

Abstract
Mechanisms underlying alcohol‐induced liver injury and its progression still remain incompletely understood. Animal models can only address some aspects of the pathophysiology that requires studies directly in humans, which are scarce. We assessed liver inflammatory and immune responses at early stages of alcoholic liver disease in a unique cohort of alcohol‐dependent patients undergoing a highly standardized alcohol withdrawal program. In active drinkers, quantitative real‐time polymerase chain reaction revealed alcohol‐induced activation of tumor necrosis factor alpha, interleukin (IL)‐1β, and nuclear factor kappa B in liver tissue already at early disease stages. Double immunofluorescence staining indicated that this proinflammatory response was restricted to activated, CD68‐positive macrophages. In parallel, down‐regulation of IL‐6, inhibition of the signal transducer and activator of transcription 3 (Stat3) pathway, as well as blunted cyclin D expression in hepatocytes, reduced proliferation and favored hepatocyte apoptosis. In addition, immunofluorescence and quantitative real‐time polymerase chain reaction of liver tissue showed that alcohol also activated the toll‐like receptor (TLR) 7–interferon (IFN) axis in hepatocytes, which was confirmed in alcohol‐stimulated primary human hepatocytes and precision‐cut liver slices in vitro. Activation of the TLR7–IFN axis strongly correlated with liver fibrosis markers and disease progression. Two weeks of abstinence attenuated the inflammatory response but did not allow recovery of the defective Stat3 pathway or effect on fibrosis‐associated factors. Conclusion: In humans, inflammation, activation of the TLR7–IFN axis, and inhibition of Stat3‐dependent repair mechanisms in early alcoholic liver disease pave the way for fibrosis development and ultimately disease progression.
Abbreviations
- α‐SMA
α‐smooth muscle actin
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- ASH
alcoholic steatohepatitis
- AST
aspartate aminotransferase
- CK
cytokeratin
- ELISA
enzyme‐linked immunosorbent assay
- HSC
hepatic stellate cell
- IFN
interferon
- IL
interleukin
- IRF3
interferon regulatory factor 3
- LBP
lipopolysaccharide‐binding protein
- LPS
lipopolysaccharide
- MCP1
monocyte chemoattractant protein 1
- mRNA
messenger RNA
- NF‐κB
nuclear factor kappa B
- PCLS
precision‐cut liver slices
- PCR
polymerase chain reaction
- PHH
primary human hepatocyte
- Stat3
signal transducer and activator of transcription 3
- TLR
toll‐like receptor
- TNF
tumor necrosis factor
Alcohol is one of the leading causes of chronic liver disease and liver‐related deaths worldwide.1 The natural history of alcoholic liver disease (ALD) cannot be separated from the natural history of alcoholism and spans a wide spectrum of disease manifestations. Up to 90% of heavy drinkers develop steatosis, but only up to 40% of those progress to alcoholic steatohepatitis (ASH), and 10%‐20% eventually develop cirrhosis.2 To date, the mechanisms that underlie alcohol‐induced liver injury and progressive ALD still remain incompletely understood. Much of our current knowledge has been generated in animal models, which have intrinsic limitations that narrow extrapolation of data to humans. In contrast to humans, animals do not develop a true addiction,3 implying that alcohol administration to rodents requires artifices (e.g., specific diets, gavage, intragastric tubes) that do not mimic human drinking patterns. Ethanol metabolism and elimination is 5 times greater in mice than in humans, which precludes achieving high blood alcohol levels.4 Even more significantly, alcohol‐induced organ damage in animals only partially reproduces the entire spectrum of the target organ damage observed in humans.5, 6 Finally, differences exist between the murine and human immune systems as well as in the capacity of immune cells to respond to damage‐associated molecular patterns, such as toll‐like receptors (TLRs) that are expressed at the cell surface in macrophages (13 in rodents, 10 in humans).7, 8, 9 For these reasons and also because alcohol‐induced inflammation, crucial in the onset and progression of ALD, implicates multiple hepatic cell types as well as innate and adaptive immune responses,10, 11 studies directly conducted in humans are mandatory. However, human studies are scarce, and those available focus on severe alcoholic hepatitis, a condition for which liver biopsies and data are more readily available than for early ALD. Moreover, they require rigorously standardized conditions for patient selection and sampling to avoid as much bias as possible.
The aim of our study was to assess liver inflammatory and immune responses at early stages of ALD in humans and to correlate them with liver damage and repair mechanisms and/or progression of fibrosis. We also assessed whether a short period of abstinence could reverse those early alcohol‐induced changes in the liver.
Materials and Methods
Clinical Setting, Patients, and Tissue and Blood Sampling
Alcohol‐dependent patients (Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition criteria) admitted to the alcohol withdrawal unit follow a highly standardized and controlled 3‐week detoxification and rehabilitation program. They are taken care of by a multidisciplinary team consisting of a gastroenterologist, psychiatrist, psychologist, dietician, social assistant, and dedicated nursing team. At admission, a complete medication and medical history is taken, and a complete physical examination is performed, including collection of basic demographic data, such as age, gender, weight and height, and self‐reported daily alcohol consumption. All patients reported long‐term (>1 year) alcohol consumption (>60 g/day) and were actively drinking until the day of admission. They routinely and prospectively underwent a large panel of investigations (Fig. 1). Transient elastography (FibroScan) was performed on the day of their admission. Fasting blood was drawn on the second day of admission during the first and second week of hospitalization and immediately centrifuged, and aliquots were stored at −80°C until use. A transjugular liver biopsy was routinely proposed to all patients with suspicion of significant liver disease (FibroScan ≥ F2 with increased aspartate aminotransferase [AST] and/or alanine aminotransferase [ALT]).
Figure 1.
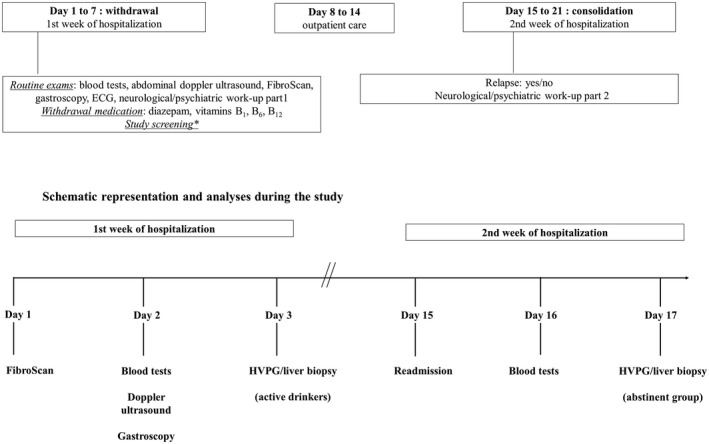
Schematic representation of the standardized working scheme of patients admitted to the alcohol withdrawal unit for a 3‐week detoxification program. Investigations and sampling are performed at predefined days as close as possible to the last drink (maximum 72‐hour delay) in active drinkers and after at least 14 days of abstinence in the abstinence group.
Patients who consented to a liver biopsy were eligible for the study and randomly assigned to undergo liver biopsy on the third day of admission (active drinking group) or during the second week of hospitalization after 14 days of abstinence (short‐term abstinence group) (Fig. 1). Additional inclusion/exclusion criteria can be found in the Supporting Materials and Methods.
Histological normal liver samples and blood samples from healthy volunteers served as controls (details in the Supporting Materials and Methods).
Ethical Aspects
The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the institution's human research and ethical committee. Written informed consent was obtained from all patients.
Reverse‐Transcription and Quantitative Real‐Time Polymerase Chain Reaction
Tissue messenger RNA (mRNA) was assessed by quantitative real‐time polymerase chain reaction (PCR) and quantified as described.12 Primers are depicted in Supporting Table S1.
RNA Sequencing
High‐throughput transcriptome profiling by RNA sequencing was performed, followed by differential expression and functional enrichment analysis. Transcriptomic footprint and canonical pathway analysis was applied to RNA sequencing data. Z score statistic was calculated (details in the Supporting Materials and Methods).
Western Blotting
Western blot analysis was performed on whole cell extracts (Nuclear Extract Kit; Active Motif, La Hulpe, Belgium) according to standard electrophoresis, transfer, and detection techniques as described.12 Membranes were stripped (Fisher Scientific, Erembodegem, Belgium) and re‐probed with several antibodies (Supporting Table S2).
Quantification of Transcription Factor Activation and Caspase 3 Activity
Signal transducer and activator of transcription 3 (Stat3) DNA‐binding and caspase 3 activity were assessed in whole cell extracts using a TransAM detection kit (Active Motif) and Caspase‐Glo‐3/7 assay (Promega, Leiden, the Netherlands), respectively, according to the manufacturer's instructions.
Histology, Immunohistochemistry, and Immunofluorescence
Liver sections were stained with hematoxylin and eosin and Masson's trichrome blue (fibrosis) or incubated with primary and secondary antibodies and quantified by morphometric analysis (Supporting Table S2).
Determination of Blood Cytokine Levels and Inflammatory Markers
Plasma cytokines, lipocalin 2, and serum amyloid A1 were assayed in duplicate with a multiplex immunoassay (Millipore, Molsheim, France) and Luminex xMap technology (Bio‐Rad Laboratories, Hercules, CA) or enzyme‐linked immunosorbent assay (ELISA) (Lipocalin‐2/NGAL Human ELISA Kit EHLCN2 and SAA Human ELISA Kit KHA0011; Invitrogen, Carlsbad, CA) following the manufacturer's instructions.
Cell Culture Experiments
Human liver tissue for cell isolation was obtained from the charitable state‐controlled foundation, Human Tissue and Cell Research, with informed patient consent and approved by the local ethics committee.
Isolation and culture of primary human hepatocytes (PHHs) and hepatic stellate cells (HSCs) were performed as described.13 In addition, we used the LX‐2 human HSC cell line. Cells were incubated with serial alcohol concentrations for up to 24 hours.
Precision‐Cut Liver Slices
Human liver tissue was obtained from patients who underwent partial hepatectomy for colorectal liver metastasis from the London Clinic (London, United Kingdom). The healthy portions of the liver specimen were harvested, and the preparation of precision‐cut liver slices (PCLS) was performed as described.14 Each slice was maintained in culture for 24 hours or 72 hours with or without the addition of 100 mM or 250 mM ethanol (details in the Supporting Materials and Methods).
Statistics
Data are presented as mean ± standard error of the mean unless otherwise indicated. The Kolmogorov‐Smirnov test was used to assess normal distribution of the data. Accordingly, the Student t test was performed for normally distributed data, and the Wilcoxon test for nonnormally distributed data. Pearson's or Spearman's correlation tests were used for correlations between data sets. A P value of less than 0.05 was considered as statistically significant.
Results
Study Population
The study population consists of a typical cohort of 88 alcohol‐dependent, middle‐aged, predominantly male subjects. Demographic, biochemical, and histology data are depicted in Table 1. Most had high transaminases and gamma‐glutamyltransferase levels. Two‐thirds of the patients had early‐stage ALD with a Metavir fibrosis score of ≤F2 and various degrees of steatosis on histology. All patients with advanced fibrosis (≥F3) had a preserved synthetic liver function and showed no clinical signs of liver decompensation.
Table 1.
Baseline Demographic and Biochemical Data of the Study Population
| Demographics | ||||||
| Controls (n = 14) | Alcoholics (n = 88) | |||||
| Gender (female/male) | 5 (35%)/9 (65%) | 26 (29.5%)/62 (70.5%) | ||||
| Mean ± Standard Deviation | ||||||
| Age (years) | 40 ± 11.7 | 49 ± 10.3 | ||||
| Height (cm) | 175 ± 8 | 171 ± 20 | ||||
| Weight (kg) | 70.3 ± 9.1 | 76.7 ± 16.4 | ||||
| BMI | 23 ± 2.9 | 26 ± 5.3 | ||||
| Biochemistry | ||||||
| Mean ± Standard Deviation (normal range) | ||||||
| AST (IU/L) | ND | 116 ± 89 (<50) | ||||
| ALT (IU/L) | ND | 80 ± 60 (<5 0) | ||||
| γ‐GT (IU/L) | ND | 476 ± 486 (<50) | ||||
| ALP (IU/L) | ND | 108 ± 77 (30‐120) | ||||
| Bilirubin (mg/dL) | ND | 1.6 ± 2.5 (0.3‐1.2) | ||||
| Albumin (g/dL) | ND | 4.55 ± 3.55 (3.5‐5.2) | ||||
| INR | ND | 1 ± 0.2 (0.8‐1.3) | ||||
| Alcoholics (n [%]) | ||||||
| Histology | ||||||
| Fibrosis Metavir | ||||||
| F0/F1 | F2 | F3 | F4 | |||
| 42 (48%) | 15 (17%) | 16 (18%) | 15 (17%) | |||
| Sinusoidal Fibrosis | ||||||
| None | Mild | Moderate | Severe | |||
| 30 (34%) | 34 (39%) | 17 (19%) | 7 (8%) | |||
| Steatosis | ||||||
| None | Mild (<33%) | Moderate (33%‐66%) | Severe (>66%) | |||
| 6 (7%) | 24 (27%) | 30 (34%) | 28 (32%) | |||
| Satelitosis | ||||||
| Absent | Present | |||||
| 71 (81%) | 17 (19%) | |||||
| Diabetes (Type 2) | ||||||
| Absent | Present | |||||
| 77 (87.5%) | 11 (12.5%) | |||||
Abbreviations: γ‐GT, gamma‐glutamyltransferase; ALP, alkaline phosphatase; BMI, body mass index; INR, international normalized ratio; ND, not done.
Early Activation of Proinflammatory Cytokines and Nuclear Factor Kappa B in Macrophages/Kupffer Cells
Alcohol elicited a proinflammatory cytokine response in the liver of active drinkers, characterized by increased tumor necrosis factor alpha (TNF‐α) and interleukin (IL)‐1β expression, down‐regulation of IL‐1 receptor antagonist expression, and activation of nuclear factor kappa B (NF‐κB) (Fig. 2A‐C). In addition, plasma levels of lipocalin‐2 and serum amyloid A1, two inflammatory molecules associated with ALD,15, 16 were elevated in active drinkers (Supporting Fig. S1). High plasma levels of macrophage inflammatory protein 1β, increased mRNA expression of the macrophage/Kupffer cell marker CD68 but not CD11b, and activated CD68‐positive macrophages co‐expressing NF‐κB were found (Fig. 2D‐F). These macrophages formed clusters around foci of steatonecrosis. Histological assessment and myeloperoxidase staining of liver sections showed only low numbers of neutrophils in early ASH (Supporting Fig. S1). These data suggest that macrophages are important for driving the proinflammatory response and likely correspond to Kupffer cells (i.e., the liver‐resident macrophages).
Figure 2.
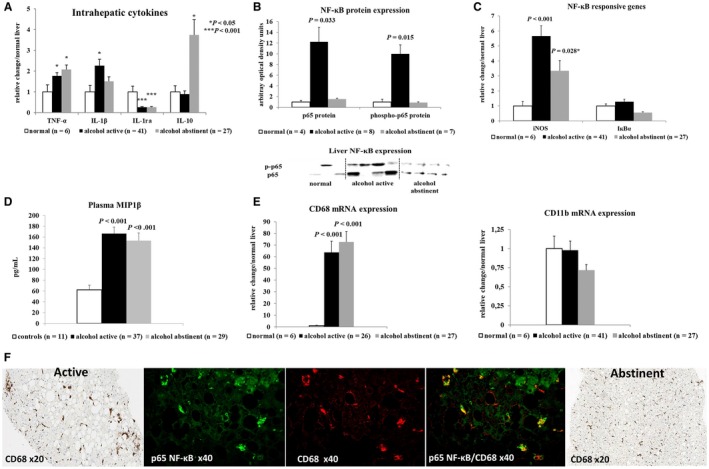
Proinflammatory response in human liver tissue of actively drinking patients with alcoholism or after 14 days of abstinence. (A) Quantitative real‐time PCR of principal pro‐inflammatory and anti‐inflammatory cytokines in liver biopsies. (B) Western blot of total and phosphorylated p65NF‐κB expression in liver tissue. (C) Quantitative real‐time PCR of NF‐κB‐regulated genes to confirm functional activation of NF‐κB. (D) Plasma levels of macrophage inflammatory protein 1β. (E) Increased expression of CD68 but not of CD11b mRNA (representing infiltrating monocytes/macrophages) on quantitative real‐time PCR. (F) Representative immunohistochemistry and double immunofluorescence staining of C68‐positive macrophages and NF‐κB. Activated macrophages in active drinkers form cell clusters around foci of hepatocyte steatonecrosis that co‐localize with NF‐κB. Normalization of the staining pattern of C68‐positive macrophages after 14 days of abstinence. Indicated P values refer to normal liver or controls. Abbreviations: IκBα, nuclear factor kappa B inhibitor alpha; IL‐1ra, interleukin‐1 receptor antagonist; iNOS, inducible nitric oxide synthase; MIP1β, macrophage inflammatory protein 1β.
Short‐term abstinence attenuated the proinflammatory response together with up‐regulation of the anti‐inflammatory cytokine IL‐10 (Fig. 2A‐C). This response was accompanied by a normalization of the CD68 staining pattern, suggesting reduced Kupffer cell activation (Fig. 2F).
Inhibition of Stat3 Signaling in Hepatocytes is Associated With Low Proliferation and High Apoptosis at Early Stages of ALD
Surprisingly, liver mRNA expression of the Stat3‐induced proinflammatory IL‐6 was suppressed by alcohol, as were Stat3, monocyte chemoattractant protein 1 (MCP1), and suppressor of cytokine signaling 3 mRNAs, the latter two being Stat3 response genes (Fig. 3A). Despite some persistent Stat 3 phosphorylation on western blotting, Stat3 DNA binding was decreased (Fig. 3B,C), which is in line with the predominant cytoplasmic localization of Stat3 in hepatocytes (Fig 4A; Supporting Fig. S2). RNA sequencing and canonical pathway analysis confirmed down‐regulation of Stat3 footprint and inhibition of the Stat3 pathway in early but not severe ALD (Fig. 3D). Short‐term abstinence did not allow for recovery of defective Stat3 signaling (Fig. 3A‐C). Inhibition of Stat3 and repression of IL‐6 and MCP1 mRNAs in the liver did not prevent plasma increase of IL‐6 and MCP1, suggesting the contribution of cells outside the liver to production and blood release of these cytokines (Fig. 3E).
Figure 3.
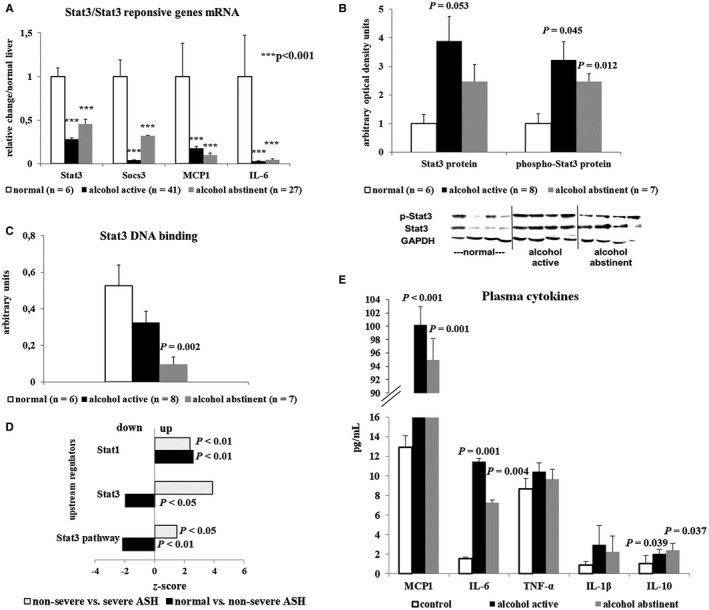
Functional inhibition of the Stat3 pathway in liver tissue at early stages of ALD. (A) Down‐regulation of Stat3 mRNA and of Stat3‐regulated genes in the liver. (B,C) Western blot of total and phosphorylated Stat3 in liver tissue lysates showing increased expression of Stat3 (B) but decreased Stat3 DNA binding in patients with alcoholism (C). (D) RNA sequencing analysis confirming down‐regulation of Stat3 footprint in early ALD in contrast to an increase with disease progression. (E) Increased blood levels of IL‐6 and MCP1 in contrast to the blunted expression of both factors in the liver. Indicated P values refer to normal liver or controls. Abbreviations: GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; Socs, suppressor of cytokine signaling.
Figure 4.
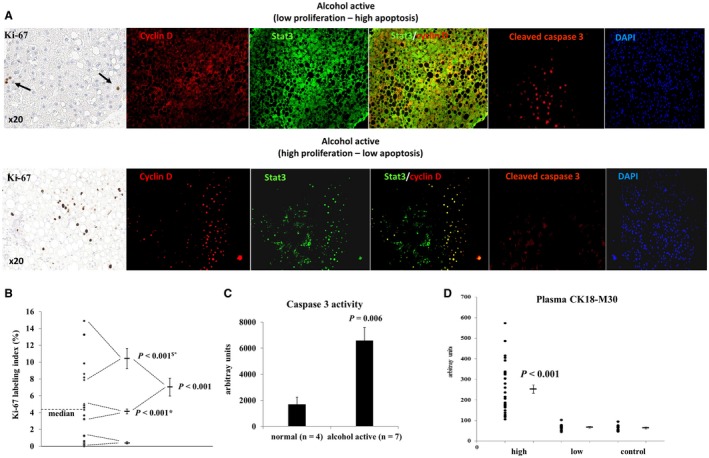
Proliferation–apoptosis balance in early‐stage ALD in actively drinking patients. (A) Representative immunohistochemistry of Ki‐67, immunofluorescence of cyclin D, Stat3, and cleaved caspase 3 showing a low proliferation, high apoptosis (upper panel), and high proliferation, low apoptosis group (lower panel). Stat3 was sequestered in the cytosol of low proliferators, but co‐localized with cyclin D in the nucleus of high proliferators. (B) Quantification of Ki‐67 expression confirming the presence of a high, intermediate, and low proliferation group. P values refer to the low (*) and intermediate ($) proliferation group. (C) Caspase 3 activity in liver lysates of controls and patients with high cleaved caspase 3 nuclear expression confirming increased apoptosis. (D) Increased blood levels of the apoptotic cytokeratin 18 fragments M30 confirming the presence of a high and low apoptosis group. P value compares to the low apoptosis group and to controls. Abbreviation: DAPI, 4′,6‐diamidino‐2‐phénylindole.
The proliferation markers Ki‐67 and cyclin D identified two subgroups in active drinkers: one with low and one with high expression of both markers. More precise quantification of Ki‐67 in hepatocytes distinguished three clusters of patients: (1) very low proliferation (Ki‐67 ≤ 1%); (2) intermediate proliferation (Ki‐67 = 4.3%; range 3.2%‐5%); and (3) high and diffuse hepatocyte proliferation (Ki‐67 ≥ 8%) (Fig. 4A,B; Supporting Fig. S3). No expression of Ki‐67 and very low expression of cyclin D (0.46% ± 0.19%) were found in normal livers. Strikingly, patients with foci of intense hepatocyte proliferation also showed nuclear staining of Stat3 co‐localizing with nuclear cyclin D, a Stat3 responsive gene. When these livers with active proliferation were stained for cleaved caspase 3, no or minimal nuclear staining of caspase 3 was observed (Fig. 4A).
By contrast, Stat3 and cyclin D were sequestered in the cytoplasm in active drinkers with no or very low proliferation. In parallel, those livers exhibited intense clusters of cleaved caspase 3 nuclear staining together with increased liver caspase 3 activity, confirming activation of apoptosis (Fig. 4A,C). Nuclear caspase 3 co‐localized with hepatocyte nuclear factor 4 on double immunofluorescence, suggesting that apoptosis principally occurred in hepatocytes (Supporting Fig. S3).
Cytokeratin (CK) 18 fragments are released following hepatocyte damage. Significantly increased levels of CK18‐M65 and the caspase‐cleaved fragment M30 were detected in the plasma of patients with alcoholism. Interestingly, one‐third (32%) of patients had M30 levels close to control values, confirming, as with cleaved caspase 3 expression, the existence of two subgroups of patients: one with increased hepatocyte apoptosis and one with minimal or no apoptosis (Fig. 4D; Supporting Fig. S4). Both markers correlated well with transaminases (Table 2), adding additional evidence for increased hepatocyte damage and apoptosis in actively drinking patients with alcoholism.
Table 2.
Correlations Between Inflammatory, Fibrosis, and Liver Damage Markers
| Fibrosis Markers | ||||
| IL‐1β | TLR7 | IFN‐β | ||
| Collagen 1 | r = 0.45; P = 0.0008 | r = 0.605; P = 0.0002 | r = 0.546; P < 0.0001 | |
| α‐SMA | r = 0.423; P = 0.0017 | r = 0.511; P = 0.002 | r = 0.500; P = 0.0002 | |
| Timp1 | r = 0.5756; P < 0.0001 | r = 0.5673; P = 0.0011 | r = 0.5979; P < 0.0001 | |
| Serum Liver Damage Markers | ||||
| IL‐1β | TLR7 | IFN‐β | ||
| AST | r = 0.162; NS | r = 0.4917; P = 0.0017 | r = 0.1839; NS | |
| ALT | r = −0.0529; NS | r = 0.2747; NS | r = −0.0207; NS | |
| CK18‐M30 | r = 0.2699; NS | r = 0.5783; P = 0.0012 | r = 0.2008; NS | |
| CK18‐M65 | r = 0.1299; NS | r = 0.4389; P = 0.01806 | r = 0.0786; NS | |
| CK18‐M30 | CK18‐M65 | |||
| AST | r = 0.476; P = 0.0013 | r = 0.529; P = 0.0003 | ||
| ALT | r = 0.508; P = 0.0003 | r = 0.555; P = 0.0001 | ||
Abbreviations: NS, not significant; Timp1, tissue inhibitor of metalloproteinase.
Alcohol Drives Induction of Intracellular TLR3 and TLR7
Given the potential role of TLRs in driving “sterile” inflammation, we assessed the expression profile of various TLRs. Intriguingly, intracellular TLR3, TLR7, and myeloid differentiation 2 mRNA expression were strongly up‐regulated in livers of active drinkers (Fig. 5A). RNA sequencing analysis showed that TLR7 footprint was already increased in early ASH and more so in severe ASH (Fig. 5B).
Figure 5.
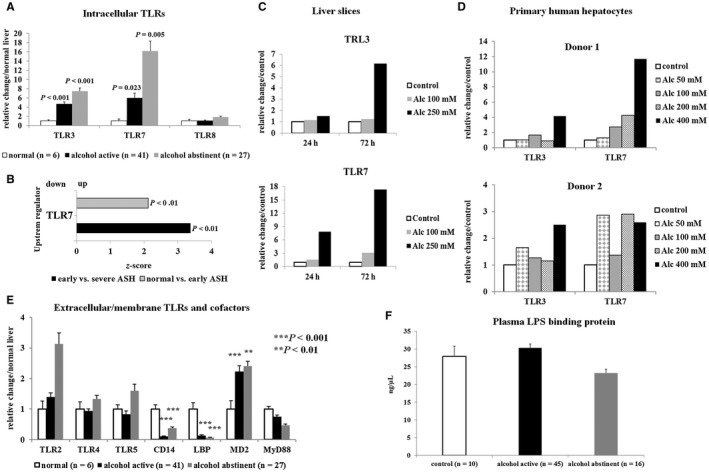
Expression of intracellular and extracellular (membrane‐bound) TLRs and their cofactors. (A) Quantitative real‐time PCR showing up‐regulation of intracellular TLR3 and TLR7 mRNA in liver tissue of patients with alcoholism who did not regress following abstinence. (B) RNA sequencing analysis indicating increased TLR7 footprint already in early AD. (C) Representative experiment showing induction of TLR3 and TLR7 mRNA in PCLS cultured for up to 72 hours with or without (control) addition of alcohol. (D) Stimulation of cultured PHHs with increasing doses of alcohol for 16 hours or 24 hours showing that already low doses of alcohol (50‐100 mM) induce TLR7 mRNA. (E) Quantitative real‐time PCR showing little variation of extracellular TLR mRNAs in liver tissue of patients with alcoholism, whereas TLR4‐associated cofactors (CD14, LBP) were significantly down‐regulated. (F) No increase of plasma LPS‐binding protein was observed in actively drinking patients with alcoholism. Indicated P values refer to normal liver. Abbreviations: Alc, alcohol; MyD88, myeloid differentiation primary response 88.
To determine whether alcohol directly induces TLR3 and TLR7 up‐regulation, we cultured human PCLS with or without ethanol for up to 72 hours. The expression of TLR3 and TLR7 rose following alcohol treatment in a time‐ and dose‐dependent manner (Fig. 5C). Similar results were obtained with alcohol‐stimulated PHHs from two different donors (Fig. 5D), whereas TLR expression did not change in alcohol‐stimulated cultured primary human HSCs or in the LX‐2 human HSC cell line (Supporting Fig. S5).
Expression of membrane‐bound TLRs, including TLR4, did not increase in livers of patients with alcoholism compared with control samples. In addition, CD14, a cofactor required for TLR4 activation, as well as lipopolysaccharide‐binding protein (LBP) mRNAs, were down‐regulated in patients with alcoholism (Fig. 5E). No increase in LBP plasma levels was found in patients with alcoholism compared with healthy controls (Fig. 5F). As a confirmation, alcohol stimulation of PHHs as well as human HSCs did not up‐regulate TLR4 expression (Supporting Fig. S5).
Alcohol Strongly Activates Interferon Signaling in Hepatocytes
Alcohol‐induced interferon (INF)‐β and INF‐γ mRNA expression and RNA sequencing analysis indicated increased INF‐γ footprint already in early ASH. Furthermore, activation of interferon signaling was suggested by canonical pathway analysis of RNA sequencing data and confirmed by quantitative real‐time PCR of the IFN responsive genes 2′‐5′ oligoadenylate synthetase 1 and IFN‐stimulated gene 6‐16 (Fig. 4A,B). In cultured human PCLS, alcohol stimulated the expression of IFN‐β and IFN‐γ mRNAs (Fig. 4C).
Alcohol also induced phosphorylation of the interferon upstream transcription factor IRF3 (Fig. 4E). IRF3 nuclear staining was found principally in hepatocytes, but cytoplasmic expression was also seen in bile ducts, as confirmed by co‐localization with CK7 (Fig. 4F). No co‐localization between IRF3 and CD68 was observed, thus excluding activation of the IFN pathway in Kupffer cells (Fig. 4F). In addition, after alcohol stimulation, a dose‐dependent increase of IFN‐γ and IFN‐β mRNA expression was only observed in PHHs (Fig. 4D), but not in primary human HSCs (Supporting Fig. S5). These observations confirm alcohol‐induced IFN pathway activation primarily in hepatocytes.
Interestingly, IFN‐β and IFN‐γ significantly correlated with TLR7 expression in liver biopsies and in alcohol‐stimulated PHHs, indicating a possible link between both pathways (Supporting Fig. S6).
IFN‐γ was also detected in two‐thirds of the plasma samples and tended to be higher in patients with alcoholism compared with controls (Supporting Fig. S6).
IL‐1β, IFN‐β, and TLR7 Are Associated With More Severe Fibrosis and Disease Progression
Patients with advanced fibrosis (F3/F4) expressed significantly higher levels of the fibrosis markers collagen 1, α‐smooth muscle actin (α‐SMA), and tissue inhibitor of metalloproteinase 1 compared with the minimal fibrosis group (F0‐F2) (Fig. 7A). No significant difference was found between the minimal fibrosis group and control livers (not shown). Collagen 1 also correlated well with quantification of fibrosis on histology assessed by Masson's trichrome staining (r = 0.5098; P = 0.0015). We next addressed which of those investigated inflammatory factors with increased expression in actively drinking patients could contribute to fibrosis progression. Intriguingly, only IL‐1β, IFN‐β, and TLR7 showed a significantly higher expression in livers with advanced fibrosis compared with the minimal fibrosis group (Fig. 7B). All three factors strongly correlated with the liver tissue fibrosis markers (Table 2). RNA sequencing showed not only that TLR7 and IFN footprints further increased with disease severity (Figs. 5B and 6B), but also were significantly (P < 0.01) associated with more severe disease stages (Fig. 7C). Importantly, those factors did not regress following short‐term abstinence (Figs. 5A and 6A). These observations suggest that they might be important players in promoting alcohol‐induced disease progression. Furthermore, TLR7 correlated with AST and serum CK18‐M30 and M65 levels, markers of liver damage and/or apoptosis (Table 2), also linking it to liver damage. Surprisingly, RNA sequencing analysis showed that Stat3 footprints as well as the Stat3 signaling pathway were activated during progression of the disease, which is in sharp contrast to the inhibition of these factors in early ASH (Fig. 7C).
Figure 7.
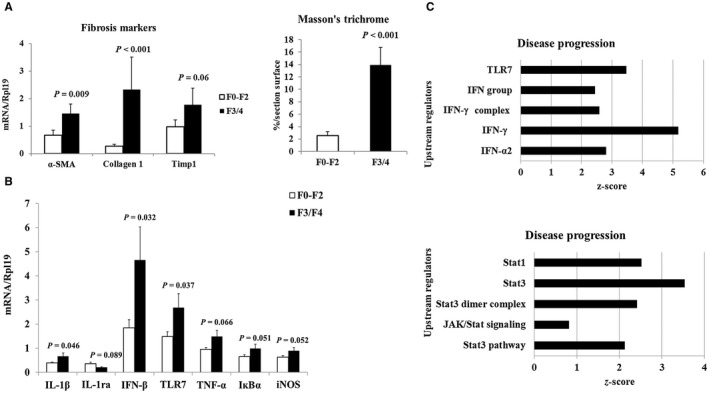
Factors associated with liver fibrosis and disease progression. (A) Quantitative real‐time PCR showing up‐regulation of liver mRNA expression of markers α‐SMA, collagen 1, and Timp1 and increased collagen deposition assessed by Masson's trichrome staining in the severe fibrosis group (F3/F4). (B) Liver mRNA expression of factors associated with more severe fibrosis. In particular, TLR7 and IFN‐β strongly increased in the advanced fibrosis group. (C) RNA sequencing showing increased footprints of TLR7, IFNs, Stat1, and Stat3 as well as activation of the Stat3 signaling pathway with disease progression. P values compare the F3/F4 group to the F0‐F2 group. Abbreviations: IκBα, nuclear factor kappa B inhibitor alpha; IL‐1ra, interleukin‐1 receptor antagonist; iNOS, inducible nitric oxide synthases factor; JAK, Janus kinase; Timp1, tissue inhibitor of metalloproteinase 1.
Figure 6.
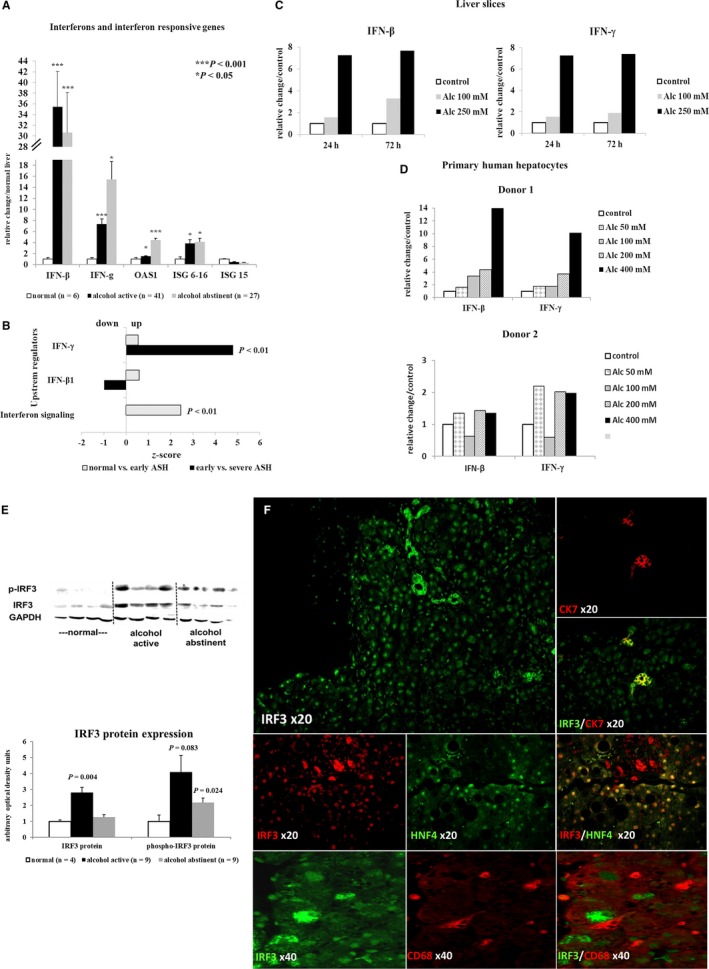
Activation of the IFN pathway in hepatocytes. (A) Quantitative real‐time PCR of INF‐β and IFN‐γ and IFN‐responsive gene mRNAs showing strong activation of IFN signaling in liver tissue of active drinkers that do not regress after 14 days of abstinence. (B) RNA sequencing analysis showing increased IFN‐γ footprint and IFN signaling early in ALD. (C) Representative experiment showing induction of IFN‐β and IFN‐γ mRNA in PCLS cultured for up to 72 hours with or without (control) addition of alcohol. (D) Stimulation of cultured primary human hepatocytes with increasing doses of alcohol for 16 hours or 24 hours, showing that already low doses of alcohol (50‐100 mM) induce IFN‐β and IFN‐γ mRNA expression. (E) Western blot of total and phosphorylated upstream IRF3, demonstrating increased levels in active drinkers. (F) Representative staining of IRF3 expression in livers of active drinkers. Immunofluorescence staining shows IRF3 nuclear staining in hepatocytes (HNF4+) as well as cytoplasmic staining in CK7‐positive bile ducts, but no co‐localization of IRF3 in CD68‐positive Kupffer cells. Indicated P values refer to normal liver. Abbreviations: Alc, alcohol; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HNF4, hepatocyte nuclear factor 4; ISG, IFN‐stimulated gene; OAS1, 2′‐5′ oligoadenylate synthetase 1.
Discussion
Our work addresses alcohol‐induced changes that occur at early stages of ALD in a unique human cohort. We show that a proinflammatory response, including activation of TNF‐α, IL‐1β, and NF‐κB, driven by liver resident macrophages, already occurred at very early stages of the disease. The early proinflammatory response was alleviated by short‐term abstinence. By contrast, the IL‐6/Stat3 axis was blunted and Stat3 sequestered in the cytoplasm of hepatocytes in livers with increased apoptosis. On the other hand, livers that were able to overcome alcohol‐induced Stat3 blockage, as indicated by preserved nuclear translocation of Stat3, retained some proliferative capacity. In contrast to animal data, intracellular TLRs, in particular TLR7, increased at early stages in humans, whereas TLR4 did not. TLR7 up‐regulation occurred concomitantly with activation of interferon signaling predominantly in hepatocytes. Recent evidence links TLR7 to alcohol‐related neuroinflammation,17, 18 whereas its role in alcohol‐induced liver damage remains unclear. TLR7 recognizes synthetic imidazoquinoline‐like molecules, guanosine analogs, and single‐stranded RNA, leading to myeloid differentiation primary response 88/IRF‐dependent induction of type I interferons.19
Two animal studies in cholestatic and nonalcoholic fatty liver disease seem in favor of a protective effect of TLR7 against disease progression and fibrosis.20, 21, 22 Expression of TLRs, including TLR7, was also elevated in a chronic ethanol‐feeding model. However, specific TLR7 stimulation did not exacerbate liver inflammation and damage.23 Animal studies concerning the IRF3 type I interferon axis yielded conflicting results. One study suggested a protective effect of IRF3 and type I interferon activation in parenchymal cells in ALD.24 By contrast, two studies linked IRF3 activation with apoptotic signaling in hepatocytes and with transactivation of proinflammatory TNF‐α in macrophages following chronic alcohol exposure.25, 26
Our results support that the TLR7 type I interferon axis has a deleterious effect in humans for several reasons. As an extension to our previous report of increased TLR7 levels in patients transplanted for end‐stage ALD,12 the current data confirm that both factors are already up‐regulated in early stages of the disease. TLR7 correlated well with AST and serum CK18‐M30 and M65 levels sustaining a link between TLR7 and liver damage. A good correlation between TLR7 and activation of the IFN cascade was found in hepatocytes in vivo. Alcohol stimulation of liver slices ex vivo as well as of cultured PHHs confirmed concomitant up‐regulation of both TLR7 and IFNs, indicating a connection between the two pathways. In addition, TLR7 and type I IFNs were the only factors that correlated with liver fibrosis markers and more advanced stages of ALD, pointing to a role in disease progression. In contrast to animal data, we did not find a predominant implication of the TLR4 system in the pathological modifications of early ALD in humans.27, 28, 29 This finding further emphasizes the prominence of the pathophysiological role for TLR7 in human ALD. It is likely that sensitization to TLR ligands is regulated by multiple mechanisms, including activation by endogenous danger signals from damaged cells in ALD.29 The latter may trigger immune reactions in the liver that are different in humans and rodents in accordance with their different immune and TLR systems.9, 10, 30
Endogenous danger signals released from damaged cells can also activate the inflammatory cascade. Although a proinflammatory response occurred in liver‐resident macrophages, the IL‐6/Stat3 pathway was surprisingly blunted in parenchymal cells. We and others have already reported decreased Stat3 activation in alcoholic cirrhosis and end‐stage ALD.31, 32 Here we show that this mechanism already operates at very early stages of ALD. Hepatocytes with deficient Stat3 activation were unable to mount a proliferative response and underwent apoptosis. On the contrary, those with preserved Stat3 nuclear translocation maintained their cell proliferation. Accumulating evidence suggests that the IL‐6/Stat3 axis plays an important role in repairing ethanol‐induced hepatocellular damage through induction of a variety of hepatoprotective genes in hepatocytes.33, 34, 35 Also, the absence of IL‐6 in mice chronically exposed to alcohol promotes liver damage.36, 37, 38 Stat3 likely acts differently depending on the cell type and the environment. Its absence in nonparenchymal cells allows unrestricted inflammation to develop, whereas the suppression of Stat3 in hepatocytes favors liver damage.39, 40 The absence of Stat3 in nonparenchymal cells and the deficient IL‐6/Stat3 axis in hepatocytes in our study were associated with impaired liver repair and up‐regulation of Kupffer cell–derived proinflammatory cytokines (TNF‐α, IL‐1β). These findings are in line with the view that activation of the IL‐6/Stat3 pathway exerts a protective effect at the early stage of ALD in humans. In more advanced fibrotic ALD, however, Stat3 is activated, which is supported by our data. In vitro evidence showed that Stat3 activation in HSCs could be deleterious and favor fibrosis progression.41, 42 Because our study focused on early ALD, we did not investigate this possibility any further.
IL‐10, an important anti‐inflammatory cytokine, acts as a brake to control inflammation by specifically targeting activated macrophages or monocytes.34, 35 Human monocytes activated by lipopolysaccharide (LPS) produce a high level of IL‐10, which in turn tempers the production of proinflammatory cytokines, such as TNF‐α and IL‐1β.43 The absence of IL‐10 in mice leads to more severe liver damage following LPS treatment or chronic ethanol feeding.44, 45 Up‐regulation of IL‐10 did not occur in human livers of active drinkers. As a consequence, the brake that is able to restrain production of proinflammatory TNF‐α and IL‐1β is not fully activated. Thus, an inadequate IL‐10 response combined with deficient liver repair mechanism driven by alcohol‐induced Stat3 inhibition could pave the way for progression of liver damage.
In absence of any effective pharmacological therapy, long‐term abstinence constitutes the cornerstone of ALD treatment. Unfortunately, frequent relapses after short periods of abstinence are common in patients with alcoholism. We therefore examined the effect of short‐term abstinence on the evolution of the putative proinflammatory, profibrotic, and pro‐liver damage factors. Importantly, the deficient IL‐6/Stat3 axis in hepatocytes did not recover following short‐term abstinence. Abstinence was also insufficient to significantly affect the TLR7/IFN axis associated with fibrosis progression.
An intrinsic limitation of this study is that data are in part descriptive and/or based on correlations, which do not formally prove a cause‐and‐effect relationship. However, given the difficulties of accessing tissue from early‐stage disease, we believe this investigation adds significant insights into the early pathogenesis of ALD. Certainly, the main strength of the study is the unique patient cohort of a high number of heavily, actively drinking subjects undergoing elective alcohol withdrawal in whom liver biopsy had been performed in a strict, standardized clinical program.
Our observations affect clinical practice because they suggest that many unfavorable factors are already present at early stages in human ALD. Short‐term abstinence does not lead to full recovery of impaired liver repair mechanisms and factors potentially associated with fibrosis progression. No biomarker allows distinguishing vulnerable patients from those with a more benign disease course; therefore, clinicians need to do their utmost to promote long‐term abstinence in patients with alcohol abuse and signs of liver disease.
Supporting information
Supported by Fond National de Recherche Scientifique Belgium (J.0146.17 and T.0217.18); and National Institute on Alcohol Abuse and Alcoholism (R01 AA024726‐01).
Potential conflict of interest: Dr. Bataller is on the speakers’ bureau for Echosens. Dr. Lanthier advises Gilead and Promethera; he received grants from MSD and Abbvie and is on the speakers’ bureau for Fresenius Kabi. Dr. Leciercq consults for Biocellvia and received grants from Genfit.
References
- 1. World Health Organization . Global status report on alcohol and health. Geneva: World Health Organization; 2014. [Google Scholar]
- 2. Mathurin P, Bataller R. Trends in the management and burden of alcoholic liver disease. J Hepatol 2015;62:S38‐S46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tabakoff B, Hoffman PL. Animal models in alcohol research. Alcohol Res Health 2000;24:77‐84. [PMC free article] [PubMed] [Google Scholar]
- 4. Cederbaum AI. Alcohol metabolism. Clin Liver Dis 2012;16:667‐685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ghosh Dastidar S, Warner JB, Warner DR, McClain CJ, Kirpich IA. Rodent models of alcoholic liver disease: role of binge ethanol administration. Biomolecules 2018;8:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gao B, Xu MJ, Bertola A, Wang H, Zhou Z, Liangpunsakul S. Animal models of alcoholic liver disease: pathogenesis and clinical relevance. Gene Expr 2017;17:173‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mestas J, Hughes CCW. Of mice and not men: differences between mouse and human immunology. J Immunol 2004;172:2731‐2738. [DOI] [PubMed] [Google Scholar]
- 8. Rehli M. Of mice and men: species variations of toll‐like receptor expression. Trends Immunol 2002;23:375‐378. [DOI] [PubMed] [Google Scholar]
- 9. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 2013;110:3507‐3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mandrekar P, Ambade A. Immunity and inflammatory signaling in alcoholic liver disease. Hepatol Int 2014;8(Suppl. 2):439‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szabo G, Petrasek J, Bala S. Innate immunity and alcoholic liver disease. Dig Dis 2012;30(Suppl. 1):55‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stärkel P, De Saeger C, Strain AJ, Leclercq I, Horsmans Y. NFkappaB, cytokines, TLR 3 and 7 expression in human end‐stage HCV and alcoholic liver disease. Eur J Clin Invest 2010;40:575‐584. [DOI] [PubMed] [Google Scholar]
- 13. Lee SM, Schelcher C, Demmel M, Hauner M, Thasler WE. Isolation of human hepatocytes by a two‐step collagenase perfusion procedure. J Vis Exp 2013;79:e50615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. de Graaf IA, Olinga P, de Jager MH, Merema MT, De KR, van de Kerkhof EG, et al. Preparation and incubation of precision‐cut liver and intestinal slices for application in drug metabolism and toxicity studies. Nat Protoc 2010;5:1540‐1551. [DOI] [PubMed] [Google Scholar]
- 15. Wieser V, Tymoszuk P, Adoph TE, Grander C, Grabherr F, Enrich B, et al. Lipocalin 2 drives neutrophilic inflammation in alcoholic liver disease. J Hepatol 2016;64:872‐880. [DOI] [PubMed] [Google Scholar]
- 16. Jiang Z, Zhou J, Zhou D, Zhu Z, Sun L, Nanji AA. The adiponectin‐SIRT1‐AMPK pathway in alcoholic fatty liver disease in the rat. Alcohol Clin Exp Res 2015;39:424‐433. [DOI] [PubMed] [Google Scholar]
- 17. Coleman LG Jr, Zou J, Crews FT. Microglial‐derived miRNA let‐7 and HMGB1 contribute to ethanol‐induced neurotoxicity via TLR7. J Neuroinflammation 2017;14:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Roberto M, Patel RR, Bajo M. Ethanol and cytokines in the central nervous system In: Grant K, Lovinger D, eds. The Neuropharmacology of Alcohol. Handbook of Experimental Pharmacology. Cham: Springer; 2017;248:397–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kawai T, Akira S. TLR signaling. Cell Death Differ 2006;13:816‐825. [DOI] [PubMed] [Google Scholar]
- 20. Chou MH, Huang YH, Lin TM, Du YY, Tsai PC, Hsieh CS, et al. Selective activation of toll‐like receptor 7 in activated hepatic stellate cells may modulate their profibrogenic phenotype. Biochem J 2012;447:25‐34. [DOI] [PubMed] [Google Scholar]
- 21. Kim S, Park S, Kim B, Kwon J. Toll‐like receptor 7 affects the pathogenesis of non‐alcoholic fatty liver disease. Sci Rep 2016;6:27849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roh YS, Park S, Kim JW, Lim CW, Seki E, Kim B. Toll‐like receptor 7‐mediated type I interferon signaling prevents cholestasis‐ and hepatotoxin‐induced liver fibrosis. Hepatology 2014;60:237‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gustot T, Lemmers A, Moreno C, Nagy N, Quertinmont E, Nicaise C, et al. Differential liver sensitization to toll‐like receptor pathways in mice with alcoholic fatty liver. Hepatology 2006;43:989‐1000. [DOI] [PubMed] [Google Scholar]
- 24. Petrasek J, Dolganiuc A, Csak T, Nath B, Hritz I, Kodys K, et al. Interferon regulatory factor 3 and type I interferons are protective in alcoholic liver injury in mice by way of crosstalk of parenchymal and myeloid cells. Hepatology 2011;53:649‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Petrasek J, Iracheta‐Vellve A, Csak T, Satishchandran A, Kodys K, Kurt‐Jones EA, et al. STING‐IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc Natl Acad Sci U S A 2013;110:16544‐16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhao XJ, Dong Q, Bindas J, Piganelli JD, Magill A, Reiser J, et al. TRIF and IRF‐3 binding to the TNF promoter results in macrophage TNF dysregulation and steatosis induced by chronic ethanol. J Immunol 2008;181:3049‐3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, et al. The critical role of toll‐like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008;48:1224‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll‐like receptor 4 is involved in the mechanism of early alcohol‐induced liver injury in mice. Hepatology 2001;34:101‐108. [DOI] [PubMed] [Google Scholar]
- 29. Petrasek J, Mandrekar P, Szabo G. Toll‐like receptors in the pathogenesis of alcoholic liver disease. Gastroenterol Res Pract 2010;2010 pii: 710381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Muralidharan S, Lim A, Catalano D, Mandrekar P. Human binge alcohol intake inhibits TLR4‐MyD88 and TLR4‐TRIF responses but not the TLR3‐TRIF pathway: HspA1A and PP1 play selective regulatory roles. J Immunol 2018;200:2291‐2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Horiguchi N, Ishac EJ, Gao B. Liver regeneration is suppressed in alcoholic cirrhosis: correlation with decreased Stat3 activation. Alcohol 2007;41:271‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stärkel P, De Saeger C, Leclercq I, Strain A, Horsmans Y. Deficient Stat3 DNA‐binding is associated with high Pias3 expression and a positive anti‐apoptotic balance in human end‐stage alcoholic and hepatitis C cirrhosis. J Hepatol 2005;43:687‐695. [DOI] [PubMed] [Google Scholar]
- 33. Miller AM, Horiguchi N, Jeong WI, Radaeva S, Gao B. Molecular mechanisms of alcoholic liver disease: innate immunity and cytokines. Alcohol Clin Exp Res 2011;35:787‐793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gao B. Hepatoprotective and anti‐inflammatory cytokines in alcoholic liver disease. J Gastroenterol Hepatol 2012;27(Suppl. 2):89‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kawaratani H, Tsujimoto T, Douhara A, Takaya H, Moriya K, Namisaki T, et al. The effect of inflammatory cytokines in alcoholic liver disease. Mediators Inflamm 2013;2013:495156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hong F, Kim WH, Tian Z, Jaruga B, Ishac E, Shen X, et al. Elevated interleukin‐6 during ethanol consumption acts as a potential endogenous protective cytokine against ethanol‐induced apoptosis in the liver: involvement of induction of Bcl‐2 and Bcl‐xL proteins. Oncogene 2002;21:32‐43. [DOI] [PubMed] [Google Scholar]
- 37. El‐Assal O, Hong F, Kim WH, Radaeva S, Gao B. IL‐6‐deficient mice are susceptible to ethanol‐induced hepatic steatosis: IL‐6 protects against ethanol‐induced oxidative stress and mitochondrial permeability transition in the liver. Cell Mol Immunol 2004;1:205‐211. [PubMed] [Google Scholar]
- 38. Zhang X, Tachibana S, Wang H, Hisada M, Williams GM, Gao B, et al. Interleukin‐6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology 2010;52:2137‐2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Horiguchi N, Wang L, Mukhopadhyay P, Park O, Jeong WI, Lafdil F, et al. Cell type‐dependent pro‐and anti‐inflammatory role of signal transducer and activator of transcription 3 in alcoholic liver injury. Gastroenterology 2008;134:1148‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miller AM, Wang H, Park O, Horiguchi N, Lafdil F, Mukhopadhyay P, et al. Anti‐inflammatory and anti‐apoptotic roles of endothelial cell STAT3 in alcoholic liver injury. Alcohol Clin Exp Res 2010;34:719‐725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kagan P, Sultan M, Tachlytski I, Safran M, Ben‐Ari Z. Both MAPK and STAT3 signal transduction pathways are necessary for IL‐6‐dependent hepatic stellate cells activation. PLoS One 2017;12:e0176173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nunez Lopez O, Bohanon FJ, Wang X, Ye N, Corsello T, Rojas‐Khalil Y, et al. STAT3 inhibition suppresses hepatic stellate cell fibrogenesis: HJC0123, a potential therapeutic agent for liver fibrosis. RSC Adv 2016;6:100652‐100663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. DeWaal Malefyt R, Abrams J, Bennett B, Figdor CG, De Vries JE. Interleukin 10 (IL‐10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL‐10 produced by monocytes. J Exp Med 1991;174:1209‐1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hill DB, D'Souza NB, Lee EY, Burikhanov R, Deaciuc IV, De Villiers WJS. A role for interleukin‐10 in alcohol‐induced liver sensitization to bacterial lipopolysaccharide. Alcoholism 2002;26:74‐82. [PubMed] [Google Scholar]
- 45. Miller AM, Wang H, Bertola A, Park O, Horiguchi N, Ki SH, et al. Inflammation‐associated interleukin‐6/signal transducer and activator of transcription 3 activation ameliorates alcoholic and nonalcoholic fatty liver diseases in interleukin‐10‐deficient mice. Hepatology 2011;54:846‐856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials