

Abstract
Chronic intermittent hypoxia and hedgehog (Hh) pathway dysregulation are associated with nonalcoholic fatty liver disease (NAFLD) progression. In this study, we determined the relationship between obstructive sleep apnea (OSA)/nocturnal hypoxia and Hh signaling in pediatric NAFLD. Adolescents with histologic NAFLD (n = 31) underwent polysomnogram testing, laboratory testing, and Sonic Hh (SHh), Indian hedgehog (IHh), glioblastoma‐associated oncogene 2 (Gli2), keratin 7 (K7), α‐smooth muscle actin (α‐SMA), and hypoxia‐inducible factor 1α (HIF‐1α) immunohistochemistry. Aspartate aminotransferase (AST) correlated with SHh, r = 0.64; Gli2, r = 0.4; α‐SMA, r = 0.55; and K7, r = 0.45 (P < 0.01), as did alanine aminotransferase (ALT) (SHh, r = 0.51; Gli2, r = 0.43; α‐SMA, r = 0.51; P < 0.02). SHh correlated with NAFLD activity score (r = 0.39), whereas IHh correlated with inflammation (r = −0.478) and histologic grade (r = −0.43); P < 0.03. Subjects with OSA/hypoxia had higher SHh (4.0 ± 2.9 versus 2.0 ± 1.5), Gli2 (74.2 ± 28.0 versus 55.8 ± 11.8), and α‐SMA (6.2 ± 3.3 versus 4.3 ± 1.2); compared to those without (P < 0.03). OSA severity correlated with SHh (r = 0.31; P = 0.09) and Gli2 (r = 0.37; P = 0.04) as did hypoxia severity, which was associated with increasing SHh (r = −0.53), Gli2 (r = −0.52), α‐SMA (r = −0.61), and K7 (r = −0.42); P < 0.02. Prolonged O2 desaturations <90% also correlated with SHh (r = 0.55) and Gli2 (r = 0.61); P < 0.05. Conclusion: The Hh pathway is activated in pediatric patients with NAFLD with nocturnal hypoxia and relates to disease severity. Tissue hypoxia may allow for functional activation of HIF‐1α, with induction of genes important in epithelial‐mesenchymal transition, including SHh, and NAFLD progression.
Abbreviations
- α‐SMA
α‐smooth muscle actin
- AHI
apnea‐hypopnea index
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BMI
body mass index
- CIH
chronic intermittent hypoxia
- Gli2
glioblastoma‐associated oncogene 2
- Hh
hedgehog
- HIF‐1α
hypoxia‐inducible factor 1α
- HPF
high‐power field
- IHh
Indian hedgehog
- K7
keratin 7
- NAFLD
nonalcoholic fatty liver disease
- NAS
NAFLD activity score
- NASH
nonalcoholic steatohepatitis
- NS
not significant
- OSA
obstructive sleep apnea
- Ptc
Patched
- SaO2
O2 saturation
- SHh
sonic hedgehog
- Smo
Smoothened
Nonalcoholic fatty liver disease (NAFLD), characterized by abnormal lipid deposition in hepatocytes in the absence of excess alcohol intake, is the most common chronic liver disease affecting both children and adults and parallels the obesity epidemic.1 Alarmingly, 9.6% of all children and 38% of children with obesity are affected by NAFLD across a spectrum of disease, including isolated hepatic steatosis, nonalcoholic steatohepatitis (NASH) (defined as steatosis, hepatocyte ballooning, and inflammation), and cirrhosis.1, 2 NASH progresses to liver fibrosis and cirrhosis in approximately 20% of cases and is associated with hepatocellular carcinoma in adults.1, 3
Mounting evidence implicates obesity‐related obstructive sleep apnea (OSA) and chronic intermittent hypoxia (CIH) in NAFLD progression. Patients with OSA/CIH are exposed to repeated cycles of hypoxia alternating with normoxia, similar to the pathophysiology of ischemia/reperfusion injury.4, 5 Obese mice fed a high‐fat high‐cholesterol diet develop hepatic steatosis. Subsequent exposure to CIH leads to significant increases in alanine aminotransferase (ALT) and histologic evidence of hepatic inflammation and fibrosis.6, 7 Adults who are morbidly obese and have moderate to severe OSA and CIH demonstrate more significant hepatic inflammation than those without hypoxia.8 Pediatric patients with NAFLD with OSA/hypoxia also have more advanced liver disease and fibrosis than those without OSA/hypoxia.9, 10 Moreover, we have recently reported that OSA/nocturnal hypoxia‐induced oxidative stress, propagated through reactive oxygen species generation, promotes pediatric NAFLD progression.10 Although these data support a role for OSA/hypoxia in NASH pathogenesis, the mechanism underlying this relationship has not been elucidated.
There is increasing evidence supporting a role for dysregulation of the hedgehog (Hh) pathway in the pathogenesis of both adult and pediatric NAFLD.11, 12 Hh, a morphogenic signaling pathway, controls organogenesis during organ development and is usually quiescent in adolescents and adults. Hh ligand activates Hh signaling by engaging Patched (Ptc), a transmembrane surface receptor on cells responsive to Hh.12, 13, 14 Hh ligands bind to Ptc, thereby preventing Ptc from inhibiting Smoothened (Smo). Activated Smo subsequently controls the cellular accumulation and nuclear localization of glioblastoma transcription factors, which in turn regulate the expression of other Hh‐regulated genes critical for the proliferation, differentiation, and survival of Hh‐responsive cells.13, 14 Although typically silent, Hh may be reactivated during injury that stimulates Hh ligand production. In the liver injury incited in both adult and pediatric NAFLD, activation of the Hh pathway is a dominant mechanism by which fibro‐inflammatory repair occurs.11, 12
In adult mice, hepatic hypoxia induces expression of the Hh ligand sonic hedgehog (SHh) and the pathway activity marker Ptc1, thereby inciting a systemic Hh response.15 The impact of hypoxia on Hh pathway activation in human liver disease, however, is unknown. This study was conducted to understand the relationship between OSA/nocturnal hypoxia and dysregulation of the Hh signaling pathway in pediatric NAFLD. We hypothesized that OSA and CIH would be associated with up‐regulation of the Hh pathway, with evidence of increasing histologic severity of pediatric NAFLD.
Patients and Methods
Eligible study subjects were children cared for at Children's Hospital Colorado Pediatric Liver Center between June 2009 and January 2014. The subjects had suspected NAFLD and were scheduled to undergo a clinically indicated liver biopsy. Inclusion criteria were male and female children, ages 8 through 18 years, and Tanner stages 2‐4. Exclusion criteria for the study were the presence of Wilson's disease, alpha‐1‐antitrypsin deficiency, viral hepatitis, autoimmune hepatitis, other known chronic liver disease, cholelithiasis or use of corticosteroids, anticonvulsants, sedatives, drugs that promote or reduce insulin resistance (including metformin, insulin sensitizers, and thiazolidinediones), or other treatments known to induce hepatic steatosis (amiodarone or parenteral nutrition) in the 2 weeks prior to the liver biopsy. Additional exclusion criteria included regular tobacco or alcohol use, insulin‐dependent diabetes, neuromuscular disorders, genetic or craniofacial abnormalities, and current use of continuous positive airway pressure. Data from a subset of these patients have been reported.9, 10 This study was approved by the Colorado Multiple Institutional Review Board, and informed written consent was obtained from parents/guardians and written assent from all subjects.
Demographic and medical history data were obtained. Heights, weights, and body mass index (BMI) z scores (based on age and sex data from the Centers for Disease Control and Prevention) were determined.16 Subjects with suspected NAFLD underwent liver biopsy for clinical indications by standard percutaneous technique. In our center, we suspect NAFLD in children who are overweight or obese (BMI >85% for age and sex), with chronically elevated aminotransferases, with negative screening for other chronic liver diseases. Liver histology (hematoxylin and eosin and Masson's trichrome stains) was reviewed and scored by a single pediatric pathologist blinded to subject information. Biopsies with histologically confirmed NAFLD (defined as ≥5% of hepatocytes containing macrovesicular fat) were assigned a grade of necro‐inflammation (0‐3) and a stage of fibrosis (0‐4) based on the standard histologic criteria described by Brunt et al.17 Each biopsy was also scored for NASH Clinical Research Network criteria18: steatosis grade 0 (<5% of hepatocytes containing macrovesicular fat), grade 1 (5%‐33%), grade 2 (34%‐66%), and grade 3 (>66%); lobular inflammation grade 0 (no foci of inflammation), grade 1 (<2 foci per high‐power field [HPF]), grade 2 (2‐4 foci per HPF), and grade 3 (>4 foci per HPF); and ballooning degeneration grade 0 (none), grade 1 (few balloon cells), and grade 2 (many/prominent balloon cells). The NAFLD activity score (NAS) was calculated by summing the individual scores for steatosis, lobular inflammation, and ballooning degeneration.18 Subjects were also classified as having definite NASH (NAS ≥5), not NASH, or borderline NASH (NAS ≤4).19 Hepatic fibrosis was scored as stage 0 (none), stage 1 (mild to moderate perisinusoidal or portal/periportal fibrosis only), stage 2 (zone 3 and periportal fibrosis), stage 3 (bridging fibrosis), or stage 4 (cirrhosis).18, 20 Furthermore, subjects were defined as either type 1 (classic adult pattern), type 2 (portal‐based histology), or an overlap of the two NASH histologic subtypes.20 Immunostaining for hypoxia‐inducible factor 1α (HIF‐1α) was carried out using the Dako Cytomation CSA system (Dako Corporation, Santa Clara, CA) for mouse primary antibodies (K1500). Antigen retrieval was performed using citrate buffer for 30 minutes. The primary antibody was Novus Biological NB 100‐123, used at a 1:8,000 dilution, and slides were counterstained with hematoxylin. HIF‐1α staining intensity was graded from 0 (no staining) to 3 (significant staining), and the location of staining was noted to be nuclear, cytoplasmic, or both.
Liver biopsy sections were stained to detect SHh, Indian hedgehog (IHh), glioblastoma‐associated oncogene 2 (Gli2), and markers of progenitors (keratin 7 [K7]) and myofibroblasts (α‐smooth muscle actin [α‐SMA]), using the Dako Envision System according to the manufacturer's protocol. Immunohistochemistry was performed as described.21 Briefly, formalin‐fixed paraffin‐embedded liver tissue was cut into 5‐μm sections and placed on glass slides. Sections were deparaffinized with xylene, dehydrated with ethanol, and incubated with 3% hydrogen peroxide to block endogenous peroxidase. Antigen retrieval was performed by heating in 10 mM sodium citrate buffer (pH 6.0). Sections were blocked in Dako protein block (X9090; Dako) and followed by incubation with primary antibodies (SHh, Abcam‐100639; IHh, Abcam‐39634; Gli2, GenWay Biotech EB‐3B44; α‐SMA, Abcam‐32575; and K7, Dako‐M7018). Horseradish peroxidase (HRP)‐conjugated anti‐rabbit (K4003; Dako) and HRP‐conjugated anti‐mouse (K4001; Dako) secondary antibodies were used to visualize target proteins. 3,3′‐diaminobenzidine reagent (K3466; Dako) was applied in the detection procedure. Tissue sections were counterstained with Aqua Hematoxylin‐INNOVEX (Innovex Biosciences). Negative controls included liver specimens exposed to 1% bovine serum albumin instead of the respective primary antibodies. The number of detectable SHh, IHh, Gli2, α‐SMA, and K7 immunoreactive cells was quantified by counting 20 randomly chosen 20× fields per section per slide. Additionally, the histologic locations of SHh and Gli2 staining were determined.
Following pathology review of initial liver biopsies, subjects with histologically confirmed NAFLD underwent a standard multichannel sleep study (polysomnogram), which was scored by a trained technician and interpreted by a single sleep medicine physician, both of whom were blinded to liver biopsy results. The following data were analyzed: total sleep time, percentage of rapid eye movement (REM) sleep, apnea‐hypopnea index (AHI), oxygen nadir, percentage of time O2 saturation (SaO2) ≤90%, and oxygen desaturation index (the number of SaO2 drops below 95% by pulse oximeter). OSA was defined as an AHI >2.0, indicating total apneas and hypopneas per hour of total sleep time.22, 23 Apnea was defined as cessation of airflow over ≥2 attempted respiratory cycles. Hypopnea was defined as a decrease in nasal pressure of ≥50%, with a corresponding decrease in SaO2 of ≥3% and/or arousal. Hypoxia was defined as SaO2 <90% for ≥1% of total sleep time.24
The morning after the polysomnogram, fasting blood specimens were collected for serum ALT, aspartate aminotransferase (AST), gamma‐glutamyl transpeptidase, ultrasensitive C‐reactive protein, total cholesterol, triglyceride, high‐density lipoprotein, glucose, and insulin. Fasting glucose and insulin levels were used to calculate insulin resistance at baseline using the homeostasis model assessment of insulin resistance.25
Statistical analyses were performed using SAS 9.3 software (SAS Institute, Inc., Cary, NC). Descriptive statistics are presented as either mean ± SD for continuous measures or percentages for categorical responses. Two‐sample t test, chi‐squared test, or Fisher's exact test were used, as appropriate, to assess differences between subjects with and without OSA/hypoxia. Pearson or Spearman correlation coefficients, as appropriate, were used to quantify the relationships between ALT, AST, and histologic parameters with polysomnographic parameters and Hh makers. P < 0.05 was considered statistically significant.
Results
Relationship of OSA/Hypoxia to NAFLD
A total of 31 subjects with liver biopsy‐confirmed NAFLD were studied. These adolescents (mean age, 13.0 ± 1.9 years) were 65% male subjects, 87% Hispanic, and had obesity (mean BMI z score, 2.2 ± 0.3). Twenty‐one subjects (68%) met our study criteria for OSA and/or hypoxia: 1 had isolated hypoxia, 12 had isolated OSA, and 8 had both OSA and hypoxia. Subjects with and without OSA/hypoxia had similarly elevated aminotransferases, inflammatory markers, triglycerides, and evidence of insulin resistance (Table 1). Although total serum cholesterol concentration was higher in those with OSA/hypoxia (P = 0.03), this did not impact NAFLD disease severity. In all subjects with NAFLD, polysomnograms were of adequate length with >12% total sleep time in rapid eye movement (REM) sleep, allowing all studies to be considered valid. Mean total sleep time and percentage of REM sleep were similar between groups. Subjects with NAFLD with OSA/hypoxia had a significantly higher (P < 0.001) mean AHI score (9.0 ± 8.2) versus those without OSA/hypoxia (0.9 ± 0.6), suggesting moderate to severe sleep‐disordered breathing in affected subjects (Table 1). Those with OSA/hypoxia also had significantly lower oxygen nadirs versus those without OSA/hypoxia (82.6% ± 6.3% versus 87.8% ± 2.0%; P = 0.004) and spent a greater percentage of sleep time with SaO2 <90% (2.2 ± 3.8 versus 0.1 ± 0.2; P = 0.02) (Table 1).
Table 1.
Demographic and Clinical Parameters of Subjects With NAFLD With and Without OSA/Hypoxia
| Clinical Parameter | NAFLD With OSA/Hypoxia (n = 21) | NAFLD Without OSA/Hypoxia (n = 10) | P Value | Normal Values |
|---|---|---|---|---|
| Mean age | 13.0 ± 1.9 | 13.1 ± 2.3 | 0.8 | – |
| Male sex (%) | 67 | 50 | 0.4 | – |
| Hispanic ethnicity (%) | 86 | 100 | 0.2 | – |
| BMI z score | 2.3 ± 0.3 | 2.1 ± 0.3 | 0.3 | – |
| ALT (IU/L) | 131 ± 109 | 93 ± 63 | 0.3 | 10‐45 IU/L |
| AST (IU/L) | 79 ± 55 | 58 ± 27 | 0.2 | 15‐40 IU/L |
| Cholesterol (mg/dL) | 157 ± 38 | 121 ± 43 | 0.03 | ≤170 mg/dL |
| Triglycerides (mg/dL) | 148 ± 66 | 139 ± 59 | 0.7 | ≤150 mg/dL |
| HOMA‐IR | 10.0 ± 6.7 | 8.1 ± 9.0 | 0.5 | <2.60 |
| Adiponectin (μg/mL) | 6.6 ± 3.4 | 6.6 ± 2.9 | 1.0 | 3.5‐14 μg/mL |
| Leptin (ng/mL) | 30.6 ± 15.5 | 29.9 ± 11.1 | 0.5 | 2.0‐5.6 ng/mL |
| CRP (mg/dL) | 3.0 ± 3.5 | 1.8 ± 1.2 | 0.2 | <0.5 mg/dL |
| Uric acid (mg/dL) | 6.6 ± 1.5 | 5.9 ± 1.3 | 0.2 | 2.3‐5.4 mg/dL |
| Sleep parameters | ||||
| Apnea‐hypopnea index | 9.0 ± 8.2 | 0.9 ± 0.6 | 0.0002 | – |
| Oxygen nadir | 82.6 ± 6.4 | 87.8 ± 2.9 | 0.004 | – |
| % Time SaO2 <90% | 2.2 ± 3.8 | 0.1 ± 0.2 | 0.02 | – |
Abbreviations: CRP, C‐reactive protein; HOMA‐IR, homeostasis model assessment of insulin resistance.
Liver Injury and OSA/Hypoxia
Both AST and ALT values were similar in subjects with NAFLD with and without OSA/CIH (P was not significant [NS]). Liver histology scores for steatosis, inflammation, ballooning degeneration, NAS summary score (mean NAS, 4.81 ± 1.29 versus 5.01 ± 1.10), and histologic grade were also similar between those with and without OSA/hypoxia. However, subjects with NAFLD with OSA/hypoxia trended toward more severe fibrosis (62% stages 0‐2; 38% stage 3) than those without OSA/hypoxia (100% stages 0‐2; P = 0.08). Increasing time with SaO2 <90% was also associated with higher hepatic steatosis (r = 0.4; P = 0.0008), histologic grade of inflammation (r = 0.36; P = 0.04), and NAS (r = 0.35; P = 0.055).
There were no differences in the distribution of histologic type 1 versus type 2 NAFLD in subjects with and without OSA/hypoxia. Subjects with NAFLD with definite NASH (NAS histologic score, ≥5; n = 20) had more severe sleep‐disordered breathing compared to those without definite NASH (NAS score, <5; n = 11). Subjects with definite NASH trended toward higher (P = 0.07) mean AHI scores (7.9 ± 9.2) than those without definite NASH (3.7 ± 2.8). Subjects with definite NASH spent significantly more time (P = 0.03) with SaO2 <90% (2.3 ± 3.9) than those without definite NASH (0.19 ± 0.35). There were no differences in oxygen nadir or oxygen desaturation index (data not shown). There were no differences noted in HIF‐1α immunohistochemistry staining intensity in subjects with NAFLD with (mean, 1.92 ± 0.86) and without OSA/hypoxia (1.78 ± 0.83) or location. There were also no associations found between HIF‐1α immunohistochemistry staining intensity or location and severity of OSA/hypoxia, as indicated by AHI, oxygen nadir, or time with SaO2 <90%.
Mutual Association of Hh Signaling Markers
The presence of SHh on immunohistochemistry was strongly correlated with the presence of Gli2 (r = 0.56; P = 0.001), K7 (r = 0.71; P = <0.001), and α‐SMA (r = 0.70; P < 0.001) but not IHh. The presence of IHh on immunohistochemistry also did not correlate with Gli2, K7, or α‐SMA. In addition, Gli2 correlated strongly with K7 (r = 0.67; P < 0 .001) and α‐SMA (r = 0.58; P = 0.001) as did K7 and α‐SMA (r = 0.74; P < 0.001).
Liver Injury and Hh Signaling
Liver injury was closely associated with markers of Hh activation. AST correlated strongly with SHh (r = 0.64; P = 0.0001), Gli2 (r = 0.47; P = 0.007), α‐SMA (r = 0.55; P = 0.002), and K7 (r = 0.45; P = 0.01). Similarly, ALT correlated strongly with SHh (r = 0.51; P = 0.003), Gli2 (r = 0.43; P = 0.02), and α‐SMA (r = 0.51; P = 0.005) but not K7 (r = 0.30; P = 0.09). SHh correlated with the NAS summary score (r = 0.39; P = 0.03) but not with steatosis, lobular inflammation, hepatocyte ballooning, or fibrosis. IHh was inversely correlated with inflammation (r = −0.478; P = 0.01) and histologic grade (r = −0.43; P = 0.02) but not with steatosis, hepatocyte ballooning, NAS summary score, or fibrosis. Neither Gli2 nor K7 correlated with steatosis, inflammation, hepatocyte ballooning, NAS summary score, or fibrosis (P was NS). Finally, α‐SMA correlated strongly with both the NAS summary score (r = 0.43; P = 0.02) and fibrosis stage (r = 0.41; P = 0.03) but not steatosis, inflammation, and hepatocyte ballooning. In addition, subjects with NAFLD inflammation grade17 3‐5 had higher α‐SMA (mean, 7.14 ± 3.32) and SHh (4.48 ± 2.75) than subjects with NAFLD inflammation grade 0‐2 (α‐SMA, 4.90 ± 2.39; P = 0.05; and SHh, 2.47 ± 2.44; P = 0.04). Subjects with type 1 NASH had slightly higher SHh (mean, 3.74 ± 2.35) compared to type 2 histologic NASH (1.88 ± 1.86; P = 0.058). There were no differences in IHh, Gli2, K7, or α‐SMA between subjects with histologic type 1 versus type 2 NAFLD (P was NS).
OSA/Hypoxia and Hh Signaling
Subjects with OSA/hypoxia had higher mean SHh (4.0 ± 2.9 versus 2.0 ± 1.5), Gli2 (74.2 ± 28.0 versus 55.8 ± 11.8), and α‐SMA immunohistochemistry scores (6.2 ± 3.3 versus 4.3 ± 1.2) but not IHh (1.1 ± 0.9 versus 0.9 ± 0.8) or K7 (25.8 ± 12.8 versus 21.5 ± 11.1) (Fig. 1). The severity of OSA, as measured by AHI, was positively correlated with both SHh (r = 0.31; P = 0.09) and Gli2 (r = 0.37; P = 0.04) (Fig. 2) but not IHh, K7, or α‐SMA. The severity of hypoxia, as indicated by worsening oxygen nadir, was also significantly associated with increasing SHh (r = −0.53; P = 0.002), Gli2 (r = −0.52; P = 0.003), α‐SMA (r = −0.61; P = 0.006), and K7(r = −0.42; P = 0.02) (Fig. 3). Prolonged duration of hypoxia was also associated with prominent Hh signaling, as demonstrated by significant correlations between increasing percentage of sleep time with SaO2 <90% and SHh (r = 0.5; P = 0.002), Gli2 (r = 0.61; P = 0.0003), α‐SMA (r = 0.7; P < 0.0001), and K7 (r = 0.36; P = 0.048) (Fig. 4). There were no associations between HIF‐1α immunohistochemistry and SHh, Gli2, or K7.
Figure 1.

Comparison of subjects with NAFLD and OSA/hypoxia. Subjects with NAFLD with OSA/hypoxia have higher (A) SHh, (B) Gli2, and (C) α‐SMA but not (D) K7. Representative immunohistochemistry (magnification ×20) demonstrates (A) SHh, (B) Gli2, (C) α‐SMA, and (D) K7 staining in subjects with NAFLD with OSA/hypoxia. Data represent mean ± SD.
Figure 2.
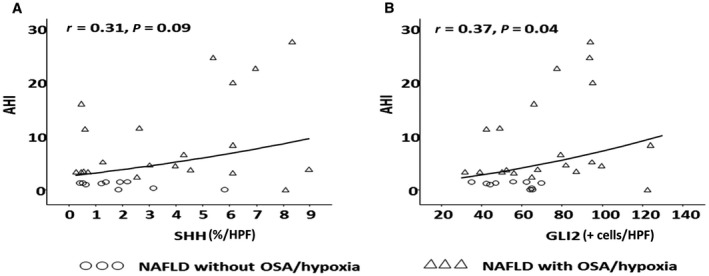
Association of obstructive sleep apnea and hedgehog signaling. The severity of obstructive sleep apnea as measured by the AHI is associated with increased (A) SHh and (B) Gli2.
Figure 3.
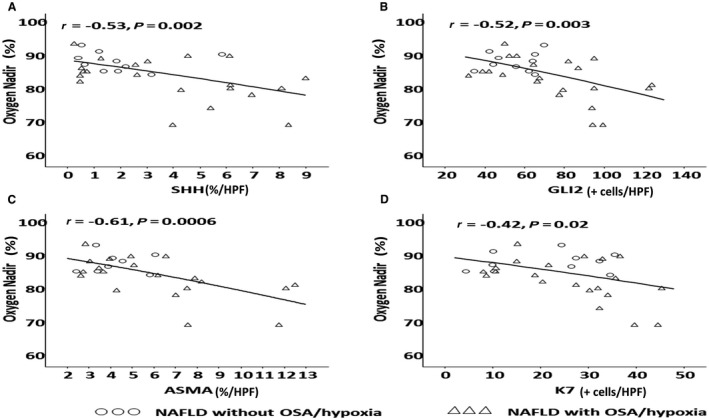
Association of hypoxia and hedgehog signaling. The severity of hypoxia as measured by worsening oxygen nadir is associated with increased Hh signaling, including (A) SHh, (B) GLi2, (C) α‐SMA, and (D) K7.
Figure 4.

Association of duration of hypoxia and hedgehog signaling. An increasing duration of hypoxia is associated with prominent Hh signaling in pediatric NAFLD, including (A) SHh, (B) Gli2, (C) α‐SMA, and (D) K7.
OSA/Hypoxia and Histologic Location of SHh and Gli2
A majority (88%) of subjects with NAFLD and OSA/hypoxia demonstrated an intense ductular reaction compared to only 40% of those without OSA/hypoxia (P = 0.007). SHh positivity was visualized in three relatively discrete histologic patterns: (1) SHh+ bile duct/ductular cells, (2) SHh+ portal/periportal hepatocytes, and (3) a combination of SHh+ bile duct/ductular cells and portal/periportal hepatocytes. Subjects with OSA/hypoxia demonstrated a predominant pattern of SHh positivity in portal/periportal hepatocytes (78%), with 11% in bile duct/ductular cells and 11% in both areas. In contrast, SHh positivity in subjects without OSA/hypoxia was predominantly in bile duct/ductular cells (60%), with only 40% in the portal/periportal hepatocytes (P = 0.01). Similarly, Gli2‐positive cells were located in the portal/periportal areas in 84% of those with OSA/hypoxia, with the remaining 16% showing Gli2 positivity diffusely across the portal/periportal, bile duct/ductular, and lobular locations. In comparison, Gli2 positivity was located in the portal/periportal areas in 50% of subjects without OSA/hypoxia and diffusely across portal/periportal, bile duct/ductular, and lobular locations in 50% of subjects (P = 0.05).
Discussion
In this study, we demonstrated that the Hh pathway is activated in pediatric NAFLD and relates not only to NAFLD disease severity but importantly to the severity of both OSA and severity and duration of chronic intermittent nocturnal hypoxia. The results of this study further implicate OSA and CIH as important factors that may promote the progression of pediatric NASH through, at least in part, the Hh pathway.
Increasing evidence implicates abnormal regulation of the Hh pathway in the pathogenesis and progression of NAFLD. Hh, a morphogenic signaling pathway quiescent in the adult liver, can be reactivated by injury that stimulates the growth of cells involved in hepatic wound healing, including immune, stellate, and progenitor cells.13, 26, 27 In adults with NASH, ballooned hepatocytes produce Hh ligands; the number of ligands and Hh‐responsive (Gli2‐positive) cells correlate with both the severity of hepatic inflammation and fibrosis.28 Similarly, children with NAFLD exhibit increasing SHh expression associated with the degree of steatosis and fibrosis.27 Inflammation and fibrosis are also related to the number of Gli2‐positive cells, and inflammation was associated with the number of K7 cells (a progenitor cell marker27). We expand on this work, showing that liver injury, as measured by aminotransferase elevation, was closely associated with markers of Hh activation. Moreover, hepatic SHh immunohistochemistry correlated strongly with the NAS summary score, although not with individual measures of steatosis, inflammation, or ballooning. These data collectively suggest that activation of the Hh pathway during liver injury in pediatric NAFLD may be a crucial mechanism driving the fibro‐inflammatory repair response.
Hypoxia is a known driver for induction of the Hh pathway in several other disease models. Hypoxia induces up‐regulation of the Hh pathway, including SHh, Smo, and Gli1, thereby contributing to the invasiveness of pancreatic, breast, and prostate cancer and neuroblastoma.29, 30, 31, 32, 33 Cultured cardiomyoblasts exposed to hypoxia also show evidence of induced gene expression of SHh.34 SHh signaling also controls human pulmonary arterial smooth muscle cell proliferation in response to hypoxia.35 Similarly, SHh expression is increased in mouse neurons and neuroprogenitor cells under hypoxic conditions.36 Children with NAFLD have been shown to produce Hh ligand in the progenitor compartment along with accumulation of Hh‐responsive (Gli2‐positive) cells.27 We now demonstrate that adolescents with NAFLD and OSA/hypoxia also produce Hh in this progenitor compartment together with accumulation of Hh‐responsive (Gli2‐positive) cells. Taken together, these findings suggest that CIH may have a significant impact on the Hh pathway and consequent hepatic fibro‐inflammatory repair response in pediatric NAFLD.
We now show in vivo in pediatric NAFLD that the Hh pathway is activated in the liver by CIH. Children with NAFLD and OSA/hypoxia have higher levels of hepatic SHh, Gli2, and α‐SMA than those without OSA/hypoxia. The severity of their OSA correlates strongly with activation of the Hh pathway in the liver, including SHh and Gli2. Both the severity and duration of their hypoxia were also associated strongly with activation of the Hh pathway. Furthermore, children with OSA and hypoxia have more severe fibrosis than those without OSA/hypoxia. Future studies in larger cohorts of children with NAFLD and OSA/hypoxia may further clarify the role of hypoxia‐driven activation of the Hh pathways in NAFLD disease progression.9, 10
In OSA and CIH, the liver experiences repeated episodes of nocturnal hypoxia, during which cells adapt to a lower than normal oxygen environment, and normoxia, with sudden increases in oxygen consumption and enhanced mitochondrial generation of reactive oxygen species.37, 38 Tissue hypoxia may allow for functional activation of HIF‐1α, with subsequent induction of genes important in epithelial‐mesenchymal transition, including SHh, thereby contributing to NAFLD disease progression. Mice exposed to CIH exhibit increased SHh protein in whole liver homogenates but require HIF‐1α accumulation to mount this response.15 Furthermore, animal models show that HIF‐1α is a proximal effector of Hh‐initiated signals that promote glycolysis‐dependent changes in hepatic stellate cells.39 Although our HIF‐1α immunohistochemistry did not demonstrate an increase in HIF‐1α protein, a potential role for HIF‐1α in up‐regulating Hh signaling in NAFLD remains a biologically plausible possibility. Given that the subjects with NAFLD evaluated in this study had intermittent nocturnal hypoxia and that liver biopsies were taken during the day, transient changes in HIF‐1α may not have been detected because although HIF‐1α responds rapidly to hypoxia, it is also rapidly degraded under normoxic conditions.40, 41 In addition, because the intermittent hypoxia was chronic in nature, it is also possible that adaptive changes in HIF‐1 α may have prevented its up‐regulation and that another hypoxia‐induced pathway was responsible for activation of Hh signaling. In the future, studies specifically designed to evaluate HIF‐1α accumulation during nocturnal conditions may help clarify its role.
Limitations of this study include the cross‐sectional study design, which precludes us from establishing causality between OSA/hypoxia, the severity of NAFLD, and Hh signaling. The relatively small sample size of this study raises the possibility of type 2 errors in detecting relationships between OSA/hypoxia and Hh signaling. This may explain why, unlike previous work,27 we found strong correlations between aminotransferases, overall NAS score, and Hh markers but not some of the individual components measuring histologic severity. Previous work has suggested changes in Hh signaling with changes in pubertal staging.27 Unfortunately, although all our subjects were Tanner stages 2‐4, their specific Tanner stage was not collected and, therefore, we cannot determine the potential impact of this in our study. In addition, our sample may be biased toward more severe liver disease because all enrolled subjects underwent liver biopsy for clinical indications that included chronic elevation of aminotransferases. Finally, our study design does not allow us to specifically examine tissue oxygenation in the liver microenvironment but will be an important aspect of future investigations.
This work contributes to the evolving evidence supporting the dysregulation of the Hh pathway in the pathogenesis of pediatric NAFLD (Fig. 5). CIH, as seen in our study cohort, may allow for activation of the Hh ligand, which we speculate may occur through HIF signaling. The now active Hh ligand triggers the Hh signaling pathway by engaging Ptc, thereby preventing Ptc from inhibiting Smo. Activated Smo allows for the cellular accumulation and nuclear localization of Gli transcription factors, which then regulate the expression of other Hh‐regulated genes critical for the proliferation, differentiation, and survival of Hh‐responsive cells. Activation of the Hh pathway, a dominant mechanism by which fibro‐inflammatory repair occurs, in patients with NAFLD and OSA/CIH may contribute to both the severity and progression of disease.
Figure 5.
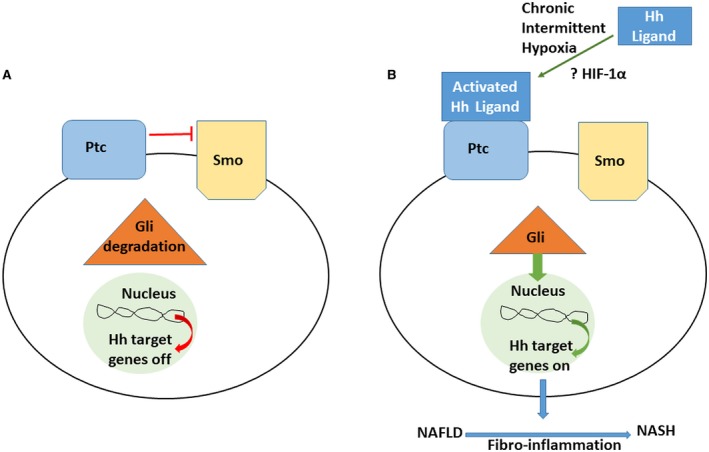
The fibro‐inflammatory response in NAFLD. (A) In the absence of Hh ligands, Ptc represses the activation of Smo, preventing it from interacting with intracellular factors that stabilize and allow for Gli transcription factor accumulation. The Gli proteins subsequently undergo phosphorylation and proteosomal degradation.12, 13, 14 (B) The effect of chronic intermittent hypoxia, which we speculate may be mediated by HIF, allows for activation of the Hh ligand, thereby engaging Ptc and preventing Smo inhibition. Activated Smo allows for nuclear localization of Gli transcription factors, which further regulate the expression of Hh‐regulated genes that are critically important in the fibro‐inflammatory response in NAFLD.
Supported by the National Institutes of Health (NIH; K23 DK085150 to S.S. and R01 DK077794, U01DK061713 and R37AA010154 to A.D.) and NIH/the National Center for Advancing Translational Sciences, Colorado Clinical and Translational Science Awards (grant UL1 TR002535).
The contents are the authors’ sole responsibility and do not necessarily represent official National Institutes of Health views.
Potential conflict of interest: Dr. Sokol consults for Alexion, Retrophin, Shire, and Albireo. The other authors have nothing to report.
References
- 1. Brunt EM. Nonalcoholic steatohepatitis: definition and pathology. Semin Liver Dis 2001;21:3‐16. [DOI] [PubMed] [Google Scholar]
- 2. Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006;118:1388‐1393. [DOI] [PubMed] [Google Scholar]
- 3. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002;346:1221‐1231. [DOI] [PubMed] [Google Scholar]
- 4. Henrion J, Colin L, Schapira M, Heller FR. Hypoxic hepatitis caused by severe hypoxemia from obstructive sleep apnea. J Clin Gastroenterol 1997;24:245‐249. [DOI] [PubMed] [Google Scholar]
- 5. Mathurin P, Durand F, Ganne N, Mollo JL, Lebrec D, Degott C, et al. Ischemic hepatitis due to obstructive sleep apnea. Gastroenterology 1995;109:1682‐1684. [DOI] [PubMed] [Google Scholar]
- 6. Savransky V, Bevans S, Nanayakkara A, Li J, Smith PL, Torbenson MS, et al. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet‐induced fatty liver. Am J Physiol Gastrointest Liver Physiol 2007;293:G871‐G877. [DOI] [PubMed] [Google Scholar]
- 7. Drager LF, Li J, Reinke C, Bevans‐Fonti S, Jun JC, Polotsky VY. Intermittent hypoxia exacerbates metabolic effects of diet‐induced obesity. Obesity (Silver Spring) 2011;19:2167‐2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Polotsky VY, Patil SP, Savransky V, Laffan A, Fonti S, Frame LA, et al. Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am J Respir Crit Care Med 2009;179:228‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sundaram SS, Sokol RJ, Capocelli KE, Pan Z, Sullivan JS, Robbins K, et al. Obstructive sleep apnea and hypoxemia are associated with advanced liver histology in pediatric nonalcoholic fatty liver disease. J Pediatr 2014;164:699‐706.e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sundaram SS, Halbower A, Pan Z, Robbins K, Capocelli KE, Klawitter J, et al. Nocturnal hypoxia‐induced oxidative stress promotes progression of pediatric non‐alcoholic fatty liver disease. J Hepatol 2016;65:560‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verdelho Machado M, Diehl AM. Role of hedgehog signaling pathway in NASH. Int J Mol Sci 2016;17:pii:E857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang JJ, Tao H, Li J. Hedgehog signaling pathway as key player in liver fibrosis: new insights and perspectives. Expert Opin Ther Targets 2014;18:1011‐1021. [DOI] [PubMed] [Google Scholar]
- 13. Omenetti A, Choi S, Michelotti G, Diehl AM. Hedgehog signaling in the liver. J Hepatol 2011;54:366‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shen X, Peng Y, Li H. The injury‐related activation of hedgehog signaling pathway modulates the repair‐associated inflammation in liver fibrosis. Front Immunol 2017;8:1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bijlsma MF, Groot AP, Oduro JP, Franken RJ, Schoenmakers SH, Peppelenbosch MP, et al. Hypoxia induces a hedgehog response mediated by HIF‐1alpha. J Cell Mol Med 2009;13:2053‐2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuczmarski RJ, Ogden CL, Grummer‐Strawn LM, Flegal KM, Guo SS, Wei R, et al. CDC growth charts: United States. Adv Data 2000:1‐27. [PubMed] [Google Scholar]
- 17. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander‐Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467‐2474. [DOI] [PubMed] [Google Scholar]
- 18. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 19. Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander‐Tetri BA; NASH Clinical Research Network (CRN) . Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology 2011;53:810‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwimmer JB, Behling C, Newbury R, Deutsch R, Nievergelt C, Schork NJ, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005;42:641‐649. [DOI] [PubMed] [Google Scholar]
- 21. Michelotti GA, Xie G, Swiderska M, Choi SS, Karaca G, Kruger L, et al. Smoothened is a master regulator of adult liver repair. J Clin Invest 2013;123:2380‐2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Halbower AC, Ishman SL, McGinley BM. Childhood obstructive sleep‐disordered breathing: a clinical update and discussion of technological innovations and challenges. Chest 2007;132:2030‐2041. [DOI] [PubMed] [Google Scholar]
- 23. Montgomery‐Downs HE, O'Brien LM, Gulliver TE, Gozal D. Polysomnographic characteristics in normal preschool and early school‐aged children. Pediatrics 2006;117:741‐753. [DOI] [PubMed] [Google Scholar]
- 24. Berry RB, Budhiraja R, Gottlieb DJ, Gozal D, Iber C, Kapur VK, et al.;American Academy of Sleep Medicine . Rules for scoring respiratory events in sleep: update of the 2007 AASM Manual for the Scoring of Sleep and Associated Events. Deliberations of the Sleep Apnea Definitions Task Force of the American Academy of Sleep Medicine. J Clin Sleep Med 2012;8:597‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412‐419. [DOI] [PubMed] [Google Scholar]
- 26. Choi SS, Omenetti A, Syn WK, Diehl AM. The role of Hedgehog signaling in fibrogenic liver repair. Int J Biochem Cell Biol 2011;43:238‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Swiderska‐Syn M, Suzuki A, Guy CD, Schwimmer JB, Abdelmalek MF, Lavine JE, et al. Hedgehog pathway and pediatric nonalcoholic fatty liver disease. Hepatology 2013;57:1814‐1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guy CD, Suzuki A, Zdanowicz M, Abdelmalek MF, Burchette J, Unalp A, et al.; NASH CRN . Hedgehog pathway activation parallels histologic severity of injury and fibrosis in human nonalcoholic fatty liver disease. Hepatology 2012;55:1711‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Onishi H, Kai M, Odate S, Iwasaki H, Morifuji Y, Ogino T, et al. Hypoxia activates the hedgehog signaling pathway in a ligand‐independent manner by upregulation of Smo transcription in pancreatic cancer. Cancer Sci 2011;102:1144‐1150. [DOI] [PubMed] [Google Scholar]
- 30. Onishi H, Morifuji Y, Kai M, Suyama K, Iwasaki H, Katano M. Hedgehog inhibitor decreases chemosensitivity to 5‐fluorouracil and gemcitabine under hypoxic conditions in pancreatic cancer. Cancer Sci 2012;103:1272‐1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Spivak‐Kroizman TR, Hostetter G, Posner R, Aziz M, Hu C, Demeure MJ, et al. Hypoxia triggers hedgehog‐mediated tumor‐stromal interactions in pancreatic cancer. Cancer Res 2013;73:3235‐3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lei J, Fan L, Wei G, Chen X, Duan W, Xu Q, et al. Gli‐1 is crucial for hypoxia‐induced epithelial‐mesenchymal transition and invasion of breast cancer. Tumour Biol 2015;36:3119‐3126. [DOI] [PubMed] [Google Scholar]
- 33. Deep G, Panigrahi GK. Hypoxia‐induced signaling promotes prostate cancer progression: exosomes role as messenger of hypoxic response in tumor microenvironment. Crit Rev Oncog 2015;20:419‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hwang JM, Weng YJ, Lin JA, Bau DT, Ko FY, Tsai FJ, et al. Hypoxia‐induced compensatory effect as related to Shh and HIF‐1alpha in ischemia embryo rat heart. Mol Cell Biochem 2008;311:179‐187. [DOI] [PubMed] [Google Scholar]
- 35. Wang G, Zhang Z, Xu Z, Yin H, Bai L, Ma Z, et al. Activation of the sonic hedgehog signaling controls human pulmonary arterial smooth muscle cell proliferation in response to hypoxia. Biochim Biophys Acta 2010;1803:1359‐1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sims JR, Lee SW, Topalkara K, Qiu J, Xu J, Zhou Z, et al. Sonic hedgehog regulates ischemia/hypoxia‐induced neural progenitor proliferation. Stroke 2009;40:3618‐3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Babior BM. Phagocytes and oxidative stress. Am J Med 2000;109:33‐44. [DOI] [PubMed] [Google Scholar]
- 38. McCord JM. Oxygen‐derived free radicals in postischemic tissue injury. N Engl J Med 1985;312:159‐163. [DOI] [PubMed] [Google Scholar]
- 39. Chen S, Zhang M, Xing L, Wang Y, Xiao Y, Wu Y. HIF‐1alpha contributes to proliferation and invasiveness of neuroblastoma cells via SHH signaling. PLoS One 2015;10:e0121115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stroka DM, Burkhardt T, Desbaillets I, Wenger RH, Neil DA, Bauer C, et al. HIF‐1 is expressed in normoxic tissue and displays an organ‐specific regulation under systemic hypoxia. FASEB J 2001;15:2445‐2453. [DOI] [PubMed] [Google Scholar]
- 41. Marxsen JH, Stengel P, Doege K, Heikkinen P, Jokilehto T, Wagner T, et al. Hypoxia‐inducible factor‐1 (HIF‐1) promotes its degradation by induction of HIF‐alpha‐prolyl‐4‐hydroxylases. Biochem J 2004;381:761‐767. [DOI] [PMC free article] [PubMed] [Google Scholar]