

Abstract
Hepatocyte estrogen receptor α (ERα) was recently recognized as a relevant molecular target for nonalcoholic fatty liver disease (NAFLD) prevention. The present study defined to what extent hepatocyte ERα could be involved in preserving metabolic homeostasis in response to a full (17β‐estradiol [E2]) or selective (selective estrogen receptor modulator [SERM]) activation. Ovariectomized mice harboring a hepatocyte‐specific ERα deletion (LERKO mice) and their wild‐type (WT) littermates were fed a high‐fat diet (HFD) and concomitantly treated with E2, tamoxifen (TAM; the most used SERM), or vehicle. As expected, both E2 and TAM prevented all HFD‐induced metabolic disorders in WT mice, and their protective effects against steatosis were abolished in LERKO mice. However, while E2 still prevented obesity and glucose intolerance in LERKO mice, hepatocyte ERα deletion also abrogated TAM‐mediated control of food intake as well as its beneficial actions on adiposity, insulin sensitivity, and glucose homeostasis, suggesting a whole‐body protective role for liver‐derived circulating factors. Moreover, unlike E2, TAM induced a rise in plasma concentration of the anorectic hepatokine growth differentiation factor 15 (Gdf15) through a transcriptional mechanism dependent on hepatocyte ERα activation. Accordingly, ERα was associated with specific binding sites in the Gdf15 regulatory region in hepatocytes from TAM‐treated mice but not under E2 treatment due to specific epigenetic modifications. Finally, all the protective effects of TAM were abolished in HFD‐fed GDF15‐knockout mice. Conclusion: We identified the selective modulation of hepatocyte ERα as a pharmacologic strategy to induce sufficient anorectic hepatokine Gdf15 to prevent experimental obesity, type 2 diabetes, and NAFLD.
Abbreviations
- AF
activation function
- Angptl
angiopoietin‐like
- BS
binding site
- CBP
cyclic adenosine monophosphate response element‐binding protein
- ChIP
chromatin immunoprecipitation
- CoA
coenzyme A
- Cyp17a1
cytochrome P450, family 17, subfamily a, polypeptide 1
- Dgat2
diacylglycerol O‐acyltransferase 2
- E2
17β‐estradiol
- Enho
energy homeostasis associated
- ERα
estrogen receptor α
- Fgf21
fibroblast growth factor 21
- Gck
glucokinase
- Gdf15
growth differentiation factor 15
- GDF15KO
growth differentiation factor 15‐deficient
- H&E
hematoxylin and eosin
- H3K27ac
twenty‐seventh amino acid in histone H3, acetylated
- HDL
high‐density lipoprotein
- HFD
high‐fat diet
- Igf
insulin‐like growth factor
- KO
knockout
- LDL
low‐density lipoprotein
- LERKO
mice with hepatocyte‐restricted ERα deletion
- mRNA
messenger RNA
- NAFLD
nonalcoholic fatty liver disease
- Nr1h
nuclear receptor subfamily 1, group H
- Pltp
phospholipid transfer protein
- Ppar
peroxisome proliferator activator receptor
- SERM
selective estrogen receptor modulator
- Srebf1
sterol regulatory element binding transcription factor 1
- TAM
tamoxifen
- VEH
vehicle
- WT
wild type
As a key regulator of energy and glucose homeostasis, estrogen receptor α (ERα) is now considered a relevant target to develop new therapeutic approaches for obesity‐related metabolic disorders, such as type 2 diabetes and nonalcoholic fatty liver disease (NAFLD).1 Understanding the mechanisms involved in ERα‐mediated metabolic protection thus remains a crucial challenge to optimize pharmacologic strategies for ERα selective modulation.2
Interestingly, recent studies highlighted the critical role of hepatic ERα signaling in the protective effects conferred by estrogens against high‐fat diet (HFD)‐induced steatosis and insulin resistance. Indeed, silencing ERα expression in the liver using adenoviral short hairpin RNA has been shown to markedly increase hepatic triglyceride accumulation in HFD‐fed C57BL/6 female mice, while overexpression of human ERα reduced the level of steatosis in obese ob/ob female mice.3 Zhu et al.4 then reported that both female and male mice with hepatocyte‐restricted ERα deletion (LERKO) are prone to steatosis and insulin resistance in response to HFD feeding. Noteworthy, 17β‐estradiol (E2) administration failed to preserve ovariectomized LERKO female mice from fatty liver and insulin resistance, although this treatment still exerted a significant protection against adipose tissue accumulation and glucose intolerance.4
As a member of the nuclear receptor superfamily, ERα acts as a transcription factor that regulates gene transcription through two activation functions (AFs), namely ERα‐AF1 and ERα‐AF2.5 ERα‐AF1 and ERα‐AF2 are both fully activated by E2, but their respective roles in gene expression regulation depend on ligands and cell types. Using mouse models with a selective ERα‐AF1 or ERα‐AF2 deletion, we demonstrated that the prevention of HFD‐induced obesity and insulin resistance by E2 required ERα‐AF2 activation, whereas ERα‐AF1 appeared dispensable.6 Besides natural estrogens, several drugs, known as selective estrogen receptor modulators (SERMs), acting as agonists or antagonists in a tissue‐specific manner have been developed for clinical use. Nevertheless, their abilities to activate or repress ERα‐AF1 or ERα‐AF2 remain largely unknown, except for tamoxifen (TAM).5 Indeed, ERα‐AF2 is the direct target of TAM antagonism, whereas its agonistic activity, which depends on both cell types and target genes, results from ERα‐AF1 activation.7
We recently showed that chronic TAM administration prevents HFD‐induced obesity, steatosis, insulin resistance, and glucose intolerance in ovariectomized female C57BL/6 mice, along with a significant reduction in food intake.8 All the protective effects of TAM were abrogated in ERα–/– and ERα‐AF1–/– mice,8 thus attesting that TAM‐induced metabolic protection is specifically mediated by ERα‐AF1. However, the specific contribution of hepatocyte ERα to the protection conferred by SERMs, such as TAM, against HFD‐induced metabolic disorders still remain to be investigated.
The present study aimed at defining to what extent hepatocyte ERα could be involved in the prevention of steatosis and in the preservation of whole‐body metabolic homeostasis in response to a selective (SERM) mode of activation compared to a full (E2) activation. To this end, we combined the use of pharmacologic tools (E2 and TAM) and transgenic mouse models to demonstrate that, in contrast to the results obtained with E2, hepatocyte ERα not only mediates the protective effect of TAM against steatosis but is also absolutely and unexpectedly required to prevent HFD‐induced obesity and glucose intolerance. Noteworthy, this overall metabolic protective action conferred by the selective modulation of hepatocyte ERα requires the anorectic hepatokine growth differentiation factor 15 (Gdf15), secretion of which is sharply enhanced by TAM.
Materials and Methods
Animals, Treatment, and Diet
Female mice with a hepatocyte‐specific deletion of ERα (LERKO, n = 40)9 and Gdf15‐deficient mice (GDF15KO, n = 23)10 (see Supporting Methods) as well as their respective wild‐type (WT) littermates (LER +/+, n = 40; GDF15 +/+, n = 14) underwent bilateral ovariectomy at 4 weeks of age to standardize the exposure to ERα ligands. All mice were then subcutaneously implanted with pellets releasing either TAM (1.2 mg/kg/day), E2 (80 µg/kg/day), or vehicle (VEH) for either 6 (GDF15KO) or 12 weeks (LERKO) and concomitantly fed an HFD (energy content: 45% fat, 20% protein, and 35% carbohydrate; 3.7 kcal/g; Research Diets, New Brunswick, NJ).
In Vivo Experimental Procedures
Mice were anesthetized with an intraperitoneal injection of ketamine (10 mg/kg; Merial, Lyon, France) and xylazine (1 mg/kg; Sigma‐Aldrich, Isle d’Abeau Chesnes, France) for ovariectomy and with exposition to 2% isoflurane for pellet implantation. Food intake and body weight were recorded weekly. One week before being killed, body composition (whole body fat and lean masses) was analyzed by EchoMRI in live animals. Intraperitoneal glucose tolerance tests were performed on the following day in overnight‐fasted mice. Blood glucose concentrations were monitored with a glucose meter (Roche Diagnostic, Meylan, France) –30, 0, 30, 60, and 90 minutes after glucose injection (1 g/kg body weight). In some experiments, basal metabolism was evaluated by indirect calorimetry 1 week before being killed and after 24 hours of acclimatization in individual cages. Oxygen consumption (VO2), carbon dioxide production (VCO2), and food and water intake were measured (Phenomaster; TSE‐Systems) in individual mice at 10‐minute intervals during a 24‐hour period at constant temperature (20°C). We calculated the respiratory exchange ratio as VCO2 /VO2 and energy expenditure (kilocalories of heat produced). Ambulatory physical activity was monitored by an infrared photocell beam interruption method. All mice were killed at 11:00 am, after 3 hours of fasting with free access to water. Blood samples were collected from the retro‐orbital venous plexus and stored at –20°C. Mice were euthanized by cervical dislocation, and organs were carefully removed, weighed, frozen in liquid nitrogen, and stored at –80°C.
Liver Tissue Histology
Liver tissues were quickly excised, fixed in 10% buffered formalin, and embedded in paraffin, and 3‐µm sections were stained with hematoxylin and eosin (H&E). Additional fresh liver samples were immersed in Tissue‐Tek optimal cutting temperature compound (Sakura, Japan) and then frozen in isopentane cooled by liquid nitrogen; cryosections (7 µm) were then stained with Oil Red O to assess neutral lipid accumulation, as described with minor modifications.11 Images of each sample were obtained at original magnifications of ×150 and ×400 with an Eclipse Ci Nikon microscope and using a DS‐FI camera driven by NIS‐AR element software (Nikon) (H&E sections) or scanned (Oil Red O sections) with a nanozoomer scanner (Hamamatsu Photonics, Hamamatsu, Japan) and analyzed with NDP view software (Hamamatsu Photonics).
Liver Lipid Content Analysis
Hepatic levels of triglycerides, free cholesterol, and cholesterol esters were determined using Bligh and Dyer methodology,12 and lipid extracts were analyzed by gas–liquid chromatography as described.13
Biochemical Analyses
Plasma samples were used to measure alanine aminotransferase and lipid profiles (triglycerides, total cholesterol, and high‐density lipoprotein (HDL) cholesterol). Plasma insulin and adipokines (resistin, leptin, adiponectin) levels were determined using the Multiplex Immunoassay Technology Xmap (MADKMAG‐71K‐05 and MADPNMAG‐70K‐01, MILLIPEX; Millipore, Saint‐Quentin‐en‐Yveline, France). Serum Gdf15 levels were determined with a commercial enzyme‐linked immunosorbent assay kit (MGD150; R&D Systems Europe, Abingdon, United Kingdom) according to the manufacturer’s instructions.
Other Methods
Methods used for ERα protein determination by western blot on isolated hepatocytes, gene expression analysis, and chromatin immunoprecipitation (ChIP) assays are detailed in Supporting Methods.
Statistical Analysis
Results are expressed as mean ± SEM. Statistical analyses were performed using GraphPad Prism, version 5.00, for Windows (GraphPad Software, San Diego, CA; www.graphpad.com). A Student t test was used to compare the respective effects of TAM and E2 with the effect of VEH within each genotype (WT or knockout [KO] mice). To test the interaction between treatments and genotypes for body weight evolution and glycemic changes during intraperitoneal glucose tolerance tests, a two‐way analysis of variance with repeated measures was carried out. In the case of interaction between treatment and genotype, Bonferroni post‐hoc tests were subsequently performed. P < 0.05 was considered statistically significant.
Results
Both E2 and TAM Prevent HFD‐INDUCED Steatosis Through Hepatocyte ERα Activation
LERKO mice were first used to determine whether ERα signaling in hepatocytes contributes to the prevention of HFD‐induced steatosis conferred by TAM and E2. Demonstrating the selective ERα deletion in this mouse model, ERα messenger RNA (mRNA) levels were decreased by 80% in total livers and primary hepatocytes from LERKO mice compared to their WT littermates, while no differences were observed in other tissues (Supporting Fig. S1A). Furthermore, ERα protein expression was not detected in the culture of primary hepatocytes from LERKO mice (Supporting Fig. S1B). Both E2 and TAM protected ovariectomized WT mice from HFD‐induced steatosis, as illustrated by histologic staining (Fig. 1A) and confirmed by a significant decrease in intrahepatic concentrations of triglycerides (Fig. 1B). Prevention of steatosis development by either TAM or E2 was abolished in LERKO mice (Fig. 1A,B). Histologic analyses found no lobular inflammation, hepatocyte ballooning, or fibrosis, irrespective of the treatment and genotype (data not shown). Accordingly, no significant changes were observed in the hepatic expression of a set of genes involved in inflammation and fibrosis (Supporting Fig. S2; data not shown). Finally, in TAM‐treated mice, we checked that hepatocyte ERα deletion does not alter the expression of genes encoding P450 enzymes that contribute to metabolize this SERM in the liver (Supporting Fig. S3). These data first confirm that hepatocyte ERα plays an important role in the hepatoprotective actions of E24 and then demonstrate that TAM‐induced prevention of steatosis8 also results from ERα activation in hepatocytes.
Figure 1.
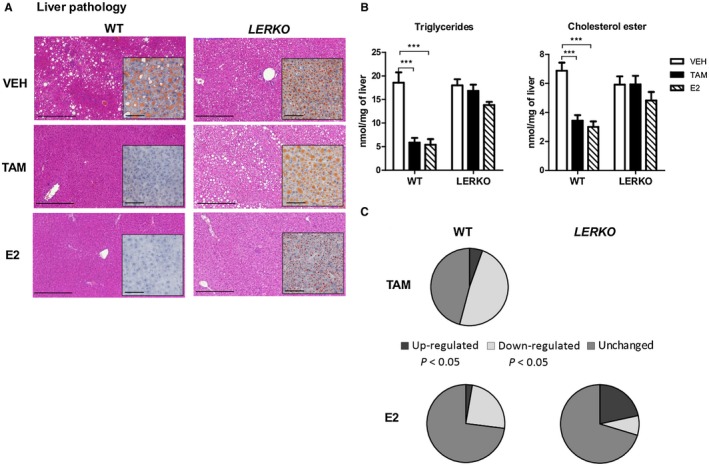
Prevention of HFD‐induced steatosis by either E2 or TAM requires hepatocyte ERα activation. LERKO mice (n = 40) and their respective WT littermates (n = 40) were fed an HFD and concomitantly treated with TAM, E2, or VEH for 12 weeks. (A) Liver histology: representative images of H&E (scale bars, 100 µm) and Oil Red O staining (insets; scale bars, 50 µm). (B) Intrahepatic lipid content. (C) Liver mRNA expression (37 genes involved in metabolic pathways). Genes were defined as up‐ or down‐regulated if the fold change compared to control was statistically significant (P < 0.05, Student t test). Data are expressed as mean ± SEM. * indicates differences between VEH and treated (TAM or E2) mice within each genotype. ***P < 0.001.
Involvement of Hepatocyte ERα in the Regulation of Hepatic Metabolic Pathways by TAM and E2
To gain further insights into the contribution of hepatocyte ERα to the protective effects of E2 and TAM against HFD‐induced steatosis, we explored the regulation of the main hepatic metabolic pathways, focusing on a set of 37 selected genes involved in lipid and glucose metabolism. In WT‐treated mice, 20/37 and 10/37 genes were significantly regulated by TAM and E2, respectively (summarized in Fig. 1C). In our experimental settings, no significant changes were observed in the mRNA expression levels of acetyl‐coenzyme A (CoA) acyltransferase 2 (Acaa2), acyl‐CoA dehydrogenase, long‐chain (Acadl), apolipoprotein B (Apob), carnitine palmitoyltransferase 1A (Cpt1a), cytochrome P450, family 17, subfamily a, polypeptide 1 (Cyp17a1), fatty acid binding protein 4, adipocyte (Fabp4), glucose‐6‐phosphatase, catalytic (G6pc), 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (Hmgcr), insulin receptor (Insr), insulin receptor substrate 2 (Irs2), lipoprotein lipase (Lpl), MLX interacting protein‐like (Mlxipl), nuclear receptor subfamily 1, group H, member 4 (Nr1h4), patatin‐like phospholipase domain containing 2 (Pnpla2), sirtuin 1 (Sirt1), and solute carrier family 2 member 4 (Slc2a4) following either TAM or E2 administration (data not shown). In TAM‐treated WT mice, genes involved in de novo lipogenesis and lipid synthesis (acetyl‐CoA carboxylase alpha [Acaca], CCAAT/enhancer binding protein, alpha [Cebpa], diacylglycerol O‐acyltransferase 2 [Dgat2], fatty acid synthase [Fasn], Nr1h3, phosphatidylethanolamine N‐methyltransferase [Pemt], peroxisome proliferator activator receptor delta [Ppard], stearoyl‐CoA desaturase 1 [Scd1], and sterol regulatory element binding transcription factor 1 [Srebf1]), cholesterol metabolism and lipoprotein assembly (Apoa1, Apoa4, Apoa5, and phospholipid transfer protein [Pltp]), lipid transport (Fabp1) and catabolism (Acacb, acyl‐CoA oxidase 1, palmitoyl (Acox1), peroxisome proliferator activated receptor alpha [Ppara]), and glucose metabolism (glucokinase [Gck]) were down‐regulated compared to VEH‐treated mice (Supporting Fig. S4). Inversely, genes encoding for gluconeogenic enzyme (phosphoenolpyruvate carboxykinase 1, cytosolic [Pck1]) and hepatic insulin sensitivity (leptin receptor [Lepr]) were up‐regulated by TAM. Noteworthy, TAM‐sensitive genes were all dependent on hepatocyte ERα because their regulations were abolished in LERKO mice (Fig. 1C; Supporting Fig. S4). In E2‐treated WT mice, some genes involved in de novo lipogenesis and lipid synthesis (Acaca, Dgat2, Fasn, Scd1, and Srebf1), cholesterol metabolism and lipoprotein assembly (Pltp), lipid catabolism (Acacb, Ppara), and glucose metabolism (Gck) were down‐regulated, while Cyp17a1, a key gene for steroidogenesis, was up‐regulated compared to VEH‐treated mice (Supporting Fig. S4). The up‐regulation of Cyp17a1 and the down‐regulation of Acaca, Acacb, Dgat2, Srebf1, Pltp, Ppara, and Gck by E2 were abrogated in LERKO mice, indicating that only 80% of E2‐regulated genes were dependent on hepatocyte ERα activation (Fig. 1C; Supporting Fig. S4). Based on the protection conferred by TAM and E2 against HFD‐induced steatosis, both molecules thus down‐regulate the expression of key lipogenic genes. Although other cellular targets could contribute to E2‐mediated control of these hepatic metabolic pathways, the present data demonstrate that their regulation by TAM entirely relies on the direct activation of hepatocyte ERα.
Hepatocyte ERα Mediates Protective Effects of TAM on Whole‐Body Energy and Glucose Homeostasis
We next considered the role of hepatocyte ERα in the preventive actions of TAM and E2 against HFD‐induced systemic disorders, namely obesity, insulin resistance, and hyperglycemia. In agreement with other reports,6, 8 both molecules protected ovariectomized WT female mice from body weight gain (Fig. 2A), but only TAM administration significantly decreased food intake (Fig. 2B). E2 and TAM reduced fat mass accumulation to a similar extent (Fig. 2C), including subcutaneous and visceral sites (Fig. 2D), without any impact on lean mass (Fig. 2C). As expected, the two treatments also prevented HFD‐induced glucose intolerance and insulin resistance in WT mice (Fig. 2E). E2 administration still protected LERKO female mice from HFD‐induced obesity (Fig. 2A,C,D), as reported,4 and from glucose intolerance, although to a lesser extent than in WT mice (Fig. 2E). In contrast, the preventive effects of TAM on body weight gain (Fig. 2A), fat mass accumulation (Fig. 2C,D), insulin resistance, and glucose intolerance (Fig. 2E) as well as its anorectic effect (Fig. 2B) were totally abrogated in LERKO mice. Plasma lipid profiles in WT mice revealed that TAM significantly reduced total and HDL‐cholesterol levels together with an increase in low‐density lipoprotein (LDL)‐cholesterol concentrations, while E2 treatment was associated with a decrease in HDL‐cholesterol and an increase in LDL‐cholesterol levels (Table 1). Adiponectin and leptin plasma concentrations were significantly decreased by both treatments in WT mice, while resistin levels were only reduced by TAM administration (Table 1). In LERKO mice, all the effects of E2 on plasma lipid and adipokine profiles were preserved, whereas those of TAM were abolished except for adiponectin concentration that was still reduced (Table 1). These results indicate that specific activation of hepatocyte ERα is dispensable for E2‐mediated prevention of obesity and associated metabolic disorders but is absolutely required for the systemic effects of TAM, including the control of food intake as well as the preservation of whole‐body energy and glucose homeostasis.
Figure 2.
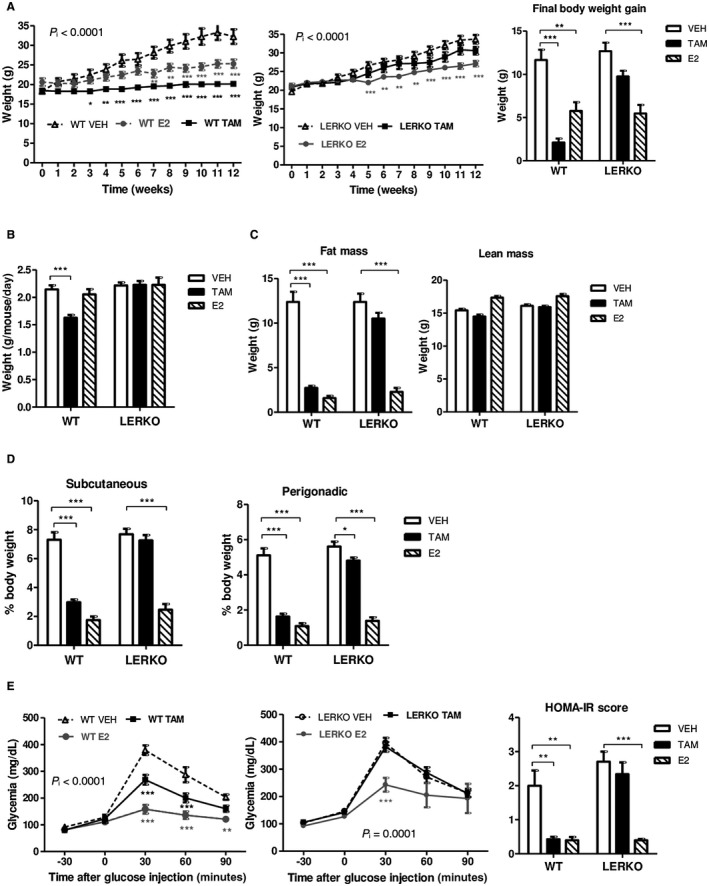
Hepatocyte ERα mediates the protective effects of TAM but not of E2 against HFD‐induced obesity, insulin resistance, and glucose intolerance. LERKO mice (n = 40) and their respective WT littermates (n = 40) were fed an HFD and treated with TAM, E2, or VEH for 12 weeks. (A) Body weight evolution and final body weight gain; (B) mean food intake; (C) body composition; (D) adipose tissue distribution; (E) intraperitoneal glucose tolerance tests and HOMA‐IR score. Data are expressed as mean ± SEM. P i < 0.0001, interaction between treatments and genotypes; *indicates differences between VEH and treated (TAM or E2) mice within each genotype. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviation: HOMA‐IR, homeostasis model assessment of insulin resistance.
Table 1.
Influence of TAM and E2 on Plasma Metabolic Parameters in HFD‐FED LERKO Mice
| WT | LERKO | |||||
|---|---|---|---|---|---|---|
| VEH | TAM | E2 | VEH | TAM | E2 | |
| ALT, U/L | 36 ± 8 | 23 ± 9 | 15 ± 10 | 28 ± 5 | 25 ± 20 | 28 ± 19 |
| Chol, mmol/L | 3.20 ± 0.20 | 2.07 ± 0.33† | 3.23 ± 0.64 | 3.57 ± 0.35 | 3.55 ± 0.22 | 3.21 ± 0.23 |
| HDL‐C, mmol/L | 1.67 ± 0.05 | 1.23 ± 0.08‡ | 1.32 ± 0.12* | 1.86 ± 0.21 | 1.83± 0.14 | 1.34 ± 0.16† |
| LDL‐C, mmol/L | 0.35 ± 0.03 | 0.43 ± 0.04* | 0.77 ± 0.25* | 0.47 ± 0.05 | 0.39 ± 0.04* | 0.64 ± 0.15* |
| TG, mmol/L | 0.88 ± 0.05 | 0.82 ± 0.06 | 0.77 ± 0.16 | 0.87 ± 0.09 | 0.98 ± 0.20 | 0.71 ± 0.05* |
| FFA, mmol/L | 0.77 ± 0.11 | 0.93 ± 0.14 | 0.63 ± 0.14 | 1.07 ± 0.14 | 0.87 ± 0.07* | 0.78 ± 0.17* |
| Adiponectin, ng/mL | 6.7 ± 1.0 | 4.7 ± 0.4† | 2.8 ± 0.5† | 7.1 ± 0.9 | 4.2 ± 0.3‡ | 3.1 ± 0.6‡ |
| Leptin, ng/mL | 20.3 ± 4.8 | 3.8 ± 1.1‡ | 2.1 ± 1.1‡ | 23.8 ± 4.7 | 19.1 ± 3.4 | 3.3 ± 2.3‡ |
| Resistin, ng/mL | 5.2 ± 0.9 | 3.1 ± 0.8† | 5.0 ± 1.6 | 4.8 ± 1.7 | 3.6 ± 0.7 | 3.9 ± 0.6 |
Ovariectomized female LERKO mice and their respective WT littermates were fed an HFD and concomitantly treated with TAM, E2, or VEH for 12 weeks. Blood samples were collected at death in 3‐hour‐fasted mice. Data are expressed as mean ± SEM.
Differences between VEH‐treated and either TAM‐ or E2‐treated mice according to the Student t test;
P < 0.05;
P < 0.01;
P < 0.001.
Abbreviations: ALT, alanine aminotransferase; C, cholesterol; Chol, total cholesterol; FFA, free fatty acids; TG, triglyceride.
Hepatocyte ERα is Required for the Effect of TAM on Food Intake and Energy Expenditure
To further characterize the involvement of hepatocyte ERα in the respective influence of TAM and E2 on energy homeostasis, HFD‐fed WT and LERKO mice were assessed in metabolic cages, including indirect calorimetry measurements. No significant change in respiratory exchange ratio was observed with either TAM or E2 (Fig. 3A). E2 did not influence energy expenditure (Fig. 3B) but tended to increase nocturnal physical activity, both in WT and in LERKO mice (Fig. 3C), suggesting that metabolic effects induced by E2 involve central mechanisms independent of hepatocyte ERα activation. Conversely, TAM tended to decrease nocturnal physical activity (Fig. 3C) in WT mice along with a significant reduction in energy expenditure during both day and night periods (Fig. 3B). TAM‐treated WT mice were also characterized by a reduction in oxygen consumption and carbon dioxide production (Supporting Fig. S3) that was associated with a significant decrease in food intake (Fig. 2B), thus suggesting that the metabolic benefit conferred by TAM probably results in a large part from its anorectic effect. Finally, all the effects of TAM on food intake (Fig. 2B) and energy expenditure (Fig. 3B) were entirely abrogated in LERKO mice, demonstrating that they are mediated through ERα activation in hepatocytes.
Figure 3.
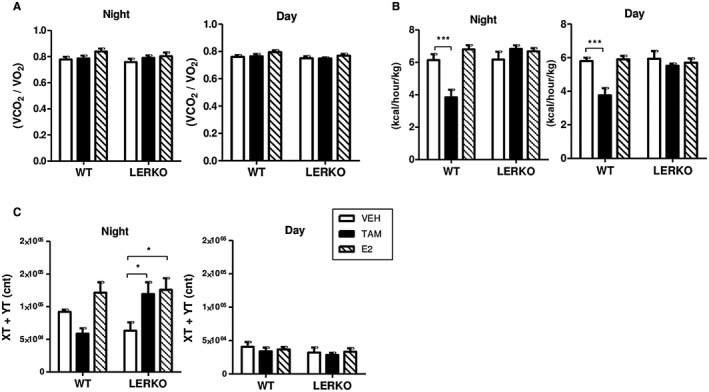
Effects of TAM and E2 and role of hepatocyte ERα on energy homeostasis. Indirect calorimetry measurements were performed in LERKO mice (n = 19) and their respective WT littermates (n = 21) fed an HFD and treated with TAM, E2, or VEH for 12 weeks. (A) Respiratory exchange ratio calculated as carbon dioxide production/oxygen consumption, (B) energy expenditure (kcal heat produced). (C) Ambulatory physical activity as measured by the average number of beambreak counts in different dimensions (XT + YT). Data are expressed as mean ± SEM. *indicates differences between VEH and treated (TAM or E2) mice within each genotype. *P < 0.05; ***P < 0.001. Abbreviations: VCO2, carbon dioxide production; VO2, oxygen consumption.
TAM Enhances Gdf15 Expression Through Hepatocyte ERα Activation
To identify the link between hepatocyte ERα activation and whole‐body metabolic protection conferred by TAM, we first explored whether TAM and E2 differently influence the expression of hepatokines known to exert specific actions on energy and glucose homeostasis. Neither E2 nor TAM altered angiopoietin‐like 4 (Angptl4), Angptl6, insulin‐like growth factor 1 (Igf1), and insulin‐like growth factor binding protein 1 (Igfbp1) hepatic expression levels in WT mice, whereas Angptl4 and Igfbp1 were significantly increased in E2‐treated LERKO mice (Fig. 4A). However, E2 enhanced fibroblast growth factor 21 (Fgf21) mRNA expression level in the liver from WT mice (Fig. 4A), as reported,14 and this positive regulation was still observed in LERKO mice (Fig. 4A). Similarly, E2 induced a significant up‐regulation of energy homeostasis associated (Enho) that codes for adropin, a hepatokine known to promote nocturnal physical activity and to prevent insulin resistance and adiposity, independently of hepatocyte ERα activation (Fig. 4A). TAM significantly increased the expression of Igfbp2 in WT but not LERKO mice (Fig. 4A). The plasma level of this hepatokine has been demonstrated to be inversely correlated with incidence of type 2 diabetes. However, the strongest effect of TAM on gene expression concerned Gdf15, which codes for a molecule described as a powerful cytokine that inhibits food intake and decreases body weight through direct action on feeding centers in the hypothalamus and brainstem.15, 16 Indeed, an 8‐fold increase in Gdf15 mRNA hepatic expression was observed in TAM‐treated WT mice, contrasting with the lack of E2 effect on this hepatokine expression (Fig. 4A). TAM‐mediated up‐regulation of Gdf15 mRNA hepatic expression was totally abolished in LERKO mice (Fig. 4A) and, importantly, serum Gdf15 concentrations were perfectly correlated with its liver expression profile in WT as in LERKO mice (Fig. 4B). We also determined Gdf15 mRNA levels in adipose tissue from WT and LERKO mice and found no influence of E2 and only a slight increase in TAM‐treated mice from both genotypes (Fig. 4C), supporting the conclusion that serum Gdf15 concentration mainly reflects liver secretion. Our data thus demonstrate that unlike E2, TAM administration induces a marked increase in Gdf15 hepatic expression and Gdf15 circulating concentration through the activation of ERα in hepatocytes.
Figure 4.
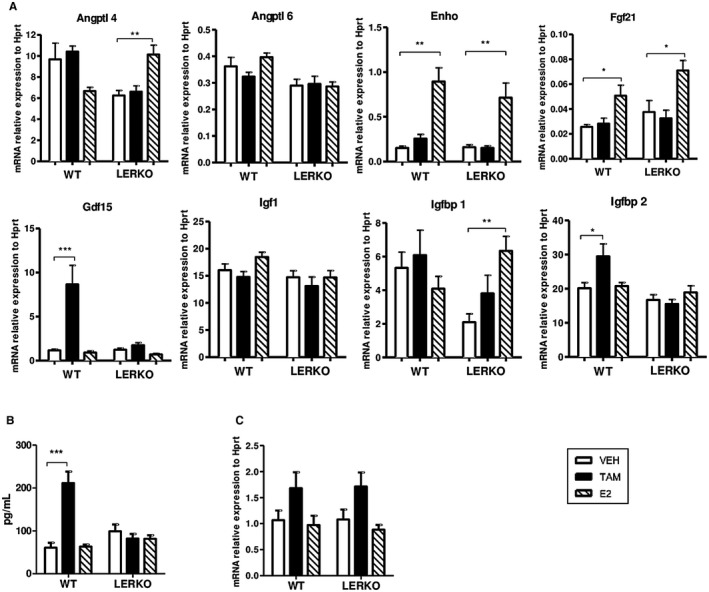
Role of hepatocyte ERα to TAM or E2 regulation of hepatokines known to improve glucose and lipid homeostasis and/or to control food intake. LERKO mice (n = 18) and their respective WT littermates (n = 17) were fed an HFD and treated with TAM, E2, or VEH for 12 weeks. (A) Hepatic mRNA expression of hepatokines (Angptl4, Angptl6, Enho, Fgf21, Gdf15, Igf1, Igfbp1, and Igfbp2) in liver tissue samples. (B) Serum Gdf15 concentration in blood samples. (C) Gdf15 mRNA expression in white adipose tissues. Data are expressed as mean ± SEM. *indicates differences between VEH and treated (TAM or E2) mice within each genotype. *P < 0.05; **P < 0.01; ***P < 0.001. Abbreviation: Hprt, hypoxanthine phosphoribosyl transferase.
Differential Regulation of Gdf15 Expression in the Liver With TAM and E2
To further explore the molecular mechanisms involved in the differential regulation of Gdf15 expression by both ERα ligands, ovariectomized female mice were treated with E2 or TAM either acutely (single subcutaneous injection) or chronically (through subcutaneous pellet over 3 weeks). After acute treatment, E2 and TAM up‐regulated Gdf15 expression at the same level (Fig. 5A), in agreement with our previous study emphasizing Gdf15 mRNA induction in the livers of female mice following oral E2 administration.17 However, no Gdf15 mRNA level induction was observed after E2 chronic treatment (Fig. 5A), as shown in HFD‐fed mice (Fig. 4A). By contrast, TAM chronic treatment still increased Gdf15 expression. This was not due to ERα protein degradation in response to E2 because neither E2 nor TAM chronic treatment altered ERα protein level (Fig. 5B). In ChIP‐quantitative polymerase chain reaction (qPCR) experiments, ERα and its coactivator CREB binding protein (CBP) were found to be associated to their binding sites (BS1 and BS2) in the Gdf15 regulatory region17 in response to TAM but not to E2 chronic administration (Fig. 5C; Supporting Fig. S6A). In liver from E2‐treated mice, the lack of ERα binding to the Gdf15 regulatory region was associated with trimethylation of the twenty‐seventh amino acid in histone H3 (H3K27) on BS1 (Fig. 5D; Supporting Fig. S6B), an epigenetic modification recently described to silence Gdf15 expression.18 These ChIP assays also demonstrated that the nucleosomes located around both BS retained acetylated H3K27 (H3K27ac) and H3K4 monomethylation (me1) and dimethylation chromatin marks (Fig. 5E). Interestingly, both BS1 and BS2 still exhibited an enhanced enrichment in H3K4me1 and H3K27ac marks in the presence of E2 compared to VEH (Supporting Fig. S6C). This is consistent with the hypothesis that these sequences were likely to have undergone activation at an earlier time point in accordance with the E2‐mediated induction of Gdf15 observed in the acute condition. Altogether, these data fit the hypothesis that at least BS1 begins to acquire features from “poised” enhancers, i.e., genomic sites that were active and underwent relative transient inactivation.19 Finally, the chronic administration of TAM appears to be less efficient than E2 in inducing the enrichment of both BS1 and BS2 in H3K4me1 and H3K27ac (Supporting Fig. S6C). This may reflect the weaker ability of ERα to recruit cofactors in the presence of TAM compared to E2, which in this precise case does not prevent ERα from modulating the transcription rate of Gdf15.
Figure 5.
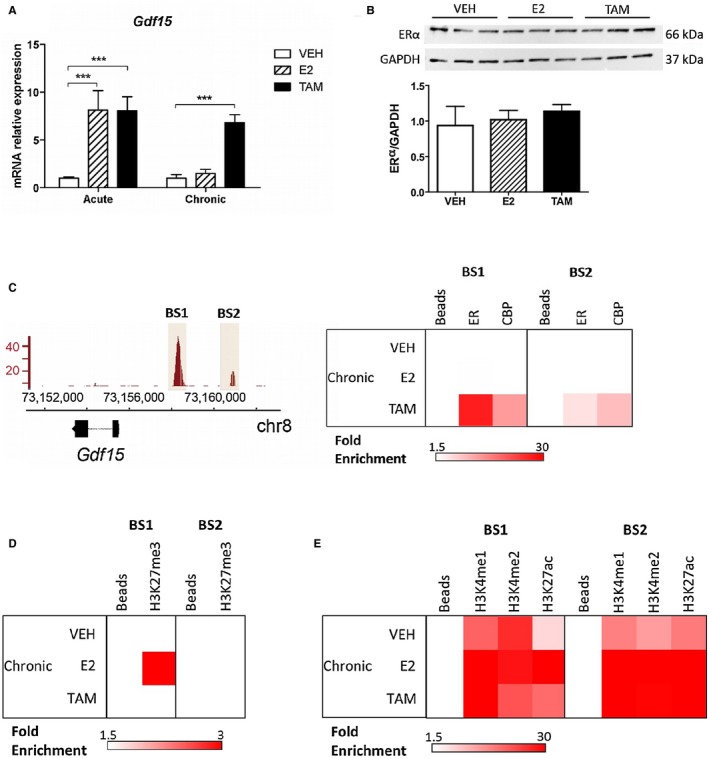
Regulation of Gdf15 expression in the liver with TAM and E2. Ovariectomized female C57Bl/6J mice (n = 12) were treated with TAM, E2, or VEH for 2 hours (acute) or for 3 weeks (chronic). (A) Hepatic Gdf15 mRNA expression was determined by RT‐qPCR in liver tissue samples. Data are expressed as mean ± SEM. ***P < 0.001. (B) Hepatic ERα protein level after chronic treatment was determined by western blot and normalized to Gapdh. (C) ERα enrichment at BS1 and BS2 on the Gdf15 promoter region (left panel) was quantified by ChIP‐qPCR experiments from livers treated as in panel B (right panel). The presence of histone marks (D) H3K27me3 and (E) H3K4me1, H3K4me2, and H3K27ac on BS1 and BS2 were assessed by ChIP‐qPCR experiments on chromatin prepared from the same livers as those used in panel C. Abbreviation: chr, chromosome; Gapdh, glyceraldehyde 3‐phosphate dehydrogenase; RT, reverse transcription.
Hepatic and Whole‐Body Metabolic Protective Effects of TAM are Abolished in HFD‐FED GDF15KO Mice
To determine whether Gdf15 contributes to the overall metabolic protective actions of TAM, ovariectomized GDF15KO mice and their WT littermates were treated with TAM, E2, or VEH for 6 weeks and concomitantly fed an HFD. The protective actions of E2 observed in WT mice in terms of body weight gain, fat mass accumulation, glucose tolerance, and steatosis were not altered by Gdf15 deficiency (Fig. 6A‐E). In striking contrast, TAM failed to protect GDF15KO mice from HFD‐induced obesity (Fig. 6A,B), glucose intolerance (Fig. 6D), and steatosis (Fig. 6E). In addition, the anorectic effect of TAM was abolished and an increase in food intake was even observed in TAM‐treated GDF15KO mice (Fig. 6C). Altogether, our results demonstrate that the selective modulation of hepatocyte ERα with TAM preserves whole‐body energy and glucose homeostasis, in a context of HFD feeding, through the up‐regulation of Gdf15 hepatic expression.
Figure 6.
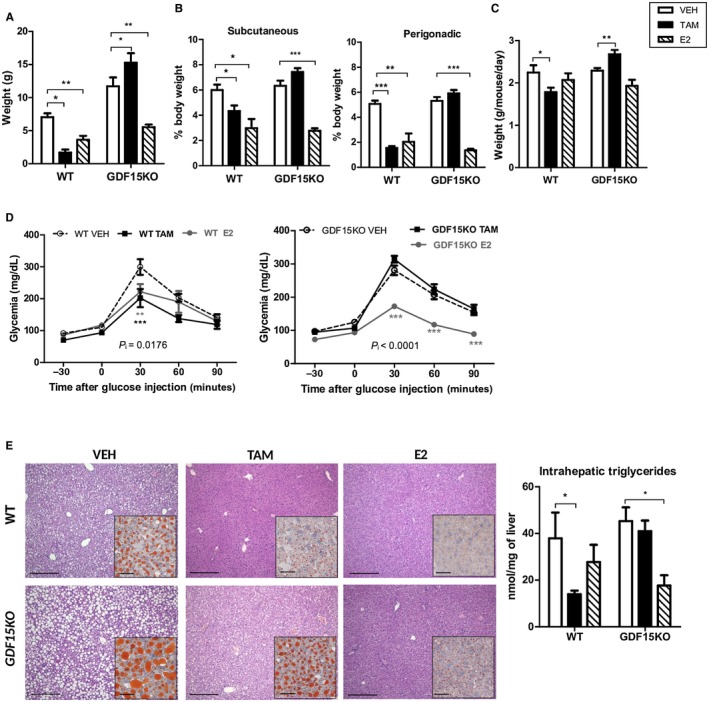
Protective metabolic effects of TAM are abolished in GDF15KO mice. GDF15KO mice (n = 23) and their respective WT littermates (n = 14) were fed an HFD and treated with TAM, E2, or VEH for 6 weeks. (A) Body weight gain; (B) food intake; (C) adipose tissue distribution; (D) intraperitoneal glucose tolerance tests; (E) liver histology, representative images of H&E (scale bars, 100 µm) and Oil Red O (insets; scale bars, 50 µm) staining and intrahepatic triglyceride content. Data are expressed as mean ± SEM. P i, interaction between treatments and genotypes. *indicates differences between VEH and treated (TAM or E2) mice. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
The present study further emphasizes the protective role of hepatocyte‐specific ERα signaling against diet‐induced steatosis. Noteworthy, in our experimental settings, such a beneficial effect on the liver was not only observed with E2 administration but also and to a similar extent following TAM treatment. This latter observation is undoubtedly more surprising because numerous clinical observations reported that TAM is able to promote or to worsen fatty liver in women prescribed with this treatment for breast cancer, although recent studies concluded that the incidence of TAM‐induced steatosis is very low.20 Studies conducted in rodent models led to divergent results depending on the experimental settings, some of them reproducing TAM‐induced steatosis21, 22 while others, in perfect agreement with our observations, reported protective effects of TAM on the liver23, 24 but also on glucose and lipid metabolism.24, 25, 26, 27, 28 Accordingly, Ceasrine et al.24 highlighted the need for caution in the interpretation of metabolic studies that require transient high‐dose TAM administration protocols for creating conditional KO in tamoxifen‐inducible estrogen receptor (ERT2)‐Cre thyroglobulin (Tg) mice, demonstrating an improved glucose tolerance in both male and female mice 1 week after the last dose and even 3 weeks later in male mice.
However, our main finding is that hepatocyte ERα, following the peculiar mode of activation of TAM, is also able to preserve whole‐body energy and glucose homeostasis from the deleterious effects of HFD feeding. Indeed, hepatocyte‐restricted ERα deletion differently alters the metabolic protection conferred by E2 and TAM, two ERα ligands respectively leading to the full activation or to the AF1‐selective modulation of the receptor.6, 8 Although E2‐treated LERKO female mice were no longer protected from steatosis, as reported,4 these mice were still preserved from HFD‐induced adiposity, insulin resistance, and glucose intolerance, underlining the involvement of targets other than hepatocytes for these latter actions of E2. In contrast, similar extensive protective actions exerted by TAM are dependent on hepatocyte ERα activation as they were all abolished in LERKO mice. Consequently, although TAM and E2 similarly protect female mice from HFD‐induced metabolic disturbances through ERα‐dependent mechanisms, our results support the conclusion that their respective effects involve distinct cellular targets.
It is now well recognized that the overall beneficial metabolic action of E2 requires ERα activation in various tissues, including the central nervous system, adipose tissue, liver, and endocrine pancreas.29 In particular, estrogens have been demonstrated to regulate different aspects of energy homeostasis through direct activation of ERα in specific types of neurons.30 ERα deletion restricted to hypothalamic steroidogenic factor‐1 neurons induces hypometabolism and abdominal obesity without associated hyperphagia, whereas ERα activation in hypothalamic pro‐opiomelanocortin neurons controls food intake without any direct influence on energy expenditure or fat distribution.30
In our experimental settings, E2 administration enhanced nocturnal physical activity level in both WT and LERKO female mice while TAM lowered energy expenditure in WT mice, probably resulting from a significant decrease in food intake as described in murine models.25, 31, 32, 33 Interestingly, although TAM anorectic effect has been attributed to Fasn inhibition in the ventromedial nucleus of the hypothalamus, leading to accumulation of malonyl‐CoA,31 no study has demonstrated that this action relies on the direct activation of ERα in hypothalamic neurons. Noteworthy, demonstrating that the influence of TAM on food intake and energy expenditure was totally abrogated in LERKO mice, the present data suggest that the control of these central regulations by TAM mainly results from peripheral signals induced by ERα selective modulation in hepatocytes.
For insight into the mechanisms leading to the discrepant metabolic consequences of hepatocyte ERα activation by E2 and TAM, we addressed their respective influence on hepatic gene expression, first focusing on a set of genes involved in the main metabolic pathways. In line with their similar protective effects against HFD‐induced steatosis, both molecules down‐regulated the transcription of genes involved in de novo lipogenesis and lipid synthesis, although with a wider influence of TAM that more specifically impacted some of them (Cebpa, Nr1h3, Pemt) in WT mice. E2 and TAM also differently alter the expression of some genes involved in lipid and/or glucose metabolism (Apoa1, ApoA4, Cyp17a1, Lepr). However, the most important observation is that TAM‐induced gene regulations were all abolished in LERKO mice whereas E2 still influenced the expression of several liver genes in these animals, thus probably through indirect mechanisms mediated by other ERα‐expressing cellular targets that remain to be identified.
We then hypothesized that the whole‐body protection conferred by TAM through hepatocyte ERα activation could be mediated by liver‐secreted circulating factors, and we decided to focus our analyses on the respective influence of E2 and TAM on the expression of hepatokines described to exert metabolic actions. It must be acknowledged that additional experiments would have been useful to definitely demonstrate that TAM acts through hepatocyte‐initiated endocrine mechanisms. From our data, we cannot exclude that neurovegetative signals and other liver‐derived factors, including other proteins or small molecules, could contribute to the systemic effects of TAM. Importantly, hepatocyte ERα is already known as a key mediator of systemic regulation as it controls Igf1 expression.9 In our experiments, E2 treatment led to a significant increase in hepatic expression of Fgf21, as reported,14 and also of Enho, which codes for adropin, an hepatokine known to increase night physical activity and to prevent insulin resistance and adiposity.34 These regulations are demonstrated to be totally independent from hepatocyte ERα and could thus have contributed, at least partially, to the preservation of energy and glucose homeostasis by E2 in HFD‐fed WT and LERKO mice. Conversely, neither Fgf21, in line with the action of another SERM (bazedoxifene),14 nor Enho expression was modified in TAM‐treated mice livers. Among the genes coding for hepatokines tested in our experimental settings, we found that Gdf15 and, to a much lesser extent Igfbp2, displays a gene expression profile strictly dependent on TAM‐induced hepatocyte ERα activation.
Noteworthy, Gdf15 gene expression in the liver perfectly correlates with plasma concentrations measured in WT and LERKO mice. Mainly produced by the liver,35 Gdf15 is a divergent member of the transforming growth factor‐β superfamily, which exerts anorectic effects and controls body weight through the activation of the glial cell line‐derived neurotrophic factor family receptor α‐like (GFRAL), recently identified as the specific receptor of Gdf15 in hindbrain neurons of the area postrema and nucleus of the solitary tract.36, 37, 38 In rodent models, even a moderate increase in Gdf15 circulating levels reduces food intake, and a sustained exposition to this hepatokine leads to cachexia, both under normal diet and HFD.15, 39 Accordingly, GDF15KO mice are characterized by accelerated weight gain and increased fat mass, mainly localized in visceral abdominal sites, but their food intake and body weight returned to the WT level following infusion with recombinant human GDF15.40 Moreover, GDF15KO mice are also prone to develop steatosis, while GDF15‐transgenic mice are protected from steatosis and associated metabolic disorders.41 Finally, in agreement with our results, prevention of obesity in HFD‐fed mice treated with recombinant GDF15 was not associated with an increase in energy expenditure, suggesting that the protective action of this hepatokine is likely driven by the reduction in food intake.36 These considerations prompted us to directly address the role of Gdf15 in whole‐body metabolic protective TAM effects. The effects of TAM were confirmed in GDF15+/+ female mice but were all totally abrogated in GDF15KO mice.
Although hepatocytes have been recognized as the main cell sources of Gdf15 in the liver,42 whether TAM‐enhanced Gdf15 expression is restricted to hepatocytes or concerns other cell types remains to be determined. However, this lack of demonstration of the cellular sources of Gdf15 in the liver of TAM‐treated mice does not challenge our main conclusion, i.e., the specific regulation of liver Gdf15 by TAM requires ERα activation in hepatocytes. GDF15 expression levels are physiologically low but rapidly increase in pathophysiological conditions, such as tissue injury, inflammation, and malignancy, as well as during dysmetabolic status, including obesity, type 2 diabetes, and NAFLD.41 Interestingly, our study demonstrates that hepatocyte ERα is also able to enhance Gdf15 expression according to its mode of activation. Contrasting with the significant increase induced by TAM, Gdf15 liver expression and Gdf15 circulating levels were not altered by E2 chronic treatment. This latter observation is surprising because we previously observed and confirmed in the present study that acute E2 administration significantly increases liver Gdf15 gene transcription.17 The discrepancy in Gdf15 transcription regulation following chronic treatments cannot be explained by ERα protein degradation in response to E2, but ChIP‐qPCR revealed that ERα and its coactivator CBP were bound to BS1 and BS2 in the Gdf15 regulatory region only in TAM‐treated mice. Accordingly, our data suggest that chronic E2 treatment induced epigenetic modifications that contribute to silent Gdf15 expression, as recently reported.18
In line with our recent work,6, 8 the present study further illustrates the redundancy between ERα‐AF1 and ERα‐AF2 in terms of metabolic protection. In contrast, both ERα‐AFs are required to mediate proliferative effects on the mammary gland and contribute to the increased risk of breast cancer elicited by estrogens, contrasting with the spectacular protection conferred by ERα‐AF2 antagonism of TAM.43 However, this ERα‐AF2 antagonism has no adverse impact on the main cellular target(s), mainly hepatocytes as demonstrated here, involved in TAM metabolic effects. This underlines the therapeutic potential of SERMs resulting in the specific activation of only ERα‐AF1. Furthermore, as discussed above, the cellular targets mediating the metabolic protective actions are different according to the nature of the ligand, although leading to a similar overall benefit.
From a therapeutic perspective, it would be very interesting to design new strategies for a preferential delivery of ERα‐AF1‐selective modulators to the liver, as recently proposed for targeting other cellular types, such as the pancreas with a glucagon‐like peptide‐1‐estrogen conjugate.44
The present work provides further evidence that hepatocyte ERα represents a promising target for the prevention of NAFLD and demonstrates for the first time that the selective modulation of ERα in hepatocytes is sufficient to preserve whole‐body energy and glucose homeostasis. The mechanism accounting for this major widespread metabolic action relies on the induction of the hepatokine Gdf15. Selective modulation of hepatocyte ERα leading to sustained increase in circulating Gdf15 could thus be sufficient to fight obesity and its related complications, such as type 2 diabetes and NAFLD.
Supporting information
Acknowledgment
We acknowledge Marc Poirot, Henrik Laurell, Alexia Vinel, Sandra Handgraaf, Marine Adlanmerini, and Professor Walter Wahli for their help in the critical interpretation of data. We thank Professor Pierre Chambon (Strasbourg, France) and Dr. Björn Spittau (Freiburg, Germany) for providing floxed ERα and GDF15KO mice, respectively. We also acknowledge the skillful technical assistance of Frédéric Boudou, Adrien Pulido, the histology platform of Institut National Polytechnique de Toulouse (INP)‐École Nationale Vétérinaire de Toulouse (Isabelle Bleuart and Isabelle Pardo), the Institut National de la Santé et de le Recherche Médicale (INSERM) Unité Médicale de Recherche (UMR) 1048 platforms (Toulouse, France) ANEXPLO‐GENOTOUL (Cédric Baudelin, Aurore Desquesnes, Sophie Le Gonidec), GeT‐TQ Genopole (Frédéric Martins, Jean‐José Maoret), MetaToul‐LIPIDOMIQUE (Justine Bertrand‐Michel), and the Institut National de la Santé et de le Recherche Médicale (INSERM) histology platform (Lucie Fontaine).
Supported by INSERM (J.F.A & P.G.), Université de Toulouse and Faculté de Médecine de Toulouse (J.F.A & P.G.), Fondation de France (C.F.), Conseil Régional Midi‐Pyrénées/Occitanie (P.G.), Fondation pour la Recherche Médicale (J.F.A.), Société Francophone du Diabète (A.M. & P.G.), and Agence Nationale de la Recherche (C.F.). The nuclear magnetic resonance facility is part of the Genotoul‐Ibisa PICT (Integrated Screening Platform of Toulouse) platform and was funded by the Centre National de la Recherche Scientifique, Région Midi‐Pyrénées, and European structural funds.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Hevener AL, Clegg DJ, Mauvais‐Jarvis F. Impaired estrogen receptor action in the pathogenesis of the metabolic syndrome. Mol Cell Endocrinol 2015;418:306‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Valera MC, Fontaine C, Dupuis M, Noirrit‐Esclassan E, Vinel A, Guillaume M, et al. Towards optimization of estrogen receptor modulation in medicine. Pharmacol Ther 2018;189:123‐129. [DOI] [PubMed] [Google Scholar]
- 3. Wang X, Lu Y, Wang E, Zhang Z, Xiong X, Zhang H, et al. Hepatic estrogen receptor alpha improves hepatosteatosis through upregulation of small heterodimer partner. J Hepatol 2015;63:183‐190. [DOI] [PubMed] [Google Scholar]
- 4. Zhu L, Brown WC, Cai Q, Krust A, Chambon P, McGuinness OP, et al. Estrogen treatment after ovariectomy protects against fatty liver and may improve pathway‐selective insulin resistance. Diabetes 2013;62:424‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Smith CL, O'Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr Rev 2004;25:45‐71. [DOI] [PubMed] [Google Scholar]
- 6. Handgraaf S, Riant E, Fabre A, Waget A, Burcelin R, Liere P, et al. Prevention of obesity and insulin resistance by estrogens requires ERalpha activation function‐2 (ERalphaAF‐2), whereas ERalphaAF‐1 is dispensable. Diabetes 2013;62:4098‐4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berry M, Metzger D, Chambon P. Role of the two activating domains of the oestrogen receptor in the cell‐type and promoter‐context dependent agonistic activity of the anti‐oestrogen 4‐hydroxytamoxifen. EMBO J 1990;9:2811‐2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guillaume M, Handgraaf S, Fabre A, Raymond‐Letron I, Riant E, Montagner A, et al. Selective activation of estrogen receptor alpha activation function‐1 is sufficient to prevent obesity, steatosis, and insulin resistance in mouse. Am J Pathol 2017;187:1273‐1287. [DOI] [PubMed] [Google Scholar]
- 9. Della Torre S, Rando G, Meda C, Stell A, Chambon P, Krust A, et al. Amino acid‐dependent activation of liver estrogen receptor alpha integrates metabolic and reproductive functions via IGF‐1. Cell Metab 2011;13:205‐214. [DOI] [PubMed] [Google Scholar]
- 10. Strelau J, Strzelczyk A, Rusu P, Bendner G, Wiese S, Diella F, et al. Progressive postnatal motoneuron loss in mice lacking GDF‐15. J Neurosci 2009;29:13640‐13648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Matic M, Bryzgalova G, Gao H, Antonson P, Humire P, Omoto Y, et al. Estrogen signalling and the metabolic syndrome: targeting the hepatic estrogen receptor alpha action. PLoS ONE 2013;8:e57458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959;37:911‐917. [DOI] [PubMed] [Google Scholar]
- 13. Barrans A, Collet X, Barbaras R, Jaspard B, Manent J, Vieu C, et al. Hepatic lipase induces the formation of pre‐beta 1 high density lipoprotein (HDL) from triacylglycerol‐rich HDL2. A study comparing liver perfusion to in vitro incubation with lipases. J Biol Chem 1994;269:11572‐11577. [PubMed] [Google Scholar]
- 14. Kim JH, Meyers MS, Khuder SS, Abdallah SL, Muturi HT, Russo L, et al. Tissue‐selective estrogen complexes with bazedoxifene prevent metabolic dysfunction in female mice. Mol Metab 2014;3:177‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnen H, Lin S, Kuffner T, Brown DA, Tsai VW, Bauskin AR, et al. Tumor‐induced anorexia and weight loss are mediated by the TGF‐beta superfamily cytokine MIC‐1. Nat Med 2007;13:1333‐1340. [DOI] [PubMed] [Google Scholar]
- 16. Tsai VW, Manandhar R, Jorgensen SB, Lee‐Ng KK, Zhang HP, Marquis CP, et al. The anorectic actions of the TGFbeta cytokine MIC‐1/GDF15 require an intact brainstem area postrema and nucleus of the solitary tract. PLoS ONE 2014;9:e100370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Palierne G, Fabre A, Solinhac R, Le Peron C, Avner S, Lenfant F, et al. Changes in gene expression and estrogen receptor cistrome in mouse liver upon acute E2 treatment. Mol Endocrinol 2016;30:709‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lu X, He X, Su J, Wang J, Liu X, Xu K, et al. EZH2‐mediated epigenetic suppression of GDF15 predicts a poor prognosis and regulates cell proliferation in non‐small‐cell lung cancer. Mol Ther Nucleic Acids 2018;12:309‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rada‐Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011;470:279‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bruno S, Maisonneuve P, Castellana P, Rotmensz N, Rossi S, Maggioni M, et al. Incidence and risk factors for non‐alcoholic steatohepatitis: prospective study of 5408 women enrolled in Italian tamoxifen chemoprevention trial. BMJ 2005;330:932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cole LK, Jacobs RL, Vance DE. Tamoxifen induces triacylglycerol accumulation in the mouse liver by activation of fatty acid synthesis. Hepatology 2010;52:1258‐1265. [DOI] [PubMed] [Google Scholar]
- 22. Lelliott CJ, Lopez M, Curtis RK, Parker N, Laudes M, Yeo G, et al. Transcript and metabolite analysis of the effects of tamoxifen in rat liver reveals inhibition of fatty acid synthesis in the presence of hepatic steatosis. FASEB J 2005;19:1108‐1119. [DOI] [PubMed] [Google Scholar]
- 23. Miyashita T, Toyoda Y, Tsuneyama K, Fukami T, Nakajima M, Yokoi T. Hepatoprotective effect of tamoxifen on steatosis and non‐alcoholic steatohepatitis in mouse models. J Toxicol Sci 2012;37:931‐942. [DOI] [PubMed] [Google Scholar]
- 24. Ceasrine AM, Ruiz‐Otero N, Lin EE, Lumelsky DN, Boehm ED, Kuruvilla R. Tamoxifen improves glucose tolerance in a delivery, sex, and strain‐dependent manner in mice. Endocrinology 2019;160:782‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lampert C, Arcego DM, Laureano DP, Diehl LA, da Costa Lima IF, Krolow R, et al. Effect of chronic administration of tamoxifen and/or estradiol on feeding behavior, palatable food and metabolic parameters in ovariectomized rats. Physiol Behav 2013;119:17‐24. [DOI] [PubMed] [Google Scholar]
- 26. Goss PE, Qi S, Hu H. Comparing the effects of atamestane, toremifene and tamoxifen alone and in combination, on bone, serum lipids and uterus in ovariectomized rats. J Steroid Biochem Mol Biol 2009;113:233‐240. [DOI] [PubMed] [Google Scholar]
- 27. Reckless J, Metcalfe JC, Grainger DJ. Tamoxifen decreases cholesterol sevenfold and abolishes lipid lesion development in apolipoprotein E knockout mice. Circulation 1997;95:1542‐1548. [DOI] [PubMed] [Google Scholar]
- 28. Ke HZ, Chen HK, Simmons HA, Qi H, Crawford DT, Pirie CM, et al. Comparative effects of droloxifene, tamoxifen, and estrogen on bone, serum cholesterol, and uterine histology in the ovariectomized rat model. Bone 1997;20:31‐39. [DOI] [PubMed] [Google Scholar]
- 29. Mauvais‐Jarvis F, Clegg DJ, Hevener AL. The role of estrogens in control of energy balance and glucose homeostasis. Endocr Rev 2013;34:309‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu Y, Nedungadi TP, Zhu L, Sobhani N, Irani BG, Davis KE, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab 2011;14:453‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lopez M, Lelliott CJ, Tovar S, Kimber W, Gallego R, Virtue S, et al. Tamoxifen‐induced anorexia is associated with fatty acid synthase inhibition in the ventromedial nucleus of the hypothalamus and accumulation of malonyl‐CoA. Diabetes 2006;55:1327‐1336. [DOI] [PubMed] [Google Scholar]
- 32. Wallen WJ, Belanger MP, Wittnich C. Sex hormones and the selective estrogen receptor modulator tamoxifen modulate weekly body weights and food intakes in adolescent and adult rats. J Nutr 2001;131:2351‐2357. [DOI] [PubMed] [Google Scholar]
- 33. Wade GN, Heller HW. Tamoxifen mimics the effects of estradiol on food intake, body weight, and body composition in rats. Am J Physiol 1993;264:R1219‐R1223. [DOI] [PubMed] [Google Scholar]
- 34. Kumar KG, Trevaskis JL, Lam DD, Sutton GM, Koza RA, Chouljenko VN, et al. Identification of adropin as a secreted factor linking dietary macronutrient intake with energy homeostasis and lipid metabolism. Cell Metab 2008;8:468‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ding Q, Mracek T, Gonzalez‐Muniesa P, Kos K, Wilding J, Trayhurn P, et al. Identification of macrophage inhibitory cytokine‐1 in adipose tissue and its secretion as an adipokine by human adipocytes. Endocrinology 2009;150:1688‐1696. [DOI] [PubMed] [Google Scholar]
- 36. Yang L, Chang CC, Sun Z, Madsen D, Zhu H, Padkjaer SB, et al. GFRAL is the receptor for GDF15 and is required for the anti‐obesity effects of the ligand. Nat Med 2017;23:1158‐1166. [DOI] [PubMed] [Google Scholar]
- 37. Mullican SE, Lin‐Schmidt X, Chin CN, Chavez JA, Furman JL, Armstrong AA, et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat Med 2017;10:1150‐1157. [DOI] [PubMed] [Google Scholar]
- 38. Emmerson PJ, Wang F, Du Y, Liu Q, Pickard RT, Gonciarz MD, et al. The metabolic effects of GDF15 are mediated by the orphan receptor GFRAL. Nat Med 2017;23:1215‐1219. [DOI] [PubMed] [Google Scholar]
- 39. Macia L, Tsai VW, Nguyen AD, Johnen H, Kuffner T, Shi YC, et al. Macrophage inhibitory cytokine 1 (MIC‐1/GDF15) decreases food intake, body weight and improves glucose tolerance in mice on normal & obesogenic diets. PLoS ONE 2012;7:e34868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tsai VW, Macia L, Johnen H, Kuffner T, Manadhar R, Jorgensen SB, et al. TGF‐b superfamily cytokine MIC‐1/GDF15 is a physiological appetite and body weight regulator. PLoS ONE 2013;8:e55174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kim KH, Kim SH, Han DH, Jo YS, Lee YH, Lee MS. Growth differentiation factor 15 ameliorates nonalcoholic steatohepatitis and related metabolic disorders in mice. Sci Rep 2018;8:6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tabula Muris Consortium; Overall coordination; Logistical coordination; Organ collection and processing; Library preparation and sequencing; Computational data analysis; Cell type annotation; Writing group; Supplemental text writing group; Principal investigators . Single‐cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018;562:367‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jordan VC. Tamoxifen: a most unlikely pioneering medicine. Nat Rev Drug Discov 2003;2:205‐213. [DOI] [PubMed] [Google Scholar]
- 44. Finan B, Yang B, Ottaway N, Stemmer K, Muller TD, Yi CX, et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat Med 2012;18:1847‐1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials