse NLRP1s differ from the human counterpart in lacking the N‐terminal PYD domain (Martinon et al, 2007). The FIIND domain, which is structurally similar to ZU5‐UPA autolytic proteolysis domain (D'Osualdo et al, 2011), undergoes post‐translational autocleavage, which is required for NLRP1 activation (Finger et al, 2012; Frew et al, 2012). Following this proteolytic processing, the N‐terminal and C‐terminal cleavage products of NLRP1 remain associated to be kept in an autoinhibited state, and when dissociated the C‐terminal fragment alone is capable of recruiting ASC for activating caspase‐1 (Frew et al, 2012).
In mouse or rat macrophages, NLRP1B can specifically respond to Bacillus anthracis lethal toxin (LT) and trigger robust caspase‐1 activation (Boyden & Dietrich, 2006). LT contains a functional metalloprotease component lethal factor (LF) and protective antigen (PA), the latter of which confers endocytosis‐mediated entry of LF into mammalian cells (Lacy & Collier, 2002). Mitogen‐activated protein kinase kinases (MEKs) are the first and most established proteolytic substrates of LT (Duesbery et al, 1998; Vitale et al, 2000). However, inhibition of the MAPK signaling does not lead to or affect NLRP1B activation, and efforts linking LF cleavage of MEKs to NLRP1B activation have been fruitless. Interestingly, only NLRP1B variants from certain mouse and rat strains are responsive to LT stimulation (Newman et al, 2010; Hellmich et al, 2012). For example, macrophages from BALB/c and 129/Sv mice are susceptible to LT‐induced caspase‐1 activation, whereas those of the C57BL/6J strain are resistant. More recent studies show that LF indeed directly targets NLRP1B and cleaves it at Lys44, and this cleavage is required for activating the NLRP1B inflammasome (Levinsohn et al, 2012). The fact that NLRP1B from both LT‐sensitive and LT‐resistant mouse strains can be similarly cleaved by LF suggests that cleavage is not sufficient to activate the downstream inflammasome responses (Hellmich et al, 2012). LF‐independent artificial cleavage of NLRP1B at Lys44 can also lead to inflammasome activation, and it has been proposed that NLRP1B may function to sense the protease activity of LF (Chavarria‐Smith & Vance, 2013).
LT‐induced NLRP1B activation can be specifically blocked by the 26S proteasome inhibitors MG132 and lactacystin (Tang & Leppla, 1999). The N‐end rule pathway—a protein‐degradation system that exists in both eukaryotic and prokaryotic cells (Tasaki et al, 2012)—has been implicated to have a possible role in LT‐induced macrophage death (Klimpel et al, 1994). This protein‐degradation system recognizes certain specific N‐terminal residues (N‐degron) of peptides or proteins by UBR box‐containing proteins (N‐recognin) that serve as E3 ligases to ubiquitinate N‐degron‐bearing protein for subsequent degradation. Preliminary data indicate that amino acid derivatives can protect mouse macrophages from LT‐triggered pyroptosis (Wickliffe et al, 2008b). However, there have been no direct or genetic evidences demonstrating the function of the N‐end rule pathway in LT activation of the NLRP1B inflammasome.
To reveal the mechanism of LT‐induced NLRP1B activation, we generated a mouse macrophage cell line stably expressing RFP‐ASC and EGFP. This robust reporter system recapitulated all known properties of LT‐induced NLRP1B inflammasome activation, which allowed for a whole‐genome siRNA screen using RFP‐ASC aggregation and pyroptotic lysis as phenotypic readouts. A genome‐wide CRISPR‐Cas9 screen for LT‐induced pyroptosis was also carried out. Both screens identified the N‐end rule E3 ligase UBR2 as an essential component in LT‐induced inflammasome activation. Further study confirmed the requirement of UBR2 and demonstrated that UBR2 functions together with an E2 ubiquitin‐conjugating enzyme UBE2O in this process. LT triggers NLRP1B activation through UBR2‐mediated ubiquitination and degradation of LT‐cleaved NLRP1B. As NLRP1B undergoes constitutive autocleavage before the C‐terminal CARD, LT‐induced degradation releases NLRP1B C‐terminal CARD domain for caspase‐1 activation. The mechanism of action for LT‐induced NLRP1B activation illustrates a unique mode of inflammasome activation in cytosolic defense against bacterial insults.
Results
A dual‐fluorescence reporter cell system for LT‐induced NLRP1B inflammasome activation
To dissect the mechanism underlying anthrax LT‐induced NLRP1B inflammasome activation, we generated mouse macrophage‐like RAW 264.7 cells stably expressing both RFP‐ASC and EGFP. This RAWRA cell line was highly sensitive to LT treatment, evident from caspase‐1 autoprocessing into the active p10 form and extensive pyroptotic cell death (Fig EV1A and B). Consistent with the previous reports (Squires et al, 2007; Wickliffe et al, 2008a), LT‐induced caspase‐1 activation and pyroptosis in the RAWRA cells could be blocked by the proteasome inhibitor MG132 as well as a high concentration of KCl (Fig EV1A and B). Neither of the two treatments affected LF cleavage of the known substrate MEK3 (Fig EV1A), verifying the intact function of the administrated toxin. In LT‐stimulated RAWRA cells, RFP‐ASC aggregated into specks structures within 1.5 h and the EGFP signal rapidly disappeared due to the lytic property of pyroptosis, which were also blocked by MG132 and KCl (Fig EV1C). Live imaging of the stimulated RAWRA cells revealed the dynamics of RFP‐ASC specks formation and EGFP disappearance, the latter of which always followed the specks formation immediately (Movie EV1). The metalloprotease‐deficient E687C mutant LT caused no such effects, and the cells showed no morphological changes (Movie EV2). When the RAWRA cells were stimulated by LT in the presence of the caspase‐1‐specific inhibitor YVAD, intact RFP‐ASC specks formation still occurred but with no loss of the EGFP signal for at least additional 1.5 h (Movie EV3). Notably, the presence of MG132 completely blocked RFP‐ASC specks formation as well as loss of the cellular EGFP signal; in the absence of inflammasome activation, the LT‐treated cells instead developed evident apoptotic morphologies (Movie EV4), consistent with that suggested in the previous study (Muehlbauer et al, 2007).
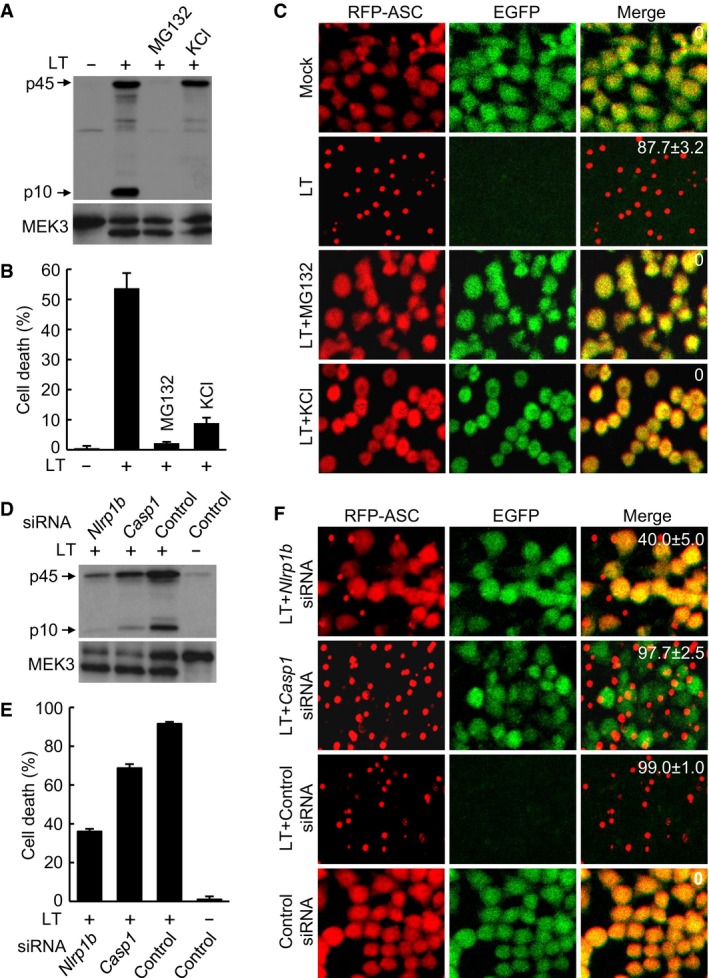
Figure EV1. Establishment of the RAWRA reporter cell system for LT‐induced NLRP1B inflammasome activation.
-
A, BEffects of MG132 and KCl on LT‐induced caspase‐1 activation and pyroptosis. RAWRA cells were treated with WT LT (+) or its protease‐deficient E687C mutant (−) in the presence of MG132 or KCl as indicated. Cell supernatants were subjected to anti‐caspase‐1 immunoblotting, and the lysates were blotted with anti‐MEK3 antibody (A). p10, mature caspase‐1; p45, caspase‐1 precursor. Cell death was measured by LDH release, and data are shown as mean values ± SD (error bar) from three replicates (B).
-
CRFP‐ASC specks formation in RAWRA cells that were treated as in (A) and imaged on a confocal microscopy. The numbers in the merged panel are the percentages of cells showing RFP‐ASC specks. Mock, DMSO.
-
D, EEffect of Nlrp1b or Casp1 knockdown on LT‐induced caspase‐1 activation and pyroptosis. Endogenous Nlrp1b or Casp1 was silenced by a mixture of four pairs of siRNA duplexes for 60 h prior to LT treatment. Culture supernatants were subjected to anti‐caspase‐1 immunoblotting, and the lysates were blotted with anti‐MEK3 antibody (D). Cell death was measured by LDH release, and data are shown as mean values ± SD from three replicates (E).
-
FRFP‐ASC specks formation in RAWRA cells that were treated as in (D) and imaged on a confocal microscopy. The numbers in the merged panel are the percentages of cells showing RFP‐ASC specks.
Source data are available online for this figure.
To determine whether the caspase‐1 activation and cell death that we observed in LT‐treated RAWRA cells were mediated by the NLRP1B inflammasome, small interference RNA (siRNA) knockdown of the inflammasome components was performed. As shown in Fig EV1D, siRNA knockdown of Nlrp1b significantly attenuated caspase‐1 activation, in which intact MEK3 cleavage was still observed. LT‐induced pyroptotic cell death and RFP‐ASC specks formation were also inhibited by siRNA knockdown of Nlrp1b (Fig EV1E and F); agreeing with the YVAD inhibitor data (Movie EV3), knockdown of Casp1 only affected the pyroptosis but showed no inhibition on RFP‐ASC specks formation (Fig EV1E and F). These analyses suggest that the RAWRA reporter cell recapitulates known properties of the NLRP1B inflammasome and represent a powerful model for investigating LT‐induced inflammasome activation in real time.
The N‐end rule pathway is involved in LT‐induced NLRP1B inflammasome activation
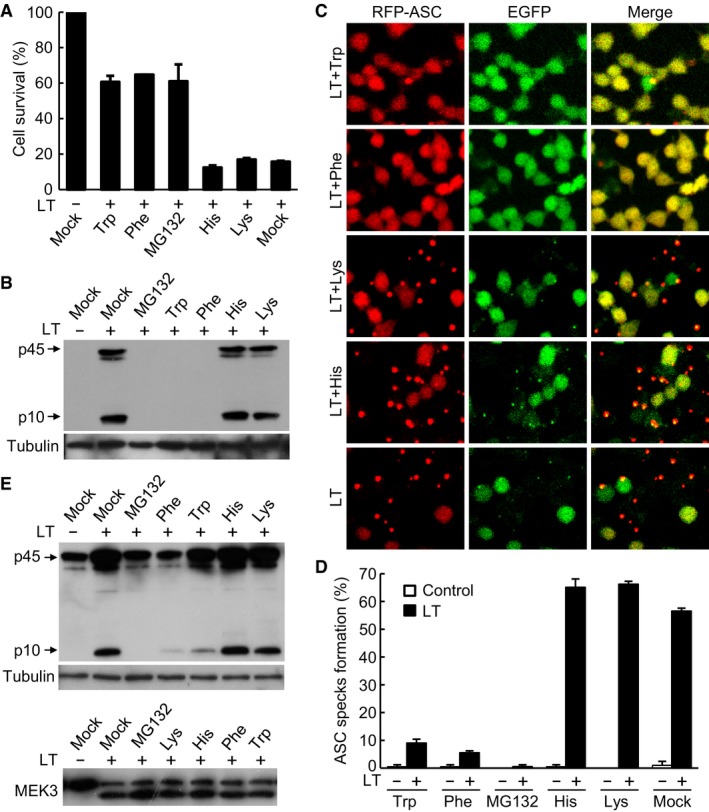
Previous analyses have indicated the involvement of the N‐end rule pathway in LT‐induced NLRP1B inflammasome activation (Gupta et al, 2008; Wickliffe et al, 2008b). In the N‐end rule pathway, two types of N‐terminal amino acids, referred to as N‐degrons, are recognized by specific ubiquitin E3 ligases that catalyze ubiquitin‐chain formation on the N‐degron‐bearing target protein, leading to its degradation by the proteasome. Type 1 N‐degron features a basic residue such as His, Lys, or Arg, while type 2 N‐degron features a bulky hydrophobic amino acid such as Leu, Ile, Phe, Trp, or Tyr. Guided by this knowledge, basic or bulky hydrophobic amino acids are shown to be effective inhibitors of the N‐end rule pathway (Varshavsky, 2011). To probe or confirm the involvement of the N‐end rule pathway in NLRP1B activation, we treated the RAWRA cells with different N‐degron inhibitory amino acids for 30 min prior to LT stimulation. As shown in Fig 1A, more than 60% of the RAWRA cells that had been treated with Trp or Phe survived the LT intoxication, phenocopying the effect of MG132, whereas for both untreated cells and cells that had been treated with basic amino acids His and Lys, less than 20% stayed alive following LT stimulation. Immunoblotting analyses showed that Trp and Phe, but not His and Lys, severely blocked caspase‐1 activation in LT‐treated RAWRA cells (Fig 1B). Results of similar nature were obtained with these amino acid inhibitors when RFP‐ASC specks formation and loss of cellular EGFP were examined (Fig 1C and D). Trp and Phe, but not His and Lys, showed a similar blocking effect on LT‐induced caspase‐1 activation without affecting MEK3 cleavage in immortalized Tlr4 −/− bone marrow‐derived macrophages (BMDMs; Shi et al, 2015; Fig 1E; TLR4 is not involved in LT‐induced NLRP1B activation and the reason to use Tlr4 −/− cells is because the cells were used in other projects carried out at the same time). These data confirm the previous proposal that the N‐end rule pathway might be important for LT‐induced NLRP1B inflammasome activation.
Figure 1. The N‐end rule pathway controls LT‐induced NLRP1B inflammasome activation.
-
A, BEffects of basic and bulky hydrophobic amino acids on LT‐induced pyroptosis and caspase‐1 activation in RAWRA cells. Cells were pre‐treated with 10 mM indicated amino acids prior to LT stimulation. Cell viability was measured by using the ATP assay, and data shown are mean values ± SD from three replicates (A). Mock, DMEM. Cell supernatants were subjected to anti‐caspase‐1 immunoblotting, and the lysates were blotted with the anti‐tubulin antibody (B). p10, mature caspase‐1; p45, caspase‐1 precursor.
-
C, DEffects of basic and bulky hydrophobic amino acids on LT‐induced RFP‐ASC specks formation in RAWRA cells. Fluorescence images were taken on a confocal microscopy (C). The percentages of cells showing RFP‐ASC specks (mean values ± SD from three replicates) are in (D).
-
EEffects of basic and bulky hydrophobic amino acids on LT‐induced caspase‐1 activation in Tlr4 −/− iBMDMs. Cells were treated as that in (A). Cell supernatants were subjected to anti‐caspase‐1 immunoblotting, and the lysates were blotted with the anti‐tubulin or anti‐MEK3 antibody.
Source data are available online for this figure.
The N‐end rule E3 ligase UBR2 is required for NLRP1B inflammasome activation
To explore the mechanism underlying LT‐induced NLRP1B inflammasome activation, we took advantage of the RAWRA reporter cell system and performed a genome‐wide siRNA screen. To increase the robustness and stringency of the screen, two readouts including the RFP‐ASC specks formation and ATP‐based cell viability were assayed, which identified Ubr2 encoding an E3 ligase in the N‐end rule ubiquitination pathway as a high‐confidence hit. The Ubr2‐targeting siRNA mixtures from the QIAGEN siRNA library (used for the screen) significantly inhibited both RFP‐ASC specks formation and cell lysis (Fig 2A). Among the four Ubr2‐targeting siRNAs, the two showing the higher knockdown efficiency were found to exhibit more apparent inhibition of LT‐induced pyroptosis (Fig EV2A and B). When the CRISPR guide RNA (gRNA) library was available in 2014 (Koike‐Yusa et al, 2014), we started to employ this powerful genetic method to identify unknown genes important for inflammasome activation including the cytosolic LPS‐induced noncanonical inflammasome pathway (Shi et al, 2015) as well as LT‐induced canonical NLRP1B inflammasome pathway. For convenience, we used the same Tlr4 −/− iBMDMs that were used in our cytosolic LPS screen for LT‐induced pyroptosis after confirming that this macrophage line had a normal and robust inflammasome activation in response to LT stimulation (Fig 1E). Among the top 30 enriched independent gRNAs recovered from the screen, three genes, Antxr2, Casp1, and Ubr2, were hit by more than one gRNAs (Fig EV2C). Casp1 and Antx2 (encodes the receptor that mediates LT entry into host cells) both are expected hits known to be required for LT‐induced pyroptosis. Of particular note is the identification of a gRNA for Nlrp1a (Fig EV2C); because the gRNA library was designed based on the genome sequence of C57BL/6J mice (Koike‐Yusa et al, 2014), the particular Nlrp1a gRNA hit in our screen indeed targets Nlrp1b in the Tlr4 −/− iBMDMs that were derived from the 129 mice (Hoshino et al, 1999) and do not express Nlrp1a (Boyden & Dietrich, 2006). All of these suggest a high confidence on the Ubr2 hit.
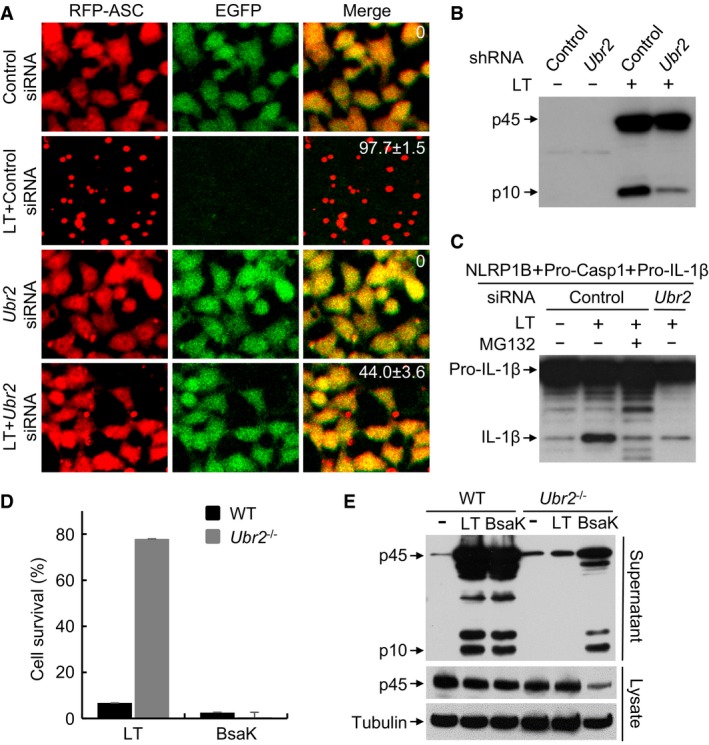
Figure 2. The ubiquitin ligase UBR2 is required for LT‐induced NLRP1B activation.
-
ARFP‐ASC specks formation assay of the effect of Ubr2 knockdown on LT‐induced inflammasome activation. RAWRA cells were transfected with a control siRNA or Ubr2‐targeting siRNA mixtures for 60 h followed by LT treatment. The numbers in the merged panel are the percentages of the cells showing RFP‐ASC specks.
-
BEffect of Ubr2 stable knockdown on caspase‐1 activation in RAWRA cells. Cells stably expressing a control or Ubr2‐targeting shRNA were incubated with WT LT (+) or its E687C mutant (−) for 3 h.
-
CEffects of Ubr2 knockdown (siRNA #05) on LT‐induced NLRP1B inflammasome activation reconstituted in 293T cells. Cells were treated with WT LT (+) or its E687C mutant (−).
-
D, EEffect of Ubr2 knockout on NLRP1B or NAIP2‐NLRC4 inflammasome activation. WT and Ubr2 −/− iBMDMs were treated with LT or BsaK for 3 h. Cell viability was measured by using the ATP assay, and data shown are mean values ± SD from three replicates (D).
Source data are available online for this figure.
Figure EV2. RNAi and CRISPR‐Cas9 screens identify Ubr2 in LT‐induced NLRP1B inflammasome activation.
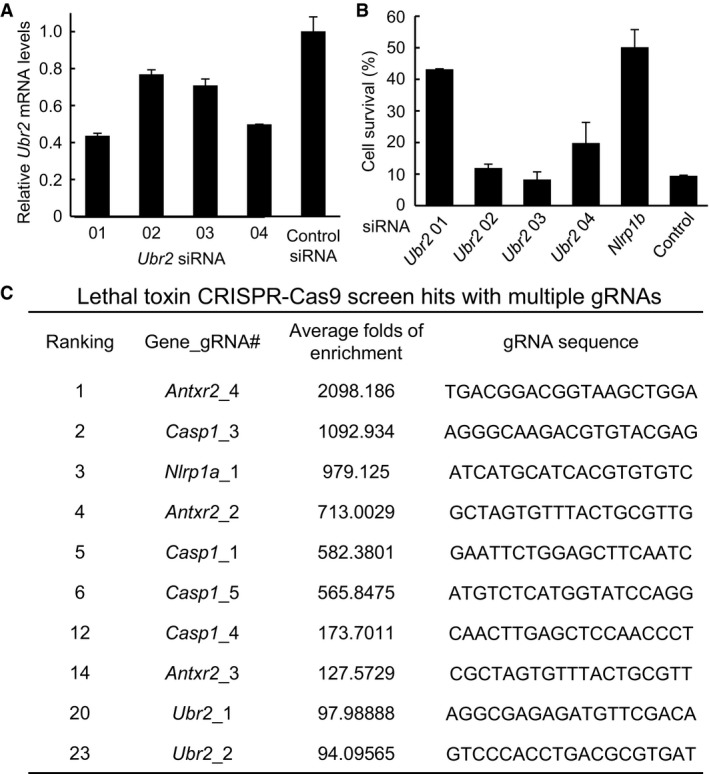
- Ubr2 siRNA knockdown efficiency measured by qPCR (mean values ± SD (error bar) from three replicates.
- Effect of Ubr2 knockdown on LT‐induced RAWRA cell death. RAWRA cells were transfected with indicated siRNA for 60 h followed by LT treatment. Cell viability was measured by using the ATP assay, and data are shown as mean values ± SD from three replicates.
- gRNA hits from a genome‐wide CRISPR‐Cas9 screen of LT‐induced pyroptosis in Tlr4 −/− iBMDMs. Shown are those genes with multiple gRNA hits. The ranking, the average fold increase, and the sequences for each gRNA are listed. Note: The gRNA library used was designed based on the genome sequence of the C57BL/6J strain. The Nlrp1a_1 gRNA in the library indeed matches the Nlrp1b sequence of iBMDMs derived from 129 mice (do not express Nlrp1a), and the five Nlrp1b gRNAs in the library all have 1–3 nucleotide mismatches, compared to the Nlrp1b sequence from the 129 mice.
To further validate the requirement of Ubr2, two additional Ubr2‐specific siRNAs (#05 and #06) and the Ubr2 siRNA mixtures from the QIAGEN library were used to knock down its expression in 129/Sv mice‐derived primary BMDMs, a natural LT‐sensitive cell system. The Ubr2‐targeting siRNAs, similar to the Nlrp1b siRNA, caused a remarkable decrease in LT‐induced caspase‐1 autoprocessing (Fig EV3A). qPCR analyses revealed an approximately 30–50% reduction of Ubr2 mRNA levels in the knockdown BMDMs (Fig EV3B). In contrast, the Ubr1‐targeting siRNAs behaved similarly as the control scramble siRNA and showed no inhibition on LT‐induced caspase‐1 activation (Fig EV3A). We also achieved short‐hairpin RNA (shRNA) stable knockdown of Ubr2 expression in the RAWRA reporter cells (Fig EV3C), in which both caspase‐1 activation and RFP‐ASC specks formation in response to LT stimulation were efficiently blocked (Figs 2B and EV3D). We also succeeded in reconstituting LT‐induced NLRP1B inflammasome activation in nonmacrophage cells by expressing pro‐caspase‐1, pro‐IL‐1β, and 129/Sv mice‐derived NLRP1B in 293T cells. In this system, LT treatment induced efficient mature IL‐1β production, which was sensitive to inhibition by MG132 (Fig EV3E). Notably, siRNA knockdown of endogenous Ubr2 expression in the reconstituted 293T cells showed a similar inhibitory effect on LT‐induced IL‐1β maturation (Fig 2C). Finally, we generated Ubr2 −/− iBMDM cells (on the Tlr4 −/− background) using CRISPR‐Cas9‐mediated targeting. The Ubr2 −/− cells were completely resistant to LT‐induced pyroptosis and caspase‐1 activation but showed a normal inflammasome activation in response to the NAIP2‐NLRC4 inflammasome agonist BsaK (Zhao et al, 2011; Fig 2D and E). These results together provide extensive and definitive genetic proofs for the requirement of the UBR2‐mediated N‐end rule pathway in LT‐induced NLRP1B inflammasome activation.
Figure EV3. The N‐end rule ubiquitin ligase UBR2 is required for LT‐mediated NLRP1B inflammasome activation.
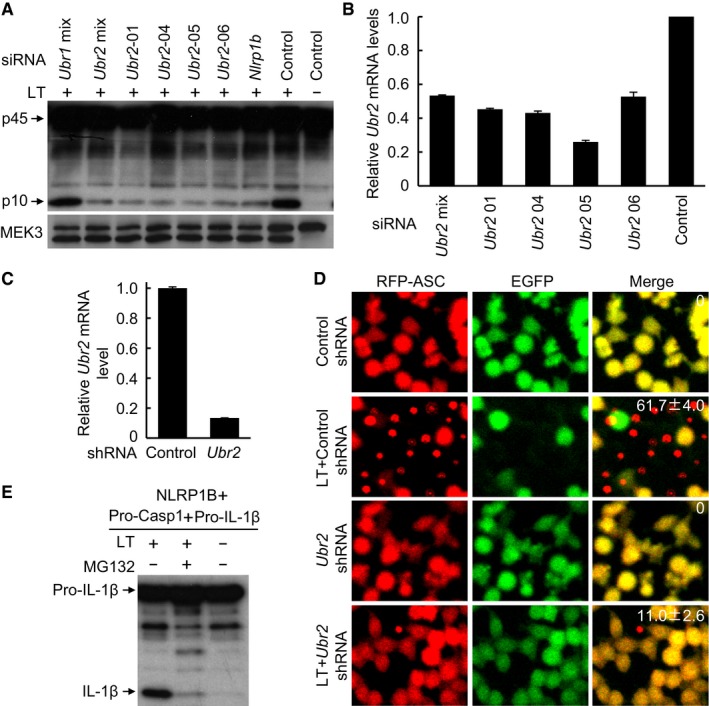
-
A, BEffect of siRNA knockdown of Ubr1, Ubr2, or Nlrp1b on LT‐induced caspase‐1 activation. Endogenous Nlrp1b or Ubrs were silenced for 60 h prior to stimulation with WT LT (+) or its E687C mutant (−). The knockdown efficiency of Ubr2 measured by qPCR is shown as mean values ± SD from three replicates in (B).
-
C, DEffect of shRNA stable knockdown of Ubr2 on LT‐induced RFP‐ASC specks formation in RAWRA cells. Shown in (C) is the knockdown efficiency measured by qPCR (mean values ± SD from three replicates). The percentages of RAWRA cells showing the RFP‐ASC specks (mean values ± SD from three replicates) are marked in the merged fluorescence images in (D).
-
EReconstitution of NLRP1B inflammasome in 293T cells and the effect of MG132 treatment. Pro‐caspase‐1, pro‐IL‐1β, and 129/Sv mice‐derived Nlrp1b expression plasmids were co‐transfected into 293T cells, and cells were stimulated with WT LT (+) or its E687C mutant (−) in the presence or absence of MG132.
Source data are available online for this figure.
UBR2 functions together with UBE2O in mediating NLRP1B inflammasome activation
The mammalian genome encodes 7 UBR box‐containing proteins (UBR1 to UBR7) that are proposed to be involved in the N‐end rule degradation. siRNA knockdown of each of the 7 Ubr genes showed that only the absence of Ubr2 could inhibit LT‐induced caspase‐1 activation in the LT‐sensitive mouse macrophages, suggesting that activation of the NLRP1B inflammasome specifically requires UBR2 among all the N‐end rule ubiquitin E3 ligases (Fig 3A). Given that LT‐induced NLRP1B inflammasome activation is sensitive to the proteasome inhibitor as well as inhibitors for the N‐end rule ubiquitination pathway, it is reasonable to conclude that UBR2 functions as a ubiquitin ligase to mediate LT‐induced inflammasome activation. We then turned to determine which E2 ubiquitin‐conjugating enzyme in the ubiquitination pathway is likely involved in mediating NLRP1B inflammasome activation. To this end, we conducted a small‐scale siRNA knockdown screen of 28 generally accepted E2s in primary BMDMs derived from the 129/Sv mice. Results of the primary screen suggested that knockdown of two E2s, Ube2o or Ube2t, appeared to be capable of suppressing LT‐stimulated caspase‐1 activation (Figs 3B and EV4A); again, proteolytic cleavage of cellular MEK3 was probed to control intact delivery of LT into the cytosol of these E2‐knockdown macrophages (Fig EV4B). As an E3 ligase generally does not work together with two E2 enzymes simultaneously, we then assayed additional Ube2o or Ube2t‐targeting siRNAs to investigate whether one of the E2 enzymes is truly involved in LT‐induced inflammasome activation. Among the seven Ube2o‐specific siRNAs assayed, four of them showed an evident inhibition on LT‐induced caspase‐1 activation, while the other three had no effect, which well correlated with the knockdown efficiency of the seven siRNAs (Fig 3C and D). For Ube2t, neither of the two highly effective siRNAs (#02 and #03) could block LT‐induced caspase‐1 activation, while the less effective siRNA (#01) instead showed an inhibitory effect (Fig EV4C and D), suggesting that Ube2t is a false positive out of the siRNA screen. Thus, UBE2O is the bona fide E2 enzyme controlling LT‐induced NLRP1B inflammasome activation. Consistently, siRNA knockdown of Ube2o expression in the engineered RAWRA cells efficiently blocked RFP‐ASC specks formation as well as release of cellular EGFP in response to LT stimulation (Fig 3E). Lastly, co‐immunoprecipitation analyses in transfected 293T cells revealed a physical association between UBR2 and UBE2O (Fig 3F). Taken together, these results suggest that the N‐end rule E3 ligase UBR2 functions together with the ubiquitin‐conjugating enzyme UBE2O in mediating LT‐induced NLRP1B inflammasome activation.
Figure 3. UBE2O functions together with UBR2 to mediate NLRP1B inflammasome activation.
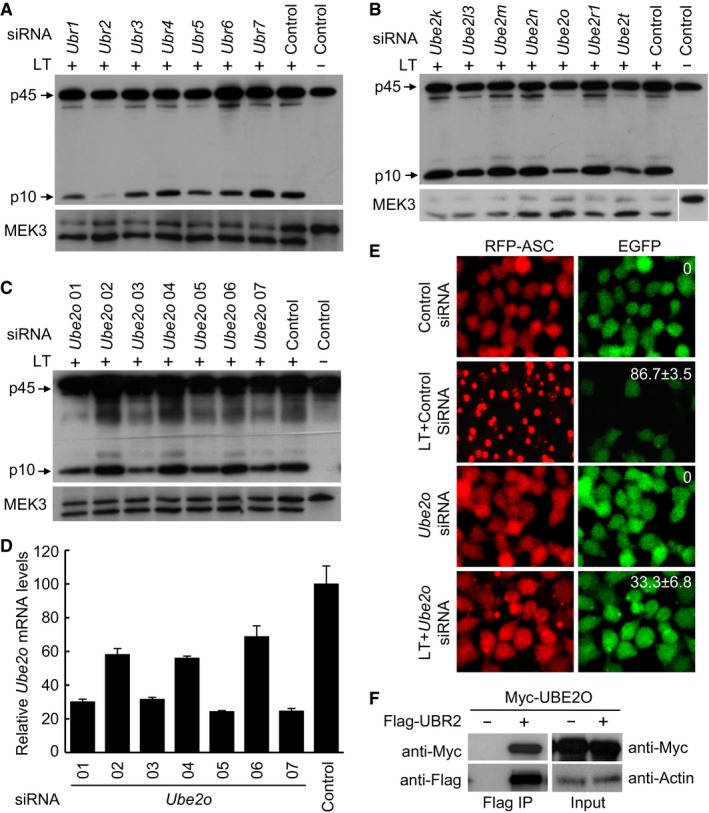
-
AImmunoblotting of caspase‐1 activation in Ubrs‐knockdown BMDMs derived from the 129/Sv mice. Cells were transfected with the indicated siRNA for 60 h, and then treated with WT LT (+) or its E687C mutant (−) for 3 h.
-
B–DImmunoblotting of caspase‐1 activation in E2s‐knockdown BMDMs derived from the 129/Sv mice. Additional Ube2o siRNAs were used to confirm the inhibitory effect of Ube2o knockdown on LT‐induced caspase‐1 activation (C). Ube2o knockdown efficiency was measured by qPCR, and data are shown as mean values ± SD from three replicates (D).
-
ERFP‐ASC specks formation assay of the effect of Ube2o knockdown (siRNA Ube2o‐01) on LT‐induced inflammasome activation in RAWRA cells. The percentages of cells showing RFP‐ASC specks (mean values ± SD from three replicates) are marked on the images.
-
FCo‐immunoprecipitation interaction of UBE2O and UBR2. Myc‐UBE2O and Flag‐UBR2 were co‐expressed in 293T cells, and cell lysates were subjected to anti‐Flag immunoprecipitation followed by immunoblotting analyses as shown.
Source data are available online for this figure.
Figure EV4. siRNA screen identifies UBE2O that is required for LT‐induced NLRP1B inflammasome activation.
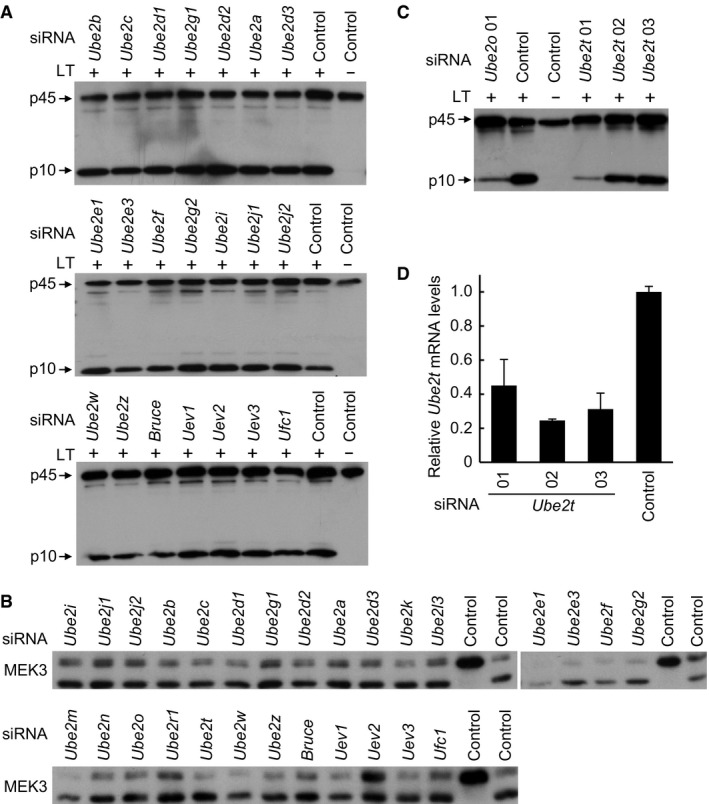
-
A, BsiRNA screen of E2s involved in LT‐induced caspase‐1 activation. 129/Sv mice‐derived BMDMs were transfected with an indicated siRNA for 60 h (control, scramble siRNA), followed by stimulation with WT LT (+) or its E687C mutant (−). Caspase‐1 activation was analyzed by anti‐caspase‐1 p10 immunoblotting (A). Total cell lysates were also blotted with the anti‐MEK3 antibody to control intact delivery of LT into the knockdown cells (B).
-
C, DEffect of siRNA knockdown of Ube2t on LT‐induced caspase‐1 activation. 129/Sv mice‐derived BMDMs were transfected with an indicated Ube2t‐targeting siRNA. Caspase‐1 activation was analyzed by anti‐caspase‐1 p10 immunoblotting. The knockdown efficiency was measured by qPCR (mean values ± SD from three replicates) in (D).
Source data are available online for this figure.
Degradation of LT‐cleaved NLRP1B leads to caspase‐1 activation by the released CARD of NLRP1B
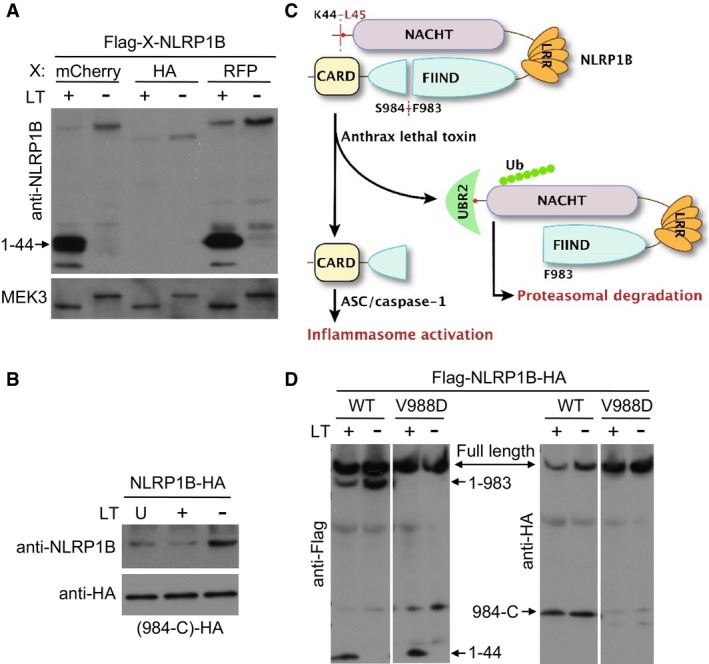
Recent studies show that LT cleaves NLRP1B at Lys44 (Fig 4A), and this cleavage is required for LT‐induced caspase‐1 activation (Hellmich et al, 2012; Levinsohn et al, 2012; Chavarria‐Smith & Vance, 2013). Interestingly, the cleavage exposes a neo‐N‐terminal Leu residue that is a classical type 2 N‐degron, raising an interesting hypothesis that the LT‐cleaved NLRP1B might be a target of UBR2‐mediated N‐end rule degradation. Supporting this idea, we found that LT stimulation in 293T cells caused an evident decrease of NLRP1B protein level although different epitopes were used to tag the transfected NLRP1B (Figs 4B, and EV5A and B). However, as these immunoblottings were performed using an antibody against the N‐terminal Flag tag or a homemade anti‐NLRP1B antibody recognizing the extreme N‐terminus of the protein, the data could not be unequivocally interpreted as LT‐induced degradation of NLRP1B. NLRP1B, like human NLRP1, contains a FIIND domain between the LRR and the C‐terminal CARD domain (Fig 4A). It has been well established that the FIIND undergoes constitutive autoproteolytic cleavage between Phe983 and Ser984, which was independent of LT stimulation (Fig 4B) but required for inflammasome activation (Finger et al, 2012; Frew et al, 2012). Following the previous study (Frew et al, 2012), we generated a NLRP1B V988D mutant that was deficient in FIIND‐mediated autocleavage; as expected, the V988D mutation abolished LT‐stimulated RFP‐ASC specks formation reconstituted in 293T cells (Fig 4C). Meanwhile, we also found that NLRP1B truncation (1–983) ending at the autocleavage site, which was still cleaved at the N‐terminus by LT, could not support LT‐stimulated RFP‐ASC specks formation, while the full‐length NLRP1B was capable of doing that, suggesting a requirement of the C‐terminal CARD‐containing fragment for inflammasome activation. Indeed, expression of NLRP1B (984‐C) alone was sufficient to induce RFP‐ASC specks formation in the absence of LT stimulation (Fig 4C). All these data implied that the NLRP1B inflammasome activation is mediated by the C‐terminal CARD domain.
Figure 4. LT cleavage of NLRP1B triggers N‐end rule‐mediated degradation of NLRP1B for inflammasome activation.
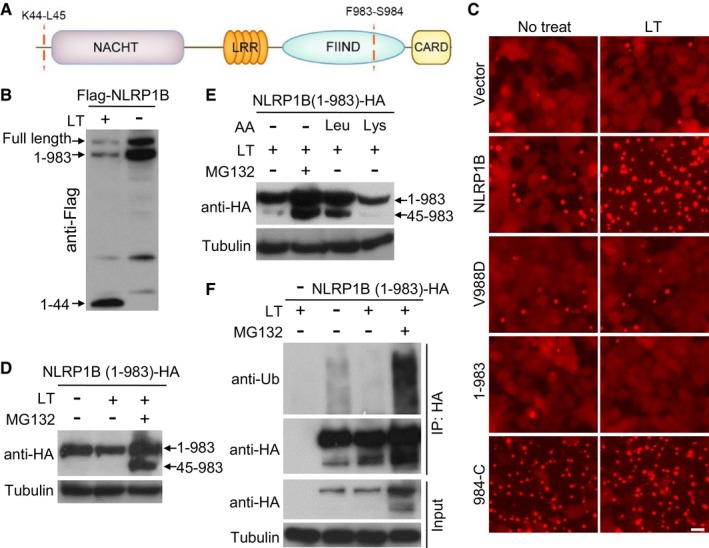
-
ADomain structure of NLRP1B. The LT cleavage site and the FIIND autocleavage site are marked.
-
BEffect of LT treatment on NLRP1B protein level. 293T cells were transfected with Flag‐NLRP1B expression plasmid for 24 h, and cells were then treated with WT LT (+) or its E687C mutant (−) for 4 h. Shown is the anti‐Flag immunoblot of the total cell lysates.
-
CRFP‐ASC specks formation in 293T cells expressing an indicated NLRP1B variant. Cells were treated with LT for 3 h. Scale bar, 20 μm. 1–983 and 984‐C (residues 984 to the C‐terminus) are the FIIND‐mediated autocleaved fragments of NLRP1B.
-
D, EEffect of MG132 and Leu on LT‐induced NLRP1B degradation. 293T cells expressing NLRP1B (1–983)‐HA were treated with LT and MG132 for 8 h. 10 mM Leu was added to cells 30 min prior to LT treatment. Shown are the anti‐HA and anti‐tubulin immunoblots of the total cell lysates.
-
FLT‐induced ubiquitination of NLRP1B. 293T cells expressing NLRP1B (1–983)‐HA were treated with LT and/or MG132 for 6 h. Cells were then harvested for anti‐HA immunoprecipitation and subjected to immunoblotting analyses using indicated antibodies.
Source data are available online for this figure.
Figure EV5. LT cleavage of NLRP1B induces degradation of the cleaved NLRP1B.
-
A, BEffect of LT treatment on NLRP1B protein level. 293T cells expressing an indicated tagged version of NLRP1B were stimulated with WT LT (+) or its E687C mutant (−) for 4 h (U: untreated). The level of cellular NLRP1B and its cleavage products were detected by anti‐NLRP1B (A) or anti‐HA immunoblotting (B). Total cell lysates were also blotted with the anti‐MEK3 antibody as shown (A). 984‐C are the NLRP1B fragment (residues 984 to the C‐terminus) generated from the FIIND‐mediated autocleavage.
-
CA tentative model for LT‐induced NLRP1B inflammasome activation.
-
DThe level of NLRP1B (WT and the V988D mutant) in LT‐stimulated cells. 293T cells expressing indicated Flag‐NLRP1B‐HA were stimulated with WT LT (+) or its E687C mutant (−) for 4 h. The level of cellular NLRP1B and its cleavage products were detected by anti‐Flag and anti‐HA immunoblotting.
Source data are available online for this figure.
Given the above results and also stimulated by the two complementary and insightful studies from the Vance and Bachovchin laboratories that appeared at bioRxiv about 10 months before the final publication (Chui et al, 2019; Sandstrom et al, 2019), an attractive hypothesis, in which the degradation of NLRP1B (residues 45–983) following LT and FIIND‐mediated cleavage releases its C‐terminal CARD for subsequent caspase‐1 activation (Fig EV5C), became apparent to us. Supporting this model, we found that LT treatment could reduce the level of both full‐length NLRP1B and the FIIND domain autocleaved NLRP1B (Fig 4B), but never observed any reduction of the C‐terminal CARD‐containing NLRP1B fragment in LT‐stimulated cells (Fig EV5B and D). Notably, the decrease of NLRP1B was not detected with the V988D mutant that blocked the FIIND‐mediated autocleavage (Fig EV5D). A possible way to interpret the data is that FIIND‐mediated autoprocessing of NLRP1B is required for LT‐induced degradation of NLRP1B, which would support the above‐proposed model. Owing to the lack of a suitable antibody that only detects NLRP1B (45–983) and also to simply the assay system, we examined the truncated NLRP1B (1–983) mutant through its C‐terminal HA tag. LT treatment induced a slight decrease in NLRP1B (1–983) protein level (Fig 4D). Importantly, NLRP1B (45–983) that in theory should be generated following LT stimulation was not detectable, but was massively accumulated in the presence of the proteasome inhibitor MG132 (Fig 4D). MG132 also increased the level of NLRP1B (1–983). These data suggest that NLRP1B (45–983) generated from LT cleavage underwent rapid degradation through the proteasome pathway. We further found that degradation of LT‐cleaved NLRP1B could be partially blocked by the amino acid Leu but not His added to the LT‐stimulated cells (Fig 4E), consistent with the notion that degradation of NLRP1B is mediated by the N‐end rule pathway. Furthermore, robust ubiquitination signal was detected on anti‐HA antibody‐immunoprecipitated NLRP1B only when LT stimulation of the cells was performed in the presence of MG132 (Fig 4F). These data together support the hypothesis that the N‐end rule‐mediated proteasomal degradation of LT‐cleaved NLRP1B drives inflammasome activation by the C‐terminal CARD of NLRP1B.
Discussion
During the final stage of completing this work, another report that also identifies UBR2 in LT‐induced NLRP1 inflammasome activation was deposited online (Chui et al, 2019). Together, identification of UBR2 uncovers the previously unknown link between the N‐end rule signaling pathway and LT‐induced NLRP1B activation, that is, the degradation of the Leu‐started NLRP1B N‐terminal fragment produced by LF cleavage through UBR2‐mediated N‐end rule pathway. This finding is consistent with the fact that UBR2 is a leucine binding protein in regulating the leucine‐mTOR signaling pathway (Kume et al, 2010). Equally importantly, the mechanism of NLRP1B inflammasome activation elucidated here and in the two complementary studies (Chui et al, 2019; Sandstrom et al, 2019), i.e., degradation‐induced release of the CARD domain for ASC/caspase‐1 activation, represents a new model of inflammasome activation distinct from those of all known inflammasomes that requires the NLR sensor to assembly a large protein complex.
To date, only two E2 enzymes, UBE2A/UBE2B and USE1 (Varshavsky, 2011), have been confirmed to participate in the N‐end rule ubiquitination pathway. Here, our study adds another one UBE2O (also termed E2‐230k) that also function in the N‐end rule pathway by partnering with UBR2 to regulate NLRP1B activation. UBE2O was initially isolated from rabbit reticulocytes as an E2 enzyme (Klemperer et al, 1989), and subsequently reported to function as an E2/E3 hybrid enzyme (Zhang et al, 2013). UBE2O can mediate the ubiquitination of many different substrates, and its high expression is associated with low survival rate of gastric, lung, breast, and prostate cancer patients (Ullah et al, 2018). Thus, UBE2O may also regulate other biological processes through UBR2‐dependent or UBR2‐independent N‐end rule pathways.
Recent studies have shown that inhibitors of DPP8/DPP9 proteases can selectively activate NLRP1 inflammasome in monocytes and macrophages of both human and mouse origins (Okondo et al, 2017, 2018). However, DPP8/9 inhibitors activate several NLRP1B alleles that are different than those by LT. Of the five Nlrp1b alleles expressed in different mice strains, those expressing Nlrp1b allele 1 or 5 are susceptible to LT‐induced pyroptosis, while alleles 1, 2, and 3, but not alleles 4 and 5, were sensitive to DPP8/9 inhibitors (Okondo et al, 2018). Both LT‐ and DPP8/9 inhibitor‐induced NLRP1 activations require the FIIND domain autoproteolysis and can be blocked by the proteasome inhibitors. Thus, it will be interesting to explore whether UBR2 and UBE2O also mediate DPP8/9 inhibitor‐induced NLRP1 degradation to trigger the inflammasome activation. Moreover, the possibility also exists that NLRP1 might be cleaved by other proteases, which does not expose an N‐terminal leucine, and under those conditions, another ubiquitin ligase may be responsible for ubiquitination and degradation of the cleaved NLRP1.
The major difference in NLRP1 domain architecture between human and nonprimate mammalian orthologs is the presence of a PYD domain at the N‐terminus of human NLRP1. Mutations in the PYD can cause several skin disorders with autoinflammation (Zhong et al, 2016), underscoring the autoinhibitory function of this domain in guarding against pathological autoactivation of human NLRP1. The Reversade group has also identified DPP9 as an interacting partner of NLRP1 in diverse human and mouse primary cells, which maintains NLRP1 in its inactive state and represses downstream inflammasome activation (Zhong et al, 2018). Moreover, NLRP1 has been proposed as the predominant inflammasome sensor in human epidermis (Zhong et al, 2016; Burian & Yazdi, 2018), and there are strong evidences suggesting a role of NLRP1 inflammasome in sensing UVB and other danger signals like nigericin in human keratinocytes (Burian & Yazdi, 2018), but the mechanism underlying NLRP1 function in these contexts is unknown. Further investigations are needed to unravel the regulation of endogenous NLRP1 and characterize its mechanism of activation in innate immune defense.
Materials and Methods
Study design
The aim of this study was to explore the mechanism of LT‐triggered NLRP1B inflammasome activation. For this, a RAW264.7 cell line that stably expressing RFP‐ASC was constructed and a whole‐genome siRNA screen was carried out, assayed by RFP‐ASC specks formation and loss of cell viability. Meanwhile, a genome‐wide CRISPR‐Cas9 screen was also performed in immortalized BMDM cells by taking advantage of LT‐induced pyroptotic cell death. siRNA and shRNA knockdown and CRISPR knockout experiments were performed to confirm the role of UBR2 in LT activation of the NLRP1B inflammasome. Further biochemical experiments reveal that the UBR2‐UBE2O ubiquitination pathway mediates degradation of LT‐cleaved NLRP1B, which releases its C‐terminal CARD domain for subsequent caspase‐1 activation.
Antibodies and reagents
Antibodies for caspase‐1 were obtained from Santa Cruz Biotechnology (SC‐515) and AdipoGen (AG‐20B‐0044). Anti‐HA beads (#26181) were purchased from Thermo Fisher. Other antibodies used in this study were anti‐HA (BioLegend, #16B12), anti‐Flag M2 (Sigma‐Aldrich, #F3165), anti‐Myc (Santa Cruz, clone # 9E10), anti‐tubulin (Sigma‐Aldrich, T5168), and anti‐MEK3 (Cell Signaling Technology, # 9238S). Homemade anti‐NLRP1B antibody was raised against the NLRP1B N‐terminal sequence (amino acids 4–20). Sequences for all siRNAs are listed in Appendix Table S1. Cell culture products were from Life Technologies; all other chemicals were from Sigma‐Aldrich unless noted otherwise.
HEK293T and RAW 264.7 macrophage cells were obtained from American Type Culture Collection (ATCC). Tlr4 −/− iBMDM cells derived from Tlr4 −/− mice (Hoshino et al, 1999) have been heavily used in our previous studies (Zhao et al, 2011; Shi et al, 2015). Cells were grown at 37°C in a 5% CO2 incubator in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 2 mM l‐glutamine. The identity of the cells was frequently checked by their morphological features but has not been authenticated by short tandem repeat (STR) profiling.
ATP cell viability and RFP‐ASC specks formation assays
For assays without siRNA silencing, 3 × 105 RAWRA cells were plated in the 24‐well format 12 h before LT treatment (1 μg/ml for both LF and PA). Cells were washed three times with serum‐free DMEM, followed by addition of 0.5 ml serum‐free DMEM into each well with or without LF plus PA or BsaK plus PA (all 1 μg/ml). When required, KCl (150 mM) or MG132 (25 μM) was added concomitantly with LT; 1.5 h later, cells and culture medium were separately collected for ATP cell viability assay (Promega) following manufacturer's instructions. To assay caspase‐1 activation, culture supernatants were subjected to trichloroacetic acid (TCA) precipitation and the precipitates were analyzed by caspase‐1 immunoblotting to detect both pro‐caspase‐1 and the autoprocessed mature p10 fragment; RFP‐ASC specks formation was monitored on a confocal microscopy (Zeiss LSM 510 Meta Confocal). To assay the effect of the N‐end rule pathway on NLRP1B inflammasome activation, RAWRA cells were pre‐treated with four different amino acids (10 mM each) as indicated for 30 min before addition of LT. For live imaging, 2 × 104 RAWRA cells were plated into the 96‐well plates 12 h before LT treatment, and DIC, EGFP, and RFP images were recorded using a PerkinElmer Ultra‐View Spinning Disc confocal microscope.
Reconstitution of NLRP1B inflammasomes in 293T cells
To reconstitute NLRP1B inflammasome, 129/Sv mice‐derived Nlrp1b, human pro‐caspase‐1, and human pro‐IL‐1β expression plasmids were co‐transfected in 293T cells plated into the 6‐well plates. About 20 h after transfection, cells were treated with WT LT or its catalytic inactive E687C mutant (1 μg/ml for each protein) for 6 h in the presence or absence of 25 μM MG132. Cell lysates were harvested in the lysis buffer containing 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1% Triton X‐100, and protease inhibitor cocktail (Roche Molecular Biochemicals). To silence Ubr2 expression, 2 × 105 293T cells were plated into the 12‐well plates and then transfected with 60 pmol siRNA using Lipofectamine 2000 (Invitrogen). Twenty‐four hours after transfection, cells from one well were re‐suspended, and then divided equally and re‐plated into three wells; about 12 h later, cells were subjected to another round of siRNA transfection. Twenty‐four hours after the second siRNA transfection, cells were co‐transfected with expression plasmids for Nlrp1b, pro‐caspase‐1, and pro‐IL‐1β. Twenty hours after transfection, cells were stimulated with LT for 6 h. Processing of pro‐IL‐1β was detected by anti‐IL‐1β immunoblotting of the cell lysates.
siRNA knockdown and siRNA screens
To knock down endogenous gene expression, 2 × 104 RAWRA cells were plated into the 24‐well plates 12 h prior to siRNA transfection. siRNAs (20 pmol each well, GenePharma or Dharmacon) were transfected into the cells using the INTERFERin reagent (Polyplus‐transfection) by following the manufacturer's instructions. Assays of ATP‐based cell viability, RFP‐ASC specks formation, and caspase‐1 activation were performed 60 h after the siRNA transfection.
A mouse genome‐wide RNAi screen (QIAGEN) in RAWRA cells was carried out in the 96‐well plates. Targeted screen for E2 ubiquitin‐conjugating enzymes was performed using BMDM cells with siRNA against 28 E2‐family gene (Appendix Table S1). Briefly, 2 × 105 differentiated 129/Sv mice‐derived BMDM cells were plated into 24‐well plates 24 h before siRNA transfection. 40 pmol siRNAs were used to knock down endogenous E2 expression in BMDM cells using the INTERFERin transfection reagents (Polyplus‐transfection). Sixty hours after siRNA transfection, cells were washed three times with pre‐warmed serum‐free DMEM and incubated with LT (1 μg/ml for both LF and PA) for 3 h, followed by collection of the media for TCA precipitation and anti‐caspase‐1 p10 immunoblotting.
Generation of stable shRNA knockdown and CRISPR knockout cells
A pLKO.1 plasmid was constructed via the insertion of a hairpin shRNA containing DNAs corresponding to the Ubr2‐05 siRNA listed in Appendix Table S1. Stable knockdown of Ubr2 in RAWRA cells was achieved following a previously developed protocol (Naldini et al, 1996; Moffat et al, 2006) using puromycin (2 μg/ml) as a selection marker. The efficiency of Ubr2 knockdown was measured by qPCR analyses. To generate knockout cells, human codon‐optimized Cas9 (hCas9) and GFP‐targeting gRNA‐expressing plasmids (gRNA_GFP‐T1) were obtained from Addgene. The 19‐bp GFP‐targeting sequence in the gRNA vector was replaced with the sequence targeting the desired gene via QuikChange site‐directed mutagenesis (Stratagene). The sequence used to target mouse Ubr2 was GCGTCGGAGATGGAGCCCG. To construct the knockout cell lines, 1 μg of gRNA‐expressing plasmid, 3 μg of hCas9 plasmid, and 1 μg of pEGFP‐C1 vector were co‐transfected into iBMDM cells. 3 days later, GFP‐positive cells were sorted into single clones via flow cytometry using the BD Biosciences FACSAria II or the Beckman Coulter MoFlo XDP cell sorter and plated into 96‐well plates. Single clones were screened using T7 endonuclease I‐cutting assay, and the candidate knockout clones were verified by sequencing of the PCR fragments and/or Western blotting analysis (Shi et al, 2014).
Genome‐wide CRISPR‐Cas9 screens
Tlr4 −/− iBMDM cells stably expressing the Cas9 protein (Shi et al, 2015) and the same gRNA library (Koike‐Yusa et al, 2014) were used for the genome‐wide CRISPR‐Cas9 screen in this study. For the large‐scale screen, cells were seeded in the 15‐cm dishes (2 × 106 cells in 20 ml media), and a total of 2 × 107 cells were infected with a gRNA lentivirus library. Sixty hours after infection, cells were re‐seeded at a density of 1 × 105 cells/ml in fresh media supplemented with 5 μg/ml puromycin to remove uninfected cells. After 6–8 days, 3 × 108 cells from five culture dishes were treated with LT (1 μg/ml for both LF and PA) for 5 h to induce pyroptosis, and another batch of 3 × 108 cells were left untreated as control samples. Surviving cells were collected after 5–6 days when the confluence reached about 90%. The cells were then lysed in the SNET buffer [20 mM Tris–HCl (pH 8.0), 5 mM EDTA, 400 mM NaCl, 400 μg/ml proteinase K, and 1% SDS]. Genomic DNAs were isolated using phenol–chloroform extraction and isopropanol precipitation method. The DNAs were dissolved in water (4–5 μg/μl) and used as the templates for amplification of the gRNA (Shi et al, 2015).
Statistics
Statistical analyses of the RFP‐ASC fluorescent images to assess the percentages of cells showing RFP‐ASC specks were conducted using the ImageJ software. At least 1,000 cells were countered for each measurement. The numbers labeled in the merged panel are the mean values of three repeat measurements, and error bars mean standard deviation (SD).
Author contributions
HX, JS, ND, and FS designed the study; HX, JS, HG, YL, and ND performed most of the experiments, data acquisition, and analysis; ZY initiated the study, and provided technical assistance and valuable intellectual inputs; HX, ND, and FS wrote the manuscript. All authors have read and approved the final manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Source Data for Expaned View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank Dr. Nicholas Duesbery for providing anthrax LF and PA, Liyan Hu for drawing the model, and John Hugh Snyder for editing the manuscript. We thank the flow cytometry, bioinformatics, and cell imaging facilities at NIBS for technical assistances. This work was supported by grants from the Basic Science Center Project of NSFC (81788104), National Key Research and Development Program of China (2017YFA0505900 and 2016YFA0501500), and the Chinese Academy of Sciences (XDB08020202) to F.S, as well as a NSFC grant (81671987) to N.D.
The EMBO Journal (2019) 38: e101996
See also: CA Lacey & EA Miao (July 2019)
Contributor Information
Feng Shao, Email: shaofeng@nibs.ac.cn.
Na Dong, Email: dongna@cau.edu.cn.
References
- Boyden ED, Dietrich WF (2006) Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38: 240–244 [DOI] [PubMed] [Google Scholar]
- Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16: 407–420 [DOI] [PubMed] [Google Scholar]
- Burian M, Yazdi AS (2018) NLRP1 is the key inflammasome in primary human keratinocytes. J Invest Dermatol 138: 2507–2510 [DOI] [PubMed] [Google Scholar]
- Chavarria‐Smith J, Vance RE (2013) Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog 9: e1003452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, Ball DP, Taabazuing CY, Orth EL, Vittimberga BA et al (2019) N‐terminal degradation activates the NLRP1B inflammasome. Science 364: 82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F (2016) Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116 [DOI] [PubMed] [Google Scholar]
- D'Osualdo A, Weichenberger CX, Wagner RN, Godzik A, Wooley J, Reed JC (2011) CARD8 and NLRP1 undergo autoproteolytic processing through a ZU5‐like domain. PLoS One 6: e27396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD et al (1998) Proteolytic inactivation of MAP‐kinase‐kinase by anthrax lethal factor. Science 280: 734–737 [DOI] [PubMed] [Google Scholar]
- Finger JN, Lich JD, Dare LC, Cook MN, Brown KK, Duraiswami C, Bertin J, Gough PJ (2012) Autolytic proteolysis within the function to find domain (FIIND) is required for NLRP1 inflammasome activity. J Biol Chem 287: 25030–25037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frew BC, Joag VR, Mogridge J (2012) Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog 8: e1002659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PK, Moayeri M, Crown D, Fattah RJ, Leppla SH (2008) Role of N‐terminal amino acids in the potency of anthrax lethal factor. PLoS One 3: e3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellmich KA, Levinsohn JL, Fattah R, Newman ZL, Maier N, Sastalla I, Liu S, Leppla SH, Moayeri M (2012) Anthrax lethal factor cleaves mouse nlrp1b in both toxin‐sensitive and toxin‐resistant macrophages. PLoS One 7: e49741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, Takeda K, Akira S (1999) Cutting edge: Toll‐like receptor 4 (TLR4)‐deficient mice are hyporesponsive to lipopolysaccharide: evidence for TLR4 as the Lps gene product. J Immunol 162: 3749–3752 [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QT et al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- Klemperer NS, Berleth ES, Pickart CM (1989) A novel, arsenite‐sensitive E2 of the ubiquitin pathway: purification and properties. Biochemistry 28: 6035–6041 [DOI] [PubMed] [Google Scholar]
- Klimpel KR, Arora N, Leppla SH (1994) Anthrax toxin lethal factor contains a zinc metalloprotease consensus sequence which is required for lethal toxin activity. Mol Microbiol 13: 1093–1100 [DOI] [PubMed] [Google Scholar]
- Koike‐Yusa H, Li Y, Tan EP, Velasco‐Herrera Mdel C, Yusa K (2014) Genome‐wide recessive genetic screening in mammalian cells with a lentiviral CRISPR‐guide RNA library. Nat Biotechnol 32: 267–273 [DOI] [PubMed] [Google Scholar]
- Kume K, Iizumi Y, Shimada M, Ito Y, Kishi T, Yamaguchi Y, Handa H (2010) Role of N‐end rule ubiquitin ligases UBR1 and UBR2 in regulating the leucine‐mTOR signaling pathway. Genes Cells 15: 339–349 [DOI] [PubMed] [Google Scholar]
- Lacy DB, Collier RJ (2002) Structure and function of anthrax toxin. Curr Top Microbiol Immunol 271: 61–85 [DOI] [PubMed] [Google Scholar]
- Levinsohn JL, Newman ZL, Hellmich KA, Fattah R, Getz MA, Liu S, Sastalla I, Leppla SH, Moayeri M (2012) Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8: e1002638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Mol Cell 10: 417–426 [DOI] [PubMed] [Google Scholar]
- Martinon F, Gaide O, Petrilli V, Mayor A, Tschopp J (2007) NALP inflammasomes: a central role in innate immunity. Semin Immunopathol 29: 213–229 [DOI] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK et al (2006) A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high‐content screen. Cell 124: 1283–1298 [DOI] [PubMed] [Google Scholar]
- Muehlbauer SM, Evering TH, Bonuccelli G, Squires RC, Ashton AW, Porcelli SA, Lisanti MP, Brojatsch J (2007) Anthrax lethal toxin kills macrophages in a strain‐specific manner by apoptosis or caspase‐1‐mediated necrosis. Cell Cycle 6: 758–766 [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gallay P, Ory D, Mulligan R, Gage FH, Verma IM, Trono D (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 272: 263–267 [DOI] [PubMed] [Google Scholar]
- Newman ZL, Printz MP, Liu S, Crown D, Breen L, Miller‐Randolph S, Flodman P, Leppla SH, Moayeri M (2010) Susceptibility to anthrax lethal toxin‐induced rat death is controlled by a single chromosome 10 locus that includes rNlrp1. PLoS Pathog 6: e1000906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okondo MC, Johnson DC, Sridharan R, Go EB, Chui AJ, Wang MS, Poplawski SE, Wu W, Liu Y, Lai JH et al (2017) DPP8 and DPP9 inhibition induces pro‐caspase‐1‐dependent monocyte and macrophage pyroptosis. Nat Chem Biol 13: 46–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okondo MC, Rao SD, Taabazuing CY, Chui AJ, Poplawski SE, Johnson DC, Bachovchin DA (2018) Inhibition of Dpp8/9 activates the Nlrp1b inflammasome. Cell Chem Biol 25: 262–267 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE (2019) Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 64: eaau1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F (2014) Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514: 187–192 [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- Squires RC, Muehlbauer SM, Brojatsch J (2007) Proteasomes control caspase‐1 activation in anthrax lethal toxin‐mediated cell killing. J Biol Chem 282: 34260–34267 [DOI] [PubMed] [Google Scholar]
- Tang G, Leppla SH (1999) Proteasome activity is required for anthrax lethal toxin to kill macrophages. Infect Immun 67: 3055–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Sriram SM, Park KS, Kwon YT (2012) The N‐end rule pathway. Annu Rev Biochem 81: 261–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp J, Martinon F, Burns K (2003) NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol 4: 95–104 [DOI] [PubMed] [Google Scholar]
- Ullah K, Zubia E, Narayan M, Yang J, Xu G (2018) Diverse roles of the E2:E3 hybrid enzyme UBE2O in the regulation of protein ubiquitination, cellular functions, and disease onset. FEBS J 10.1111/febs.14708 [DOI] [PubMed] [Google Scholar]
- Varshavsky A (2011) The N‐end rule pathway and regulation by proteolysis. Protein Sci 20: 1298–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C (2000) Susceptibility of mitogen‐activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J 352(Pt 3): 739–745 [PMC free article] [PubMed] [Google Scholar]
- Wickliffe KE, Leppla SH, Moayeri M (2008a) Anthrax lethal toxin‐induced inflammasome formation and caspase‐1 activation are late events dependent on ion fluxes and the proteasome. Cell Microbiol 10: 332–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickliffe KE, Leppla SH, Moayeri M (2008b) Killing of macrophages by anthrax lethal toxin: involvement of the N‐end rule pathway. Cell Microbiol 10: 1352–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang J, Bauer A, Zhang L, Selinger DW, Lu CX, Ten Dijke P (2013) Fine‐tuning BMP7 signalling in adipogenesis by UBE2O/E2‐230K‐mediated monoubiquitination of SMAD6. EMBO J 32: 996–1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F (2011) The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477: 596–600 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Shao F (2016) Diverse mechanisms for inflammasome sensing of cytosolic bacteria and bacterial virulence. Curr Opin Microbiol 29: 37–42 [DOI] [PubMed] [Google Scholar]
- Zhong FL, Mamai O, Sborgi L, Boussofara L, Hopkins R, Robinson K, Szeverenyi I, Takeichi T, Balaji R, Lau A et al (2016) Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 167: 187–202 e17 [DOI] [PubMed] [Google Scholar]
- Zhong FL, Robinson K, Teo DET, Tan KY, Lim C, Harapas CR, Yu CH, Xie WH, Sobota RM, Au VB et al (2018) Human DPP9 represses NLRP1 inflammasome and protects against auto‐inflammatory diseases via both peptidase activity and FIIND domain binding. J Biol Chem 293: 18864–18878 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Source Data for Expaned View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4