

Abstract
In recent years, ellagic acid (EA), a naturally-occurring phenolic compound richly contained in some of the human food sources such as Longan and Litchi, was reported to have a number of biological effects. Based on our earlier 3D-QSAR/CoMFA models for cyclooxygenase (COX) I and II, we hypothesize that EA may have the potential to modulate the catalytic activity of COX enzymes, and this hypothesis is examined in the present study. The results from both in vitro and in vivo experiments show that EA is an activator of COX enzyme-catalyzed production of prostaglandin E2, a representative prostaglandin tested. Mechanistically, EA can activate the peroxidase active site of COX enzymes by serving as a co-substrate, presumably for the reduction of protoporphorin IX with FeIV inside. The effect of EA is abrogated by the co-presence of galangin, which is known to bind to COX's peroxidase active site and thereby blocks the effect of the reducing co-substrates. In view of the known physiological functions of COX enzymes in the body, it is suggested that some of the pharmacological and/or toxicological effects of EA may result from an increased production of certain prostaglandins and their related derivatives in the body.
Keywords: ellagic acid, cyclooxygenase, peroxidase activity, mechanism of catalytic activation
Introduction
Ellagic acid (EA4) is a naturally-occurring phenolic compound (structure shown in Fig. 1) found in certain oak species (1), medicinal mushroom Phellinus linteus (2), and macrophyte Myriophyllum spicatum (3). It is also richly contained in some human food sources (4–12). High levels of EA are found in Longan (also known as Dimocarpus longan), Litchi (Litchi chinensis), walnuts, pecans, cranberries, raspberries, strawberries, grapes, and peach (4–12). EA has been reported to have a number of biological activities, including antioxidant and antiproliferative properties as observed in some of the in vitro and animal models (10,13–16). As with other polyphenol antioxidants, it has been suggested that EA may have a chemoprotective effect in cellular models by inhibiting reactive chemical carcinogens [e.g., nitrosamines (17,18) and polycyclic aromatic hydrocarbons (19)] from covalently modifying DNA (17–19). It is noteworthy that in recent years, EA has been controversially marketed as a dietary supplement with a number of assumed benefits against cancer, heart disease, as well as other medical issues, and these claims have received warnings from the US Food and Drug Administration (FDA) (20).
Figure 1.

Chemical structure of EA, a natural phenolic compound. Note that EA is a symmetric compound, and its A- and B-rings are equivalent, and the C- and D-rings are equivalent. For instance, after ionization (i.e., deprotonation) of C-4 hydroxyl group, its oxygen atom carries a negative charge, with an additional electron retained. Similar ionization can also occur with the C-3-OH group as well as with the C-4′-OH and C-3′-OH groups. EA, ellagic acid.
Cyclooxygenase I and II (COX I and II) are key enzymes that catalyze the metabolism of arachidonic acid (AA), resulting in the formation of important biological mediators including prostaglandins (PGs), prostacyclins, thromboxanes, and others (21–24). Since these mediators affect many pathological and physiological processes, COX enzymes have become important targets in pharmacology and toxicology. Pharmacological modulation of the COX enzyme activity has become an effective approach in treating many medical conditions (25–28).
We have recently shown that certain natural phenolics, such as quercetin and myricetin, can activate the catalytic activity of COX I and II in enzymatic assays by functioning as reducing co-substrates for these enzymes (29). This phenomenon was further confirmed when they were tested in cultured cells (29) and animal models (30). Notably, these compounds are effective in activating COX enzyme activity for PG biosynthesis in intact cells with effective concentrations in the nM range (29). Additional mechanistic studies showed that some of the flavonoids can bind inside the peroxidase active site of the enzymes and directly interact with protoporphorin IX with FeIV inside (P+FeIV) to facilitate the electron transfer from these reducing compounds to the Fe ion (31).
Based on our earlier three dimensional (3D)-QSAR/CoMFA models for COX I and II that were derived from experimental study of representative flavonoids (29), we predicted that EA may share the COX enzyme-modulating activity. In the present study, we aimed to experimentally examine the ability of EA to modulate PG production using cultured cells and intact animals. The possible mechanism for its modulating effect was explored using computational modeling approach by studying their binding interaction with the COX-1 and COX-2 enzymes.
Materials and methods
Chemicals and reagents
EA (purity >99%), galangin, AA, lipopolysaccharide (LPS; from Escherichia coli, serotype 055:b5), and Dulbecco's modified Eagle's medium (DMEM) were purchased from Sigma-Aldrich (St. Louis, ΜO, USA). The anti-COX I and anti-COX II antibodies were obtained from Abcam (Cambridge, UK), and the anti-GAPDH antibody was obtained from Cell Signaling Technology (Danvers, MA, USA). Fetal bovine serum (FBS) was obtained from Gibco-Thermo Fisher Scientific, Inc. (Waltham, MA, USA), and the enzymatic immunoassay (EIA) kit for detecting PGE2 was obtained from Cayman Chemical (Ann Arbor, MI, USA).
Cell culture experiments and compound screening
The murine macrophage RAW264.7 cell line was obtained from the American Type Culture Collection (Manassas, VA, USA), and maintained in DMEM containing L-glutamine, glucose and sodium bicarbonate supplemented with 10% FBS at 37°C under 5% CO2. In the experiments that were designed to determine the effect of these phenolic compounds on the formation of PGE2 in cultured RAW264.7 cells, the cells were first stimulated with 1 µg/ml LPS for 2 h to induce the expression of COX enzymes. Then the medium was removed and replaced with 300 µl serum-free DMEM with or without different concentrations (0.01, 0.1, 1, 10, and 100 µM) of a phenolic compound of interest. After an additional 2-h incubation, the culture media were collected for measurement of PGE2 level by using an EIA kit obtained from Cayman Chemical.
In vivo animal experiments
All the procedures involving the use of live animals as described in this study were approved by the Institutional Animal Care and Use Committee of the Southern University of Science and Technology (approval number: SUSTC-G-2014009), and the guidelines for humane treatment of animals accepted by the National Institutes of Health (NIH) of the USA were followed. The male Sprague-Dawley rats (4 to 5-week-old, specific pathogen-free) were obtained from Guangdong Medical Laboratory Animal Center (Guangdong, China), and they were maintained in our institute's central animal facility. After arrival, the animals were allowed to acclimatize for one week prior to being used for experimentation. The animals were housed under constant conditions of temperature (20±1°C) and 12-h light/dark cycle, and had free access to food and water.
Male rats were divided into the following two groups: The control group (receiving vehicle treatment only) and the EA group (treated with 6 mg/kg body weight EA, dissolved in 1.5 ml of 1% methyl cellulose). Blood samples were collected through tail bleeding at different time points (0, 3, 6, 12, and 24 h) following administration, and stored in small vials containing heparin. Plasma was prepared from the collected blood by centrifugation at 1,000 × g for 10 min at 37°C. The plasma level of PGE2 was determined using an EIA kit (Cayman Chemical) according to the manufacturer's instructions.
Molecular docking analysis of the binding interaction of EA with COX II. Energy minimization and molecular docking were performed on a Dell PowerEdge R730 Server with the Discovery Studio modeling software (version 2007; Accelrys, San Diego, CA, USA).
Protein processing
Since the X-ray structure of sheep COX I protein [PDB code: 1q4g (32)] and mouse COX II protein [(DB code: 3nt1 (33)] in complex with P+FeIV are available, we used these structures as templates for computational docking analysis. All small molecules except P+FeIV that are non-covalently attached to the COX protein were removed, and then the amino acid residues in the protein structure were re-numbered according to the correct known sequences. The Clean Protein module in Discovery Studio was used to complete the side chains for amino acid residues, correct bonding and bond orders, and add hydrogens back. Notably, P+FeIV in the sheep COX I protein [PDB code: 1q4g (32)] and mouse COX II structure (PDB code: 3nt1 (33)] are already contained in the structure as complex, and the ion atom is set as FeIV. Lastly, the Prepare Protein module in Discovery Studio was used for protein preparation along with the CHARMm force field.
Ligand processing
The structure of EA was downloaded from the Protein Data Bank and minimized with the CHARMm force field. In addition, we used the Prepare Ligands module to generate EA in a non-ionizing state and two partially-ionizing states. The non-ionizing state has all hydrogens of the four phenolic hydroxyl groups retained, whereas the ionizing states each have one proton removed (i.e., deprotonation) from a different hydroxyl group in EA, which include the C-4-OH in the A-ring (equivalent to the C-4′-OH in the B-ring) and the C-3-OH in the A-ring (equivalent to the C-3′-OH in the B-ring) (Fig. 1).
Flexible docking
For flexible docking, we used the Find Sites from Receptor Cavities module to identify the binding site in the prepared 1q4g COX I and 3nt1 COX II structures. According to our earlier study, the target site is the peroxidase active site in these two COX proteins (31). We selected all amino acid residues within a 5-Å reach of the target site and allow them to have flexible side chains. The SBD Site Sphere is centered at the target site and then expanded to a 13-radius size. Under the Flexible Docking mode with conformation method set to BEST, the Simulated Annealing docking method was then applied to dock EA into the target sites of COX I and COX II. Notably, two flexible docking modes were separately executed for COX I and II, corresponding to the two different ionizing states of EA. The whole structure of each COX protein was further minimized with the CHARMm force field.
Calculation of binding energy
The Calculate Binding Energies module in Discovery Studio is used to find the complexes with the lowest binding energy values. According to Discovery Studio, the free energy for the binding interaction between a protein and its ligand is estimated according to the free energies of the complex, the protein, and the ligand. These free energy values are separately calculated using the CHARMm force field and the generalized Born with smooth switching (GBSW) method (34). In this approach, a van der Waals-based surface with a smooth dielectric boundary is used in the calculation of the self-electrostatic solvation energy. The ligand conformational entropy is also considered during the free binding energy calculation. The following equation is used to calculate the binding energy (ΔGbinding) between EA and the COX I or COX II protein: ΔGbinding = Gcomplex - (GCOX + Gligand), where Gcomplex is the absolute free energy of the complex, GCOX is the absolute free energy of the COX protein, and Gligand is the absolute free energy of the ligand (35,36). The ΔGbinding value is used to reflect the relative interaction affinity between the COX enzyme and EA.
Statistical analysis. Data were determined as mean ± SD of triple determinations.
Results
Effect of EA on PGE2 production in vitro and in vivo
In vitro studies
To determine whether EA can modulate PG production in cultured RAW264.7 cells, the cells were first stimulated with 1 µg/ml LPS for 2 h to induce the expression of COX proteins as well as PG production (Fig. 2). We found that LPS pretreatment mostly induced COX II expression in these cells as confirmed by western blotting (Fig. 2A), which is in agreement with an earlier report (21). The increased expression of COX II also correlated with increased production of PGE2, a representative PG selected for testing in this study (Fig. 2B).
Figure 2.
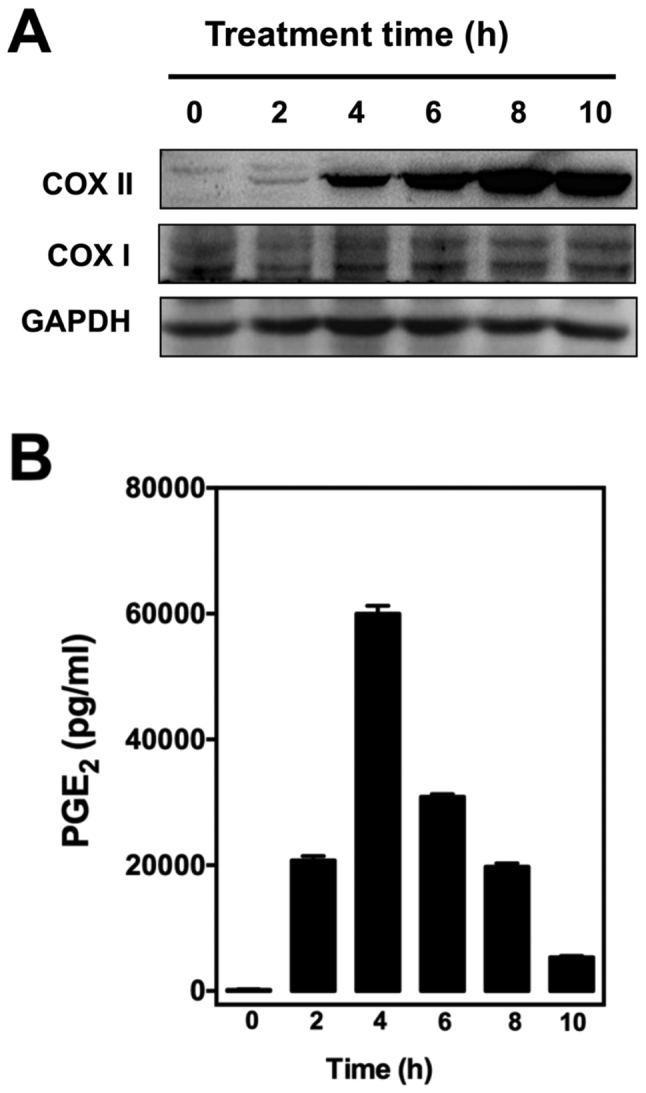
Effect of different length of treatment with LPS on (A) COX I/II protein levels in cultured RAW265.7 cells and (B) PGE2 levels in cell culture media. After treatment with 1 µg/ml LPS for 0, 2, 4, 6, 8, and 10 h, RAW264.7 cells were incubated with serum-free medium for an additional 2 h, and the supernatants were collected for measurement of PGE2 levels by using an enzymatic immunoassay kit. Western blot analysis of cell lysates was performed with antibodies COX I or II, coupled with a secondary antibody conjugated with horseradish peroxidase. LPS, lipopolysaccharide; COX I and COX II, cyclooxygenase I and II, respectively.
Using LPS-pretreated RAW264.7 cells as an in vitro model, we then tested the modulating effect of EA on PGE2 production. Following LPS pretreatment, the medium was removed and replaced with 300 µl serum-free DMEM with or without different concentrations (0.01, 0.1, 1, 10, and 100 µM) of EA. After an additional 2-h incubation, the culture media were collected for measurement of PGE2. We found that EA at 10 nM showed a weak stimulatory effect on PGE2 production, and this stimulation reached a plateau when the concentration of EA reached 100–1,000 nM. The maximal stimulation of COX-mediated PGE2 production by EA was approximately 140% above the control in these cells (Fig. 3, left panel). Notably, when EA concentration further increased to 10 µM, PGE2 production is slightly reduced. It is noteworthy that this phenomenon was also observed in our earlier in vitro and in vivo studies with other reducing co-substrates such as quercetin and myricetin (29,30). For comparison, we also included for testing the effect of morin (an analog of quercetin) on PGE2 production. We found that morin modulates the production of PGE2 in a similar manner as EA (Fig. 3, right panel).
Figure 3.

Effect of EA on the release of PGE2 from LPS-pretreated RAW264.7 cells. The cells were pretreated with 1 µg/ml LPS for 2 h to induce COX II expression, and then the culture media were removed and replaced with 300 µl serum-free medium containing EA for another 2 h. The following concentrations of the test compound were used: 0.01, 0.1, 1, and 10 µM. The levels of PGE2 were measured using an enzymatic immunoassay kit. Each point was the mean ± SD of triple determinations. EA, ellagic acid; LPS, lipopolysaccharide; COX I and COX II, cyclooxygenase I and II, respectively.
In this study, we confirmed that galangin (a known competitive inhibitor of the COX peroxidase active site) (37) does not have a significant stimulatory effect on PGE2 production when it is added alone to LPS-pretreated RAW264.7 cells in culture. However, when it is added along with EA, it can inhibit EA-stimulated PGE2 production in a concentration-dependent manner, with an IC50 value of <1 µM (Fig. 4, left panel). Notably, when galangin was added alone to the LPS-pretreated RAW264.7 cells, it also inhibited the baseline production of PGE2 in a similar manner (Fig. 4, right panel).
Figure 4.

Effect of galangin on ethyl gallate-stimulated PGE2 release from LPS-pretreated RAW264.7 cells. Cells were pretreated with 1 µg/ml LPS for 2 h to induce COX II expression, and then the culture media were removed and replaced with 300 µl serum-free medium containing 1 µM ethyl gallate plus different concentrations (0.01, 0.1, 1 and 10 µM) of galangin for another 2 h. The levels of PGE2 were measured using an enzymatic immunoassay kit (Cayman Chemical, Ann Arbor, MI, USA). Each point was the mean ± SD of triple determinations. LPS, lipopolysaccharide; COX I and COX II, cyclooxygenase I and II, respectively; EA, ellagic acid.
In vivo studies
In the present study, we also determined the effect of EA on the plasma levels of PGE2 by using normal male Sprague-Dawley rats as an in vivo model. The reason for use of this animal model is because it was successfully used earlier to study the effect of other representative phenolic compounds on plasma and tissue levels of several PG products (30). We found that administration (oral route or injection) of these phenolic compounds to normal male rats can significantly increase the tissue and blood levels of PG products in vivo (30).
In this experiment, the animals receive a single oral dose of EA alone (at 6 mg/kg body weight). Blood samples were collected through tail bleeding at different time points, and plasma samples were prepared for PGE2 measurement. We found that oral administration of EA alone markedly increased the plasma level of PGE2 in a time-dependent manner (Fig. 5). Plasma PGE2 level started to increase significantly at 3 h after administration, and peaked at approximately 6 h after administration, with a maximal increase of the plasma PGE2 level by approximately 3.5-fold (Fig. 5). This observation is very similar to the observations made in our earlier study with two other COX activators (quercetin and myricetin) (30).
Figure 5.
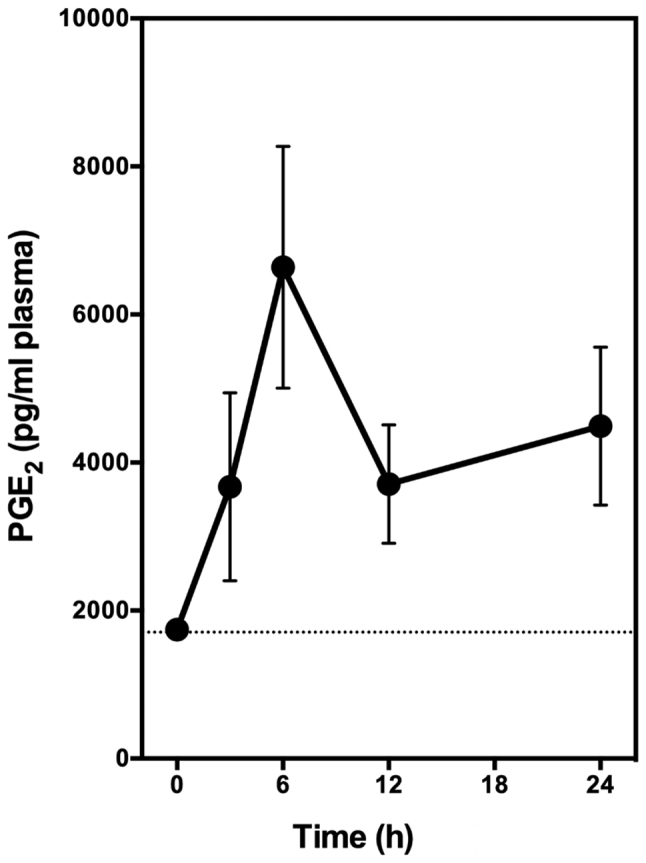
Time-dependent effect of EA on plasma levels of PGE2 in rats. Male Sprague-Dawley rats were given oral administration of EA (6 mg/kg body weight) or vehicle alone. Blood samples were collected from tail bleeding at 3, 6, 9, 12, 24, and 48 h after oral administration. The plasma was immediately prepared and stored at −80°C. The plasma levels of PGE2 were measured using an enzymatic immunoassay kit. Each data point is the mean ± SD (n=5). EA, ellagic acid.
In summary, in vitro experiments using LPS-pretreated RAW264.7 cells and in vivo experiments using rats both showed that EA is an activator of COX-mediated production of PGE2. This effect is abrogated by the co-presence of galangin, an inhibitor of the peroxidase activity of COX, presumably by blocking the effect of the reduction of co-substrates.
Computational docking analysis of EA binding inside the peroxidase active sites of COX I and II
We employed sheep COX I [PDB code: 1q4g (32)] and mouse COX II [PDB code: 3nt1 (33)] proteins as templates to model the docking interaction between EA and COX I/II. The 3D structural models of these two proteins were prepared using Discovery Studio. Using these structural models, we docked EA in three different ionizing states (one non-ionizing state vs. two partially-ionizing states) into the peroxidase active sites of COX I/II (Figs. 6 and 7). The results are summarized below.
Figure 6.

Molecular docking analysis of the binding interaction of COX I with non-ionizing EA (A-C), C4-OH-ionizing EA (D-F), and C-3-OH-ionizing EA (G-I). (A, D and G) The dominant docking result for non-ionizing EA (A), C4-OH-ionizing EA (D) and C-3-OH-ionizing EA (G) inside the peroxidase active site of COX I. The protein structure is shown in a flat ribbon format. In P+FeIV, carbon is colored in orange, nitrogen in blue, oxygen in red, hydrogen in white, and iron in navy blue. In EA, carbon is colored in green, oxygen in red, and hydrogen in white. (B, E and H) The same structure as in respective panels (A, D and G) with a white dash line added to indicate the distance between Fe4+ ion and O in one of EA's OHs. (C, F and I) Suggested potential hydrogen bonds (green dash lines) between the amino acid residues and the non-ionizing EA (C), C-4-OH-ionizing EA (F), and C-3-OH-ionizing EA (I). The amino acid residues are colored in light blue. EA, ellagic acid; COX I and COX II, cyclooxygenase I and II, respectively.
Figure 7.

Molecular docking analysis of the binding interaction of COX II with non-ionizing EA (A-C), C4-OH-ionizing EA (D-F), and C-3-OH-ionizing EA (G-I). (A, D and G) The dominant docking result for non-ionizing EA (A), C4-OH-ionizing EA (D) and C-3-OH-ionizing EA (G) inside the peroxidase active site of COX II. The protein structure is shown in a flat ribbon format. In P+FeIV, carbon is colored in orange, nitrogen in blue, oxygen in red, hydrogen in white, and iron in navy blue. In EA, carbon is colored in green, oxygen in red, and hydrogen in white. (B, E and H) The same structure as in respective panels (A, D and G) with a white dash line added to indicate the distance between Fe4+ ion and O in one of EA's OHs. (C, F and I) Suggested potential hydrogen bonds (green dash lines) between the amino acid residues and the non-ionizing EA (C), C-4-OH-ionizing EA (F), and C-3-OH-ionizing EA (I). The amino acid residues are colored in light blue. EA, ellagic acid; COX I and COX II, cyclooxygenase I and II, respectively.
COX I
Docking analysis of EA in a non-ionizing state suggests that it can bind inside the peroxidase active site in two possible binding modes: One with its A-ring structure inside the peroxidase site facing P+FeIV, and the other one with its B-ring structure inside the peroxidase site. Based on binding energy ΔGbinding values (Table I), it is predicted that the binding mode with its B-ring inside is the dominant binding pose (ΔGbinding of −3.180 kcal/mol). However, in this pose, all hydroxyl groups of EA are not too close to the Fe ion of P+FeIV (Fig. 6A and B), suggesting that this binding pose is an inactive pose, and would not be able to transfer its electrons to the Fe ion of P+FeIV for reduction. For poses ranked 2–10, all hydroxyl groups of EA are even slightly farther away from the Fe ion of P+FeIV than the dominant binding pose.
Table I.
Computed binding energy values (ΔGbinding, kcal/mol) for the molecular docking analysis of the best binding poses between EA (partially-ionized vs. non-ionized) and COX I/II proteins.
| Binding energy value ΔGbinding (kcal/mol) | |||
|---|---|---|---|
| Type of protein | No ionization | C-4-OH ionization | C-3-OH ionization |
| COX I protein | −3.180 | −78.952 | −15.470 |
| COX II protein | −3.061 | −76.923 | −47.614 |
EA, ellagic acid; COX I and COX II, cyclooxygenase I and II, respectively.
Potential hydrogen bonds between COX I and EA in its dominant binding pose as suggested by the Receptor-Ligand Hydrogen Bonds module are shown in Fig. 6C, and they involve five amino acid residues: two with His207 (1.314 and 2.289 Å), two with Phe210 (1.928 and 1.949 Å), and one with Gln289 (2.219 Å).
It is estimated that under physiological conditions, a small fraction of the hydroxyl groups in EA's A-ring would undergo ionization (deprotonation), i.e., removal of a proton. Results from our recent study (38) suggest that the binding interaction of a reducing substrate (such as quercetin) under partial ionization is dramatically enhanced in comparison with the non-ionizing state. Therefore, we also performed docking analysis using the partially-ionizing EA. Predicted by the Discovery Studio, C-4-OH has a higher tendency to deprotonate than C-3-OH under physiological conditions. In the present study, we chose to determine the docking conformation when deprotonation occurs only with one hydroxyl group at any given moment, because simultaneous deprotonation of multiple protons in the same molecule is considered nearly impossible to occur under physiological pH conditions.
We found that when each of the hydroxyl groups in A-ring is individually deprotonated, the dominant poses (based on ΔGbinding values; Table I) all have its A-ring inside (Fig. 6D and G). Under C-4-OH deprotonation (Fig. 6E; ΔGbinding of −78.7952 kcal/mol), the distance between Fe and O− is 2.259 Å. Under C-3-OH deprotonation (Fig. 6H; ΔGbinding of −15.470 kcal/mol), the distance between the Fe ion and O− is 3.067 Å.
Potential hydrogen bonds between COX I and EA in two ionizing states as suggested by the receptor-ligand hydrogen bonds are shown in Fig. 6F and I. Under C-4-OH deprotonation, EA in its dominant binding pose only forms one hydrogen bond with Gln203 (2.297 Å), and under C-3-OH deprotonation, it forms two hydrogen bonds with His207 (1.494 and 1.786 Å).
COX II
As predicted according to the binding energy ΔGbinding value (ΔGbinding of −3.061 kcal/mol), the dominant binding pose of EA in a non-ionizing state has its B-ring inside COX II's peroxidase site (Fig. 7A). All hydroxyl groups in this pose are very far away from the Fe ion of P+FeIV (shortest distance at 8.515 Å; Fig. 7B). For poses ranked 2–10, the hydroxyl groups of EA are even slightly farther away from the Fe ion of P+FeIV than the dominant pose.
We also analyzed the docking conformations when deprotonation occurs individually with EA's C-4-OH and C-3-OH hydroxyl groups (data shown in Fig. 7D and E, respectively). We found that when C-4-OH is deprotonated, the dominant pose has its A-ring closer to the Fe ion of P+FeIV (ΔGbinding of −76.923 kcal/mol), particularly the O− ion in EAs C-4-OH (distance of 2.206 Å; Fig. 7G). Under C-3-OH deprotonation, the dominant pose (Fig. 7H) also has its A-ring inside (ΔGbinding of −47.614 kcal/mol), with the distance of 2.607 Å between Fe ion and O- ion in EA's C-3-OH. The suggested hydrogen bonds in the dominant poses in three different states are shown in Fig. 7C, F and I.
Discussion
The results from both in vitro and in vivo experiments in this study showed to the best of our knowledge, for the first time, that EA is an activator of the COX enzyme-catalyzed production of PGE2. Mechanistically, EA likely exerts this effect through activating the peroxidase active site of COX enzymes by serving as a reducing co-substrate for the Fe ion of P+FeIV. The effect of EA is very similar to the effect of some naturally-occurring flavonoids, such as quercetin and myricetin, that were reported earlier by our group, which can activate the catalytic activity of the COX I and II enzymes by functioning as reducing co-substrates (29).
Our earlier study revealed that the hydroxyl groups of the B-ring of quercetin play a critical role in re-activating the COX I/II catalytic activity (29,31). Recently, we have further shown that galangin, a flavonoid that has the same A/C-ring structure as quercetin but does not have any hydroxyl group in its B-ring, can function as a COX inhibitor, by competitively blocking the binding of those flavonoids that can serve as reducing co-substrates for the COX enzymes (37). In this study, we confirmed that galangin does not have a stimulatory effect on PGE2 production when it is added to LPS-pretreated RAW264.7 cells in culture, but it can inhibit EA-stimulated PGE2 production in a concentration-dependent manner, with an IC50 value of <1 µM (Fig. 4, left panel). Notably, when galangin is added alone to the LPS-pretreated RAW264.7 cells in culture, it also inhibits the baseline production of PGE2 in a similar manner (Fig. 4, right panel). This phenomenon was also observed in our recent study (37), which likely is due to the presence of other reducing substrates either indigenously produced by the cells or contained in the cell culture medium, and these compounds can support the basal COX activity as detected in cultured cells. In support of this explanation, we observed earlier that when galangin is tested in the in vitro biochemical enzyme assays involving COX I and II proteins where no other unknown chemicals are introduced, it does not have any meaningful stimulatory or inhibitory effect (37). The observed modulating effect of galangin on EA mirrors the effect of galangin on quercetin-induced PG production as observed in our recent study (37), providing support for the concept that EA has a similar mechanism of action as quercetin.
Computational docking analysis provides insight into the mechanism of the COX-activating action of EA at the molecular level. Comparison of EA in both non-ionizing and partially-ionizing states indicates that ionization of C-4-OH and C-3-OH shortens the distance between Fe4+ and the respective O- (from 6.903 to 2.259 and 3.067 Å, respectively) and increases the binding infinity (from −3.180 to −78.952 and −15.470 kcal/mol, respectively). These data suggest that deprotonation would facilitate the transfer of electron from EA to P+FeIV for peroxidase reduction. In addition, when deprotonation of C-4-OH and C-3-OH is compared, the former shows a shorter distance and higher binding infinity than the latter, suggesting that C-4-OH can more readily transfer its electron to P+FeIV than C-3-OH.
When a partially-ionizing EA is bound inside the peroxidase active site, its estimated shortest distance is 2.259 Å, and this interaction distance is expected to enable a facile transfer of an electron from its hydroxyl group to P+FeIV. Notably, while the best binding poses of ionizing EA in COX I and II proteins are very different, the distances between the Fe ion and oxygen ion are very similar, as are their overall binding energy values. This observation provides additional support for the suggestion that ionic interaction between Fe ion and the respective O− is the dominant force that determines the binding energy level and binding affinity.
It appears that the number of suggested hydrogen bonds does not correlate with the overall binding energy values. The reasons for this apparent discrepancy might be: First, the strong ionic interaction between ionized EA (which contains a negatively-charged O− ion) and the positively-charged Fe ion plays a more important role than hydrogen bonds (39). This suggestion is consistent with the fact that hydrogen bonds are far weaker than the ionic interactions. Second, some of the suggested hydrogen bonds may be of negligible significance in strength due to their rather long bond distance.
Earlier studies have shown that EA is richly present in Longan and Litchi (Lychee) at high concentrations (4–9,14). Longan and Litchi are members of the soapberry family (Sapindaceae). These plants are grown extensively in China and South East Asia, as well as in Australia, Florida (USA), southern Europe, and southern Africa (40,41). In traditional Chinese medicine, Longan and Litchi are fruits with health beneficial functions, but they are also best known for their ‘hot’ properties when overdosed, i.e., overingestion of Longan and Litchi is known to promote inflammatory-type responses. In recent years, there are increasing reports in Southeast Asia regions of Litchi-associated acute encephalitis syndrome among children (42–45). However, the mechanism for some of their beneficial as well as their pro-inflammatory effects is poorly understood at present. The results of the present study showed that EA, a natural phenolic compound richly contained in Longan and Litchi, can stimulate the catalytic activity of the COX I and II enzymes in vitro by functioning as a reducing co-substrate for these enzymes. This unique effect may help partially account for some of the beneficial as well as pro-inflammatory effects of Litchi and Longan.
Lastly, it is noteworthy that most of the pharmacological design and strategies aim to inhibit the COX I/II enzymes, because of their well-known roles in some of the pathogenic processes. However, it is of note that abnormally-low levels of COX I activity are also associated with some serious pathogenic conditions, such as gastrointestinal ulceration and bleeding and cardiovascular diseases (28,46–48). Thus, too low basal levels of the COX activity (particularly COX I) are not beneficial for optimal health. Our finding that some of the natural phenolics can be used in the body as reducing co-substrates of COX enzymes to support their normal catalytic activity for biosynthesis of PG-related mediators may offer a new mechanistic explanation for some of their health beneficial functions in the body.
In summary, both in vitro and in vivo experiments showed that EA is an activator of PGE2 production. Mechanistically, it is suggested that EA can activate the peroxidase active site of COX enzymes by serving as a reducing co-substrate for the reduction of P+FeIV in the catalytic site. The effect of EA is abrogated by the co-presence of galangin, which is known to bind to COX's peroxidase active site and thereby blocks the effect of the reducing co-substrates.
Acknowledgements
Not applicable
Glossary
Abbreviations
- EA
ellagic acid
- COX I and COX II
cyclooxygenase I and II
- AA
arachidonic acid
- PG
prostaglandin
- P+FeIV
protoporphorin IX with FeIV inside
Funding
The present study was supported by research grants from the National Natural Science Foundation of China (NSFC nos. 81473224 and 81630096), Shenzhen City Basic Science Project (no. JCYJ20140714151402768), and Shenzhen Peacock Plan (no. KQTD2016053117035204).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Authors' contributions
HRW conducted the cell culture and animal experiments, analyzed the data, and prepared part of the initial draft of the manuscript; HCS performed the computational analysis, analyzed the data, and prepared part of the initial draft of the manuscript; BTZ had the initial ideas and designed all the experiments, analyzed the data, and prepared and finalized the manuscript.
Ethics approval and consent to participate
All procedures involving the use of live animals as described in the present study were approved by the Institutional Animal Care and Use Committee of the Southern University of Science and Technology (approval number: SUSTC-G-2014009), and the guidelines for the humane treatment of animals accepted by the National Institutes of Health (USA) were followed.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests..
References
- 1.Mämmelä P, Savolainen H, Lindroos L, Kangas J, Vartiainen T. Analysis of oak tannins by liquid chromatography-electrospray ionisation mass spectrometry. J Chromatogr A. 2000;891:75–83. doi: 10.1016/S0021-9673(00)00624-5. [DOI] [PubMed] [Google Scholar]
- 2.Nierenstein M. The formation of ellagic acid from galloyl-glycine by penicillium. Biochem J. 1915;9:240–244. doi: 10.1042/bj0090240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakai S. Myriophyllum spicatum-released allelopathic polyphenols inhibiting growth of blue-green algae Microcystis aeruginosa. Water Res. 2000;34:3026–3032. doi: 10.1016/S0043-1354(00)00039-7. [DOI] [Google Scholar]
- 4.Prasad KN, Yang B, Yang S, Chen Y, Zhao M, Ashraf M, Jiang Y. Identification of phenolic compounds and appraisal of antioxidant and antityrosinase activities from litchi (Litchi sinensis Sonn.) seeds. Food Chem. 2009;116:1–7. doi: 10.1016/j.foodchem.2009.01.079. [DOI] [Google Scholar]
- 5.Estela de Rezende Q, Patto de Abreu CM, Kelly da Silva O, Vinicius de Oliveira R, Fráguas RM. Bioactive phytochemicals and antioxidant activity in fresh and dried lychee fractions. Rev Ciênc Agron. 2015;46:163–169. doi: 10.1590/S1806-66902015000100019. [DOI] [Google Scholar]
- 6.Soong YY, Barlow PJ. Isolation and structure elucidation of phenolic compounds from longan (Dimocarpus longan Lour.) seed by high-performance liquid chromatography-electrospray ionization mass spectrometry. J Chromatogr A. 2005;1085:270–277. doi: 10.1016/j.chroma.2005.06.042. [DOI] [PubMed] [Google Scholar]
- 7.Zheng G, Xu L, Wu P, Xie H, Jiang Y, Chen F, Wei X. Polyphenols from longan seeds and their radical-scavenging activity. Food Chem. 2009;116:433–436. doi: 10.1016/j.foodchem.2009.02.059. [DOI] [Google Scholar]
- 8.Rangkadilok N, Sitthimonchai S, Worasuttayangkurn L, Mahidol C, Ruchirawat M, Satayavivad J. Evaluation of free radical scavenging and antityrosinase activities of standardized longan fruit extract. Food Chem Toxicol. 2007;45:328–336. doi: 10.1016/j.fct.2006.08.022. [DOI] [PubMed] [Google Scholar]
- 9.Tseng HC, Wu WT, Huang HS, Wu MC. Antimicrobial activities of various fractions of longan (Dimocarpus longan Lour. Fen Ke) seed extract. Int J Food Sci Nutr. 2014;65:589–593. doi: 10.3109/09637486.2014.886181. [DOI] [PubMed] [Google Scholar]
- 10.Vattem DA, Shetty K. Biological function of ellagic acid: A review. J Food Biochem. 2005;29:234–266. doi: 10.1111/j.1745-4514.2005.00031.x. [DOI] [Google Scholar]
- 11.Usta C, Ozdemir S, Schiariti M, Puddu PE. The pharmacological use of ellagic acid-rich pomegranate fruit. Int J Food Sci Nutr. 2013;64:907–913. doi: 10.3109/09637486.2013.798268. [DOI] [PubMed] [Google Scholar]
- 12.Infante R, Contador L, Rubio P, Aros D, Peña-Neira Á. Postharvest sensory and phenolic characterization of ‘Elegant Lady’ and ‘Carson’ peaches. Chil J Agric Res. 2011;71:445–451. doi: 10.4067/S0718-58392011000300016. [DOI] [Google Scholar]
- 13.Seeram NP, Adams LS, Henning SM, Niu Y, Zhang Y, Nair MG, Heber D. In vitro antiproliferative, apoptotic and antioxidant activities of punicalagin, ellagic acid and a total pomegranate tannin extract are enhanced in combination with other polyphenols as found in pomegranate juice. J Nutr Biochem. 2005;16:360–367. doi: 10.1016/j.jnutbio.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 14.Vattem DA, Shetty K. Biological function of ellagic acid: A Review. J Food Biochem. 2005;29:234–266. doi: 10.1111/j.1745-4514.2005.00031.x. [DOI] [Google Scholar]
- 15.Emanuele S, Lauricella M, Calvaruso G, D'Anneo A, Giuliano M. Litchi chinensis as a functional food and a source of antitumor compounds: An overview and a description of biochemical pathways. Nutrients. 2017;9:E992. doi: 10.3390/nu9090992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narayanan BA, Geoffroy O, Willingham MC, Re GG, Nixon DW. p53/p21(WAF1/CIP1) expression and its possible role in G1 arrest and apoptosis in ellagic acid treated cancer cells. Cancer Lett. 1999;136:215–221. doi: 10.1016/S0304-3835(98)00323-1. [DOI] [PubMed] [Google Scholar]
- 17.Mandal S, Shivapurkar NM, Galati AJ, Stoner GD. Inhibition of N-nitrosobenzylmethylamine metabolism and DNA binding in cultured rat esophagus by ellagic acid. Carcinogenesis. 1988;9:1313–1316. doi: 10.1093/carcin/9.7.1313. [DOI] [PubMed] [Google Scholar]
- 18.Mandal S, Stoner GD. Inhibition of N-nitrosobenzylmethylamine-induced esophageal tumorigenesis in rats by ellagic acid. Carcinogenesis. 1990;11:55–61. doi: 10.1093/carcin/11.1.55. [DOI] [PubMed] [Google Scholar]
- 19.Teel RW, Babcock MS, Dixit R, Stoner GD. Ellagic acid toxicity and interaction with benzo[a]pyrene and benzo[a]pyrene 7,8-dihydrodiol in human bronchial epithelial cells. Cell Biol Toxicol. 1986;2:53–62. doi: 10.1007/BF00117707. [DOI] [PubMed] [Google Scholar]
- 20.Casewatch, corp-author. https://www.casewatch.net/fdawarning/prod/2008/best_on_earth.shtml. [Dec 2;2018 ];
- 21.Marnett LJ. Cyclooxygenase mechanisms. Curr Opin Chem Biol. 2000;4:545–552. doi: 10.1016/S1367-5931(00)00130-7. [DOI] [PubMed] [Google Scholar]
- 22.Williams CS, Mann M, DuBois RN. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene. 1999;18:7908–7916. doi: 10.1038/sj.onc.1203286. [DOI] [PubMed] [Google Scholar]
- 23.Fitzpatrick FA. Cyclooxygenase enzymes: Regulation and function. Curr Pharm Des. 2004;10:577–588. doi: 10.2174/1381612043453144. [DOI] [PubMed] [Google Scholar]
- 24.Mitchell JA, Kirkby NS. Eicosanoids, prostacyclin and cyclooxygenase in the cardiovascular system. Br J Pharmacol. 2018 Feb 21; doi: 10.1111/bph.14167. (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duggan KC, Walters MJ, Musee J, Harp JM, Kiefer JR, Oates JA, Marnett LJ. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J Biol Chem. 2010;285:34950–34959. doi: 10.1074/jbc.M110.162982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blobaum AL, Marnett LJ. Structural and functional basis of cyclooxygenase inhibition. J Med Chem. 2007;50:1425–1441. doi: 10.1021/jm0613166. [DOI] [PubMed] [Google Scholar]
- 27.Marnett LJ, Rowlinson SW, Goodwin DC, Kalgutkar AS, Lanzo CA. Arachidonic acid oxygenation by COX-1 and COX-2. Mechanisms of catalysis and inhibition. J Biol Chem. 1999;274:22903–22906. doi: 10.1074/jbc.274.33.22903. [DOI] [PubMed] [Google Scholar]
- 28.Kurumbail RG, Kiefer JR, Marnett LJ. Cyclooxygenase enzymes: Catalysis and inhibition. Curr Opin Struct Biol. 2001;11:752–760. doi: 10.1016/S0959-440X(01)00277-9. [DOI] [PubMed] [Google Scholar]
- 29.Bai HW, Zhu BT. Strong activation of cyclooxygenase I and II catalytic activity by dietary bioflavonoids. J Lipid Res. 2008;49:2557–2570. doi: 10.1194/jlr.M800358-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bai HW, Zhu BT. Myricetin and quercetin are naturally occurring co-substrates of cyclooxygenases in vivo. Prostaglandins Leukot Essent Fatty Acids. 2010;82:45–50. doi: 10.1016/j.plefa.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang P, Bai HW, Zhu BT. Structural basis for certain naturally occurring bioflavonoids to function as reducing co-substrates of cyclooxygenase I and II. PloS One 2010, 2010;5:e12316. doi: 10.1371/journal.pone.0012316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta K, Selinsky BS, Kaub CJ, Katz AK, Loll PJ. The 2.0 Å resolution crystal structure of prostaglandin H2 synthase-1: Structural insights into an unusual peroxidase. J Mol Biol. 2004;335:503–518. doi: 10.1016/j.jmb.2003.10.073. [DOI] [PubMed] [Google Scholar]
- 33.Duggan KC, Walters MJ, Musee J, Harp JM, Kiefer JR, Oates JA, Marnett LM. Molecular basis for cyclooxygenase inhibition by the non-steroidal anti-inflammatory drug naproxen. J Biol Chem. 2010;285:34950–34959. doi: 10.1074/jbc.M110.162982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Im W, Lee MS, Brooks CL., III Generalized born model with a simple smoothing function. J Comput Chem. 2003;24:1691–1702. doi: 10.1002/jcc.10321. [DOI] [PubMed] [Google Scholar]
- 35.Uciechowska U, Schemies J, Scharfe M, Lawson M, Wichapong K, Jung M, Sippl W. Binding free energy calculations and biological testing of novel thiobarbiturates as inhibitors of the human NAD+ dependent histone deacetylase Sirt2. Med Chem Comm. 2012;3:167–173. doi: 10.1039/C1MD00214G. [DOI] [Google Scholar]
- 36.Pouplana R, Lozano JJ, Ruiz J. Molecular modelling of the differential interaction between several non-steroidal anti-inflammatory drugs and human prostaglandin endoperoxide H synthase-2 (h-PGHS-2) J Mol Graph Model. 2002;20:329–343. doi: 10.1016/S1093-3263(01)00133-4. [DOI] [PubMed] [Google Scholar]
- 37.Zhu BT, Bai HW, Rao S, Sui HC. Galangin inhibits cyclooxygenase by blocking the function of the reducing cosubstrate at the peroxidase site. FASEB J. 2018 doi: 10.1016/j.ejphar.2021.174036. submitted. [DOI] [PubMed] [Google Scholar]
- 38.Sui HC, Zhu BT. Catalytic mechanism of the peroxidase activity of human cyclooxygenase and the role of phenol as a reducing co-substrate. Sci Rep. 2018 doi: 10.1038/s41598-020-71237-x. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anslyn EV, Dougherty DA. University Science; Sausalito, CA: 2004. Modern Physical Organic Chemistry. [Google Scholar]
- 40.Chen H. The production and uses of litchis in China. http://ir4.rutgers.edu/GMUS/presentation%20pdf/day1Chen.pdf. [Apr;2018 ];
- 41.Menzel CM, Waite GK, editors. Botany, Production and Uses. CABI Publishing; Wallingford, UK: 2005. Litchi and Longan. [Google Scholar]
- 42.Spencer PS, Palmer VS. The enigma of litchi toxicity: An emerging health concern in southern Asia. Lancet Glob Health. 2017;5:e383–e384. doi: 10.1016/S2214-109X(17)30046-3. [DOI] [PubMed] [Google Scholar]
- 43.Paireau J, Tuan NH, Lefrançois R, Buckwalter MR, Nghia ND, Hien NT, Lortholary O, Poirée S, Manuguerra JC, Gessain A, et al. Litchi-associated acute encephalitis in children, Northern Vietnam, 2004–2009. Emerg Infect Dis. 2012;18:1817–1824. doi: 10.3201/eid1811.111761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shrivastava A, Kumar A, Thomas JD, Laserson KF, Bhushan G, Carter MD, Chhabra M, Mittal V, Khare S, Sejvar JJ, et al. Association of acute toxic encephalopathy with litchi consumption in an outbreak in Muzaffarpur, India, 2014: A case-control study. Lancet Glob Health. 2017;5:e458–e466. doi: 10.1016/S2214-109X(17)30035-9. [DOI] [PubMed] [Google Scholar]
- 45.Islam MS, Sharif AR, Sazzad HMS, Khan AKMD, Hasan M, Akter S, Rahman M, Luby SP, Heffelfinger JD, Gurley ES. Outbreak of sudden death with acute encephalitis syndrome among children associated with exposure to lychee orchards in Northern Bangladesh, 2012. Am J Trop Med Hyg. 2017;97:949–957. doi: 10.4269/ajtmh.16-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krumholz HM, Ross JS, Presler AH, Egilman DS. What have we learnt from Vioxx? BMJ. 2007;334:120–123. doi: 10.1136/bmj.39024.487720.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGettigan P, Henry D. Cardiovascular risk and inhibition of cyclooxygenase: A systematic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase 2. JAMA. 2006;296:1633–1644. doi: 10.1001/jama.296.13.jrv60011. [DOI] [PubMed] [Google Scholar]
- 48.White WB. Cardiovascular risk, hypertension, and NSAIDs. Curr Rheumatol Rep. 2007;9:36–43. doi: 10.1007/s11926-007-0020-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.