

Abstract
Hepatocellular carcinoma (HCC) is the second leading cause of cancer‐related deaths worldwide. Treatment options for patients with advanced‐stage disease are limited. A major obstacle in drug development is the lack of an in vivo model that accurately reflects the broad spectrum of human HCC. Patient‐derived xenograft (PDX) tumor mouse models could overcome the limitations of cancer cell lines. PDX tumors maintain the genetic and histologic heterogeneity of the originating tumors and are used for preclinical drug development in various cancers. Controversy exists about their genetic and molecular stability through serial passaging in mice. We aimed to establish PDX models from human HCC biopsies and to characterize their histologic and molecular stability during serial passaging. A total of 54 human HCC needle biopsies that were derived from patients with various underlying liver diseases and tumor stages were transplanted subcutaneously into immunodeficient, nonobese, diabetic/severe combined immunodeficiency gamma‐c mice; 11 successfully engrafted. All successfully transplanted HCCs were Edmondson grade III or IV. HCC PDX tumors retained the histopathologic, transcriptomic, and genomic characteristics of the original HCC biopsies over 6 generations of retransplantation. These characteristics included Edmondson grade, expression of tumor markers, tumor gene signature, tumor‐associated mutations, and copy number alterations. Conclusion: PDX mouse models can be established from undifferentiated HCCs, with an overall success rate of approximately 20%. The transplanted tumors represent the entire spectrum of the molecular landscape of HCCs and preserve the characteristics of the originating tumors through serial passaging. HCC PDX models are a promising tool for preclinical personalized drug development.
Abbreviations
- AFP
alpha‐fetoprotein
- BCLC
Barcelona Clinic Liver Cancer
- CD
clusters of differentiation
- CNA
copy number alternation
- EBV
Epstein‐Barr virus
- H&E
hematoxylin and eosin
- HCC
hepatocellular carcinoma
- ID
identification number
- PDX
patient‐derived xenograft
- PDX‐1/6
patient‐derived xenograft first/sixth generation
- TCGA
The Cancer Genome Atlas
- WES
whole‐exome sequencing
Hepatocellular carcinoma (HCC) is the most common primary liver cancer and the second most frequent cause of cancer‐related mortality worldwide.1 Current treatment options are limited, with less than 30% of patients with HCC in Europe qualifying for curative treatments.2 Sorafenib and lenvatinib are the only first‐line treatments for advanced HCC.3, 4 These drugs moderately improve survival of patients with advanced HCC but have adverse effects that preclude their use in patients with advanced cirrhosis.3, 4, 5 Novel therapies are urgently needed to improve treatment options for patients with advanced HCCs.
Current in vitro cell culture models of HCC are based on conventional hepatoma cell lines that fail to recapitulate key features of tumor tissues, such as three‐dimensional tumor architecture, cellular heterogeneity, and cell–cell interactions. Chemically induced HCC mouse models or genetically engineered mice better reflect in vivo tumor biology6; however, they do not recapitulate the different genetic alterations present in HCCs of different patients. Patient‐derived xenograft (PDX) models have the potential to overcome these limitations and are considered valuable tools in the field of anticancer drug development and prediction of response to cancer therapy.7, 8 However, a recent publication described widespread mouse‐specific tumor evolution and seriously challenged the usefulness of serially passaged PDXs.9 The authors analyzed more than 1,000 PDX samples across 24 cancer types and observed rapid accumulation of copy number alterations (CNAs) during PDX passaging.9 However, the available data on liver cancers were very limited.9 Another limitation of the published HCC PDX models is that all of these have been generated from surgically resected HCCs.10, 11, 12, 13, 14, 15 Because surgical resections are predominantly performed in patients with early stage tumors (typically Barcelona Clinic Liver Cancer [BCLC] stages 0/A), PDXs derived from resected tumors are heavily biased against advanced‐stage HCCs. Furthermore, from the published HCC PDX reports, it is not clear how much of the original tumor characteristics are preserved in the transplanted tumors. It is also not clear if successful engraftment is restricted to a subgroup of HCCs with specific molecular characteristics. These questions need to be resolved before further developing PDX models for preclinical drug development.
In the present study, we report the results of a comprehensive comparative analysis at the histopathologic, transcriptomic, and genomic levels of 11 newly generated PDX models serially passaged over 6 generations and the originating human HCCs.
Materials and Methods
Human HCC Tissues and Biopsy Procedure
A total of 54 human HCC needle biopsies were obtained at the University Hospital Basel, Basel, Switzerland, from 44 patients who were treatment naive and undergoing diagnostic liver biopsy (Supporting Table S1; Supporting Methods). Written informed consent was obtained from all patients. The study was approved by the ethics committee of the northwestern part of Switzerland (protocol #EKNZ 2014‐099). One biopsy cylinder was formalin fixed and paraffin embedded for diagnosis and staging. Additional cylinders were immediately snap frozen in liquid nitrogen for later use in DNA/RNA extraction or embedded in O.C.T. compound (Tissue‐Tek) and frozen using standard procedures. For PDX model generation, biopsy pieces were placed in phosphate‐buffered saline. An additional biopsy from nontumor liver parenchyma was also obtained from all patients.
Mice and Xenotransplantation
HCC needle biopsies were transplanted subcutaneously into 6‐ to 10‐week‐old, nonobese, diabetic/severe combined immunodeficiency gamma‐c mice (Supporting Methods). Experiments were conducted with the approval of the ethics committee of the northwestern part of Switzerland (protocol #EKNZ 2014‐099) and the animal care committee of Canton Basel‐Stadt, Switzerland.
Histology and Immunohistochemistry
Paraffin‐embedded tumor biopsies and PDX tumors were processed for hematoxylin and eosin (H&E) or immunohistochemical staining using standard procedures (Supporting Methods). Histopathology evaluation was carried out blindly by two board‐certified hepatopathologists (Dr. Luigi Tornillo and Dr. Matthias S. Matter) at the Institute of Pathology of the University Hospital Basel. Disease stage and histopathologic grading were classified according to the BCLC staging system16 and the Edmondson grading system,17 respectively.
RNA Sequencing and Analysis
Total RNA was extracted from 45 HCC biopsies derived from 37 patients (Supporting Table S2), from 16 first‐, 8 fourth‐ and 20 sixth‐generation HCC PDX tumors and 3 first‐generation lymphoma PDX tumors. All RNA samples were sequenced on an Illumina HiSeq 2500 system (Supporting Methods).
Sequence reads were aligned by STAR (version 2.5.2a)18 (Supporting Methods). Transcript quantification for human genes was performed using RSEM (version 1.2.31),19 differential expression analysis using the edgeR R package (version 3.18.1),20 pathway enrichment analyses using the hypergeometric test, and single‐sample gene set enrichment analysis21 using the GSVA package (version 1.24.2)22 for the hallmark gene sets in the Molecular Signatures Database.23 Cluster analysis was performed using hierarchical clustering. Batch effects associated with the xenografting process were removed using a linear model20 prior to clustering. The expression of somatic mutations in the RNA sequencing data was determined by GATK Unified Genotyper,24 given the somatic mutations identified in HCC biopsies by whole‐exome sequencing (WES). For statistical comparisons, only missense and synonymous mutations were considered because truncating, insertions, and deletions and splice site mutations are likely to affect transcript stability. CNA profiles were derived with the CNVkit (version 0.9.5)25 using 15 unrelated normal livers as control. Correlation between samples was calculated as the Spearman’s rho of the gene‐level segmented log2 ratio. Transcriptomic classification was performed according to Hoshida et al.26 (see Supporting Methods for further details).
WES and Analysis
Paired‐end 101‐base pair sequencing was performed for the DNA extracted from 14 engrafted HCC biopsies (derived from 12 patients) and the 12 matched nontumoral biopsies (Supporting Table S2) using the SureSelectXT Clinical Research Exome (Agilent) platform and the Illumina HiSeq 2500 according to the manufacturers’ guidelines. WES data were analyzed essentially as described27 (Supporting Methods).
The Cancer Genome Atlas Data Analysis
The Cancer Genome Atlas (TCGA) HCC RNA sequencing data retrieved from the Genomics Data Commons28 were merged with the RNA sequencing data of our cohort for clustering (Supporting Methods). Histologic grading was performed according to the Edmondson grading system17 as described29 (Supporting Methods).
Data Availability
Sequencing data are available at the European Genome‐phenome Archive under accession EGAS00001003396.
Results
Fresh Human HCC Needle Biopsies Generate Xenograft Tumors in Immunocompromised Mice
PDX models were generated from human HCC needle biopsies as described in Materials and Methods. A total of 54 needle biopsies from 44 patients with HCC were included in this study (Supporting Tables S1 and S2). The patient cohort covered all major HCC risk factors and the entire spectrum of Edmondson grades (I‐IV)17 (Supporting Table S1). Additionally, in contrast to the use of resected HCC tissue, which is limited to early stage disease,11, 14, 15 our cohort comprised all four classes of the BCLC staging system.16 Although we typically obtained a single biopsy sample per patient, there were 9 patients that were simultaneously biopsied at two locations because of the presence of multiple lesions in the liver (Supporting Table S1). One patient was biopsied at two different locations within the same large tumor nodule (Supporting Table S1). In total, we successfully established 11 HCC‐derived PDX models from 10 patients (Supporting Table S1).
The time from subcutaneous introduction of the HCC biopsy material to onset of tumor growth ranged from 4 to 28 weeks (Fig. 1A,B). Time to onset of PDX tumor growth of Edmondson grade IV‐derived biopsies appeared to be shorter than that of grade III biopsies, but the difference did not reach statistical significance (P = 0.072; Mann‐Whitney U test) (Fig. 1). Interestingly, PDX C078b derived from subcutaneous injection of a biopsy cell suspension showed the latest tumor growth onset (Fig. 1A,B), suggesting that the presence of an intact tumor architecture may accelerate tumor engraftment and growth onset. Finally, the onset of tumor growth varied between mice that received a transplant with tumor tissue derived from the same biopsy cylinder (Supporting Fig. S1A), suggesting that, among other factors, the tumor cell content may also influence the lag time to tumor growth. Indeed, the lag time to tumor growth was similar when a homogeneous cell suspension generated from one biopsy cylinder was subcutaneously injected into two mice (Supporting Fig. S1B).
Figure 1.
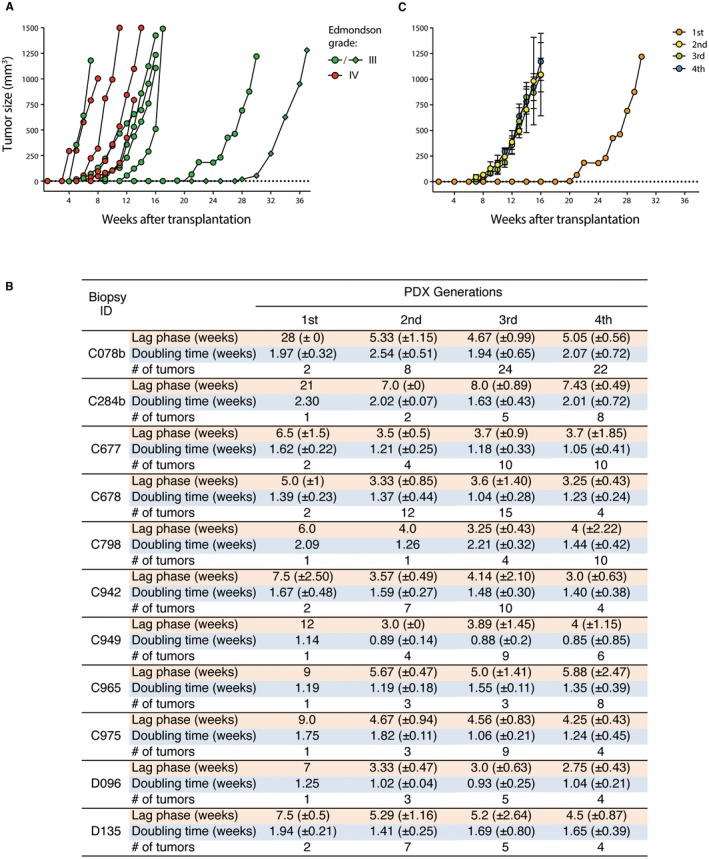
Xenograft tumor growth patterns. (A) Lag phase and tumor growth rate of first‐generation xenografts. Edmondson grades III and IV are shown in green and red, respectively. Circles and diamonds represent transplanted solid pieces and cell suspensions, respectively. (B) Growth characteristics of PDX models. Lag phase until detectable tumor formation, doubling time of tumors from first‐, second‐, third‐, and fourth‐generation PDX tumors and number of transplanted tumors are indicated. Results are shown as mean ± SD. (C) Tumor growth rate over several generations (first to fourth) in one representative PDX model (C284b). Transplantation generations are color coded.
Taken together, these results demonstrate that human HCC tissue derived from fresh needle biopsies can generate xenografts following subcutaneous transplantation and injection.
Growth Kinetics of HCC Xenograft Tumors Remain Stable Over Subsequent Passages
Having successfully established 11 PDX models, we next investigated the retransplantation capacity of the initial xenografts over at least 6 generations. Retransplanted tumors showed a shortened lag phase until onset of tumor growth compared to the xenograft tumor derived from the biopsy tissue and remained stable in subsequent PDX generations (Fig. 1B,C). The doubling time of tumor growth differed between PDX models. There were three models (C677, C798, D135) with a >20% shorter and one model (C078b) with a >20% longer doubling time in one or more of the subsequent generations compared with the first generation. In the other seven models, growth rates remained stable among generations (Fig. 1B,C). Furthermore, Ki‐67 staining of first‐ and fourth‐generation PDX tumors showed similar proliferation rates (Supporting Fig. S2). Taken together, these data indicate that, once established, PDX tumors can be expanded for several generations with reasonable stable growth kinetics over time.
PDX Tumors Recapitulate the Histopathologic Features of the Original Human HCC Tumors
We next investigated whether the individual PDX tumors retained the histologic characteristics of the original human tumors. HCC PDX tumors maintained the differentiation grade,17 growth pattern, and cytologic subtype30, 31 of the originating human HCC biopsies over at least 4 generations (Table 1; Fig. 2; Supporting Fig. S3A‐J). As an example, the PDX tumors originating from tumor biopsy C942 displayed a solid‐trabecular growth pattern with an Edmondson grade III, as observed in the original tumor (Fig. 2; Table 1). Of note, only in one case (PDX model C284b) did we observe the loss of the trabecular aspect of the growth pattern that was present in the original biopsy; however, the solid growth pattern and differentiation grade were maintained over subsequent passages from mouse to mouse (Table 1; Supporting Fig. S3B).
Table 1.
Edmondson Grade, Growth Pattern, and Cytologic Subtype of Biopsies and Corresponding PDX Tumors
| Biopsy ID | Edmondson Grade | Growth Pattern | Cytologic Subtype | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Biopsy | PDX‐1 | PDX‐4 | Biopsy | PDX‐1 | PDX‐4 | Biopsy | PDX‐1 | PDX‐4 | |
| C078b | III | III | III | Solid | Solid | Solid | ‐ | ‐ | ‐ |
| C284b | III | III | III | Solid‐trabecular | Solid | Solid | ‐ | ‐ | ‐ |
| C677 | IV | IV | IV | Solid | Solid | Solid | Giant cells | Giant cells | Giant cells |
| C678 | IV | IV | IV | Solid | Solid | Solid | Giant cells | Giant cells | Giant cells |
| C798 | III | III | III | Solid‐trabecular | Solid‐trabecular | Solid‐trabecular | ‐ | ‐ | ‐ |
| C942 | III | III | III | Solid‐trabecular | Solid‐trabecular | Solid‐trabecular | ‐ | ‐ | ‐ |
| C949 | III | III | III | Solid‐trabecular | Solid‐trabecular | Solid‐trabecular | ‐ | ‐ | ‐ |
| C965 | III | III | III | Solid | Solid | Solid | ‐ | ‐ | ‐ |
| C975 | III | III | III | Solid | Solid | Solid | ‐ | ‐ | ‐ |
| D096 | IV | IV | IV | Solid | Solid | Solid | ‐ | ‐ | ‐ |
| D135 | IV | IV | IV | Solid‐trabecular | Solid‐trabecular | Solid‐trabecular | Clear cell, giant cells | Clear cell, giant cells | Clear cell, giant cells |
Abbreviation: PDX‐4, fourth‐generation tumor.
Figure 2.
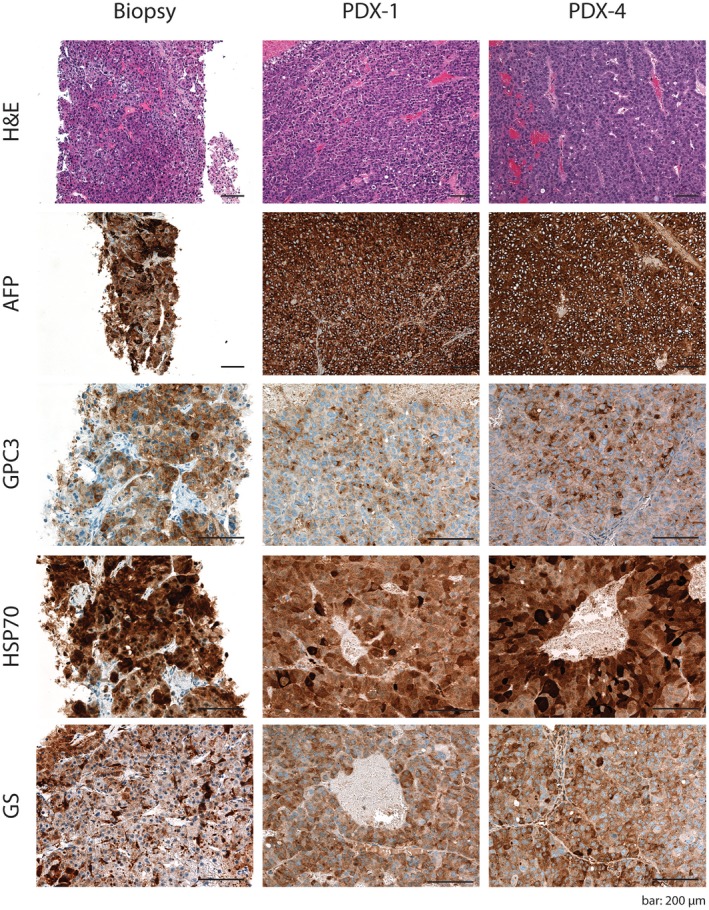
HCC PDX mice recapitulate the histologic characteristics as well as the expression patterns of HCC markers of the original tumor. Histologic sections of the original HCC biopsies and their derivative PDX tumors (PDX‐1 and PDX‐4) stained with H&E for AFP expression as well as GPC3, HSP70, and GS detected by immunohistochemistry. Scale bar, 200 μm. Representative stainings are shown for PDX model C942. Abbreviations: GPC3, glypican 3; GS, glutamine synthetase; HSP70, heat shock protein 70; PDX‐1, first‐generation PDX; PDX‐4, fourth‐generation PDX.
Immunohistochemical analysis demonstrated that the expression and distribution of the tumoral marker alpha‐fetoprotein (AFP)32 were maintained from the original tumor biopsies to the HCC xenografts over several generations (Fig. 2; Supporting Fig. S3A‐J). Likewise, the expression pattern of the three most widely used markers for HCC diagnosis,33, 34 glypican 3, heat shock protein 70, and glutamine‐synthetase, confirmed the histologic stability of the xenograft tumors compared to the original HCC biopsy (Fig. 2; Supporting Fig. S3A‐J). Taken together, these results show that the HCC PDX tumors retain the histologic features of the original human tumors through serial passaging.
PDX Tumors Recapitulate the Transcriptomic Profile of the Original Human HCC Tumors
To investigate the transcriptomic changes induced by PDX establishment and whether the PDX models recapitulate the gene expression pattern of the tumors from which they originated, we performed RNA sequencing of all PDX models at multiple time points (Supporting Fig. S4) and of the originating HCC biopsies. Differential expression analysis revealed that 2,102 genes were up‐regulated and 3,576 genes were down‐regulated in the PDX tumors (first generation) compared to their matched biopsies (Fig. 3A; Supporting Table S3). Compared to the HCC tumors, first‐generation PDX tumors showed down‐regulation of pathways of inflammatory response and angiogenesis (Fig. 3B; Supporting Table S4), suggesting the loss of cells of the human immune system and vasculature. Indeed, the xenograft tumors were positive for mouse‐specific clusters of differentiation (CD)31, suggesting that the human vascular system in the HCC tumors was replaced by mouse vessels in the xenografts (Supporting Fig. S5). In contrast, pathways related to the cell cycle, such as Myc and E2F target genes and mammalian target of rapamycin complex C1 signaling, were up‐regulated in PDX tumors (Fig. 3B; Supporting Table S4), likely reflecting the enrichment of tumor cells in the PDX tumors compared to the biopsies. Importantly, unsupervised clustering after correcting for the systematic effects introduced by xenografting using a linear model demonstrated that first‐generation PDX tumors stably clustered with their corresponding HCC tumors (Fig. 3C; stability as assessed by bootstrap resampling), suggesting that, aside from the loss of the human immune and vasculature systems, the PDX tumors maintained the tumor‐specific transcriptomic features of their originating tumors. By contrast, gene expression over PDX generations remained markedly stable, with only three and 43 up‐ or down‐regulated genes, respectively, between the first‐ and sixth‐generation PDX tumors (Fig. 3A,B), and the expression profile was maintained over at least 6 generations (Fig. 3D; Supporting Fig. S6A). Taken together, these data indicate that the PDX tumors recapitulate the transcriptomic features of the original HCC biopsies over several generations.
Figure 3.
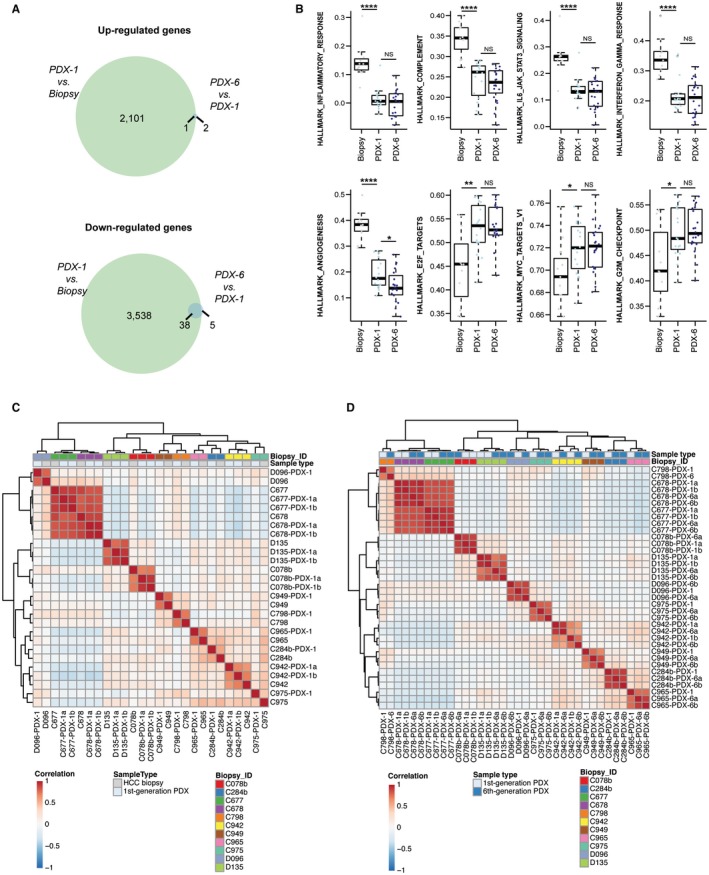
HCC PDX mice recapitulate the transcriptomic features of their original tumors. (A) Venn diagrams illustrate the number of up‐ and down‐regulated genes between HCC biopsies and the first‐generation PDX tumors and between the first‐ and sixth‐generation PDX tumors. (B) Box plots of gene set enrichment scores from selected pathways differentially expressed between HCC biopsies and first‐generation PDX tumors across all biopsies and PDX tumors. Each dot represents the enrichment score from single‐sample Gene Set Enrichment Analysis of a given pathway in a given sample. The boxes represent the first and third quartile with the line representing the median. The whiskers extend to 1.5 times the length of the box. *: P < 0.05; **: P < 0.01; ****: P < 0.0001. (C) Unsupervised clustering of HCC biopsies and first‐generation PDX tumors, corrected for systemic biases between the two groups. (D) Unsupervised clustering of first‐ and sixth‐generation PDX tumors. Abbreviations: IL6, interleukin‐6; JAK‐STAT3, Janus kinase‐signal transducer and activator of transcription 3; NS, not significant.
PDX Tumors Retain the Genomic Characteristics of the Original HCC Tumors
To assess whether the PDX tumors retained the expression of the somatic mutations found in their originating tumors, we identified the repertoire of somatic mutations by performing WES of the original tumor biopsies and their nontumoral counterparts. We then assessed the expression of these somatic mutations by RNA sequencing in the HCC biopsies and the PDX tumors, focusing on missense and synonymous point mutations because truncating mutations are likely to affect transcript stability. Based on WES, we identified a median of 121 (range, 49‐8,015) somatic missense and synonymous point mutations, of which 40% (range, 33%‐48%) were expressed in the corresponding HCC biopsies (Supporting Table S5). Of the expressed mutations in the HCC biopsies, a median of 85% (range, 65%‐100%) was also expressed in the respective PDX tumors (Fig. 4A). Importantly, all missense mutations in HCC cancer genes, such as tumor protein p53 (TP53), catenin beta 1 (CTNNB1), albumin (ALB), and lysine methyltransferase 2B (KMT2B), were expressed in the corresponding PDX tumors, and their expression was maintained over at least 6 generations (Fig. 4A; Supporting Fig. S6B).
Figure 4.
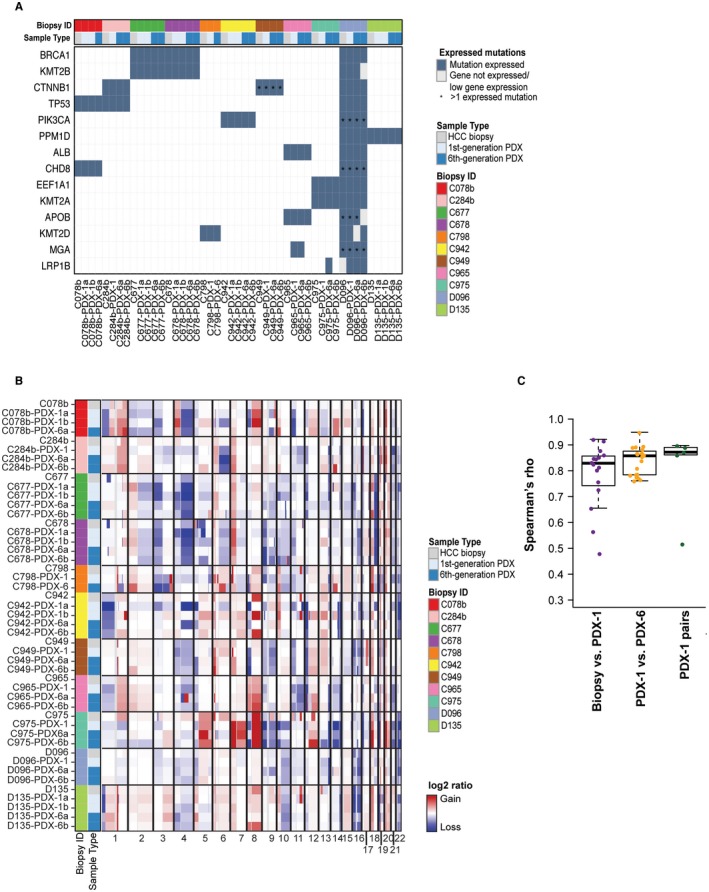
HCC PDX mice recapitulate the genomic features of their original tumors. (A) The repertoire of expressed somatic missense mutations affecting cancer genes28, 42, 43, 44 in HCC biopsies and their corresponding PDX tumors. Nonexpressed genes and genes with low expression are indicated with a gray box. Multiple expressed mutations in the same gene are indicated by an asterisk. (B) Heatmap of copy number alterations (adjusted segmented log2 ratios) in HCC biopsies and the corresponding PDX tumors (first and sixth generation). Samples are presented in rows and chromosomal positions on the x axis (columns). Red, CNA gains; white, neutral; blue, CNA losses. (C) Box plots of the Spearman’s rho correlation coefficient between HCC biopsies and first‐generation tumors, first‐ and their corresponding derivative sixth‐generation tumors, and pairs of first‐generation tumors. The boxes represent the first and third quartile with the line representing the median. The whiskers extend to 1.5 times the length of the box. Abbreviations: ALB, albumin; APOB, apolipoprotein B; BRCA1, breast cancer 1; CHD8, chromodomain‐helicase‐DNA‐binding protein 8; CTNNB1, catenin beta‐1 ; EEF1A1, eukaryotic translation elongation factor 1 alpha 1; KMT2A, lysine methyltransferase 2A; KMT2B, lysine methyltransferase 2B; KMT2D, lysine methyltransferase 2D; LRP1B, low density lipoprotein receptor‐related protein 1B; MGA, MAX dimerization protein; PIK3CA, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase; PPM1D, protein phosphatase 1D.
The overall pattern of CNAs remained largely similar between the original HCC biopsies and the corresponding PDX tumors (Fig. 4B,C). CNAs were found in only two cases. In HCC biopsy C942, copy number gain of chromosome 8q found in the HCC biopsy was observed in the derivative first/sixth(b)‐generation PDX but not in first/sixth(a)‐generation PDX, whereas first/sixth(a)‐generation PDX but not first/sixth(b)‐generation PDX acquired copy number loss of chromosome 4q (Fig. 4B; Supporting Fig. S7A). In C975, copy number gain of chromosome 1q was not seen in the PDX tumors (Fig. 4B). Of note, there were no significant changes in CNAs throughout the first‐ to sixth‐generation tumors (Fig. 4; Supporting Fig. S7), demonstrating genomic stability over several generations.
Taken together, these results demonstrate that the PDX tumors largely maintain the mutational landscape and genetic alterations observed in their originating tumors. Importantly, genomic stability was observed over at least 6 PDX generations.
Engrafted HCC Biopsies are Broadly Representative of the Spectrum of Poorly Differentiated HCCS
The success rate of engraftment was 20% (10/50 biopsies) and 25% (1/4 biopsies) of all transplanted and injected biopsy tissues, respectively (Supporting Table S1). This success rate is in accordance with published data on subcutaneously injected PDX models derived from resected specimens.11, 13, 14, 15 The use of HCC needle biopsies that encompass all clinical stages offered the unique opportunity to investigate the determinants of successful engraftment capacity into mice. When we compared the clinicopathologic characteristics of the engrafted and nonengrafted biopsies, we observed a clear correlation with histopathologic grading; all HCC PDX tumors were derived from poorly differentiated tumors (Edmondson grades III/IV) (Fig. 5A; Supporting Table S1). Indeed, differential expression analysis between engrafted and nonengrafted biopsies revealed 2,401 up‐regulated and 1,440 down‐regulated genes. The up‐regulated genes were enriched for pathways associated with cell‐cycle progression, epithelial‐to‐mesenchymal transition, hypoxia, and angiogenesis, whereas the down‐regulated genes were enriched for liver metabolic functions (Supporting Table S6). Furthermore, molecular subtyping based on Hoshida subclasses26 revealed that all engrafted HCC biopsies were subclasses S1 and S2 (Fig. 5A), characterized by poor differentiation, high proliferation rate, and poor survival compared to subclass S3. Besides Edmondson grading, no other available clinicopathologic characteristics seemed to influence the engraftment success rate (Supporting Table S1; Fig. 5A). When restricting the differential expression analysis to Edmondson grade III biopsies (the only histologic grade that had a variable successful engraftment rate), only two genes were differentially expressed (false discovery rate for both, 0.04; data not shown), suggesting that there was no systematic difference between engrafted and nonengrafted biopsies after accounting for histologic grading.
Figure 5.
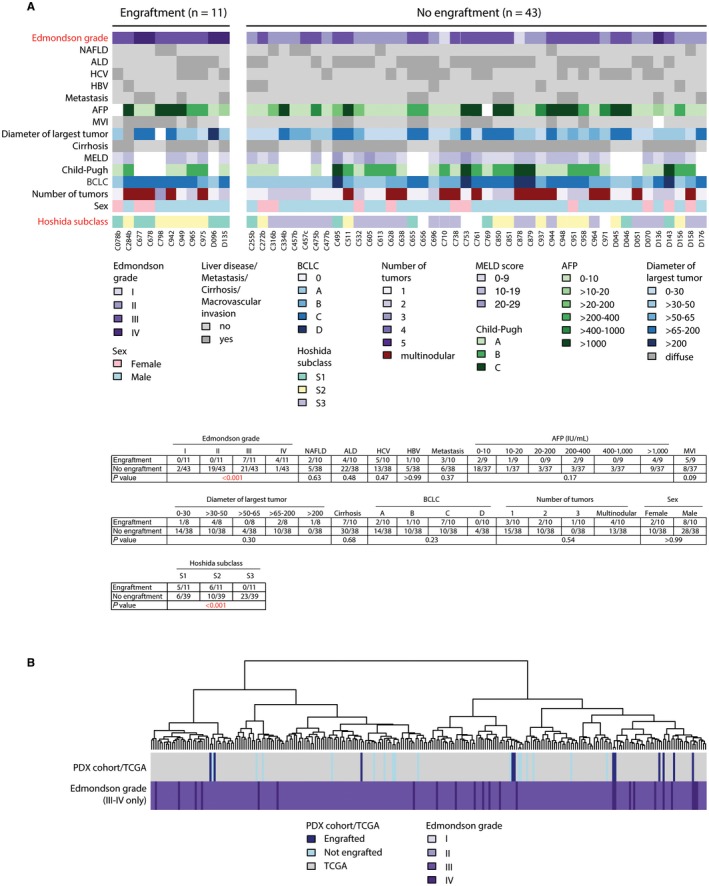
Clinical, histopathologic, and molecular features of engrafted and not engrafted HCC biopsies. (A) Patient and biopsy characteristics of all transplanted biopsies, grouped by engraftment status. Calculations of significant differences (indicated in red) between engrafted versus not engrafted biopsies are shown underneath. Two‐sided Fisher’s exact tests for categorical data and two‐sided Mann‐Whitney U tests for ordinal data. (B) Unsupervised hierarchical clustering analysis. Biopsy (PDX) cohort (this study) combined with high‐grade (Edmondson III to IV) HCCs from TCGA cohort. Abbreviations: ALD, alcoholic liver disease; BCLC, Barcelona Clinic Liver Cancer; HBV, hepatitis B virus; HCV, hepatitis C virus; MELD, Model for End‐Stage Liver Disease; MVI, macrovascular invasion; NAFLD, nonalcoholic fatty liver disease.
To assess the distribution of our samples within a reference set from TCGA,28 we performed an unsupervised hierarchical clustering analysis of the gene expression, combining our HCC biopsy cohort with TCGA.28 We observed that the 11 engrafted biopsies preferentially clustered together in a subclass located at the left end of the clustering tree (Supporting Fig. S8). Because all our PDX models originated from poorly differentiated HCCs, we repeated the unsupervised clustering analysis using only the subset of poorly differentiated HCCs from TCGA. In this scenario, both the 11 engrafted samples and the nonengrafted samples broadly represented the spectrum of poorly differentiated HCCs (Fig. 5B). These results demonstrate that poorly differentiated tumor cells engraft more efficiently in mice and that the HCC biopsies that engrafted are broadly representative of the diversity of poorly differentiated HCCs.
Transformation Into Human B‐Cell Lymphoid Neoplasms of PDX Tumors
During histologic analyses of the engrafted tumors, we observed that three PDX tumors histologically differed from the originating tumors. Specifically, they primarily contained a population of middle‐sized and large lymphoid mononuclear cells with a diffuse growth pattern and high mitotic index that were not present in the corresponding HCC biopsies (Fig. 6A). Transcriptome‐based unsupervised clustering showed that these PDX tumors did not cluster with their originating HCCs (Fig. 6B) and did not express any of the mutations expressed in the corresponding biopsy (Fig. 6C; Supporting Table S5). Compared to the biopsies and the HCC PDX tumors, these three PDX tumors overexpressed genes consistent with immune infiltration, interferon‐alpha and gamma responses, and the nuclear factor kappa B pathway and reduced expression of genes involved in liver metabolism and liver cancer (Supporting Table S7). CNA analyses further confirmed that first‐generation PDX tumors largely differed from the original HCC tumors (Fig. 6D). These results suggest the development of lymphomas in the xenografts as has been reported in PDX studies of various cancers.35, 36, 37, 38 These three PDX tumors stained positive for the human B‐cell marker CD20 (Fig. 6E), consistent with the presence of a B‐cell lymphoma. Importantly, species‐specific alignment demonstrated that CD20 messenger RNA expression was exclusively of human origin (Supporting Table S8). Of note, Chen et al.35 suggested that the reactivation of latent Epstein‐Barr virus (EBV) of intratumoral passenger B lymphocytes led to the generation of lymphoid tumors in immunocompromised mice. Consistent with this hypothesis, EBV transcripts were detected in all three of these PDX tumors but were absent from the corresponding HCC and nontumor biopsies and were virtually absent from all the other PDX models (Supporting Table S8). Taken together, these results suggest that reactivation of latent EBV may lead to lymphomagenesis in some cases using our biopsy‐derived xenograft system.
Figure 6.
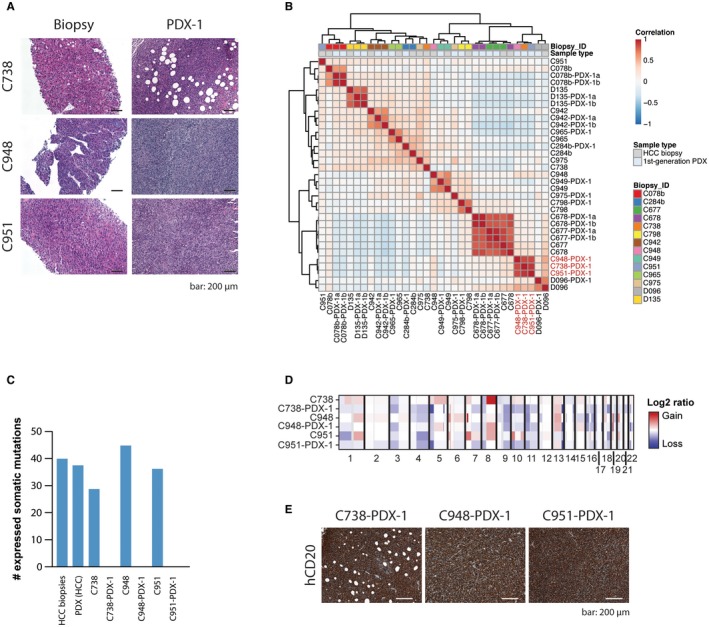
Unintended lymphoid neoplasm transformation in PDX models. (A) H&E‐stained sections of original HCC biopsies and their derivative lymphoma PDX tumors (PDX‐1). (B) Unsupervised clustering of first‐generation PDX and corresponding HCC biopsies. Lymphoma PDX tumors indicated in red. (C) Bar chart illustrating the number of expressed somatic missense and synonymous mutations for the nine HCC biopsies (mean) and the 10 corresponding HCC PDX tumors (mean) and for C738, C948, and C951 and their corresponding lymphoma PDX tumors (C738‐PDX‐1, C948‐PDX‐1, and C951‐PDX‐1). (D) Heatmap of copy number alterations (adjusted segmented log2 ratios) in HCC biopsies and their derivative lymphoma PDX tumors (first generation). Samples are presented in rows and chromosomal positions on the x axis (columns). Red, CNA gains; white, neutral; blue, CNA losses. (E) PDX tumors of the lymphoma models (C738‐PDX‐1, C948‐PDX‐1, and C951‐PDX‐1) stained with hCD20. Scale bar, 200 μm. Abbreviation: hCD20, human CD20.
Discussion
During the last 10 years since the introduction of sorafenib as a first‐line therapy for advanced HCC, a number of additional drugs have failed in phase 2 or phase 3 trials.39 A major obstacle for the development of new therapies is the lack of suitable animal models, specifically animal models that reflect the heterogeneity of HCCs. PDX mice derived from human tumors offer a tool for developing anticancer therapies and personalized medicine for patients with cancer.8 PDX models have also been established for HCCs10, 11, 12, 13, 14, 15; however, these previously published HCC PDX mice were established from resected tumor specimens and therefore predominantly from early stage HCCs. In the present study, we expand the spectrum of HCC PDX models to advanced HCCs.
As in published reports,11, 13, 14, 15 we had a limited overall success rate of approximately 20%. From 54 transplanted HCCs, 11 were successfully grown and retransplanted as xenografts. The reasons for this low success rate remain to be fully elucidated; however, we observed that none of the well‐differentiated (Edmondson grades I/II) HCCs could be engrafted in the mice. Of note, we stopped to monitor the mice for tumor growth 8 months after transplantation. Because low‐grade tumors tend to grow slowly, we cannot exclude the possibility that they would have started growing beyond 8 months. However, we think that tumor cell intrinsic biological properties of undifferentiated HCCs, such as growth factor‐independent proliferation or resistance to hypoxia, are more likely reasons for the successful engraftment of poorly differentiated HCCs. Another factor favoring engraftment might be an intact tumor tissue architecture providing three‐dimensional oriented cell–cell contacts. Compared to transplantations of small tumor pieces, we observed that the injection of a cell suspension showed a delayed tumor growth onset. Interestingly, time to tumor growth onset was invariably shorter following retransplantation of all PDX tumors and then remained constant over several passages. It is possible that the presence of a mouse‐derived vascular system in the PDX tumors facilitated the accelerated engraftment following retransplantation. Once tumors started growing, most PDX models maintained consistent doubling time over generations, which may reflect the intrinsic tumor growth rate once a murine vasculature system is established.
The HCC biopsy‐derived PDX models maintained a striking similarity with the original human HCCs with respect to differentiation grade, growth pattern, cytologic subtype, and the expression pattern of typical HCC markers. Of note, only one PDX model (C284b) lost part of the histologic characteristics (i.e., trabecular growth pattern) observed in the originating human HCC. This may be due to heterogeneous growth patterns in different regions of the same tumor. RNA sequencing of the HCC PDX tumors and the corresponding human HCC biopsies revealed that pathways related to angiogenesis and immune cells were generally down‐regulated in the PDX models, likely a consequence of the loss of human immune cells and the presence of murine instead of human vasculature in these PDX models. By contrast, cell‐cycle‐related genes were up‐regulated in the PDX models compared to the biopsies, suggesting an enrichment of cancer cells in the PDX tumors. Importantly, after accounting for these systematic differences, all PDX tumors clustered with their corresponding human HCC, demonstrating that they recapitulate the tumor‐specific gene expression profiles. In C284b, the correlation between the human HCC and the PDX tumors was weaker than in other cases. This is likely explained by the loss of the trabecular growth pattern in the PDX tumor and the lower tumor content in the biopsy (<30% according to WES‐based CNA analysis; data not shown40, 41). Furthermore, HCC‐specific expression of somatic mutations in cancer genes was maintained in the corresponding PDX models. Of note, transcriptome‐based CNA analysis revealed that the PDX tumors are stable over several generations. A recent study questioned the ability of cancer PDX models to faithfully recapitulate the molecular changes present in the original tumors because CNA landscapes of cancer PDX models diverge from those of the originating tumors during initial transplantation and, to a lesser extent, during passaging.9 However, HCC‐derived data were limited in their study. In the present study we show that the overall pattern of CNAs is largely preserved from HCC biopsy to first‐generation tumors and that the CNA landscape remains stable over 6 generations. We observed two cases that showed clear differences from HCC biopsy to first‐generation tumors. Is this evidence for mouse‐specific tumor evolution? We think that these genetic differences resulted from intratumor genetic heterogeneity within the original HCC tumors rather than the xenografting process. Taken together, our results demonstrate that the PDX models faithfully recapitulate the histopathologic, transcriptomic, and genomic characteristics of the HCC tumors from which they were derived.
The diversity of our patient cohort allowed us to identify potential determinants of tumor engraftment in mice. Differentiation grade was the major determinant for successful PDX development because only poorly differentiated biopsies (Edmondson grades III/IV) and those of molecular subclasses S1 and S226 formed tumors in mice. Accordingly, engrafted tumors overexpressed genes related to proliferation and epithelial‐to‐mesenchymal transition. However, within the group of poorly differentiated HCCs, we could not identify further clinical or transcriptomic characteristics affecting engraftment.
Three HCC biopsies resulted in PDX models that did not resemble the corresponding HCC. The histologic appearance of lymphocytes, strong positivity for the human B‐cell marker CD20, transcriptional up‐regulation of immune cell signatures, loss of expression of the HCC‐specific somatic mutations, and distinct CNA profiles suggest that human B‐cell lymphomas were formed in these PDX models. This phenomenon has been described for PDX models derived from a number of human cancer tissues.35, 36, 37, 38 It has been reported that treatment with rituximab, an anti‐CD20 monoclonal antibody, prevented lymphomatous outgrowth in early passage ovarian xenografts.38 Whether a similar strategy would improve the success rate of HCC PDX generation and prevent lymphomagenesis remains to be investigated.
In conclusion, using needle biopsies instead of resected specimens allows the generation of PDX models from advanced stages of HCC. Our model system has a success rate of approximately 20% and is currently restricted to Edmondson grades III and IV tumors, but within this group, it is representative of the entire molecular spectrum of poorly differentiated HCCs. We demonstrated that the PDX tumors recapitulate the histopathologic, transcriptomic, and genomic features of the original human HCCs, and these features remain stable over 6 generations of retransplantation. Serial passaging over 6 generations allows for the expansion of the tumors from small pieces of biopsies into several hundred xenograft‐carrying mice. Although not suitable for the testing of immunotherapy due to the use of immunodeficient mice, we conclude that PDX models from HCC needle biopsies fulfill the requirements for preclinical drug development strategies that account for the diversity of anticancer drug responses in HCCs.
Supporting information
Acknowledgment
We thank all the patients who participated in this study, Helena Antoniadis for teaching the subcutaneous transplantation, Petra Hirschmann for performing histopathologic staining, Christian Beisel (Genomics Facility Basel) for performing whole‐exome and RNA sequencing, and Sylvia Ketterer for excellent technical assistance. Calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing center at the University of Basel.
Supported by the European Research Council (Synergy grant 609883 and SystemsX.ch grant to M.H.H.), Swiss National Science Foundation (grants 310030B_14708 and 310030_166202 to M.H.H. and Ambizione PZ00P3_168165 to S.P.), and Swiss Cancer League (grants KLS‐3639‐02‐2015 to L.M.T. and KFS‐3995‐08‐2016 to S.P.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Llovet JM, Zucman‐Rossi J, Pikarsky E, Sangro B, Schwartz M, Sherman M, et al. Hepatocellular carcinoma. Nat Rev Dis Primers 2016;2:16018. [DOI] [PubMed] [Google Scholar]
- 2. European Association for the Study of the Liver; European Organisation for Research and Treatment of Cancer . EASL‐EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol 2012;56:908‐943. Erratum. In: J Hepatol 2012;56:1430. [DOI] [PubMed] [Google Scholar]
- 3. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al.; SHARP Investigators Study Group . Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378‐390. [DOI] [PubMed] [Google Scholar]
- 4. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first‐line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non‐inferiority trial. Lancet 2018;391:1163‐1173. [DOI] [PubMed] [Google Scholar]
- 5. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, et al. Efficacy and safety of sorafenib in patients in the Asia‐Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double‐blind, placebo‐controlled trial. Lancet Oncol 2009;10:25‐34. [DOI] [PubMed] [Google Scholar]
- 6. Santos NP, Colaco AA, Oliveira PA. Animal models as a tool in hepatocellular carcinoma research: a review. Tumour Biol 2017;39:1010428317695923. [DOI] [PubMed] [Google Scholar]
- 7. Tentler JJ, Tan AC, Weekes CD, Jimeno A, Leong S, Pitts TM, et al. Patient‐derived tumour xenografts as models for oncology drug development. Nat Rev Clin Oncol 2012;9:338‐350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Siolas D, Hannon GJ. Patient‐derived tumor xenografts: transforming clinical samples into mouse models. Cancer Res 2013;73:5315‐5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ben‐David U, Ha G, Tseng YY, Greenwald NF, Oh C, Shih J, et al. Patient‐derived xenografts undergo mouse‐specific tumor evolution. Nat Genet 2017;49:1567‐1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Armengol C, Tarafa G, Boix L, Sole M, Queralt R, Costa D, et al. Orthotopic implantation of human hepatocellular carcinoma in mice: analysis of tumor progression and establishment of the BCLC‐9 cell line. Clin Cancer Res 2004;10:2150‐2157. [DOI] [PubMed] [Google Scholar]
- 11. Gu Q, Zhang B, Sun H, Xu Q, Tan Y, Wang G, et al. Genomic characterization of a large panel of patient‐derived hepatocellular carcinoma xenograft tumor models for preclinical development. Oncotarget 2015;6:20160‐20176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huynh H, Soo KC, Chow PK, Panasci L, Tran E. Xenografts of human hepatocellular carcinoma: a useful model for testing drugs. Clin Cancer Res 2006;12:4306‐4314. [DOI] [PubMed] [Google Scholar]
- 13. Jiang Z, Jiang X, Chen S, Lai Y, Wei X, Li B, et al. Anti‐GPC3‐CAR T cells suppress the growth of tumor cells in patient‐derived xenografts of hepatocellular carcinoma. Front Immunol 2017;7:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xin H, Wang K, Hu G, Xie F, Ouyang K, Tang X, et al. Establishment and characterization of 7 novel hepatocellular carcinoma cell lines from patient‐derived tumor xenografts. PLoS ONE 2014;9:e85308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yan M, Li H, Zhao F, Zhang L, Ge C, Yao M, et al. Establishment of NOD/SCID mouse models of human hepatocellular carcinoma via subcutaneous transplantation of histologically intact tumor tissue. Chin J Cancer Res 2013;25:289‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Llovet JM, Bru C, Bruix J. Prognosis of hepatocellular carcinoma: the BCLC staging classification. Semin Liver Dis 1999;19:329‐338. [DOI] [PubMed] [Google Scholar]
- 17. Edmondson HA, Steiner PE. Primary carcinoma of the liver: a study of 100 cases among 48,900 necropsies. Cancer 1954;7:462‐503. [DOI] [PubMed] [Google Scholar]
- 18. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013;29:15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA‐Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nikolayeva O, Robinson MD. edgeR for differential RNA‐seq and ChIP‐seq analysis: an application to stem cell biology. Methods Mol Biol 2014;1150:45‐79. [DOI] [PubMed] [Google Scholar]
- 21. Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, et al. Systematic RNA interference reveals that oncogenic KRAS‐driven cancers require TBK1. Nature 2009;462:108‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1:417‐425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010;20:1297‐1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Talevich E, Shain AH, Botton T, Bastian BC. CNVkit: genome‐wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput Biol 2016;12:e1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hoshida Y, Nijman SM, Kobayashi M, Chan JA, Brunet JP, Chiang DY, et al. Integrative transcriptome analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res 2009;69:7385‐7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ng CKY, Piscuoglio S, Geyer FC, Burke KA, Pareja F, Eberle CA, et al. The landscape of somatic genetic alterations in metaplastic breast carcinomas. Clin Cancer Res 2017;23:3859‐3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cancer Genome Atlas Research Network . Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017;169:1327‐1341.e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kancherla V, Abdullazade S, Matter MS, Lanzafame M, Quagliata L, Roma G, et al. Genomic analysis revealed new oncogenic signatures in TP53‐mutant hepatocellular carcinoma. Front Genet 2018;9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schlageter M, Terracciano LM, D’Angelo S, Sorrentino P. Histopathology of hepatocellular carcinoma. World J Gastroenterol 2014;20:15955‐15964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schlageter M, Quagliata L, Matter M, Perrina V, Tornillo L, Terracciano L. Clinicopathological features and metastatic pattern of hepatocellular carcinoma: an autopsy study of 398 patients. Pathobiology 2016;83:301‐307. [DOI] [PubMed] [Google Scholar]
- 32. Bruix J, Sherman M; American Association for the Study of Liver Diseases . Management of hepatocellular carcinoma: an update. Hepatology 2011;53:1020‐1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Di Tommaso L, Franchi G, Park YN, Fiamengo B, Destro A, Morenghi E, et al. Diagnostic value of HSP70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology 2007;45:725‐734. [DOI] [PubMed] [Google Scholar]
- 34. Di Tommaso L, Destro A, Seok JY, Balladore E, Terracciano L, Sangiovanni A, et al. The application of markers (HSP70 GPC3 and GS) in liver biopsies is useful for detection of hepatocellular carcinoma. J Hepatol 2009;50:746‐754. [DOI] [PubMed] [Google Scholar]
- 35. Chen K, Ahmed S, Adeyi O, Dick JE, Ghanekar A. Human solid tumor xenografts in immunodeficient mice are vulnerable to lymphomagenesis associated with Epstein‐Barr virus. PLoS ONE 2012;7:e39294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujii E, Kato A, Chen YJ, Matsubara K, Ohnishi Y, Suzuki M. Characterization of EBV‐related lymphoproliferative lesions arising in donor lymphocytes of transplanted human tumor tissues in the NOG mouse. Exp Anim 2014;63:289‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bondarenko G, Ugolkov A, Rohan S, Kulesza P, Dubrovskyi O, Gursel D, et al. Patient‐derived tumor xenografts are susceptible to formation of human lymphocytic tumors. Neoplasia 2015;17:735‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Butler KA, Hou X, Becker MA, Zanfagnin V, Enderica‐Gonzalez S, Visscher D, et al. Prevention of human lymphoproliferative tumor formation in ovarian cancer patient‐derived xenografts. Neoplasia 2017;19:628‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Llovet JM, Hernandez‐Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res 2014;20:2072‐2079. [DOI] [PubMed] [Google Scholar]
- 40. Nuciforo S, Fofana I, Matter MS, Blumer T, Calabrese D, Boldanova T, et al. Organoid models of human liver cancers derived from tumor needle biopsies. Cell Rep 2018;24:1363‐1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen R, Seshan VE. FACETS: allele‐specific copy number and clonal heterogeneity analysis tool for high‐throughput DNA sequencing. Nucleic Acids Res 2016;44:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fujimoto A, Furuta M, Totoki Y, Tsunoda T, Kato M, Shiraishi Y, et al. Whole‐genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat Genet 2016;48:500‐509. Erratum. In: Nat Genet 2016;48:700. [DOI] [PubMed] [Google Scholar]
- 43. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502:333‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014;505:495‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are available at the European Genome‐phenome Archive under accession EGAS00001003396.