

Abstract
Microbial pathogens can be detected by inflammasomes that induce inflammation and programmed cell death. Inflammasomes are sensors that survey cells for signs of compromise. One of these sensors, NLRP1, detects anthrax lethal toxin; however, the mechanism of NLRP1 activation has remained unknown. Here, Xu et al discover NLRP1 cleavage by lethal toxin induces the N‐end rule, which targets NLRP1 for degradation. Surprisingly, the active inflammasome fragment escapes the proteasome and becomes an activate inflammasome itself.
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction
Host immune responses attempt to counteract pathogen strategies by recognizing bacterial virulence factors, triggering an immune response that eliminates the pathogen. As bacteria evolve new virulence factors to evade the immune system, the host must also evolve in response. Inflammasomes, which often belong to the NLR family, are a class of innate immune sensor that can identify signs of bacterial infection. Upon activation, inflammasomes oligomerize in order to cluster their signaling domains (either CARD or PYD). Inflammasomes that contain a CARD can directly activate caspase‐1 via CARD‐CARD interactions, but are also amplified by the ASC adaptor (which is composed of a PYD and a CARD). PYD‐containing inflammasomes, on the other hand, rely on the ASC adaptor in order to activate caspase‐1. Recruitment of ASC triggers ASC polymerization, resulting in the formation of an ASC speck, which intensifies caspase‐1 activation. Caspase‐1 then triggers maturation of the pro‐inflammatory cytokines IL‐1β and IL‐18, and cleaves gasdermin D to cause pyroptosis. Although the general activation of inflammasomes has been extensively investigated, the mechanism by which the NLRP1 inflammasome detects pathogens had remained a mystery until recent work by Xu et al in this issue of EMBO (Xu et al, 2019) and two similar papers recently published in Science (Chui et al, 2019; Sandstrom et al, 2019).
NLRP1 has a unique structure (Fig 1; Yu et al, 2018). Human NLRP1 contains a PYD on its N‐terminus followed by a nucleotide‐binding oligomerization domain (NOD; also called NACHT) and an LRR domain. Following these lies a function to find domain (FIIND) which undergoes auto‐proteolysis while remaining non‐covalently associated with NLRP1. This FIIND auto‐proteolysis is required for activation of the NLRP1 inflammasome. Subsequent to FIIND, a CARD is located on the C‐terminus, which is opposite to other NLRs where signaling domains are N‐terminal. Another way NLRP1 is different from all other NLR inflammasomes is that the ATPase function of the NOD domain is not required for NLRP1 signaling. Contrary to human NLRP1, mouse NLRP1b does not contain a PYD yet the inflammasome is still functional (Yu et al, 2018). Thus, it is perplexing that the N‐terminal signaling domain is dispensable for NLRP1 activation in mice.
Figure 1. The NLRP1 inflammasome contains decoy domains to detect virulence factor attack.
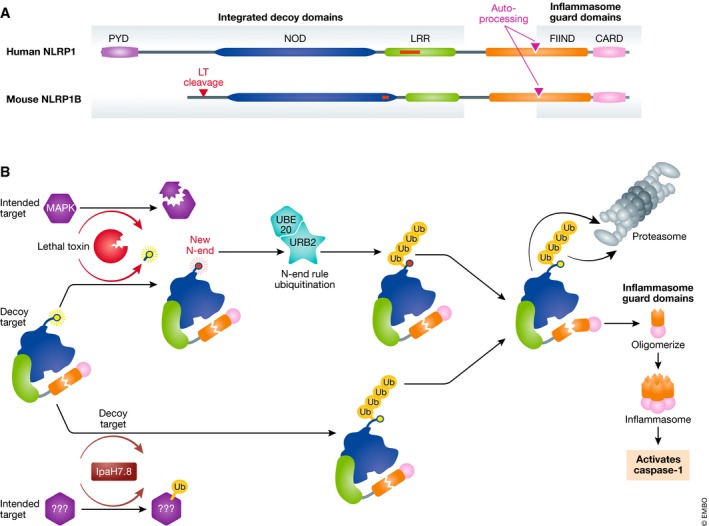
(A) Domain structure of human NLRP1 and mouse NLRP1b. Red boxes are present in humans and absent in mice, or vice versa. Lethal toxin cleavage site (red triangle) and FIIND auto‐processing site (purple triangle) are indicated. (B) Murine NLRP1b activation pathways. Virulence factors degrade proteins that are useful to the host immune response. However, they may also be lured into attacking the NLRP1b decoy domains. Lethal toxin‐cleaving NLRP1b triggers UBR2/UBE2O‐mediated N‐end rule degradation, whereas IpaH7.8 ubiquitinates NLRP1b directly. Both target NLRP1b to the proteasome, but as the proteasome approaches the FIIND‐CARD auto‐processing site, the FIIND‐CARD fragment escapes and forms the inflammasome.
In some mice, the NLRP1b inflammasome has long been known to activate caspase‐1 in response to anthrax lethal toxin (LT) (Boyden & Dietrich, 2006). LT directly cleaves NLRP1 between amino acids 44 and 45. Perplexingly, the proteasome is also required for activation of NLRP1. This created an enigma in that the proteasome seemed to be required for NLRP1, but not other inflammasome activation.
Xu et al in this issue of EMBO (Xu et al, 2019) as well as Chui et al (2019), Sandstrom et al (2019) solve these mysteries. Xu et al began by generating a RAW264.7 macrophage cell line that expresses RFP‐ASC and EGFP (RAWRA). Thus, when LT activates the NLRP1 inflammasome, RFP‐ASC polymerizes and ASC specks can be visualized; simultaneously, EGFP is released from the cell upon pyroptotic membrane rupture (Xu et al, 2019). Agreeing with other investigations (Chui et al, 2019; Sandstrom et al, 2019), the proteasome was required for speck formation and pyroptosis in LT‐treated cells (Xu et al, 2019).
Previous studies revealed that a process known as the N‐end rule is involved in LT‐mediated killing of host cells (Wickliffe et al, 2008), but the mechanism by which the N‐end rule intersects with NLRP1 remained nebulous. The N‐end rule detects when the N‐terminus is an amino acid which is not the normal N‐terminus of the protein. This new N‐terminus, referred to as a degron, is a marker that the protein primary amino acid sequence is disrupted. Degrons are detected by E3 ubiquitin ligases, which form a polyubiquitin chain on the protein in order to target it to the proteasome for degradation (Lucas & Ciulli, 2017). RAWRA cells whose N‐end rule pathway was functionally inhibited resisted LT‐induced pyroptosis, a phenotype which was mimicked in cells treated with a proteasome inhibitor (Xu et al, 2019).
To identify which N‐end rule E3 ubiquitin ligase was involved in activating NLRP1, the authors performed genomewide screens using both siRNA‐ and CRISPR‐Cas9‐mediated targeting. Both identified the E3 ubiquitin ligase, UBR2, as a high‐confidence candidate. Ultimately, the authors generated Ubr2 −/− immortalized bone marrow‐derived macrophages using CRISPR‐Cas9‐mediated targeting. These Ubr2 −/− macrophages were entirely resistant to LT‐induced caspase‐1 activation and pyroptosis. The authors further elucidated that the E2 ligase, UBE2O, interacted with and was required upstream of UBR2, but exactly how UBR2 was important for the NLRP1 pathway remained unknown at this point (Xu et al, 2019).
The authors then made the observation that LT treatment, which cleaves off only 44 amino acids, caused loss of the entire large molecular weight NLRP1b band (amino acid 1–983, which contained the NOD and LRR). In contrast, no change was observed in the C‐terminal FIIND‐CARD fragment (amino acids 984–1,233). The loss of the large NLRP1 fragment could be recapitulated using cells expressing only the 1–983 fragment of NLRP1, indicating the FIIND‐CARD fragment did not participate in degradation of the rest of the protein. The author's key insight was connecting NLRP1 degradation to the N‐end rule. They hypothesized that the N‐end rule drove degradation of the large LT‐cleaved 45–983 fragment. Indeed, addition of a proteasome inhibitor or an N‐end rule inhibitor prevented degradation of the 45–983 NLRP1 band. Finally, a pulldown of NLRP1 demonstrated it is robustly ubiquitinated upon LT stimulation (Xu et al, 2019). Collectively, these data demonstrate NLRP1 activation is initiated by LT cleavage which triggers N‐end rule ubiquitination and proteasome degradation. Once the proteasome reaches the FIIND auto‐processing site, the FIIND‐CARD fragment dissociates and escapes the proteasome. The FIIND‐CARD fragment then oligomerizes to form the active inflammasome.
LT also cleaves MAP kinase kinases in order to dampen the host immune response (Yu et al, 2018). Thus, it is widely believed that anthrax LT cleavage of NLRP1 is an unintended target. The host may have evolved NLRP1 to act as a decoy in order to bait LT into activating it, allowing some mice to be resistant to anthrax infection (Yu et al, 2018). Sandstrom et al (2019) also discovered that the Shigella IpaH7.8 virulence factor, which includes a bacterial novel E3 ubiquitin ligase domain, directly ubiquitinates NLRP1, targeting it for degradation by the host proteasome, thereby activating it. IpaH7.8 may intend to ubiquitinate and degrade other innate immune proteins, perhaps other NLR inflammasomes, but instead accidentally activates the NLRP1 decoy.
Hosts have evolved strategies to combat virulence factors; these host anti‐microbial mechanisms sometimes directly recognize bacterial proteins. In other cases, innate immune sensors, called guards, can monitor a critical cellular protein, ensuring that it is functioning normally. When the guarded protein is degraded or otherwise compromised, the guard can recognize the perturbation and induce an innate immune response, often programmed cell death. The host can also deploy decoys that mimic the protein that is monitored. Decoy domains can be integrated within the innate immune sensor itself. We propose that this is the case with NLRP1, where integrated decoy domains, such as the LT cleavage site, are guarded by the FIIND‐CARD inflammasome. Decoys and integrated decoys have been well established in the plant field as a way to combat infection (Jones et al, 2016); however, NLRP1 is the first integrated decoy to be described in mammalian systems.
The integrated decoy and guard model explain why NLRP1 has an LT cleavage site as well as FIIND and CARD domains but does not explain the presence of the PYD, NOD, and LRR, as these domains are dispensable for inflammasome activity. It is interesting to speculate that these domains evolved to entice pathogens into degrading them with protease or ubiquitinase virulence factors. NOD and LRR domains are also encoded in many innate immune sensors, including NOD1, NOD2, NLRC4, and NLRP3; thus, bacterial virulence factors may intend to degrade one of these NLRs but additionally attack the NLRP1 decoy. NLRP1 genes are polymorphic both between species and within a species (Yu et al, 2018). This versatility most likely evolved due to different pathogenic threats and could reflect the addition of new or modified integrated decoy domains within NLRP1. Other mammalian proteins, including CARD8 and PIDD, also contain a FIIND. This suggests that this functional degradation may be a conserved mechanism for activating other innate immune responses.
The EMBO Journal (2019) 38: e102494
See also: H Xu et al (July 2019)
References
- Boyden ED, Dietrich WF (2006) Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 38: 240–244 [DOI] [PubMed] [Google Scholar]
- Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, Ball DP, Taabazuing CY, Orth EL, Vittimberga BA et al (2019) N‐terminal degradation activates the NLRP1B inflammasome. Science 364: 82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JDG, Vance RE, Dangl JL (2016) Intracellular innate immune surveillance devices in plants and animals. Science 354: aaf6395 [DOI] [PubMed] [Google Scholar]
- Lucas X, Ciulli A (2017) Recognition of substrate degrons by E3 ubiquitin ligases and modulation by small‐molecule mimicry strategies. Curr Opin Struct Biol 44: 101–110. [DOI] [PubMed] [Google Scholar]
- Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE (2019) Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 364: eaau1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickliffe KE, Leppla SH, Moayeri MJ (2008) Killing of macrophages by anthrax lethal toxin: involvement of the N‐end rule pathway. Cell Microbiol 10: 1352–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Jianjin S, Hang G, Ying L, Zhenxiao Y, Feng S, Na D (2019) The N‐end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. EMBO J 38: e101996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C‐H, Moecking J, Geyer M, Masters SL (2018) Mechanisms of NLRP1‐mediated autoinflammatory disease in humans and mice. J Mol Biol 430: 142–152 [DOI] [PubMed] [Google Scholar]