

Abstract
The human pathogen Staphylococcus aureus produces saturated fatty acids, but can incorporate both exogenous saturated and unsaturated fatty acids into its lipid membrane. S. aureus encounters unsaturated fatty acids in the host skin where they serve as an innate immune defence due to their toxicity. Previously, we identified a fatty acid kinase in S. aureus that is necessary for the utilization of exogenous fatty acids. The goal of this study was to determine the effects of fatty acids on mutants deficient in the exogenous fatty acid utilization machinery. We have demonstrated that mutants lacking a functional fatty acid kinase (fakA) or both fatty acid carrier proteins (fakB1 fakB2) are more resistant to unsaturated fatty acids. Previous studies suggested a role for ammonia-producing enzymes in resistance to unsaturated fatty acids, but these enzymes do not contribute to the resistance of the fakA mutant, despite increased urease transcription and protein activity in the mutant. Additionally, while pigment is altered in mutants unable to use exogenous fatty acids, staphyloxanthin does not contribute to fatty acid resistance of an fakA mutant. Because exposure to unsaturated fatty acids probably initiates a stress response, we investigated the role of the alternative sigma factor σB and determined if it is necessary for the fatty acid resistance observed in the fakA mutant. Collectively, this study demonstrates that the inability to incorporate unsaturated fatty acids leads to increased resistance to those fatty acids, and that resistance requires a σB stress response.
Keywords: VfrB, FakA, fatty acid kinase, fatty acid resistance
Introduction
Staphylococcus aureus is a major burden on human healthcare due to high morbidity and mortality rates resulting from a broad range of infections. Most cases of skin and soft tissue infections are caused by S. aureus [1] and are dominated by the USA300 lineage in the United States [2]. The skin provides multiple environmental challenges to invading pathogens, including antimicrobial peptides and antimicrobial fatty acids (reviewed in [3–5]). The major unsaturated fatty acids in human skin are palmitoleic acid (C16 : 1), oleic acid (C18 : 1) and linoleic acid (C18 : 2) [6], while skin saturated fatty acids include palmitic acid (C16 : 0), stearic acid (C18 : 0) and myristic acid (C14 : 0). In addition, humans secrete the unsaturated fatty acid sapienic acid (C16 : 1) as part of the sebum. For comparison, because mice are the model of choice for skin infection studies, the fatty acid composition of BALB/c mice consists of unsaturated fatty acids oleic acid, palmitoleic acid and linoleic acid and saturated fatty acids stearic acid, arachidic acid (C20 : 0), palmitic acid and myristic acid [7].
Fatty acids are important cellular metabolites, and serve as the building blocks needed for cellular components, including phospholipids. S. aureus produces saturated fatty acids through the endogenous FASII system, but will also uptake and utilize exogenous fatty acids (exoFAs). While S. aureus is tolerant to saturated fatty acids, unsaturated fatty acids, such as those found on mammalian skin, have been shown to be toxic to the bacterium [8–10]. The mechanisms for this phenomenon are not completely understood. However, previous transcriptomic and proteomic studies have examined the response of S. aureus to unsaturated fatty acids, and have identified potential pathways involved in resistance [11–13]. While these studies have provided insight into the global changes found in response to unsaturated fatty acids, it is not clear how the ability, or inability, to use unsaturated fatty acids influences resistance.
The ability of S. aureus to uptake and use exoFAs is thought to be a primary means to bypass the inhibitory effects of FASII inhibitors. Recently, the pathway for exoFA incorporation was found to be important for utilizing fatty acids derived from low-density lipoproteins to resist the FASII inhibitor triclosan [14]. Currently, three proteins make up the exoFA utilization pathway. FakA, previously referred to as VfrB, is a recently identified fatty acid kinase in S. aureus [15], thought to phosphorylate fatty acids so that they can be used for phospholipid generation. FakA works in conjunction with two fatty acid binding proteins, FakB1 and FakB2. In the same study by Parsons et al., preference of fatty acid saturation for each fatty acid binding protein was demonstrated in vitro, wherein FakB1 has a higher affinity for saturated fatty acids and FakB2 preferentially binds unsaturated fatty acids. One function of the Fak proteins is to incorporate exoFAs into the cellular membrane. In addition to its role in fatty acid metabolism, we have previously demonstrated that FakA is a key activator of the virulence regulator SaeRS [16] and the absence of FakA leads to changes in cellular metabolism [17]. Moreover, we have also previously demonstrated that fakA mutants are hyper-virulent in a murine model of dermonecrosis [18]. Regarding FakB1 and FakB2, despite an apparent specificity for different fatty acids, these proteins appear to be redundant when examining virulence factor regulation [15]. The use of exoFAs could be viewed as a way to conserve energy needed for membrane generation or could serve as a host signal. Regardless of their role, exoFAs are abundant in the natural niche that S. aureus occupies and FakA, FakB1 and FakB2 are key to their uptake and use. Here, we report that the inactivation of the exoFA utilization machinery, through either deletion of fakA or the combined inactivation of fakB1 and fakB2, leads to enhanced resistance to unsaturated fatty acids.
Methods
Strains, media and growth conditions
Strains used in this study are detailed in Table 1. Escherichia coli strains were grown in lysogeny broth (LB) and S. aureus strains were grown in tryptic soy broth (TSB). When necessary, the following antibiotics were used: 100 µg ampicillin ml−1, 10 µg chloramphenicol ml−1 or 5 µg erythromycin ml−1. Overnight cultures were grown in test tubes with shaking at 250 r.p.m. at 37 °C. Mutants from the Nebraska Transposon Mutant Library (designated with prefix 'NE') were obtained from BEI Resources. The mutations were transferred into recipient strains using Φ11-mediated transduction [19]. To make the arcA1 arcA2 combination mutants, the erythromycin-resistant transposon insertion in arcA2 of strain JLB149 was exchanged for the tetracycline resistance gene using pTET [20]. The exchange was confirmed based on antibiotic resistance profile and restriction digest of a PCR product across the insert. This arcA2::tetR mutation (JLB263) was then introduced into JLB151 and JLB152 to make the arcA1 arcA2 and fakA arcA1 arcA2 mutants.
Table 1. Selected strains and plasmids used in this study.
| Strain or plasmid | Relevant characteristics*,† | Source or reference |
|---|---|---|
| Strains | ||
| AH1263 | USA300 CA-MRSA strain LAC lacking LAC-p03 | [32] |
| JLB2 | AH1263 ΔfakA | [18] |
| JLB15 | AH1263 saeR::NΣ | [16] |
| JLB16 | AH1263 ΔfakA saeR::NΣ | [16] |
| JLB28 | AH1263 fakB2::NΣ | This study |
| JLB30 | AH1263 ΔfakB2 | [15] |
| JLB31 | AH1263 fakB1::NΣ ΔfakB2 | [15] |
| JLB112 | AH1263 crtM::NΣ | This study |
| JLB129 | AH1263 ΔfakA crtM::NΣ | This study |
| JLB130 | AH1263 sigB::NΣ | This study |
| JLB131 | AH1263 ΔfakA sigB::NΣ | This study |
| JLB132 | AH1263 ureB::NΣ | [17] |
| JLB133 | AH1263 ΔfakA ureB::NΣ | [17] |
| JLB148 | AH1263 fakB1::NΣ | This study |
| JLB149 | AH1263 arcA2::NΣ | This study |
| JLB150 | AH1263 ΔfakA arcA2::NΣ | This study |
| JLB151 | AH1263 arcA1::NΣ | This study |
| JLB152 | AH1263 ΔfakA arcA1::NΣ | This study |
| JLB263 | AH1263 arcA2::tetR | This study |
| JLB266 | AH1263 arcA1::NΣ arcA2::tetR | This study |
| JLB267 | AH1263 ΔfakA arcA1::NΣ arcA2::tetR | This study |
| NE1109 | Strain containing sigB::NΣ (SAUSA300_2022) | [33] |
| NE623 | Strain containing arcA1::NΣ (SAUSA300_2570) | [33] |
| NE1444 | Strain containing crtM::NΣ (SAUSA300_2499) | [33] |
| NE1540 | Strain containing fakB1::NΣ (SAUSA300_0733) | [33] |
| NE1594 | Strain containing arcA2::NΣ (SAUSA300_0065) | [33] |
| RN4220 | Highly transformable S. aureus | [34] |
| Plasmids | ||
| pCM28 | E. coli–S. aureus shuttle vector | [35] |
| pJB165 | fakA complementation plasmid | [18] |
| pJB1031 | fakB1 complementation plasmid | This study |
| pJB1033 | fakB2 complementation plasmid | This study |
Fatty acid plates were made using 0.01 % (v/v) (314.0 µM) oleic acid or 25 µg myristic acid ml−1 (109.5 µM) in tryptic soy agar (TSA). Bacteria cultures were grown overnight with shaking at 37 °C. Optical density at 600 nm was measured using a cuvette with 1 cm pathlength in a GENESYS 10S UV-Vis spectrophotometer (ThermoFisher). The cultures were then equalized to an OD600 of 0.1 and 1 µl of each strain was added to the plate and incubated overnight at 37 °C.
Construction of fakB1 and fakB2 complementation plasmids
Complementation plasmids were generated using pCM28. The fakB1 gene and upstream sequence were amplified by PCR from the AH1263 chromosome using primers JBKU69 (CCGGATCCTTAATTCATAAGCTTAAGATTATTTAATCTTC) and JBKU70 (CACCTGCAGCTTTAGCATTTTGGCTTTCAAATGATGAAC). The resulting product was digested with BamHI and PstI and cloned into the same sites of pCM28. The fakB2 gene appears to be co-transcribed with folA because the genes are separated by 14 bp. Therefore, a complementation plasmid was generated that included the presumed folA promoter and fakB2, with folA deleted. The upstream fragment was amplified using primers JBKU71 (CAGAATTCACACCTTATTGCCAAAGAATGTG) and JBKU72 (CTGGATCCTTCTTCTATCATTTCATTTTTTATTACTAAG) while the downstream fragment including fakB2 was amplified with primers JBKU73 (CAGGATCCTAAGGGGGAAAACGACCATGACAAAACAG) and JBKU74 (GGTCTGCAGTCTATAAAGGATTGAAATGGAAGTAATTAAC). The upstream fragment was digested with BamHI and PstI and ligated into the same sites of pCM28 to produce pJB1032. The downstream fragment containing fakB2 was digested with EcoRI and BamHI and ligated into the same sites of pJB1032 to generate pJB1033. The respective complementation plasmids were then introduced into fakB1 or fakB2 mutants by Φ11-mediated transduction.
Plate reader growth assays
Overnight cultures were standardized to an OD600 of 0.005 in wells of a 96-well plate in TSB or TSB containing fatty acids to a volume of 200 µl. TSB was supplemented with glucose to 14 mM and fatty acids were added just prior to inoculation of cultures. Myristic acid and stearic acid were resuspended in methanol and diluted to a final concentration of 25 µg ml−1, which equals 109.5 and 87.9 µM, respectively. Unsaturated fatty acid concentration was chosen based on similar studies and adjusted to close to the MIC for AH1263. Linoleic acid was added at final concentration of 0.005 % (v/v) (160 µM), oleic acid to 0.01 % (v/v) (314 µM) and palmitoleic acid was used at 0.02 % (v/v) (703 µM). Cultures were grown in a Spark 10M plate reader (Tecan Group) with orbital shaking at 37 °C, and OD600 measurements were collected at 15 min intervals. Only 30 min intervals are reported in the graphs. For some figures, the area under the curve (AUC) was calculated for each individual well using GraphPad Prism v6.07 and averaged.
Fatty acid killing assay
Wild-type, fakA mutant and fakA complement strains were grown to exponential phase in 12.5 ml TSB in 125 ml flasks at 37 °C with shaking at 250 r.p.m. Cells were then harvested by centrifugation, and washed once with PBS. Following this, cells were re-suspended in 12.5 ml PBS and 0.1 % (v/v) (3.14 mM) oleic acid or 250 µg myristic acid mL−1 (1.09 mM), and incubated with shaking at 37 °C for 15 min. Cells were plated on TSA at 0 and 15 min post-exposure to fatty acids to determine percent recovery.
Reverse transcription-quantitative real-time PCR (qPCR)
qPCR was performed as previously described with modifications [16]. Briefly, cultures were grown as detailed above, and duplicate biological wells were pooled in triplicate to extract RNA. Primers CNK66 (CATTTTACACAACGAGAGCAAGAC) and CNK67 (GCTGATTAAAGCTAATGCCTCAG) were used for ureA. The primer pair RT-sigA-f and RT-sigA-r [21] was used to amplify housekeeping sigma factor sigA as an internal control.
Urease activity assay
Strains were grown overnight in a 96-well plate with or without fatty acids as detailed in the ‘Plate reader growth assays’ section. The final OD600 values of the cultures were read before centrifuging the 96-well plate at 200 g for 10 min to sediment cells. Supernatants were removed, and cell pellets were then re-suspended in 230 µl Stuart’s broth [22], and incubated statically at 37 °C until the first sample developed a pink colour, typically after 1–2 h. Following incubation, the 96-well plate was centrifuged to sediment cells, and supernatants were transferred into a new 96-well plate. The 96-well plate containing the clarified supernatants was read at A560.
Statistical analysis
All statistical analyses were performed using GraphPad Prism v6.07 using either a Student’s t-test or a two-way ANOVA as indicated.
Results
The fakA mutants display a growth advantage in the presence of unsaturated fatty acids.
To investigate the effects of fatty acids on fak mutants, mutant strains of each one of the Fak proteins (FakA, FakB1 and FakB2) along with respective complement strains were incubated in media with or without unsaturated fatty acids. We first examined growth of the fakA mutant in TSB and found that under these conditions, the fakA mutant grows similarly to the parent strain, AH1263 (Fig. 1a). By contrast, the fakA mutant displayed enhanced growth when compared to the wild-type strain in the presence of unsaturated fatty acids (Fig. 1b). Specifically, in TSB containing linoleic acid, fakA is able to grow whereas the wild-type and fakA complement strains fail to proliferate (Fig. 1b). The fakB1 fakB2 double mutant strain phenocopies the fakA mutant and is consistent with our previous results showing that deletion of both fakB1 and fakB2 results in a strain resembling the fakA mutant [18]. Additionally, all other strains including fakB1 mutant, fakB2 mutant, fakA complement, fakB1 complement and fakB2 complement strains are unable to grow in the presence of linoleic acid (Figs 1b and S1, available in the online version of this article). To statistically analyse the data, we calculated the AUC for each strain and found that only the fakA and fakB1 fakB2 mutants were statistically different from the wild-type strain (Fig. 1c). A similar trend was observed in the presence of oleic acid, another unsaturated fatty acid that is less toxic than linoleic acid. Despite the decreased toxicity of oleic acid, the fakA mutant again displays a growth advantage compared to the wild-type and fakA mutant complement strains (Fig. 1d, e). Moreover, growth of the fakB1 fakB2 double mutant strain more similarly resembles that of the fakA mutant strain. Both fakB1 and fakB2 single mutant strains display an intermediate growth phenotype compared to the wild-type strain (Fig. S1). Complementation of each fatty acid binding protein restores growth to wild-type levels. Finally, we examined resistance to the unsaturated fatty acid palmitoleic acid. Consistent with results for other fatty acids, the fakA mutant displayed increased resistance to palmitoleic acid (Fig. S2). Collectively, these results demonstrate that the abrogation of FakA functionality, by deletion of either fakA by itself or the two fatty acid carrier proteins, leads to increased tolerance of unsaturated fatty acids.
Fig. 1.

fakA mutants are more resistant to unsaturated fatty acids. (a) Growth of wild-type (WT), fakA mutant and fakA complement strains (pfakA) in TSB. (b) Growth of wild-type, fakA mutant, fakB1 mutant, fakB2 mutant, fakB1 fakB2 double mutant and fakA complement strains in TSB containing 160 µM linoleic acid. (c) Data from (b) plotted as area under the curve (AUC). (d) Growth of wild-type, fakA mutant, fakB1 fakB2 double mutant and fakA complement strains in TSB containing 314 µM oleic acid. (e) Data from (d) plotted as AUC. Data are the average (n=3) of a representative experiment. All points have error bars (sd) which may be smaller than symbols. An asterisk indicates significantly different (P<0.01) from the wild type using a one-way ANOVA.
FakA deletion does not confer increased resistance to saturated fatty acids
Although S. aureus is generally tolerant to saturated fatty acids [23, 24], we sought to examine the effects of saturated fatty acids on growth of the fakA mutant using either myristic acid or stearic acid. When grown in TSB containing myristic acid, the fakA mutant, fakB1 mutant, fakB2 mutant and fakB1 fakB2 double mutant strains grow similarly to the wild-type strain (Fig. 2a). Likewise, when grown in TSB containing stearic acid, the fakA mutant and fakB1 fakB2 double mutant strains grow similarly to the wild-type strain until the post-exponential growth phase when there is a minor decrease to final growth yield compared to the wild-type (Fig. 2b). In contrast to what is observed for unsaturated fatty acids, fakA mutants do not display a growth advantage when grown in the presence of all saturated fatty acids tested. fakA mutants are more resistant to killing by unsaturated fatty acids. As an alternative approach to the growth experiments, we examined survivability to lethal doses of unsaturated fatty acids. To this end, wild-type, fakA mutant, and fakA complement strains were exposed to 10-fold higher concentrations of oleic acid (3.14 mM) or myristic acid (1.09 mM) and plated for colony forming units to determine percentage recovery. Similar to the experiments above, when exposed to lethal concentrations of oleic acid, the fakA mutant is able to withstand the challenge better than the wild-type strain (Fig. 3a). Specifically, after 15 min of incubation with 3.14 mM oleic acid, fakA mutant recovery is 11.4–fold higher than that of the wild-type strain exposed to the same stress. By contrast, when myristic acid was tested no difference was observed between the fakA mutant and wild-type strain (Fig. 3b). The fakA complement strain displayed a slight, but reproducible, decrease in survival in response to myristic acid. The same experiment was performed with 2.18 mM myristic acid and no difference was observed between the wild-type and fakA mutant (Fig. S3). Taking this evidence together, fakA mutants are more resistant to killing by lethal dosages of unsaturated fatty acids.
Fig. 2.

fakA does not improve growth in saturated fatty acids. Growth of wild-type (WT), fakA mutant, fakB1 mutant, fakB2 mutant and fakB1 fakB2 double mutant strain in TSB containing (a) 109 µM myristic or (b) 89 µM stearic acid. Data are the average (n=3) of a representative experiment. All points have error bars (sd) and may be smaller than symbols.
Fig. 3.

fakA mutants are more resistant to lethal concentrations of oleic acid. Percentage recovery was determined from wild-type (WT), fakA mutant and fakA complement (pfakA) strains incubated in PBS containing 3.14 mM oleic acid (a) or 1.09 mM myristic acid (b) for 15 min. Data are the average (n=3) with sem of a representative experiment. An asterisk indicates significantly different (P<0.01) relative to WT using a Student’s t-test.
The two-component system SaeRS is not involved in FakA-dependent unsaturated fatty acid resistance
We and another group have recently demonstrated that FakA positively regulates the two-component system SaeRS and that this system responds to the addition of fatty acids [16, 25]. We therefore wanted to observe whether SaeRS is involved in resistance of the fakA mutant to unsaturated fatty acids. To this end, we tested growth of saeR and fakA saeR mutants in the prescence of linoleic acid. We found that the fakA saeR double mutant grows similarly to the fakA single mutant, and the saeR single mutant grows similarly to the wild-type (Fig. S4). These data demonstrate that SaeRS does not contribute to the enhanced resistance of the fakA mutant to unsaturated fatty acids.
Ammonia-producing enzymes do not contribute to increased resistance of fakA mutant
A previous transcriptomic study focused on analysing transcriptional changes in S. aureus and Staphylococcus epidermidis after treatment with the unsaturated fatty acid sapienic acid [26]. These studies revealed that genes involved in ammonia and ammonium production (arginine deiminase) are up-regulated in response to this unsaturated fatty acid. Specifically, in S. aureus strain Newman, the operon encoding urease was up-regulated in response to sapienic acid challenge, while in S. epidermidis other ammonia-producing enzymes were up-regulated. Recently, during a metabolic analysis of an fakA mutant, we reported increased expression and activity of urease [17]. Therefore, we hypothesized that one of the mechanisms that fakA mutants utilize to resist unsaturated fatty acid challenge is to increase transcription of the urease operon and/or ammonia-producing enzymes. Consistent with our other study, ureA expression is 3.5-fold higher in the fakA mutant than in the wild-type strain when grown in TSB under these growth conditions (Fig. 4a). Much like sapienic acid [26], growth in TSB containing oleic acid increased expression of the urease operon, which was observed in the wild-type, fakA mutant, and fakA complement strains. Next, we confirmed that this increased transcription resulted in enhanced urease activity. We first examined the mutants when grown in the absence of added fatty acids and found increased urease activity in the fakA and fakB1 fakB2 mutants, but not fakB1 and fakB2 single mutants, compared to the wild-type strain (Fig. S5a). In agreement with the transcription studies, urease activity is higher in strains that have been grown in the presence of oleic acid compared to TSB alone (Fig. S5b). An intermediate increase was also observed in the fakA mutant in response to myristic acid.
Fig. 4.

Urease expression and activity is increased an fakA mutant. (a) Transcript levels of ureA were measured by qPCR for wild-type (WT), fakA mutant and fakA complement (pfakA) strains in TSB and TSB containing 314 µM oleic acid. An asterisk indicates a significant difference (P<0.01) relative to WT using Student’s t-test. Data are representative of three or more individual experiments. (b) Growth, represented as the area under the curve (AUC) for WT, fakA, ureB and fakA ureB in TSB containing 160 µM linoleic acid. Data are calculated from the growth curve in Fig. S5 and are the average (n=3) with sd. An asterisk indicates a significant difference (P<0.01) from the wild-type using a one-way ANOVA.
Considering the previous report with sapienic acid and our results showing increased urease activity in the absence of FakA, we tested whether urease contributes to fakA mutant resistance to unsaturated fatty acids. First, ureB and fakA ureB mutants were challenged with linoleic acid. As shown in Figs 4b and S5c, the presence or absence of ureB did not impact growth of the fakA mutant in the presence of linoleic acid. In the study with sapienic acid, it was demonstrated that inactivation of arginine deiminase led to a two-fold decreased MIC in sapienic acid [26]. Our strain encodes two arginine deiminase enzymes, ArcA1 and ArcA2; therefore, mutations were made in arcA1 and arcA2 individually in the fakA mutant. Upon exposure to linoleic acid, we identified no difference in growth for fakA with or without arcA1 or arcA2 (Figs 5 and S6). However, we did observe a slight improvement in growth for the single arcA1 mutant compared to the wild-type strain. We considered whether ArcA1 and ArcA2 could provide redundant function in this phenotype and, therefore, constructed an fakA arcA1 arcA2 triple mutant. This mutant is equally resistant as the fakA only mutant (Figs 5c and S6d). Based on these results, ammonia-producing enzymes do not contribute to the resistance of the fakA mutant to linoleic acid.
Fig. 5.
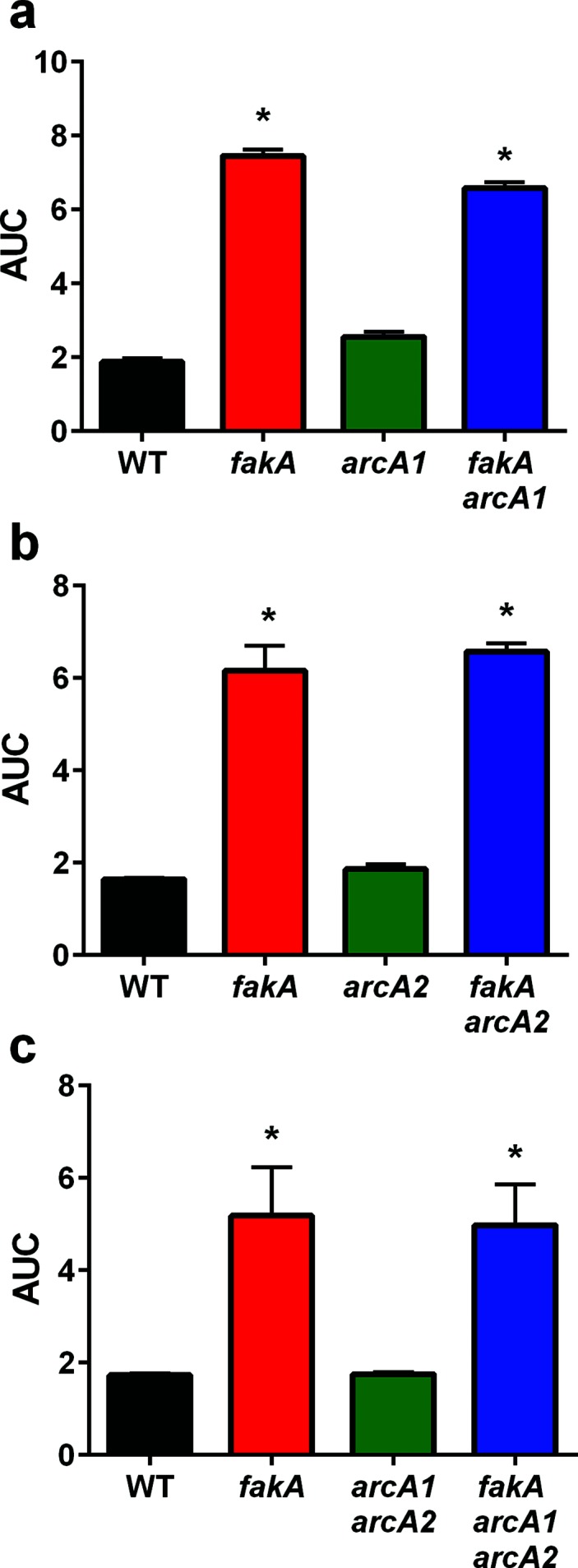
Arginine deiminase does not contribute to fakA mutant resistance to unsaturated fatty acids. (a) Growth of wild-type (WT), fakA mutant, arcA1 and fakA arcA1 mutants in TSB supplemented with 160 µM linoleic acid. (b) Growth of wild-type, fakA mutant, arcA2 and fakA arcA2 mutants in TSB supplemented with 160 µM linoleic acid. (c) Growth of wild-type, fakA mutant, arcA1 arcA2 and fakA arcA1 arcA2 mutants in TSB supplemented with 160 µM linoleic acid. Data are the average (n=5) with sd of the area under the curve (AUC) of a representative experiment. Full growth curves are provided in the Fig. S6b–d. An asterisk indicates significantly different (P<0.01) compared to WT using a one-way ANOVA.
Staphyloxanthin does not play a role in the ability of an fakA mutant to resist fatty acids
Throughout our studies with fakA mutants, we noted decreased pigmentation compared to the wild-type. Because staphyloxanthin has been shown to affect membrane fluidity [27, 28], we considered whether a change in pigmentation may cause resistance to fatty acids. We first looked at pigmentation of wild-type, fakA, fakB1, fakB2 and fakB1 fakB2 mutant strains on TSA plates containing oleic acid or myristic acid. On TSA, only the fakA and fakB1 fakB2 mutants showed decreased pigmentation (Fig. 6a). The fakA mutant pigmentation could be restored by expression of fakA from a plasmid (Fig. S7). Growth on TSA with myristic acid did not alter the pigmentation phenotypes and all strains appeared comparable to TSA alone. By contrast, growth on oleic acid led to decreased pigmentation in all strains, but most prominently seen in the wild-type, fakB1 and fakB2 mutant strains. Considering the pigmentation alterations, we tested whether staphyloxanthin directly affected resistance to fatty acids. To achieve this, we inactivated the staphyloxanthin biosynthesis pathway by generation of a crtM mutant and incubated crtM and crtM fakA mutants in the presence of linoleic acid. The absence of crtM did not alter the resistance of the fakA mutant (Figs 6b and S6); therefore, while staphyloxanthin production is altered by fakA, it is not a contributing factor to the resistance of the fakA mutant to unsaturated fatty acids.
Fig. 6.

Staphyloxanthin does not contribute to the resistance of the fakA mutant to fatty acids. (a) TSA plates containing 109 µM myristic acid or 314 µM oleic acid were spotted with wild-type, fakA, fakB1, fakB2 and fakB1 fakB2 cultures that were diluted to an OD600 of 0.1 and incubated at 37 °C for 24 h. (b) Growth of wild-type, fakA, crtM and fakA crtM in TSB containing 160 µM linoleic acid calculated as area under the curve (AUC) growth curve in Fig. S6. Data are the average (n=3) with sd. An asterisk denotes a significant difference from WT (P<0.01) using a one-way ANOVA.
Sigma factor B contributes to fatty acid resistance in the fakA mutant
Staphyloxanthin production is used as a visual indicator of the activity of the stress-response alternative sigma factor σB. As described above, the fakA mutant has decreased pigmentation, but staphyloxanthin does not play a role in fakA resistance to fatty acids. In addition, we envisaged that the presence of toxic fatty acids would probably elicit a stress response. To this end, we tested whether σB is important for the resistance to unsaturated fatty acids in the fakA mutant. As before, we grew the strains in the presence and absence of linoleic acid. We saw that the fakA mutant grew better than the wild-type strain, while the fakA sigB double mutant grew to an intermediate maximal optical density (Fig. S6e). However, when calculating the AUC, only the fakA single mutant was statistically different from the wild-type strain (Fig. 7). Together, these data demonstrate that the σB stress response is at least partially responsible for resistance of the fakA mutant to unsaturated fatty acids.
Fig. 7.
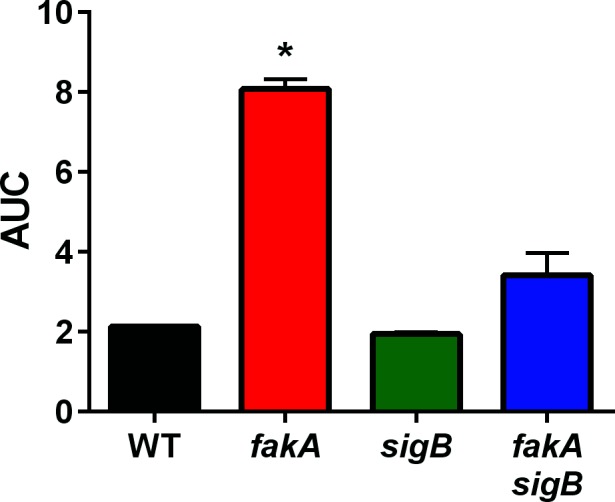
Alternative sigma factor σB contributes to fakA mutant resistance to fatty acids. Growth of wild-type (WT), fakA, sigB and fakA sigB in TSB containing 160 µM linoleic acid calculated as area under the curve (AUC) growth curve in Fig. S6. Data are the average (n=3) with sd. An asterisk denotes a significant difference from wild-type (P<0.01) using a one-way ANOVA.
Discussion
The discovery of a fatty acid kinase and fatty acid carrier proteins in S. aureus is a relatively new finding and, as such, much is left to elucidate about the role of this machinery in staphylococcal biology. Until now, the relationship between toxic fatty acids and fak mutants of S. aureus has remained largely unexplored. This facet is of particular interest as one of the hallmark roles elucidated for this complex is the binding of unsaturated and saturated fatty acids coupled with final incorporation into the lipid bilayer. Thus, the focus of this study was to determine if the ability to use exoFAs impacts resistance to unsaturated fatty acids.
The studies included herein demonstrate that mutants lacking Fak proteins are more resistant to unsaturated fatty acids. Specifically, either deletion of the fatty acid kinase (fakA) or mutation of both fatty acid binding proteins (fakB1 and fakB2) in combination allows for increased growth capabilities with linoleic acid, oleic acid or palmitoleic acid. Although previous studies have demonstrated fatty acid saturation specificity for the fatty acid binding proteins [15], single mutations of either fakB1 or fakB2 do not provide a growth advantage in the presence of either saturated or unsaturated fatty acids. This resembles our previous studies showing that α-haemolysin production is absent in an fakA mutant as well as the fakB1 fakB2 double mutant, but is relatively unaffected by either fakB single mutant strain [15]. These findings suggest that despite a previous demonstration of substrate preference [15], the fatty acid binding proteins share some phenotypic redundancy in function.
Unsaturated fatty acids are known to be toxic to bacteria; however, the mechanism behind this is not completely clear. If fatty acids disrupt the cellular membrane, this could be envisaged to occur at two primary points: (1) passive entrance into the lipid bilayer as a free fatty acid or (2) final incorporation into the membrane as a lipid sidechain. The FakB proteins are thought to be able to exchange fatty acids in the membrane, which are then phosphorylated by FakA for membrane incorporation [15]. The finding that the fakA and fakB1 fakB2 mutants resist the toxicity of unsaturated fatty acids is consistent with the model in which unsaturated fatty acids inflict damage upon incorporation into the membrane. However, confirmation of this is difficult because both exogenous and endogenous fatty acids feed through the same enzymatic pathway, making those enzymes essential. Similarly, the intricate details by which bacteria resist fatty acid toxicity are not fully understood.
One mechanism that has been attributed to unsaturated fatty acid resistance in staphylococci is the altered expression of genes that lead to the production of ammonia or ammonium [11, 26]. Specifically, genes encoding proteins for the arginine deiminase pathway (arcC), urease metabolism (ureC) and nitrate reductase pathways (nreABC, narGHIJ and nirBD) are all up-regulated in response to sapienic acid challenge in either S. aureus or S. epidermidis [26]. The same study noted that inactivation of the ammonia-producing arginine deiminase pathway increased the sensitivity of two S. aureus strains to sapienic acid. Increased expression of UreC and UreE proteins has also been observed in response to cis-6-hexadecenoic acid [13]. However, none of those studies tested whether urease contributes to resistance. In a recent metabolic study of an fakA mutant, we observed changes in urease expression and activity [17]. This was also observed under the growth conditions used here (Figs 4 and S6). Thus, we considered that ammonia production through either urease or arginine deiminase may have been involved in fakA mutant fatty acid resistance. However, inactivation of these pathways did not affect growth of the fakA mutant in linoleic acid.
Mutation of fakA mutants has been shown to lead to the accumulation of fatty acids in the cytoplasm [25]. Some of these fatty acids are toxic and therefore would be expected to elicit a stress response. In S. aureus, the alternative sigma factor B (σB) initiates a stress response by inducing the transcription of genes that will aid the bacterium in high-stress situations [29, 30]. One well-known operon controlled by σB is the crtOPQMN operon that encodes the staphyloxanthin biosynthesis pathway [31]. Staphyloxanthin contributes to multiple roles in the cell, including alteration of membrane fluidity. In addition, this pathway was previously shown to impart a protective effect to strain SH1000 when challenged with 1 mM linoleic acid [11]. Under our experimental conditions, our LAC-derived wild-type strain does not grow; therefore, our goal was to test if staphyloxanthin contributes to enhanced resistance in the fakA mutant and we found that it is not a contributing factor (Fig. 6b). In the same study mentioned above, sigB was also found to be important for survival of SH1000. Considering that σB controls the global stress response, and the sensitivity of an SH1000 sigB mutant to linoleic acid, it is not surprising that we identified that σB is important for fakA mutant resistance to unsaturated fatty acids. Since σB is a global regulator, we were unable to identify the specific process that σB alters that leads to resistance, and this will be one focus of future studies.
Supplementary Data
Funding information
This study was supported by funding from the National Institute of Allergy and Infectious Diseases award R01AI121073 to JLB. In addition, CNK was supported, in part, by a University of Kansas Medical Center Biomedical Research Training Program fellowship.
Acknowledgements
We would like to thank Dr. Kelly Rice at the University of Florida for her input on the manuscript. Mutants containing the bursa aurealis transposon were acquired from BEI Resources.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
No human subjects or animals were used in this study.
Footnotes
Abbreviations: AUC, area under the curve; exoFA, exogenous fatty acid; qPCR, quantitative PCR.
Seven supplementary figures are available with the online version of this article.
Edited by: A. Grundling and C. Dahl
References
- 1.Moet GJ, Jones RN, Biedenbach DJ, Stilwell MG, Fritsche TR. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998-2004) Diagn Microbiol Infect Dis. 2007;57:7–13. doi: 10.1016/j.diagmicrobio.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JK, Khoie T, Shurland S, Kreisel K, Stine OC, et al. Skin and soft tissue infections caused by methicillin-resistant Staphylococcus aureus USA300 clone. Emerg Infect Dis. 2007;13:1195–1200. doi: 10.3201/eid1308.061575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kiezel-Tsugunova M, Kendall AC, Nicolaou A. Fatty acids and related lipid mediators in the regulation of cutaneous inflammation. Biochem Soc Trans. 2018;46:119–129. doi: 10.1042/BST20160469. [DOI] [PubMed] [Google Scholar]
- 4.Dawgul MA, Greber KE, Sawicki W, Kamysz W. Human host defense peptides - role in maintaining human homeostasis and pathological processes. Curr Med Chem. 2016 [PubMed] [Google Scholar]
- 5.Gallo RL, Hooper LV. Epithelial antimicrobial defence of the skin and intestine. Nat Rev Immunol. 2012;12:503–516. doi: 10.1038/nri3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lampe MA, Burlingame AL, Whitney J, Williams ML, Brown BE, et al. Human stratum corneum lipids: characterization and regional variations. J Lipid Res. 1983;24:120–130. [PubMed] [Google Scholar]
- 7.Wilkinson DI, Karasek MA. Skin lipids of a normal and mutant (asebic) mouse strain. J Invest Dermatol. 1966;47:449–455. doi: 10.1038/jid.1966.168. [DOI] [PubMed] [Google Scholar]
- 8.Heczko PB, Lütticken R, Hryniewicz W, Neugebauer M, Pulverer G. Susceptibility of Staphylococcus aureus and group A, B, C, and G streptococci to free fatty acids. J Clin Microbiol. 1979;9:333–335. doi: 10.1128/jcm.9.3.333-335.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller SJ, Aly R, Shinefeld HR, Elias PM. In vitro and in vivo antistaphylococcal activity of human stratum corneum lipids. Arch Dermatol. 1988;124:209–215. doi: 10.1001/archderm.1988.01670020027012. [DOI] [PubMed] [Google Scholar]
- 10.Wille JJ, Kydonieus A. Palmitoleic acid isomer (C16:1delta6) in human skin sebum is effective against gram-positive bacteria. Skin Pharmacol Appl Skin Physiol. 2003;16:176–187. doi: 10.1159/000069757. [DOI] [PubMed] [Google Scholar]
- 11.Kenny JG, Ward D, Josefsson E, Jonsson IM, Hinds J, et al. The Staphylococcus aureus response to unsaturated long chain free fatty acids: survival mechanisms and virulence implications. PLoS One. 2009;4:e4344. doi: 10.1371/journal.pone.0004344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moran JC, Alorabi JA, Horsburgh MJ. Comparative transcriptomics reveals discrete survival responses of S. aureus and S. epidermidis to sapienic acid. Front Microbiol. 2017;8:33. doi: 10.3389/fmicb.2017.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumann Y, Ohlsen K, Donat S, Engelmann S, Kusch H, et al. The effect of skin fatty acids on Staphylococcus aureus. Arch Microbiol. 2015;197:245–267. doi: 10.1007/s00203-014-1048-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Delekta PC, Shook JC, Lydic TA, Mulks MH, Hammer ND. Staphylococcus aureus utilizes host-derived lipoprotein particles as sources of exogenous fatty acids. J Bacteriol. 2018 doi: 10.1128/JB.00728-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parsons JB, Broussard TC, Bose JL, Rosch JW, Jackson P, et al. Identification of a two-component fatty acid kinase responsible for host fatty acid incorporation by Staphylococcus aureus. Proc Natl Acad Sci USA. 2014;111:10532–10537. doi: 10.1073/pnas.1408797111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krute CN, Rice KC, Bose JL. VfrB Is a Key Activator of the Staphylococcus aureus SaeRS Two-Component System. J Bacteriol. 2017;199 doi: 10.1128/JB.00828-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Demars ZR, Bose JL. Redirection of metabolism in response to fatty acid kinase in Staphylococcus aureus. J Bacteriol. 2018 doi: 10.1128/JB.00345-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bose JL, Daly SM, Hall PR, Bayles KW. Identification of the Staphylococcus aureus vfrAB operon, a novel virulence factor regulatory locus. Infect Immun. 2014;82:1813–1822. doi: 10.1128/IAI.01655-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krausz KL, Bose JL. Bacteriophage Transduction in Staphylococcus aureus: Broth-Based Method. Methods Mol Biol. 2016;1373:63–68. doi: 10.1007/7651_2014_185. [DOI] [PubMed] [Google Scholar]
- 20.Bose JL, Fey PD, Bayles KW. Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl Environ Microbiol. 2013;79:2218–2224. doi: 10.1128/AEM.00136-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lehman MK, Bose JL, Sharma-Kuinkel BK, Moormeier DE, Endres JL, et al. Identification of the amino acids essential for LytSR-mediated signal transduction in Staphylococcus aureus and their roles in biofilm-specific gene expression. Mol Microbiol. 2015;95:723–737. doi: 10.1111/mmi.12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Onal Okyay T, Frigi Rodrigues D. High throughput colorimetric assay for rapid urease activity quantification. J Microbiol Methods. 2013;95:324–326. doi: 10.1016/j.mimet.2013.09.018. [DOI] [PubMed] [Google Scholar]
- 23.Kitahara T, Koyama N, Matsuda J, Aoyama Y, Hirakata Y, et al. Antimicrobial activity of saturated fatty acids and fatty amines against methicillin-resistant Staphylococcus aureus. Biol Pharm Bull. 2004;27:1321–1326. doi: 10.1248/bpb.27.1321. [DOI] [PubMed] [Google Scholar]
- 24.Parsons JB, Yao J, Frank MW, Jackson P, Rock CO. Membrane disruption by antimicrobial fatty acids releases low-molecular-weight proteins from Staphylococcus aureus. J Bacteriol. 2012;194:5294–5304. doi: 10.1128/JB.00743-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ericson ME, Subramanian C, Frank MW, Rock CO. Role of fatty acid kinase in cellular lipid homeostasis and SaeRS-dependent virulence factor expression in Staphylococcus aureus. MBio. 2017;8 doi: 10.1128/mBio.00988-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moran JC, Alorabi JA, Horsburgh MJ. Comparative transcriptomics reveals discrete survival responses of S. aureus and S. epidermidis to sapienic acid. Front Microbiol. 2017;8:33. doi: 10.3389/fmicb.2017.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishra NN, Liu GY, Yeaman MR, Nast CC, Proctor RA, et al. Carotenoid-related alteration of cell membrane fluidity impacts Staphylococcus aureus susceptibility to host defense peptides. Antimicrob Agents Chemother. 2011;55:526–531. doi: 10.1128/AAC.00680-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tiwari KB, Gatto C, Wilkinson BJ. Interrelationships between fatty acid composition, staphyloxanthin content, fluidity, and carbon flow in the Staphylococcus aureus membrane. Molecules. 2018;23:1201. doi: 10.3390/molecules23051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan PF, Foster SJ, Ingham E, Clements MO. The Staphylococcus aureus alternative sigma factor sigmaB controls the environmental stress response but not starvation survival or pathogenicity in a mouse abscess model. J Bacteriol. 1998;180:6082–6089. doi: 10.1128/jb.180.23.6082-6089.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gertz S, Engelmann S, Schmid R, Ziebandt AK, Tischer K, et al. Characterization of the sigma(B) regulon in Staphylococcus aureus. J Bacteriol. 2000;182:6983–6991. doi: 10.1128/JB.182.24.6983-6991.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pelz A, Wieland KP, Putzbach K, Hentschel P, Albert K, et al. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. J Biol Chem. 2005;280:32493–32498. doi: 10.1074/jbc.M505070200. [DOI] [PubMed] [Google Scholar]
- 32.Boles BR, Thoendel M, Roth AJ, Horswill AR. Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One. 2010;5:e10146. doi: 10.1371/journal.pone.0010146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, et al. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio. 2013;4:e00537. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kreiswirth BN, Löfdahl S, Betley MJ, O'Reilly M, Schlievert PM, et al. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature. 1983;305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 35.Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, et al. agr-Dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. J Innate Immun. 2010;2:546–559. doi: 10.1159/000319855. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.