

Abstract
The accumulation of excess fat in the liver (hepatic steatosis) in the absence of heavy alcohol consumption causes nonalcoholic fatty liver disease (NAFLD), which has become a global epidemic. Identifying metabolic risk factors that interact with the genetic risk of NAFLD is important for reducing disease burden. We tested whether serum glucose, insulin, insulin resistance, triglyceride (TG), low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol, body mass index (BMI), and waist‐to‐hip ratio adjusted for BMI interact with genetic variants in or near the patatin‐like phospholipase domain containing 3 (PNPLA3) gene, the glucokinase regulatory protein (GCKR) gene, the neurocan/transmembrane 6 superfamily member 2 (NCAN/TM6SF2) gene, and the lysophospholipase‐like 1 (LYPLAL1) gene to exacerbate hepatic steatosis, estimated by liver attenuation. We performed association analyses in 10 population‐based cohorts separately and then meta‐analyzed results in up to 14,751 individuals (11,870 of European ancestry and 2,881 of African ancestry). We found that PNPLA3‐rs738409 significantly interacted with insulin, insulin resistance, BMI, glucose, and TG to increase hepatic steatosis in nondiabetic individuals carrying the G allele. Additionally, GCKR‐rs780094 significantly interacted with insulin, insulin resistance, and TG. Conditional analyses using the two largest European ancestry cohorts in the study showed that insulin levels accounted for most of the interaction of PNPLA3‐rs738409 with BMI, glucose, and TG in nondiabetic individuals. Insulin, PNPLA3‐rs738409, and their interaction accounted for at least 8% of the variance in hepatic steatosis in these two cohorts. Conclusion: Insulin resistance, either directly or through the resultant elevated insulin levels, more than other metabolic traits, appears to amplify the PNPLA3‐rs738409‐G genetic risk for hepatic steatosis. Improving insulin resistance in nondiabetic individuals carrying PNPLA3‐rs738409‐G may preferentially decrease hepatic steatosis.
Abbreviations
- AA
African ancestry
- AGES
Age, Gene/Environment Susceptibility‐Reykjavik
- Amish
Old Order Amish
- BMI
body mass index
- CARDIA
Coronary Artery Risk Development in Young Adults
- CI
confidence interval
- EA
European ancestry
- FamHS
Family Heart Study
- FHS
Framingham Heart Study
- GCKR
glucokinase regulatory protein
- GENOA
Genetic Epidemiology Network of Arteriopathy
- HDL
high‐density lipoprotein
- HOMA‐IR
homeostatic model of insulin resistance
- HU
Hounsfield units
- LA
liver attenuation
- LAivn
inverse normal‐transformed residuals of liver attenuation
- LDL
low‐density lipoprotein
- LYPLAL1
lysophospholipase‐like 1
- MBOAT7/TMCA
membrane bound O‐acyltransferase domain containing 7/Transmembrane channel‐like 4
- MESA
Multi‐Ethnic Study of Atherosclerosis
- MTivn
inverse normal‐transformed residuals of metabolic trait
- NAFLD
nonalcoholic fatty liver disease
- NCAN
neurocan
- OR
odds ratio
- PNPLA3
patatin‐like phospholipase domain‐containing protein 3
- SD
standard deviation
- SE
standard errors
- SNP
single nucleotide polymorphism
- TG
triglyceride
- TM6SF2
transmembrane 6 superfamily member 2
- WHRadjBMI
waist‐to‐hip ratio adjusted for body mass index
Nonalcoholic fatty liver disease (NAFLD) is a result of the excess accumulation of lipids in hepatocytes (hepatic steatosis) in the absence of heavy alcohol consumption.1 Hepatic steatosis is also associated with the risk of developing dyslipidemia or dysglycemia2 as well as cardiovascular disease, which is the number one cause of death in individuals with NAFLD.3, 4 Hepatic steatosis may progress to advanced liver disease in the form of nonalcoholic steatohepatitis, fibrosis (cirrhosis), and cancer (hepatocellular carcinoma).5, 6, 7 In the United States, the prevalence of hepatic steatosis in the adult population is between 10% and 30%; worldwide it is 25%‐45%.8 While the pathogenesis of NAFLD is not entirely understood, both genetic factors and metabolic traits increase the risk of hepatic steatosis.
Heritability of hepatic steatosis ranges from 22% to 38% across all ancestries, suggesting that specific genotypes may predispose individuals to NAFLD.1 Previously, the Genetics of Obesity‐Related Liver Disease Consortium conducted a genome‐wide association study in 7,176 individuals of European ancestry (EA), with replication in histology‐based samples.9 This study identified that rs738409 (in patatin‐like phospholipase domain containing 3 [PNPLA3]), a missense single nucleotide polymorphism (SNP) first associated with hepatic fat content a decade ago10; the missense variant rs2228603 (in neurocan/transmembrane 6 superfamily member 2 [NCAN/TM6SF2]); and intronic variants rs12137855 (in lysophospholipase‐like 1 [LYPLAL1]) and rs780094 (in glucokinase regulatory protein [GCKR]) were significantly associated with hepatic steatosis.9 We and others have replicated the association of these common variants with hepatic steatosis in other populations and ethnicities,11, 12, 13 and the associations are consistent between those of EA and African ancestry (AA) (direction of effect is similar).11 Further, the G allele for rs738409 was associated with susceptibility to nonalcoholic steatohepatitis (odds ratio [OR], 2.64; 95% confidence interval [CI], 1.85‐3.75; P < 1.0E–04), nonalcoholic steatohepatitis severity (OR, 1.85; 95% CI, 1.05‐3.26; P < 3.5E–02), and fibrosis (OR, 1.95; 95% CI, 1.17‐3.26; P < 1.3E–02) in EA individuals.14
Traits that predispose to metabolic syndrome, i.e., higher body mass index (BMI),15 dyslipidemia, hyperglycemia, and insulin resistance, are associated with hepatic steatosis.2, 3, 16 Approximately 80%‐90% of adults with obesity (BMI ≥ 30 kg/m2) have hepatic steatosis,17 while 20%‐80% of individuals with hepatic steatosis also have higher levels of triglyceride (TG) and low‐density lipoprotein (LDL) cholesterol but lower levels of high‐density lipoprotein (HDL) cholesterol.18 Diabetes is also commonly associated with hepatic steatosis.19 How these modifiable metabolic traits interact with genetic variation to influence the risk for hepatic steatosis is not known.
In this cross‐sectional study, we tested whether several metabolic traits interact with the four genetic variants associated with hepatic steatosis9 to affect liver attenuation (LA), a computed tomographic quantitative measure that is inversely related to histologically measured liver fat.20 The metabolic traits tested were insulin resistance (as homeostatic model of insulin resistance [HOMA‐IR]), fasting insulin, fasting glucose, BMI, centralized fat deposition measured by waist‐to‐hip ratio adjusted for BMI (WHRadjBMI), fasting TG, fasting HDL, and fasting LDL. We first carried out interaction analyses between each of these traits and each of the genetic variants in 10 separate population‐based cohorts from seven different studies. We then meta‐analyzed results by ancestry (EA, n = 11,870; AA, n = 2,881) and across cohorts in up to 14,751 individuals. We then carried out conditional analyses in the two largest EA cohorts in the study to determine the driving metabolic factor.
Participants and Methods
Ethics Statement
This study was approved by the Icelandic National Bioethics Committee (VSN 00‐063) and the institutional review boards or equivalent committees of all participating studies. The principal investigator of each institution obtained written consent from participants.
Study Description
The study was comprised of up to 14,751 individuals (EA, n = 11,870; AA, n = 2,881), and 56% of participants were women. The sample derived from seven population‐based studies participating in the Genetics of Obesity‐Related Liver Disease Consortium: Age, Gene/Environment Susceptibility‐Reykjavik (AGES), Old Order Amish (Amish), Coronary Artery Risk Development in Young Adults (CARDIA), Family Heart Study (FamHS), Framingham Heart Study (FHS), Genetic Epidemiology Network of Arteriopathy (GENOA), and Multi‐Ethnic Study of Atherosclerosis (MESA). In total, 10 cohorts were included in the analysis as three studies contributed two ancestry groups (AA, EA). Each ancestry group was analyzed separately. CARDIA, MESA, and AGES have unrelated individuals, while FHS, Amish, GENOA, and FamHS are family‐based studies. Detailed information about the characteristics and design of each study is provided in Supporting Table S1.
Outcome Variable and Metabolic Traits
The outcome variable was LA, measured noninvasively with computed tomography in Hounsfield units (HU).21 LA is inversely proportional to liver fat, i.e., lower LA values indicate a higher fat content in the liver (more hepatic steatosis).2 The procedures followed by each cohort to measure LA are described in Supporting Table S2. Individuals with active malignancies, focal lesions, or other incidental findings on computed tomography were excluded from the studies.
Metabolic traits of interest were harmonized across cohorts following standard clinical definitions. Overall adiposity was characterized by BMI (kg/m2) and abdominal adiposity by WHRadjBMI (cm). Because waist‐to‐hip ratio is correlated with both BMI and visceral fat, we chose to use WHRadjBMI to have a measure that is independent of overall fatness (i.e., BMI) but reflects visceral adiposity and is easily measured in the clinic. Fasting insulin (mU/L) and fasting glucose (mmol/L) were measured from plasma or serum using standard laboratory techniques detailed in Supporting Table S2. When fasting glucose was measured from whole blood, it was converted to plasma glucose using a correction factor of 1.13.22 HOMA‐IR was assessed using fasting glucose (mmol/L) × fasting insulin (mU/L) divided by 22.5.23 Each cohort assayed fasting TG (mg/dL) and fasting HDL (mg/dL), using methods described in Supporting Table S2. If fasting LDL (mg/dL) was assayed, it was used; otherwise, LDL was calculated using the Friedewald formula LDLF = (total cholesterol [mg/dL] – HDL [mg/dL] – TG [mg/dL]/5.0), only if TG <400 mg/dL.24
Alcohol consumption, history of diabetes, and use of lipid‐lowering medications were acquired by questionnaire. Total alcohol consumption, defined in drinks per week, was calculated from daily intake of beer, wine, and spirits. One drink was defined as a serving of 14 g ethanol, the same as a 12‐ounce (354.88 mL) bottle or can of beer, 5‐ounce (147.87 mL) glass of wine, or 1.5‐ounce (44.36 mL) shot of 80‐proof spirits, such as gin, vodka, or whiskey.25 Heavy drinking was defined as ≥8 drinks per week for women and ≥15 drinks per week for men.26 Diabetes (type 1, type 2) was defined as having fasting plasma glucose levels ≥7 mmol/L (126 mg/dL), or self‐reporting the use of insulin or oral antidiabetic medications, or having a physician’s diagnosis of diabetes. The use of statins was assessed from medication questionnaires.
Genotyping and Imputation
Four common variants were included in the analyses: rs738409, a missense variant in the PNPLA3 gene; rs780094, an intronic variant within the GCKR gene that is in high linkage disequilibrium (r 2 = 0.93) with rs1260326, a likely functional missense variant in this gene; rs2228603, a missense variant in the NCAN gene that is in high linkage disequilibrium (r 2 = 0.798) with rs585422926, a likely functional missense variant in the TM6SF2 gene; and rs12137855, an intronic variant in the LYPLAL1 gene. These variants were either directly genotyped (allele counts were coded 0, 1, or 2) or dosages were imputed from HapMap II or 1000G. Genotype calling algorithms and imputation methods are detailed in Supporting Table S3.
Statistical Analysis
Cohort‐Specific Analyses
Analyses were performed separately in each ancestry group (EA, AA). LA and metabolic traits, used as continuous variables in all analyses, were adjusted for sex, age, principal component estimates of ancestry, and study‐specific covariates, using linear regression as detailed in Supporting Table S2. LA was also adjusted for alcohol consumption, a continuous variable (drinks/week), and for scan penetrance using phantom or spleen density. Residuals from adjusted LA and metabolic traits were transformed using inverse normal transformation to reduce the influence of outliers and to standardize the phenotypes across cohorts. Inverse normal‐transformed residuals of LA (LAivn) and each metabolic trait (MTivn) were used to fit the interaction models.
Each cohort tested for statistical interactions between each variant and each metabolic trait, using multivariable linear regression or mixed linear modeling. LAivn was the dependent variable. The independent variables were each SNP and MTivn, plus their interaction: LAivn = α + β1(SNP) + β2(MTivn) + β3(SNP × MTivn) + є. An additive model of inheritance was assumed. Studies with family data (FHS, GENOA, Amish, and FamHS) used linear mixed models to account for family relatedness among participants and computed robust standard errors (SE). Participants with diabetes (type1 and type 2) were excluded from the insulin, glucose and HOMA‐IR models, and those taking statins were excluded from the LDL model. As a secondary analysis, BMI was included as a covariate in the models to investigate whether the effect of the interaction between each SNP and each metabolic trait on LAivn occurred independent of overall adiposity. Associations were carried out using MMAP,27 R,28 and SAS29 software.
Meta‐Analyses
We conducted fixed‐effects meta‐analyses by ancestry and overall on the parameter estimates (β coefficient and SE) for the main effects and interaction effects. We used the inverse variance weighting method implemented in METAL.30 Using Cochran’s Q test,31 we tested for heterogeneity of effects across all analyses. Within ancestries and focusing on interactions, we found evidence of heterogeneity only for the interaction between TG and GCKR in the EA cohorts. We did not find any heterogeneity for the interaction in the meta‐analyses between the two ancestry groups (EA versus AA); thus, we report the combined ancestry meta‐analyses. To determine the level of statistical significance while accounting for multiple testing, we applied a Bonferroni correction that consisted of grouping correlated traits into three metabolic domains: insulin‐glucose, adiposity, and lipids. The critical P value α = 0.05 was divided by 12 (4 variants × 3 metabolic domains) to obtain a corrected P value. Meta‐analyses results and heterogeneity tests were considered significant if P ≤ 4.17E–03 (two‐tailed). As a secondary analysis to investigate whether the statistically significant interactions were consistent between sexes, we fit the interaction models in men and women separately and meta‐analyzed results within each sex.
Conditional Analyses in FamHS and FHS
To determine whether the interaction of BMI, glucose, or TG with PNPLA3‐rs738409 was independent of insulin, we analyzed each trait’s interaction effect before and after including insulin in the model. The analyses were performed with EA individuals in FamHS and replicated in FHS. We chose these two cohorts because they are the two largest cohorts in the study; together they represent more than one third of our total sample. Individuals with diabetes and/or missing information for the metabolic traits of interest were excluded, resulting in a sample of 2,280 individuals in FamHS and 2,581 in FHS. After adjusting LA for phantom in both cohorts and for field centers in FamHS, LA residuals were transformed using inverse normal transformation to approximate normality. LAivn were used as the dependent variable. Using linear mixed models, we first regressed LAivn on either BMI, glucose, or TG and their interaction with PNPLA3‐rs738409 (Supporting Text). We then added insulin to the models and its interaction with PNPLA3‐rs738409 and the metabolic trait (either BMI, glucose, or TG). Insulin and TG were log‐transformed due to the presence of influential outliers. Models were adjusted for age, sex, and alcohol consumption (drinks/week) and for genotype batch effects in FamHS. Results from conditional analyses in each cohort were then meta‐analyzed.
The conditional models included principal components to adjust for population stratification. Because the principal components were not associated with LAivn in either cohort and their inclusion in the conditional models did not change the inferences, we present the models without them. We also performed conditional analyses after excluding individuals from FamHS (n = 231) and FHS (n = 371) who reported heavy alcohol use (≥8 drinks per week for women and ≥15 drinks per week for men) (Supporting Tables S10‐S12).26 Because the inferences were unchanged, we included all individuals to increase power and adjusted for alcohol as a covariate. Additionally, we conducted the conditional analyses with log‐transformed HOMA‐IR instead of log‐transformed insulin (Supporting Tables S13‐S15). Insulin and HOMA‐IR provided similar inferences. Because glucose explained significantly less of the variation in LAinv, we focused on insulin over HOMA‐IR because there was no added benefit of measuring glucose on variance explained by HOMA‐IR than with just measuring insulin.
Illustration in FamHS of the Interaction Between Insulin and PNPLA3‐rs738409 in Individuals Without Diabetes
To assess the interaction effect of insulin with PNPLA3‐rs738409 on hepatic steatosis prevalence in FamHS, we plotted the percentage of individuals with LA ≤60 HU per PNPLA3‐rs738409 genotype by the lowest and highest quartile of insulin. Individuals with diabetes and/or missing information for insulin were excluded and ancestries were combined to obtain a sample of n = 2,725, which was divided into quartiles of insulin. LA and insulin were not adjusted or transformed. The LA cut‐off point of ≤60 HU, which corresponds to a liver:spleen ratio of 1.1, has been shown to identify individuals with moderate to severe macrovesicular steatosis (≥30% of the liver parenchyma with fat) at histology with a high diagnostic accuracy.32 In the literature, ≥30% liver fat suggests moderate to severe hepatic steatosis.33
Results
Demographics and clinical characteristics across the study cohorts are presented in Table 1. The mean age ± standard deviation (SD) across cohorts ranged from 49.47 ± 3.86 to 76.38 ± 5.46 years old. All cohorts included more women than men. The mean ± SD of LA across cohorts ranged from 55.05 ± 12.28 HU to 65.40 ± 9.83 HU. In those without diabetes, mean ± SD of fasting insulin levels ranged from 8.30 ± 5.73 to 13.02 ± 10.22 mU/L and fasting blood glucose levels ranged from 4.90 ± 0.58 to 5.49 ± 0.50 mmol/L. The lowest mean ± SD for HOMA‐IR in those without diabetes was 1.99 ± 1.27 and the highest was 3.14 ± 2.69. The mean ± SD of BMI ranged from 27.00 ± 4.49 to 32.71 ± 7.37 kg/m2. Several cohorts reported mean fasting TG >100 mg/dL. Mean ± SD for fasting LDL cholesterol in nonstatin users was borderline high in Amish (141.31 ± 38.66 mg/dL) and AGES (146.84 ± 35.73 mg/dL). Across cohorts, the range of fasting HDL cholesterol was within the recommended limit of ≥40 mg/dL. Heavy drinking varied among studies, with GENOA having the lowest percentage (0%) and CARDIA the highest (37%).
Table 1.
Demographic and Characteristics of Study Participants in Each Cohort by Ancestry
| Demographic | AGES | Amish | CARDIA | FamHS | FHS | MESA | CARDIA | FamHS | GENOA | MESA |
|---|---|---|---|---|---|---|---|---|---|---|
| European Ancestry (11,870) | African Ancestry (2,881) | |||||||||
| N = 14,751 | 2,865 | 541 | 1,282 | 2,684 | 2,966 | 1,532 | 642 | 620 | 560 | 1,059 |
| Age | 76.38 ± 5.46 | 56.84 ± 12.81 | 50.74 ± 3.33 | 57.14 ± 13.28 | 50.54 ± 10.14 | 63.05 ± 10.49 | 49.47 ± 3.86 | 53.35 ± 10.82 | 68.86 ± 8.01 | 63.17 ± 10.00 |
| Men (6,444) | 1,139 (40%) | 252 (47%) | 595 (46%) | 1,207 (45%) | 1,454 (49%) | 746 (49%) | 233 (36%) | 212 (34%) | 141 (25%) | 465 (44%) |
| Women (8,307) | 1,726 (60%) | 289 (53%) | 687 (54%) | 1,477 (55%) | 1,512 (51%) | 786 (51%) | 409 (64%) | 408 (66%) | 419 (75%) | 594 (56%) |
| Characteristics | ||||||||||
| Liver attenuation (HU) || | 59.22 ± 8.64 | 63.05 ± 7.76 | 55.05 ± 12.28 | 59.14 ± 11.19 | 65.40 ± 9.83 | 59.33 ± 12.43 | 56.38 ± 10.86 | 59.52 ± 9.23 | 60.10 ± 9.39 | 61.18 ± 9.06 |
| Insulin (mU/L) | 9.22 ± 6.39 | 11.75 ± 6.22 | 9.26 ± 6.77 | 9.88 ± 7.16 | 9.05 ± 7.38 | 8.90 ± 4.94 | 11.60 ± 8.29 | 13.02 ± 10.22 | 8.30 ± 5.73 | 9.61 ± 5.53 |
| HOMA‐IR‡ | 2.31 ± 1.76 | 2.69 ± 1.63 | 2.20 ± 1.77 | 2.37 ± 1.87 | 2.32 ± 2.31 | 1.99 ± 1.27 | 2.75 ± 2.14 | 3.14 ± 2.69 | 2.02 ± 1.46 | 2.19 ± 1.41 |
| Glucose (mmol/L) | 5.49 ± 0.50 | 4.94 ± 0.52 | 5.18 ± 0.50 | 5.25 ± 0.53 | 5.47 ± 1.12 | 4.90 ± 0.58 | 5.17 ± 0.54 | 5.25 ± 0.57 | 5.37 ± 0.50 | 5.03 ± 0.59 |
| BMI (kg/m2) | 27.00 ± 4.49 | 27.72 ± 4.85 | 28.50 ± 6.18 | 28.86 ± 5.69 | 27.51 ± 5.22 | 28.06 ± 5.05 | 31.94 ± 7.48 | 32.71 ± 7.37 | 32.71 ± 7.27 | 29.95 ± 5.77 |
| Obese† | 618 (22%) | 155 (29%) | 425 (33%) | 972 (36%) | 769 (26%) | 449 (29%) | 352 (55%) | 377 (61%) | 332 (59%) | 465 (44%) |
| WHR (cm)§ | nval | 0.87 ± 0.07 | 0.85 ± 0.10 | 0.91 ± 0.10 | 0.94 ± 0.08 | 0.93 ± 0.09 | 0.85 ± 0.08 | 0.92 ± 0.07 | 0.89 ± 0.08 | 0.92 ± 0.08 |
| TG (mg/dL) | 106.48 ± 59.06 | 90.42 ± 57.45 | 121.64 ± 85.07 | 144.03 ± 94.05 | 126.11 ± 88.07 | 136.55 ± 99.31 | 101.55 ± 73.24 | 111.82 ± 80.09 | 100.28 ± 62.67 | 103.82 ± 60.61 |
| LDL (mg/dL) | 146.84 ± 35.73 | 141.31 ± 38.66 | 116.27 ± 30.15 | 112.90 ± 34.22 | 117.70 ± 31.71 | 120.24 ± 30.42 | 112.59 ± 33.83 | 115.39 ± 36.05 | 123.85 ± 33.59 | 118.39 ± 32.87 |
| HDL (mg/dL) | 61.75 ± 17.31 | 57.05 ± 15.37 | 58.43 ± 18.42 | 48.82 ± 14.37 | 54.16 ± 16.77 | 51.68 ± 15.59 | 57.59 ± 16.70 | 53.55 ± 15.41 | 57.31 ± 16.52 | 52.39 ± 15.14 |
| Alcohol (drinks/week) | 1.09 ± 2.37 | nval | 5.73 ± 10.07 | 2.98 ± 7.10 | 5.39 ± 7.88 | 5.06 ± 8.40 | 3.86 ± 10.60 | 3.24 ± 9.45 | 0.28 ± 1.18 | 3.86 ± 8.89 |
| Heavy drinkers* | 17 (0.59%) | nval | 470 (37%) | 152 (6%) | 424 (14.3%) | 335 (22%) | 144 (22%) | 69 (11%) | 0 | 139 (13%) |
Statistics are presented as mean ± SD or as n (%). The table shows summary statistics for individuals with LA and genetic information from each cohort who were included in analyses. LA and metabolic traits were not adjusted for covariates. The sample size for each metabolic trait varied from N depending on the data available. Summary statistics for fasting insulin, HOMA‐IR, and fasting glucose excludes individuals with diabetes; fasting LDL excludes statin users.
Defined as ≥8 drinks per week for women and ≥15 drinks per week for men; the Amish do not consume alcohol;
Defined as BMI ≥30 kg/m2;
calculated as (fasting insulin [mU/L] × fasting glucose [mmol/L]/22.5);
not adjusted for BMI; ||Raw LA measured in Hounsfield units.
Abbreviations: nval, not available in cohort; SD, standard deviation.
PNPLA3‐rs738409 and GCKR‐rs780094 Interact With Several Metabolic Traits
We found significant interactions for PNPLA3‐rs738409 and GCKR‐rs780094 with several metabolic traits in combined ancestries after adjusting for multiple comparisons (Table 2; Supporting Table S4). PNPLA3‐rs738409 interacted with insulin (P = 4.79E–14), HOMA‐IR (P = 4.68E–15), glucose (P = 1.26E–03), BMI (P = 8.13E–08), and TG (P = 2.95E–03). As each of these metabolic traits increased, a decrease in LAivn (i.e., higher fat content in the liver) became more pronounced in the presence of the G allele at PNPLA3‐rs738409 compared to the presence of the C allele. Additionally, GCKR‐rs780094 interacted with insulin (P = 4.57E–04), HOMA‐IR (P = 1.32E–03), and TG (P = 4.17E–03). As levels of insulin, HOMA‐IR, and TG increased, a decrease in LAivn (i.e., higher fat content in the liver) became more pronounced in the presence of the T allele at GCKR‐rs780094 compared to the C allele. All interactions remained significant after adjusting for BMI (Supporting Table S5), suggesting that overall adiposity did not alter these effects. We did not find evidence of significant interactions between any of the four genetic variants and WHRadjBMI, LDL, or HDL. Although the interaction between WHRadjBMI and PNPLA3‐rs738409 did not reach the Bonferroni significance level, it was borderline significant. This suggests that a larger sample size may be needed to detect an interaction. Alternatively, the lack of statistical significance could be because WHRadjBMI does not represent overall fatness to the extent that BMI or other anthropometric measurements do.
Table 2.
Meta‐Analyses Results for Interactions Between Four SNPs and Inverse Normal‐Transformed Residuals of Metabolic Traits on LAivn in Combined Ancestries
| Metabolic traits | rs738409 | rs780094* | rs2228603† | rs12137855 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Chr | Alleles (Ref/O) | Ref AF | Gene | Chr | Alleles (Ref/O) | Ref AF | Gene | Chr | Alleles (Ref/O) | Ref AF | Gene | Chr | Alleles (Ref/O) | Ref AF | |
| PNPLA3 | 22 | G/C | 0.22 | GCKR | 2 | T/C | 0.38 | NCAN/TM6SF2 | 19 | T/C | 0.09 | LYPLAL1 | 8 | C/T | 0.79 | |
| (SNP × Metabolic Traits) | ||||||||||||||||
| βint | SE | P Value | n | βint | SE | P Value | n | βint | SE | P Value | n | βint | SE | P Value | n | |
| Insulin | –0.11 | 0.02 | 4.79E–14 | 12,651 | –0.04 | 0.01 | 4.57E–04 | 12,651 | –0.06 | 0.03 | 4.37E–02 | 12,651 | –0.02 | 0.02 | 1.55E–01 | 12,651 |
| HOMA‐IR | –0.12 | 0.02 | 4.68E–15 | 12,554 | –0.04 | 0.01 | 1.32E–03 | 12,554 | –0.06 | 0.03 | 3.63E–02 | 12,554 | –0.02 | 0.02 | 1.38E–01 | 12,554 |
| Glucose | –0.05 | 0.02 | 1.26E–03 | 12,742 | –0.01 | 0.01 | 4.37E–01 | 12,742 | –0.06 | 0.03 | 7.41E–02 | 12,742 | –0.02 | 0.02 | 1.91E–01 | 12,742 |
| BMI | –0.08 | 0.01 | 8.13E–08 | 14,693 | –0.03 | 0.01 | 6.31E–03 | 14,693 | –0.05 | 0.03 | 5.89E–02 | 14,693 | –0.02 | 0.01 | 8.18E–01 | 14,693 |
| WHRadjBMI | –0.05 | 0.02 | 7.59E–03 | 10,051 | –0.04 | 0.02 | 1.32E–02 | 10,051 | –0.08 | 0.03 | 1.26E–02 | 10,051 | 0.01 | 0.02 | 7.76E–01 | 10,051 |
| TG | –0.05 | 0.02 | 2.95E–03 | 14,551 | –0.04 | 0.01 | 4.17E–03 | 14,551 | 0.00 | 0.03 | 9.77E–01 | 14,551 | –0.03 | 0.02 | 5.75E–02 | 14,551 |
| LDL | 0.00 | 0.02 | 7.94E–01 | 12,123 | 0.00 | 0.01 | 9.33E–01 | 12,123 | –0.06 | 0.03 | 5.50E–02 | 12,123 | 0.02 | 0.02 | 2.29E–01 | 12,123 |
| HDL | 0.04 | 0.02 | 1.41E–02 | 14,543 | 0.03 | 0.01 | 3.72E–02 | 14,543 | 0.00 | 0.03 | 9.55E–01 | 14,543 | –0.01 | 0.02 | 3.72E–01 | 14,543 |
P ≤ 4.17E–03 is considered significant; n is the highest sample size in meta‐analyses.
rs780094 is in LD (r 2 = 0.93) with rs1260326, a functional missense variant in GCKR;
rs2228603 is in LD (r 2 = 0.79) with rs58542926, a functional missense variant in TM6SF2.
Abbreviations: βint, interaction effect size; Chr, chromosome; LD, linkage disequilibrium; Ref AF, reference allele frequency; Ref/O, reference/other allele (reference allele is the effect allele of each SNP); SE, standard error; SNP, single nucleotide polymorphism.
We also carried out meta‐analyses in men and women separately to investigate possible sex differences, focusing only on the statistically significant interactions with PNPLA3‐rs738409 and GCKR‐rs780094 (Supporting Table S6). Women made up 56% of our study sample. The interaction effects of insulin and HOMA‐IR with PNPLA3‐rs738409 did not differ between men and women, and both reached statistical significance (for insulin, women, P = 3.24E–11; men, 7.24E–05; for HOMA‐IR, women, P = 1.62E–11; men, P = 2.88E–05). For glucose, the interaction effect was slightly less in men than in women (smaller beta) and did not reach significance in men. These results suggest that sex did not alter the interactions between PNPLA3‐rs738409 and insulin/HOMA‐IR and that the interaction effect of glucose was still present only in women in the present study. Further, the interaction effects of BMI with PNPLA3‐rs738409 were similar between men and women and reached significance in both (P = 1.20E–03 and P = 3.39E–05, respectively). The interaction effect of TG with PNPLA3‐rs738409 did not reach statistical significance in either sex. Moreover, the interaction effects of both insulin and HOMA‐IR with GCKR‐rs780094 reached significance only in women (P = 1.02E–03 and P = 6.46E–04, respectively). Similarly, the interaction of TG with GCKR‐rs780094 was significant only in women (P = 8.71E–04). Stratifying by sex substantially reduced our sample size and as a result, power.
Conditional Analyses Suggest that Insulin May Mediate the Interaction Effect of BMI, TG, and Glucose on LAivn in Individuals Without Diabetes
We observed that the interaction of insulin with PNPLA3‐rs738409 had a greater effect on LAivn (hepatic steatosis) than that of BMI, TG, or glucose. To determine if the interaction of BMI, TG, or glucose with PNPLA3‐rs738409 was independent of insulin, we carried out conditional analyses in FamHS and FHS and meta‐analyzed results. We found that the interaction of BMI (P = 7.57E–02), TG (P = 3.49E–01), or glucose (P = 9.09E–01) with PNPLA3‐rs738409 was no longer statistically significant after including insulin as a main effect and interactor with PNPLA3‐rs738409 and the respective metabolic trait in the models (Supporting Tables S7‐S9). In contrast, the interaction of insulin with PNPLA3‐rs738409 remained significant after controlling for BMI, TG, or glucose (P insulin–BMI = 4.04E–04; P insulin–TG = 3.24E–06; P insulin–glucose = 8.40E–08), although the effect sizes and P values were attenuated. These results suggest that insulin may account for most of the interaction effect of BMI, glucose, and TG with PNPLA3‐rs738409 on LAivn. Previously, we reported that PNPLA3‐rs738409 explained 2.4% of the variance in hepatic steatosis, estimated by LA, in EA individuals.11 In the present study, PNPLA3‐rs738409, insulin, and their interaction together explain as much as 8% of the variance in hepatic steatosis in the two largest EA cohorts, excluding individuals with diagnosed diabetes. This suggests that insulin levels/insulin resistance may be a key contributor to NAFLD. Excluding heavy drinkers from the conditional analyses did not change our inferences regarding PNPLA3‐rs738409 (Supporting Tables S10‐S12). We were not powered to carry out these analyses for GCKR‐rs780094.
Interaction Effect of Insulin With PNPLA3 on Hepatic Steatosis Prevalence in FamHS
We assessed the interaction effect of insulin with PNPLA3‐rs738409 on hepatic steatosis prevalence in individuals without diabetes (Fig. 1). In the lowest quartile of insulin levels (≤5.20 mU/L), the percentage of individuals with ≥30% liver fat (i.e., moderate to severe hepatic steatosis) was 23.42%, 35.81%, and 39.47% for CC, CG, and GG individuals, respectively. In the highest quartile of insulin levels (≥13.06 mU/L), the percentage of individuals with ≥30% liver fat was 54.44%, 76.32%, and 94.29% for CC, CG, and GG individuals, respectively. The data show that as insulin levels increase, the percentage of individuals with moderate to severe hepatic steatosis increases. However, among those with the GG genotype, this effect is magnified. The difference in the percentage of individuals with moderate to severe hepatic steatosis increases by 55 percentage points between the lowest and highest insulin quartiles among those with the GG genotype and increases by 41 percentage points among heterozygotes, while that difference increases only by 31 percentage points among those with the CC genotype. These data suggest that insulin has a strong effect on exacerbating the accumulation of liver fat in individuals without diabetes who have one or two G alleles at PNPLA3‐rs738409.
Figure 1.
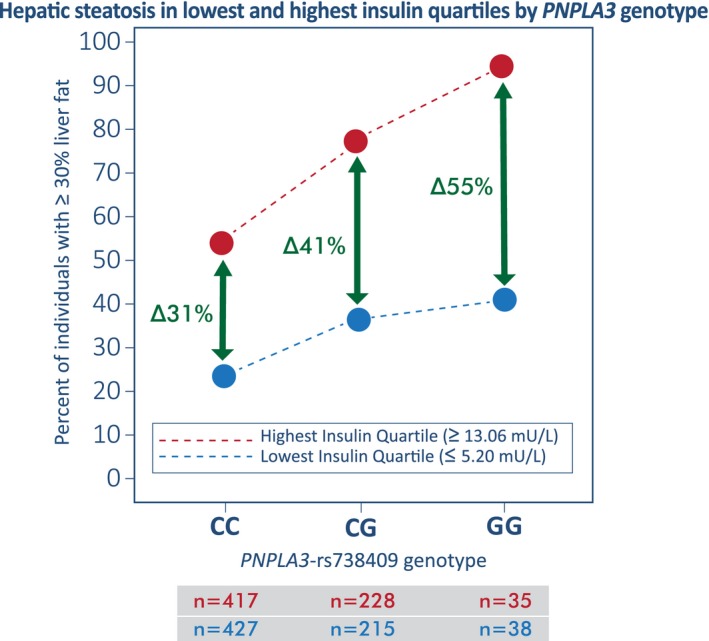
Percentage of individuals without diabetes in FamHS with ≥30% fat in the liver (moderate to severe hepatic steatosis) is shown per PNPLA3‐rs738409 genotype in the lowest and highest quartile of insulin levels. The number of individuals (n) in the lowest (blue circle) and highest (red circle) insulin quartiles are shown by genotype. As the level of insulin increases, the percentage of individuals with ≥30% fat in the liver increases more markedly with increasing copies of the G risk allele (nonparallel lines show interaction). Among those with the GG genotype, the difference (∆) in the percentage of individuals with moderate to severe liver fat increases by 55 percentage points between the lowest and highest insulin quartiles. In contrast, this difference is lower among those with the CG genotype (41%) and CC genotype (31%).
Discussion
In a sample of 14,751 EA and AA individuals, we found interactions between PNPLA3‐rs738409 and insulin, HOMA‐IR, BMI, glucose, and TG on LAinv (hepatic steatosis) after adjusting for differences in age, sex, and alcohol consumption. We also found interactions between GCKR‐rs780094 and insulin, HOMA‐IR, and TG on LAinv. Conditional analyses in more than 5,000 EA individuals suggest that insulin, more than glucose, BMI, or TG, drives the interaction with PNPLA3‐rs738409 to affect LAinv in those without diabetes. We did not see significant interactions between PNPLA3‐rs738409 and BMI, TG, or glucose once insulin was accounted for, whereas the reverse was not true. That is, there was still evidence for an interaction between PNPLA3‐rs738409 and insulin even after accounting for the other metabolic traits. These results persist after accounting for alcohol intake, sex, and overall adiposity. We estimated in FamHS and FHS that as much as 8% of the variance in hepatic steatosis is explained by PNLPA3‐rs738409, insulin, and their interaction in EA individuals without diabetes. In our previous study, PNPLA3‐rs738409 alone explained only 2.4% of hepatic steatosis variance in EA individuals.11
Our findings suggest that individuals without diabetes with PNPLA3‐rs738409‐G and high insulin levels may have a particularly high risk for hepatic steatosis. The PNPLA3 gene encodes adiponutrin, an enzyme found on the membrane of lipid droplets within hepatocytes.34 Its function may be to break down TG stored in the droplets, helping regulate hepatic TG content.34, 35 The missense polymorphism rs738409 (C>G) in PNPLA3 substitutes the amino acid isoleucine for methionine at residue 148 (I148M), changing the configuration of adiponutrin’s catalytic site and rendering the enzyme inactive.10, 36 The accumulation of the inactive enzyme on lipid droplets is associated with TG buildup in hepatocytes.36 Humans and mice carrying one or two copies of the I148M mutation (rs738409 CG or GG genotype) accumulate excess TG in lipid droplets and show more pronounced hepatic steatosis and NAFLD than those without the mutation.35, 36
It is possible that having high insulin levels in addition to the PNPLA3‐rs738409‐G allele may result in a strong synergistic effect that exacerbates the accumulation of fat in the liver of individuals without diabetes, predisposing them to NAFLD. Insulin resistance stimulates the hydrolysis of TG in adipose tissue, releasing fatty acids in the bloodstream, which are taken up by the liver in an unregulated manner, promoting the accumulation of TG in hepatocytes.37 Higher insulin levels also activate fatty acid synthesis in the liver, further driving the formation and storage of TG.34 In addition, insulin resistance elevates plasma glucose, which is sequestered by the liver, phosphorylated, and metabolized to make glycerol and acetyl‐coenzyme A, the building blocks for the synthesis of TG.34, 38 In this context, it is possible that increased lipid synthesis and fatty acid delivery to the liver may combine with the inability of hepatocytes to dispose of TG from lipid droplets due to the presence of PNPLA3‐rs738408‐G and lead to increased hepatic steatosis. High insulin levels and PNPLA3‐rs738409‐G may also be involved in molecular feedback loops that increase hepatic steatosis. Insulin resistance and increased insulin levels augment the activity of transcription factors, such as sterol regulatory element binding protein 1c.39 These transcription factors may promote TG synthesis in the liver and up‐regulate the expression of PNPLA3 I148M by binding to its promoter in a positive‐feedback loop.39 In this way, insulin and PNPLA3 I148M may synergize to promote hepatic steatosis. This conjecture is also consistent with the enhanced risk of steatosis and liver damage, as evident by elevated liver enzymes and liver fat content seen with liver‐directed long‐acting insulin analogues in type 2 diabetics carrying the PNPLA3 variant.40
When taken together, results show evidence that insulin and PNPLA3‐rs738409 interact to have an important role in hepatic steatosis and as a result, NAFLD. Consequently, lowering the risk of hepatic steatosis and its liver complications in individuals with PNPLA3‐rs738409‐G may be achieved by reducing insulin resistance and concomitant high levels of insulin. One way to accomplish this could be through lifestyle changes that include increased exercise, weight loss, and better nutrition.41 For example, decreasing exposure to carbohydrate‐rich diets, which adversely increase insulin levels, may mitigate risk.42, 43 Also, treatments that target insulin resistance may be of greater benefit for preventing or treating hepatic steatosis than drugs that simply lower glucose. For example, insulin‐sensitizing medications, such as pioglitazone, may be an option; it has already been shown to improve NAFLD, although at the expense of weight gain.44 More studies are warranted to better understand the effect of the relationship between insulin levels and PNPLA3‐rs738409‐G on hepatic steatosis in different populations.
We also observed significant interactions of PNPLA3‐rs738409 with BMI, glucose, and TG. Our results support the findings of Stender et al.45 who reported that high BMI augmented the effect of PNPLA3‐rs738409‐G on hepatic steatosis, conferring susceptibility to NAFLD. Graff et al. 46 also showed an interaction effect between PNPLA3‐rs738409 and visceral fat content, a measure of metabolic dysfunction. However, we found that the effect of BMI in exacerbating hepatic steatosis in the presence of PNPLA3‐rs738409‐G is attenuated by controlling for insulin levels in the model. We made the same observation for glucose and TG, suggesting that insulin/insulin resistance in the presence of PNPLA3‐rs738409‐G may confer most of the risk for hepatic steatosis on its own or through other metabolic intermediates.
Studies have reported an association between LDL and hepatic steatosis.47, 48 However, our study did not find an interaction between any of the genetic variants considered and LDL. This suggests that for individuals carrying PNPLA3‐rs738409‐G, reducing insulin levels or insulin resistance may have a greater effect on reducing the risk of hepatic steatosis than reducing LDL.
In addition to PNPLA3, we found that GCKR interacts with insulin resistance to increase susceptibility to hepatic steatosis. GCKR encodes the glucokinase regulatory protein, which has an important role in glucose metabolism.49 The glucokinase regulatory protein binds to the glucose metabolizing enzyme glucokinase to inhibit its role in the uptake and storage of dietary glucose through stimulating de novo lipogenesis.49 The variant rs780094/rs12060326 in the glucokinase regulatory protein reduces its ability to inhibit glucokinase.49 This results in increased activity of glucokinase in the liver, which promotes de novo lipogenesis. When this mutation is combined with insulin resistance, it may amplify de novo lipogenesis to promote hepatic steatosis. We did not replicate the interaction between TM6SF2 and BMI reported by Stender et al.45; however, our results show a similar trend. The interaction was borderline nonsignificant in the combined ancestry meta‐analyses (interaction effect size, βint = –0.05; P = 5.89E–02). Some differences between Stender et al. and this study may explain why we did not detect a statistically significant interaction. First, Stender et al. used proton magnetic resonance spectrometry to measure steatosis, which is a more sensitive measure than computed tomography. Second, they used the genotyped missense variant rs58542926; we used the proxy imputed variant rs2228603. The two variants are in high‐linkage disequilibrium (D’ = 0.926, r 2 = 0.798). Third, Stender et al. combined the heterozygotes (EK) and homozygotes (KK) and compared them to those without the risk allele (EE). These three differences may have increased their power to see the weak effect they reported.
Our study has several limitations. It is a cross‐sectional design that cannot prove temporal causality of insulin exposure on increasing hepatic steatosis. Because we used population‐based cohorts that lacked biopsy information, we do not know whether we included individuals with advanced stages of NAFLD, such as nonalcoholic steatohepatitis, fibrosis, or cirrhosis. We also could not differentiate peripheral insulin resistance from hepatic insulin resistance with our data. Moreover, even though HOMA‐IR was highly correlated to a single value of insulin (r 2 = 0.98) in individuals who were euglycemic, we do not have direct measures of dynamic glucose regulation. Therefore, functional studies are needed to gain more insight into the biological processes driving our observations. Finally, our study did not include the missense variant, rs641738, at the membrane bound O‐acyltransferase domain containing 7/Transmembrane channel‐like 4 (MBOAT7/TMC4) locus. It has been associated with hepatic fat accumulation.50 In our prior association analyses,11 we did not see an association between MBOAT7/TMC4 and LA (β = –0.03; P = 0.15). Because our inclusion criterion for variants was that they needed to be associated with LA and we could not substantiate the association of MBOAT7/TMC4 in our sample, we excluded it.
In conclusion, to our knowledge, this is the largest study examining the interaction between multiple metabolic traits and four genetic variants on hepatic steatosis in multiple cohorts representing two different ancestry groups. Our findings suggest that insulin levels/insulin resistance more than other correlated metabolic traits, including glucose, TG, and BMI, interact with genetic variants in PNPLA3 to promote hepatic steatosis. Through conditional analyses, we show that insulin levels explain the interactions observed between PNPLA3‐rs738409 and BMI as well as the interactions between PNPLA3‐rs738409 and glucose and TG in almost 5,000 EA individuals without diabetes. Our work suggests that improving insulin resistance and reducing insulin levels in individuals who are prediabetic and who carry fatty liver‐promoting alleles at PNPLA3‐rs738409 may offer preferential benefit and mitigate their risk of developing NAFLD. Although PNPLA3 genotype information is not currently used to make clinical decisions, it may be helpful in the future not only to risk stratify individuals but also to tailor their treatment. Our work contributes to the understanding of the pathophysiology of NAFLD and informs further interventional research to better diagnose and/or treat individuals with increased risk of NAFLD.
Supporting information
Acknowledgment
We are indebted to the participants for their willingness to participate in the study. The Amish study gratefully acknowledges our Amish liaisons, research volunteers, field workers, and Amish Research Clinic staff and the extraordinary cooperation and support of the Amish community without which these studies would not have been possible. Data from the FHS came from the database of Genotypes and Phenotypes. This manuscript has been reviewed by CARDIA for scientific content. We thank all anonymous reviewers for their helpful suggestions, which have improved this paper. We also thank Allison Picinotti from the University of Michigan for her graphic design contributions. This work was performed under the auspices of the Genetics of Obesity‐Related Liver Disease Consortium.
Supported by the National Institutes of Health (NIH) through the Age, Gene/Environment Susceptibility‐Reykjavik study (contracts N01‐AG‐1‐2100 and 271201200022C), the National Institute of Aging (NIA) Intramural Research Program, Hjartavernd (the Icelandic Heart Association), and the Althingi (the Icelandic Parliament). The Amish studies are supported by grants and contracts from NIH (including U01 HL072515, U01 HL84756, U01 HL137181, and P30 DK72488). Funding for the Coronary Artery Risk Development in Young Adults Study (CARDIA) is supported by the National Heart, Lung, and Blood Institute (NHLBI) in collaboration with the University of Alabama at Birmingham (HHSN268201300025C and HHSN268201300026C), Northwestern University (HHSN268201300027C), University of Minnesota (HHSN268201300028C), Kaiser Foundation Research Institute (HHSN268201300029C), Johns Hopkins University School of Medicine (HHSN268200900041C), and Vanderbilt University Medical Center (R01 HL 098445). CARDIA is also partially supported by the Intramural Research Program of NIA and an intra‐agency agreement between NIA and NHLBI (AG0005). The National Human Genome Research Institute (NHGRI) supported genotyping of CARDIA participants (grants U01‐HG‐004729, U01‐HG‐004446, and U01‐HG‐004424). The Family Heart Study (FamHS) was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (grant R01‐DK‐089256) and NHLBI (grant R01HL117078). L.B.VW is supported by the NIH (grant K23 HL136891), and E.K.S and B.H. are supported by the NIH (grants R01 DK106621, R01 DK107904) and the University of Michigan Department of Internal Medicine. L.F.B. and P.A.P. are supported, in part, by NIH grants R01 DK106621 and R01 DK107904. Support for the Genetic Epidemiology Network of Arteriopathy (GENOA) study was provided by NHLBI (HL054457, HL054464, HL054481, HL087660, and HL085571). The Multi‐Ethnic Study of Atherosclerosis (MESA) and the MESA SHARe project are conducted and supported by NHLBI in collaboration with MESA investigators. This research was supported by R01 HL071739, and MESA was supported by contracts HHSN268201500003I, N01‐HC‐95159, N01‐HC‐95160, N01‐HC‐95161, N01‐HC‐95162, N01‐HC‐95163, N01‐HC‐95164, N01‐HC‐95165, N01‐HC‐95166, N01‐HC‐95167, N01‐HC‐95168, and N01‐HC‐95169 from the NHLBI and by grants UL1‐TR‐000040, UL1‐TR‐001079, and UL1‐TR‐001420 from the National Center for Research Resources. The provision of exome chip genotyping data was supported in part by the NHLBI (contract N02‐HL‐64278), National Center for Advancing Translational Sciences, (CTSI grant UL1TR001881), and the NIDDK Diabetes Research Center (DRC) (grant DK063491 to the Southern California Diabetes Endocrinology Research Center). Funding support for MESA's NAFLD dataset was provided by the NHLBI (grant HL071739‐05A2).
The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the NHLBI, NIA, NIDDK or NIH.
Potential conflict of interest: Dr. Yerges‐Armstrong is employed by and owns stock in GlaxoSmithKline. Dr. Ingrid B. Borecki owns stock in Regeneron Pharmaceuticals. Dr. Jeffrey R. O’Connell was a consultant for Regeneron Pharmaceuticals for a period of time during this study. The other authors have nothing to report.
Contributor Information
Llilda Barata, Email: barata@wustl.edu.
Elizabeth K. Speliotes, Email: espeliot@med.umich.edu
Michael A. Province, Email: mprovince@wustl.edu
References
Author names in bold designate shared co‐first authorship.
- 1. Kahali B, Halligan B, Speliotes EK. Insights from genome‐wide association analyses of nonalcoholic fatty liver disease. Semin Liver Dis 2015;35:375‐391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Speliotes EK, Massaro JM, Hoffmann U, Vasan RS, Meigs JB, Sahani DV, et al. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: the Framingham Heart Study. Hepatology 2010;51:1979‐1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non‐alcoholic fatty liver disease (NAFLD) and its connection with insulin resistance, dyslipidemia, atherosclerosis and coronary heart disease. Nutrients 2013;5:1544‐1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hassan K, Bhalla V, El Regal ME, A‐Kader HH . Nonalcoholic fatty liver disease: a comprehensive review of a growing epidemic. World J Gastroenterol 2014;20:12082‐12101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing‐steatohepatitis using paired biopsies: implications for prognosis and clinical management. J Hepatol 2015;62:1148‐1155. [DOI] [PubMed] [Google Scholar]
- 6. Pais R, Charlotte F, Fedchuk L, Bedossa P, Lebray P, Poynard T, et al.; LIDO Study Group . A systematic review of follow‐up biopsies reveals disease progression in patients with non‐alcoholic fatty liver. J Hepatol 2013;59:550‐556. [DOI] [PubMed] [Google Scholar]
- 7. Wong VW, Wong GL, Choi PC, Chan AW, Li MK, Chan HY, et al. Disease progression of non‐alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut 2010;59:969‐974. [DOI] [PubMed] [Google Scholar]
- 8. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐Meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 9. Speliotes EK, Yerges‐Armstrong LM, Wu J, Hernaez R, Kim LJ, Palmer CD, et al.; NASH CRN; GIANT Consortium; MAGIC Investigators; GOLD Consortium . Genome‐wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet 2011;7:e1001324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Palmer ND, Musani SK, Yerges‐Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, et al. Characterization of European ancestry nonalcoholic fatty liver disease‐associated variants in individuals of African and Hispanic descent. Hepatology 2013;58:966‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hernaez R, McLean J, Lazo M, Brancati FL, Hirschhorn JN, Borecki IB, et al.; Genetics of Obesity‐Related Liver Disease (GOLD) Consortium . Association between variants in or near PNPLA3, GCKR, and PPP1R3B with ultrasound‐defined steatosis based on data from the third National Health and Nutrition Examination Survey. Clin Gastroenterol Hepatol 2013;11:1183‐1190.e1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin YC, Chang PF, Chang MH, Ni YH. Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am J Clin Nutr 2014;99:869‐874. [DOI] [PubMed] [Google Scholar]
- 14. Liu YL, Patman GL, Leathart JB, Piguet AC, Burt AD, Dufour JF, et al. Carriage of the PNPLA3 rs738409 C >G polymorphism confers an increased risk of non‐alcoholic fatty liver disease associated hepatocellular carcinoma. J Hepatol 2014;61:75‐81. [DOI] [PubMed] [Google Scholar]
- 15. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al.; ADIPOGen Consortium; AGEN‐BMI Working Group; CARDIOGRAMplusC4D Consortium; CKDGen Consortium; GLGC; ICBP; MAGIC Investigators; MuTHER Consortium; MIGen Consortium; PAGE Consortium; ReproGen Consortium; GENIE Consortium; International Endogene Consortium . Genetic studies of body mass index yield new insights for obesity biology. Nature 2015;518:197‐206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chatrath H, Vuppalanchi R, Chalasani N. Dyslipidemia in patients with nonalcoholic fatty liver disease. Semin Liver Dis 2012;32:22‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non‐alcoholic fatty liver disease. Dig Dis 2010;28:155‐161. [DOI] [PubMed] [Google Scholar]
- 18. Zhang QQ, Lu LG. Nonalcoholic fatty liver disease: dyslipidemia, risk for cardiovascular complications, and treatment strategy. J Clin Transl Hepatol 2015;3:78‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petaja EM, Yki‐Jarvinen H. Definitions of normal liver fat and the association of insulin sensitivity with acquired and genetic NAFLD‐a systematic review. Int J Mol Sci 2016;17:pii.E633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Limanond P, Raman SS, Lassman C, Sayre J, Ghobrial RM, Busuttil RW, et al. Macrovesicular hepatic steatosis in living related liver donors: correlation between CT and histologic findings. Radiology 2004;230:276‐280. [DOI] [PubMed] [Google Scholar]
- 21. Graffy PM, Pickhardt PJ. Quantification of hepatic and visceral fat by CT and MR imaging: relevance to the obesity epidemic, metabolic syndrome and NAFLD. Br J Radiol 2016;89:20151024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. D’Orazio P, Burnett RW, Fogh‐Andersen N, Jacobs E, Kuwa K, Kulpmann WR, et al.; International Federation of Clinical Chemistry Scientific Division Working Group on Selective Electrodes and Point of Care Testing . Approved IFCC recommendation on reporting results for blood glucose (abbreviated). Clin Chem 2005;51:1573‐1576. [DOI] [PubMed] [Google Scholar]
- 23. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985;28:412‐419. [DOI] [PubMed] [Google Scholar]
- 24. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low‐density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972;18:499‐502. [PubMed] [Google Scholar]
- 25. National Institute of Alcohol Abuse and Alcoholism . What is a standard drink? https://www.niaaa.nih.gov/alcohol-health/overview-alcohol-consumption/what-standard-drink. Accessed March, 2016. [Google Scholar]
- 26. U.S. Department of Agriculture. 2015–2020 Dietary Guidelines . Appendix 9. Alcohol. https://health.gov/dietaryguidelines/2015/guidelines/appendix-9/. Accessed May, 2018. [Google Scholar]
- 27. O’Connell J. MMAP: mixed model analysis for pedigrees and populations. https://mmap.github.io/. Published 2017. Accessed April 2017. [Google Scholar]
- 28. The R Foundation . The R project for statistical computing. http://www.R-project.org/. Accessed June, 2016. [Google Scholar]
- 29. SAS Institute Inc . SAS Software. Version 9.4. Cary, NC:SAS Institute Inc.; 2011. [Google Scholar]
- 30. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics 2010;26:2190‐2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cochran WG. The combination of estimates from different experiments. Biometrics 1954;10:101‐129. [Google Scholar]
- 32. Park SH, Kim PN, Kim KW, Lee SW, Yoon SE, Park SW, et al. Macrovesicular hepatic steatosis in living liver donors: use of CT for quantitative and qualitative assessment. Radiology 2006;239:105‐112. [DOI] [PubMed] [Google Scholar]
- 33. Zeb I, Li D, Nasir K, Katz R, Larijani VN, Budoff MJ. Computed tomography scans in the evaluation of fatty liver disease in a population based study: the multi‐ethnic study of atherosclerosis. Acad Radiol 2012;19:811‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chamoun Z, Vacca F, Parton RG, Gruenberg J. PNPLA3/adiponutrin functions in lipid droplet formation. Biol Cell 2013;105:219‐233. [DOI] [PubMed] [Google Scholar]
- 35. Li JZ, Huang Y, Karaman R, Ivanova PT, Brown HA, Roddy T, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest 2012;122:4130‐4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smagris E, BasuRay S, Li J, Huang Y, Lai KM, Gromada J, et al. Pnpla3I148M knockin mice accumulate PNPLA3 on lipid droplets and develop hepatic steatosis. Hepatology 2015;61:108‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson AM, Olefsky JM. The origins and drivers of insulin resistance. Cell 2013;152:673‐684. [DOI] [PubMed] [Google Scholar]
- 38. Saponaro C, Gaggini M, Gastaldelli A. Nonalcoholic fatty liver disease and type 2 diabetes: common pathophysiologic mechanisms. Curr Diab Rep 2015;15:607. [DOI] [PubMed] [Google Scholar]
- 39. Dubuquoy C, Robichon C, Lasnier F, Langlois C, Dugail I, Foufelle F, et al. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J Hepatol 2011;55:145‐153. [DOI] [PubMed] [Google Scholar]
- 40. Pillai S, Duvvuru S, Bhatnagar P, Foster W, Farmen M, Shankar S, et al. The PNPLA3 I148M variant is associated with transaminase elevations in type 2 diabetes patients treated with basal insulin peglispro. Pharmacogenomics J 2018;18:487‐493. [DOI] [PubMed] [Google Scholar]
- 41. Maglio C, Pirazzi C, Pujia A, Valenti L, Romeo S. The PNPLA3 I148M variant and chronic liver disease: when a genetic mutation meets nutrients. Food Res Int 2014;63:293‐243. [Google Scholar]
- 42. Davis JN, Le KA, Walker RW, Vikman S, Spruijt‐Metz D, Weigensberg MJ, et al. Increased hepatic fat in overweight Hispanic youth influenced by interaction between genetic variation in PNPLA3 and high dietary carbohydrate and sugar consumption. Am J Clin Nutr 2010;92:1522‐1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stojkovic IA, Ericson U, Rukh G, Riddestrale M, Romeo S, Orho‐Melander M. The PNPLA3 Ile148Met interacts with overweight and dietary intakes on fasting triglyceride levels. Genes Nutr 2014;9:388. Erratum in: Genes Nutr 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al.; NASH CRN . Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362:1675‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Stender S, Kozlitina J, Nordestgaard BG, Tybjaerg‐Hansen A, Hobbs HH, Cohen JC. Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci. Nat Genet 2017;49:842‐847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Graff M, North KE, Franceschini N, Reiner AP, Feitosa M, Carr JJ, et al. PNPLA3 gene‐by‐visceral adipose tissue volume interaction and the pathogenesis of fatty liver disease: the NHLBI family heart study. Int J Obes (Lond) 2013;37:432‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun DQ, Liu WY, Wu SJ, Zhu GQ, Braddock M, Zhang DC, et al. Increased levels of low‐density lipoprotein cholesterol within the normal range as a risk factor for nonalcoholic fatty liver disease. Oncotarget 2016;7:5728‐5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Papandreou D, Karabouta Z, Rousso I. Are dietary cholesterol intake and serum cholesterol levels related to nonalcoholic fatty liver disease in obese children? Cholesterol 2012;2012:572820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Curr Opin Lipidol 2015;26:88‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7‐TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016;150:1219‐1230.e1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials