

Abstract
Cryptolepine, neocryptolepine and isocryptolepine are naturally occurring indoloquinoline alkaloids with various spectrum of biological properties. Structural modification is an extremely effective means to improve their bioactivities. This review enumerates several neocryptolepine and isocryptolepine analogues with potent antiproliferative activity against MV4-11 (leukemia), A549 (lung cancer), HCT116 (colon cancer) cell lines in vitro. Its activity towards normal mouse fibroblasts BALB/3T3 was also evaluated. Furthermore, structure activity relationships (SAR) are briefly discussed. The anticancer screening of neocryptolepine derivatives was performed in order to determine their cytotoxic and growth inhibitory activities across the JFCR39 cancer cell line panel.
Keywords: cryptolepine, neocryptolepine, isocryptolepine, antiproliferative activity, structure activity relationships
1. Introduction
1.1. Antitumoral Activity of Cryptolepine
Cryptolepine (1), neocryptolepine (2), and isocryptolepine (3) are typical indoloquinoline alkaloids isolated from the roots of Cryptolepis sanguinolenta [1,2].
These alkaloids are composed of the tetracyclic indoloquinoline ring system, which only differ with respect to the orientation and site of their indole and quinoline ring junctures (Figure 1). By introducing the appropriate motif at the certain positions, their derivatives can increase better biological activity than the mother core [3].
Figure 1.

Structures of typical indoloquinolines from Cryptolepis sanguinolenta.
Many researchers have achieved the structural modification of cryptolepine scaffold for the purpose of improving antitumor activity, while the pharmacological properties of the analogues are being studied deeply.
The bioactivities of many antitumoral agents are related to their interactions with the DNA molecule, which is regarded as a classical target for these drugs in clinical use. The basic mechanism of antitumoral activity of these drugs is to affect the replication, expression, transcription and other physiological functions of the DNA, which causes the tumor cell death [4].
In 1990, Yamato et al. synthesized the indoloquinoline derivatives 4 (Figure 2), and screened its biological properties in vitro and in vivo. The compounds 4 showed potential antitumor activity (P388 leukemia in mice), DNA intercalative property, and ability to induce topoisomerase II dependent DNA cleavage [5].
Figure 2.

Structures of cryptolepine derivatives.
In 1997, Deady et al. studied a series of cryptolepine derivatives 5 and evaluated their antitumoral activity in a series of murine and human tumor cell lines such as the mice lung cancer cells (LLC), mice leukemia cells (P388), human leukocyte cells (JL). These compounds appear to be mixed topoisomerase I/II inhibitors in the human leukemia cell lines studied [6].
In 1998, Bonjean et al. verified the cryptolepine alkaloids bound tightly to DNA as a typical intercalating agent by various means of absorption, such as fluorescence, circular, and linear dichroism, as well as by a relaxation assay using DNA topoisomerases. At the same time, they provided direct evidence that DNA is the primary target of cryptolepine. The mechanism of the compounds inhibiting tumor cell proliferation is mainly based on the synthesis of DNA inhibition, not the inhibition of proteins and RNA [7].
In 2002, Lisgarten, John N., reported that cryptolepine interacts with the DNA fragment d(CCTAGG)2 in a base-stacking intercalation mode by using X-ray crystallography. It was found that cryptolepine intercalated between pyrimidine bases of the fragment in the form of π-π accumulation. This is the first single crystal structure of DNA intercalator complex, which is the small molecule to bind a non-alternating (pyrimidine-pyrimidine) DNA sequence [8].
In 2012, Boddupally et al. have synthesized a series of 11-substituted cryptolepine derivatives 6 (Figure 2). The compound 6 showed the most potent anticancer activity with IC50 = 0.97 μM against HCT-116 colon cancer cell line and IC50 = 2.33 μM against Raji lymphoma cells in further cytotoxic test in vitro. At the same time, this compound showed a strong inhibition of c-MYC expression [9].
Gu and Lu demonstrated the binding of aniline-substituted cryptolepine derivatives with calf-thymus DNA presumably via an intercalation mechanism and studied the binding mode of these derivatives with duplex DNA by Surflex-dock software. They reported that these derivatives intercalated into the base-pairs, and reacted with DNA via mainly π-π interaction with medium, moreover the functional groups substituted on aniline ring affected the binding abilities [10,11].
The aim of this review is to present an overview of the potential of neocryptolepine and isocryptolepine as scaffolds for the design and development of new anticancer drugs. Both compounds have also the linearly arranged tetracyclic plane as same as cryptolepine, so they can be expected as candidates of antitumoral agent, although they have a slightly weaker capability to intercalate into DNA and inhibit human topoisomerase II [12].
1.2. Antitumoral Activity of Neocryptolepines and Isocryptolepines
In our group, we have synthesized an array of novel neocryptolepine derivatives 7, and their congeners 8, 11-aminoalkylamino-substituted chromeno[2,3-b]indoles 9, and isocryptolepine derivatives 10 (Figure 3). Then the antiproliferative activities of these compounds were tested in vitro against MV4-11 (human leukemia), HCT116 (human colon cancer), and A549 (human non-small cell lung cancer) and BALB/3T3 (normal murine fibroblasts) cell lines.
Figure 3.

Structure of neocryptolepine derivatives 7, 8, chromeno[2,3-b]indoles 9, and isocryptolepine derivatives 10.
We focused our attention on modifying the neocryptolepine structure at four positions: (1) the side chain at C11, introducing the various amino groups, e.g., -NH(CH2)nNH2 (n = 2,3,4), -NH(CH2)3N(C2H5)2, -NH(CH2)3OH, -NHCH2CH(OH)CH2NH2, -NHCH2CH(CH3)CH2NH2, -NHCH2CH(CH3)NH2, -NHCH2C(CH3)2NH2, and so on; (2) replacing the H atom at C2 with MeO, Br, Cl, Me, and replacing H at C9 with COOMe; (3) altering the Me from N5 to N6, (4) changing the N5 to O atom and (5) incorporating metal ion coordinated with diamine side chain [13,14,15,16,17,18].
In addition, we have also engaged in modifying isocryptolepine derivatives in three ways: the amino substituent effect at C6, and N11 methyl localization effect, and the substituent group effect at C2 of quinoline moiety [19].
Some compounds with higher antiproliferative activity are listed in Table 1 and Table 2. The SAR (structure activity relationships) studies reveal that the most necessary strategy is introducing ω-aminoalkylamino group in the side chain. Among the tested 84-neocryptolepine derivatives and 44-isocryptolepine derivatives, the most potent compounds with higher activity contain the -NH(CH2)3NH2 side chain.
Table 1.
| No. | Substituent | IC50 μM | |||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | R4 | R5 | BALB/3T3 | MV4-11 | A549 | HCT116 | |
| 7a | H | NH(CH2)3NH2 | H | Me | - | 0.884 ± 0.115 | 0.066 ± 0.023 | 0.205 ± 0.079 | 0.302 ± 0.056 |
| 7b | Cl | NH(CH2)3NH2 | H | Me | - | 0.401 ± 0.015 | 0.068 ± 0.018 | 0.761 ± 0.169 | 0.195 ± 0.044 |
| 7c | Br | NH(CH2)3NH2 | H | Me | - | 0.869 ± 0.018 | 0.012 ± 0.002 | 0.543 ± 0.256 | 0.274 ± 0.050 |
| 7d | OMe | NH(CH2)3NH2 | H | Me | - | 0.978 ± 0.021 | 0.102 ± 0.021 | 0.407 ± 0.117 | 0.155 ± 0.042 |
| 7e | Cl | NHCH2CH(OH)CH2NH2 | H | Me | - | 0.896 ± 0.042 | 0.042 ± 0.014 | 0.197 ± 0.028 | 0.138 ± 0.050 |
| 7f | Br | NHCH2CH(OH)CH2NH2 | H | Me | - | 0.864 ± 0.015 | 0.057 ± 0.015 | 0.190 ± 0.027 | 0.117 ± 0.055 |
| 7g | H | NH(CH2)3NH2 | COOMe | Me | - | 0.933 ± 0.047 | 0.044 ± 0.011 | 0.820 ± 0.456 | 0.176 ± 0.055 |
| 7h | Br | NH(CH2)3NH2 | COOMe | Me | - | 0.773 ± 0.023 | 0.050 ± 0.016 | 0.302 ± 0.138 | 0.164 ± 0.070 |
| 7i | Cl | NH(CH2)3NH2 | COOMe | Me | - | 0.833 ± 0.030 | 0.056 ± 0.035 | 0.194 ± 0.063 | 0.116 ± 0.078 |
| 7j | Cl | NH(CH2)3NH2 | Br | Me | - | 0.834 ± 0.067 | 0.076 ± 0.034 | 0.464 ± 0.011 | 0.284 ± 0.142 |
| 8 | Cl | NH(CH2)2NH2 | H | - | Me | 0.437 ± 0.400 | 0.456 ± 0.123 | 8.282 ± 0.585 | 6.989 ± 1.416 |
| 9 | OMe | NH(CH2)2NH2 | H | - | - | 6.87 ± 1.74 | 0.12 ± 0.07 | 9.29 ± 1.10 | 1.93 ± 0.24 |
| Cisplatin | 8.53 ± 3.53 | 2.36 ± 0.68 | 7.83 ± 2.60 | 3.47 ± 0.77 | |||||
| Doxorubicin HCl | 1.08 ± 0.03 | 0.006 ± 0.002 | 0.33 ± 0.10 | 0.39 ± 0.10 | |||||
Table 2.
Cytotoxic effect of isocryptolepine derivatives [17].
| No. | Substituent | IC50 μM | |||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | BALB/3T3 | MV4-11 | A549 | HCT116 | |
| 10a | H | NH(CH2)3NH2 | H | 1.05 ± 0.13 | 0.12 ± 0.01 | 0.17 ± 0.05 | 0.26 ± 0.11 |
| 10b | OMe | NH(CH2)3NH2 | H | 0.31 ± 0.03 | 0.12 ± 0.05 | 0.16 ± 0.03 | 0.07 ± 0.03 |
| 10c | NO2 | NH(CH2)3NH2 | H | 0.36 ± 0.15 | 0.15 ± 0.06 | 0.35 ± 0.22 | 0.10 ± 0.02 |
| 10d | H | NH(CH2)3NH2 | Me | 0.95 ± 0.24 | 0.11 ± 0.04 | 0.18 ± 0.03 | 0.06 ± 0.02 |
| 10e | NO2 | NH(CH2)3NH2 | Me | 0.08 ± 0.02 | 0.05 ± 0.01 | 0.11 ± 0.07 | 0.01 ± 0.00 |
| 10f | H | NH(CH2)3NH(CH3)2 | H | 1.18 ± 0.20 | 0.41 ± 0.31 | 0.87 ± 0.27 | 0.55 ± 0.17 |
| 10g | Cl | NH(CH2)3NH(CH3)2 | Me | 0.82 ± 0.08 | 0.08 ± 0.03 | 0.57 ± 0.31 | 0.10 ± 0.02 |
Among them, 11-(3-amino-2-hydroxy)propylamino derivatives 7e and 7f were the most cytotoxic with a mean IC50 value of 0.042 μM and 0.057 μM against the MV4-11 cell line, 0.197 μM and 0.190 μM against the A549 cell line, and 0.138 μM and 0.117 μM against the HCT116 cell line.
We propose the reason that the amino terminus domain in the side chain would react with negative charged phosphate groups in DNA, which increases the insertion ability of the complex to DNA.
The methyl localization effect is an important additional contributor to increase the activity. The activity is greatly reduced (10~30 fold lower) by changing the Me group from N5 to N6 (i.e., 8) of neocryptolepine derivatives. The Me group can increase the activity of isocryptolepine derivatives, as found by a comparison of the compounds 10e and 10c.
Replacing H atom at the C2 with MeO, Br, Cl, Me, or replacing H at C9 by COOMe can improve the antiproliferactive activity of neocryptolepine derivatives. Each group contributes differently to the antiproliferactive activity of different tumor cells. For example, 7c with Br at C2 shows the best antiproliferactive activity against MV4-11 leukemia cells, and this modification did not increase antiproliferactive activity against normal BALB/3T3 cells, but unexpectedly it decreases 2-fold the antiproliferactive activity against A549 lung cancer cells. It can be suggested that this modification increases the selectivity of the tested compound, improving their effect on leukemic cells, without increasing possible adverse effects on normal cells. The substituent effect at C2 of isocryptolepine core also can change the activity; NO2 is the effective group to increase the activity against cancer but unfortunately also normal cells (10c and 10e). Therefore, we must examine our best to find an effective substituent for further improving the activity.
We boldly tried to replace the 5-nirtogen atom with oxygen atom, forming 9, which significantly drop the antiproliferactive activity [16].
From all the assay data, isocryptolepine analogues are more potential as the anticancer drug candidates in comparison with the neocrytolepine analogues, compound 10e (or 10a) shows the higher antiproliferactive activity against A549 and HCT116 cancer cell, and lower antiproliferactive activity against normal cells comparing 7a, when amino-substituent is NH(CH2)3NH2 and substituent group is H at C2.
The mode of neocryptolepine and isocryptolepine binding to DNA was studied using UV-VIS absorption spectroscopy with salmon fish sperm DNA. From the DNA binding studies, it can be proven that the methyl localization effect and the substituent group effect at C2 of quinoline moiety influence the capability to intercalate into DNA. Two effects improve the activity of isocryptolepine to interact with DNA, the binding constant of 10f-DNA was 1.05 × 106 L/mol and 10g-DNA was 4.84 × 106 L/mol [19]. The activity of neocryptolepine to interact with DNA varied; the binding constant of 7c-DNA was 2.93 × 105 and 8-DNA was 3.28 × 105 L/mol [13].
In a series of neocrytolepine analogues, 7c shows the best antiproliferactive activity against MV4-11 leukemia with IC50 0.012 ± 0.002 μM. Compounds 7f and 7i show almost the same highest antiproliferactive activity against A549 lung cancer cells with IC50 0.190 ± 0.027 μM and 0.194 ± 0.063 μM, and the antiproliferative activity against HCT116 colon cancer cells with IC50 0.117 ± 0.055 μM and 0.116 ± 0.078 μM. Analogue 7b unfortunately shows 2-fold higher cytotoxic activity against normal BALB/3T3 cells with the IC50 0.401 ± 0.015 μM than other studied neocrytolepine analogues and 20-fold higher than cisplatin [13].
In series of isocryptolepine analogues, 10e shows the best antiproliferactive activity against cancer cell lines with IC50 0.05 ± 0.01 μM (MV4-11), IC50 0.11 ± 0.07 μM (A549), and IC50 0.01 ± 0.00 μM (HCT116). Unfortunately, this compound had also 4~15 fold higher cytotoxic activity against normal BALB/3T3 cells with the IC50 0.08 ± 0.02 μM. These two simultaneous modifications at C2-NO2 and at N11-Me showed 15- and 100-fold increased cytotoxicity of 10e in comparison to doxorubicin and cisplatin, cytostatic used as a control.
In 2016, in vitro antiproliferative activities of neocryptolepine derivatives were evaluated by Okada (Table 3). The compounds 7a and 7d showed the highest anticancer activity, having IC50 values of 0.50 μM and 0.20 μM against the breast cancer MDA-MB-453 cell line, at the same time 7a having 0.64 μM and 2.7 μM against WiDr (colon adenocarcinoma) and SKOv3 (ovarian cancer) cell lines, 7d having 0.37 μM and 1.3 μM against WiDr (colon adenocarcinoma) and SKOv3 (ovarian cancer) cell lines.
Table 3.
Antiproliferative activity of neocryptolepine derivatives against cell lines of the breast cancer MDA-MB-453, WiDr (colon adenocarcinoma) and SKOv3 (ovarian cancer).
| Compound | MDA-MB-453 IC50(μM) | WiDr IC50(μM) | SKOv3 IC50(μM) |
|---|---|---|---|
| Neocryptolepine | 7.48 ± 4.42 | N.T. | N.T. |
| 7a | 0.50 ± 0.24 | 0.64 ± 0.13 | 2.7 ± 0.42 |
| 7d | 0.20 ± 0.09 | 0.37 ± 0.07 | 1.3 ± 0.18 |
| Cisplatin | 7.6 ± 0.7 a | 8.84 ± 0.52 c | 18.2 ± 0.1 d |
| Doxorubicin | 0.086 ± 0.006 b | 0.089 e | 0.52 |
N.T.: not tested, a Rakic et al. (2012), b Sandhu et al. (2012), c Kuo et al. (2010), d Ganta et al. (2014), e Molphy et al. (2014).
Recently, we reported the in vitro antiproliferative activity of 11-substituted neocryptolepines with branched ω-aminoalkylamino chain. These 2-substituted 5-methyl-indolo[2,3-b]quinoline derivatives were prepared by nucleophilic aromatic substitution (SNAr) of 11-chloroneocryptolepines 2 with appropriate 1,2- and 1,3-diamines (Scheme 1). Many of the prepared neocryptolepine derivatives showed submicromolar antiproliferative activity of less than μM against the human leukemia MV4-11 cell line (Table 4).
Scheme 1.

Synthesis of 11-aminoalkylamino-5-methylindolo[2,3-b]quinolines.
Table 4.
Antiproliferative activity of 11-aminoalkylamino-5-methyl-indolo[2,3-b]quinolines against cell lines of MV4-11, A549 and HCT116, and cytotoxicity against normal mice fibroblast BALB/3T3 [15].
| No. | Substituent | IC50 μM | ||||
|---|---|---|---|---|---|---|
| Compounds | R1 | C11-Substituent | MV4-11 | BALB/3T3 | A549 | HCT116 |
| Cisplatin | - | 2.820 ± 0.450 | 8.700 ± 0.097 | 9.870 ± 2.400 | 8.500 ± 0.540 | |
| Doxorubicin HCl | - | 0.006 ± 0.002 | 1.078 ± 0.033 | 0.329 ± 0.097 | 0.390 ± 0.098 | |
| 2a | H | Cl | 1.312 ± 0.262 | - | - | - |
| 2b | Br | Cl | 0.810 ± 0.145 | - | - | - |
| 7l | H |
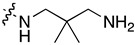
|
0.150 ± 0.060 | 4.789 ± 2.018 | 1.512 ± 0.198 | 1.262 ± 0.361 |
| 7m | Cl |
|
0.119 ± 0.043 | 9.131 ± 0.844 | 1.455 ± 0.168 | 1.373 ± 0.351 |
| 7n | Me |
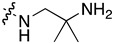
|
0.105 ± 0.027 | 10.558 ± 0.330 | 1.795 ± 0.270 | 2.370 ± 0.481 |
| 7o | Cl |
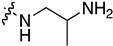
|
0.103 ± 0.014 | 1.018 ± 0.017 | 1.269 ± 0.118 | 1.204 ± 0.283 |
| 7p | Br |
|
0.078 ± 0.020 | 0.939 ± 0.018 | 0.988 ± 0.164 | 0.842 ± 0.367 |
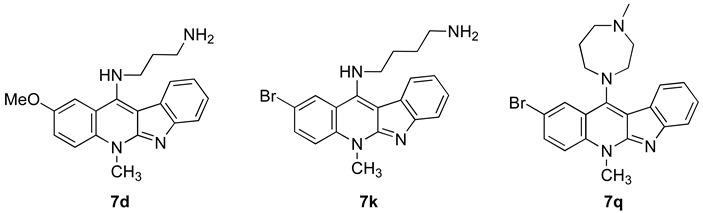
For further studying cytotoxic and growth inhibitory activities of neocryptolepine derivatives, the anticancer screening was performed across the JFCR39 cancer cell line panel, evaluated at five concentration levels (100, 10, 1.0, 0.1, and 0.01 µM) (Table 5). The results (the means of GI50, TGI, and LC50 values) showed that 7d have potent anticancer activity against the melanoma (LOX-IMVI) and lung (NCIH522, A549, and DMS273) cancer cell lines in the JFCR39 panels. Compound 7d shows a notable cytotoxicity against the breast (BSY-1, LC50 = 0.82 µM), CNS (SF-539, LC50 = 0.60 µM, SNB-75, LC50 = 0.91 µM), colon (HCC2998, LC50 = 0.74 µM), lung (NCI-H522, LC50 = 0.71 µM, A549, LC50 = 0.85, DMS273, LC50 = 0.70, DMS114, LC50 = 0.72 µM), and ovarian (VCAR-4, LC50 = 0.48 µM) cancer cell lines [20].
Table 5.
Growth inhibitory (GI50) and cytotoxic (LC50) activities of compounds 7d, 7k and 7q across the JFCR 39 panel.
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Tissue of Origin | Cell Line | 7d | 7k | 7q | ||||||
| GI 50 (µM) | TGI (µM) | IC50 (µM) | GI 50 (µM) | TGI (µM) | IC50 (µM) | GI 50 (µM) | TGI (µM) | IC50 (µM) | ||
| Breast | HBC-4 | 0.06 | 0.80 | 9.5 | 0.08 | 0.61 | 6.1 | 1.3 | 4.3 | 19 |
| BSY-1 | 0.11 | 0.30 | 0.82 | 0.12 | 0.35 | 3.5 | 2.0 | 5.2 | 20 | |
| HBC-5 | 0.06 | 0.33 | 3.4 | 0.09 | 0.41 | 4.1 | 0.86 | 10 | 53 | |
| MCF-7 | 0.04 | 0.24 | 17 | 0.03 | 0.31 | 3.1 | 0.87 | 7.9 | 44 | |
| MDA-MB-231 | 0.08 | 0.33 | 6.9 | 0.07 | 0.34 | 3.4 | 0.50 | 4.1 | 44 | |
| CNS | U251 | 0.04 | 0.47 | 17 | 0.05 | 0.55 | 7.9 | 0.7 | 13 | 58 |
| SF-268 | 0.05 | 0.59 | 26 | 0.06 | 0.75 | 4.7 | 0.94 | 14 | 76 | |
| SF-295 | 0.10 | 0.81 | 14 | 0.11 | 0.55 | 2.9 | 1.3 | 14 | 49 | |
| SF-539 | 0.05 | 0.20 | 0.60 | 0.07 | 0.23 | 0.63 | 0.7 | 3.9 | 22 | |
| SNB-75 | 0.13 | 0.34 | 0.91 | 0.15 | 0.38 | 0.93 | 3.3 | 15 | 42 | |
| SNB-78 | 0.12 | 0.78 | 25 | 0.20 | 0.98 | 5.2 | 2.9 | 20 | 62 | |
| Colon | HCC2998 | 0.05 | 0.22 | 0.74 | 0.07 | 0.23 | 0.69 | 1.0 | 2.9 | 8.0 |
| KM-12 | 0.06 | 0.64 | 23 | 0.09 | 0.59 | 4.6 | 0.72 | 7.0 | 64 | |
| HT-29 | 0.04 | 0.54 | 24 | 0.06 | 0.64 | 28 | 0.56 | 5.6 | 46 | |
| HCT-15 | 0.72 | 6.9 | 50 | 1.0 | 3.7 | 100 | 0.58 | 5.8 | 49 | |
| HCT-116 | 0.04 | 0.57 | 22 | 0.04 | 0.80 | 5.1 | 0.43 | 2.3 | 15 | |
| Lung | NCI-H23 | 0.06 | 0.28 | 1.8 | 0.07 | 0.36 | 2.5 | 1.2 | 4.8 | 98 |
| NCI-H226 | 0.08 | 0.49 | 25 | 0.09 | 0.63 | 6.2 | 0.74 | 4.8 | 44 | |
| NCI-H522 | 0.04 | 0.20 | 0.71 | 0.04 | 0.18 | 0.57 | 0.42 | 1.8 | 5.5 | |
| NCI-H460 | 0.05 | 0.27 | 3.4 | 0.05 | 0.23 | 1.7 | 0.45 | 2.6 | 20 | |
| A549 | 0.06 | 0.23 | 0.85 | 0.05 | 0.26 | 1.9 | 0.68 | 2.5 | 8.3 | |
| DMS273 | 0.04 | 0.18 | 0.70 | 0.05 | 0.18 | 0.61 | 0.42 | 1.6 | 4.7 | |
| DMS114 | 0.08 | 0.26 | 0.72 | 0.10 | 0.33 | 1.10 | 1.1 | 3.7 | 18 | |
| Melanoma | LOX-IMVI | 0.03 | 0.11 | 1.1 | 0.03 | 0.11 | 0.75 | 0.31 | 1.5 | 7.6 |
| Ovarian | OVCAR-3 | 0.06 | 0.28 | 8.0 | 0.05 | 0.30 | 42 | 0.64 | 4.6 | 82 |
| OVCAR-4 | 0.04 | 0.15 | 0.48 | 0.04 | 0.16 | 59 | 0.52 | 2.7 | 19 | |
| OVCAR-5 | 0.05 | 0.35 | 9.5 | 0.05 | 0.26 | 42 | 0.56 | 3.1 | 32 | |
| OVCAR-8 | 0.05 | 0.33 | 23 | 0.05 | 0.43 | 100 | 0.48 | 3.4 | 100 | |
| SK-OV-3 | 0.13 | 1.0 | 20 | 0.13 | 1.2 | 54 | 1.2 | 7.4 | 51 | |
| Renal | RXF-631L | 0.27 | 1.4 | 7.4 | 0.19 | 0.67 | 3.0 | 1.1 | 3.0 | 18 |
| ACHN | 0.27 | 2.1 | 20 | 0.25 | 2.2 | 10 | 0.7 | 7.1 | 51 | |
| Stomach | St-4 | 0.06 | 0.43 | 3.8 | 0.12 | 0.60 | 3.7 | 0.72 | 4.9 | 44 |
| MKN1 | 0.09 | 0.38 | 12 | 0.11 | 0.47 | 3.1 | 1.1 | 3.6 | 14 | |
| MKN7 | 0.04 | 0.34 | 23 | 0.06 | 0.48 | 36 | 0.9 | 8.0 | 69 | |
| MKN28 | 0.05 | 0.9 | 37 | 0.05 | 1.1 | 58 | 0.55 | 12 | 100 | |
| MKN45 | 0.05 | 1.0 | 28 | 0.05 | 1.1 | 8.0 | 0.44 | 6.2 | 100 | |
| MKN74 | 0.05 | 1.0 | 52 | 0.06 | 0.94 | 8.0 | 0.56 | 6.8 | 100 | |
| Prostate | DU145 | 0.1 | 0.34 | 17 | 0.09 | 0.43 | 3.3 | 1.2 | 4.0 | 22 |
| PC-3 | 0.16 | 1.3 | 70 | 0.16 | 1.3 | 22 | 2.0 | 12 | 56 | |
CNS: Central nervous system; GI50: 50% growth inhibition concentration (μM); TGI: Total Growth inhibition concentration (μM); LC50: Lethal concentration (μM).
Table 5 shows that all of the compounds are highly potent against the melanoma (LOX-IMVI) and lung (NCI-H522, A549, and DMS273) cancer cell lines in the JFCR39 panels. Compounds 7d and 7k are more potent than compound 7q towards the cancer cell lines in the JFCR39 panel. Compounds 7d and 7k show a similar growth inhibitory activity (GI50 > 0.01 µM) against the 31 and 28 cell lines, respectively, of the JFCR39 panel. Compound 7k shows a notable cytotoxicity against the breast (BSY-1, LC50 = 0.82 µM), CNS (SF-539, LC50 = 0.60 µM, SNB-75, LC50 = 0.91 µM), colon (HCC2998, LC50 = 0.74 µM), lung (NCI-H522, LC50 = 0.71 µM, A549, LC50 = 0.85, DMS273, LC50 = 0.70, DMS114, LC50 = 0.72) and ovarian (VCAR-4, LC50 = 0.48 µM) cancer cell lines. Compound 7q shows a mentionable cytotoxicity for the CNS (SF-539, LC50 = 0.63 µM, SNB-75, LC50 = 0.93 µM), colon (HCC2998, LC50 = 0.69 µM), lung (NCI-H522, IC50 = 0.57 µM, DMS273, LC50 = 0.61 µM), and melanoma (LOX-IMVI, LC50 = 0.75 µM) cancer cell lines. Compound 7q displayed a strong growth inhibitory activity (GI50 < 1 µM) for 28 panels in the JFCR39 panel of the various cell lines. DMS273 (lung cancer) and NCI-H522 (lung cancer) are the most sensitive cell lines of the 39 cell lines (GI50 = 0.42 µM for both and LC50 = 4.7 µM and 5.5 µM respectively). Compound 7q also showed a strong antitumor effect on LOX-IMVI (melanoma, GI50 = 0.3 µM, LC50 = 7.6 µM).
In our group, we achieved the synthesis of the artemisinin-indoloquinoline hybrids and studied their antiproliferative activity (Figure 4). The artemisinin-neocryptolepine hybrids 11a, 11b and the artemisinin-isocryptolepine hybrids 12 showed the potent antiproliferative activity, based on the data in Table 6.
Figure 4.

Structures of artemisinin-neocryptolepine hybrids.
Table 6.
Antiproliferative activity of the artemisinin-indoloquinoline hybrids against cell line of MV4-11, A549 and HCT116, and cytotoxicity against normal mice fibroblast BALB/3T3 [21].
| Compound | R1 | R2 | R3 | MV4-11 | A549 | HCT116 | BALB/3T3 |
|---|---|---|---|---|---|---|---|
| 11a | Cl | CH3 | CH3 | 0.072 ± 0.022 | 4.555 ± 2.086 | 0.893 ± 0.397 | 6.423 ± 0.996 |
| 11b | CO2Me | CH3 | CH3 | 0.075 ± 0.001 | 5.060 ± 0.911 | 2.206 ± 0.687 | 5.945 ± 1.163 |
| 12 | CO2Me | H | H | 0.086 ± 0.020 | 0.649 ± 0.080 | 0.130 ± 0.014 | 0.768 ± 0.155 |
In 2017, Li Wang synthesized the novel tacrine-neocryptolepine hybrids, and evaluated as inhibitors of human AChE (hAChE) and human BuChE (hBuChE) (Figure 5). Compound 13 was a highly potent hAChEI having IC50 = 0.95 ± 0.04 nM, and a highly potent hBuChE having IC50 = 2.29 ± 0.14 nM [22].
Figure 5.

Structure of tacrine-neocryptolepine hybrids.
2. Compare Study
The COMPARE analysis assesses the correlation coefficient between the fingerprints of the test compounds and those of the various reference compounds [23]. This system provides an information intensive approach to identify the molecular targets of new compounds. The JFCR39 COMPARE analysis-guided assay is a successful means to find new anticancer drug candidates. The COMPARE analysis is carried out by calculation of the Pearson correlation coefficient (r value) between the fingerprints of compounds X and Y. The r value is then used to determine the degree of similarity, that is, the higher the r value, the greater the similarity of X to Y. Generally, an r value of 0.5 < r < 0.75 between two agents suggests they might have a similar mechanism of action (Figure 6).
Figure 6.


Growth inhibitory- and cytotoxic activities of compounds (A) 7d, (B) 7k, and (C) 7q across a panel of the JFCR39 cell lines. The mean graph was produced by computer processing of the 50% growth inhibition (GI50) and the 50% lethal concentration (LC50) values. The logarithm of the GI50 and the LC50 values for each cell line is indicated. The X-axis shows the difference on a logarithmic scale between the mean of Log GI50/Log LC50 values for all 39-cell lines (MG-MID, expressed as 0 in the fingerprint) and the Log GI50/Log LC50 for each cell line in the JFCR39 panel. Columns to the right of 0 indicate the sensitivity of the cell lines to a given compound, and columns to the left indicate their resistance. The MG-MID mean of the Log GI50/Log LC50 values for all 39 cell lines; delta difference between the MG-MID and the Log GI50/Log LC50 value for the most sensitive cell line; range difference between the Log GI50/Log LC50 values for the most resistant cell line and the most sensitive cell line.
The COMPARE analysis revealed that compounds 7d and 7k have a very good match to actinomycin D (r = 0.7 for both). Similarly, compound 7k has a slight similarity to paclitaxel (r = 0.64). Compound 7q shows some resemblance to vindesine sulfate (r = 0.58) and aclarubicinHCl (r = 0.57).
3. Conclusions
Indoloquinoline alkaloids are important scaffolds for antitumoral drug development. This review discussed the SAR of neocryptolepine and isocryptolepine, and presents the useful methods for improving the antitumoral activity of neocryptolepine and isocryptolepine analogues. The amino substituent effect and methyl localization effect are now available strategies, but the substituent group effect at the benzene ring of the quinoline moiety has no effect on regular SAR. Thus, the antitumoral activity is highly related to the activity of interacting with DNA. The computer-assisted database analysis, COMPARE, suggested that 7d and 7k have a mode of action similar to actinomycin D. It also suggested that 7l has a mode of action similar to vindesine sulfate or aclarubicin HCl. However, the new compounds may have other unique modes of action since the correlation coefficients (r) were at relatively low levels, which present an interesting possibility to examine in further studies.
Funding
This study was partially supported by the Adaptable and Seamless Technology Transfer Program from Japan Science and Technology Agency (JST), No. AS242Z02199Q, and Menoufia University, No. Ib-C2013.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Oliver-Bever B. Medicinal Plants in Tropical West Africa. Cambridge University Press; Cambridge, UK: 1986. p. 41. [Google Scholar]
- 2.Cimanga K., Bruyne T., Pieters L., Claeys M., Vlietinck A. New alkaloids from Cryptolepis sanguinolenta. Tetrahedron Lett. 1996;37:1703–1706. doi: 10.1016/0040-4039(96)00112-8. [DOI] [Google Scholar]
- 3.Bailly C., Laine W., Baldeyrou B., De Pauw-Gillet M.C., Colson P., Houssier C., Cimanga K., Van Miert S., Vlietinck A.J., Pieters L. DNA intercalation topoisomerase II inhibition and cytotoxic activity of the plant alkaloid neocryptolepine. Anti-Cancer Drug Des. 2000;15:191–201. [PubMed] [Google Scholar]
- 4.Hurley L. DNA and its associated processes as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:188–200. doi: 10.1038/nrc749. [DOI] [PubMed] [Google Scholar]
- 5.Yamato M., Hashigaki K., Takeuchi Y., Ikeda Y. Synthesis and antitumor activity of fused tetracyclic quinoline derivatives. 1. J. Med. Chem. 1989;32:1295–1300. doi: 10.1021/jm00126a025. [DOI] [PubMed] [Google Scholar]
- 6.Deady L.W., Kaye A.J., Finlay G.J., Baguley B.C., Denny W.A. Synthesis and antitumor properties of N-[2-(Dimethylamino)ethyl] carboxamide derivatives of fused tetracyclic quinolines and quinoxalines: A new class of putative Topoisomerase inhibitors. J. Med. Chem. 1997;40:2040–2046. doi: 10.1021/jm970044r. [DOI] [PubMed] [Google Scholar]
- 7.Bonjean K., De Pauw-Gillet M.C., Defresne M.P., Colson P., Houssier C., Dassonneville L., Bailly C., Greimers R., Wright C., Quetin-Leclercq J., et al. The DNA intercalating alkaloid cryptolepine interferes with Topoisomerase II and inhibits primarily DNA synthesis in B 16 melanoma cells. Biochemistry. 1998;37:5136–5146. doi: 10.1021/bi972927q. [DOI] [PubMed] [Google Scholar]
- 8.Lisgartent J.N., Coll M., Portugal J., Wright C.W., Aymami J. The antimalarial and cytotoxic drug cryptolepine intercalates into DNA at cytosine-cytosine sites. Nat. Struct. Biol. 2002;9:57–60. doi: 10.1038/nsb729. [DOI] [PubMed] [Google Scholar]
- 9.Boddupally P.V.L., Hahn S., Beman C., De B., Brooks T.A., Gokhale V., Hurley L.H. Anticancer Activity and Cellular Repression of c-MYC by the GQuadruplex-Stabilizing 11-Piperazinylquindoline is Not Dependent on Direct Targeting of the G-Quadruplex in the c-MYC Promoter. J. Med. Chem. 2012;55:6076–6086. doi: 10.1021/jm300282c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou J.L., Lu Y.J., Ou T.-M., Zhou J.-M., Huang Z.-S., Zhu X.-F., Du C.-J., Bu X.-Z., Ma L., Gu L.-Q., et al. Synthesis and evaluation of quindoline derivatives as G-quadruplex inducing and stabilizing ligands and potential inhibitors of telomerase. J. Med. Chem. 2005;48:7315–7321. doi: 10.1021/jm050041b. [DOI] [PubMed] [Google Scholar]
- 11.Lu Y.J., Ou T.M., Tan J.H., Hou J.Q., Shao W.Y., Peng D., Sun N., Wang X.D., Wu W.B., Bu X.Z., et al. 5-N-methylated quindoline derivatives as telomeric G-quadruplex stabilizing ligands: Effects of 5-N positive charge on quadruplex binding affinity and cell proliferation. J. Med. Chem. 2008;51:6381–6392. doi: 10.1021/jm800497p. [DOI] [PubMed] [Google Scholar]
- 12.Guittat L., Alberti P., Rosu F., Van Miert S., Thetiot E., Pieters L., Gabelica V., De Pauw E., Ottaviani A., Riou J.-F., et al. Interaction of cryptolepine and neocryptolepine with unusual DNA structures. Biochimie. 2003;85:535–547. doi: 10.1016/S0300-9084(03)00035-X. [DOI] [PubMed] [Google Scholar]
- 13.Wang L., Switalska M., Mei Z.-W., Lu W.-J., Takahara Y., Feng X.-W., El-Sayed I.E.-T., Wietrzyk J., Inokuchi T. Synthesis and in vitro antiproliferative activity of new 11-aminoalkylamino-substituted 5H- and 6H-indolo[2,3-b]quinolines; structure–activity relationships of neocryptolepines and 6-methyl congeners. Bioorg. Med. Chem. 2012;20:4820–4829. doi: 10.1016/j.bmc.2012.05.054. [DOI] [PubMed] [Google Scholar]
- 14.Lu W., Switalska M., Wang L., Yonezawa M., El-Sayed I.E.-T., Wietrzyk J., Inokuchi T. In vitro antiproliferative activity of 11-aminoalkylaminosubstituted 5H-indolo[2,3-b]quinolines; improving activity of neocryptolepines by installation of ester substituent. Med. Chem. Res. 2013;22:4492–4504. doi: 10.1007/s00044-012-0443-x. [DOI] [Google Scholar]
- 15.Shaban E., Switalska M., Wang L., Wang N., Xiu F., Hayashi I., Ngoc T.A., Nagae S., El-Ghlban S., Shimoda S., et al. Synthesis and In Vitro Antiproliferative Activity of 11-Substituted Neocryptolepines with a Branched ω-Aminoalkylamino Chain. Molecules. 2017;22:1954. doi: 10.3390/molecules22111954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Peng W., Świtalska M., Wang L., Mei Z.-W., Edazawa Y., Pang C.-Q., El-Sayed I.E.-T., Wietrzyk J., Inokuchi T. Synthesis and in vitro antiproliferative activity of new 11-aminoalkylaminosubstituted chromeno[2,3-b]indoles. Eur. J. Med. Chem. 2012;58:441–451. doi: 10.1016/j.ejmech.2012.10.023. [DOI] [PubMed] [Google Scholar]
- 17.Emam S., El Sayed I., Ayad M., Hathout H. Synthesis, characterization and anticancer activity of new Schiff bases bearing neocryptolepine. J. Mol. Struct. 2017;1146:600–619. doi: 10.1016/j.molstruc.2017.06.006. [DOI] [Google Scholar]
- 18.Emam S., El Sayed I., Nassar N. Transition metal complexes of neocryptolepine analogues Part I: Synthesis, spectroscopic characterization, and in vitro anticancer activity of Copper(II) Complexes, Spectrochim. Mol. Biomol. Spectrosc. 2015;138:942–953. doi: 10.1016/j.saa.2014.03.114. [DOI] [PubMed] [Google Scholar]
- 19.Wang N., Świtalska M., Wu M.-Y., Imai K., Ngoc T.A., Pang C.-Q., Wang L., Wietrzyk J., Inokuchi T. Synthesis and in vitro cytotoxic effect of 6-amino-substituted 11H- and 11Me-indolo[3,2-c]quinolines. Eur. J. Med. Chem. 2014;78:314–323. doi: 10.1016/j.ejmech.2014.03.038. [DOI] [PubMed] [Google Scholar]
- 20.Okada M., Mie Z.-W., Hossain M.I., Wang L., Tominaga T., Takebayashi T., Murakami M., Yasuda M., Shigehiro T., Kasai T., et al. Synthesis and in vitro cancer cell growth inhibition evaluation of 11-amino-modified 5-Me-indolo[2,3-b]quinolines and their COMPARE analyses. Med. Chem. Res. 2016;25:879–892. doi: 10.1007/s00044-016-1508-z. [DOI] [Google Scholar]
- 21.Wang L., Świtalska M., Wang N., Du Z.-J., Fukumoto Y., Diep N.K., Kiguchi R., Nokami J., Wietrzyk J., Inokuchi T., et al. Design, Synthesis, and Biological Evaluation of Artemisinin-Indoloquinoline Hybrids as Potent Antiproliferative Agents. Molecules. 2014;19:19021–19033. doi: 10.3390/molecules191119021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L., Moraleda I., Iriepa I., Romero A., López-Muñoz F., Chioua M., Inokuchi T., Bartolini M., Marco-Contelles J. 5-Methyl-N-(8-(5,6,7,8-tetrahydroacridin-9-ylamino)octyl)-5H-indoloij2,3-b]quinolin-11-amine: A highly potent human cholinesterase inhibitor. Med. Chem. Commun. 2017;8:1307–1317. doi: 10.1039/C7MD00143F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong D., Yamori T. JFCR39, a panel of 39 human cancer cell lines, and its application in the discovery and development of anticancer drugs. Bioorg. Med. Chem. 2012;20:1947–1951. doi: 10.1016/j.bmc.2012.01.017. [DOI] [PubMed] [Google Scholar]