

Abstract
In cirrhosis, liver microvascular dysfunction is a key factor increasing hepatic vascular resistance to portal blood flow, which leads to portal hypertension. De‐regulated inflammatory and pro‐apoptotic processes due to chronic injury play important roles in the dysfunction of liver sinusoidal cells. The present study aimed at characterizing the effects of the pan‐caspase inhibitor emricasan on systemic and hepatic hemodynamics, hepatic cells phenotype, and underlying mechanisms in preclinical models of advanced chronic liver disease. We investigated the effects of 7‐day emricasan on hepatic and systemic hemodynamics, liver function, hepatic microcirculatory function, inflammation, fibrosis, hepatic cells phenotype, and paracrine interactions in rats with advanced cirrhosis due to chronic CCl4 administration. The hepato‐protective effects of emricasan were additionally investigated in cells isolated from human cirrhotic livers. Cirrhotic rats receiving emricasan showed significantly lower portal pressure than vehicle‐treated animals with no changes in portal blood flow, indicating improved vascular resistance. Hemodynamic improvement was associated with significantly better liver function, reduced hepatic inflammation, improved phenotype of hepatocytes, liver sinusoidal endothelial cells, hepatic stellate cells and macrophages, and reduced fibrosis. In vitro experiments demonstrated that emricasan exerted its benefits directly improving hepatocytes’ expression of specific markers and synthetic capacity, and ameliorated nonparenchymal cells through a paracrine mechanism mediated by small extracellular vesicles released by hepatocytes. Conclusion: This study demonstrates that emricasan improves liver sinusoidal microvascular dysfunction in cirrhosis, which leads to marked amelioration in fibrosis, portal hypertension and liver function, and therefore encourages its clinical evaluation in the treatment of advanced chronic liver disease.
Abbreviations
- α‐SMA
α–smooth muscle actin
- ACLD
advanced chronic liver disease
- cGMP
cyclic guanosine monophosphate
- col1α1
collagen 1α
- eNOS
endothelial nitric oxide synthase
- ET‐1
endothelin 1
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- HMϕ
hepatic macrophage
- HSC
hepatic stellate cell
- HVR
hepatic vascular resistance
- lEV
large extracellular vesicle
- IL
interleukin
- LSEC
liver sinusoidal endothelial cell
- MAP
mean arterial pressure
- mRNA
messenger RNA
- NO
nitric oxide
- PBF
portal blood flow
- PBS
phosphate‐buffered saline
- PP
portal pressure
- sEV
small extracellular vesicle
Advanced chronic liver disease (ACLD) is characterized by the presence of regenerative nodules surrounded by fibrous septa, causing a marked architectural distortion, eventually leading to portal hypertension and liver failure.1, 2 Progression of cirrhosis encompasses multiple coordinated cellular de‐regulations, including initial injuries to parenchymal and endothelial cells, which lead to hepatocyte cell death and sinusoidal dysfunction, activation of hepatic stellate cells and resident macrophages, and recruitment of other immune cells.3, 4 Intercellular communication between parenchymal and nonparenchymal cells further contributes to progressive fibrogenesis and aggravates ACLD. Indeed, it has been described that, in response to injury, hepatocytes release damage‐associated molecular patterns and other soluble and microencapsulated components that are able to affect neighboring cells.5, 6 Such crosstalk is important for both cirrhosis progression and improvement. In fact, previous studies have demonstrated that amelioration of a specific liver cell type, such as the sinusoidal endothelium, paracrinally improves other cells such as hepatic stellate cells.7, 8, 9 Nevertheless, little is known about the possibility that the improvement in hepatocytes may exert beneficial effects on other liver cells in the context of ACLD.
Improvement of ACLD might be accomplished with the cessation of injury10 or by drug‐induced amelioration of hepatic cells.11 Unfortunately, current options to improve ACLD and its main clinical complications are limited,3, 11 and although statins have shown beneficial effects on portal hypertension and survival, the risk reduction afforded by these and other agents is limited,12, 13 indicating that new therapeutic strategies are warranted.
Emricasan (IDN‐6556) is an irreversible pan‐caspase inhibitor that primarily targets the liver when orally administered,14 and is being investigated in clinical trials for chronic liver disease and portal hypertension after pilot studies in patients with chronic hepatitis C suggested a beneficial effect on liver function as well as on portal pressure in patients with severe portal hypertension.15, 16, 17, 18 Previous preclinical work has shown that inhibition of hepatocyte cell death using caspase inhibitors protects the liver from acute injury,19 in scenarios of chronic liver disease with mild fibrosis20 or in cholestatic liver injury.21 Nevertheless, the possible beneficial effects of this therapeutic option in models of ACLD and portal hypertension are mostly unknown.
The main objective of the present study was to characterize the effects of emricasan on liver hemodynamic and fibrosis in animals with ACLD. Secondary aims included the understanding of the underlying mechanisms of emricasan in experiments using liver cells both from rodent and human cirrhotic livers.
Materials and Methods
Animal Model of Advanced Chronic Liver Disease
Cirrhosis of the liver was induced in male Wistar rats by chronic inhalation of CCl4 (Sigma‐Aldrich, St. Louis, MO), together with phenobarbital in the drinking water. When rats developed ascites, approximately after 12‐14 weeks, toxicants administration was stopped and treatment started 1 week later.22 Animals were kept in environmentally controlled animal facilities. All procedures were approved by the Laboratory Animal Care and Use Committee of the University of Barcelona and were conducted in accordance with the European Community guidelines for the protection of animals used for experimental and other scientific purposes (EEC Directive 86/609).
The personnel who prepared and administered treatments and those who performed the experimental studies were different. For maintaining investigator blinding during assessment of the results, the treatment’s codes were not open until completion of the study.
Characterization of Chronic Liver Disease Rats Treated With Emricasan
Emricasan Administration
Cirrhotic rats received either emricasan (10 mg/kg/day; per os; n = 20) or vehicle (0.9% dimethylcarboxycellulose; per os; n = 20) for 7 days. The dose was selected based on a conversion calculation starting from the dose used in humans and in previous preclinical publications.20, 21
In Vivo Hemodynamics
Rats (n = 12 per group) were anesthetized with ketamine hydrochloride (100 mg/kg; Merial Laboratories, Duluth, GA) plus midazolam (5 mg/kg; Laboratorios Reig Jofré, Toledo, Spain) intraperitoneally. A tracheostomy was performed and a polyethylene tube PE‐240 was inserted into the trachea to ensure a patent airway. PE‐50 catheters were introduced into the femoral artery to measure mean arterial pressure (MAP; mmHg) and into the ileocolic vein to measure portal pressure (PP; mmHg). A perivascular ultrasonic flow probe (Transonic System, Ithaca, NY) was placed around the portal vein, as close as possible to the liver to avoid portal‐systemic collaterals to measure portal blood flow (PBF; mL∙min−1). Hepatic vascular resistance (HVR; mmHg·min·mL−1) was calculated as PP/PBF. Blood pressures and flows were registered on a multichannel computer‐based recorder (Power Lab; ADInstruments, Sydney, Australia). Body temperature of the animals was maintained at 37 ± 0.5°C, and hemodynamic data were collected after 20 minutes of stabilization.22, 23 Blood serum and plasma samples were stored for biochemical analysis.
Liver Microvascular Function
Immediately after recording in vivo hemodynamics, rat livers (n = 8 per group) were isolated and perfused with Krebs buffer as previously described.23, 24 The perfused rat liver preparation was allowed to stabilize for 20 minutes before vasoactive substances were added. Intrahepatic microcirculation was preconstricted by adding the α1‐adrenergic agonist methoxamine (10‐4 M; Sigma‐Aldrich) to the reservoir, and liver microvascular function was assessed as concentration–response curves to cumulative doses of acetylcholine (10−7‐10−5 M; Sigma‐Aldrich). Bile production was monitored through the entire ex vivo perfusion experiment and expressed as μL/min g of liver. At the end of the experiment, liver tissue was snap‐frozen for subsequent molecular analysis.
Evaluation of Hepatic Fibrosis and Cell Death
Cirrhotic rat livers were fixed in 10% formalin, embedded in paraffin, and sectioned. For fibrosis quantification, samples were stained with 0.1% sirius red, photographed, and analyzed using a microscope equipped with a digital camera. The proportion of red‐stained area was measured using Axiovision software.25 For cell death analysis, deparaffined sections were probed with an In Situ Cell Death Detection Kit (TUNEL; Roche Diagnostics, Basel, Switzerland) according to the manufacturer’s instructions. Values are expressed as the mean of eight fields per sample.
Sinusoidal Characterization Using Scanning Electron Microscopy
In a subgroup of animals (n = 3 per group), after obtaining in vivo hemodynamics, livers were perfused through the portal vein with a solution containing 2.5% glutaraldehyde and 2% paraformaldehyde and fixed overnight at 4ºC. Samples were washed 3 times with 0.1M cacodylate buffer. Liver sections were fixed with 1% osmium in cacodylate buffer, dehydrated in ethanol, and dried with hexamethyldisilazane. Six randomly selected blocks from each animal were mounted onto stubs, and sputter coated with gold. Ten images per animal were acquired at a resolution of ×15,000 using a Jeol 6380 scanning electron microscope (JEOL Ltd, Tokyo, Japan). Liver sinusoidal fenestrations were quantified using ImageJ Software (National Institutes of Health, New York, NY).26
Nitric Oxide Bioavailability
Levels of cyclic guanosine monophosphate (cGMP), a marker of nitric oxide (NO) bioavailability, were analyzed in liver homogenates using an enzyme immunoassay (Cayman Chemical Co., Ann Arbor, MI) as previously described.27
Liver Cells Isolation and Treatments
Rat
Hepatocytes, liver sinusoidal endothelial cells (LSECs), hepatic stellate cells (HSCs), and hepatic macrophages (HMϕs) were isolated from cirrhotic animals treated with emricasan or vehicle following well‐standardized protocols.28 Highly pure (Hep 97% purity; LSEC 96%; HSC 98%; HMϕ 95%) and viable cells (all above 90%) were used. No differences in cell viability were observed comparing both groups of rats.
In additional mechanistic experiments, primary rat cirrhotic hepatocytes were pretreated for 24 hours with emricasan (50 μM) or vehicle (0.01% DMSO), washed twice with phosphate‐buffered saline (PBS), and incubated with fresh media. After 24 hours, the preconditioned media was centrifuged at 2000 g to remove apoptotic bodies and added to primary rat cirrhotic HSCs, LSECs, and HMϕs for 24 hours. The phenotype of nonparenchymal cells was determined in five independent experiments.
To further understand the intercellular crosstalk in response to emricasan, the main subfractions of the cell secretome (i.e., large extracellular vesicles [lEVs] and small extracellular vesicles [sEVs]) were purified from hepatocyte preconditioned media and exposed to HSCs. Briefly, primary hepatocytes isolated from CCl4‐cirrhotic rats were cultured for 24 hours in vesicle‐depleted media supplemented with emricasan (50 μM) or vehicle, washed twice with PBS, and then incubated with fresh microvesicle‐free media for 24 hours. Supernatants were collected, centrifuged at 2000 g to remove apoptotic bodies, and microvesicle‐enriched subfractions were obtained by subsequent ultracentrifugations as described.29 Preparations were routinely tested for purity, size, and concentration using cryo‐electron microscopy and nanoparticle tracking analysis.30 A total of 30 μg of each subfraction was added to primary cirrhotic HSCs for 24 hours (n = 3 independent experiments).
Human
Hepatocytes and HSCs were isolated from liver tissue remnants from cirrhotic liver explants (all of alcoholic etiology) following standardized protocols.31 Human hepatic cells were treated in vitro with increasing concentrations of emricasan (10‐50 μM) or vehicle (0.01% DMSO) in conventional culture plates or in the ExoLiver platform31 (n = 5 independent experiments).
The ethics committee of the Hospital Clinic de Barcelona approved the study protocol (HCB/2018/0028), and sample manipulation and isolation procedures were carried out following good laboratory practices. In all cases, patients signed an informed consent.
Protein and Messenger RNA Expression Analysis
Protein expression was determined by western blot in hepatic tissue and cell samples using antibodies described in Supporting Table S1. Blots were revealed by chemiluminescence, and digital images were taken using a luminescent image analyzer LAS‐4000 (General Electric, Little Chalfont, United Kingdom). Protein expression was determined by densitometric analysis using the Science Lab 2001, Multi Gauge V2.1 (Fuji Photo Film, Düsseldorf, Germany). Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used for the normalization of quantitative densitometry values.
Immunostaining of paraffin‐embedded liver sections was performed with antibodies described in Supporting Table S1 or with PBS as a negative control. Ten fields from each slide at ×400 magnification were randomly selected, and photographs were done using a fluorescence microscope (Olympus, Tokyo, Japan) and quantified with Image J 1.33u software (National Institutes of Health). Two blinded independent researchers performed immunostaining quantifications.
Gene expression was analyzed in total RNA from all hepatic tissue and cell samples according to the manufacturer's protocol using primers described in Supporting Table S2. Values were reported relative to the endogenous control GAPDH. All amplification reactions were performed in duplicate, and nuclease‐free water was used as no template control in the reaction sets.
Results
Emricasan Inhibits Hepatic Cell Death and Improves Liver Function in Rats With Advanced Cirrhosis
Oral administration of emricasan resulted in significant inhibition of hepatic cell death, as demonstrated by marked reductions in caspase‐3 activity and overall hepatic cell death as measured by TUNEL (Fig. 1). Amelioration of hepatic function by emricasan was confirmed by improvement in blood tests and a marked increase in bile production (Table 1).
Figure 1.
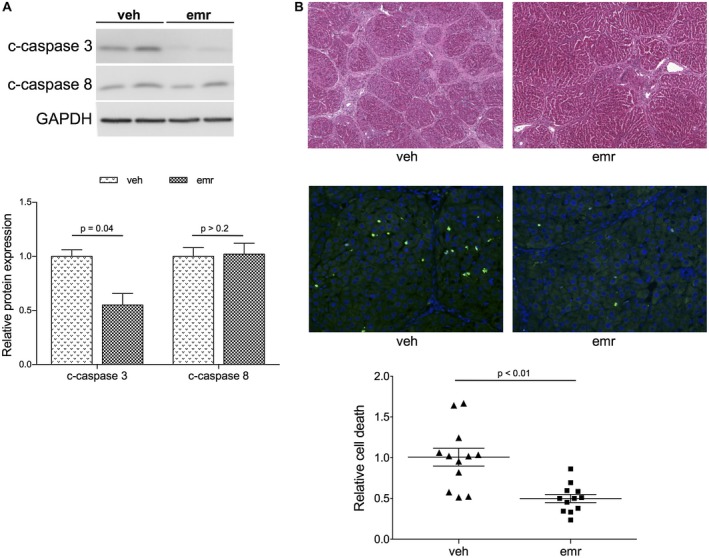
Effects of emricasan on hepatic cell death. (A) Protein expression of caspases in livers from cirrhotic rats treated with emricasan or vehicle (n = 12 per group). (B) Top: Representative images of hematoxylin and eosin staining in livers from (A) (magnification ×5). Bottom: Representative images and quantification of hepatic cell death using TUNEL analysis in animals described in (A) (magnification ×20). Values represent mean ± standard error of the mean. Abbreviations: emr, emricasan; veh, vehicle.
Table 1.
Effects of Emricasan on Morphometric Descriptors, Biochemical Parameters, and Bile Production in Rats With Advanced Chronic Liver Disease
| Vehicle | Emricasan | P Value | |
|---|---|---|---|
| Body weight pretreatment (g) | 353 ± 6 | 367 ± 7 | >0.20 |
| Body weight post treatment (g) | 367 ± 11 | 367 ± 12 | >0.20 |
| Liver weight (g) | 10.6 ± 0.3 | 10.3 ± 0.5 | >0.20 |
| Spleen weight (g) | 1.04 ± 0.03 | 0.99 ± 0.08 | >0.20 |
| AST (U/L) | 251 ± 51 | 123 ± 24 | 0.05 |
| ALT (U/L) | 110 ± 29 | 51 ± 5 | 0.1 |
| Bilirubin (mg/dL) | 0.58 ± 0.17 | 0.20 ± 0.06 | 0.1 |
| Albumin (g/L) | 22.7 ± 0.7 | 24.5 ± 1.0 | >0.20 |
| Bile (μL·min−1·g−1) | 3.4 ± 0.4 | 7.5 ± 0.5 | 0.01 |
Values represent mean ± standard error of the mean (n = 12 per group for all parameters except n = 8 per group for bile production). Italicized values are those statistically significant.
Abbreviations: ALT, alanine aminotransferase; and AST, aspartate aminotransferase.
Emricasan Reduces Portal Hypertension and Hepatic Microvascular Dysfunction in Rats With Advanced Cirrhosis
As shown in Fig. 2A, rats treated with emricasan exhibited lower PP than those receiving vehicle (12.7 ± 0.6 versus 14.9 ± 0.3 mmHg; −14%; P < 0.01) without changes in PBF, thus pointing to an improved HVR: 0.88 ± 0.09 versus 1.14 ± 0.12 mmHg·min·mL−1; −23%; P = 0.1). Importantly, emricasan did not modify MAP or heart rate. Supporting an improved intrahepatic vascular tone, ex vivo perfusion experiments revealed that emricasan‐treated animals exhibited significantly lower portal perfusion pressure and ameliorated liver microcirculatory dysfunction, as demonstrated by enhanced vasodilatory response to incremental concentrations of acetylcholine (Fig. 2B).
Figure 2.
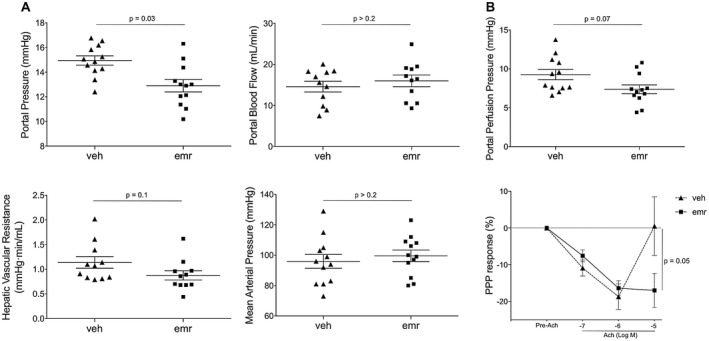
Emricasan improves portal hypertension and liver microvascular dysfunction in cirrhosis. (A) PP, PBF, HVR, and MAP measured in vivo in cirrhotic rats treated with emricasan or vehicle (n = 12 per group). (B) Portal perfusion pressure and sinusoidal vasorelaxation in response to acetylcholine determined in a subpopulation of animals included in (A) (n = 8 per group). Values represent mean ± SEM. Abbreviations: PPP, portal perfusion pressure.
Underlying Mechanisms of Emricasan‐Mediated Improvement in Cirrhotic Portal Hypertension
As shown in Fig. 3, cirrhotic animals that were treated with emricasan showed significantly lower liver fibrosis, measured as the percentage of sirius red–positive area, as well as expression of collagen 1α (col1α1). Improvement in fibrosis was associated with a reduced activation of HSCs, demonstrated by a lower expression of several activation markers, including α–smooth muscle actin (α‐SMA), p‐moesin, PDGFRβ, matrix metalloproteinases, and tissue inhibitor of metalloproteinases. Moreover, a reduction in desmin content was observed, suggesting a diminished number of HSCs.
Figure 3.
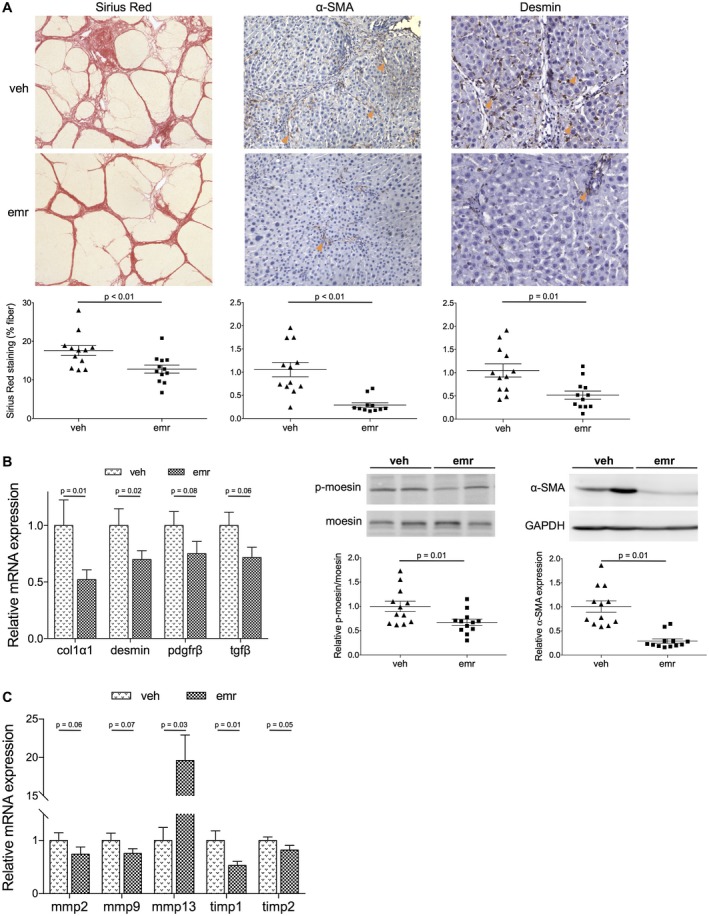
Analysis of the HSC phenotype and liver fibrosis in cirrhotic rats treated with emricasan. (A) Representative images and corresponding quantifications of sirius red staining (magnification ×5), and α‐SMA ( ×10) and desmin ( ×20) immunohistochemistry in livers from cirrhotic rats treated with emricasan or vehicle (n = 12 per group). (B) Messenger RNA (mRNA) and protein expression of HSC activation markers in liver tissue from animals described in (A). (C) mRNA expression of extracellular matrix remodeling genes in tissues described in (A). Values represent mean ± SEM. Abbreviations: emr, emricasan; mmp, matrix metalloproteinase; tgf β, transforming growth factor β; timp, tissue inhibitor of metalloproteinase; veh, vehicle.
Additionally, emricasan markedly improved the hepatic endothelium. Livers from rats receiving emricasan showed improved endothelial phenotype, demonstrated by increased endothelial fenestration, reduction in sinusoidal von Willebrand factor (vWF) expression, and improved NO availability, shown by higher phospho‐endothelial nitric oxide synthase (eNOS) expression and higher cGMP content (Fig. 4A).
Figure 4.
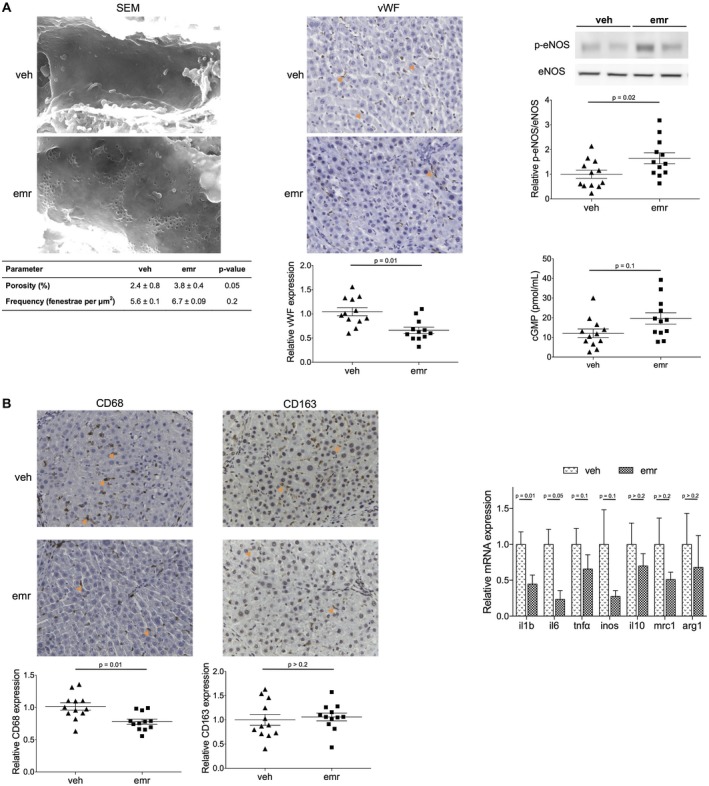
Effects of emricasan on the liver sinusoidal endothelium and hepatic inflammation in cirrhosis. (A) Analysis of sinusoidal fenestration in terms of porosity and frequency, sinusoidal expression of vWF, expression of phospho‐eNOS/eNOS, and cGMP levels in livers from cirrhotic rats treated with emricasan or vehicle. (B) Analysis of inflammatory markers in livers described in (A). Values represent mean ± standard error of the mean. All images magnification ×20. Abbreviations: arg, arginase 1; emr, emricasan; SEM, scanning electron microscopy; tnfα, tumor necrosis factor α; veh, vehicle.
Finally, and considering the importance of inflammation driving both the progression and regression of cirrhosis, we characterized the hepatic inflammatory status in cirrhotic animals receiving emricasan or vehicle. As shown in Fig. 4B, livers from emricasan‐treated rats had significantly lower expression of diverse pro‐inflammatory molecules (such as interleukin [IL] 1, IL‐6, and tumor necrosis factor α) as well as a reduced number of CD68 positive cells, without modifying the expression of anti‐inflammatory genes or CD163 positive cells, altogether suggest that the drug was able to shift the intrahepatic cellular environment from a disease progression toward a disease resolution phenotype.
Emricasan Directly Improves Hepatocytes Phenotype, Which Leads to Paracrine Improvement in Nonparenchymal Cells
Analysis of hepatocytes, LSECs, HSCs, and HMϕs freshly isolated from emricasan‐treated or vehicle‐treated cirrhotic rats revealed that this pan‐caspase inhibitor was able to improve all cell populations. In fact, hepatocytes isolated from emricasan‐treated animals exhibited significant improvement in the expression of different markers and master regulators of hepatocyte status, which was accompanied by re‐gain of function, demonstrated by significantly higher cyp4503a4 activity and protein synthesis capacity (Fig. 5A). In addition, nonparenchymal cells also exhibited improvement in their phenotype, as shown by differential expression of HSC markers including α‐SMA collagen I and PDGFRβ, eNOS and endothelin 1 (ET‐1) in LSECs, and inducible nitric oxide synthase in HMϕs (Fig. 5B).
Figure 5.

Phenotype of primary liver cells isolated from cirrhotic animals receiving emricasan. (A) Analysis of hepatocyte phenotype in terms of expression of key transcription factors and cellular transporters, CYP450 activity, and protein synthesis determined in cells isolated from cirrhotic rats treated with emricasan or vehicle. (B) Characterization of nonparenchymal cells isolated from animals described in (A). mRNA expression of key cellular markers in HSCs (left), LSECs (middle), and HMϕs (right) (n = 5 independent isolations per group). Values represent mean ± standard error of the mean. Abbreviations: emr, emricasan; iNOS, inducible nitric oxide synthase; klf, Kruppel‐like factor; tgf β, transforming growth factor β; tnfα, tumor necrosis factor α; veh, vehicle.
In vitro experiments confirmed that cirrhotic hepatocytes directly treated with emricasan exhibited improved phenotype in comparison to cells receiving vehicle (Fig. 6A). Interestingly, and although emricasan had no direct effect on the phenotype of nonparenchymal cells (Supporting Fig. S1), crosstalk experiments disclosed a significant improvement in the expression of a variety of specific markers when incubated with preconditioned medium obtained from emricasan‐treated hepatocytes (Fig. 6B).
Figure 6.
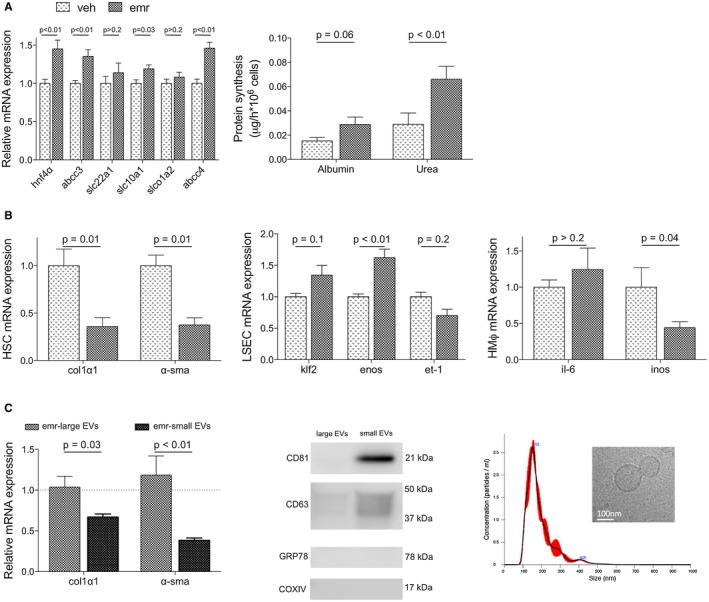
Effects of emricasan on hepatic cell phenotype and underlying paracrine mechanisms. (A) Phenotype of hepatocytes isolated from cirrhotic rats and treated in vitro with emricasan or vehicle. (B) Phenotype of HSCs (left), LSECs (middle), and HMϕs (right) isolated from cirrhotic rats and treated in vitro with preconditioned media from vehicle‐treated or emricasan‐treated cirrhotic hepatocytes. (C) Left: Analysis of cirrhotic rat HSC phenotype after incubation with lEVs‐enriched or sEVs‐enriched secretome subfractions purified from cirrhotic rat hepatocytes treated with emricasan or vehicle. Dotted line corresponds to mRNA expression in HSCs incubated with vehicle‐treated hepatocyte secretome subfractions. Right: Protein expression of specific markers of EVs and negative controls, and nanoparticle tracking analysis and electron microscopy images from small EVs‐enriched preparations (n = 3‐5 independent isolations). Values represent mean ± standard error of the mean. Abbreviations: emr, emricasan; veh, vehicle.
Analysis of the intercellular crosstalk in response to emricasan revealed that the sEVs‐enriched subfraction of the secretome was the mediator of the hepatocyte‐derived beneficial effects to nonparenchymal cells (Fig. 6C). Indeed, HSCs treated with sEVs from emricasan‐treated hepatocytes exhibited significantly reduced expression of the activation markers α‐SMA and collagen I, in comparison to HSCs treated with the same amount of sEVs from vehicle‐treated hepatocytes. No effects were observed when incubating cells with lEVs‐enriched fractions.
Hepatoprotective Effects of Emricasan Are Confirmed in Human Liver Cells
The effects of emricasan modulating the phenotype of human hepatocytes were investigated in primary cells cultured under sinusoidal‐like conditions using the Exoliver platform, a recently developed liver‐on‐a‐chip microfluidic device,31 or in conventional platforms. Figure 7 shows that human cirrhotic hepatocytes cultured in vitro and treated with emricasan exhibit a marked amelioration in their phenotype when compared with vehicle‐treated cells, with improved expression of HNF4α, abcc3 and slc22a1, and higher albumin and urea synthesis, without signs of hepatotoxicity. The number of hepatocytes at the end of the experiment were not different between groups, as indicated by the amount of RNA obtained in each experimental condition. Interestingly, the beneficial effects of the drug were not observed in hepatocytes undergoing spontaneous de‐differentiation due to culture in conventional methods (data not shown).
Figure 7.
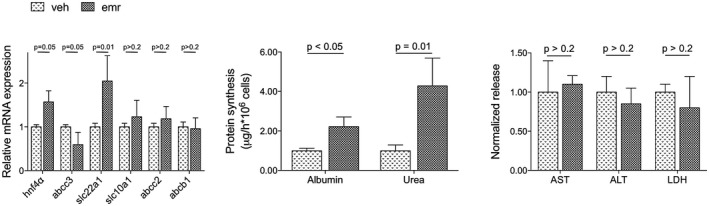
Effects of emricasan on human cirrhotic hepatocytes. Phenotype of hepatocytes isolated from human cirrhotic livers and treated in vitro with emricasan or vehicle (n = 5 independent isolations). Values represent mean ± standard error of the mean. Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; emr, emricasan; LDH, lactate dehydrogenase; veh, vehicle.
Discussion
In ACLD, increased hepatic vascular resistance caused by the development of hepatic microvascular dysfunction, fibrosis, and architectural remodeling is the main player in the development of portal hypertension. Different preclinical studies have evaluated the possibility of reducing hepatic vascular resistance using various strategies32, 33, 34, 35; nevertheless, none of them have successfully moved to standard bedside applications. Therefore, novel strategies that target the major pathophysiological abnormalities of ACLD without causing systemic adverse effects are required to improve treatments for patients with portal hypertension.
The present study demonstrates that 1‐week emricasan treatment leads to significant improvement in cirrhotic portal hypertension in a widely accepted preclinical model of ACLD, the induction of decompensated cirrhosis by prolonged CCl4 administration. The decrease in PP was not associated with modifications in PBF, pointing to decreased hepatic vascular resistance. Importantly, emricasan administration showed no apparent hepatic or systemic adverse effects. Interestingly, our results are in alignment with a previous study, demonstrating benefits of this pan‐caspase inhibitor by decreasing PP in patients with cirrhosis and severe portal hypertension.17
Underlying mechanisms explaining the improvement in portal hypertension were diverse and included benefits on both vascular and architectural components of the increased hepatic vascular resistance.3 Liver microvascular dysfunction leads to increased hepatic vascular tone, a key factor in PP elevation in cirrhosis, and was significantly improved in animals treated with emricasan. In addition, cirrhotic liver architectural distortion, which mechanically increases liver vascular resistance, was also ameliorated as shown by a significant diminution in liver fibrosis.
To understand the global improvement in liver hemodynamics and fibrosis due to emricasan, we evaluated the changes in the phenotype of the three major cellular components of the sinusoid (i.e., HSCs, LSECs, and HMϕs). Analysis performed in liver tissue from emricasan‐treated cirrhotic rats demonstrated partial de‐activation of HSC, and improvement in LSEC and HMϕ. The amelioration in HSC was evidenced by a decrease in pro‐activation markers, extracellular matrix components, and proliferative indicators. These data are in agreement with previous reports demonstrating antifibrotic effects of pan‐caspase inhibitors in preclinical models of mild injury.36, 37, 38 Our current study, however, further demonstrates the ability of emricasan to promote fibrosis regression in a model of very advanced liver disease, as exemplified by cirrhotic rats with ascites. In addition to HSCs, LSECs represent an important component of the hepatic sinusoid with essential roles modulating hepatic vascular tone, inflammation, and hemostasis.3 Herein we describe global improvement in the LSEC phenotype and function, as demonstrated by increased NO bioavailability, which has important vasodilatory effects. In addition, emricasan treatment increased sinusoidal porosity, which may lead to improved overall liver function due to enhanced communication between hepatocytes and the blood stream.6 HMϕ analysis further demonstrated the protective effects of emricasan, which promoted a change in the phenotype of cirrhotic HMϕ, leading to a reduction in M1‐like macrophages, with no changes in M2‐like cells. Importantly, restoration of HMϕ balance was accompanied by significant reduction in intrahepatic inflammation, in which the improved LSEC function may also contribute. Finally, and although our results and previous data do not support possible interfering effects of long‐term emricasan on fibrosis resolution or carcinogenesis,20, 39 future studies should unveil these possibilities.
To further analyze the mechanisms by which emricasan improves the phenotype of parenchymal and nonparenchymal cells in the cirrhotic liver, we evaluated whether the drug was directly affecting cells or required paracrine interactions between the different cellular subpopulations. This information is relevant, as it has been demonstrated that pathways involving caspases share molecular intermediates with pathways related with intercellular communication.40 Interestingly, emricasan administration to primary cells isolated from cirrhotic rat livers improved the expression of different markers of hepatocytes functionality, but had no clear direct effects on cultured nonparenchymal cells. Subsequent experiments evaluating possible paracrine effects from emricasan‐treated hepatocytes evidenced a positive crosstalk, leading to marked de‐activation of HSC, together with partial improvements in LSECs and HMϕs. These observations are in agreement with previous works demonstrating the modulation of paracrine effectors in response to pan‐caspase inhibitors.14, 36, 41 Nevertheless, our study adds a significant piece of knowledge in this process. Although previous work attributed the beneficial paracrine effect mediated by pan‐caspase inhibitors to a decrease in the release of apoptotic bodies, we demonstrate herein that hepatocytes receiving emricasan modulate the intracellular secretome machinery to beneficially communicate with neighboring cells. Importantly, hepatocyte‐derived sEVs appear to be the main mechanism responsible for the paracrine communications in response to emricasan. Nevertheless, we cannot discard that other soluble mediators not associated with extracellular vesicles may also contribute. Future studies should unravel the specific molecular mediator(s) responsible of this cellular crosstalk. In this regard, the sinusoidal‐protective effects of emricasan may be partly explained by the capability of the drug to modulate pro‐coagulant mediators,42 thus blunting one of the mechanisms in the pathophysiology of cirrhosis and portal hypertension.43, 44
Finally, and aiming at validating our findings in the preclinical model into an experimental setting as close as possible to human cirrhosis, we evaluated the effects of emricasan modulating the phenotype of primary cells isolated from cirrhotic human livers cultured in a liver‐on‐a‐chip device. These experiments demonstrated a marked improvement in hepatocyte phenotype, without signs of toxicity, altogether indicating that the hepatoprotective effects of emricasan are also observed in human cirrhosis.
In conclusion, the present study provides information in the field of chronic liver disease, showing that emricasan administration to cirrhotic animals decreases PP and liver fibrosis, thus ameliorating portal hypertension. Underlying mechanisms included direct hepatocyte protection and subsequent beneficial paracrine effects on sinusoidal cells. Emricasan hepato‐protection was further confirmed in human cirrhotic liver cells, altogether suggesting that emricasan may represent an effective therapeutic approach for the treatment of patients with advanced cirrhosis and severe portal hypertension.
Supporting information
Acknowledgments
The authors are indebted with Sergi Vila, Montse Monclús, and Giusi Marroni for the excellent assistance, and to Drs. Fondevila and Molina for providing human liver tissues.
Supported by Conatus Pharmaceuticals and Instituto de Salud Carlos III (FIS PI17/00012).
Potential conflict of interest: Drs. Gracia‐Sancho and Bosch consults for and received grants from Conatus. Drs. Contreras and Spada own stock in and are employed by Conatus.
[Correction made 23 April, 2019. Dr. Gracia‐Sancho's degree was incorrectly listed as M.D.]
References
Author names in bold designate shared co‐first authorship.
- 1. Tsochatzis EA, Bosch J, Burroughs AK. Liver cirrhosis. Lancet 2014;383:1749‐1761. [DOI] [PubMed] [Google Scholar]
- 2. Fernández‐Iglesias A, Gracia‐Sancho J. How to face chronic liver disease: the sinusoidal perspective. Front Med 2017;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gracia‐Sancho J, Marrone G, Fernández‐Iglesias A. Hepatic microcirculation and mechanisms of portal hypertension. Nat Rev Gastroenterol Hepatol 2019;16:221‐234. [DOI] [PubMed] [Google Scholar]
- 4. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 2017;17:306‐321. [DOI] [PubMed] [Google Scholar]
- 5. Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147:765‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marrone G, Shah VH, Gracia‐Sancho J. Sinusoidal communication in liver fibrosis and regeneration. J Hepatol 2016;65:608‐617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Marrone G, Russo L, Rosado E, Hide D, García‐Cardeña G, García‐Pagán JC, et al. The transcription factor KLF2 mediates hepatic endothelial protection and paracrine endothelial‐stellate cell deactivation induced by statins. J Hepatol 2013;58:98‐103. [DOI] [PubMed] [Google Scholar]
- 8. Di Pascoli M, Diví M, Rodríguez‐Vilarrupla A, Rosado E, Gracia‐Sancho J, Vilaseca M, et al. Resveratrol improves intrahepatic endothelial dysfunction and reduces hepatic fibrosis and portal pressure in cirrhotic rats. J Hepatol 2013;58:904‐910. [DOI] [PubMed] [Google Scholar]
- 9. Xie G, Wang X, Wang L, Wang L, Atkinson RD, Kanel GC, et al. Role of differentiation of liver sinusoidal endothelial cells in progression and regression of hepatic fibrosis in rats. Gastroenterology 2012;142:918‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lens S, Alvarado‐Tapias E, Marino Z, Londono M‐C, LLop E, Martinez J, et al. Effects of all‐oral anti‐viral therapy on HVPG and systemic hemodynamics in patients with hepatitis C virus‐associated cirrhosis. Gastroenterology 2017;153:1273‐1283. [DOI] [PubMed] [Google Scholar]
- 11. Vilaseca M, Guixé‐Muntet S, Fernández‐Iglesias A, Gracia‐Sancho J. Advances in therapeutic options for portal hypertension. Therap Adv Gastroenterol 2018;11:1756284818811294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo‐controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 2016;375:631‐643. [DOI] [PubMed] [Google Scholar]
- 14. Ueno Y, Ohmi T, Yamamoto M, Kato N, Moriguchi Y, Kojima M, et al. Orally‐administered caspase inhibitor PF‐03491390 is retained in the liver for prolonged periods with low systemic exposure, exerting a hepatoprotective effect against alpha‐fas‐induced liver injury in a mouse model. J Pharmacol Sci 2007;105:201‐205. [DOI] [PubMed] [Google Scholar]
- 15. Pockros PJ, Schiff ER, Shiffman ML, McHutchison JG, Gish RG, Afdhal NH, et al. Oral IDN‐6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology 2007;46:324‐329. [DOI] [PubMed] [Google Scholar]
- 16. Shiffman ML, Pockros P, McHutchison JG, Schiff ER, Morris M, Burgess G. Clinical trial: the efficacy and safety of oral PF‐03491390, a pancaspase inhibitor—a randomized placebo‐controlled study in patients with chronic hepatitis C. Aliment Pharmacol Ther 2010;31:969‐978. [DOI] [PubMed] [Google Scholar]
- 17. Garcia‐Tsao G, Fuchs M, Shiffman M, Borg BB, Pyrsopoulos N, Shetty K, et al. Emricasan (IDN‐6556) lowers portal pressure in patients with compensated cirrhosis and severe portal hypertension. Hepatology 2019;69:717‐728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frenette CT, Morelli G, Shiffman ML, Frederick RT, Rubin RA, Fallon MB, et al. Emricasan improves liver function in patients with cirrhosis and high model for end‐stage liver disease scores compared with placebo. Clin Gastroenterol Hepatol 2018;8:224‐234. [DOI] [PubMed] [Google Scholar]
- 19. Jaeschke H, Farhood A, Cai SX, Tseng BY, Bajt ML. Protection against TNF‐induced liver parenchymal cell apoptosis during endotoxemia by a novel caspase inhibitor in mice. Toxicol Appl Pharmacol 2000;169:77‐83. [DOI] [PubMed] [Google Scholar]
- 20. Barreyro FJ, Holod S, Finocchietto PV, Camino AM, Aquino JB, Avagnina A, et al. The pan‐caspase inhibitor Emricasan (IDN‐6556) decreases liver injury and fibrosis in a murine model of non‐alcoholic steatohepatitis. Liver Int 2015;35:953‐966. [DOI] [PubMed] [Google Scholar]
- 21. Eguchi A, Koyama Y, Wree A, Johnson CD, Nakamura R, Povero D, et al. Emricasan, a pan‐caspase inhibitor, improves survival and portal hypertension in a murine model of common bile‐duct ligation. J Mol Med 2018;96:575‐583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gracia‐Sancho J, Russo L, García‐Calderó H, García‐Pagán JC, García‐Cardeña G, Bosch J. Endothelial expression of transcription factor Kruppel‐like factor 2 and its vasoprotective target genes in the normal and cirrhotic rat liver. Gut 2011;60:517‐524. [DOI] [PubMed] [Google Scholar]
- 23. Hide D, Ortega‐Ribera M, Garcia‐Pagan JC, Peralta C, Bosch J, Gracia‐Sancho J. Effects of warm ischemia and reperfusion on the liver microcirculatory phenotype of rats: underlying mechanisms and pharmacological therapy. Sci Rep 2016;6:22107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gracia‐Sancho J, Laviña B, Rodríguez‐Vilarrupla A, Brandes RP, Fernández M, Bosch J, et al. Evidence against a role for NADPH oxidase modulating hepatic vascular tone in cirrhosis. Gastroenterology 2007;133:959‐966. [DOI] [PubMed] [Google Scholar]
- 25. Marrone G, Maeso‐Díaz R, García‐Cardena G, Abraldes JG, García‐Pagán JC, Bosch J, et al. KLF2 exerts antifibrotic and vasoprotective effects in cirrhotic rat livers: behind the molecular mechanisms of statins. Gut 2015;64:1434‐1443. [DOI] [PubMed] [Google Scholar]
- 26. Hilmer SN, Cogger VC, Fraser R, McLean AJ, Sullivan D, Le Couteur DG. Age‐related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology 2005;42:1349‐1354. [DOI] [PubMed] [Google Scholar]
- 27. Hide D, Ortega‐Ribera M, Fernández‐Iglesias A, Fondevila C, Salvadó MJ, Arola L, et al. A novel form of the human manganese superoxide dismutase protects rat and human livers undergoing ischaemia and reperfusion injury. Clin Sci 2014;127:527‐537. [DOI] [PubMed] [Google Scholar]
- 28. Gracia‐Sancho J, Laviña B, Rodríguez‐Vilarrupla A, García‐Calderó H, Bosch J, García‐Pagán JC. Enhanced vasoconstrictor prostanoid production by sinusoidal endothelial cells increases portal perfusion pressure in cirrhotic rat livers. J Hepatol 2007;47:220‐227. [DOI] [PubMed] [Google Scholar]
- 29. Thierry Théry C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants. Curr Protoc Cell Biol 2006;3:1‐29. [DOI] [PubMed] [Google Scholar]
- 30. Royo F, Moreno L, Mleczko J, Palomo L, Gonzalez E, Cabrera D, et al. Hepatocyte‐secreted extracellular vesicles modify blood metabolome and endothelial function by an arginase‐dependent mechanism. Sci Rep 2017;7:42798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ortega‐Ribera M, Fernández‐Iglesias A, Illa X, Moya A, Molina V, Maeso‐Díaz R, et al. Resemblance of the human liver sinusoid in a fluidic device with biomedical and pharmaceutical applications. Biotechnol Bioeng 2018;18:2023‐2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Trebicka J, Hennenberg M, Laleman W, Shelest N, Biecker E, Schepke M, et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho‐kinase and activation of endothelial nitric oxide synthase. Hepatology 2007;46:242‐253. [DOI] [PubMed] [Google Scholar]
- 33. Vilaseca M, García‐Calderó H, Lafoz E, Ruart M, López‐Sanjurjo CI, Murphy MP, et al. Mitochondria‐targeted antioxidant mitoquinone deactivates human and rat hepatic stellate cells and reduces portal hypertension in cirrhotic rats. Liver Int 2017;37:1002‐1012. [DOI] [PubMed] [Google Scholar]
- 34. Steib CJ, Hennenberg M, Beitinger F, Hartmann AC, Bystron M, De Toni EN, et al. Amiloride reduces portal hypertension in rat liver cirrhosis. Gut 2010;59:827‐836. [DOI] [PubMed] [Google Scholar]
- 35. Abraldes JG, Rodriguez‐Vilarrupla A, Graupera M, Zafra C, Garcia‐Caldero H, Garcia‐Pagan JC, et al. Simvastatin treatment improves liver sinusoidal endothelial dysfunction in CCl4 cirrhotic rats. J Hepatol 2007;46:1040‐1046. [DOI] [PubMed] [Google Scholar]
- 36. Canbay A, Feldstein A, Baskin‐Bey E, Bronk SF, Gores GJ. The caspase inhibitor IDN‐6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther 2004;308:1191‐1196. [DOI] [PubMed] [Google Scholar]
- 37. Roychowdhury S, Chiang DJ, Mandal P, McMullen MR, Liu X, Cohen JI, et al. Inhibition of apoptosis protects mice from ethanol‐mediated acceleration of early markers of CCl4‐induced fibrosis but not steatosis or inflammation. Alcohol Clin Exp Res 2012;36:1139‐1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Witek RP, Stone WC, Karaca FG, Syn WK, Pereira TA, Agboola KM, et al. Pan‐caspase inhibitor VX‐166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 2009;50:1421‐1430. [DOI] [PubMed] [Google Scholar]
- 39. Elbekai RH, Paranjpe MG, Contreras PC, Spada A. Carcinogenicity assessment of the pan‐caspase inhibitor, emricasan, in Tg.rasH2 mice. Regul Toxicol Pharmacol 2015;72:169‐178. [DOI] [PubMed] [Google Scholar]
- 40. Yuana Y, Sturk A, Nieuwland R. Extracellular vesicles in physiological and pathological conditions. Blood Rev 2013;27:31‐39. [DOI] [PubMed] [Google Scholar]
- 41. Deaciuc IV, D’Souza NB, De Villiers WJS, Burikhanov R, Sarphie TG, Hill DB, et al. Inhibition of caspases in vivo protects the rat liver against alcohol‐induced sensitization to bacterial lipopolysaccharide. Alcohol Clin Exp Res 2001;25:935‐943. [PubMed] [Google Scholar]
- 42. Kopec AK, Spada AP, Contreras PC, Mackman N, Luyendyk JP. Caspase inhibition reduces hepatic tissue factor‐driven coagulation in vitro and in vivo. Toxicol Sci 2018;162:396‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tripodi A. Hemostasis in acute and chronic liver disease. Semin Liver Dis 2017;37:28‐32. [DOI] [PubMed] [Google Scholar]
- 44. Vilaseca M, Garcia‐Caldero H, Lafoz E, Garcia‐Irigoyen O, Avila M, Reverter JC, et al. The anticoagulant Rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology 2017;65:2031‐2044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials