

Abstract
Chronic hepatitis C virus (HCV) infection often leads to end‐stage liver disease, including hepatocellular carcinoma (HCC). We have previously observed reduced expression of microRNA‐30e (miR‐30e) in the liver tissues and sera of patients with HCV‐associated HCC, although biological functions remain unknown. In this study, we demonstrated that HCV infection of hepatocytes transcriptionally reduces miR‐30e expression by modulating CCAAT/enhancer binding protein β. In silico prediction suggests that autophagy‐related gene 5 (ATG5) is a direct target of miR‐30e. ATG5 is involved in autophagy biogenesis, and HCV infection in hepatocytes induces autophagy. We showed the presence of ATG5 in the miR‐30e–Argonaute 2 RNA‐induced silencing complex. Overexpression of miR‐30e in HCV‐infected hepatocytes inhibits autophagy activation. Subsequent studies suggested that ATG5 knockdown in Huh7.5 cells results in the remarkable inhibition of sterol regulatory element binding protein (SREBP)‐1c and fatty acid synthase (FASN) level. We also showed that overexpression of miR‐30e decreased lipid synthesis‐related protein SREBP‐1c and FASN in hepatocytes. Conclusion: We show new mechanistic insights into the interactions between autophagy and lipid synthesis through inhibition of miR‐30e in HCV‐infected hepatocytes.
Abbreviations
- Ago
Argonaute
- ATG5
autophagy related gene 5
- C/EBP‐β
CCAAT/enhancer binding protein β
- DAPI
4´,6‐diamidino‐2‐phenylindole
- FASN
fatty acid synthase
- GFP
green fluorescent protein
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- Ig
immunoglobulin
- LC3B
microtubule‐associated protein 1 light chain 3 beta
- LD
lipid droplet
- miR
microRNA
- mRNA
messenger RNA
- mTOR
mammalian target of rapamycin
- p‐mTOR
phosphorylated‐mammalian target of rapamycin
- RT‐qPCR
reverse‐transcription real‐time polymerase chain reaction
- siRNA
small interfering RNA
- SREBP‐1c
sterol regulatory element binding protein 1c
- UPR
unfolded protein response
- UTR
untranslated region
Chronic hepatitis C virus (HCV) infection is one of the etiologic causes of developing cirrhosis and eventually hepatocellular carcinoma (HCC).1 Approximately 55% of patients infected with HCV develop steatosis because HCV uses host lipid metabolism for its life cycle.2 Abnormal lipid metabolism influences metabolic disorders, cirrhosis, and liver carcinogenesis.3, 4, 5 Histologically, HCV infection induces lipid droplet (LD) formation in hepatocytes.6 We and others have shown that HCV infection induces autophagy7, 8 and modulates signaling pathways involved in lipogenesis, resulting in the accumulation of LDs,9, 10 although the precise mechanism is not fully understood.
MicroRNAs (miRNAs) are small noncoding RNAs that modulate the translation and stability of messenger RNAs (mRNAs) through a mechanism involving specific binding to mRNA based on base pair complementarity, controlling protein production. Most human miRNAs are processed into mature 20‐26 nucleotide duplexes through endonucleolytic cleavage by ribonuclease (RNase) III enzymes and Argonaute (Ago) proteins. Dicer and Ago cooperate to form the RNA‐induced silencing complex (RISC), consisting of Ago‐bound single‐strand miRNAs, which target host RNAs and induce a combination of translational repression.11 miRNAs have a widespread impact on the etiologies of disease in viral infections12 either directly or indirectly through regulation of virus‐associated host pathways.11 Moreover, several miRNAs interact directly with host lipid metabolism.13, 14
In the present study, we observed that miR‐30e expression is inhibited at the transcriptional level following HCV infection by down‐regulating the transcription factor CCAAT/enhancer binding protein β (C/EBP‐β). miR‐30e targets autophagy‐related gene 5 (ATG5), a key molecule of autophagy signaling, and inhibits de novo lipogenesis‐related protein, implicating its role in HCV‐mediated pathogenesis.
Materials and Methods
Cell Culture, HCV Infection, and Transfection
Huh7.5 cells and Huh7.5 cells harboring the HCV genotype 2a genome‐length replicon (Rep2a cells; kindly provided by Hengli Tang, Florida State University, FL, USA) were used. Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10 % fetal bovine serum and penicillin–streptomycin at 37°C in a 5% CO2 atmosphere.
HCV genotype 2a (clone JFH1) was grown in Huh7.5 cells as described.15 Virus released into cell culture supernatant was filtered through a 0.45‐μm pore cellulose acetate membrane and quantitated in standard IU/mL. For infection, cells were incubated with HCV genotype 2a (clone JFH1) (multiplicity of infection, 0.1) in a minimum volume of medium as described.16 The cellular RNA was extracted 3 days postinfection.
Transfection of ATG5 small interfering RNA (siRNA) into Huh7.5 cells was performed using lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA). Briefly, Huh7.5 cells were plated at a density of approximately 1 × 105 cells/well in a 12‐well plate and transfected with 50 nM ATG5 siRNA (sc‐41445; Santa Cruz, Dallas, TX) or control siRNA and lysed for western blot analysis at 48 hours after transfection. Transfection of an miR‐30e‐5p mimic (002223; ThermoFisher Scientific, Waltham, MA) into Huh7.5 cells was performed using lipofectamine (Invitrogen). Briefly, Huh7.5 cells were plated at a density of approximately 1 × 105 cells/well in a 12‐well plate and transfected with 20 nM of miR‐30e‐5p mimic or control miR. Cells were lysed for western blot analysis 48 hours after transfection. Total RNA was prepared in a separate transfection.
RNA Quantitation and Reverse‐Transcription Real‐Time Polymerase Chain Reaction
Total RNA was isolated by using a TRIzol reagent (Invitrogen). RNA was quantified by using a NanoDrop ND‐1000 spectrophotometer (Thermo Fisher Scientific). Complementary DNA was synthesized using miR‐30e‐ or a U6‐specific primer (Thermo Fisher Scientific) with a TaqMan miRNA reverse transcription (RT) kit or random hexamers and a Superscript III RT kit (Invitrogen). Real‐time polymerase chain reaction (qPCR) was performed with a 7500 real‐time PCR system (Applied Biosystems, Foster City, CA). TaqMan universal PCR master mix and a 6‐carboxyfluorescein (FAM)‐minor groove binder (MGB) probe for ATG5 (Hs00169468_m1; Thermo Fisher Scientific) and 18S ribosomal RNA (rRNA; Hs03928985_g1; Thermo Fisher Scientific) were used. Relative expression level was calculated by normalizing with U6 or 18S rRNA, using the 2−ΔΔCT formula (ΔΔCT = ΔCT of the sample − ΔCT of the untreated control).
Western Blot Analysis
Cells were lysed by using a sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) sample loading buffer. The lysates were subjected to PAGE and transferred onto a nitrocellulose membrane. The membranes were blocked with 5% nonfat dried milk and probed with the following specific primary antibodies: C/EBP‐β, sterol regulatory element binding protein (SREBP)‐1 (2A4), and fatty acid synthase (FASN) (A‐5; Santa Cruz); ATG5 (Proteintech, IL); and mammalian target of rapamycin (mTOR; 7C10) and phosphorylated‐mTOR (p‐mTOR; D9C2) (Cell Signaling Technology, Danvers, MA). After washing, the blots were incubated with secondary antibody for 1 hour. Proteins were detected by using an enhanced chemiluminescence western blot substrate (Pierce; ThermoFisher Scientific). Membranes were reprobed with antibody to actin (Santa Cruz) as an internal control for normalization of the protein load. ImageJ software (National Institutes of Health [NIH]) was used for densitometric scanning of western blot images.
Luciferase Reporter Assays
The miR‐30e promoter (nucleotides −1813 to +1; P0) or promoter fragment of the deletion mutant of miR‐30e (P1) were PCR amplified from Huh7.5 genomic DNA by using specific primers (Table 1). The fragments were digested with MluI and HindIII and cloned into the pGL3‐Basic luciferase vector (Promega, Madison, WI). Huh7.5 cells and Rep2a cells were transfected with a luciferase reporter plasmid containing the miR‐30e promoter (1 µg/mL) by using Lipofectamine 3000 (Invitrogen). Luciferase activity was determined as described.17
Table 1.
Primer Sequences Used in This Study
| Primers | Sequence |
|---|---|
| miR‐30e‐full promoter_For | 5′‐ATTAGCCTTCTGACGCGTGTGTTCTCTTAC‐3′ |
| miR‐30e‐F1 promoter_For | 5′‐ATTCAATACGCGTGAACCAAAGG‐3′ |
| miR‐30e promoter_Rev | 5′‐TGACAGAAGCTTGCTAATCC‐3′ |
Huh7.5 cells were plated at a density of 0.8 × 105 cells/well in a 24‐well plate and cotransfected with 25 nM C/EBP‐β siRNA (Santa Cruz) or control siRNA and the promoter construct of miR‐30e using Endofectin (GeneCopoeia, Rockville, MD). A luciferase reporter assay was performed 48 hours after transfection.
Ago2‐RNA Co‐Immunoprecipitation
The hepatocyte RNA–miR‐30e complex was precipitated as described.18 Briefly, miR‐30e transfected Huh7.5 cells were lysed with lysis buffer (150 mM KCl, 25 mM Tris‐HCl [pH 7.4], 5 mM ethylene diamine tetraacetic acid, 1% Triton X‐100, 5 mM dithiothreitol, protease inhibitor mixture, and 100 U/mL RNase‐OUT [Invitrogen]). Lysates were clarified, incubated with an anti‐Ago2 monoclonal antibody (11A9; Sigma‐Aldrich, St. Louis, MO) or isotype control immunoglobulin (Ig)G2a at 4°C for 7 hours, and mixed with protein G‐Sepharose (GE Healthcare, Piscataway, NJ) overnight. The beads were washed 4 times, and RNA was isolated by using TRIzol reagent (Invitrogen). Target RNA was quantitated by RT‐qPCR using specific primers.
Immunofluorescence
Huh7.5 cells were transfected with miR‐30e mimic and infected with green fluorescent protein (GFP)‐tagged HCV. Cells were fixed for 30 minutes with 4% paraformaldehyde after 72 hours of infection and then treated with 0.2% Triton X‐100 for 5 minutes. After blocking with 3% bovine serum albumin, cells were incubated with primary antibody rabbit polyclonal microtubule‐associated protein light chain 3B (LC3B; Sigma‐Aldrich) and then conjugated to Alexa Fluor 594 (Molecular Probes, Eugene, OR) with anti‐rabbit Ig. 4´,6‐Diamidino‐2‐phenylindole (DAPI) was incubated for nuclear staining. Three‐channel optical images (green for HCV, red for LC3, and blue for nucleus) were collected using the sequential scanning mode of the Olympus FV1000 confocal system (Olympus, Center Valley, PA). Images were superimposed digitally for fine comparisons.
Huh7.5 cells were transfected with ATG5 siRNA (sc‐41445; Santa Cruz) or control siRNA for 48 hours. Cells were stained by boron‐dipyrromethene (BODIPY) 493/503 (Invitrogen) for 1 hour, and DAPI was incubated for nuclear staining. Lipid accumulation was observed by using the Olympus FV1000 confocal system (Olympus) in cells with DAPI‐stained nuclei. Images were superimposed digitally for fine comparisons. ImageJ software (NIH) was used for densitometric scanning of immunofluorescence images.
Statistical Analysis
Results are presented as mean ± SD. Data were analyzed by the Student t test with a two‐tailed distribution. P < 0.05 was considered statistically significant.
Results
HCV Infection Down‐Regulates miR‐30e Expression
We have previously shown that miR‐30e expression was significantly lower (P < 0.003) in the sera of patients with HCV‐infected HCC compared with healthy volunteers.19 We also observed down‐regulation of miR‐30e in liver samples from patients with HCV‐associated HCC, although the mechanism of miR‐30e down‐regulation remains unknown. We examined the expression level of miR‐30e in HCV‐infected human hepatocytes. The level of miR‐30e was significantly reduced following HCV infection in Huh7.5 cells (Fig. 1, panel A). HCV RNA was measured from HCV‐infected Huh7.5 cells by RT‐qPCR (Fig. 1, panel B). A similar result was obtained when we examined the level of miR‐30e in Huh7.5 cells harboring the genome‐length HCV replicon (Rep2a cells) compared to parental Huh7.5 cells (Fig. 1, panel C). Together, our results indicate that HCV infection suppresses miR‐30e expression in hepatocytes.
Figure 1.
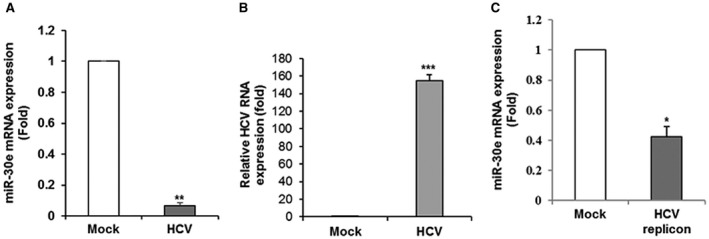
HCV inhibits miR‐30e expression in hepatocyte and replicon (genotype 2a) cells. (A) Huh7.5 cells were left uninfected (mock) or infected with JFH1 for 48 hours. Expression of the miR‐30e gene was examined by RT‐qPCR. U6 RNA was used as an internal control. miR‐30e was down‐regulated in JFH1‐infected Huh7.5 cells. (B) Expression of HCV RNA from virus‐infected Huh7.5 cells was measured by RT‐qPCR. (C) miR‐30e was down‐regulated in Huh7.5 cells harboring the HCV genome‐length replicon cells. Values represent data from three independent experiments, mean ± SD. Statistical significance was analyzed using the two‐tailed Student t test; *P < 0.05, **P < 0.01, ***P < 0.001.
HCV Infection Down‐Regulates C/EBP‐β‐Mediated miR‐30e Promoter Activation
We next examined how HCV modulates miR‐30e expression. For this, an miR‐30e promoter sequence (nucleotides −1,813 to +1; P0) was cloned into pGL3 luciferase reporter plasmid DNA, using specific primers (Table 1). Huh7.5 cells and Huh7.5 cells harboring the HCV genome‐length replicon cells were transfected with the P0 construct to examine promoter activity. miR‐30e promoter activity was significantly down‐regulated in HCV replicon‐expressing cells compared to control parental cells (Fig. 2, panel A). To determine the cis‐regulatory elements present in the miR‐30e promoter region (nucleotides −1,813 to +1), we searched PROMO, using version 8.3 of TRANSFAC. Several regulatory elements were found in the designated consensus sequence of the miR‐30e promoter, including binding sites for C/EBP‐β. C/EBP‐β, which belongs to the leucine zipper family, is an important transcriptional regulator.20 Moreover, we have previously shown that HCV down‐regulates promoter activity of complement C3 and miR‐181c by C/EBP‐β.21, 22
Figure 2.
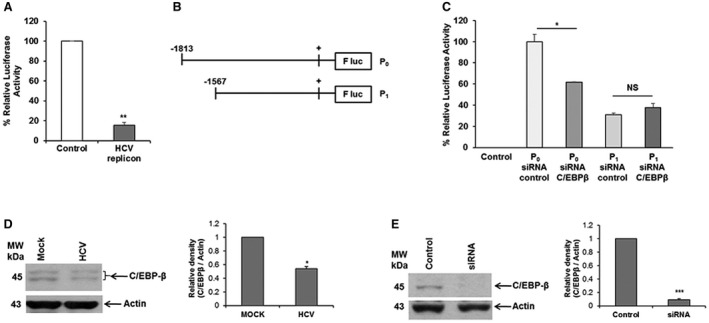
HCV transcriptionally suppresses miR‐30e promoter activity. (A) Huh7.5 cells (control) and Huh7.5 cells harboring the HCV genome‐length replicon (HCV replicon) were transfected with the miR‐30e‐luciferase reporter construct (P0), and promoter activity was measured by a relative luciferase assay after 48 hours of transfection. Data are presented as mean ± SD from at least three independent experiments. (B) Schematic diagram of the miR‐30e promoter region (nucleotides −1813 to +1; P0) cloned into the luciferase reporter plasmid (P0) and a deletion mutant of the miR‐30e promoter construct (nucleotides −1567 to +1 [P1]). (C) Huh7.5 cells transfected with the miR‐30e‐luciferase reporter constructs (P0 or P1) knocked down C/EBP‐β using a specific siRNA, and promoter activity was measured by a relative luciferase assay after 48 hours of transfection. Data are presented as mean ± SD from three independent experiments. (D) Huh7.5 cells were left uninfected (mock) or infected with HCV for 48 hours. Cell lysates were analyzed for C/EBP‐β expression by western blot using specific antibodies. The blot was reprobed with antibody to actin for protein load. Densitometric scanning results are presented. (E) Huh7.5 cells transfected to control or siRNA to C/EBP‐β. Cell lysates were prepared 48 hours after transfection and analyzed for C/EBP‐β expression by western blot using specific antibodies. The blot was reprobed with antibody to actin for protein load. Densitometric scanning results are presented. Statistical significance was analyzed using the two‐tailed Student t test; *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: F luc, firefly luciferase; NS, not significant.
We next identified the specific region on the miR‐30e promoter responsible for HCV‐mediated repression. We generated deletion mutants of the miR‐30e promoter constructs (nucleotides −1567 to +1 [P1]) and cloned these into pGL3 luciferase reporter plasmid DNA (Fig. 2, panel B). We knocked down C/EBP‐β using a specific siRNA and examined the activity of the P0 or P1 construct of the miR‐30e promoter in Huh7.5 cells to analyze the role of C/EBP‐β in the regulation of HCV‐mediated miR‐30e expression. Knockdown of C/EBP‐β inhibited the activity of the miR‐30e P0 promoter, but the P1 fragment did not exert an effect (Fig. 2, panel C), suggesting that the trans‐suppressor element may be located between regions –1,813 to –1,565. We further confirmed that HCV infection in Huh7.5 cells reduces C/EBP‐β expression (Fig. 2, panel D), in agreement with previous reports.21, 22 Transfection of siRNA to C/EBP‐β in Huh7.5 cells reduced protein expression (Fig. 2, panel E). Together, these results suggest that HCV‐mediated C/EBP‐β suppression is an important mechanism for miR‐30e down‐regulation.
ATG5 IS a Direct Target of miR‐30e
In silico prediction suggests that ATG5 is a direct target of miR‐30e (Fig. 3, panel A). A recent report showed that miR‐30e targets the 3′‐untranslated region (UTR) of ATG5 and inhibits its translation in gastric cancer cells.23 We examined the interaction between miR‐30e and ATG5 in hepatocytes. For this, miR‐30e‐transfected Huh7.5 cell lysates were immunoprecipitated with an Ago2‐specific monoclonal antibody or an unrelated isotype control. RNA was isolated from the immunoprecipitated lysates and analyzed by RT‐qPCR. Higher ATG5 level was noted in the miR‐30e–Ago2 complex compared to immunoprecipitated control, suggesting its presence in their complex (Fig. 3, panel B). Overexpression of miR‐30e inhibits ATG5 expression (Fig. 3, panel C). siRNA to ATG5 was used as a positive control. As reported,23 we did not observe the inhibition of ATG5 mRNA by miR‐30e in our experimental system. We and others have shown that HCV induces autophagy.17, 24 ATG5 is an important molecule in the autophagy signaling pathway. We therefore predicted that overexpression of miR‐30e in HCV‐infected hepatocytes will reduce ATG5 expression and may impair the induction of autophagy. For this, we overexpressed miR‐30e in GFP‐tagged HCV‐infected cells and stained these for LC3, a marker for autophagy. Our results demonstrated that overexpression of miR‐30e inhibited HCV‐induced LC3 lipidation compared to control miR‐treated cells (Fig. 3, panel D). We also observed that overexpression of miR‐30e inhibited HCV‐induced LC3 lipidation by western blot analysis (Fig. 3, panel E).
Figure 3.
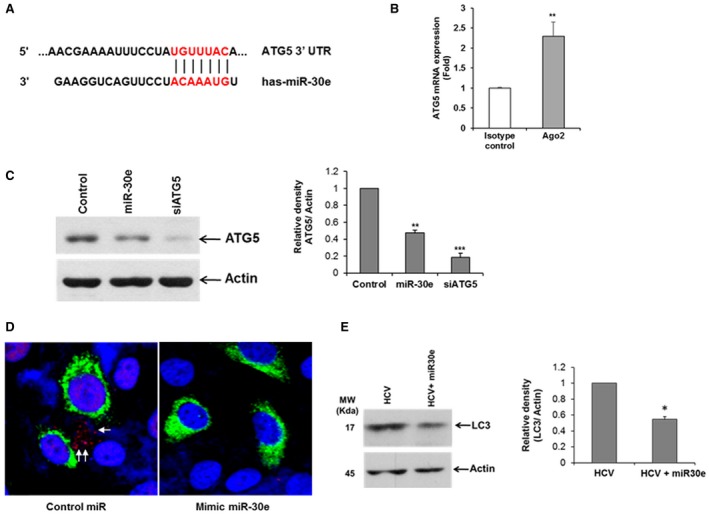
miR‐30e targets the ATG5 3′ UTR. (A) Schematic representation of miR‐30e‐predicted binding sites in the 3′ UTR of ATG5 mRNAs. (B) Huh7.5 cells transfected with miR‐30e mimic cell lysates were immunoprecipitated with Ago2‐specific monoclonal antibody or an unrelated IgG2a isotype control. RNA was isolated from immunoprecipitates, and ATG5 expression was analyzed by RT‐qPCR. Statistical significance was analyzed using the two‐tailed Student t test; **P < 0.01. (C) Huh7.5 cells were transfected to control, mimic miR‐30e, or siRNA to ATG5. Cell lysates were prepared 48 hours after transfection and analyzed for ATG5 expression by western blot using specific antibodies. The blot was reprobed with antibody to actin for protein load. Densitometric scanning results are presented. Statistical significance was analyzed using the two‐tailed Student t test; *P < 0.05, **P < 0.01, ***P < 0.001. (D) Hepatocytes were infected with GFP‐HCV, and transfected miR‐30e or control miR was used as the negative control. Cells were fixed and stained with LC3 antibody 72 hours after infection. Confocal microscopy examination for subcellular localization of LC3 visualized by LC3 (red), GFP‐HCV (green), and nucleus (DAPI) in merged image panels. Representative image (60×) is shown, and punctate localization of LC3 on autophagic vacuoles (arrows) was clearly visible in HCV‐infected hepatocytes. (E) HCV‐infected Huh7.5 cells were treated with miR‐30e, and cell lysates were subjected to western blot analysis using the LC3B‐specific antibody. The blot was reprobed with antibody to actin for normalization. Densitometric scanning results are presented. Statistical significance was analyzed using the two‐tailed Student t test; *P < 0.05.
Depletion of ATG5 Inhibits Lipid Synthesis in Hepatocytes
LDs have been implicated as an essential link for efficient autophagy in the yeast system,25 and autophagy is inhibited following depletion of free fatty acids. In HCV infection, both autophagy and LDs are induced in hepatocytes. Because ATG5 is a direct target of miR‐30e, we wanted to understand the interplay between ATG5 and lipid synthesis. ATG5 and ATG7 are essential molecules for the induction of autophagy; ATG7 conjugates ATG5 to ATG12 for autophagosome formation.26 ATG7‐deficient mice suppress LD formation,27 and SREBP‐1c forms LDs through de novo lipogenesis.28 To investigate the relationship between ATG5 and SREBP‐1c expression, we knocked down ATG5 using a specific siRNA and performed western blot analysis using specific antibodies. A significant down‐regulation of SREBP‐1c and FASN expression level was observed compared to that of the control cell after normalization with actin (Fig. 4, panel A). A similar result was obtained when we examined the expression of SREBP‐1c and FASN in Huh7.5 cells harboring the genome‐length HCV replicon (Rep2a cells) compared to parental cells (Fig. 4, panel B). We also observed a significant down‐regulation of p‐mTOR, an intermediate between ATG5 and SREBP‐1c expression level, compared to that of the control cell (Fig. 4, panel C). We further showed an inhibition of LDs in ATG5‐depleted cells (Fig. 4, panel D). Together, these data suggest that depletion of ATG5 inhibits SREBP‐1c and FASN expression.
Figure 4.
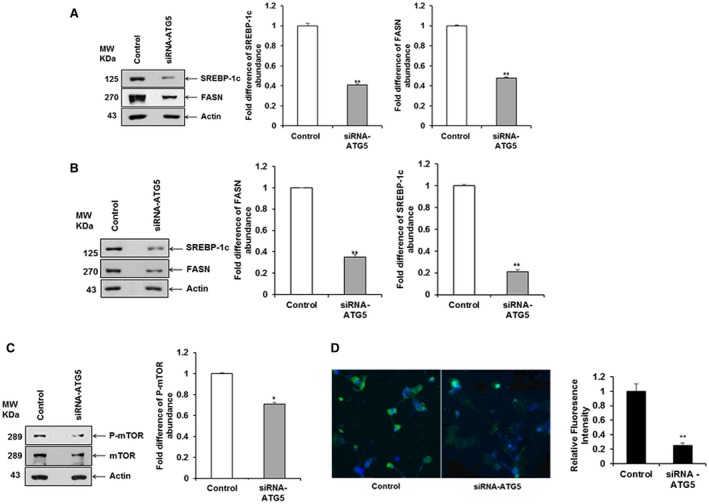
Knockdown of ATG5 inhibits SREBP‐1c and FASN in hepatocytes. (A) Huh7.5 cells were transfected with siRNA to ATG5 (siRNA–ATG5) or siRNA negative control, and cell lysates were prepared after 48 hour post‐transfection. SREBP‐1c and FASN were detected by western blot analysis using specific antibodies. The blot was reprobed with antibody to actin for loading control. (B) Huh7.5 cells harboring the HCV genome‐length replicon cells were transfected for 48 hours with siRNA–ATG5 or siRNA negative control, and cell lysates were prepared. SREBP‐1c and FASN were down‐regulated in siRNA–ATG5‐transfected hepatocytes. (C) A significant down‐regulation of p‐mTOR expression level in Huh7.5 cells transfected with siRNA–ATG5 compared to that of the control cells was observed after normalization with actin. A densitometry scan is shown on the right in all panels. (D) Hepatocytes were transfected for 48 hours with siRNA–ATG5 or siRNA negative control, and cells were stained 48 hours after transfection. Confocal microscopy examination for LDs visualized by LDs (BODIPY, green) and nucleus (DAPI, blue) in merged image panels (20×). A representative image is shown. Quantitation of the BODIPY staining is shown in the bar graph on the right. Statistical significance was analyzed using the two‐tailed Student t test; *P < 0.05, **P < 0.01. Abbreviation: BODIPY, boron‐dipyrromethene.
Overexpression of miR‐30e Inhibits Lipid Synthesis in Hepatocytes
SREBP‐1 and FASN are important players in lipid metabolism, and SREBPs are key transcriptional factors in lipogenesis and lipid uptake.29, 30 We observed that HCV infection in Huh7.5 cells increased expression of SREBP‐1c and its downstream target FASN (Fig. 5, panel A). We also observed ATG5 depletion inhibits SREBP‐1c and FASN expression. We therefore examined whether miR‐30e overexpression modulates lipid synthesis in hepatocytes. For this, we performed western blot analysis in Huh7 cells transfected with an miR‐30e mimic or control miR. A significant down‐regulation of SREBP‐1c and FASN expression level compared to that of the control parental cell was observed after normalization with the housekeeping protein actin (Fig. 5, panel B). A similar result was obtained when we examined the expression of SREBP‐1c and FASN in Huh7.5 cells harboring the genome‐length HCV replicon (Rep2a cells) compared to parental cells (Fig. 5, panel C). Together, our data suggest that mir‐30e inhibits ATG5 and eventually SREBP‐1c and FASN in hepatocytes.
Figure 5.
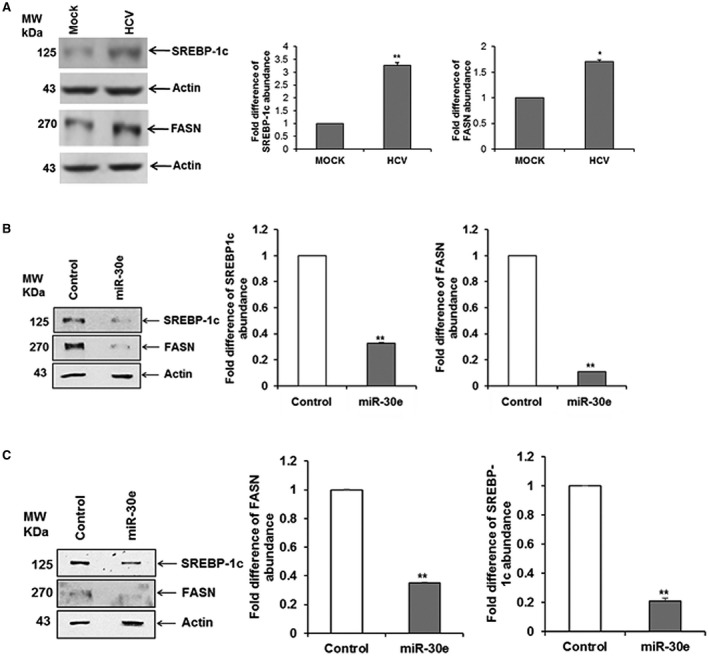
miR‐30e inhibits SREBP‐1c and FASN in hepatocyte and replicon (genotype 2a) cells. (A) Huh7.5 cells were left uninfected (mock) or infected with HCV, and cell lysates were analyzed for SREBP‐1c and FASN expression by western blot using specific antibodies. The blots were reprobed with antibody to actin for protein load. (B) Huh7.5 cells were transfected with miR‐30e mimic or miRNA mimic negative control, and cell lysates were prepared after 48 hour post‐transfection. SREBP‐1c and FASN were detected by western blot analysis using specific antibodies. The blot was reprobed with antibody to actin for loading control. (C) SREBP‐1c and FASN were down‐regulated in Huh7.5 cells harboring the HCV genome‐length replicon cells. A densitometry scan is shown on the right in all panels. Statistical significance was analyzed using the two‐tailed Student t test; *P < 0.05, **P < 0.01.
Discussion
In the present study, we demonstrated that (i) HCV infection transcriptionally down‐regulates miR‐30e, (ii) down‐regulation of miR‐30e targets ATG5 and modulates induction of autophagy, and (iii) overexpression of miR‐30e or ATG5 knockdown inhibits de novo lipogenesis.
We previously showed that HCV infection increased lipid accumulation through metabolic gene regulation.9 Increased accumulation of LDs frequently causes liver steatosis in patients chronically infected with HCV.2, 31 In this report, we observed that HCV infection modulates lipogenesis processes primarily by regulating miR‐30e transcription. Overexpression of miR‐30e inhibits the expression of SREBP‐1c and FASN, which are known to be involved in de novo lipogenesis and an increased expression of LDs, promoting lipid accumulation. Together, these results suggest that HCV increases de novo lipogenesis in hepatocytes by inhibiting miR‐30e expression.
miRNAs impact the regulation of many cellular processes, and viruses modulate the host miRNA expression either directly or indirectly to accelerate pathogenesis.12, 32 In reaction to viral infection, signaling pathways in hepatocytes direct a variety of intracellular events that generate de novo lipogenesis in the infected cells.13, 14 In earlier reports, miR‐30e was implicated as a subtype‐specific prognostic marker in breast cancer and ovarian cancer33, 34 and is inhibited in chronic myeloid leukemia, lung cancer, and patients with HCC.19, 35, 36 miR‐30 has been implicated as a tumor suppressor and inhibits epithelial‐to‐mesenchymal transition in HCC through metastasis‐associated protein 1 modulation.37, 38 In cancer development, fatty acids are understood to be the most relevant because lipid metabolism participates in the regulation of many cellular processes, such as cell growth, proliferation, differentiation, and survival.39 Moreover, miR‐30e has been implicated in the regulation of fatty acid metabolism in goat mammary epithelial cells.40
We observed inhibition of miR‐30e expression by HCV infection or HCV replicon in human hepatocytes, suggesting direct evidence for an association with HCV infection. C/EBP‐β is an important transcriptional factor belonging to the leucine zipper family and is involved in several cellular functions, such as cell proliferation and differentiation.20 We previously showed that C/EBP‐β is down‐regulated following HCV infection.21 In our experiments, we demonstrated that HCV inhibits miR‐30e promoter activity by down‐regulating C/EBP‐β expression. Deletion of the C/EBP‐β binding site on an miR‐30e promoter (P1 fragment) or knockdown of C/EBP‐β showed suppression of miR‐30e promoter activity.
We demonstrated an interaction between miR‐30e and ATG5 in our complex by RISC assay; this concurs with a report that ATG5 is a direct target of miR‐30e.23 ATG5 and ATG7 are autophagy‐specific genes involved in the initiation of autophagosome formation and completion through a ubiquitin‐like conjugation system.41 Interestingly, ATG7‐deficient mice suppressed LD formation,27 and liver‐specific ATG5‐knockout mice displayed impaired LD biogenesis.42 HCV infection induces autophagy. We postulate that HCV‐mediated inhibition of miR‐30e facilitates ATG5 for autophagy induction following HCV infection. We showed previously that autophagy acts as an upstream positive regulator of mTOR complex 1 (mTORC1),17 which regulates SREBP‐1c at the transcriptional level43; however, further study is needed to dissect this mechanism. SREBP‐1c forms LDs through de novo lipogenesis.28 Furthermore, unfolded protein response (UPR) activates autophagy in HCV‐infected hepatocytes,44 and UPR activates SREBP maturation in cultured cells.45, 46 Moreover, the activation of multiple UPR responses and the SREBP pathway significantly overlapped.47 Processing of SREBP‐1c has been reported to depend on mTORC1.48 In our study, SREBP‐1c and FASN were inhibited by knockdown of ATG5 in hepatocytes. These results demonstrated that de novo lipogenesis is regulated through autophagy in hepatocytes following HCV infection. HCV causes steatosis, with the reported mean prevalence of HCV‐related NAFLD being 55% and nonalcoholic steatohepatitis being 4%‐10% of cases.49 We previously showed that HCV‐infected hepatocytes increased expression of SREBP‐1c and its downstream target FASN, both of which are involved in lipogenesis.9 We showed here HCV‐mediated down‐regulation of miR‐30e is responsible, in part, for up‐regulating SREBP‐1c and FASN expression.
We observed that expression levels of de novo lipogenesis‐related protein SREBP‐1c and FASN are significantly inhibited in miR‐30e‐overexpressed hepatocytes. miR‐30e also directly targets the 3′ UTR of ATG5. Moreover, knockdown of ATG5 expression negatively regulates SREBP‐1c and FASN protein expression level. In summary, we have demonstrated that HCV infection transcriptionally down‐regulates miR‐30e expression by targeting C/EBP‐β, which in turn enhances the expression of de novo lipogenesis‐related protein SREBP‐1c and FASN through autophagy. Our observations contribute to the understanding of the mechanistic role of miR‐30e in HCV–host interactions toward pathogenesis.
Supported by the National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK081817) and Saint Louis University Liver Center Seed Grants.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Goossens N, Hoshida Y. Hepatitis C virus‐induced hepatocellular carcinoma. Clin Mol Hepatol 2015;21:105‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Modaresi Esfeh J, Ansari‐Gilani K. Steatosis and hepatitis C. Gastroenterol Rep (Oxf) 2016;4:24‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Powell EE, Jonsson JR, Clouston AD. Steatosis: co‐factor in other liver diseases. Hepatology 2005;42:5‐13. [DOI] [PubMed] [Google Scholar]
- 4. Clark JM, Diehl AM. Hepatic steatosis and type 2 diabetes mellitus. Curr Diab Rep 2002;2:210‐215. [DOI] [PubMed] [Google Scholar]
- 5. Ibrahim AA, Abdel Aleem MH, Abdella HM, Helmy A. Study of the role of insulin resistance as a risk factor in HCV related hepatocellular carcinoma. J Egypt Soc Parasitol 2015;45:107‐113. [DOI] [PubMed] [Google Scholar]
- 6. Martins AS, Martins IC, Santos NC. Methods for lipid droplet biophysical characterization in Flaviviridae infections. Front Microbiol 2018;9:1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ait‐Goughoulte M, Kanda T, Meyer K, Ryerse JS, Ray RB, Ray R. Hepatitis C virus genotype 1a growth and induction of autophagy. J Virol 2008;82:2241‐2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang L, Ou JJ. Regulation of autophagy by hepatitis C virus for its replication. DNA Cell Biol 2018;37:287‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bose SK, Kim H, Meyer K, Wolins N, Davidson NO, Ray R. Forkhead box transcription factor regulation and lipid accumulation by hepatitis C virus. J Virol 2014;88:4195‐4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yim SA, Lim YS, Kim JW, Hwang SB. Nonstructural 5A protein of hepatitis C virus interacts with pyruvate carboxylase and modulates viral propagation. PLoS ONE 2013;8:e68170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singaravelu R, Russell RS, Tyrrell DL, Pezacki JP. Hepatitis C virus and microRNAs: miRed in a host of possibilities. Curr Opin Virol 2014;7:1‐10. [DOI] [PubMed] [Google Scholar]
- 12. Sarnow P, Jopling CL, Norman KL, Schütz S, Wehner KA. MicroRNAs: expression, avoidance and subversion by vertebrate viruses. Nat Rev Microbiol 2006;4:651‐659. [DOI] [PubMed] [Google Scholar]
- 13. Xu M, Zheng XM, Jiang F, Qiu WQ. MicroRNA‐190b regulates lipid metabolism and insulin sensitivity by targeting IGF‐1 and ADAMTS9 in non‐alcoholic fatty liver disease. J Cell Biochem 2018;119:5864‐5874. [DOI] [PubMed] [Google Scholar]
- 14. Tao YF, Qiang J, Bao JW, Li HX, Yin GJ, Xu P, et al. miR‐205‐5p negatively regulates hepatic acetyl‐CoA carboxylase β mRNA in lipid metabolism of Oreochromis niloticus. Gene 2018;660:1‐7. [DOI] [PubMed] [Google Scholar]
- 15. Kanda T, Basu A, Steele R, Wakita T, Ryerse JS, Ray R, et al. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J Virol 2006;80:4633‐4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. Knockdown of autophagy enhances the innate immune response in hepatitis C virus‐infected hepatocytes. Hepatology 2011;53:406‐414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shrivastava S, Bhanja Chowdhury J, Steele R, Ray R, Ray RB. Hepatitis C virus upregulates Beclin1 for induction of autophagy and activates mTOR signaling. J Virol 2012;86:8705‐8712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mukherjee A, Di Bisceglie AM, Ray RB. Hepatitis C virus‐mediated enhancement of microRNA miR‐373 impairs the JAK/STAT signaling pathway. J Virol 2015;89:3356‐3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhattacharya S, Steele R, Shrivastava S, Chakraborty S, Di Bisceglie AM, Ray RB. Serum miR‐30e and miR‐223 as novel noninvasive biomarkers for hepatocellular carcinoma. Am J Pathol 2016;186:242‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo L, Li X, Tang QQ. Transcriptional regulation of adipocyte differentiation: a central role for CCAAT/enhancer‐binding protein (C/EBP) β. J Biol Chem 2015;290:755‐761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mazumdar B, Kim H, Meyer K, Bose SK, Di Bisceglie AM, Ray RB, et al. Hepatitis C virus proteins inhibit C3 complement production. J Virol 2012;86:2221‐2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukherjee A, Shrivastava S, Bhanja Chowdhury J, Ray R, Ray RB. Transcriptional suppression of miR‐181c by hepatitis C virus enhances homeobox A1 expression. J Virol 2014;88:7929‐7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ye Y, Fang Y, Xu W, Wang Q, Zhou J, Lu R. 3,3′‐Diindolylmethane induces anti‐human gastric cancer cells by the miR‐30e‐ATG5 modulating autophagy. Biochem Pharmacol 2016;115:77‐84. [DOI] [PubMed] [Google Scholar]
- 24. Chan ST, Ou JJ. Hepatitis C virus‐induced autophagy and host innate immune response. Viruses 2017;9:pii:E224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shpilka T, Welter E, Borovsky N, Amar N, Mari M, Reggiori F, et al. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J 2015;34:2117‐2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banduseela VC, Chen YW, Kultima HG, Norman HS, Aare S, Radell P, et al. Impaired autophagy, chaperone expression, and protein synthesis in response to critical illness interventions in porcine skeletal muscle. Physiol Genomics 2013;45:477‐486. [DOI] [PubMed] [Google Scholar]
- 27. Shibata M, Yoshimura K, Furuya N, Koike M, Ueno T, Komatsu M, et al. The MAP1‐LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun 2009;382:419‐423. [DOI] [PubMed] [Google Scholar]
- 28. Go GW, Mani A. Low‐density lipoprotein receptor (LDLR) family orchestrates cholesterol homeostasis. Yale J Biol Med 2012;85:19‐28. [PMC free article] [PubMed] [Google Scholar]
- 29. Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP‐1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res 2012;10:133‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Menendez JA, Decker JP, Lupu R. In support of fatty acid synthase (FAS) as a metabolic oncogene: extracellular acidosis acts in an epigenetic fashion activating FAS gene expression in cancer cells. J Cell Biochem 2005;94:1‐4. [DOI] [PubMed] [Google Scholar]
- 31. Iqbal J, Sarkar‐Dutta M, McRae S, Ramachandran A, Kumar B, Waris G. Osteopontin regulates hepatitis C virus (HCV) replication and assembly by interacting with HCV proteins and lipid droplets and by binding to receptors αVβ3 and CD44. J. Virol 2018;92:pii:e02116‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Singaravelu R, Chen R, Lyn RK, Jones DM, O'Hara S, Rouleau Y, et al. Hepatitis C virus induced up‐regulation of microRNA‐27: a novel mechanism for hepatic steatosis. Hepatology 2014;59:98‐108. [DOI] [PubMed] [Google Scholar]
- 33. D'Aiuto F, Callari M, Dugo M, Merlino G, Musella V, Miodini P, et al. miR‐30e* is an independent subtype‐specific prognostic marker in breast cancer. Br J Cancer 2015;113:290‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y, Li L, Qu Z, Li R, Bi T, Jiang J, et al. The expression of miR‐30a* and miR‐30e* is associated with a dualistic model for grading ovarian papillary serious carcinoma. Int J Oncol 2014;44:1904‐1914. [DOI] [PubMed] [Google Scholar]
- 35. Hershkovitz‐Rokah O, Modai S, Pasmanik‐Chor M, Toren A, Shomron N, Raanani P, et al. MiR‐30e induces apoptosis and sensitizes K562 cells to imatinib treatment via regulation of the BCR‐ABL protein. Cancer Lett 2015;356:597‐605. [DOI] [PubMed] [Google Scholar]
- 36. Zhu WY, Luo B, An JY, He JY, Chen DD, Xu LY, et al. Differential expression of miR‐125a‐5p and let‐7e predicts the progression and prognosis of non‐small cell lung cancer. Cancer Invest 2014;32:394‐401. [DOI] [PubMed] [Google Scholar]
- 37. Mao J, Hu X, Pang P, Zhou B, Li D, Shan H. miR‐30e acts as a tumor suppressor in hepatocellular carcinoma partly via JAK1/STAT3 pathway. Oncol Rep 2017;38:393‐401. [DOI] [PubMed] [Google Scholar]
- 38. Deng L, Tang J, Yang H, Cheng C, Lu S, Jiang R, et al. MTA1 modulated by miR‐30e contributes to epithelial‐to‐mesenchymal transition in hepatocellular carcinoma through an ErbB2‐dependent pathway. Oncogene 2017;36:3976‐3985. [DOI] [PubMed] [Google Scholar]
- 39. Huang C, Freter C. Lipid metabolism, apoptosis and cancer therapy. Int J Mol Sci 2015;16:924‐949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen Z, Qiu H, Ma L, Luo J, Sun S, Kang K, et al. miR‐30e‐5p and miR‐15a synergistically regulate fatty acid metabolism in goat mammary epithelial cells via LRP6 and YAP1. Int J Mol Sci 2016;17:pii:E1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Luo S, Rubinsztein DC. Atg5 and Bcl‐2 provide novel insights into the interplay between apoptosis and autophagy. Cell Death Differ 2007;14:1247‐1250. [DOI] [PubMed] [Google Scholar]
- 42. Li Y, Chao X, Yang L, Lu Q, Li T, Ding WX, et al. Impaired fasting‐induced adaptive lipid droplet biogenesis in liver‐specific Atg5‐deficient mouse liver is mediated by persistent nuclear factor‐like 2 activation. Am J Pathol 2018;188:1833‐1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Norrmén C, Figlia G, Lebrun‐Julien F, Pereira JA, Trötzmüller M, Köfeler HC, et al. mTORC1 controls PNS myelination along the mTORC1‐RXRγ‐SREBP‐lipid biosynthesis axis in Schwann cells. Cell Rep 2014;9:646‐660. [DOI] [PubMed] [Google Scholar]
- 44. Dash S, Chava S, Aydin Y, Chandra PK, Ferraris P, Chen W, et al. Hepatitis C virus infection induces autophagy as a prosurvival mechanism to alleviate hepatic ER‐stress response. Viruses 2016;8:pii:E150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jeon TI, Osborne TF. SREBPs: metabolic integrators in physiology and metabolism. Trends Endocrinol Metab 2012;23:65‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee JN, Ye J. Proteolytic activation of sterol regulatory element‐binding protein induced by cellular stress through depletion of Insig‐1. J Biol Chem 2004;279:45257‐45265. [DOI] [PubMed] [Google Scholar]
- 47. Osborne TF, Espenshade PJ. Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it's been. Genes Dev 2009;23:2578‐2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol 2015;16:678‐689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adinolfi LE, Rinaldi L, Guerrera B, Restivo L, Marrone A, Giordano M, et al. NAFLD and NASH in HCV infection: prevalence and significance in hepatic and extrahepatic manifestations. Int J Mol Sci 2016;17:pii:E803. [DOI] [PMC free article] [PubMed] [Google Scholar]